

Summary
The COVID-19 pandemic has caused over 220 million infections and 4.5 million deaths worldwide. Current risk factor cannot fully explain the diversity in disease severity. Here, we present a comprehensive analysis of a broad range of patients' laboratory and clinical assessments to investigate the genetic contributions to COVID-19 severity. By performing GWAS analysis, we discovered several concrete associations for laboratory traits and used Mendelian randomization (MR) analysis to further investigate the causality of traits on disease severity. Two causal traits, WBC counts and cholesterol levels, were identified based on MR study, and their functional genes are located at genes MHC complex and ApoE, respectively. Our gene-based analysis and GSEA revealed four interferon pathways, including type I interferon receptor binding and SARS coronavirus and innate immunity. We hope that our work will contribute to studying the genetic mechanisms of disease and serve as a useful reference for COVID-19 diagnosis and treatment.
Subject areas: Virology, Biological sciences, Genetics
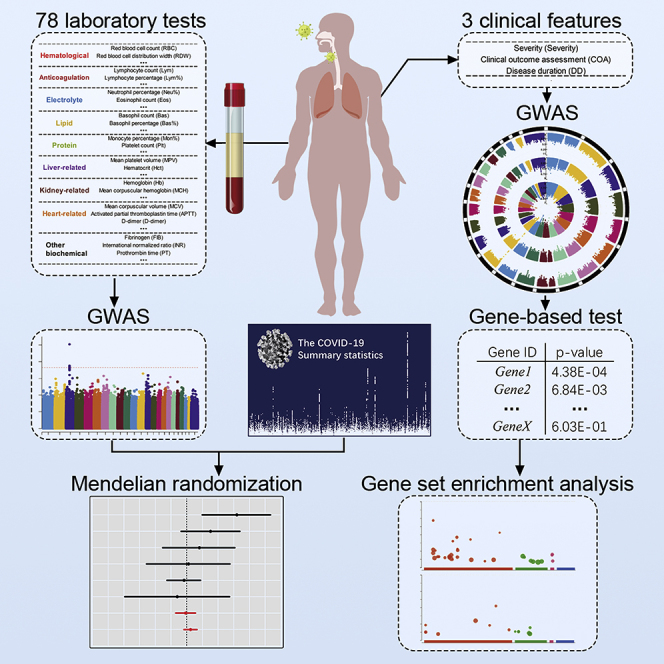
Graphical abstract
Highlights
-
•
Identification of GWAS associations for 81 phenotypes in COVID-19 patients
-
•
GSEA reveals IFNs pathways including SARS coronavirus and innate immunity
-
•
Discover causality of WBC and LDL-C on COVID-19 functioned by MHC system and ApoE gene
-
•
Insights into the host genetic background of COVID-19 in the Chinese population
Virology; Biological sciences; Genetics
Introduction
The coronavirus disease 2019 (COVID-19) is a contagious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Since the late December of 2019, COVID-19 has spread rapidly worldwide, leading to an ongoing pandemic. As of early September 2021, over 220 million confirmed cases of COVID-19 were reported to the World Health Organization, including over 4.5 million deaths. Common symptoms include fever, cough, and fatigue. Meanwhile, the symptoms could be largely variable; for example, about a third of patients do not develop noticeable symptoms; of patients who develop noticeable symptoms, 81% develop mild to moderate symptoms, whereas 14% develop severe symptoms and 5% have critical symptoms (Jordan et al., 2020). Many key factors have been reported to be associated with COVID-19 severity, such as age, sex, and comorbidities. Specifically, older people, male patients, and patients with comorbidities are more likely to be infected by SARS-CoV-2 and experience more severe symptoms. However, these risk factors cannot fully explain the clinical variability among the patients. Many recent studies turn their attention to the host genetic backgrounds and believe that the genetic factor may play an essential role in determining the host responses to SARS-CoV-2 (Wang et al., 2020b; Ellinghaus et al., 2020b; Pairo-Castineira et al., 2021; Shelton et al., 2021). By performing large-scale genome-wide association studies (GWAS) of COVID-19 clinical phenotypes, several disease-associated variants and genes were identified and summarized by the Host Genetics Initiative (HGI) (COVID-19 Host Genetics Initiative, 2020), such as the rs11385942 (SLC6A20), rs657152 (ABO), and rs2236757 (IFNAR2) (Ellinghaus et al., 2020a; Pairo-Castineira et al., 2021). However, most of the existing GWASs are based on European (EUR) populations or meta-analyses with multiple populations. It is a pity that the genomic studies based on East Asian (EAS) populations, especially Chinese (CHN) population, are relatively few. Wang et al. (2020a, 2020b, 2020c) reported the first host genetic study in the CHN population of 332 patients with COVID-19 and suggested some relatively significant genetic loci as candidate variants associated with severity status (Wang et al., 2020b). However, their study did not identify any significant genetic variants or functional pathways in explaining the genetic background of COVID-19 in the CHN population.
In this study, we identified nearly 500 patients with COVID-19 from the Wuhan Union Hospital from the first half year of 2020 and investigated the genetic mechanisms underlying COVID-19 disease. For each patient, the disease severity and clinical outcome were recorded at the admission to the hospital and in the end, respectively. A wide range of laboratory traits were measured at different time points to trace the quantity change and disease progress. Based on these phenotypes and patients' genomic data, we carried out four major analyses: (1) logistic regressions and two-sample t tests between laboratory traits and clinical assessments to uncover disease-related features; (2) GWAS analyses for all laboratory traits to identify genome-wide significant associations; (3) one-sample and two-sample Mendelian randomization (MR) analyses to test causality of laboratory traits and discover novel potential genetic pathways of candidate SNPs influencing COVID-19; and (4) gene-based analysis and gene set enrichment analysis (GSEA) based on single-SNP tests of disease severity to identify disease functional pathways. From the first two analyses, we detected many laboratory traits that were associated with disease status and several concrete genome-wide significant associations, for example, rs7412 and LDL-C. In recent years, MR has rapidly gained popularity in epidemiology and medical research, and it uses genetic variants as instrumental variables to determine whether an observational association between a risk exposure and an outcome disease is also a causality (Verduijn et al., 2010). By performing MR analysis, we uncovered the causal associations of white blood cells (WBCs) and LDL-C on the disease severity. The used instrumental variants are the MHC (major histocompatibility complex) complex and ApoE gene for WBC and LDL-C, respectively. The gene-based analysis and GSEA discovered four functional pathways: regulation of IFNA signaling, SARS coronavirus and innate immunity, type I interferon receptor binding, and overview of interferons-mediated signaling pathway. In March 2020, the National Health Commission and the National Administration of Traditional Chinese Medicine issued the COVID-19 diagnosis and treatment plan: IFN-I is one of the main antiviral drugs (National Health Commission, 2020). To the best of our knowledge, this is the first time that the interferons-related pathways are uncovered from the genetic studies of patients with COVID-19 in CHN population. These findings provide new insights in studying the genetic mechanisms of COVID-19 susceptibility and severity. We hope that our work will serve as a useful reference for the academic field and contribute to investigating the COVID-19 disease and finally stop the pandemic.
Results
Basic information of the enrolled patients
After quality control (Method details), there were 466 patients for analysis, of which 229 were men (49.1%) and 237 were women (50.9%) (Figure 1A). The age of patients ranged from 23 to 97 years, composing with 20–39 (8.5%), 40–59 (31.1%), 60–79 (51.1%), and 80–99 (9.2%) years (Figure 1A). According to the patients' severity of illness at the time of admission to the hospital, they were classified into four categories as mild (N = 6, 1.29%), moderate (N = 164, 35.19%), severe (N = 227, 48.71%), and critical (N = 69, 14.81%). The method of classifying the severity followed the criteria made by the National Health Commission of the People's Republic of China (Wu and Mcgoogan, 2020). We further broadly defined the mild group as mild and moderate patients (N = 170) and the severe group as severe and critical patients (N = 296) (Figures 1B and 1C). We then fitted a single factor linear regression model and statistically proved that age was a risk factor for severe symptoms of COVID-19 (z-score = 4.146, p value = 3.38E-05). Besides, we performed a Fisher's exact test to test the independence of patients' gender and severity and found a significant correlation (odds ratio [OR] = 1.59, p value = 0.016), revealing a higher propensity for severity in men with COVID-19. Global data also indicate higher COVID-19 fatality rates among men than women. Most countries reported that the male case fatality is more than 1.0 higher than that of female (Jin et al., 2020; Haitao et al., 2020).
Figure 1.

Basic clinical information of patients with COVID-19
(A) Pie diagrams for sex ratio and age distribution of 466 samples.
(B) Bar charts for severity category and histogram of hospitalized days. In the severity chart, the blue and green bars indicate the mild group, orange and red bars indicate the severe group.
