

Abstract
High grade gliomas are among the deadliest of all cancers despite standard treatments, and new therapeutic strategies are needed to improve patient outcome. Targeting the altered metabolic state of tumors with traditional chemotherapeutic agents has a history of success, and our increased understanding of cellular metabolism in the past two decades has reinvigorated the concept of novel metabolic therapies in brain tumors. Here we highlight metabolic alterations in advanced gliomas and their translation into clinical trials using both novel agents and already established drugs repurposed for cancer treatment in an effort to improve outcome for these deadly diseases.
High grade gliomas (HGGs) are the predominant advanced malignancies of the central nervous system and among the deadliest of all cancer types. Historically, HGG referred to astrocytomas or oligodendrogliomas that were grade 3 (anaplastic) or grade 4 (glioblastoma) according to conventional histopathology 1,2. As our molecular understanding of these tumors has improved, the term HGG has persisted, but changed with respect to the tumor entities it encompasses 3. For the purposes of this review, we will use HGG to describe IDH mutant astrocytomas (grade 3 and 4), IDH mutant oligodendrogliomas (grade 3) and IDH wild type glioblastomas (grade 4).
HGGs are estimated to have afflicted 25,000 Americans in 2020 with near uniform lethality and an average patient survival time of only six months 4,5. Standard therapies for gliomas include surgical resection, radiation, and alkylating chemotherapy, yet HGGs still have particularly poor prognoses due to near-universal recurrence following treatment 6–9. Despite our growing understanding of tumor biology and a host of new technologies and pharmaceutical approaches, HGGs remain exceedingly difficult to eradicate even with current therapies 10,11.
Large-scale genomic analyses have identified numerous genetic aberrations in HGGs that disrupt normal functioning of growth signaling 12–20, cell cycle 21–24, autophagy 25, and cell death 26,27. These findings have significant potential for molecularly targeted cancer-specific therapeutics, although attempts to target precise molecular alterations in HGGs have not yet yielded clinical benefit. This lack of success is likely to due to the extensive intratumoral heterogeneity characteristic of HGGs 28. Indeed, the only therapies that improve survival in HGGs do not require specific oncogenic mutations for efficacy 8,29–31. Standard therapies (temozolomide, TMZ; and radiation therapy, RT) exert their beneficial effects through the induction of DNA damage, and additional pro-tumorigenic processes are under intense investigation as candidate targets for the treatment of HGG 32,33.
Altered cellular metabolism is a hallmark of cancer and a promising therapeutic vulnerability in the clinic 34. Since distinct genetic alterations can lead to common metabolic adaptations within a tumor 35, targeting metabolism in genomically heterogeneous tumors such as HGGs may be more successful than targeting specific genomic alterations. Metabolism influences virtually all cellular phenotypes, and targeting metabolism has a long history of success in a variety of diseases. Notable examples include methotrexate, gemcitabine and asparaginase for the treatment of cancer 36–38, statins for hypercholesterolemia 39, and metformin for diabetes 40. Moreover, antimetabolites against nearly every known pathway are already known and characterized, many of which are currently used for other purposes in patients and hold potential for use in cancer.
A general map of central carbon metabolism and metabolic targets relevant to current and emerging therapies for HGGs is shown in Figure 1. Most common metabolic processes can be traced to the central carbon backbone pathway known as glycolysis. Glycolysis converts glucose to pyruvate through a series of consecutive enzymatic reactions generating ATP and NADH. Glycolytic intermediates can also be used for biosynthetic processes via the pentose phosphate pathway. Pyruvate can enter the tricarboxylic acid (TCA) cycle (alternatively known as the citric acid cycle or Krebs cycle), which is a central metabolic hub in the mitochondria for a variety of pathways including breakdown and synthesis of amino acids and lipids, as well as the production of the redox cofactors NADH and FADH2. These cofactors are often used to fuel electron transport (ETC) and oxidative phosphorylation (OXPHOS) for mitochondrial ATP production. In the context of cancer, cells require both ATP and macromolecule biosynthesis to survive and fuel growth and proliferation. The biologically delicate act of balancing these fundamentally opposing processes makes cancer cells particularly sensitive to metabolic perturbations.
Figure 1. Overview of tumor metabolism and therapeutic strategies to target metabolic activity in HGGs.
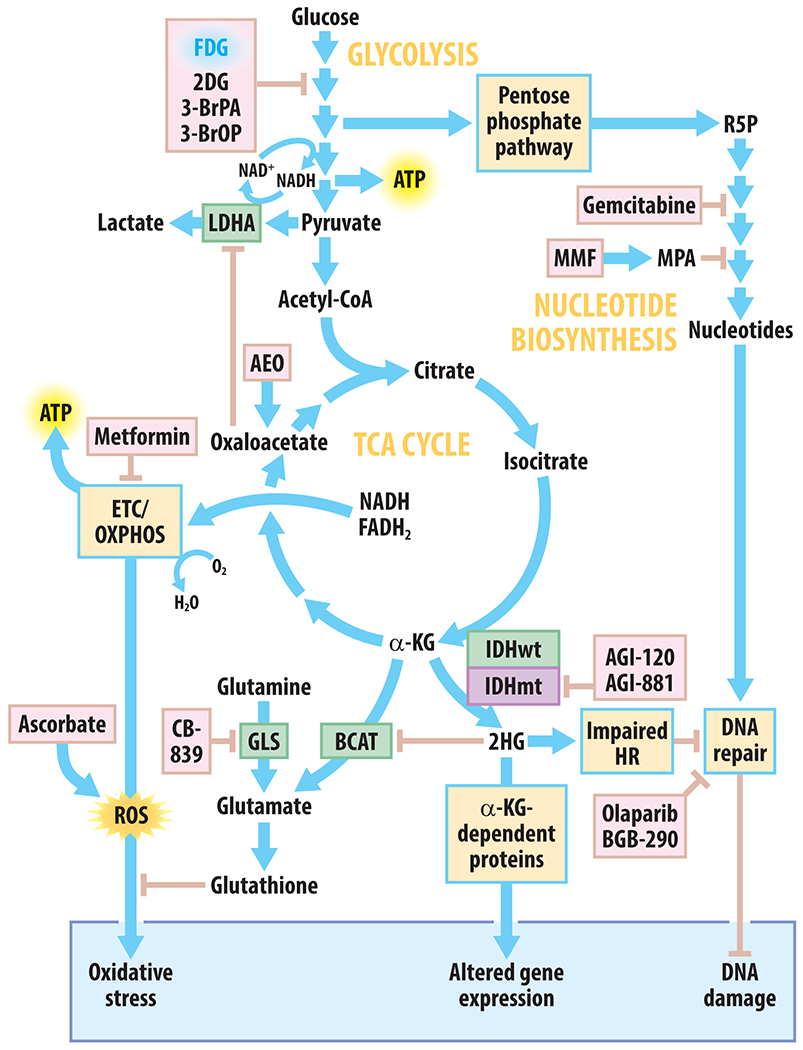
Blue arrows indicate biochemical conversions, metabolic pathway steps, or positive regulation; peach arrows with flat heads indicate inhibition or negative regulation; green boxes indicate metabolic enzymes; violet boxes indicate mutant enzymes; yellow boxes indicate general processes; pink boxes indicate chemical agents that can disrupt metabolism and are used clinically or undergoing preclinical studies. Abbreviations used: 2DG, 2-deoxyglucose; 3-BrOP, 3-bromo-2-oxopropionate-1-propyl ester; 3-BrPA, 3-bromopyruvate; AEO, anhydrous enol-oxaloacetate; ATP, adenosine triphosphate; BCAT, branched-chain amino acid aminotransferase; ETC, electron transport chain; FDG, 2-deoxy-2-[18F]-fluoro-glucose; GLS, glutaminase; HR, homologous recombination; IDHwt/mt, isocitrate dehydrogenase wildtype/mutant; LDHA, lactate dehydrogenase A; MMF, mycophenolate mofetil; MPA, mycophenolic acid; NAD, nicotinamide adenine dinucleotide; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species; α-KG, α-ketoglutarate.
