

Summary
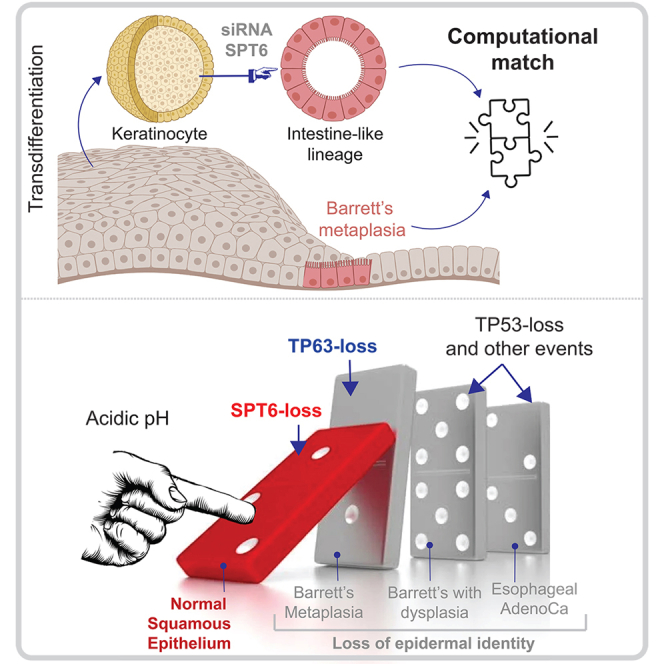
Transient depletion of the transcription elongation factor SPT6 in the keratinocyte has been recently shown to inhibit epidermal differentiation and stratification; instead, they transdifferentiate into a gut-like lineage. We show here that this phenomenon of transdifferentiation recapitulates Barrett’s metaplasia, the only human pathophysiologic condition in which a stratified squamous epithelium that is injured due to chronic acid reflux is trans-committed into an intestinal fate. The evidence we present here not only lend support to the notion that the keratinocytes are potentially the cell of origin of Barrett’s metaplasia but also provide mechanistic insights linking transient acid exposure, downregulation of SPT6, stalled transcription of the master regulator of epidermal fate TP63, loss of epidermal fate, and metaplastic progression. Because Barrett’s metaplasia in the esophagus is a pre-neoplastic condition with no preclinical human models, these findings have a profound impact on the modeling Barrett’s metaplasia-in-a-dish.
Subject areas: Molecular biology, Cell biology, Bioinformatics
Graphical abstract
Highlights
-
•
Keratinocytes transdifferentiate into a gut lineage upon depletion of SPT6
-
•
Such transdifferentiation resembles Barrett’s metaplasia not the healthy gut
-
•
Acid downregulates SPT6, which inhibits the expression and functions of TP63
-
•
Such downregulation precedes the metaplasia-dysplasia-neoplasia cascade
Molecular biology; Cell biology; Bioinformatics
Introduction
The stratified squamous epithelium, which is comprised mainly of keratinocytes, acts as a physical barrier and is replaced every few weeks by resident stem cells residing in the basal layer (Gonzales and Fuchs, 2017; Watt, 2014). Besides our skin, stratified squamous epithelia form barriers to antigens in the oral cavity and oral pharynx including the palatine and lingual tonsils, the anal canal, the male foreskin, and the female vagina and ectocervix. Recently, it has been shown (Li et al., 2021) that in epidermal stem and progenitor cells, approximately a third of the genes that are induced during differentiation already contain stalled Pol II at the promoters which is then released into productive transcription elongation upon differentiation. Using a combination of Pol II ChIP Seq and RNAi screen, SPT6 was identified as one of the critical mediators of such elongation (Li et al., 2021). SPT6-depleted keratinocytes fail to differentiate into stratified squamous epithelium; instead, they transdifferentiate into an “intestine-like” lineage (by morphology and gene expression analysis; Figures 1A and 1B). This claim of transdifferentiation was supported in part by morphological characteristics in 3D growth and in a more definitive way by transcriptomic studies (GSE153129) (Li et al., 2021). The list of genes that were upregulated ≥10-fold in small interfering RNA (siRNA)-depleted SPT6 samples (SPT6i) compared to controls (CTLi). The resultant SPT6-depleted 472-gene signature was used to query the Human Gene Atlas and ARCHS4; the latter is a web-based resource that provides access to human and mouse transcriptomic data sets from gene expression omnibus (GEO) and sequence read archive (SRA) (Li et al., 2021). Mechanistically, depletion of SPT6 resulted in stalled transcription of the master regulator of epidermal fate p63 (Truong et al., 2006; Yang et al., 1999; Senoo et al., 2007, Crum and Mckeon, 2010). Studies in SPT6-depleted keratinocytes that were subsequently rescued with exogenous expression of p63 suggested that SPT6 favors the differentiation into stratified squamous epithelium and arrests the intestinal phenotype through the control of transcriptional elongation of p63 and its targets. Despite the mechanistic insights into how SPT6 regulates keratinocyte fate, the translational relevance of the observed transdifferentiation of keratinocytes into an intestinal fate remained unknown.