(C) Bar chart for the counts of severity in each age range.
(D) Bar charts for the distributions of comorbidities and symptoms. For the comorbidities, HTN, CHD, BRN, TUM, SMK, CHB, COPD, HBV, ASM, TUB, and CKD indicate hypertension, coronary heart disease, brain infarction, tumor, smoking history, chronic bronchitis, chronic obstructive pulmonary disease, hepatitis B virus, asthma, tuberculosis, and chronic kidney disease, respectively. For the symptoms, COU, FTG, CHT, APP, SPU, MUS, DIA, VOM, HDC, DIZ, THR, DYS, PAL, RHI, NEW, CHP, HEMO, ABD, and HEMO indicate cough, fatigue, chest tightness, poor appetite, sputum, muscle ache, diarrhea, vomiting, headache, dizziness, sore throat, dyspnea, palpitation, rhinorrhea, night sweating, chest pain, hemoptysis, abdominal pain, and hematemesis, respectively.
More than 50% of the patients (N = 288) had at least one comorbidity prior to admission to the hospital, and the most frequent ones were hypertension (N = 180, 38.63%), diabetes (N = 95, 20.38%), and coronary heart disease (N = 63, 13.52%). The distribution of comorbidities among mild and severe patients is provided in Figure 1D. We then tested whether the presence or absence of comorbidities would affect the patients' severity by performing a Fisher's exact test. We found that having comorbidities is a risk determinant to develop severe symptoms (OR = 1.86, p = 2.09E-03). Many studies have supported that comorbidities have a critical role in poor outcomes, severity of disease, and high fatality rate of COVID-19 cases (Leung, 2020; Wang et al., 2020a; Ejaz et al., 2020). Most of the patients experienced various COVID-19 symptoms, including cough (N = 302, 64.81%), fatigue (N = 200, 42.92%), and chest tightness (N = 188, 40.34%). We also reported the distribution of symptoms among mild and severe patients (Figure 1D).
Time-series laboratory features
The laboratory measurements were grouped into 10 distinct categories (Table 1): hematological (n = 22), anticoagulation (n = 7), electrolyte (n = 7), lipid (n = 7), protein (n = 4), liver-related (n = 12), kidney-related (n = 3), heart-related (n = 8), inflammation (n = 3), and other biochemical (n = 5). We evaluated the correlation between laboratory traits and disease severity and clinical outcome, separately, with a logistic regression after adjusting for age and sex (Figures 2A, 2B, and S1).
Table 1.
Overview of the tested laboratory assessments
| Category | Trait | Abbreviation | N |
|---|---|---|---|
| Hematological | Red blood cell count | RBC | 420 |
| Red blood cell distribution width | RDW | 420 | |
| White blood cell count | WBC | 420 | |
| Lymphocyte count | Lym | 420 | |
| Lymphocyte percentage | Lym% | 420 | |
| Neutrophil count | Neu | 420 | |
| Neutrophil percentage | Neu% | 420 | |
| Eosinophil count | Eos | 420 | |
| Eosinophil percentage | Eos% | 420 | |
| Basophil count | Bas | 420 | |
| Basophil percentage | Bas% | 420 | |
| Monocyte count | Mon | 420 | |
| Monocyte percentage | Mon% | 420 | |
| Platelet count | Plt | 420 | |
| Platelet distribution width | PDW | 420 | |
| Mean platelet volume | MPV | 420 | |
| Hematocrit | Hct | 420 | |
| Plateletcrit | PCT | 420 | |
| Hemoglobin | Hb | 420 | |
| Mean corpuscular hemoglobin | MCH | 420 | |
| Mean corpuscular hemoglobin concentration | MCHC | 420 | |
| Mean corpuscular volume | MCV | 420 | |
| Anticoagulation | Activated partial thromboplastin time | APTT | 410 |
| D-dimer | D-dimer | 410 | |
| Erythrocyte sedimentation rate | ESR | 184 | |
| Fibrinogen | FIB | 410 | |
| International normalized ratio | INR | 410 | |
| Prothrombin time | PT | 410 | |
| Thrombin time | TT | 410 | |
| Electrolyte | Sodium | NA | 420 |
| Potassium | K | 420 | |
| Calcium | Ca | 420 | |
| Magnesium | Mg | 420 | |
| Chloride | Cl | 420 | |
| Phosphorus | P | 420 | |
| Anion gap | AG | 420 | |
| Lipid | Triglyceride | TG | 406 |
| Apoprotein A | apoA | 402 | |
| Apoprotein B | apoB | 402 | |
| Lipoprotein(a) | LpA | 402 | |
| Total cholesterol | TC | 406 | |
| High-density lipoprotein cholesterol | HDL-C | 406 | |
| Low-density lipoprotein cholesterol | LDL-C | 406 | |
| Protein | Total protein | TP | 420 |
| Albumin | Alb | 420 | |
| Globulin | Glb | 420 | |
| Albumin/globulin ratio | A/G | 420 | |
| Liver-related | Aspartate aminotransferase | AST | 420 |
| Alanine aminotransferase | ALT | 420 | |
| Aspartate aminotransferase/alanine aminotransferase ratio | AST/ALT | 420 | |
| Total bilirubin | Tbil | 420 | |
| Direct bilirubin | Dbil | 420 | |
| Indirect bilirubin | Ibil | 420 | |
| Acetylcholinesterase | AChE | 350 | |
| Alkaline phosphatase | AKP | 420 | |
| Lactate dehydrogenase | LDH | 420 | |
| γ-Glutamyl transferase | GGT | 420 | |
| Prealbumin | PA | 420 | |
| Total bile acids | TBA | 420 | |
| Kidney related | Blood urea nitrogen | BUN | 420 |
| Serum creatinine | Cre | 420 | |
| Uric acid | UA | 420 | |
| Heart related | α-Hydroxybutyric dehydrogenase | HBDH | 414 |
| Myoglobin | Mb | 343 | |
| High-sensitivity cardiac troponin | hscTn | 347 | |
| Homocysteine | Hcy | 350 | |
| Brain natriuretic peptide | BNP | 309 | |
| Creatine kinase | CK | 414 | |
| Creatine kinase-MB active | CK-MBa | 414 | |
| Creatine kinase-MB quality | CK-MBq | 343 | |
| Inflammation | C-reactive protein | CRP | 418 |
| Interleukin-6 | IL6 | 351 | |
| Procalcitonin | PCTN | 394 | |
| Other biochemical | Cystatin C | CysC | 420 |
| Osmotic pressure | Osm | 420 | |
| Ferritin | FER | 203 | |
| Blood glucose | BG | 420 | |
| Total carbon dioxide | TCO2 | 420 |
Figure 2.

The time-series laboratory features
(A and B) Show results of logistic regression analyses between laboratory features at five time points and disease severity and clinical outcome. The y axis denotes -log10(p value) multiplied by the effect direction (positive effect is 1 and negative effect is −1). We used red dashed line to denote y = 0. A line below (above) the red dashed line indicates a negative (positive) correlation. The blue dashed line denotes the horizontal line y = ±-log10(0.05) = ±1.3.
(C–I) Boxplots of three patients' group: mild (red), recovery (light green), and death (pink). Patients in the mild group were diagnosed as mild and recovered, patients in the recovery group were diagnosed as severe and recovered, and patients in the death group were diagnosed as severe and dead. The light blue backgrounds represent the normal range of each laboratory trait. The y axis denotes the quantities of each trait. We performed two-sample t test for each pair of groups at different significances of 0.05 (∗), 0.01 (∗∗), and 0.001 (∗∗∗).
The results based on one-time measured traits may be sensitive to the possible data entry errors and also cannot make full use of the valuable time-series features; meanwhile, regressing response variable on laboratory traits at mismatched phases may cause biased results. Therefore, we regrouped the patients as mild, recovery, and death based on their severity status and clinical outcome and provided boxplots for each group at different phases (Figures 2C–2I and S2). We then performed two-sample t tests for testing the means for every two groups at each phase. We added statistical annotations for only significant results at significance levels of 0.05 (∗), 0.01 (∗∗), and 0.001 (∗∗∗). For most of the traits, the two-sample t tests showed mostly significant difference between mild and death groups at all phases; the mean differences between recovery and death groups increased from the early to late phases; and the mild and recovery groups were gradually close to each other. Several trait categories, including liver-related, hematological features, lipids, and proteins, were significantly correlated with the disease severity.