The profoundly altered metabolic state of cancer cells was first posited by Otto Warburg in the 1920s 41, who observed dramatically increased glycolytic activity in cancer cells despite abundant oxygen 42. This aerobic glycolysis is now thought to sustain the biosynthesis required for continued proliferation 43. The intense dependence of cancer cells upon increased metabolic activities for growth and invasion makes targeting metabolism an appealing therapeutic strategy.
Increased sensitivity of cancer cells to metabolic targeting is highlighted by the successes of metabolic agents used as anti-cancer therapeutics as early as the 1940s. Antagonists of folate metabolism (required for cell proliferation and nucleotide synthesis) were among the first antimetabolites used in patients, with methotrexate causing remission in children with leukemia in 1948 44. In the 1950s, the purine analogs 6-mercaptopurine and 6-thioguanine, and the pyrimidine analog 5-fluorouracil, were first used to treat cancer patients 45–48. Targeting these pathways has expanded to include other antimetabolites such as gemcitabine and pemetrexed 49. In addition to their efficacy as single agents, these anti-metabolites can potentiate the efficacy of other treatments in a variety of cancer types 50.
As our understanding of metabolism in normal physiology and disease has expanded over the decades, so too has our treatment arsenal against diseases characterized by aberrant metabolism. In this review, we highlight the metabolically targeted therapeutic strategies that are under clinical investigation for patients with HGG.
Glycolysis as a diagnostic tool and metabolic target
Warburg’s observation of increased glucose uptake in cancer cells is routinely observed clinically when tumors incorporate higher levels of the radioactive glucose analog 2-deoxy-2-[18F]-fluoro-glucose (FDG) than normal tissues in patients. FDG contains an 18F atom attached to the 2-carbon of glucose, whereas normal glucose contains an oxygen atom at the 2-position (2-OH). Hexokinase phosphorylates FDG to generate [18F]FDG-6-phosphate, which is trapped intracellularly but cannot undergo further glycolytic metabolism due to the absence of the hydroxyl group on the 2-position. Therefore, the absence of 2-OH in FDG allows [18F]FDG-6-phosphate to accumulate to high, detectable levels in many highly glycolytic cancerous tissues. FDG-PET imaging has therefore emerged as an important imaging tool for numerous cancers. In HGG, the utility of FDG-PET is somewhat limited by the glucose-avidity of normal cortex, however this modality can be useful for some applications such as distinguishing progressive HGG from treatment-related necrosis 51.
Removing the 2-OH from glucose also has therapeutic potential. 2-Deoxyglucose (2DG) has the 2-OH group replaced with a hydrogen instead of [18F]. Like FDG, 2DG is phosphorylated by hexokinase but cannot be further catabolized through glycolysis. 2DG-6-phosphate exerts product inhibition on hexokinase and thus can inhibit glycolysis. Treatment with 2DG can enhance RT in preclinical models of HGG 52 and has been tested with RT in patients. Initial clinical trials using 2DG with RT in GBM patients suggested that 2DG was safe, reasonably well tolerated and could be efficacious 53–55. These findings prompted a randomized trial of RT alone in comparison to RT with 2DG for patients with GBM that was performed in India. While the results of this trial have never been published, an abstract from 2014 indicated that 2DG treatment did not confer a survival benefit 56. These disappointing results could in part be due to the poor drug-like properties of 2DG, which require millimolar concentrations for efficacy 55. While 2DG analogs could have some therapeutic promise in the future, as of 2021 there are no listed trials on ClincalTrials.gov utilizing 2DG as a therapeutic intervention.
Nitrosoureas are a class of alkylating agents used in GBM patients as an alternative to TMZ 57 but some have also been reported to inhibit glycolysis (Figure 1). The brominated derivative of pyruvate, 3-bromopyruvate (3-BrPA) is an alkylating agent that also inhibits glycolysis and depletes ATP 58. Using a single drug to induce DNA damage and simultaneously inhibit glycolysis is an attractive therapeutic strategy, and while 3-BrPA has been used in patients, it has not been studied in rigorous clinical trials and has serious safety concerns 57. A potential alternative and more stable 3-BrPA derivative known as 3-bromo-2-oxopropionate-1-propyl ester (3-BrOP) has been shown to sensitize TMZ- and carmustine-resistant GBM stem cells (GSCs) to both agents 59. Moreover, 3-BrPA analogs can reverse hypoxia-induced nitrosurea resistance 60 and further suggest potential opportunities to inhibit glycolysis for improved therapeutic responses.
Recent data have shown that oxaloacetate, a naturally occurring metabolite in the TCA cycle, suppresses the Warburg effect in GBM 61. This may be due to the ability of oxaloacetate to inhibit the glycolytic enzyme lactate dehydrogenase (LDHA, Figure 1) 62. Increasing oxaloacetate levels reduces tumor growth and improves survival in GBM animal models 63,64, and a phase 2 clinical trial with a proprietary oxaloacetate pro-drug called Anhydrous Enol-Oxaloacetate (AEO) in combination with standard therapy for GBM patients is now underway (NCT04450160).
TCA Cycle and IDH mutations
The TCA cycle utilizes pyruvate (derived from glycolysis) as an electron source for the mitochondrial respiratory chain and is a central metabolic hub for a variety of synthetic pathways. The isocitrate dehydrogenase (IDH) enzymes, which function in the TCA cycle and other metabolic processes, hold particular relevance to gliomas. In normal tissues, IDH1 catalyzes the NADP-dependent reversible conversion of isocitrate to α-ketoglutarate (α-KG) in the cytosol, while IDH2 catalyzes the same reaction in the mitochondria. IDH3 is a mitochondrial enzyme that is linked to NAD+ instead of NADP+ and thus is an important producer of NADH in the TCA cycle. Subsequently, IDH-produced α-ketoglutarate be used to produce the amino acids glutamate and glutamine (Figure 1). IDH1 and IDH2 can also run in the “reverse” direction to produce isocitrate, which can be used for lipid synthesis.
