

Abstract
Nonstructural protein 1 (nsp1) is a coronavirus (CoV) virulence factor that restricts cellular gene expression by inhibiting translation through blocking the mRNA entry channel of the 40S ribosomal subunit and by promoting mRNA degradation. We perform a detailed structure-guided mutational analysis of severe acute respiratory syndrome (SARS)-CoV-2 nsp1, revealing insights into how it coordinates these activities against host but not viral mRNA. We find that residues in the N-terminal and central regions of nsp1 not involved in docking into the 40S mRNA entry channel nonetheless stabilize its association with the ribosome and mRNA, both enhancing its restriction of host gene expression and enabling mRNA containing the SARS-CoV-2 leader sequence to escape translational repression. These data support a model in which viral mRNA binding functionally alters the association of nsp1 with the ribosome, which has implications for drug targeting and understanding how engineered or emerging mutations in SARS-CoV-2 nsp1 could attenuate the virus.
Keywords: SARS-CoV-2, SARS, coronavirus, nsp1, translation, RNA, ribosome, nuclease
Graphical abstract
The coronavirus Nsp1 protein binds the 40S ribosomal subunit to induce host translational suppression and mRNA cleavage. Mendez et al. reveal that mutation of specific residues in the Nsp1 N terminus destabilizes its interaction with the 40S subunit and eliminates the protection of viral-leader-sequence-containing transcripts from translational repression.
Introduction
Viral infections frequently result in massive remodeling of the gene expression landscape within the cell due to a combination of populating the cell with viral transcripts, inducing innate immune pathways, and the activity of viral proteins that hijack or restrict key gene expression machinery. Protein synthesis is a focal point of control, as all viruses rely on cellular ribosomes for their protein synthesis and thus compete with endogenous mRNA for access to the translation machinery. A common viral strategy to shift translational resources toward viral mRNA is to restrict host gene expression, for example by inhibiting cap-dependent translation or encoding nucleases that degrade the cellular mRNA pool (de Breyne et al., 2020; Glaunsinger, 2015; Lloyd, 2006; Rivas et al., 2016; Stern-Ginossar et al., 2019; Walsh et al., 2013). This phenotype, termed host shutoff, both increases viral transcript access to ribosomes and promotes innate immune evasion (Abernathy and Glaunsinger, 2015).
Host shutoff is a prominent feature of coronavirus (CoV) infection and has been shown to contribute significantly to the suppression of innate immune responses in multiple pathogenic CoVs, including severe acute respiratory syndrome (SARS)-CoV, Middle East respiratory syndrome (MERS) CoV and the pandemic SARS-CoV-2 (Hartenian et al., 2020; Nakagawa and Makino, 2021; Narayanan et al., 2015). SARS-CoV-2-induced host shutoff is multi-faceted and involves inhibition of host mRNA splicing by the nonstructural protein 16 (nsp16), restriction of cellular cytoplasmic mRNA accumulation and translation by nsp1, and disruption of protein secretion by nsp8 and nsp9 (Banerjee et al., 2020; Hillen et al., 2020; Littler et al., 2020; Zhang et al., 2021). Among them, the contribution and mechanism of action of nsp1 are best understood, in part because nsp1 was previously characterized as a key virulence factor in SARS-CoV and related betacoronaviruses like mouse hepatitis virus (MHV) (Brockway and Denison, 2005; Züst et al., 2007). Indeed, an MHV mutant lacking functional nsp1 is severely attenuated in infected mice, and the mutation of nsp1 has thus been explored as a strategy for the development of a live attenuated vaccine for SARS-CoV (Lei et al., 2013; Wathelet et al., 2007; Züst et al., 2007).
Foundational work with SARS-CoV nsp1 together with recent structural insights for the highly homologous SARS-CoV-2 nsp1 established that it engages in a bifunctional mechanism of shutoff of cytoplasmic mRNA that is unique among all characterized viral proteins (Huang et al., 2011; Kamitani et al., 2006, 2009; Lokugamage et al., 2012; Narayanan et al., 2008). Nsp1 binds directly to the 40S ribosomal subunit and positions its carboxy-terminal (C-terminal) domain in the 40S mRNA entry channel, thereby blocking transcript access to the ribosome (Schubert et al., 2020; Shi et al., 2020; Thoms et al., 2020; Yuan et al., 2020). Structural studies revealed that the C-terminal helices of nsp1 dock within the entry channel through interactions with the RPS2, RPS3, and RPS30A ribosomal proteins as well as 18S rRNA (Schubert et al., 2020; Shi et al., 2020; Thoms et al., 2020; Yuan et al., 2020). These interactions are likely allosterically enhanced by the initiation factor eIF1, perhaps because eIF1 induces an “open head” conformation of the 40S subunit mRNA entry channel that is favorable for nsp1 binding (Lapointe et al., 2021). In addition to blocking translation, SARS-CoV nsp1 also promotes cleavage of mRNA near the transcript 5′ end and SARS-CoV-2 nsp1 similarly reduces mRNA levels in cells (Huang et al., 2011; Kamitani et al., 2009; Lei et al., 2020; Lokugamage et al., 2012). Unlike other virally encoded host shutoff factors that deplete mRNA, nsp1 itself has no apparent ribonuclease activity, and thus, the mechanism underlying mRNA cleavage remains unknown (Huang et al., 2011; Kamitani et al., 2009; Lokugamage et al., 2012; Nakagawa and Makino, 2021).
SARS-CoV and SARS-CoV-2 nsp1 are 20-kilodalton (kDa) proteins with three general domains. The well-characterized helical C-terminal domain is connected to a globular amino terminal (N-terminal) domain through an unstructured central region. Mutations within the C-terminal domain that disrupt 18S ribosomal RNA (and thus 40S subunit) binding also block RNA cleavage, suggesting that mRNA cleavage is linked to translational repression (Huang et al., 2011; Kamitani et al., 2009; Lokugamage et al., 2012). Furthermore, the identification of a double point mutant at the border of the N-terminal and central domains of SARS-CoV nsp1 that disrupts mRNA cleavage but retains translational repression activity has led to the hypothesis that mRNA cleavage occurs subsequent to translational repression and is a functionally separable phenotype (Lokugamage et al., 2012). The structure of the N-terminal domain in isolation has been solved, but neither it nor the unstructured central region was resolved in the cryoelectron microscopy structures of SARS-CoV-2 nsp1 bound to the 40S ribosomal subunit, and thus, their role in host shutoff is unclear (Clark et al., 2021; Semper et al., 2021).
Notably, CoV mRNAs contain a common 5′ leader sequence that protects them against nsp1-imposed host shutoff (Huang et al., 2011). Studies with SARS-CoV nsp1 and SARS-CoV-2 nsp1 have implicated the N-terminal domain in binding the first stem loop (SL1) in the viral leader sequence to somehow facilitate the continued translation of viral mRNA (Shi et al., 2020; Tanaka et al., 2012; Tidu et al., 2020). Thus, deciphering the mechanistic contributions of domains outside the C-terminal region in nsp1-induced host shutoff and viral escape will be key to ultimately understanding CoV virulence. Indeed, a recent SARS-CoV-2 genomic monitoring study reported a recurrent viral variant found in 37 countries containing an 11-amino acid (aa) deletion in the N terminus of nsp1 that is associated with lower viral load, lower serum interferon B (IFN-β), and enrichment of less severe disease (Benedetti et al., 2020; Lin et al., 2021).
Here, we define the functional contributions of each of the three nsp1 domains by performing a structure-function analysis of SARS-CoV-2 nsp1 by using a combination of purified proteins and cell-based activity assays. Our results demonstrate that regions outside the defined 40S interaction domain, including conserved residues in the N-terminal and central domains of nsp1 contribute to interactions with the 40S ribosomal subunit and with mRNA. Nsp1 mRNA binding and cleavage appear to occur only in the context of ribosome binding. Although mRNA containing the SARS-CoV-2 leader sequence escapes repression by wild-type (WT) nsp1, we identify mutations in nsp1 that render these transcripts susceptible to potent translational inhibition. We hypothesize that an nsp1-40S-mRNA complex positions cellular mRNA for cleavage when the mRNA cannot engage the ribosome entry tunnel, whereas viral mRNA escapes cleavage because its association with nsp1 triggers remodeling of the complex to enable translation. However, if viral mRNA is not properly engaged by nsp1, it becomes susceptible to repression. It is therefore likely that mutations within the N-terminal domain of nsp1 could decrease SARS-CoV-2 virulence both by decreasing host shutoff and by restricting viral gene expression. In this regard, drugs that target the interaction between the nsp1 N terminus and mRNA may be good candidates for antiviral therapy.
Results
SARS-CoV-2 nsp1 promotes translational shutoff and mRNA decay
SARS-CoV nsp1 and SARS-CoV-2 nsp1 are 84.4% identical at the amino acid level, including the conservation of residues characterized in SARS-CoV nsp1 as required for nsp1 binding to the 40S subunit (K164/H165) and for promoting mRNA turnover (R124/K125) (Figure S1A; Kamitani et al., 2009; Lokugamage et al., 2012; Min et al., 2020). To confirm that SARS-CoV-2 nsp1 functions in a similarly bifunctional manner as SARS-CoV nsp1, we measured its translation repression and RNA cleavage activity in vitro and in cells. We assume that the changes in protein output in these assays are a combined reflection of changes to translational efficiency and mRNA abundance. As expected, when added to HEK293T in vitro translation extracts, purified SARS-CoV-2 nsp1 markedly suppressed translation of a nanoluciferase reporter containing the 5′ UTR of human β-globin (HBB-nLuc) in a dose-dependent manner (Figure 1 A). Introducing K164A/H165A mutations, which inhibit 40S binding, also abolishes translation inhibition (Figure 1A), whereas mutant R124A/K125A retained the ability to suppress nLuc translation at high concentrations but was partially defective at lower concentrations (Figure 1A). Similar results were obtained with rabbit reticulocyte lysates (Figure S1B). Nsp1 also induced HBB-nLuc mRNA cleavage in translation extracts, as measured by primer extension assays (Figure 1B). Cleavage required nsp1 binding to ribosomes, as the mRNA was uncleaved in the absence of translation extracts or by the mutant K164A/H165A (Figure 1B). Nsp1 R124A/K125A showed reduced mRNA cleavage relative to the WT protein, confirming that these residues within the central nsp1 domain are involved in its mRNA turnover activity (Figure 1B).
Figure 1.

CoV-2 nsp1 promotes translational suppression and mRNA decay in vitro and in cells
(A) HBB-nLuc reporter RNA was incubated with HEK293T translation extracts alone or in the presence of increasing concentrations of purified WT, R124A/K125A, or K164A/H165A nsp1. Translation of the reporter was then evaluated by luciferase assay and normalized to a glutathione S-transferase (GST) protein control. Technical triplicate measurements were taken for each biological replicate. A total of at least three biological replicates were taken for each measurement. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001; one-sample t test versus hypothetical value of 1. The bars represent the mean value of the replicates and error bars represent standard deviation.