Genome-wide association analysis of laboratory features
We first evaluated the imputation accuracy of genetic variants by two measurements: imputation score and correlation with chip array sequencing. After quality control (Method details), a total of 6,349,370 variants were selected for further analysis, and 99.6% of these variants had imputation score over 0.8 based on the reference panel as EAS population from the 1KGP. In addition, 214 patients were sequenced with high depth and high coverage. We took the overlap of variants between their chip array genotypes and imputed genotypes, and it yielded 479,823 sites. Over 98.1% patients had correlation coefficients above 0.8 across these genetic sites. With a mean sequencing depth of 17.8x, we finally tested a total of 6,185,321 autosomal variants and 164,049 X-chromosome variants for association with 78 quantitative laboratory traits in 466 patients with COVID-19. The study workflow is designed as in Figure 3. When we applied a multiple-testing correction to the number of the studied traits, five variant-trait associations were significant signals (p value < 5E-08/78 = 6.41E-10), four of which were previously identified in EUR, EAS, or both populations (Table 2). These associations include rs1801020 (F12, p value = 4.13E-16) with activated partial thromboplastin time (APTT), rs56393506 (LPA, p value = 1.97E-14) with lipoprotein-A (LpA), rs28946889 (UGT1A complex, p value = 5.08E-14) with total bilirubin levels (Tbil), and rs28946889 (UGT1A complex, p value = 1.51E-16) with indirect bilirubin levels (Ibil). The Manhattan plots and QQ-plots were drawn for APTT, LpA, and Ibil with the CMplot package in R (Yin et al., 2021) and provided in Figure 4. A novel association was rs11032789 (EHF, p value = 6.40E-10) with apoprotein A (apoA). Even though the association between rs7412 (ApoE, p value = 2.30E-08) and the LDL-C levels did not reach the study-wide significance threshold, it had been widely identified in EUR, EAS, and CHN populations. The association between rs9268517 (BTNL2, p value = 4.05E-08) with the WBC counts did not pass the threshold either. The gene BTNL2 encoded an MHC class II protein and was reported to be associated with WBC; thus we considered this identified association a worth-investigating signal. The Manhattan plots for LDL-C and WBC were provided in Figures 5A and 5B, respectively.
Figure 3.

The workflow of the main analyses performed in this study
We performed SNP-based GWAS analyses for 78 laboratory traits and employed significant SNP-trait associations for further MR analyses to investigate the causality of laboratory traits on COVID-19 disease. Based on the single SNP case-control GWAS of COVID-19 severity, we conducted gene-based and pathway analyses to uncover functional pathways.
Table 2.
The concrete associations identified from single-variant GWAS analysis
| Trait | SNP | CHR | POS | REF | ALT | Mapped/Closest gene | AF | R2 | Beta | se | p value | N | EUR | EAS | CHN |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| APTT | rs1801020 | 5 | 177409531 | A | G | F12 | 0.254 | 1.000 | −0.606 | 0.071 | 4.13E−16 | 410 | X | √ | X |
| LpA | rs56393506 | 6 | 160668275 | C | T | LPA | 0.114 | 0.998 | 0.817 | 0.103 | 1.97E−14 | 402 | √ | X | X |
| Tbil | rs28946889 | 2 | 233762816 | G | T | UGT1A Complex | 0.400 | 0.996 | −0.521 | 0.067 | 5.08E−14 | 420 | X | √ | X |
| Ibil | rs28946889 | 2 | 233762816 | G | T | UGT1A Complex | 0.400 | 0.996 | −0.585 | 0.068 | 1.51E−16 | 420 | X | √ | X |
| apoA | rs11032789 | 11 | 34624907 | T | G | EHF | 0.040 | 1.000 | 0.960 | 0.151 | 6.40E−10 | 402 | X | X | X |
| LDL-C | rs7412 | 19 | 44908822 | C | T | ApoE | 0.092 | 0.998 | −0.652 | 0.114 | 2.30E−08 | 406 | √ | √ | √ |
| WBC | rs9268517 | 6 | 32411963 | C | T | BTNL2, HLA-DRA | 0.067 | 1.000 | 0.721 | 0.129 | 4.05E−08 | 420 | X | X | X |
AF indicates the allele frequency for the effect/alternate allele; R2 indicates the imputation score based on EAS population from the 1KGP; N is the sample size used in GWAS analysis; “√” and “X” indicate the corresponding associations were previously reported and not reported in a population based on genomic studies, respectively.
Figure 4.

The Manhattan plots and QQ plots of three strong signals
(A–F) A, C, and E are Manhattan plots and B, D, F are QQ plots for APTT, LpA, and Ibil, respectively. In the Manhattan plots, the dashed horizontal line denotes 1E-6, and the solid horizontal line denotes 5E-8.
Figure 5.

The Manhattan plots of WBC and LDL-C, and the genetic mechanisms of MHC complex and ApoE influence COVID-19 by acting on WBC and LDL-C
(A and B) Manhattan plot of the GWAS single-variant test results of WBC and LDL-C. The red dashed line indicates the genome-wide significance threshold 5E-8.
(C) Genetic mechanisms of how the MHC complex and ApoE genotype influences the COVID-19 susceptibility and severity through WBC counts and cholesterol levels. For WBC, the MHC complex encoded HLA to activate T lymphocyte cells and natural killer cells against SARS-CoV-2, reflecting on increased numbers of WBC. A causal effect could be considered as a risk predictor of body experiencing acute immune response. As the response abilities were different, people experienced different symptoms. For cholesterol level, people with ApoE e4 have an increased risk of high cholesterol levels. When they are exposed to SARS-CoV-2, the accumulation of cholesterol in alveolar epithelial cells increased the density of lipid rafts, from which the virus binds to its target receptor ACE2. Therefore, a higher density of lipid rafts facilitates the bindings in cell membranes and eventually raised the susceptibility to SARS-CoV-2 infection and severity of COVID-19.
We additionally illustrated more details on the detected associations. Specifically, the rs1801020 (F12)-APTT association was previously identified in GWAS analysis from the BioBank Japan Project (BBJ), one of the largest EAS biobanks with over 160,000 subjects (Kanai et al., 2018). The gene F12 encodes coagulation factor XII that participates in the initiation of blood coagulation, and mutation of F12 will cause prolonged coagulation time and poor thromboplastin production (Zou et al., 2018). The rs56393506 (LPA)-LpA association was previously identified by GWAS in the EUR population with over 13,781 individuals (Mack et al., 2017) but not in the EAS population based on genomic studies. The gene LPA encodes a serine proteinase that constitutes a substantial portion of lipoprotein(a) (Mclean et al., 1987). The rs28946889 (UGT1A complex)-Tbil and UGT1A complex-Ibil associations were identified from the BBJ database (Kanai et al., 2018). The UGT1A complex represents a complex locus that encodes several UDP-glucuronosyltransferases. The mutation of UGT1A1 gene is the only enzyme involved in bilirubin glucuronidation in hepatocytes, which can reduce the activity of the enzyme and cause insufficient bilirubin glucuronidation, thus increasing the level of serum bilirubin. The rs7412 (ApoE)-LDL-C association was previously identified by GWAS analysis in EUR, EAS, and CHN populations. The gene ApoE is a type of apolipoprotein that participates in lipid metabolism, and particular ApoE genotype results in a higher risk of elevated LDL-C levels. The rs9268517-WBC is a novel genetic association identified by our GWAS analysis. However, its closest gene BTNL2 was previously identified to be associated with WBC by a GWAS analysis with 408,112 EUR individuals (Vuckovic et al., 2020). The gene BTNL2 encodes MHC II type I transmembrane protein, and binding to its receptor can inhibit T cell activation and cytokine production.
The one-sample and two-sample MR analyses
In the genome-wide association analysis of laboratory features section, we identified many laboratory traits that were correlated to the disease status. A natural question to ask was whether this correlation was also a causality. To answer this question, we performed one-sample and two-sample MR analyses to examine whether the traits had causal effects on COVID-19 disease. The one-sample MR results were provided in Table S1. After SNPs clumping and pruning, there was one SNP left in the analysis for each trait. The Wald test p values were all above 0.05, indicating no significant causal relationships. We then performed two-sample MR study based on summary statistics calculated from our datasets (Table S1). The results were similar to those of one-sample MR. Note that the one-sample MR analyses cannot control for confounding factors very well, and compared with quantitative GWAS analysis, the case-control studies based on only a few hundred subjects have low powers.