With the dawn of advanced genomics in the 2000s, exome sequencing studies identified a mutation in IDH1 and IDH2 in many cancers including about 10% of GBM patients 65. The majority of IDH mutations occur at a residue essential for isocitrate binding (R132 in IDH1; R172 and R140 in IDH2) and are most prevalent in grade II-III gliomas and secondary GBMs 65–67. IDH mutant (IDHmt) gliomas contain one wildtype IDH allele, catalyzing the conversion of isocitrate to α-KG, while the mutant allele loses normal catalytic activity and undergoes a gain of function to catalyze the production of 2-hydroxyglutarate (2HG) from α-KG 68. Notably, 2HG is considered an oncometabolite due to its ability to directly regulate tumorigenesis. Due to its structural resemblance to α-KG, 2HG can act as a substrate inhibitor of α-KG-dependent epigenetic regulators, in particular TET enzymes and histone demethylases 69,70. IDHmt-mediated inhibition of these proteins blocks differentiation of non-transformed cells, which is thought to promote gliomagenesis 71.
Inhibitors of mutant IDH have been developed, are FDA-approved for patients with IDHmt leukemias, and are under clinical investigation for patients with IDHmt gliomas. The IDHmt inhibitor AGI-5198 delays growth and promotes differentiation of IDHmt but not wtIDH glioma cells 72, and two additional IDHmt inhibitors, AGI-120 (ivosidenib) and AGI-881 (vorasidenib) have shown acceptable safety and potential efficacy in cancer patients. Both agents inhibit 2HG production in human gliomas 73. Ivosidenib is an inhibitor of the mutant IDH1 isoform (IDH1mt) and approved by the FDA as a first-line treatment for acute myeloid leukemia (AML), an additional tumorigenic context in which IDH is frequently mutated 74–76. Ivosidenib has shown safety and potential efficacy in patients with IDH1mt advanced gliomas 77. Vorasidenib is an inhibitor of both mutant IDH1 and mutant IDH2 that penetrates the brain in multiple species and blocks 2HG production in glioma tissue by >90% 78. A randomized phase 3 trial (NCT04164901) to determine single agent efficacy of vorasidenib vs. placebo in patients with IDH1/2 mutant gliomas is ongoing 79. Some studies suggest that 2HG may also promote DNA repair and RT resistance, especially when the IDH mutation occurs alongside common mutations present in IDHmt astrocytomas 80. These findings suggest that vorasidenib may have utility in combination with genotoxic agents such as RT and temozolomide
Targeting vulnerabilities conferred by IDH mutations
The most efficacious strategy to exploit the IDH mutation may not be through its catalytic inhibition. IDH mutations in glioma are associated with improved survival compared to IDHwt tumors 65, but whether this is due to the mutation itself or the tumor’s natural history is an open question. IDHmt and 2HG appear particularly important for tumor growth early in tumorigenesis 67,81,82. This is matched by clinical data showing that inhibition of mutant IDH may slow the growth of less aggressive non-enhancing IDHmt low grade gliomas but be less effective for higher grade contrast-enhancing IDHmt gliomas 77. These findings suggest that inhibition of mutant IDH may be initially beneficial but become less effective once the tumor is established 83.
IDH mutation sensitizes tumors to PARP inhibitors
Rather than directly targeting the IDH mutation itself, several groups have instead defined and targeted the downstream vulnerabilities conferred by 2HG. A handful of novel IDH-mediated therapeutic vulnerabilities have been recently identified and targeted in clinical trials. In a variety of genetic contexts, the 2HG produced by mutant IDH inhibits DNA repair due to defective homologous recombination (HR) 84,85. When HR is defective, for example when BRCA is inactive, cells become reliant on alternative mechanisms of DNA repair such as non-homologous end joining or single strand base repair. Inhibitors of poly-ADP ribose polymerase (PARP) have increased efficacy in cells lacking HR, including both BRCA and IDH mutant cells 86,87. This concept is now being tested in IDHmt brain tumor patients with the PARP inhibitor BGB-290, which is being given in combination with temozolomide for patients with IDHmt brain tumors (NCT03914742). Preliminary trials treating IDHmt mesenchymal sarcoma patients with monotherapy olaparib, another PARP inhibitor, have suggested therapeutic benefit in individuals with IDHmt chondrosarcoma and pulmonary epithelioid hemangioendothelioma 88. Early results from a separate trial combining olaparib with TMZ, also an inducer of DNA damage, in recurrent GBM indicates that olaparib reliably penetrates many HGGs 89, and this agent could be useful in IDHmt brain tumors as well.
2HG inhibits metabolic enzymes BCAT1 and BCAT2 and sensitizes tumors to glutaminase inhibition
Increased levels of 2HG produced by IDHmt can also directly inhibit metabolic enzymes that require the structurally similar molecule α-KG as a cofactor. This biology raises the possibility of IDHmt-dependent metabolic vulnerabilities. This is particularly true of glutamate production in IDHmt tumors. Glutamate is a precursor for the antioxidant molecule glutathione, which protects cells against oxidative stress (Figure 1). Glutamate can be produced from one of several mechansims. Among these are the deamidation of glutamine via glutaminase (GLS) or branched chain amino acid transamination via branched chain amino acid transaminase 1 (BCAT1) and 2 (BCAT2). BCAT1 and BCAT2 are α-KG-dependent and are competitively inhibited by 2HG 90. Glutaminase inhibition thus selectively depletes glutamate, and consequently the glutamate contaminating tripeptide antioxidant glutathione, levels in IDHmt cells and sensitizes IDHmt gliomas to RT in laboratory models 91. In light of these therapeutically significant findings, a clinical trial combining the glutaminase inhibitor CB-839 with RT in IDHmt anaplastic astrocytoma and IDHmt diffuse astrocytoma patients is underway (NCT03528642).
Modulators of mitochondrial activity and oxidative stress
In healthy tissues, redox cofactors such as NADH generated in the TCA cycle are used to fuel the mitochondrial electron transport chain (ETC). The ETC is a series of protein complexes that builds an electrochemical gradient across the inner mitochondrial membrane. The stored free energy across this gradient is then used to power ATP synthesis during the process of oxidative phosphorylation. Electron leak from the ETC makes mitochondria the main source of cellular reactive oxygen species (ROS, Figure 1), which are potentially toxic at high levels. At low levels, ROS stimulate pro-survival and proliferation signaling pathways, while high levels cause cell death. ROS are normally kept in check by antioxidant molecules such as glutathione and thioredoxin, but this delicate balance is dysregulated in tumors. In the context of cancer, tumor cells often exploit mitochondrial activity for ROS signaling. However, this also makes cancer cells more vulnerable to mitochondrial targeting and inducers of oxidative stress 92.
The biguanide metformin has been used clinically for over 60 years and is the first line treatment for type 2 diabetes. Metformin inhibits glucose production in the liver and improves insulin signaling to increase glucose uptake in skeletal myocytes. Both mechanisms reduce hyperglycemia and associated clinical symptoms. Metformin has been extensively studied and reviewed elsewhere 93,94, but new mechanistic insights and therapeutic uses continue to be discovered. Recent epidemiological data has suggested that it also holds the potential for use in a variety of cancers. Metformin inhibits complex I of the ETC, which depletes ATP and NAD+ and causes activation of AMP-activated protein kinase (AMPK) 93. Metformin therapy is associated with prolonged progression free survival in diabetic GBM patients 95, suggesting its anti-tumorigenic metabolic activity may be useful clinically. Metformin is currently being studied in a 33-patient phase 2 trial in combination with TMZ and RT in GBM patients (NCT02780024). Metformin treatment appears to be safe and feasible and promising survival results, including a 3-year overall survival over 50% have been presented in abstract form 96. Whether these encouraging results are due to systemic changes in glucose and insulin or direct action of metformin on glioma cells is not certain.