(B) A primer extension assay was used to measure cleavage of the HBB-nLuc RNA in the presence of purified WT or mutant nsp1. Lane 1 (no IVT) shows nsp1 and HBB-nLuc incubation in primer extension buffer only, whereas lanes 2–4 show reactions incubated in the presence of translation extracts. Hash marks denote cleavage intermediates.
(C and D) HEK293T cells were transfected with a GFP reporter plasmid alone or together with the indicated nsp1-expressing plasmids and then harvested for protein or RNA. GFP and nsp1 protein levels were measured by ⍺-GFP and ⍺-FLAG western blots, respectively, with vinculin used as a protein loading control (C). GFP mRNA was quantified by qRT-PCR and normalized to 18S rRNA, with the level of GFP mRNA in cells lacking nsp1 then set to 1 (D). Each dot represents an independent experiment. ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001; one-way ANOVA followed by Dunnett’s multiple comparisons test versus WT nsp1. For (D), the bars represent the mean value of the replicates and error bars represent standard deviation.
(E) HEK293T cells transfected with a GFP reporter plasmid alone or together with the indicated nsp1-expressed plasmids were subsequently treated with 5-μg/mL actinomycin D (ActD) and harvested at the time points indicated after ActD treatment. GFP mRNA was quantified by qRT-PCR and normalized to 18S rRNA, and the changes in GFP mRNA abundance are relative to the time point immediately before ActD treatment. Each dot represents an independent experiment. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001; two-way ANOVA with Geisser-Greenhouse correction followed by Tukey’s multiple comparisons test versus “0”-h time point. See also Figures S1 and S2. The points represent the mean values of the replicates and error bars represent standard deviation.
Finally, we monitored SARS-CoV-2 nsp1 activity in cells by co-transfecting HEK293T cells with plasmids expressing WT or mutant nsp1 together with a GFP reporter plasmid and then evaluating levels of GFP protein by western blot and GFP mRNA by qRT-PCR. WT nsp1 prevented GFP protein expression and caused a 40-fold reduction of GFP transcript levels (Figures 1C and 1D). This mRNA depletion is more profound than what we observed in vitro, suggesting that factors involved in nsp1-induced mRNA decay may be limiting in translation extracts. In line with the in vitro assays, the K164A/H165A mutant had no affect on GFP protein or mRNA, whereas the R124A/K125A mutant caused a modest reduction in GFP protein and a 2-fold reduction of GFP mRNA (Figures 1C and 1D). To verify that the reduction in GFP mRNA in nsp1-expressing cells was due to increased mRNA decay, we measured the decline in GFP mRNA abundance over time after treatment with actinomycin D to halt transcription. Indeed, WT nsp1 significantly decreased the stability of GFP mRNA, whereas the K164A/H165A mutant had a limited effect and resembled the empty vector control (Figure 1E). Note that the nsp1 used here lacks the viral leader sequence and can target its own transcript for repression; so, in these and subsequent cell-based assays, the level of the nsp1 protein generally reflects its activity (i.e., WT nsp1 accumulates to lower levels than functionally defective mutants). We confirmed that the differences in nsp1 protein levels were not due to uneven transfection efficiency by including co-transfection controls of RNA polymerase III (Pol III)-transcribed RNAs (B2 SINE and VAI) (Figures S2A and S2B), which are not targeted by nsp1 (Gaglia et al., 2012).
The N-terminal and central domains of nsp1 are required for host shutoff
The above results and recent published data have confirmed the essential role of the C-terminal domain of SARS-CoV-2 nsp1 in translational suppression and have shown that residues R124 and K125 in the central domain are important for promoting efficient mRNA decay. However, little is known about the contribution of the N-terminal domain to host shutoff activity or the mechanistic role of the central domain. To address these questions, we first constructed a series of deletion mutants in which we removed either the N-terminal domain (Δ1–117) or the C-terminal and central domains (Δ118–180) (Figure 2 A). We also tested whether the region containing the R124/K125 “RNA destabilization” residues had a sequence-dependent role or a sequence-independent spacing role by deleting residues encompassing this region (Δ122–130) and then replacing them with a size-matched glycine linker (G-linker). We evaluated these mutants by using the cell-based assays described above to measure protein and mRNA levels of the GFP reporter. The WT and mutant nsp1 proteins were tagged with a 3xFLAG-Halo tag for detection, which we confirmed does not interfere with nsp1 function (Figures S2C and S2D).
Figure 2.

The N-terminal and central domains of nsp1 are required for translational suppression and mRNA depletion
(A) Schematic of the N-terminal 3xFLAG-Halo-tagged versions of WT and mutant nsp1. Amino acids 122–130 encompass the RNA destabilization domain, which was either deleted (Δ122–130) or replaced with a size-matched glycine linker (G-linker). Mutant Δ118–180 lacks the central and C-terminal domains, whereas Δ1–117 lacks the N-terminal domain.
(B and C) HEK293T cells were transfected with a GFP reporter plasmid alone or together with plasmids containing WT or the indicated mutant nsp1 and then harvested for protein or RNA. GFP, and nsp1 protein levels were measured by ⍺-GFP and ⍺-FLAG western blots, respectively, with vinculin used as a protein loading control (B). GFP mRNA was quantified by qRT-PCR and normalized to 18S rRNA, with the level of GFP mRNA in cells lacking nsp1 then set to 1 (C). Each dot represents an independent experiment. ∗∗∗∗p ≤ 0.0001; one-way ANOVA followed by Dunnett’s multiple comparisons test versus WT nsp1. See also Figure S2. The bars represent the mean value of the replicates and error bars represent standard deviation.
Notably, we found that in the absence of its N-terminal domain, nsp1 induced shutoff of both the GFP protein and mRNA to reduced levels similar to the R124A/K125A mutant (Figures 2B and 2C). The N-terminal domain alone showed no translational suppression or mRNA turnover activity, which is not surprising given that it lacks the K164/H165 residues essential for 40S subunit binding (Figures 2B and 2C). The Δ122–130 and G-linker mutants behaved similarly to the R124A/K125A point mutant, suggesting that this region has a more specific function and is not simply a flexible spacer separating the N-terminal and C-terminal domains (Figures 2B and 2C). Together, these data establish that residues within the N terminus of nsp1 play key roles in host shutoff.
Residue R99 in the N-terminal domain of nsp1 contributes to its mRNA destabilization function and translational shutoff
We next sought to better define which residues within the N-terminal domain are required for suppressing gene expression. Guided by existing structural data for the N terminus (Clark et al., 2021; Semper et al., 2021), we mutated a series of conserved, surface exposed, and charged residues (E36A/E37A, E55A/E57A/K58A, R99A, and R119A/K120A) (Figure S3A). The mutants were then screened using our GFP reporter assay alongside the R124A/K125A and K164A/H165A controls to determine the effects on protein translation and mRNA decay. Most of the N-terminal domain mutants behaved similarly to WT nsp1 in their ability to suppress GFP protein and mRNA levels, although E36A/E37A was modestly impaired for mRNA depletion (Figures 3A and 3B). The one exception was mutant R99A, which displayed a moderate defect in restricting GFP protein accumulation and a severe defect in mRNA targeting (Figures 3A, 3B, S2A, and S2B).
Figure 3.

Residue R99 located in the N-terminal domain plays key roles in nsp1-induced host shutoff
(A and B) HEK293T cells were transfected with a GFP reporter plasmid alone or together with plasmids containing WT or the indicated mutant nsp1 and then harvested for protein or RNA. GFP and nsp1 protein levels were measured by ⍺-GFP and ⍺-FLAG western blots, respectively, with vinculin used as a protein loading control (A). GFP mRNA was quantified by qRT-PCR and normalized to 18S rRNA, with the level of GFP mRNA in cells lacking nsp1 then set to 1 (B). Each dot represents an independent experiment. ∗p ≤ 0.05, ∗∗∗∗p ≤ 0.0001; one-way ANOVA followed by Dunnett’s multiple comparisons test versus WT nsp1. For (B), the bars represent the mean value of the replicates and error bars represent standard deviation.
(C) HBB-nLuc reporter RNA was incubated with HEK293T translation extracts in the presence of 80 nM of purified WT or the indicated mutant nsp1 protein. Translation of the reporter was then evaluated by luciferase assay and normalized to levels from lysates incubated with 80 nM of a control GST protein. Technical triplicate measurements were taken for each biological replicate. A total of at least three biological replicates were taken for each measurement. ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001; one-way ANOVA followed by Dunnett’s multiple comparisons test versus WT nsp1. The bars represent the mean value of the replicates and error bars represent standard deviation.
(D) Primer extension assay to measure degradation of the HBB-nLuc RNA in the presence and absence of purified WT or mutant nsp1. Lanes 1 and 2 are controls lacking translation extract (no IVT) or nsp1, respectively. See also Figure S3.
The R99A defect was also observed when we purified the protein and tested its activity in the HEK293T translation extracts (Figure S3B). Compared to WT nsp1, which suppressed translation of HBB-nLuc by 5-fold, the R99A mutant suppressed translation by only 1.2-fold, similar to the 1.7-fold suppression seen with the R124A/K125A mutant (Figure 3C). Even at high protein concentrations, R99A did not markedly reduce translation of the reporter (Figure S3C). Primer extensions revealed that R99A showed a modest reduction in nsp1-induced RNA cleavage, similar to R124A/K125A (Figure 3D). We note that although R99A, R124A/K125A, and K164A/H165A mutants all show pronounced defects in mRNA depletion in cells, only the 40S binding mutant strongly disrupts RNA cleavage in the in vitro assay.
The nsp1 N terminus and central domain are involved in ribosome binding
To identify the mechanism(s) underlying the defects of the various SARS-CoV-2 nsp1 domain mutants, we first evaluated their ability to bind the 40S ribosomal subunit both in cells and using quantitative in vitro measurements. Binding in cells was evaluated by immunoprecipitation (IP) of nsp1 followed by western blotting to monitor co-purification of the 40S subunit proteins RPS2, RPS3, RPS24, and RACK1, which are enriched as nsp1 interaction partners in published mass spectrometry datasets (Gordon et al., 2020; Zhang et al., 2021; Figure 4 A). Notably, all mutants we tested in this assay showed reduced levels of ribosomal protein interactions, including the point mutations R124A/K125A, E36A/E37A, and (to a lesser extent) R99A located in the N terminus and the deletion mutant Δ1–117 lacking the N-terminal domain (Figure 4A). As expected, no interaction with 40S proteins was observed in the absence of the C-terminal domain (Δ118–180) or with the K164A/H165A 40S binding mutant.
Figure 4.