We instead conducted two-sample MR analyses with SNP-outcome summary statistics obtained from the large-scale HGI database. For the SNP-exposure results, we used our dataset as explore study and publicly available consortiums in EAS and EUR as replication studies. With a significance level of 0.05, we identified four causal associations in the explore design (Table S2), including WBC-B2 (p value = 0.009), WBC-C2 (p value = 0.024), LDL.C-B1 (p value = 0.034), and apoA-B1 (p value = 0.047). The valid instrumental variants for WBC, LDL-C, and apoA were rs9268517 (BTNL2, HLA-DRA), rs7412 (ApoE), and rs11032789 (EHF), respectively. A positive causal effect of WBC on disease susceptibility (HGI B2&C2) might represent an acute stage of body immune response, indicating that WBC could be a risk predictor for COVID-19. For LDL-C and apoA, causal effects were observed on COVID-19 severity (B1). After controlling the FDR with 12 multiple testing in terms of 12 HGI phenotypes, the smallest q-value (WBC-B2) is 0.1 and other p values were greater than 0.1 showing no test-wide significant causalities. Despite this, we thought these findings suggested potential genetic mechanisms of COVID-19 through laboratory traits. Therefore, we performed replication studies with SNP-exposure associations from publicly available large-scale consortiums in the EAS and EUR populations.
We then performed the first replication design where SNP-exposure results were from EAS populations and the HGI database as SNP-outcome association. We selected EAS studies with at least one variant that was mapped to the functional genes from the explore design and first performed MR analysis based on only these variants, then all trait-associated variants in the study. To investigate the causal effects of WBC counts, we downloaded a large EAS study with sample size of 151,807 (Chen et al., 2020b). First, we tested the causality of WBC counts based on SNPs mapped to the MHC family, gene HLA-C (rs2524084, p value = 1.260E-53). The results were provided in Table S3 showing four causal associations on COVID-19 susceptibility (HGI C2). These four causal effects were still significant after controlling the FDR at ≤5% and suggested a candidate pathway that the MHC family affects disease susceptibility by regulating WBC counts. Second, we did the MR analysis based on all WBC-associated SNPs in the consortium. After SNPs clumping and harmonization with the HGI database, a total of 51 SNPs were used in MR analysis based on different causal effect estimation methods (Table S4). By using the inverse variance weighted (IVW) method, WBC had two causal effects on COVID-19 susceptibility (HGI phenotype B2). The scatter plot of SNP-WBC effects versus SNP-B2 effects, the forest plot of MR causal effect for each SNP, the funnel plot from single SNP analyses, and the leave-one-SNP-out plot from leave-one-out analysis were provided in Figure S3. The MR-Egger p value for testing heterogeneity is 0.365 > 0.05 (Q-statistic = 48.69), showing no heterogeneity. The MR-PRESSO p value for testing horizontal pleiotropy is 0.413 > 0.05, meaning no pleiotropy. The results of no heterogeneity and no pleiotropy enhanced the validity of MR results.
To investigate the causal effects of LDL-C, we downloaded the significant summary statistics for LDL-C from the large-scale EAS database BBJ with a sample size of 72,866 (Kanai et al., 2018). First, we tested on SNPs mapped to the ApoE gene (rs769446, p value = 2.977E-322). Based on the Wald ratio method, LDL-C had causal effects on HGI phenotypes B2&C2 (Table S5). However, the effect directions were not consistently positive or negative, implying complex genetic mechanisms of how the SNPs affected COVID-19 through LDL-C. The q-values of these associations are also less than 0.1 with two less than 0.05. Second, we did the MR analysis based on all the LDL-C associated SNPs. After SNP clumping and harmonization with the HGI database, 12 SNPs were used in MR with different causal effect estimation methods (Table S6). The IVW method suggested two causal effects on COVID-19 susceptibility (HGI C2). The scatter plot of SNP-LDL.C effects versus SNP-C2 effects, the forest plot, the funnel plot, and the leave-one-out plot were provided in Figure S4. The MR-Egger p value for testing heterogeneity is 0.464 > 0.05 (Q-statistic = 8.71) showing no heterogeneity. The MR-PRESSOR p value for testing pleiotropy is 0.539 > 0.05, meaning no direct effect of the analyzed SNPs on outcome severity.
We then performed replication studies where the SNP-exposure results were obtained from the EUR population in UKBB dataset, and the SNP-outcome association were from the HGI database. First, we tested the causality of WBC and LDL-C based on SNPs mapped to MHC family and ApoE, respectively, and then based on all trait-related SNPs. For WBC, the summary statistics were reported by a study with sample size 350,470 (Liam Abbott et al., 2018). By using SNPs mapped to HLA-DPA1/HLA-DPB1 (rs3135024, p value = 3.66E-10), WBC had causal effects on four HGI susceptibility phenotypes B2 (Table S7), matching with the genetic mechanisms in EAS population. After SNPs clumping and harmonization, 235 WBC-related variants were used but no causal associations were significant based on the IVW method (Table S8). The LDL-C summary results were obtained from a study with 431,167 subjects (Price et al., 2008). There was one SNP mapped to gene ApoE (rs1081105, p value = 1.00E-200) and the Wald ratio test did not identify causal effects (Table S9). With all 243 LDL.C-related SNPs after clumping and harmonization, one causal association (HGI B2) was significant (p value = 0.026) (Table S10). From these replication studies in the EUR population, we identified similar functional pathways that the MHC family influenced COVID-19 disease by controlling WBC counts. We did not replicate the effects of ApoE on disease severity through LDL-C, but the MR analysis based on all SNPs also suggested causality of LDL-C on COVID-19.
Finally, we tried to explain the potential genetic mechanisms of how the MHC family and ApoE influenced COVID-19 susceptibility and severity by associating with the WBC counts and LDL-C levels, respectively. From the explore and replication studies, we observed causal effects of WBC based on instrumental SNPs mapped to the MHC complex. In humans, the MHC complex encoded the human leukocyte antigen (HLA), a group of related proteins to activate T lymphocyte cells and natural killer cells. Previous studies identified the relationship between HLA and susceptibility to COVID-19 (Nguyen et al., 2020). The SARS-CoV-2 was found to restrain antigen presentation and suppress immune reaction by regulating the expression of MHC class in COVID-19 cases (Paces et al., 2020). A causal effect could be considered as a risk predictor of body experiencing acute immune response and the number of WBC rapidly increased against the virus. Previous studies showed that, as the disease progresses, mHLA-DR levels and lymphocyte cell counts varied in patients with COVID-19 (Benlyamani et al., 2020; Zheng et al., 2020). For the genetic pathways of how ApoE influenced disease status through LDL-C, we raised a possible mechanism observed in our study and also reported by other studies. Patients who carried ApoE ε4/ε4 genotype tend to be infected by SARS-CoV-2 and experience severe symptoms from COVID-19 (Goldstein et al., 2020; Kuo et al., 2020). For example, a study concluded that, among older people, patients with ApoE ε4/ε4 genotype had a much higher risk of developing severe symptoms compared with those with ApoE ε3/ε3 (OR = 2.31, p value = 1.19E-06) (Kuo et al., 2020). By investigating the ApoE genotypes in all 466 patients with COVID-19, we found 7 patients who carried ApoE ε4/ε4 in total, of which 5 patients were severe. Specifically, people with ApoE ε4/ε4 have an increased risk of high cholesterol levels. When they are exposed to SARS-CoV-2, the accumulation of cholesterol in alveolar epithelial cells increased the density of lipid rafts, from which the virus binds to its target receptor ACE2. Therefore, a higher density of lipid rafts facilitates the bindings in cell membranes and eventually raised the susceptibility to SARS-CoV-2 infection and severity of COVID-19 (Goldstein et al., 2020; Wang et al., 2020c; Gkouskou et al., 2021). The genetic mechanism is illustrated in Figure 5C.
Reverse MR analysis
In the one-sample and two-sample MR analyses section, we identified two potential genetic pathways that the MHC complex and ApoE gene affected the COVID-19 vulnerability and severity by mediating WBC counts and cholesterol levels, respectively. We were also interested in whether there existed functional mechanism that some instrumental SNPs could alter laboratory traits through disease severity. In this section, we did not focus on one COVID-19-related gene but used all associated SNPs. First, we used the HGI database as exposure variable and laboratory traits as outcome from our dataset. The valid instrumental variants corresponding to each exposure phenotype were provided, and the MR results did not suggest potential causal effects of COVID-19 on WBC (Table S11), indicating their phenotypic correlations were not causalities triggered by genetic SNPs. For LDL-C, we did not obtain causal effects of COVID-19 based on its associated SNPs (Table S12). Then, we repeated the examination by using UKBB dataset as outcome variable and had similar results (Table S13 for WBC; Table S14 for LDL-C). These results implied that the COVID-19-related variants might not be potential genetic factors that caused the correlation between disease severity and laboratory traits.