Recent studies of ascorbate (vitamin C) in glioma cells have found that pharmacological levels of ascorbate dramatically elevate ROS, inhibit glycolysis, and increase labile iron 97–99. Ascorbate also induces double strand DNA breaks and increases sensitivity of GBM cells to RT due to excessive ROS and oxidative stress 100,101. A phase 1 trial (NCT01752491) with high-dose ascorbic acid infusions in GBM patients receiving standard chemoradiation has shown minimal toxicity and favorable progression-free and overall survival 97,102,103 compared to historical controls 30. A phase 2 trial (NCT02344355) assessing efficacy of ascorbate with chemoradiation in GBM patients is ongoing. The anticancer mechanisms and therapeutic potential of high-dose ascorbate are reviewed in detail in reference 104.
Metabolic Nutritional Therapies for HGGs
The links between metabolic inputs, or nutrition, and wellbeing have been long recognized. However, understanding the complex relationships among the numerous genetic and environmental factors of diet and metabolism and their potential for intervention in diseases such as cancer has been challenging 105. Altering tumor biology using “precision nutrition,” or the restriction of specific nutrients in food 106, may be a feasible dietary intervention strategy when based on metabolic activities of both the tumor and systemic environment within individual patients.
The ketogenic diet (KD) is a potential precision nutrition therapy that has recently received substantial interest in oncology. The KD is based on restricted carbohydrate intake to rewire systemic metabolism through the decreased level of circulating glucose and insulin, and consequently the production of ketones as an alternative fuel source. This may produce an unfavorable metabolic environment for tumors that rely on insulin as a growth factor and glucose as a fuel 107,108. While the preclinical rationale for the KD is strong, especially in combination with other anti-cancer agents 109, results from clinical trials investigating feasibility and efficacy of KD in cancer patients are mixed or limited to case studies and numerous clinical trials are ongoing 107. Initial studies of the ketogenic diet in patients with GBM have shown that the diet is reasonably well-tolerated though logistically difficult to administer 110. Indeed, a recent phase 1 study of the KD (NCT02046187) attempted to determine if KD could enhance standard chemoradiation in newly diagnosed GBM patients; however, this was terminated due to excessive protocol deviations due to the strict nature of dietary requirements. More recently, a modified dietary intervention termed the GLAD diet (Glioma modified Atkins Diet) was developed as a less restrictive diet that was still ketogenic, but also involved periods of intermittent fasting111. HGG patients (both IDHwt and IDHmt astrocytoma) adhered to the GLAD diet reasonably well and most achieved ketosis. However, GLAD was initiated in patients with stable brain tumors after completion of most RT and chemotherapy, which likely improved patient compliance. Whether these dietary interventions will be feasible in patients with newly diagnosed HGG undergoing chemoradiation, and whether they provide therapeutic benefit, remain unanswered questions.
Purine synthesis is a targetable vulnerability that promotes therapy resistance in IDHwt GBM
Nucleotide metabolism has been successfully targeted in numerous cancers and recent data suggest its inhibition could provide therapeutic benefit in HGG. Nucleotides are a class of biomolecules that are the building blocks of nucleic acids (DNA, RNA, ribosomes) but also play a wide variety of cellular functions including roles as signaling molecules and comprising chemically accessible energy (e.g., ATP, GTP). Structurally, nucleotides contain a nitrogenous base attached to a sugar unit (ribose or deoxyribose) that contains a mono-, di-, or triphosphate group. Nucleotide species can be classified as either purines (containing a double ring nitrogenous base structure) or pyrimidines (containing a single ring), which are synthesized through distinct pathways.
Glioma cells synthesize nucleotides using different pathways than non-malignant cortical cells, which provides a potential therapeutic window for targeting these metabolic pathways (Figure 2). In non-malignant cells, nucleotides are typically generated through salvage pathways in which pre-formed nitrogenous bases from the diet or breakdown of nucleic acids are directly conjugated to activated ribose (phosphoribosyl pyrophosphate, which is generated from glucose in the pentose phosphate pathway, Figure 1). By contrast, proliferating cells typically generate nucleotides from scratch using the de novo synthetic pathways. In de novo pyrimidine synthesis, the nitrogenous base is constructed from carbamoyl phosphate and aspartate, and then conjugated to PRPP. In contrast, de novo purine synthesis occurs through the building of the nitrogenous base directly on the PRPP using several different amino acids, one-carbon carbon units, and a substantial quantity of free energy from ATP. Both de novo purine and pyrimidine synthesis are activated by numerous oncogenic abnormalities including AKT/mTOR activation, receptor tyrosine kinase signaling and MYC activation 112–117.
Figure 2. De novo purine synthesis is increased by oncogene activation, promotes pro-tumor cellular activities, and represents a therapeutic liability in HGG.
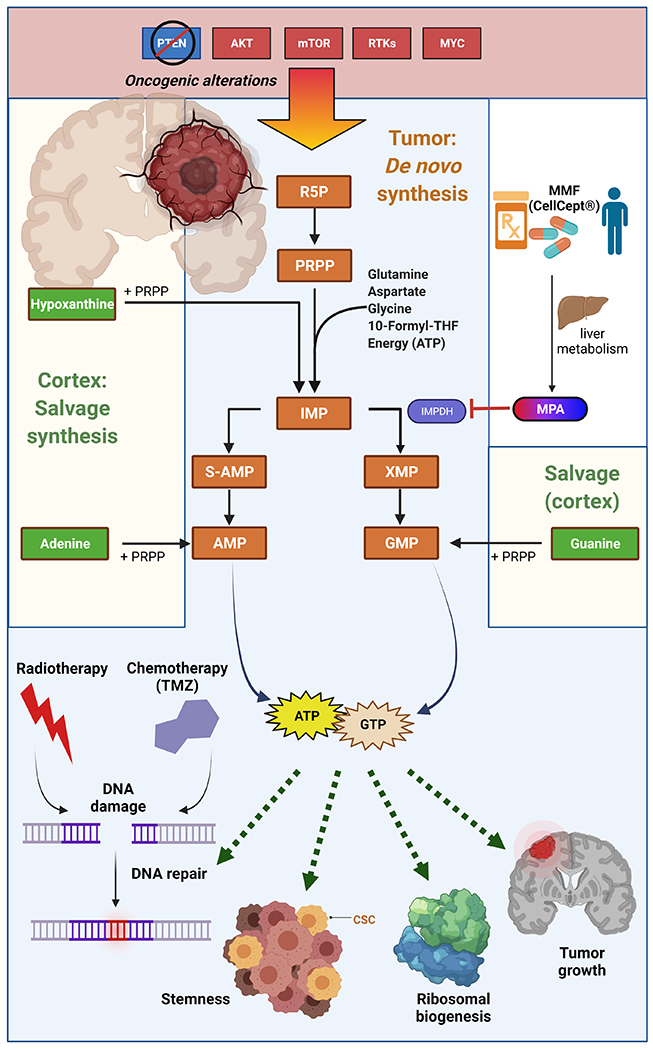
De novo purine synthesis is activated by oncogenic alterations including PTEN deletions and upregulation or mutations in AKT, mTOR, receptor tyrosine kinases (RTKs), and MYC. In the de novo purine synthetic pathway, ribose 5-phosphate (R5P) is activated to phosphoribosyl pyrophosphate (PRPP), allowing the biochemical construction of a purine ring upon the ribose unit to form inosine monophosphate (IMP). IMP can be converted to adenylosuccinate (S-AMP) and then adenosine monophosphate (AMP) and adenosine triphosphate (ATP). IMP can also be converted to xanthosine monophosphate (XMP) via IMP dehydrogenase (IMPDH) and then guanosine monophosphate (GMP) and guanosine triphosphate (GTP). In non-malignant brain tissue, purines are produced through salvage synthesis, in which free nitrogenous bases are conjugated to PRPP. Increased purine levels in HGGs promote stemness, tumor growth, ribosomal biogenesis, and DNA repair and subsequent therapeutic resistance. Purine synthesis can be pharmacologically targeted using mycophenolate mofetil (MMF/CellCept®), which is converted to mycophenolic acid (MPA) in the liver. MPA inhibits IMPDH to suppress purine synthesis and gliomagenesis in preclinical models. Figure created with BioRender.