Nsp1 N-terminal and central domain mutants are defective for ribosome and mRNA binding
(A) HEK293T cells were transfected with plasmids expressing WT or the indicated mutant 3xFLAG-Halo-tagged nsp1. Nsp1 was immunoprecipitated (IP) using ⍺-FLAG beads and coIP of ribosomal proteins RACK1, RPS2, RPS3, and RPS24 was monitored by western blotting, with vinculin serving as a loading control. Input lanes contain 1/10 of the amount of protein used for the IPs.
(B) Equilibrium binding measurements of fluorescently labeled WT (blue), R124A,K125A (red), and R99A (green) nsp1 to purified ribosomes. Data represent a total of 3 biological replicates.
(C) HEK293T cells were co-transfected with HBB-nLuc and either a control plasmid or the indicated 3xFLAG-Halo-tagged nsp1 constructs. Technical triplicate measurements were taken for each biological replicate. A total of at least three biological replicates were taken for each measurement. Nsp1 was immunoprecipitated using ⍺-FLAG beads, whereupon the co-immunoprecipitating RNAs were extracted and nLuc mRNA was quantified by qRT-PCR. The mRNA values were then normalized to the values obtained from the empty vector control. Each dot represents an independent experiment. ∗p ≤ 0.05, ∗∗p ≤ 0.01; one-way ANOVA followed by Dunnett’s multiple comparisons test versus WT nsp1. The bars represent the mean value of the replicates and error bars represent standard deviation.
(D) HEK293T cells were co-transfected with a 3xFLAG-Halo-tagged nsp1 plasmid or empty vector control, together with a plasmid expressing either GFP with a 5′ stem loop (GFP+SL) or a control GFP lacking the stem loop (GFP). Nsp1 was immunoprecipitated using ⍺-FLAG beads, whereupon the co-immunoprecipitating GFP+SL or GFP mRNAs were quantified by qRT-PCR. The mRNA values were then normalized to those obtained from the empty vector control. The bars represent the mean value of the replicates and error bars represent standard deviation.
(E) The levels of GFP+SL and GFP mRNA present in the input samples from (D) were quantified by qRT-PCR and normalized to 18S rRNA, with the level of GFP mRNA in cells lacking nsp1 (empty vector control) set to 1. Each dot represents an independent experiment. ∗∗p ≤ 0.01; unpaired t test. See also Figures S4, S5, and S6. The bars represent the mean value of the replicates and error bars represent standard deviation.
We next quantified ribosome affinity directly by using fluorescent polarization (FP) equilibrium binding experiments in the presence of an increasing concentration of purified ribosomes. We site-specifically labeled WT nsp1 and nsp1 mutants at an engineered internal position with a TAMRA dye for these ribosome binding measurements. Raw polarization units confirmed minimal binding of the K164A/H165A to ribosomes compared with that of WT nsp1, which bound with a dissociation constant (KD) of 29.36 nM (Figures 4B and S4). In agreement with the reduced co-purification of 40S proteins from cells, R124A/K125A and R99A showed a 2.2-fold and a 1.8-fold increase in their KD values, respectively (Figure 4B). Thus, although the C-terminal domain is critical for ribosome binding, both the N-terminal and central regions contribute to the stability of the interaction.
Multiple nsp1 domains contribute to binding mRNA in association with the ribosome
In addition to their contribution to ribosome binding, we also considered the possibility that the nsp1 N-terminal and central domains may be involved in binding mRNA. We were unable to detect a direct interaction between purified nsp1 and RNA in vitro (Figure S5), perhaps suggesting that nsp1 binds RNA only in the context of the 40S ribosomal subunit in cells. To test this hypothesis, we evaluated the ability of WT and mutant nsp1 to bind the HBB-nLuc reporter mRNA in transfected HEK293T cells by using RNA IP (RIP) experiments. Indeed, we observed an 85-fold enrichment of HBB-nLuc mRNA upon IP of the FLAG-tagged WT nsp1, but no binding to the reporter by the 40S binding mutant K164A/H165A, and a 15-fold decrease in binding by the nsp1 Δ1–117 mutant lacking the N-terminal domain (Figures 4C and S6A). RIP experiments with the R124A/K125A and R99A point mutants showed a 5-fold and 2-fold decrease in mRNA binding, respectively (Figure 4C). Importantly, qRT-PCR measurements of co-purifying 18S rRNA in each RIP correlated with the level of 40S binding seen by western blotting (Figure S6B), suggesting that the observed nsp1-mRNA interactions are likely to be bridged by the 40S ribosomal subunit.
To more directly test whether an mRNA must be bound to 40S to associate with nsp1, we performed RIPs to evaluate the ability of FLAG-nsp1 to associate with a GFP mRNA containing a cap-proximal stem loop structure (GFP+SL) that blocks 40S binding and thus cannot be translated (Figure S6C; Gaglia et al., 2012; Kozak, 1989). Indeed, nsp1 binding to the GFP+SL was reduced 4.5-fold relative to the translation-competent version of GFP, and nsp1 was incapable of degrading GFP+SL (Figures 4D and 4E). Collectively, these data suggest that the nsp1 N terminus contributes to a stable nsp1-mRNA-40S subunit complex, which is needed to efficiently induce mRNA cleavage.
SARS-CoV-2 leader-sequence-mediated escape requires nsp1 residues R124/R125 and R99
The 5′ leader sequence of SARS-CoV and SARS-CoV-2 interact with nsp1 to somehow protect viral transcripts from its cleavage activity (Huang et al., 2011; Shi et al., 2020; Tanaka et al., 2012). To evaluate the contribution of the nsp1 domains toward CoV-2 leader sequence binding, we generated an nLuc reporter mRNA containing the SARS-CoV-2 leader sequence at its 5′ end (CoV2L-nLuc). As expected, RIPs with WT FLAG-nsp1 were significantly enriched for CoV2L-nLuc compared with the empty vector control (Figure S6D), whereas the 40S binding mutant K164A/H165A showed no enrichment for the CoV2L-nLuc reporter (Figure 5 A). Similar to the results with the mRNA lacking the SARS-CoV-2 leader, the truncation mutant lacking the N-terminal domain (Δ1-117), the R124A/K125A mutant, and the R99A mutant all displayed reduced binding to the SARS-CoV-2 leader mRNA (a 17-fold, 13-fold, and 7-fold reduction, respectively) (Figure 5A). Thus, like cellular mRNA, the transcript containing the SARS-CoV-2 leader does not readily associate with nsp1 in the absence of 40S binding.
Figure 5.

Protection from translational repression conferred by the CoV-2-leader sequence is selectively eliminated by nsp1 N-terminal and central domain mutants
(A) HEK293T cells were co-transfected with a plasmid expressing CoV-2 leader-nLuc and either a control plasmid or the indicated 3xFLAG-Halo-tagged nsp1 construct. Nsp1 was immunoprecipitated using ⍺-FLAG beads, whereupon the co-immunoprecipitating RNAs were quantified by qRT-PCR. The mRNA values were then normalized to the mRNA values obtained from the empty vector control. Each dot represents an independent experiment. ∗p ≤ 0.05, ∗∗p ≤ 0.01; one-way ANOVA followed by Dunnett’s multiple comparisons test versus WT nsp1. The bars represent the mean value of the replicates and error bars represent standard deviation.
(B) HEK293T cells were transfected with either HBB-nLuc or CoV2L-nLuc together with control empty vector or the indicated nsp1 construct. Translation of HBB-nLuc or CoV2L-nLuc was measured by luciferase assay, and the fold change in luciferase activity was calculated relative to the empty vector control. Technical triplicate measurements were taken for each biological replicate. A total of at least three biological replicates were taken for each measurement. ∗p ≤ 0.05, ∗∗p ≤ 0.01; one-sample t test versus hypothetical value of 1. The bars represent the mean value of the replicates and error bars represent standard deviation.
(C) CoV2L-nLuc mRNA was quantified from the above experiment by qRT-PCR and normalized to 18S rRNA, with the level of CoV2L-nLuc mRNA in cells lacking nsp1 then set to 1. Each dot represents an independent experiment. ∗∗p ≤ 0.01; one-way ANOVA followed by Dunnett’s multiple comparisons test versus WT nsp1. See also Figure S6. The bars represent the mean value of the replicates and error bars represent standard deviation.
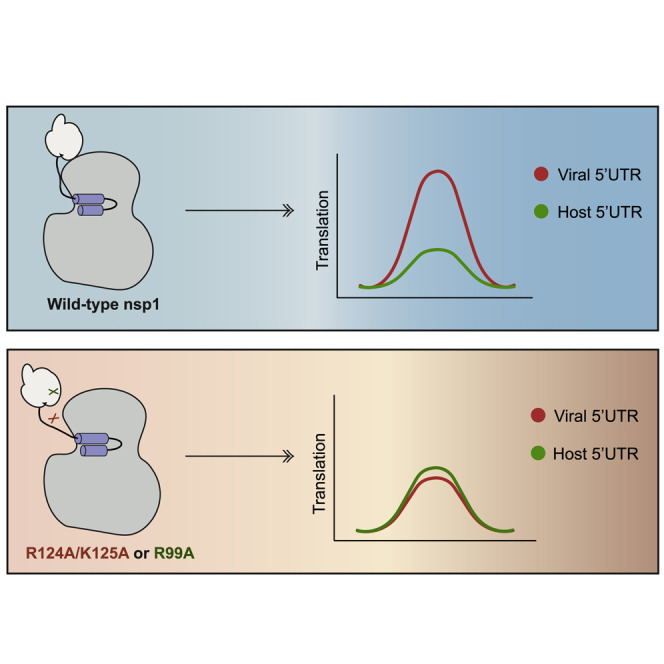
Although the N-terminal and central domains of nsp1 participate in binding both cellular and viral mRNA, the consequences of this binding are presumably different. Indeed, the CoV2L-nLuc mRNA remained fully translationally competent in the presence of nsp1, under conditions that suppressed translation of HBB-nLuc (Figures 5B and 5C). Remarkably, however, both the R124A/K125A and R99A mutants gained the ability to translationally repress CoV2L-nLuc (3-fold and 2.4-fold, respectively), although they remained impaired for the translational suppression of HBB-nLuc (Figure 5B). Nsp1 K164A/H165A had no effect on the translation of either reporter, as expected (Figure 5B).
Finally, we performed qRT-PCR to determine whether the translational suppression of CoV2L-nLuc also resulted in degradation of the mRNA. Surprisingly, there was no decrease in CoV2L-nLuc mRNA abundance in the presence of these mutants, indicating that the decrease in protein expression was solely due to translational repression (Figure 5C). Collectively, these data show that the interaction of the viral leader sequence with nsp1 residues R124, K125, and R99 in an mRNA-nsp1-40S subunit ternary complex is critical for mediating escape from translational suppression, underscoring their functional importance for both host shutoff and efficient viral gene expression.