Gene-based analysis and GSEA of clinical measurements
We analyzed three clinical features, including severity, clinical outcome, and disease duration of first performing single-variant GWAS. The Circular-Manhattan plot and QQ-plot were provided in Figures 6A and 6B. No genetic variants reach the genome-wide significance threshold (p value < 5E-08) owing to the current small sample size (N = 466), and thus the effect sizes of single variants tend to be small. To aggregate the single-variant effects, we further performed VEGAS gene-based test (Mishra and Macgregor, 2015) and g:GOST GSEA analysis (Reimand et al., 2007) for clinical severity. With a window size of 50 kb, 25,345 genes were mapped, and the average number of SNPs on each gene is 251. For the window of 10 kb, 24,640 genes were mapped, and the average number of SNPs is 119. Then, we selected only genes with a p value less than 0.05. A number of 1,170 genes passed the significance threshold for window size 50 kb and 1,099 genes for 10 kb. We obtained an intersection of 705 genes from the two sets of significant genes for further GSEA.
Figure 6.

The genome-wide association studies of COVID-19 severity: a case-control study
(A) Circular Manhattan plots for clinical diagnoses. The inner circle is for severity status (mild versus severe), the middle circle is for clinical outcome assessments (survival versus death), and the outer circle is for disease duration (hospitalized days).
(B) The QQ plot for three clinical diagnoses.
(C) Bubble plot of GSEA based on the single-variant and gene-based studies on severity status. The red dashed line is the threshold of 0.05.
The GSEA results identified four significant pathways with p value less than 0.05 (Figure 6C). These pathways include regulation of IFNA signaling (REAC:R-HSA-912694, p value = 6.42E-04), SARS coronavirus and innate immunity (WP:WP4912, p value = 2.54E-03), overview of interferons-mediated signaling pathways (WP:WP4558, p value = 8.64E-03), and type I interferon receptor binding (GO:0005132, p value = 4.38E-02). All the four pathways belong to the IFNA family, a member of the alpha interferon gene cluster that encodes the type I interferon (IFN) family produced in response to viral infection. The IFNA family is a key part of the innate immune response with potent antiviral, antiproliferative, and immunomodulatory properties. Insufficient virus-induced type I IFN production is characteristic of SARS-CoV-2 infection since SARS-CoV-2 suppresses the IFN response by interacting with essential IFN signaling pathways (Lin and Shen, 2020). Blunted amounts of IFNs have been detected in the peripheral blood or lungs of patients with severe COVID-19 (Acharya et al., 2020). We note that since VEGAS is based on a simulation procedure to calculate the gene-based p values, its results may vary slightly every time we rerun the analysis. We examined the effect of running the analysis multiple times and found that the results of gene-based association and the subsequent GSEA study were robust and reliable. We also investigated the effect of varying the window sizes around each gene and found that the results are robust to the choice of window sizes. In summary, based on the single variant associations, VEGAS gene-based tests, and GSEA analysis, we identified four IFNs pathways whose imbalanced responses may cause the pathology of COVID-19 based on genomic studies in the CHN population.
As we mentioned in the Introduction section, several genetic loci have been identified to be associated with the critical illness in COVID-19 (Pairo-Castineira et al., 2021). We summarized eight genome-wide significant associations in Table 3, including the lead SNP in each locus, the p values of these SNPs for testing severity status in our dataset, and their allele frequencies in EAS and EUR populations from 1KGP and in our case subjects. Among these eight SNPs, one SNP (rs74956615, 19:10427721) does not exist in our imputed genotype and two SNPs (rs73064425, 3:45901089; rs3131294, 6:32180146) were removed from analysis owing to low allele frequencies. For four of the other five SNPs, their allele frequencies in EUR populations are much higher than in the EAS population (average difference is 0.16), showing that these significant SNPs are more prominent in the EUR population than in the EAS population. The eighth SNP, rs9380142 (6:29798794), is mapped to gene HLA-G. The HLA-G gene belongs to the MHC region that plays a critical role in immune responses and regulations. We believe that a large-scale COVID-19 case-control study in the CHN population can potentially uncover the MHC region.
Table 3.
The summary of eight reported COVID-19 illness-associated loci
| SNP | Chr:BP | Locus | p value | A1 | 1KGP EAS Freq | 1KGP EUR Freq | A1 Freq | Mild Freq | Severe Freq |
|---|---|---|---|---|---|---|---|---|---|
| rs73064425 | 3:45901089 | LZTFL1 | / | T | 0.005 | 0.0795 | 0.005365 | 0.005882 | 0.005068 |
| rs9380142 | 6:29798794 | HLA-G | 0.846804 | G | 0.3492 | 0.3439 | 0.43133 | 0.4206 | 0.4375 |
| rs143334143 | 6:31121426 | CCHCR1 | 0.966751 | A | 0.0347 | 0.1123 | 0.059013 | 0.06176 | 0.05743 |
| rs3131294 | 6:32180146 | NOTCH4 | / | A | 0.0079 | 0.1133 | 0.006438 | 0.008824 | 0.005068 |
| rs10735079 | 12:113380008 | OAS1–OAS3 | 0.433044 | G | 0.252 | 0.3638 | 0.233906 | 0.2265 | 0.2382 |
| rs2109069 | 19:4719443 | DPP9 | 0.788196 | A | 0.1399 | 0.3211 | 0.143777 | 0.1382 | 0.147 |
| rs74956615 | 19:10427721 | TYK2 | / | / | / | / | / | / | / |
| rs2236757 | 21:34624917 | IFNAR2 | 0.4025 | G | 0.4345 | 0.7058 | 0.381974 | 0.3618 | 0.3936 |
A1 denotes the effect/alternate allele. 1KGP EAS Freq indicates the allele frequency in the EAS population from the 1KGP. Mild Freq indicates the allele frequency in patients from mild and moderate (grouped into mild) groups. Severe Freq indicates the allele frequency in patients from severe and critical (grouped into severe) groups.
Discussion
The SARS-CoV-2 virus is a new coronavirus that causes the ongoing COVID-19 pandemic. Patients with COVID-19 experience largely various clinical and laboratory assessments, from no symptoms to exhausted respiratory system, and even death. Many clinical and experimental studies have concluded that several significant determinants are responsible for the disease variability, including old age, male gender, and having comorbidities at admission to the hospital. However, these factors still cannot fully account for the diverse symptoms among patients. Recent studies have turned more attention to the host genetic background. The HGI database has reported many candidate loci by performing large-scale GWAS analysis with thousands of cases and up to millions of controls.
In this study, we analyzed 466 patients with COVID-19 hospitalized in the Wuhan Union Hospital. A broad range of clinical information, such as age, gender, comorbidities, and laboratory tests were collected for each patient. We performed GWAS analysis for the numerous laboratory features and discovered seven concrete genome-wide variant-trait associations, five of which were previously uncovered by large-scale genomic studies. Our results were either the first replication or the first identification study in the CHN population based on the GWAS. With these well-established genetic associations, we conducted MR analyses to uncover important laboratory traits that have causal effects on the susceptibility and severity of COVID-19 disease. Our analyses highlighted two fundamental pathways. One is the WBC counts with functional gene MHC complex, and the other is the cholesterol levels with functional gene ApoE. We further researched and explained the genetic mechanisms of how genes ApoE and MHC family influenced the disease status by acting on cholesterol levels and WBC counts.
We additionally carried out the gene-based analysis and GSEA based on the single-SNP GWAS results of severity case-control study. Interestingly, we, for the first time, revealed four interferons-related functional pathways based on host genetic studies in the CHN population, including regulation of IFNA signaling, SARS coronavirus and innate immunity, overview of interferons-mediated signaling pathway, and type I interferon receptor binding. Most of these studies were based on bulk RNA-seq, scRNA-seq, or experimental designs, whereas our analysis is built on genomic data, supporting this solid conclusion from a new perspective.
Limitations of the study
Despite the many compelling discoveries of our work, there are still a few limitations. First, the single-variant GWAS analysis of severity status did not identify any genome-wide signals due to the current sample size (N = 466) and thus small genetic effect sizes. We believe that large-scale case-control studies have potentials to uncover genome-wide significant variants. Second, even though we identified two potential genetic mechanisms of how genomic effects influenced COVID-19 through laboratory traits, there was merely one valid instrumental variant after SNPs clumping and pruning, while the tested traits were often known as polygenic. Larger studies that identified multiple independent trait-related SNPs on the functional gene (i.e., MHC complex, ApoE) should be applied in the MR analysis. Especially, SNPs with the same direction of genetic effects on traits were favored to produce appropriate causal effects. Third, we raised some possible explanations of how the genetic mechanisms worked on COVID-19 by mediating traits; our explanations were not enough to elucidate the complex genetic background, deeper investigations and more reasonable interpretations are still needed to uncover the complicated genetic impacts in COVID-19 disease susceptibility and severity. Fourth, many genetic variants influenced complex diseases by modulating gene expression and thus altering the abundance and structure of proteins; these variants were also called eQTL (expression quantitative trait loci) and pQTL (proteomic QTL), respectively (Gusev et al., 2016; Chick et al., 2016). In recent years, the transcriptome-wide association studies (TWAS) and proteome-wide association studies (PWAS) were developed and used to identify candidate genes whose regulated gene expressions and proteins were associated with complex diseases (Gusev et al., 2016; Gamazon et al., 2015; Hu et al., 2019; Yuan et al., 2020; Wingo et al., 2021). The TWAS/PWAS analysis was essentially an MR analysis with the exposure variable being gene expression/proteins. As the gene expression/protein levels of patients with COVID-19 in the CHN population were available, TWAS/PWAS analyses were worth investigating to uncover functional genes that influence COVID-19 by regulating gene expression/proteins.