While nucleotide salvage does occur in HGG 118, glioma cells, especially glioma stem-like cells, appear to primarily rely on de novo pyrimidine and purine synthesis 112,119,120. Both de novo purine and pyrimidine synthesis in HGG regulate tumor stemness 112,119. De novo purine synthesis also mediates other key oncogenic features of HGG including tumor growth and ribosomal biogenesis 121. Purine metabolism also regulates double strand DNA break repair, and thus mediates resistance to both radiation and temozolomide, the primary treatments used for HGG 122,123. Despite these key links between purine metabolism and HGG growth and treatment resistance, anti-folate therapy, which inhibits purine metabolism, has largely been ineffective for the treatment of HGG. These findings may be due to relatively high concentrations of hypoxanthine in the brain, which could allow HGG to refill purine pools by generating inosine monophosphate through salvage pathways when the proximal steps of de novo purine synthesis are inhibited (Figure 2) 124.
Inhibiting purine synthesis downstream of the hypoxanthine salvage step may be more efficacious for the treatment of HGG. Mycophenolate mofetil (MMF) is an FDA-approved agent that is metabolized by the liver into mycophenolic acid (MPA) which directly inhibits the purine synthetic enzyme inosine monophosphate dehydrogenase 125 (Figure 2). MMF is routinely used as an immunosuppressant for organ transplant patients, is well tolerated and appears to cross the blood-brain barrier 126,127. While MMF is an immunosuppressant, which may raise concerns for use in cancer patients, preclinical studies in orthotopic patient-derived GBM models demonstrate that short-term MMF treatment can overcome both RT and temozolomide resistance without compromising animal health 122,123. Given these promising findings, our research group is now performing a phase I clinical trial combining MMF with RT for patients with GBM (NCT04477200).
Other inhibitors of nucleotide metabolism may prove clinically beneficial for HGG as well. Gemcitabine is a cytidine analog used in other cancers that can be incorporated into DNA or impair deoxynucleotide production via inhibition of ribonucleotide reductase. Gemcitabine can cross the blood brain barrier and accumulate into tumors such as GBM at active concentrations 128,129. A clinical trial combining gemcitabine and radiation for patients with HGGs demonstrated acceptable safety and promising clinical outcomes, though further investigation was halted due to the success of the cytotoxic chemotherapy temozolomide 130. Nucleotide biosynthesis is an established therapeutic target in multiple disease states, and we are optimistic that increasing our understanding of altered nucleotide metabolism in GBM will lead to therapeutic advances.
Conclusions and Future of Metabolic Therapy in HGG
Advances in therapy for HGG have stalled due, in part, to the plasticity of glioma cells and the genomic and epigenomic heterogeneity of these tumors. Targeting metabolism, which is the level of biology closest to phenotype, may be a promising strategy to improve HGG outcomes despite this heterogeneity. Over the past decade we have developed a tremendous understanding of how many of the oncogenic molecular drivers of HGG activate common metabolic pathways, which in turn promote key oncogenic phenotypes. New methods of measuring metabolism including in vivo stable isotope tracing in HGG patients 131,132, spatial mass spectrometry 133 and single cell RNA sequencing 134 will enable additional discoveries regarding cell-type specific metabolic alterations and cross-talk between malignant and non-malignant cells in HGG. As we work to translate our understanding of these metabolic alterations into clinical implementation through metabolic inhibitors and dietary modulation, we are optimistic that the proximity of metabolism to phenotype may allow metabolically targeted therapies to move the needle and further improve outcomes for these deadly diseases.
Acknowledgments
We thank Steven Kronenberg for assistance producing illustrations. A.J.S. was supported by the NCI (F32CA260735). D.R.W. was supported by the NCI (K08 CA234416, R37 CA258346), the Damon Runyon Foundation, the Ivy Glioblastoma Foundation and the Taubman Institute. C.A.L. was supported by the NCI (R01CA248160, R01CA244931).
Sources of support:
A.J.S. was supported by the NCI (F32CA260735). D.R.W. was supported by the NCI (K08 CA234416, R37 CA258346), the Damon Runyon Foundation, the Ivy Glioblastoma Foundation and the Taubman Institute. C.A.L. was supported by the NCI (R01CA248160, R01CA244931).
Footnotes
Conflict of Interest
A.J.S. has no conflicts of interest to disclose. D.R.W. has received consulting fees from Agios (now Servier) Pharmaceuticals and is an inventor on a patent related to methods of determining optimal treatments for patients with glioblastoma. C.A.L. has received consulting fees from Astellas Pharmaceuticals and Odyssey Therapeutics and is an inventor on patents pertaining to Kras regulated metabolic pathways, redox control pathways in cancer, and targeting the GOT1-pathway as a therapeutic approach.
Contributor Information
Andrew J. Scott, Department of Radiation Oncology, University of Michigan, Ann Arbor, MI 48109; Rogel Cancer Center, University of Michigan, Ann Arbor, MI 48109.
Costas A. Lyssiotis, Department of Molecular & Integrative Physiology, University of Michigan, Ann Arbor, MI 48109; Department of Internal Medicine, Division of Gastroenterology and Hepatology, University of Michigan, Ann Arbor, MI 48109; Rogel Cancer Center, University of Michigan, Ann Arbor, MI 48109.
Daniel R. Wahl, Department of Radiation Oncology, University of Michigan, Ann Arbor, MI 48109; Rogel Cancer Center, University of Michigan, Ann Arbor, MI 48109; University of Michigan, Ann Arbor, MI 48109.