Discussion
Virus-induced host shutoff has profound effects on viral pathogenesis, as has been demonstrated for pathogens ranging from influenza virus to CoV to herpesviruses (Richner et al., 2011; Strelow and Leib, 1995; Sun et al., 2020; Zhang et al., 2015; Züst et al., 2007). The mechanisms by which viral transcripts retain robust expression during host shutoff are generally tailored to the specific host shutoff strategy and include the use of alternative RNA processing or ribosome recruitment mechanisms, as well as kinetic regulation of the host shutoff factor (Gaucherand et al., 2019; Levene et al., 2021; Sarnow et al., 2005). Despite numerous studies on the dual translational repression and mRNA cleavage functions of nsp1, significant knowledge gaps exist for how it mechanistically coordinates both activities against cellular mRNA while sparing viral transcripts. Here, we report a detailed structure-guided mutational analysis of nsp1. Using a combination of in vitro and cell-based assays to measure translation, ribosome binding, RNA binding, and mRNA degradation by WT and mutant SARS-CoV-2 nsp1, we find that its host shutoff activities all appear to occur in the context of an nsp1-mRNA-40S complex. We also made two additional notable new findings that are summarized in the model shown in Figure 6 . First, the SARS-CoV-2 nsp1 N terminus and neighboring residues that do not contact the mRNA entry channel nonetheless stabilize the nsp1-40S subunit interaction and in doing so enhance its host shutoff functions. Second, we identify specific residues in nsp1 whose mutation abrogates the translational escape of viral leader-containing mRNA. Collectively, these findings provide insight into the functional contribution of nsp1 regions outside the 40S docking domain and have implications for understanding why alterations to these regions may impair viral pathogenesis.
Figure 6.

Model for how the N-terminal and central domains of nsp1 are critical for 40S ribosome association and preservation of leader-containing transcripts
(A) All three nsp1 domains contribute to its interaction with the 40S ribosome. While the C-terminal domain interjects into the mRNA entry channel of the ribosome to block mRNA access, the N-terminal and central domains stabilize the interaction. When cellular mRNA encounters an nsp1-bound ribosome, it is translationally blocked and undergoes degradation. However, mRNA containing the CoV-2 leader sequence engages the N-terminal and central domains of 40S-bound nsp1 in a manner involving nsp1 residues R124, K125, and R99, leading to relief from translational repression.
(B) Nsp1 mutants R124A/K125A and R99A have reduced affinity for the 40S ribosome, which alleviates the translational repression of cellular transcripts. However, CoV-2 leader-containing transcripts instead become translationally repressed, perhaps due to a “nonproductive” interaction with nsp1 in the absence of proper engagement with residues R124/K125 or R99.
Mutation of SARS-CoV-2 nsp1 N-terminal and central domain residues R99, R124, and K125 compromises its binding to the 40S ribosomal subunit, as observed by the reduced association with ribosomal proteins and 18S rRNA in IP experiments, as well as increased dissociation constants with purified ribosomes. Although the nsp1 C-terminal domain docks in the 40S mRNA entry channel, if and where these other nsp1 domain interactions occur on the ribosome are unknown. It is possible that mRNA stabilizes the nsp1-40S complex through interactions with the N-terminal and central domains, as nsp1 mRNA binding decreases in accordance with reduced nsp1-40S binding. In this regard, a stable interaction between nsp1 and mRNA presumably does not occur prior to 40S subunit binding, as shown by the markedly reduced nsp1 binding to an mRNA with a 40S blocking sequence. Also, no cellular or SARS-CoV-2 leader sequence mRNA binding was detected using nsp1 K164A/H165, which fails to engage the 40S subunit. However, recent in vitro data also argue against nsp1-binding ribosomes that have already engaged mRNA in their entry channel or eIF3j-bound ribosomes that have a closed head conformation (Lapointe et al., 2021; Yuan et al., 2020). Thus, mRNA binding by nsp1 presumably occurs subsequent to nsp1-40S binding and/or with mRNA that is associated with the 40S subunit but has not yet engaged the entry channel. Either cellular mRNA binding could occur indirectly by bridging interactions with the 40S subunit or nsp1 could possess weak intrinsic mRNA binding activity that is significantly stabilized by 40S binding.
Our results indicate that weakening the nsp1-mRNA-40S subunit interaction rescues cellular mRNA from degradation, as does adding a 40S subunit blocking sequence to the mRNA. How the nsp1-mRNA interaction is stabilized by the 40S subunit and how this interaction leads to cleavage of the mRNA are open central questions. Given that the nsp1 C-terminal domain occludes the 40S subunit mRNA entry channel, the mRNA present in the tripartite nsp1-mRNA-40S subunit complex is presumably held elsewhere on the ribosome, perhaps through interactions with translation initiation factors. This notion is supported by the observation that mRNAs containing certain internal ribosome entry site (IRES) elements can escape cleavage by SARS-CoV nsp1, depending on their initiation factor requirements (Kamitani et al., 2009; Lokugamage et al., 2012; Narayanan et al., 2008; Yang and Wang, 2019). For example, mRNAs bearing a cricket paralysis virus IRES (which recruits the 40S ribosomal subunit in the absence of any initiation factors) or a hepatitis C virus IRES are not cleaved, whereas mRNAs with picornavirus type I and type II IRES elements are susceptible to SARS-CoV nsp1-induced cleavage (Kamitani et al., 2009; Lokugamage et al., 2012; Tidu et al., 2020). In the future, it could be informative to test whether susceptible versus uncleaved IRES elements have different abilities to bind the nsp1-40S complex.
Unlike cellular mRNA, viral mRNA remains robustly translated in the presence of nsp1 (Finkel et al., 2021; Lin et al., 2021; Narayanan et al., 2015; Tanaka et al., 2012). As shown for both SARS-CoV and SARS-CoV-2, preservation of viral gene expression in the presence of nsp1 requires a conserved stem loop (termed SL1) within the 5′ leader sequence present on viral mRNAs (Narayanan et al., 2015; Shi et al., 2020; Tanaka et al., 2012; Tidu et al., 2020). Two main models have been put forth to explain viral mRNA escape. One posits that nsp1 can target both cellular and viral transcripts equally, but leader-containing transcripts have higher intrinsic translational efficiency than cellular mRNA and can thus preferentially engage free 40S ribosomes (Lapointe et al., 2021; Schubert et al., 2020; Thoms et al., 2020; Yuan et al., 2020). The other model proposes that viral mRNA is directly refractory to suppression by nsp1, perhaps because it interacts with nsp1 in a manner that causes an allosteric change in nsp1 that removes its C-terminal domain from the 40S entry channel (Sakuraba et al., 2021; Shi et al., 2020; Tanaka et al., 2012; Tidu et al., 2020).
Although we agree that viral mRNA may have intrinsically high translational efficiency, several of our observations favor the second model. First, we found that mRNA bearing the SARS-CoV-2 leader was readily associated with nsp1-40S subunit complexes but was not subject to degradation. Even if a fraction of the SARS-CoV-2 leader mRNA was more efficiently engaged by non-nsp1-bound 40S subunits, in the absence of protection, we would still have expected to see cleavage of those bound by the nsp1-40S ribosomes. Second, and more telling, are the observations that specific mutations within and bordering the N-terminal domain of nsp1 (R99A and R124A/K125A) have negative rather than neutral effects on the translation of SARS-CoV-2 leader mRNA. Both of these mutants have a weaker association with ribosomes and mRNA and thus would be expected to free up even more 40S ribosomes for efficient translation of viral transcripts. Instead, R99A and R124A/K125A gain the ability to suppress the translation of SARS-CoV-2 leader-containing transcripts. We therefore propose that these residues are crucial for viral leader-sequence-triggered conformational changes to nsp1 that enable viral mRNA translation.
Our data argue against the direct binding of the SARS-CoV-2 leader sequence to nsp1 and indicate that viral or host mRNA binding instead is facilitated by the ribosome. However, the observation that leader sequence escape from nsp1 repression is not observed in in vitro translation extracts (Lapointe et al., 2021; Schubert et al., 2020) suggests that one or more additional cellular factors are required to potentiate the translation of viral leader-containing mRNA on nsp1-bound ribosomes. One possibility is that there is a multi-component RNA-protein complex that forms on the ribosome between viral leader mRNA, cellular factors, and the nsp1 N terminus that triggers a conformational change in nsp1 that exposes the mRNA entry channel. In this scenario, the R99A and R124A/K125A mutations may prevent proper assembly of this complex. Alternatively, they could “lock” nsp1 in a translationally repressive conformation. Intriguingly, these mutants translationally repress SARS-CoV-2 leader mRNA translation without reducing its mRNA abundance, which may have implications for understanding how mRNA cleavage is activated. Notably, the above model is supported by the observation that mutating the central domain residue R124 in SARS-CoV nsp1 reduces viral gene expression (Tanaka et al., 2012), suggesting conservation across betacoronaviruses in the functional role for regions outside the C terminus in promoting viral escape.
The finding that nsp1 point mutants like R99A can abrogate translational protection of viral leader-containing mRNA while having reduced host shutoff activity against cellular mRNA has important implications for understanding the role of nsp1 in pathogenesis. In vivo experiments with the model betacoronavirus MHV showed that the deletion of residues within the nsp1 central domain results in reduced viral load and heightened survival rates, and this virus showed promise as a live attenuated vaccine platform (Lei et al., 2013). Additionally, mutation of the nsp1 R124 residue in the SARS-CoV replicon system decreased viral gene expression and replication (Tanaka et al., 2012). It is therefore possible that specific nsp1 mutations such as R99, R124, and R125 could impair viral pathogenesis because of the translational suppression of viral transcripts in addition to the impaired shutoff of host genes such as those involved in IFN signaling. It will be of interest to determine whether this underlies the lower viral load and decreased pathogenesis reported for the recurrent SARS-CoV-2 variant that contains an 11-aa nsp1 N-terminal domain deletion or other variants that may emerge with changes to these regions of nsp1 (Lin et al., 2021). Likewise, small molecule drugs that phenocopy these mutations could serve as therapies or prophylactics for treating viral infection.