STAR★Methods
Key resources table
Resource availability
Lead contact
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Subjects
All the subjects enrolled in this study were recruited by the Wuhan Union Hospital (Union hospital of Tongji Medical College of Huazhong University of Science and Technology). These subjects had been diagnosed with COVID-19 respiratory disease and hospitalized in Wuhan Union Hospital between January 15 and April 4, 2020. Written informed consent was obtained from all participants, as approved by the Medical Ethics Committee of Union Hospital, Tongji Medical College, Huazhong University of Science and Technology.
Phenotype
There are two types of phenotypes: laboratory tests and clinical diagnoses. A broad range of laboratory tests were measured at different time points during hospitalization. The clinical diagnoses include three traits: severity status (mild, moderate, severe, and critical) collected at the time of admission to the hospital, clinical outcome assessments (survival versus death), and disease duration (i.e., hospitalized days) at the time of eventual treatment and prevention of disease.
Time-series laboratory features
The patients had different dates for in- and out-hospital, it was difficult to present all the dates. We instead divided the hospitalization days into several phases for each patient. We only kept patients with more than two non-missing records and divided these records into two equal-length groups named early and late phases. At each phase, we took the average of all available values for each feature and treated the mean as the patient's phase-wide measurement. We also defined the first and last non-missing measurements as an initial and end record. In addition, we took the average of all non-missing records as the overall average. By doing so, for each patient, we obtained trait values at five phases: initial, early, average, late, and end. We first investigated the correlation between laboratory traits and disease status and outcome by building logistic regressions between each laboratory feature with the COVID-19 severity and clinical outcome, respectively, with adjustment of age and sex. However, note that, the laboratory traits were measured multiple times, while the disease severity and clinical outcome were only recorded once during the patients' hospitalization period. There might be some bias when regressing the response variable measured at one timepoint on the laboratory traits measured several times. Thus, we reclassified the patients into three groups based on both the disease severity and clinical outcome as: mild + survival (mild), severe + survival (recovery), and severe + death (death). At each phase, we performed two-sample t-tests to test whether population means were significantly different between each group pair.
Method details
Genotyping and imputation
We sequenced samples with the DNBSEQ platform (MGI, Shenzhen, China) to generate 100bp paired-end reads. The mean sequencing depth was 17.8×. We excluded samples with (i) sample call rate <0.99, (ii) closely related individuals identified by identity-by-descent (IBD >0.1) calculated in KING (Manichaikul et al., 2010), and (iii) outliers identified by principal component analysis based on three-sigma rules. We then applied standard quality control criteria for genetic variants by removing those with (i) SNP call rate <0.99, (ii) minor allele frequency (MAF) < 0.01, and (iii) Hardy-Weinberg equilibrium p value < 1E-06. Based on the VCF files after VQSR with biallelic variants, imputation was performed with Beagle v4.0 (Browning and Browning, 2007), taking GL as input in EAS population of 1,000 Genomes Project (1KGP) as reference panel.
Quantification and statistical analysis
Genome-wide association studies
We used PLINK v2.0 (Chang et al., 2015) to perform single-variant GWAS analyses using a linear regression model for the quantitative laboratory features under the assumption of additive allelic effects of the SNP dosage. For each trait, we adjusted for age, sex, and the top six principal components (PCs) of genetic ancestry and normalized the resulting residuals by applying a Z-score normal transformation. The number of PCs was chosen by using EIGENSTRAT software (Price et al., 2006; Patterson et al., 2006). We set a genome-wide significance threshold at the level of 5E-08 and a study-wide significance threshold at the level of 6.41E-10 (=5E-08/78) by applying Bonferroni correction based on the number of laboratory traits (n = 78).
The host genetics initiative datasets
We used the HGI round 5 GWAS meta-analysis results. There are four types of phenotypes: very severe respiratory confirmed covid versus population (A2), hospitalized covid versus not hospitalized covid (B1), hospitalized covid versus population (B2), and covid versus population (C2). We selected B2, C2, and B1 phenotypes to study the susceptibility and critical illness of COVID-19. For each type of phenotype, there are four different sets of populations: all populations but not 23andme, all populations but not UKBB, all EURs, and all EURs but not UKBB.
One-sample and two-sample MR analyses
In the Time-series laboratory features section, we tested the phenotypic correlation between laboratory traits and COVID-19 disease status, then we examined whether this correlation is also a causality. We performed MR analyses to examine causal effects between them and uncover genetic variants that determined disease status by acting on the laboratory traits. Note that the causal interpretation of the exposure variable on the disease outcome requires three standard assumptions to hold: (i) relevance: instrumental variants are highly associated with the exposure; (ii) no unmeasured confounders: variants are not associated with any confounding factors that may be associated with both exposure and outcome; and (iii) exclusion restriction: variants influence the outcome only through the path of exposure, i.e., no horizontal pleiotropic effects of variants on the outcome. We performed both one-sample and two-sample MR analyses. In the one-sample MR analysis, we used the individual-level genotypic data, laboratory traits, and clinical severity from our own datasets. Specifically, we used the R package AER (Kleiber and Zeileis, 2008) to conduct two-stage least squares (2SLS) method (Burgess et al., 2017). In 2SLS, the instrumental variables were used to obtain the predicted exposure with least squares estimates of effect sizes and the binary disease status was regressed on the predicted values to estimate the causal effects based on the Wald test (Ludwig Fahrmeir et al., 2013).
The two-sample MR analyses require summary statistics from SNP-exposure and SNP-outcome association. We used the genetic variants that were strongly associated with laboratory traits (p value < 5E-08) to ensure the relevance assumption. We also removed the genome-wide significant SNPs based on the SNP-outcome summary results to ensure the exclusion restriction assumption. We first performed explore studies to discover potential causal relationships, and then replication studies to validate our findings. The explore studies included: (1) the SNP-exposure and SNP-outcome studies were based on our datasets, and (2) the SNP-exposure study was based on our datasets and the SNP-outcome was based on the HGI database. The replication studies included: (1) the SNP-exposure was from public large-scale consortium in EAS and the SNP-outcome was from the HGI database, and (2) the SNP-exposure study was from UK Biobank (UKBB) datasets and the SNP-outcome was from HGI. Yet, the EAS-EUR designs involved two different populations; we harmonized the effect of each SNP on the exposure and outcome so that variants share the same allele pair and have matched effect estimates and allele frequencies between datasets, which ensures reasonable results with different populations. Specifically, we used R (version 4.0.2) with the TwoSampleMR package (Hemani et al., 2017, 2018) and set the significance threshold at the level of 0.05. The standard two-sample MR methods require independent instrumental variables, thus we performed clumping and pruning based on their linkage disequilibrium in the 1KGP reference panel with appropriate sub-populations. Specifically, pairs of SNPs in a window of 10,000 kb with squared correlation greater than 0.001 are noted and the SNP with the larger p value is pruned. We used the function clump_data in the TwoSampleMR package to perform this procedure. When there is only one valid instrumental variant, we used Wald ratio method to estimate the causal effects. The most-commonly used method is inverse variance weighted (IVW) (Burgess et al., 2016). Other methods included weighted median (Bowden et al., 2016), simple mode, weighted mode (Hartwig et al., 2017), and MR-Egger (Bowden et al., 2015). For causal effects identified by IVW method, we also tested the heterogeneity and horizontal pleiotropy effects to examine whether the MR results were valid. In details, we used MR-Egger method to test heterogeneity (Bowden et al., 2015) and MR-PRESSO global test for horizontal pleiotropy (Verbanck et al., 2018) by running the functions mr_heterogeneity and run_mr_presso in TwoSampleMR, respectively. We also calculated the adjusted p values (i.e., q-values) by controlling the false discovery rate (FDR) for each trait. In details, we used R (version 4.0.2) with the p.adjust function from stats package (Benjamini and Yekutieli, 2001) to obtain the q-values and declared more stringently significant associations based on an FDR of 0.1 and 0.05.