References
- 1.Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta neuropathologica 2016;131:803–20. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Brada M, van den Bent MJ, Tonn JC, Pentheroudakis G. High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014;25Suppl 3:iii93–101. [DOI] [PubMed] [Google Scholar]
- 3.Louis DN, Perry A, Wesseling P, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro-oncology 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin 2021;71:7–33. [DOI] [PubMed] [Google Scholar]
- 5.deSouza RM, Shaweis H, Han C, et al. Has the survival of patients with glioblastoma changed over the years? Br J Cancer 2016;114:146–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown TJ, Brennan MC, Li M, et al. Association of the Extent of Resection With Survival in Glioblastoma: A Systematic Review and Meta-analysis. JAMA Oncol 2016;2:1460–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weller M, van den Bent M, Hopkins K, et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol 2014;15:e395–403. [DOI] [PubMed] [Google Scholar]
- 8.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 2009;10:459–66. [DOI] [PubMed] [Google Scholar]
- 9.Gebhardt BJ, Dobelbower MC, Ennis WH, Bag AK, Markert JM, Fiveash JB. Patterns of failure for glioblastoma multiforme following limited-margin radiation and concurrent temozolomide. Radiation Oncology (London, England) 2014;9:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med 2008;359:492–507. [DOI] [PubMed] [Google Scholar]
- 11.Cloughesy TF, Cavenee WK, Mischel PS. Glioblastoma: from molecular pathology to targeted treatment. Annu Rev Pathol 2014;9:1–25. [DOI] [PubMed] [Google Scholar]
- 12.Felsberg J, Hentschel B, Kaulich K, et al. Epidermal Growth Factor Receptor Variant III (EGFRvIII) Positivity in EGFR-Amplified Glioblastomas: Prognostic Role and Comparison between Primary and Recurrent Tumors. Clin Cancer Res 2017;23:6846–55. [DOI] [PubMed] [Google Scholar]
- 13.Francis JM, Zhang CZ, Maire CL, et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov 2014;4:956–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell 2013;155:462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Stefano AL, Fucci A, Frattini V, et al. Detection, Characterization, and Inhibition of FGFR-TACC Fusions in IDH Wild-type Glioma. Clin Cancer Res 2015;21:3307–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh D, Chan JM, Zoppoli P, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012;337:1231–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu X, Bao Z, Liu Y, et al. PBX3/MEK/ERK1/2/LIN28/let-7b positive feedback loop enhances mesenchymal phenotype to promote glioblastoma migration and invasion. J Exp Clin Cancer Res 2018;37:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galanis E, Anderson SK, Lafky JM, et al. Phase II study of bevacizumab in combination with sorafenib in recurrent glioblastoma (N0776): a north central cancer treatment group trial. Clin Cancer Res 2013;19:4816–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wen PY, Touat M, Alexander BM, et al. Buparlisib in Patients With Recurrent Glioblastoma Harboring Phosphatidylinositol 3-Kinase Pathway Activation: An Open-Label, Multicenter, Multi-Arm, Phase II Trial. J Clin Oncol 2019;37:741–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pitz MW, Eisenhauer EA, MacNeil MV, et al. Phase II study of PX-866 in recurrent glioblastoma. Neuro Oncol 2015;17:1270–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michaud K, Solomon DA, Oermann E, et al. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res 2010;70:3228–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cen L, Carlson BL, Schroeder MA, et al. p16-Cdk4-Rb axis controls sensitivity to a cyclin-dependent kinase inhibitor PD0332991 in glioblastoma xenograft cells. Neuro Oncol 2012;14:870–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willems E, Dedobbeleer M, Digregorio M, et al. Aurora A plays a dual role in migration and survival of human glioblastoma cells according to the CXCL12 concentration. Oncogene 2019;38:73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gobin M, Nazarov PV, Warta R, et al. A DNA Repair and Cell-Cycle Gene Expression Signature in Primary and Recurrent Glioblastoma: Prognostic Value and Clinical Implications. Cancer Res 2019;79:1226–38. [DOI] [PubMed] [Google Scholar]
- 25.Talukdar S, Pradhan AK, Bhoopathi P, et al. MDA-9/Syntenin regulates protective autophagy in anoikis-resistant glioma stem cells. Proc Natl Acad Sci U S A 2018;115:5768–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Costa B, Bendinelli S, Gabelloni P, et al. Human glioblastoma multiforme: p53 reactivation by a novel MDM2 inhibitor. PLoS One 2013;8:e72281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verreault M, Schmitt C, Goldwirt L, et al. Preclinical Efficacy of the MDM2 Inhibitor RG7112 in MDM2-Amplified and TP53 Wild-type Glioblastomas. Clin Cancer Res 2016;22:1185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qazi MA, Vora P, Venugopal C, et al. Intratumoral heterogeneity: pathways to treatment resistance and relapse in human glioblastoma. Ann Oncol 2017;28:1448–56. [DOI] [PubMed] [Google Scholar]
- 29.Keime-Guibert F, Chinot O, Taillandier L, et al. Radiotherapy for glioblastoma in the elderly. N Engl J Med 2007;356:1527–35. [DOI] [PubMed] [Google Scholar]
- 30.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–96. [DOI] [PubMed] [Google Scholar]
- 31.Stupp R, Taillibert S, Kanner A, et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. Jama 2017;318:2306–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan AC, Ashley DM, Lopez GY, Malinzak M, Friedman HS, Khasraw M. Management of glioblastoma: State of the art and future directions. CA Cancer J Clin 2020;70:299–312. [DOI] [PubMed] [Google Scholar]
- 33.Zhou W, Wahl DR. Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers (Basel) 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 35.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011;11:85–95. [DOI] [PubMed] [Google Scholar]
- 36.Huennekens FM. The methotrexate story: a paradigm for development of cancer chemotherapeutic agents. Adv Enzyme Regul 1994;34:397–419. [DOI] [PubMed] [Google Scholar]
- 37.de Sousa Cavalcante L, Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur J Pharmacol 2014;741:8–16. [DOI] [PubMed] [Google Scholar]
- 38.Shrivastava A, Khan AA, Khurshid M, Kalam MA, Jain SK, Singhal PK. Recent developments in L-asparaginase discovery and its potential as anticancer agent. Crit Rev Oncol Hematol 2016;100:1–10. [DOI] [PubMed] [Google Scholar]
- 39.Collins R, Reith C, Emberson J, et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 2016;388:2532–61. [DOI] [PubMed] [Google Scholar]
- 40.Pernicova I, Korbonits M. Metformin--mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol 2014;10:143–56. [DOI] [PubMed] [Google Scholar]
- 41.Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol 1927;8:519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Warburg O On the origin of cancer cells. Science 1956;123:309–14. [DOI] [PubMed] [Google Scholar]
- 43.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324:1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farber S, Diamond LK. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med 1948;238:787–93. [DOI] [PubMed] [Google Scholar]
- 45.Burchenal JH, Murphy ML, Ellison RR, et al. Clinical evaluation of a new antimetabolite, 6-mercaptopurine, in the treatment of leukemia and allied diseases. Blood 1953;8:965–99. [PubMed] [Google Scholar]
- 46.Philips FS, Sternberg SS, Hamilton S, Clarke DA. The toxic effects of 6-mercaptopurine and related compounds. Ann N Y Acad Sci 1954;60:283–96. [DOI] [PubMed] [Google Scholar]