Limitations of the study
A limitation of our study is that it was conducted on nsp1 expressed in uninfected cells, and thus, future work should be geared toward validating the phenotypes of these mutations in the context of SARS-CoV-2 infection, including in primary cell types relevant to the in vivo biology of CoV replication. Furthermore, our study relies on reporter mRNAs that function as a proxy for cellular or viral transcripts. Extending this work to broadly evaluate endogenous mRNAs, for example through genomics-based approaches, would further strengthen the conclusions.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-GFP | Clontech | Cat# 632381; RRID: AB_2313808 |
| Rabbit anti-Vinculin | Abcam | Cat# GR268234-50; RRID: AB_2241513 |
| Mouse anti-FLAG M2 | Sigma-Aldrich | Cat# SLBT654; RRID:AB_262044 |
| Rabbit anti-RPS2 | Bethyl labs | Cat# A303-794A-M; RRID:AB_2781471 |
| Rabbit anit-RPS3 | Proteintech | Cat# 11990-1-AP; RRID:AB_2180758 |
| Rabbit anti-RPS24 | Bethyl labs | Cat# A303-545A; RRID:AB_2620193 |
| Rabbit anit-RACK1 | Bethyl labs | Cat# A302-545A; RRID:AB_1999012 |
| Rabbit anit-FLAG M2 (magnetic beads) | Sigma-Aldrich | Cat# M8823-5mL; RRID:AB_2637089 |
| HRP goat anit-mouse IgG | SouthernBiotech | Cat# 1031-05; RRID:AB_2794307 |
| HRP goat anti-rabbit IgG | SouthernBiotech | Cat# 4030-05; RRID:AB_2687483 |
| Enzymes | ||
| EcoRI | New England Biolabs | R3101S (10,000U) |
| In-Fusion® snap assembly (master mix) | Clontech (TaKaRa) | 638943 |
| TURBO DNase | Invitrogen | AM2239 (5,000U) |
| AMV Reverse Transcriptase | Promega | M5101 (300U) |
| SuperScript IV RT | ThermoFisher | 18090050 (10,000U) |
| PreScission protease | Cytiva | 27084301 |
| T7 RNA polymerase | New England Biolabs | M0251L (25,000U) |
| 2′-O-Methyltransferase | New England Biolabs | M0366S (50,000U) |
| Vaccinia D1/D2 (Capping enzyme) | New England Biolabs | M2080S (400U) |
| Cell lines | ||
| BL21-CondonPlus (DE3)-RIPL | Agilent | 230240 |
| HEK293T cells | UC, Berkeley TC Core | N/A |
| HELA cells | UC, Berkeley TC Core | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Overnight ExpressTM instant TB medium | Novagen | 71300-4 |
| cOmpleteTM, EDTA-free protease inhibitor cocktail | Roche | 5056489001 |
| Halt Protease and Phosphatase inhibitor single-use | ThermoFisher | 78444 |
| Trichloroacetic acid (TCA) | Sigma-Aldrich | T0699-100ML |
| 3XFLAG peptide | Sigma-Aldrich | F4799-4MG |
| TRIzol Reagent | ThermoFisher | 15596026 |
| GMP-PNP trisodium salt hydrate | Sigma-Aldrich | G0635-5MG |
| Cycloheximide (CHX) | Sigma-Aldrich | C4859-1ML |
| Glutathione Sepharose 4 (fast flow) | Cytiva | 17513202 |
| Kaposi’s sarcoma associated herpesvirus (ORF37) | This lab | PMID: 30321376 |
| TAMRA maleimide, 6-isomer | Lumiprobe life science | 28180 (5mg) |
| Murine RNase inhibitor | New England Biolabs | M0312L (15,000U) |
| Actinomycin D | Sigma-Aldrich | A1410-5MG |
| SUPERase⋅In RNase inhibitor | Invitrogen | AM2696 (10,00U) |
| Critical commercial assays | ||
| PolyJet (transfection reagent) | SignaGen® Labs | SL100688 |
| Nano-Glo® Luciferase Assay kit | Promega | N1110 |
| iTaq universal SYBR® green supermix | Bio-Rad | 1725121 |
| Nuclease-treated rabbit reticulocyte lysate (RRL) | Promega | L4960 |
| Oligonucleotides | ||
| Primers for qPCRs, primer extension assay, and cloning | This paper | See Table S1 |
| Recombinant DNA | ||
| pCDNA4TO-3XFLAG Halo WT nsp1 | This paper | 175412 (Addgene) |
| pCDNA4TO-3XFLAG Halo R124A/K125A nsp1 | This paper | 175417 (Addgene) |
| pCDNA4TO-3XFLAG Halo K164A/H165A nsp1 | This paper | 175421 (Addgene) |
| pCDNA4TO-3XFLAG Halo R99A nsp1 | This paper | 175422 (Addgene) |
| pCDNA4TO-3XFLAG Halo delta 1-117 nsp1 | This paper | 175423 (Addgene) |
| pCDNA4TO-3XFLAG Halo delta 118-180 nsp1 | This paper | 175424 (Addgene) |
| pCDNA4TO-3XFLAG Halo delta 122-130 nsp1 | This paper | 175425 (Addgene) |
| pCDNA4TO-3XFLAG Halo G-Linker nsp1 | This paper | 175426 (Addgene) |
| pCDNA4TO-3XFLAG Halo E55A E57A K58A nsp1 | This paper | 175427 (Addgene) |
| pCDNA4TO-3XFLAG Halo R119A K120A nsp1 | This paper | 175428 (Addgene) |
| pCDNA4TO-3XFLAG Halo E36A E37A nsp1 | This paper | 175429 (Addgene) |
| pJP-CoV2leader-nLuc-TSS | This paper | 175430 (Addgene) |
| pJP-HBB-nLuc | This paper | 175431 (Addgene) |
| pUC57-HBB-nLuc | This paper | 175432 (Addgene) |
| pGEX nsp1 CoV2 | This paper | 175512 (Addgene) |
| pGEX nsp1 R124A/K125A CoV2 | This paper | 175513 (Addgene) |
| pGEX nsp1 K164A/H165A CoV2 | This paper | 175514 (Addgene) |
| pGEX nsp1 R99A CoV2 | This paper | 175515 (Addgene) |
| pGEX C_K nsp1 CoV2 | This paper | 175516 (Addgene) |
| pGEX C_K nsp1 K164A/H165A | This paper | 175517 (Addgene) |
| pUC Ad VAI | This paper | 175518 (Addgene) |
| pCDNA4 CoV-2 nsp1 3xFLAG | This paper | 176057 (Addgene) |
| pCDNA4TO R124A K125A CoV-2 nsp1 3xFLAG | This paper | 176059 (Addgene) |
| pCDNA4TO K164A H165A CoV-2 nsp1 3xFLAG | This paper | 176060 (Addgene) |
| pCDNA4TO R99A CoV-2 nsp1 3xFLAG | This paper | 176061 (Addgene) |
| Pd2EGFP-N1_plusSL | This lab | PMID: 22740404 |
| Software and algorithms | ||
| GraphPad (Prism) | This paper | Version 9.2.0 (283) |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Britt Glaunsinger (glaunsinger@berkeley.edu).
Materials availability
Plasmids generated in this study have been deposited to Addgene (see Key resources table).
Experimental model and subject details
Cell lines
HEK293T cells were grown in Dulbecco’s modified Eagle’s medium (GIBCO) supplemented with 10% fetal bovine serum (Peak Serum) at 37°C and 5% CO2. HeLa cells were grown in Dulbecco’s modified Eagle’s medium (Life Technologies) supplemented with 10% fetal bovine serum (Seradigm) and 1% penicillin/streptomycin (Life Technologies) at 37°C and 5% CO2. The sex of both cell lines is female, and they were both authenticated by UC Berkeley TC Core.
Method details
Cloning and mutagenesis
All plasmids generated for this study have been deposited in Addgene (see Key resources table). Sequences for the oligos and gene blocks used in this study are listed in Table S1. Full-length nsp1 was synthesized as a gene fragment from Integrated DNA technologies (IDT) and cloned into the EcoR1 restriction site of pGEX-6P-2 (GE healthcare) using in-fusion cloning (TakaraBio). Single primer-based mutagenesis was used to generate R124A/K125A, K164A/H165A, all N-terminal domain mutants (E36A/E37A, E55A/E56A/K57A, R99A, R119A/K120A) and the N-terminal cysteine/lysine containing nsp1 (nsp1 C-K) used in the fluorescence polarization assays was generated as mentioned above (Mendez et al., 2018). A T7 promoter upstream of the human β-globin 5′ UTR or the SARS-CoV-2 leader fused to a nano luciferase (HBB-nLuc and CoV-2 leader-nLuc, respectively) was synthesized by Twist Bioscience then subcloned by in-fusion cloning into an EcoRI site in a pUC57 destination vector. The leader sequence was derived from the SARS-CoV-2 isolate Wuhan-Hu-1 reference genome (Chan et al., 2020). pCDNA4-3X-FLAG-Halo was used as a destination vector for mammalian cell transfection experiments for immunoprecipitations and GFP assays. SARS-CoV-2 nsp1 was subcloned into the NotI site of pCDNA4-3X-FLAG-Halo using in-fusion cloning. Single primer mutagenesis was used to generate R124A/K125A, K164A/H165A and all N-terminal mutants (E36A/E37A, E55A/E56A/K57A, R99A, R119A/K120A). Truncation mutants Δ118-180 and Δ1-117 were PCR amplified from the parental vector pCDNA4-3x-FLAG-Halo- nsp1 and cloned as described above. For the luciferase and RIP assays, HBB-nLuc was PCR amplified from the pUC57 vector (ThermoFisher) and cloned into AfeI and EcoRI digested pLJM1 vector (Addgene #19319) with In-Fusion. The SARS-CoV-2-leader nLuc sequence was synthesized by IDT and similarly cloned into the pLJM1 vector. All constructs were sequence verified by sanger sequencing.
Protein expression and purification
WT or mutant nsp1 was expressed using BL21-CodonPlus (DE3)-RIPL cells (Agilent) using Overnight ExpressTM instant TB medium (Novagen). Cells were grown at 37°C to an OD600 of 0.6 then transferred to 18°C for 24 hr before being spun down at 4704 x g for 10 min. Cells were washed with PBS containing cOmpleteTM, EDTA-free protease inhibitor cocktail (Roche) and spun down once more. Wash solution was then decanted and pellets were resuspended in a buffer containing 500mM NaCl (Millipore sigma), 5mM MgCl2 (Millipore Sigma), 20mM HEPES (Millipore Sigma), 0.5% Triton x-100 (Sigma-Aldrich), 5% glycerol (Sigma-Aldrich) and 1mM Tris(2-Carboxyethyl)phosphine hydrochloride (Sigma-Aldrich) (Buffer A). The buffer pH was brought to 7.5 using concentrated hydrochloric acid solution (Sigma-Aldrich). The cell suspension was lysed at 4°C using a macrotip sonicator set at 80 A with a 3 s pulse and 17 s rest for 12 min. Lysates were cleared by centrifuging at 50,000 x g for 30 min at 4°C. The cleared lysate was incubated for 2 hr at 4°C on a rotating wheel with GST beads (Cytiva) that were pre-washed twice with buffer A. After incubation, beads were pelleted at 40 x g for 5 min at 4°C then washed 3x with Buffer A before resuspension in a buffer containing 250mM NaCl, 5mM MgCl2, 5% glycerol, 20mM HEPES pH 7.5, and 1mM TECEP (Buffer B). Beads were then loaded onto a polypropylene disposable column (QIAGEN), and washed with 20 column volumes of Buffer B. After the final wash, beads were resuspended in one column volume of Buffer B. PreScission protease (Cytiva) was then added to the bead slurry and incubated on a rotating wheel overnight at 4°C. The elution was then collected, and the beads were washed with an additional column volume of Buffer B. Both elutions were pooled and concentrated using a 10K Centriprep (EMD Millipore) before being loaded onto a HiLoad Superdex 200pg size exclusion column (Cytiva). Proteins eluted at expected masses as determined by protein standards and purity was determined using SDS-PAGE gel electrophoresis. Nsp1-containing fractions were then pooled and concentrated to 5 mg/ml and aliquots were stored at −80°C.