Reverse Mendelian randomization
The reverse MR is also an MR analysis by switching the exposure variable (i.e., laboratory traits) and outcome variable (i.e., COVID-19 severity), while the instrumental variables are related to the disease outcome. We performed the inverse MR analyses based on SNP-exposure summary statistics from the HGI database and the SNP-WBC/LDL-C results from our datasets and the UKBB dataset. The causal effect estimation methods included IVW, weighted median, weighted mode, simple mode, and MR-Egger.
Gene-based analysis and GSEA
For the patients' clinical features, we performed GWAS single-variant analysis in PLINK 2.0 based on a logistic (for severity status and clinical outcome assessments) or linear (disease duration) regression model. For the severity status, we further conducted gene-based tests and GSEA to aggregate the effects of multiple genetic variants from the single tests. The gene-based test is VEGAS method (Mishra and Macgregor, 2015) that combines the p values of the single variants. A list of selected genes from the gene-based results was taken for further GSEA analysis to uncover functional pathways based on the g:GOST toolset (Reimand et al., 2007). We used six existing gene set databases, including GO (gene ontology) molecular function (Ashburner et al., 2000), GO cellular component (Ashburner et al., 2000), GO biological process (Ashburner et al., 2000), KEGG (Kyoto encyclopedia of genes and genomes) (Kanehisa and Goto, 2000), Reactome (Joshi-Tope et al., 2005), and WikiPathways (Pico et al., 2008).
Acknowledgments
This study was supported by National Natural Science Foundation of China (32000398), Natural Science Foundation of Guangdong Province, China (2017A030306026), Guangdong-Hong Kong Joint Laboratory on Immunological and Genetic Kidney Diseases (2019B121205005), The Innovative Major Emergency Project Funding against the COVID-19 in Hubei Province (No. 2020FCA041), the Innovative Major Emergency Project Funding against the COVID-19, HUST (No. 2020kfyXGYJ039), Guangdong Provincial Key Laboratory of Genome Read and Write (No. 2017B030301011), and China National GeneBank. We would like to acknowledge Juehan Liu's and Daniel Wu's contribution on literature search and grammatical errors correction in this study.
Author contributions
F.C. and X.J. conceived the study, designed the research program, and managed the project. F.Z., Y.J., Zhen L., L.T., N.X., D.H., X.Z., R.X., Y.C., and W.L. collected the samples. Y.L. finished the laboratory processing and data acquisition. Zilong L. and P.L. preprocessed the data and finished the quality control. H.Z., L.Li., and J.Z. performed the statistical analyses. Y.S., S.L., and X.S. advised on statistical methods. H.Z, F.Z, Linxuan L., Y.J., Y.L., Zhen L., and J.Z. wrote the manuscript. All authors participated in revising the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: October 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103186.
Contributor Information
Xin Jin, Email: jinxin@genomics.cn.
Fanjun Cheng, Email: chengfanjun001@sina.com.
Supplemental information
6
Data and code availability
The data that support the findings of this study have been deposited into CNGB Sequence Archive (CNSA) (Guo et al., 2020) of China National GeneBank DataBase (CNGBdb) (Chen et al., 2020a) with accession number CNSA: CNP0001876. A summary of analysis software and tools were provided in key resources table. Additional Supplemental Tables are available from Mendeley Data at https://doi.org/10.17632/3wr9mgm2b6.1. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Acharya D., Liu G., Gack M.U. Dysregulation of type I interferon responses in COVID-19. Nat. Rev. Immunol. 2020;20:397–398. doi: 10.1038/s41577-020-0346-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001;29:1165. [Google Scholar]
- Benlyamani I., Venet F., Coudereau R., Gossez M., Monneret G. Monocyte HLA-DR measurement by flow cytometry in COVID-19 patients: an interim review. Cytometry A. 2020;97:1217–1221. doi: 10.1002/cyto.a.24249. [DOI] [PubMed] [Google Scholar]
- Bowden J., Davey Smith G., Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015;44:512–525. doi: 10.1093/ije/dyv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden J., Davey Smith G., Haycock P.C., Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 2016;40:304–314. doi: 10.1002/gepi.21965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning S.R., Browning B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess S., Dudbridge F., Thompson S.G. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat. Med. 2016;35:1880–1906. doi: 10.1002/sim.6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess S., Small D.S., Thompson S.G. A review of instrumental variable estimators for Mendelian randomization. Stat. Methods Med. Res. 2017;26:2333–2355. doi: 10.1177/0962280215597579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.Z., You L.J., Yang F., Wang L.N., Guo X.Q., Gao F., Hua C., Tan C., Fang L., Shan R.Q. CNGBdb: China national GeneBank database. Yi Chuan. 2020;42:799–809. doi: 10.16288/j.yczz.20-080. [DOI] [PubMed] [Google Scholar]
- Chen M.H., Raffield L.M., Mousas A., Sakaue S., Huffman J.E., Moscati A., Trivedi B., Jiang T., Akbari P., Vuckovic D. Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell. 2020;182:1198–1213.e14. doi: 10.1016/j.cell.2020.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chick J.M., Munger S.C., Simecek P., Huttlin E.L., Choi K., Gatti D.M., Raghupathy N., Svenson K.L., Churchill G.A., Gygi S.P. Defining the consequences of genetic variation on a proteome-wide scale. Nature. 2016;534:500–505. doi: 10.1038/nature18270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COVID-19 Host Genetics Initiative The COVID-19 host genetics initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 2020;28:715–718. doi: 10.1038/s41431-020-0636-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejaz H., Alsrhani A., Zafar A., Javed H., Junaid K., Abdalla A.E., Abosalif K.O.A., Ahmed Z., Younas S. COVID-19 and comorbidities: deleterious impact on infected patients. J. Infect Public Health. 2020;13:1833–1839. doi: 10.1016/j.jiph.2020.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinghaus D., Degenhardt F., Bujanda L., Buti M., Albillos A., Invernizzi P., Fernández J., Prati D., Baselli G., Asselta R. Genomewide association study of severe Covid-19 with respiratory failure. N. Engl. J. Med. 2020;383:1522–1534. doi: 10.1056/NEJMoa2020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinghaus D., Degenhardt F., Bujanda L., Buti M., Albillos A., Invernizzi P., Fernández J., Prati D., Baselli G., Asselta R. The ABO blood group locus and a chromosome 3 gene cluster associate with SARS-CoV-2 respiratory failure in an Italian-Spanish genome-wide association analysis. medRxiv. 2020 doi: 10.1101/2020.05.31.20114991. [DOI] [Google Scholar]
- Gamazon E.R., Wheeler H.E., Shah K.P., Mozaffari S.V., Aquino-Michaels K., Carroll R.J., Eyler A.E., Denny J.C., Nicolae D.L., Cox N.J., Im H.K. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015;47:1091–1098. doi: 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkouskou K., Vasilogiannakopoulou T., Andreakos E., Davanos N., Gazouli M., Sanoudou D., Eliopoulos A.G. COVID-19 enters the expanding network of apolipoprotein E4-related pathologies. Redox Biol. 2021;41:101938. doi: 10.1016/j.redox.2021.101938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein M.R., Poland G.A., Graeber A.C.W. Does apolipoprotein E genotype predict COVID-19 severity? Qjm. 2020;113:529–530. doi: 10.1093/qjmed/hcaa142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X., Chen F., Gao F., Li L., Liu K., You L., Hua C., Yang F., Liu W., Peng C. Database; 2020. CNSA: A Data Repository for Archiving Omics Data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A., Ko A., Shi H., Bhatia G., Chung W., Penninx B.W., Jansen R., de Geus E.J., Boomsma D.I., Wright F.A. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016;48:245–252. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haitao T., Vermunt J.V., Abeykoon J., Ghamrawi R., Gunaratne M., Jayachandran M., Narang K., Parashuram S., Suvakov S., Garovic V.D. COVID-19 and sex differences: mechanisms and biomarkers. Mayo Clin. Proc. 2020;95:2189–2203. doi: 10.1016/j.mayocp.2020.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig F.P., Davey Smith G., Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 2017;46:1985–1998. doi: 10.1093/ije/dyx102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemani G., Tilling K., Davey Smith G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. Plos Genet. 2017;13:e1007081. doi: 10.1371/journal.pgen.1007081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemani G., Zheng J., Elsworth B., Wade K.H., Haberland V., Baird D., Laurin C., Burgess S., Bowden J., Langdon R. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408. doi: 10.7554/eLife.34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Li M., Lu Q., Weng H., Wang J., Zekavat S.M., Yu Z., Li B., Gu J., Muchnik S. A statistical framework for cross-tissue transcriptome-wide association analysis. Nat. Genet. 2019;51:568–576. doi: 10.1038/s41588-019-0345-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J.M., Bai P., He W., Wu F., Liu X.F., Han D.M., Liu S., Yang J.K. Gender differences in patients with COVID-19: focus on severity and mortality. Front Public Health. 2020;8:152. doi: 10.3389/fpubh.2020.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan R.E., Adab P., Cheng K.K. Covid-19: risk factors for severe disease and death. Bmj. 2020;368:m1198. doi: 10.1136/bmj.m1198. [DOI] [PubMed] [Google Scholar]