- 47.Munshi PN, Lubin M, Bertino JR. 6-thioguanine: a drug with unrealized potential for cancer therapy. Oncologist 2014;19:760–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Curreri AR, Ansfield FJ, Mc IF, Waisman HA, Heidelberger C. Clinical studies with 5-fluorouracil. Cancer Res 1958;18:478–84. [PubMed] [Google Scholar]
- 49.Guchelaar HJ, Richel DJ, van Knapen A. Clinical, toxicological and pharmacological aspects of gemcitabine. Cancer Treat Rev 1996;22:15–31. [DOI] [PubMed] [Google Scholar]
- 50.Galmarini CM, Mackey JR, Dumontet C. Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol 2002;3:415–24. [DOI] [PubMed] [Google Scholar]
- 51.Langleben DD, Segall GM. PET in Differentiation of Recurrent Brain Tumor from Radiation Injury*. Journal of Nuclear Medicine 2000;41:1861–7. [PubMed] [Google Scholar]
- 52.Gupta S, Farooque A, Adhikari JS, Singh S, Dwarakanath BS. Enhancement of radiation and chemotherapeutic drug responses by 2-deoxy-D-glucose in animal tumors. J Cancer Res Ther 2009;5Suppl 1:S16–20. [DOI] [PubMed] [Google Scholar]
- 53.Singh D, Banerji AK, Dwarakanath BS, et al. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther Onkol 2005;181:507–14. [DOI] [PubMed] [Google Scholar]
- 54.Dwarakanath BS, Singh D, Banerji AK, et al. Clinical studies for improving radiotherapy with 2-deoxy-D-glucose: present status and future prospects. J Cancer Res Ther 2009;5Suppl 1:S21–6. [DOI] [PubMed] [Google Scholar]
- 55.Pajak B, Siwiak E, Soltyka M, et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int J Mol Sci 2019;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharma AD, Patnaik A, Dhawan S. A randomized multicenteric controlled comparative trial to evaluate the efficacy and safety of 2-deoxy-D-glucose (2-DG) as a radiomodifier in the treatment of glioblastoma multiforme. India: Institute of Nuclear Medicine and Allied Sciences; 2014. [Google Scholar]
- 57.Yadav S, Pandey SK, Goel Y, Temre MK, Singh SM. Diverse Stakeholders of Tumor Metabolism: An Appraisal of the Emerging Approach of Multifaceted Metabolic Targeting by 3-Bromopyruvate. Front Pharmacol 2019;10:728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lis P, Dylag M, Niedzwiecka K, et al. The HK2 Dependent "Warburg Effect" and Mitochondrial Oxidative Phosphorylation in Cancer: Targets for Effective Therapy with 3-Bromopyruvate. Molecules 2016;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yuan S, Wang F, Chen G, et al. Effective elimination of cancer stem cells by a novel drug combination strategy. Stem Cells 2013;31:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou Y, Zhou Y, Shingu T, et al. Metabolic alterations in highly tumorigenic glioblastoma cells: preference for hypoxia and high dependency on glycolysis. J Biol Chem 2011;286:32843–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ijare O, Conway D, Cash A, Baskin D, Pichumani K. CBMT-49. OXALOACETATE ALTERS GLUCOSE METABOLISM IN GLIOBLASTOMA: 13C ISOTOPOMER STUDY. Neuro-Oncology 2019;21:vi43–vi4. [Google Scholar]
- 62.Wiese EK, Hitosugi S, Loa ST, et al. Enzymatic activation of pyruvate kinase increases cytosolic oxaloacetate to inhibit the Warburg effect. Nat Metab 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ruban A, Berkutzki T, Cooper I, Mohar B, Teichberg VI. Blood glutamate scavengers prolong the survival of rats and mice with brain-implanted gliomas. Invest New Drugs 2012;30:2226–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Augur ZM, Doyle CM, Li M, Mukherjee P, Seyfried TN. Nontoxic Targeting of Energy Metabolism in Preclinical VM-M3 Experimental Glioblastoma. Front Nutr 2018;5:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008;321:1807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hartmann C, Meyer J, Balss J, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 2009;118:469–74. [DOI] [PubMed] [Google Scholar]
- 67.Lai A, Kharbanda S, Pope WB, et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol 2011;29:4482–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009;462:739–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011;19:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010;18:553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012;483:474–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rohle D, Popovici-Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013;340:626–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mellinghoff IK, Cloughesy TF, Wen PY, et al. A phase I, open label, perioperative study of AG-120 and AG-881 in recurrent IDH1 mutant, low-grade glioma: Results from cohort 1. Journal of Clinical Oncology 2019;37:2003-. [Google Scholar]
- 74.Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009;361:1058–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010;17:225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gross S, Cairns RA, Minden MD, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med 2010;207:339–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mellinghoff IK, Ellingson BM, Touat M, et al. Ivosidenib in Isocitrate Dehydrogenase 1-Mutated Advanced Glioma. J Clin Oncol 2020;38:3398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Konteatis Z, Artin E, Nicolay B, et al. Vorasidenib (AG-881): A First-in-Class, Brain-Penetrant Dual Inhibitor of Mutant IDH1 and 2 for Treatment of Glioma. ACS Med Chem Lett 2020;11:101–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mellinghoff IK, Bent MJVD, Clarke JL, et al. INDIGO: A global, randomized, double-blind, phase III study of vorasidenib (VOR; AG-881) vs placebo in patients (pts) with residual or recurrent grade II glioma with an isocitrate dehydrogenase 1/2 (IDH1/2) mutation. Journal of Clinical Oncology 2020;38:TPS2574-TPS. [Google Scholar]
- 80.Núñez FJ, Mendez FM, Kadiyala P, et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Science translational medicine 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 2009;174:1149–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Juratli TA, Kirsch M, Robel K, et al. IDH mutations as an early and consistent marker in low-grade astrocytomas WHO grade II and their consecutive secondary high-grade gliomas. J Neurooncol 2012;108:403–10. [DOI] [PubMed] [Google Scholar]
- 83.Johannessen TA, Mukherjee J, Viswanath P, et al. Rapid Conversion of Mutant IDH1 from Driver to Passenger in a Model of Human Gliomagenesis. Mol Cancer Res 2016;14:976–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sulkowski PL, Oeck S, Dow J, et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 2020;582:586–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sulkowski PL, Sundaram RK, Oeck S, et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat Genet 2018;50:1086–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sulkowski PL, Corso CD, Robinson ND, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Science translational medicine 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Turk AA, Wisinski KB. PARP inhibitors in breast cancer: Bringing synthetic lethality to the bedside. Cancer 2018;124:2498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Eder JP, Doroshow DB, Do KT, et al. Clinical Efficacy of Olaparib in IDH1/IDH2-Mutant Mesenchymal Sarcomas. JCO Precision Oncology 2021:466–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hanna C, Kurian KM, Williams K, et al. Pharmacokinetics, safety, and tolerability of olaparib and temozolomide for recurrent glioblastoma: results of the phase I OPARATIC trial. Neuro-oncology 2020;22:1840–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McBrayer SK, Mayers JR, DiNatale GJ, et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018;175:101–16 e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Losman JA, Kaelin WG Jr. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev 2013;27:836–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer 2014;14:709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab 2014;20:953–66. [DOI] [PubMed] [Google Scholar]