In vitro transcription
RNA constructs were made by in vitro transcription with T7 RNA polymerase (New England Biolabs) as previously described (Lee et al., 2015), with the following modifications. DNA templates were amplified from a plasmid containing the corresponding 5′ HBB UTR or SARS-CoV-2 leader sequence and the NanoLuc Luciferase coding sequence. Primers used for this amplification added a 60T sequence at the 3′ end to form a poly(A) tail after transcription. Transcription was then performed using gel-extracted PCR products and the T7 RNA polymerase New England Biolabs protocol. RNA was then capped using Vaccinia D1/D2 (Capping enzyme) (New England Biolabs) and 2′O-methylated using Vaccinia VP39 (2′O Methyltransferase) (New England Biolabs), then purified by phenol-chloroform extraction and ethanol precipitation.
In vitro translation assays in rabbit reticulocyte lysate
In vitro translation reactions were performed using nuclease-treated rabbit reticulocyte lysate (RRL) (Promega) following the manufacturer’s protocol for non-radioactive luciferase reactions, with the following modifications. In vitro translation reactions were pre-incubated with the corresponding recombinant nsp1 variant for 10 min at 4°C before addition of 40nM of the corresponding mRNA. Reactions were then incubated at 30°C for 30 min, whereupon luciferase assays were performed using the NanoLuc luciferase assay kit (Promega). Luminescence was measured using the Spark multimode microplate reader (TECAN). Technical triplicate measurements were taken for each biological replicate. These technical triplicates were averaged to plot for a given biological replicate and normalized to independent luminescence measurements from in vitro translation reactions containing the corresponding concentration of GST in place of nsp1.
Preparation of HEK293T translation extracts
In vitro translation extracts were made from HEK293T cells using a previously described protocol (Rakotondrafara and Hentze, 2011). Cells were scraped and collected by centrifugation for 2 minutes at 376 x g at 4°C. Cells were washed once with cold PBS (137mM NaCl, 2.7mM KCl, 100mM Na2HPO4, 2mM KH2PO4) then homogenized with an equal volume of freshly made cold hypotonic lysis buffer (10mM HEPES-KOH pH 7.6, 10mM KOAc, 0.5mM Mg(OAc)2, 5mM dithiothreitol (DTT), and 1 Complete EDTA-free Proteinase Inhibitor Cocktail tablet (Roche) per 10 mL of buffer). After hypotonic-induced swelling for 45 min on ice, cells were homogenized using a syringe attached to a 27G needle until 95% of cells burst as determined by trypan blue staining. Lysate was then centrifuged at 14,000 x g for 1 min at 4°C. The resulting supernatant was moved to a new tube, avoiding the top lipid layer. Lysate aliquots were quickly frozen with liquid nitrogen and stored at −80°C.
In vitro translation assays
In vitro translation reactions were performed using HEK293T translation-competent cell lysate, as previously described, with modifications (Lee et al., 2015). Translation reactions contained 50% HEK293T translation-competent cell extract, 2mM ATP, 0.42mM GTP, 7mM tris(2-carboxyethyl)phosphine, 28mM HEPES pH 7.5, 2mM creatine phosphate (Roche), 0.01 μg/μl creatine kinase (Roche), 2mM Mg(OAc)2, 60mM KOAc, 10μM amino acids (Promega), 0.21mM spermidine, 0.6mM putrescine and 0.8U/ml murine RNase inhibitor (NEB). Translation reactions were pre-incubated with the corresponding recombinant nsp1 variant for 10 min at 4°C before addition of the mRNA. 40nM of the corresponding RNA was then added to the reaction. Translation reactions were then incubated at 30°C for 30 min. Luciferase assays were then performed using the NanoLuc luciferase assay kit (Promega), following the manufacturer’s protocol. Luminescence was measured using the Spark multimode microplate reader (TECAN). Technical triplicate measurements were taken for each biological replicate. These technical triplicates were averaged to plot for a given biological replicate and normalized to independent luminescence measurements from in vitro translation reactions containing the corresponding concentration of GST in place of nsp1.
Primer extension assays
The 40μL reactions contained 50% HEK293T translation extract, 2mM ATP, 0.05mM GTP, 7mM tris(2-carboxyethyl)phosphine, 28mM HEPES pH 7.5, 2mM creatine phosphate (Roche), 0.01μg/μl creatine kinase (Roche), 2mM Mg(OAc)2 and 10μM of the amino acid mixture (Promega). Reactions were incubated with 5μM of the indicated nsp1 protein for 30 min on ice. 5μM of 5′-Guanylyl imidodiphosphate (GMPPNP) (Sigma-Aldrich) and 500μM of cycloheximide (CHX) (Sigma-Aldrich) was then added and incubated at RT for 10 min, whereupon 1μg of the HBB-nLuc mRNA was added and reactions were incubated at 30°C for 15 min. Reactions were stopped with the addition of 400μL of RNase free water and equal amounts of TRIzol reagent (ThermoFisher). RNA was extracted and resuspended in RNase free water then added to primer extension buffer containing 7mM MgOAc (Sigma-Aldrich), 100mM KOAc (Sigma-Aldrich), 50mM Tris-HCl, 1mM DTT, 1 μg/μL of SuperScript IV RT (Ambion), 550μM of DNTP mixture (ThermoFisher) and 500nM of Cy5 labeled primer (IDT). The reaction was incubated at 30°C for 15 minutes then ethanol precipitated. DNA pellets were washed twice with 70% ethanol, dried, and resuspended in 95% formamide solution containing 10mM EDTA. Samples were loaded onto a pre-run 6% UREA-PAGE gel then imaged using an Amersham Typhoon 5 laser-scanner (Cytiva).
Transfections
Transfections were carried out using PolyJet™ (SignaGen labs) according to the manufacturer’s DNA transfection protocol and cells were harvested 24 hr post transfection. For the GFP co-transfection experiments, 1x106 cells were seeded into each well of a 6-well plate and transfected the next day with 100 ng of pCAGEN-pEF-GFP and 900 ng of the indicated pCDNA4-3X-FLAG-Halo-nsp1 construct. For GFP mRNA half-life measurements, the cells were treated with 5 μg/mL actinomycin D for 0-8h as indicated and GFP mRNA was quantified at each time point by RT-qPCR. For experiments that included B2 SINE or adenovirus VAI transfection controls, cells were transfected with 100 ng pCAGEN-pEF-GFP, 400 ng pCDNA4-3X-FLAG-Halo-Nsp1 and 500 ng of B2 SINE or VAI. For the luciferase assays, 4.5x104 cells were seeded into each well of a 96-well plate and transfected the next day with 10 ng of HBB-nLuc or CoV2L-nLuc and 25 ng of the indicated C-terminally 3xFLAG tagged nsp1 (pCDNA4-nsp1-3xFLAG) construct. For the immunoprecipitation experiments, a 150 mm plate of cells were transfected with 25μg of the indicated pCDNA4-3X-FLAG-Halo-nsp1 construct. Cells were then harvested and washed with PBS (gibco) and snap frozen in liquid nitrogen and stored at −80°C until further processing.
Luciferase assays
The media was aspirated off in the 96-well plate, and PBS was added onto the cells. The nanoluciferase luminescence was activated following the Nano-Glo Luciferase Assay System (Promega) protocol. The raw luminescence values were measured by TECAN SPARKCONTROL.
RNA extraction and RT-qPCR
Cells were lysed in TRIzol reagent and RNA was extracted following the manufacturer’s protocol. The RNA was subsequently treated with TURBO DNase (ThermoFisher) and subjected to reverse transcription using AMV Reverse Transcripase (Promega) with a random 9-mer primer. cDNA was then used in qPCR following the iTaq Universal SYBR Green Supermix protocol (Bio-Rad laboratories) and gene-specific qPCR primers.
Western blot analysis and nsp1 immunoprecipitation
For western blots on direct cell lysates, cell pellets were lysed in 2X laemmli buffer (Bio-Rad laboratories) then homogenized using a microtip sonicator set at 20 A, boiled for 10 min and resolved by SDS-PAGE. For immunoprecipitations, cell pellets were lysed in 1 mL of lysis buffer (Buffer A) containing 20mM Tris-KOH pH 7.5, 150mM KOAc, 5mM MgCl2, 5% glycerol, 1X HaltTM protease inhibitor cocktail (ThermoFisher), 1mM DTT (Danville Scientific) and 0.5% NP-40 (Danville Scientific) and placed on a rotating wheel for 30 min. Lysates were then passed through a 25G syringe needle ten times then spun at 21130 x g for 25 min to clear cell debris. Cleared lysates were combined with 20μL of ANTI-FLAG M2 (Sigma-Aldrich) magnetic beads that had been pre-washed 2x with Buffer A then incubated for 2 hr on a rotating wheel at 4°C. Beads were then washed 4x with Buffer A containing only 0.1% NP-40 and bound protein was eluted by 3 sequential incubations in PBS containing 0.1% triton x-100 (Sigma-Aldrich) and 0.5mg/ml of 3x FLAG peptide (Sigma-Aldrich) at 30°C for 20 min under vigorous shaking. The eluted protein was then precipitated using 100% trichloroacetic acid (Sigma-Aldrich) added drop wise to 20% of the final elution followed by vortexing and incubation at 4°C overnight prior to pelleting at 21130 x g for 20 min at 4°C. The protein pellet was washed with 100% cold acetone then dried at RT for 10 min, resuspended in 2X Laemmli buffer and boiled for 5 min prior to resolution by SDS-PAGE.
The following antibodies were used for western blotting: mouse anti-GFP (1:5000; Clontech 632381), rabbit anti-Vinculin (1:1000, Abcam GR268234-50), mouse anti-FLAG M2 (1:1000, Sigma-Aldrich SLBT7654), rabbit anti-RPS2 (1:2000, Bethyl labs A303-794A-M), rabbit anti-RPS3 (1:500, Proteintech 11990-1-AP), rabbit anti-RPS24 (1:1000, Bethyl labs A303-842A-T), rabbit anti-RACK1 (1:1000, Bethyl labs A302-545A), HRP goat anti-mouse IgG (1:10,000 SouthernBiotech 1031-05), and HRP goat anti-rabbit IgG (1:10,000 SouthernBiotech 4030-05).