- Joshi-Tope G., Gillespie M., Vastrik I., D'eustachio P., Schmidt E., de Bono B., Jassal B., Gopinath G.R., Wu G.R., Matthews L. Reactome: a knowledgebase of biological pathways. Nucl. Acids Res. 2005;33:D428–D432. doi: 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai M., Akiyama M., Takahashi A., Matoba N., Momozawa Y., Ikeda M., Iwata N., Ikegawa S., Hirata M., Matsuda K. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet. 2018;50:390–400. doi: 10.1038/s41588-018-0047-6. [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiber C., Zeileis A. New York; 2008. Applied Econometrics with R. Springer-Verlag. ISBN 978-0-387-77316-2, https://CRAN.R-project.org/package=AER. [Google Scholar]
- Kuo C.L., Pilling L.C., Atkins J.L., Masoli J.A.H., Delgado J., Kuchel G.A., Melzer D. APOE e4 genotype predicts severe COVID-19 in the UK Biobank community cohort. J. Gerontol. A. Biol. Sci. Med. Sci. 2020;75:2231–2232. doi: 10.1093/gerona/glaa131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung C. Risk factors for predicting mortality in elderly patients with COVID-19: a review of clinical data in China. Mech. Ageing Dev. 2020;188:111255. doi: 10.1016/j.mad.2020.111255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liam Abbott S.B., Claire C., Andrea G., Daniel H., Duncan P., Ben N., Raymond W., Caitlin C., The Hail Team . 2018. UK Biobank - Neale Lab [Online]http://www.nealelab.is/uk-biobank/ [Google Scholar]
- Lin F., Shen K. Type I interferon: from innate response to treatment for COVID-19. Pediatr. Investig. 2020;4:275–280. doi: 10.1002/ped4.12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig Fahrmeir T.K., Lang S., Marx B.D. Springer-Verlag; 2013. Regression: Models, Methods and Applications. [Google Scholar]
- Mack S., Coassin S., Rueedi R., Yousri N.A., Seppälä I., Gieger C., Schönherr S., Forer L., Erhart G., Marques-Vidal P. A genome-wide association meta-analysis on lipoprotein (a) concentrations adjusted for apolipoprotein (a) isoforms. J. Lipid Res. 2017;58:1834–1844. doi: 10.1194/jlr.M076232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichaikul A., Mychaleckyj J.C., Rich S.S., Daly K., Sale M., Chen W.M. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mclean J.W., Tomlinson J.E., Kuang W.J., Eaton D.L., Chen E.Y., Fless G.M., Scanu A.M., Lawn R.M. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature. 1987;330:132–137. doi: 10.1038/330132a0. [DOI] [PubMed] [Google Scholar]
- Mishra A., Macgregor S. VEGAS2: software for more flexible gene-based testing. Twin Res. Hum. Genet. 2015;18:86–91. doi: 10.1017/thg.2014.79. [DOI] [PubMed] [Google Scholar]
- National Health Commission Diagnosis and treatment protocol for novel coronavirus pneumonia (trial version 7) Chin Med. J. (Engl) 2020;133:1087–1095. doi: 10.1097/CM9.0000000000000819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A., David J.K., Maden S.K., Wood M.A., Weeder B.R., Nellore A., Thompson R.F. Human leukocyte antigen susceptibility map for severe acute respiratory syndrome coronavirus 2. J. Virol. 2020;94:e00510. doi: 10.1128/JVI.00510-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paces J., Strizova Z., Smrz D., Cerny J. COVID-19 and the immune system. Physiol. Res. 2020;69:379–388. doi: 10.33549/physiolres.934492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pairo-Castineira E., Clohisey S., Klaric L., Bretherick A.D., Rawlik K., Pasko D., Walker S., Parkinson N., Fourman M.H., Russell C.D. Genetic mechanisms of critical illness in COVID-19. Nature. 2021;591:92–98. doi: 10.1038/s41586-020-03065-y. [DOI] [PubMed] [Google Scholar]
- Patterson N., Price A.L., Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pico A.R., Kelder T., van Iersel M.P., Hanspers K., Conklin B.R., Evelo C. WikiPathways: pathway editing for the people. PLoS Biol. 2008;6:e184. doi: 10.1371/journal.pbio.0060184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Price R.A., Li W.D., Zhao H. FTO gene SNPs associated with extreme obesity in cases, controls and extremely discordant sister pairs. BMC Med. Genet. 2008;9:4. doi: 10.1186/1471-2350-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J., Kull M., Peterson H., Hansen J., Vilo J. g:Profiler--a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucl. Acids Res. 2007;35:W193–W200. doi: 10.1093/nar/gkm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton J.F., Shastri A.J., Ye C., Weldon C.H., Filshtein-Sonmez T., Coker D., Symons A., Esparza-Gordillo J., Aslibekyan S., Auton A. Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nat. Genet. 2021;53:801. doi: 10.1038/s41588-021-00854-7. [DOI] [PubMed] [Google Scholar]
- Verbanck M., Chen C.Y., Neale B., Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018;50:693–698. doi: 10.1038/s41588-018-0099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verduijn M., Siegerink B., Jager K.J., Zoccali C., Dekker F.W. Mendelian randomization: use of genetics to enable causal inference in observational studies. Nephrol. Dial Transpl. 2010;25:1394–1398. doi: 10.1093/ndt/gfq098. [DOI] [PubMed] [Google Scholar]
- Vuckovic D., Bao E.L., Akbari P., Lareau C.A., Mousas A., Jiang T., Chen M.H., Raffield L.M., Tardaguila M., Huffman J.E. The polygenic and monogenic basis of blood traits and diseases. Cell. 2020;182:1214–1231.e11. doi: 10.1016/j.cell.2020.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Li R., Lu Z., Huang Y. Does comorbidity increase the risk of patients with COVID-19: evidence from meta-analysis. Aging (Albany NY) 2020;12:6049–6057. doi: 10.18632/aging.103000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Huang S., Gao R., Zhou Y., Lai C., Li Z., Xian W., Qian X., Li Z., Huang Y. Initial whole-genome sequencing and analysis of the host genetic contribution to COVID-19 severity and susceptibility. Cell Discov. 2020;6:83. doi: 10.1038/s41421-020-00231-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Yuan Z., Pavel M.A., Hansen S.B. The role of high cholesterol in age-related COVID19 lethality. bioRxiv. 2020 doi: 10.1101/2020.05.09.086249. [DOI] [Google Scholar]
- Wingo T.S., Liu Y., Gerasimov E.S., Gockley J., Logsdon B.A., Duong D.M., Dammer E.B., Lori A., Kim P.J., Ressler K.J. Brain proteome-wide association study implicates novel proteins in depression pathogenesis. Nat. Neurosci. 2021;24:810–817. doi: 10.1038/s41593-021-00832-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., Mcgoogan J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the Chinese center for disease control and prevention. Jama. 2020;323:1239–1242. doi: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- Yin L., Zhang H., Tang Z., Xu J., Yin D., Zhang Z., Yuan X., Zhu M., Zhao S., Li X., Liu X. rMVP: a memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genomics Proteomics Bioinform. 2021 doi: 10.1016/j.gpb.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z., Zhu H., Zeng P., Yang S., Sun S., Yang C., Liu J., Zhou X. Testing and controlling for horizontal pleiotropy with probabilistic Mendelian randomization in transcriptome-wide association studies. Nat. Commun. 2020;11:3861. doi: 10.1038/s41467-020-17668-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M., Gao Y., Wang G., Song G., Liu S., Sun D., Xu Y., Tian Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol. Immunol. 2020;17:533–535. doi: 10.1038/s41423-020-0402-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou A., Wang M., Jin Y., Cheng X., Su K., Yang L. Genetic analysis of a novel missense mutation (Gly542Ser) with factor XII deficiency in a Chinese patient of consanguineous marriage. Int. J. Hematol. 2018;107:436–441. doi: 10.1007/s12185-017-2393-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
6
Data Availability Statement
The data that support the findings of this study have been deposited into CNGB Sequence Archive (CNSA) (Guo et al., 2020) of China National GeneBank DataBase (CNGBdb) (Chen et al., 2020a) with accession number CNSA: CNP0001876. A summary of analysis software and tools were provided in key resources table. Additional Supplemental Tables are available from Mendeley Data at https://doi.org/10.17632/3wr9mgm2b6.1. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.