- 94.Drzewoski J, Hanefeld M. The Current and Potential Therapeutic Use of Metformin-The Good Old Drug. Pharmaceuticals (Basel) 2021;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Adeberg S, Bernhardt D, Ben Harrabi S, et al. Metformin influences progression in diabetic glioblastoma patients. Strahlenther Onkol 2015;191:928–35. [DOI] [PubMed] [Google Scholar]
- 96.Shenouda G, Souhami L, Petrecca K, et al. A Phase 2 Study of Neo-adjuvant Metformin and Temozolomide followed by Hypofractionated Accelerated RadioTherapy (HART) with Concomitant and Adjuvant Metformin and Temozolomide (TMZ) in Patients with Glioblastoma. International journal of radiation oncology, biology, physics 2020;108:S21. [DOI] [PubMed] [Google Scholar]
- 97.Schoenfeld JD, Sibenaller ZA, Mapuskar KA, et al. O2(-) and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 2017;31:487–500 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ma Y, Chapman J, Levine M, Polireddy K, Drisko J, Chen Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci Transl Med 2014;6:222ra18. [DOI] [PubMed] [Google Scholar]
- 99.Yun J, Mullarky E, Lu C, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015;350:1391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Castro ML, McConnell MJ, Herst PM. Radiosensitisation by pharmacological ascorbate in glioblastoma multiforme cells, human glial cells, and HUVECs depends on their antioxidant and DNA repair capabilities and is not cancer specific. Free Radic Biol Med 2014;74:200–9. [DOI] [PubMed] [Google Scholar]
- 101.Castro ML, Carson GM, McConnell MJ, Herst PM. High Dose Ascorbate Causes Both Genotoxic and Metabolic Stress in Glioma Cells. Antioxidants (Basel) 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Allen BG, Bodeker KL, Smith MC, et al. First-in-Human Phase I Clinical Trial of Pharmacologic Ascorbate Combined with Radiation and Temozolomide for Newly Diagnosed Glioblastoma. Clin Cancer Res 2019;25:6590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cushing CM, Petronek MS, Bodeker KL, et al. Magnetic resonance imaging (MRI) of pharmacological ascorbate-induced iron redox state as a biomarker in subjects undergoing radio-chemotherapy. Redox Biol 2021;38:101804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ngo B, Van Riper JM, Cantley LC, Yun J. Targeting cancer vulnerabilities with high-dose vitamin C. Nature Reviews Cancer 2019;19:271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mayne ST, Playdon MC, Rock CL. Diet, nutrition, and cancer: past, present and future. Nat Rev Clin Oncol 2016;13:504–15. [DOI] [PubMed] [Google Scholar]
- 106.Tajan M, Vousden KH. Dietary Approaches to Cancer Therapy. Cancer Cell 2020;37:767–85. [DOI] [PubMed] [Google Scholar]
- 107.Weber DD, Aminzadeh-Gohari S, Tulipan J, Catalano L, Feichtinger RG, Kofler B. Ketogenic diet in the treatment of cancer - Where do we stand? Mol Metab 2020;33:102–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tan-Shalaby J Ketogenic Diets and Cancer: Emerging Evidence. Fed Pract 2017;34:37S–42S. [PMC free article] [PubMed] [Google Scholar]
- 109.Hopkins BD, Pauli C, Du X, et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018;560:499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schwartz KA, Noel M, Nikolai M, Chang HT. Investigating the Ketogenic Diet As Treatment for Primary Aggressive Brain Cancer: Challenges and Lessons Learned. Front Nutr 2018;5:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schreck KC, Hsu FC, Berrington A, et al. Feasibility and Biological Activity of a Ketogenic/Intermittent-Fasting Diet in Patients With Glioma. Neurology 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang X, Yang K, Xie Q, et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat Neurosci 2017;20:661–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013;339:1323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016;351:728–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ali ES, Sahu U, Villa E, et al. ERK2 Phosphorylates PFAS to Mediate Posttranslational Control of De Novo Purine Synthesis. Molecular Cell 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Makinoshima H, Takita M, Matsumoto S, et al. Epidermal Growth Factor Receptor (EGFR) Signaling Regulates Global Metabolic Pathways in EGFR-mutated Lung Adenocarcinoma. Journal of Biological Chemistry 2014;289:20813–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liu YC, Li F, Handler J, et al. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One 2008;3:e2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nikaki A, Angelidis G, Efthimiadou R, et al. (18)F-fluorothymidine PET imaging in gliomas: an update. Ann Nucl Med 2017;31:495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Laks DR, Ta L, Crisman TJ, et al. Inhibition of Nucleotide Synthesis Targets Brain Tumor Stem Cells in a Subset of Glioblastoma. Mol Cancer Ther 2016;15:1271–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Garrett M, Sperry J, Braas D, et al. Metabolic characterization of isocitrate dehydrogenase (IDH) mutant and IDH wildtype gliomaspheres uncovers cell type-specific vulnerabilities. Cancer Metab 2018;6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kofuji S, Hirayama A, Eberhardt AO, et al. IMP dehydrogenase-2 drives aberrant nucleolar activity and promotes tumorigenesis in glioblastoma. Nat Cell Biol 2019;21:1003–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhou W, Yao Y, Scott AJ, et al. Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat Commun 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shireman JM, Atashi F, Lee G, et al. De novo purine biosynthesis is a major driver of chemoresistance in glioblastoma. Brain 2021;144:1230–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Spector R Hypoxanthine transport and metabolism in the central nervous system. J Neurochem 1988;50:969–78. [DOI] [PubMed] [Google Scholar]
- 125.Allison AC. Mechanisms of action of mycophenolate mofetil. Lupus 2005;14Suppl 1:s2–8. [DOI] [PubMed] [Google Scholar]
- 126.Wang J, Figurski M, Shaw LM, Burckart GJ. The impact of P-glycoprotein and Mrp2 on mycophenolic acid levels in mice. Transpl Immunol 2008;19:192–6. [DOI] [PubMed] [Google Scholar]
- 127.Androdias G, Maillet D, Marignier R, et al. Mycophenolate mofetil may be effective in CNS sarcoidosis but not in sarcoid myopathy. Neurology 2011;76:1168–72. [DOI] [PubMed] [Google Scholar]
- 128.Apparaju SK, Gudelsky GA, Desai PB. Pharmacokinetics of gemcitabine in tumor and non-tumor extracellular fluid of brain: an in vivo assessment in rats employing intracerebral microdialysis. Cancer Chemother Pharmacol 2008;61:223–9. [DOI] [PubMed] [Google Scholar]
- 129.Sigmond J, Honeywell RJ, Postma TJ, et al. Gemcitabine uptake in glioblastoma multiforme: potential as a radiosensitizer. Ann Oncol 2009;20:182–7. [DOI] [PubMed] [Google Scholar]
- 130.Kim MM, Camelo-Piragua S, Schipper M, et al. Gemcitabine Plus Radiation Therapy for High-Grade Glioma: Long-Term Results of a Phase 1 Dose-Escalation Study. Int J Radiat Oncol Biol Phys 2016;94:305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Marin-Valencia I, Yang C, Mashimo T, et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell metabolism 2012;15:827–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mashimo T, Pichumani K, Vemireddy V, et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014;159:1603–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Randall EC, Lopez BGC, Peng S, et al. Localized Metabolomic Gradients in Patient-Derived Xenograft Models of Glioblastoma. Cancer research 2020;80:1258–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Garofano L, Migliozzi S, Oh YT, et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat Cancer 2021;2:141–56. [DOI] [PMC free article] [PubMed] [Google Scholar]