RNA immunoprecipitation (RIP)
Cells were lysed in IP buffer (100mM KCl, 0.1mM EDTA, 20mM HEPES-KOH pH 7.6, 0.4% NP-40, 10% glycerol; supplemented with fresh 1mM DTT, SUPERaseIN RNase Inhibitors (Ambion), and EDTA-free protease inhibitors (Roche)) then rotated at 4°C for 30 min and subsequently clarified by centrifugation at 21130 x g for 10 min at 4°C. The supernatant was brought up to 1 mL volume using IP buffer, and 90% of the sample was used for the RIP while 10% was saved as input samples. For each RIP, 20 μL of anti-FLAG M2 Magnetic beads (Sigma-Aldrich) were used, and the samples were rotated overnight at 4°C. The next day, samples were washed three times using 1 mL IP buffer with 5 min of rotation for each wash. The beads were then split into two fractions and one fraction was treated with TRIzolTM (ThermoFisher) for RNA extraction and the other was resuspended in Laemmli buffer (Bio-Rad laboratories) for western blot analysis. For the RNA extraction and RT-qPCR, the RIP samples were normalized to their respective input samples.
Electrophoretic mobility shift assay (EMSA)
Leader sequence RNA was 5′ labeled with Cy5 (Dharmacon Horizon Inc.). Reactions were incubated at RT for 30 min in buffer containing 20mM HEPES pH 7.8, 30mM KCl, 5mM CaCl2, 30mM NaCl, 0.01% Tween-20, 0.5 TCEP, 0.2 mg/ml BSA (Sigma-Aldrich) and the indicated amount of purified nsp1 protein or 5 μM of the Kaposi’s sarcoma associated herpesvirus RNA binding protein ORF37 as a positive control. Reaction volumes were kept at 20 μL and stopped with 3 μL 7x EMSA loading dye (70mM HEPES pH 8.0, 420mM KCl, 35% glycerol). Reactions were resolved by 8% native PAGE, and gels were imaged on a Typhoon 5 multivariable imager (Cytiva Amersham) and quantified using GelQuant software package (Molecular Dynamics).
TAMRA labeling of nsp1
Nsp1 C-K (3 mg/mL) was desalted using a 7K MWCO, 0.5 mL ZebaTM spin column (ThermoFisher) into a buffer containing 20mM HEPES-KOH pH 7.5, 120mM KOAc, 5mM Mg(OAc)2. A 1:5 mole ratio of protein to TAMRA maleimide, 6-isomer (lumiprobe Life science solutions) was used for each reaction. The reaction was gently mixed and placed at RT for 1 hr protected from light. The protein solution was then desalted once more to remove any free probe into a buffer containing 20mM HEPES-KOH pH 7.5, 120mM KOAc, 5mM Mg(OAc)2, 5% glycerol and 1mM TCEP. The dye:protein ratio for each labeled protein was between 0.84-0.91. Final protein concentration was determined by A280 and generally yielded a ∼90 percent recovery.
Ribosome purification
HeLa cell extract was prepared as described previously (Khatter et al., 2014). A frozen HeLa cell pellet was thawed and suspended with an equal volume of lysis buffer (20mM HEPES pH 7.5, 10mM KOAc, 1.8mM Mg(OAc)2 and 1mM DTT). After incubation on ice for 20 min, the cells were lysed with a Dounce homogenizer 150 times, followed by centrifugation twice at 1,200 x g for 5 min. Supernatant was then aliquoted, flash-frozen with liquid nitrogen and stored at −80°C.
Crude 80S solutions were prepared from HeLa cell lysate using a previously described protocol (Jan et al., 2014), with the following modifications. Lysate was loaded on 50% sucrose cushion prepared in Buffer A (20mM Tris pH 7.5, 2mM Mg(OAc)2, 150mM KCl), with the addition of 1mM dithiothreitol (DTT). Sucrose cushions were then centrifuged at 100,000 rpm using an MLA-130 rotor (Beckman Coulter) for 60 min at 4°C. The resulting pellet was resuspended in cold Buffer A to homogeneity. Resuspended ribosome pellet was then centrifuged at 21130 x g for 10 min at 4°C to remove any remaining cell debris. Supernatant was then aliquoted, flash-frozen with liquid nitrogen and stored at −80°C. 80S concentration was calculated as described previously (Algire et al., 2002; Jan et al., 2014).
Fluorescence polarization experiments
Binding experiments with TAMRA-labeled nsp1 were conducted using a Spark multimode microplate reader (TECAN). The final concentration of labeled nsp1 was limiting (5-10nM), and the concentration of ribosomes was varied in each reaction. Binding reactions of 20 μL were set up containing 20mM HEPES-KOH, 100mM KOAc, 5mM Mg(OAc)2, 5% glycerol, 0.2 mg/mL BSA (Ambion). Reactions were allowed to reach equilibrium at room temperature before the anisotropy was measured. Total fluorescence was also measured in order to account for quantum yield effects. The average change in anisotropy between free and bound nsp1 (and mean deviation) over three replicate experiments were: 0.078 ± 0.009. Multiple repeated-measurements showed that equilibrium had been reached. Plotting and fitting the data to obtain Kd values was conducted assuming a “tight-binding” regime. This was based on recent publications where binding affinities of nsp1 to 40S subunits were determined (Lapointe et al., 2021). Furthermore, saturation of binding was reached by 5 μM of ribosome, so the maximum anisotropy of the complex could be directly measured. The fraction bound was then calculated (incorporating quantum yield changes between the free and bound fluorophore) and fit to the solution of a quadratic equation describing an equilibrium reaction as previously reported (Fraser et al., 2007, 2009; Lapointe et al., 2021). The Kd values reported are the averages and the errors reported are the mean deviations.
Quantification and statistical analysis
All statistical analyses were performed in GraphPad Prism v9.1.2 using the tests indicated in the figure legends. When comparing two samples, unpaired t test was used, unless the comparison is to an “empty vector” control, in which case one-sample t test was performed instead to account for the unequal standard deviation. When more than two samples were compared, one-way ANOVA followed by Dunnett’s multiple comparison test was used.
Acknowledgments
We thank David Morgens and Lucas Ferguson for helpful discussions throughout this project as well as for providing critical feedback on the manuscript. This project was funded by a COVID19 Excellence in Research Award from the Laboratory of Genomics Research to B.A.G., J.H.C., and N.T.I. and an Emergent Ventures COVID19 Fast Grant (2155) to B.A.G., who is also an investigator of the Howard Hughes Medical Institute. M.L. is funded by NSERC Predoctoral Fellowship PGSD3-516787-2018.
Author contributions
Performed experiments, including ASM-primer extension assays, western blotting, immunoprecipitations, fluorescence polarization assays, ML-RNA stability measurements, RT-qPCR, western blotting, RNA immunoprecipitations, luciferase assays, AMGS-in vitro translation assays, assisted with fluorescence polarization assays, EH-RT-qPCR, luciferase assays, XXX; generated key reagents, A.S.M., M.L., A.M.G.-S., and E.H.; assisted with experimental design and analyzing data, A.S.M., M.L., E.H., A.M.G.-S., N.T.I., J.H.C., and B.A.G.; participated in writing the original paper draft, B.A.G., A.S.M., and M.L.; assisted with editing the manuscript, A.M.G.-S., N.T.I., J.H.C., E.H., B.A.G., A.S.M., and M.L.; supervised the project, B.A.G., N.T.I., and J.H.C.
Declaration of interests
B.A.G. is a member of the Vaccine Advisory Board for Celsion Corporation.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
Published: September 30, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109841.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Abernathy E., Glaunsinger B. Emerging roles for RNA degradation in viral replication and antiviral defense. Virology. 2015;479–480:600–608. doi: 10.1016/j.virol.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Algire M.A., Maag D., Savio P., Acker M.G., Tarun S.Z., Jr., Sachs A.B., Asano K., Nielsen K.H., Olsen D.S., Phan L., et al. Development and characterization of a reconstituted yeast translation initiation system. RNA. 2002;8:382–397. doi: 10.1017/s1355838202029527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A.K., Blanco M.R., Bruce E.A., Honson D.D., Chen L.M., Chow A., Bhat P., Ollikainen N., Quinodoz S.A., Loney C., et al. SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell. 2020;183:1325–1339.e21. doi: 10.1016/j.cell.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti F., Snyder G.A., Giovanetti M., Angeletti S., Gallo R.C., Ciccozzi M., Zella D. Emerging of a SARS-CoV-2 viral strain with a deletion in nsp1. J. Transl. Med. 2020;18:329. doi: 10.1186/s12967-020-02507-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockway S.M., Denison M.R. Mutagenesis of the murine hepatitis virus nsp1-coding region identifies residues important for protein processing, viral RNA synthesis, and viral replication. Virology. 2005;340:209–223. doi: 10.1016/j.virol.2005.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F., Kok K.H., Zhu Z., Chu H., To K.K., Yuan S., Yuen K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020;9:221–236. doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark L.K., Green T.J., Petit C.M. Structure of Nonstructural Protein 1 from SARS-CoV-2. J. Virol. 2021;95:e02019-20. doi: 10.1128/JVI.02019-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Breyne S., Vindry C., Guillin O., Condé L., Mure F., Gruffat H., Chavatte L., Ohlmann T. Translational control of coronaviruses. Nucleic Acids Res. 2020;48:12502–12522. doi: 10.1093/nar/gkaa1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel Y., Mizrahi O., Nachshon A., Weingarten-Gabbay S., Morgenstern D., Yahalom-Ronen Y., Tamir H., Achdout H., Stein D., Israeli O., et al. The coding capacity of SARS-CoV-2. Nature. 2021;589:125–130. doi: 10.1038/s41586-020-2739-1. [DOI] [PubMed] [Google Scholar]
- Fraser C.S., Berry K.E., Hershey J.W., Doudna J.A. eIF3j is located in the decoding center of the human 40S ribosomal subunit. Mol. Cell. 2007;26:811–819. doi: 10.1016/j.molcel.2007.05.019. [DOI] [PubMed] [Google Scholar]
- Fraser C.S., Hershey J.W., Doudna J.A. The pathway of hepatitis C virus mRNA recruitment to the human ribosome. Nat. Struct. Mol. Biol. 2009;16:397–404. doi: 10.1038/nsmb.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaglia M.M., Covarrubias S., Wong W., Glaunsinger B.A. A common strategy for host RNA degradation by divergent viruses. J. Virol. 2012;86:9527–9530. doi: 10.1128/JVI.01230-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaucherand L., Porter B.K., Levene R.E., Price E.L., Schmaling S.K., Rycroft C.H., Kevorkian Y., McCormick C., Khaperskyy D.A., Gaglia M.M. The Influenza A Virus Endoribonuclease PA-X Usurps Host mRNA Processing Machinery to Limit Host Gene Expression. Cell Rep. 2019;27:776–792.e7. doi: 10.1016/j.celrep.2019.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaunsinger B.A. Modulation of the Translational Landscape During Herpesvirus Infection. Annu. Rev. Virol. 2015;2:311–333. doi: 10.1146/annurev-virology-100114-054839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartenian E., Nandakumar D., Lari A., Ly M., Tucker J.M., Glaunsinger B.A. The molecular virology of coronaviruses. J. Biol. Chem. 2020;295:12910–12934. doi: 10.1074/jbc.REV120.013930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillen H.S., Kokic G., Farnung L., Dienemann C., Tegunov D., Cramer P. Structure of replicating SARS-CoV-2 polymerase. Nature. 2020;584:154–156. doi: 10.1038/s41586-020-2368-8. [DOI] [PubMed] [Google Scholar]
- Huang C., Lokugamage K.G., Rozovics J.M., Narayanan K., Semler B.L., Makino S. SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: viral mRNAs are resistant to nsp1-induced RNA cleavage. PLoS Pathog. 2011;7:e1002433. doi: 10.1371/journal.ppat.1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan C.H., Williams C.C., Weissman J.S. Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science. 2014;346:1257521. doi: 10.1126/science.1257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani W., Narayanan K., Huang C., Lokugamage K., Ikegami T., Ito N., Kubo H., Makino S. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl. Acad. Sci. USA. 2006;103:12885–12890. doi: 10.1073/pnas.0603144103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani W., Huang C., Narayanan K., Lokugamage K.G., Makino S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol. 2009;16:1134–1140. doi: 10.1038/nsmb.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatter H., Myasnikov A.G., Mastio L., Billas I.M., Birck C., Stella S., Klaholz B.P. Purification, characterization and crystallization of the human 80S ribosome. Nucleic Acids Res. 2014;42:e49. doi: 10.1093/nar/gkt1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Circumstances and mechanisms of inhibition of translation by secondary structure in eucaryotic mRNAs. Mol. Cell. Biol. 1989;9:5134–5142. doi: 10.1128/mcb.9.11.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe C.P., Grosely R., Johnson A.G., Wang J., Fernández I.S., Puglisi J.D. Dynamic competition between SARS-CoV-2 NSP1 and mRNA on the human ribosome inhibits translation initiation. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2017715118. e2017715118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.S., Kranzusch P.J., Cate J.H. eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature. 2015;522:111–114. doi: 10.1038/nature14267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L., Ying S., Baojun L., Yi Y., Xiang H., Wenli S., Zounan S., Deyin G., Qingyu Z., Jingmei L., Guohui C. Attenuation of mouse hepatitis virus by deletion of the LLRKxGxKG region of Nsp1. PLoS One. 2013;8:e61166. doi: 10.1371/journal.pone.0061166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei X., Dong X., Ma R., Wang W., Xiao X., Tian Z., Wang C., Wang Y., Li L., Ren L., et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020;11:3810. doi: 10.1038/s41467-020-17665-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levene R.E., Shrestha S.D., Gaglia M.M. The influenza A virus host shutoff factor PA-X is rapidly turned over in a strain-specific manner. J. Virol. 2021;95:e02312-20. doi: 10.1128/JVI.02312-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.W., Tang C., Wei H.C., Du B., Chen C., Wang M., Zhou Y., Yu M.X., Cheng L., Kuivanen S., et al. Genomic monitoring of SARS-CoV-2 uncovers an Nsp1 deletion variant that modulates type I interferon response. Cell Host Microbe. 2021;29:489–502.e8. doi: 10.1016/j.chom.2021.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littler D.R., Gully B.S., Colson R.N., Rossjohn J. Crystal Structure of the SARS-CoV-2 Non-structural Protein 9, Nsp9. iScience. 2020;23:101258. doi: 10.1016/j.isci.2020.101258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd R.E. Translational control by viral proteinases. Virus Res. 2006;119:76–88. doi: 10.1016/j.virusres.2005.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokugamage K.G., Narayanan K., Huang C., Makino S. Severe acute respiratory syndrome coronavirus protein nsp1 is a novel eukaryotic translation inhibitor that represses multiple steps of translation initiation. J. Virol. 2012;86:13598–13608. doi: 10.1128/JVI.01958-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez A.S., Vogt C., Bohne J., Glaunsinger B.A. Site specific target binding controls RNA cleavage efficiency by the Kaposi’s sarcoma-associated herpesvirus endonuclease SOX. Nucleic Acids Res. 2018;46:11968–11979. doi: 10.1093/nar/gky932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min Y.Q., Mo Q., Wang J., Deng F., Wang H., Ning Y.J. SARS-CoV-2 nsp1: Bioinformatics, Potential Structural and Functional Features, and Implications for Drug/Vaccine Designs. Front. Microbiol. 2020;11:587317. doi: 10.3389/fmicb.2020.587317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa K., Makino S. Mechanisms of Coronavirus Nsp1-Mediated Control of Host and Viral Gene Expression. Cells. 2021;10:300. doi: 10.3390/cells10020300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K., Huang C., Lokugamage K., Kamitani W., Ikegami T., Tseng C.T., Makino S. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J. Virol. 2008;82:4471–4479. doi: 10.1128/JVI.02472-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K., Ramirez S.I., Lokugamage K.G., Makino S. Coronavirus nonstructural protein 1: Common and distinct functions in the regulation of host and viral gene expression. Virus Res. 2015;202:89–100. doi: 10.1016/j.virusres.2014.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakotondrafara A.M., Hentze M.W. An efficient factor-depleted mammalian in vitro translation system. Nat. Protoc. 2011;6:563–571. doi: 10.1038/nprot.2011.314. [DOI] [PubMed] [Google Scholar]
- Richner J.M., Clyde K., Pezda A.C., Cheng B.Y., Wang T., Kumar G.R., Covarrubias S., Coscoy L., Glaunsinger B. Global mRNA degradation during lytic gammaherpesvirus infection contributes to establishment of viral latency. PLoS Pathog. 2011;7:e1002150. doi: 10.1371/journal.ppat.1002150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas H.G., Schmaling S.K., Gaglia M.M. Shutoff of Host Gene Expression in Influenza A Virus and Herpesviruses: Similar Mechanisms and Common Themes. Viruses. 2016;8:102. doi: 10.3390/v8040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuraba S., Qilin X., Kasahara K., Iwakiri J., Kono H. Modeling the SARS-CoV-2 nsp1–5′-UTR complex via extended ensemble simulations. bioRxiv. 2021 doi: 10.1101/2021.02.24.432807. [DOI] [Google Scholar]
- Sarnow P., Cevallos R.C., Jan E. Takeover of host ribosomes by divergent IRES elements. Biochem. Soc. Trans. 2005;33:1479–1482. doi: 10.1042/BST0331479. [DOI] [PubMed] [Google Scholar]
- Schubert K., Karousis E.D., Jomaa A., Scaiola A., Echeverria B., Gurzeler L.A., Leibundgut M., Thiel V., Mühlemann O., Ban N. Author Correction: SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 2020;27:1094. doi: 10.1038/s41594-020-00533-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semper C., Watanabe N., Savchenko A. Structural characterization of nonstructural protein 1 from SARS-CoV-2. iScience. 2021;24:101903. doi: 10.1016/j.isci.2020.101903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M., Wang L., Fontana P., Vora S., Zhang Y., Fu T.M., Lieberman J., Wu H. SARS-CoV-2 Nsp1 suppresses host but not viral translation through a bipartite mechanism. bioRxiv. 2020 doi: 10.1101/2020.09.18.302901. [DOI] [Google Scholar]
- Stern-Ginossar N., Thompson S.R., Mathews M.B., Mohr I. Translational Control in Virus-Infected Cells. Cold Spring Harb. Perspect. Biol. 2019;11:a033001. doi: 10.1101/cshperspect.a033001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strelow L.I., Leib D.A. Role of the virion host shutoff (vhs) of herpes simplex virus type 1 in latency and pathogenesis. J. Virol. 1995;69:6779–6786. doi: 10.1128/jvi.69.11.6779-6786.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Hu Z., Zhang X., Chen M., Wang Z., Xu G., Bi Y., Tong Q., Wang M., Sun H., et al. An R195K Mutation in the PA-X Protein Increases the Virulence and Transmission of Influenza A Virus in Mammalian Hosts. J. Virol. 2020;94:e01817-19. doi: 10.1128/JVI.01817-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T., Kamitani W., DeDiego M.L., Enjuanes L., Matsuura Y. Severe acute respiratory syndrome coronavirus nsp1 facilitates efficient propagation in cells through a specific translational shutoff of host mRNA. J. Virol. 2012;86:11128–11137. doi: 10.1128/JVI.01700-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoms M., Buschauer R., Ameismeier M., Koepke L., Denk T., Hirschenberger M., Kratzat H., Hayn M., Mackens-Kiani T., Cheng J., et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science. 2020;369:1249–1255. doi: 10.1126/science.abc8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidu A., Janvier A., Schaeffer L., Sosnowski P., Kuhn L., Hammann P., Westhof E., Eriani G., Martin F. The viral protein NSP1 acts as a ribosome gatekeeper for shutting down host translation and fostering SARS-CoV-2 translation. RNA. 2020;27:253–264. doi: 10.1261/rna.078121.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D., Mathews M.B., Mohr I. Tinkering with translation: protein synthesis in virus-infected cells. Cold Spring Harb. Perspect. Biol. 2013;5:a012351. doi: 10.1101/cshperspect.a012351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wathelet M.G., Orr M., Frieman M.B., Baric R.S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J. Virol. 2007;81:11620–11633. doi: 10.1128/JVI.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Wang Z. IRES-mediated cap-independent translation, a path leading to hidden proteome. J. Mol. Cell Biol. 2019;11:911–919. doi: 10.1093/jmcb/mjz091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S., Peng L., Park J.J., Hu Y., Devarkar S.C., Dong M.B., Shen Q., Wu S., Chen S., Lomakin I.B., Xiong Y. Nonstructural Protein 1 of SARS-CoV-2 Is a Potent Pathogenicity Factor Redirecting Host Protein Synthesis Machinery toward Viral RNA. Mol. Cell. 2020;80:1055–1066.e6. doi: 10.1016/j.molcel.2020.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R., Li Y., Cowley T.J., Steinbrenner A.D., Phillips J.M., Yount B.L., Baric R.S., Weiss S.R. The nsp1, nsp13, and M proteins contribute to the hepatotropism of murine coronavirus JHM.WU. J. Virol. 2015;89:3598–3609. doi: 10.1128/JVI.03535-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Miorin L., Makio T., Dehghan I., Gao S., Xie Y., Zhong H., Esparza M., Kehrer T., Kumar A., et al. Nsp1 protein of SARS-CoV-2 disrupts the mRNA export machinery to inhibit host gene expression. Sci. Adv. 2021;7:eabe7386. doi: 10.1126/sciadv.abe7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Züst R., Cervantes-Barragán L., Kuri T., Blakqori G., Weber F., Ludewig B., Thiel V. Coronavirus non-structural protein 1 is a major pathogenicity factor: implications for the rational design of coronavirus vaccines. PLoS Pathog. 2007;3:e109. doi: 10.1371/journal.ppat.0030109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.