Figure 1.

Keratinocyte stem cells depleted of SPT6 trans-differentiates into gut lineage that resembles Barrett’s metaplasia
(A) Schematic summarizes the chromosomal location of SPT6 and its known functions in transcriptional elongation and mRNA processing. SPT6 coordinates nucleosome dis- and re-assembly, transcriptional elongation, and mRNA processing. SPT6 is a conserved factor that controls transcription and chromatin structure across the genome.
(B) Schematic summarizing the key findings in gene expression and epithelial morphology observed and reported earlier (Li et al., 2021) upon depletion of SPT6 in keratinocyte stem cells by siRNA (Li et al., 2021). While control keratinocytes formed stratified squamous epithelium, siRNA-mediated transient depletion of SPT6 in keratinocytes (SPT6i) grew as “intestine-like” monolayers.
(C) Schematic showing the only known human pathophysiologic context in which stratified squamous epithelium is known to be replaced by “intestine-like” epithelium.
(D) Summary of computational approach used in (E-J).
(E–G) Differentially expressed genes (DEGs; see Table S1) in Barrett’s metaplasia vs. normal esophagus were used to rank order control (CTLi) and SPT6-depleted samples (SPT6i), either using UP genes alone (F), DOWN-genes alone (G) or both UP and DOWN signatures together (E). Results are presented as bar (top) and violin (bottom) plots. ROC-AUC in all cases reflects a perfect strength of classification (1.00). Welch’s two sample unpaired t test is performed on the composite gene signature score to compute the p values.
(H–J) Differentially expressed genes (DEGs; see Data S1) between control (CTLi) and SPT6-depleted (SPT6i) samples were used to rank order normal (N) from Barrett’s esophageal (BE) samples across 9 publicly available independent cohorts (8 human, 1 mouse), either using UP genes alone (I), DOWN-genes alone (J), or both UP and DOWN signatures together (H). See also Figure S1 for violin plots for each dataset. ROC-AUC in each case is annotated on the right side of the corresponding bar plots.
(K) Pearson correlation matrix showing clustering of control (CTLi) and SPT6-depleted (SPT6i) gene expression signatures with the brain, colon, BE, adipocyte, trachea, and skeletal muscle. Two distinct RNA-Seq samples of adult origin are shown for each tissue (see Table S2 for the list of datasets used to generate the matrix).
Among the various organs that are protected by stratified squamous epithelium, the only human pathophysiologic condition in which a stratified squamous epithelium in adults transdifferentiates into intestinal fate is that described in the foregut, a phenomenon termed Barrett’s metaplasia (Mcdonald et al., 2015) of the esophagus (BE). BE develops when the non-keratinized stratified squamous epithelium in the lower esophagus is replaced by a single layer of “intestine-like” cells after a prolonged phase of injury due to chronic acid reflux (Figure 1C). The origin of BE remains widely debated; theories include a direct origin from the esophageal stratified squamous epithelium or by proximal migration and subsequent intestinalization of the gastric epithelium (Zhang and Wang, 2018; Que et al., 2019; Xian et al., 2012; Gindea et al., 2014, Mcdonald et al., 2015). Alternative proposals include a niche cell at the squamocolumnar junction, or cells lining the esophageal gland ducts, or circulating bone-marrow-derived cells (Que et al., 2019). Much of these theories originate from experimental models, and to date, there are no models that recapitulate the process of transdifferentiation of the epithelial lining that is the pathognomonic feature of BE. In fact, our inability to observe the process of metaplastic conversion in vivo and the lack of reliable physiological models (Fitzgerald, 2006) are cited as the factors limiting our ability to trace the cell of origin for BE. Despite the lack of models, or dispute surrounding the origin of BE, what is undisputed is that it represents a bona fide preneoplastic state; patients with BE have approximately 40–125 times higher risk of esophageal adenocarcinoma than the general population (Wang and Canto, 2010). We hypothesized that the phenomenon of transdifferentiation from stratified squamous to an intestinal fate in SPT6-depleted keratinocytes may resemble and recapitulate the fundamental molecular and cellular aspects of keratinocyte transcommitment in BE.
Results
SPT6 loss resembles Barrett’s esophagus
We carried out a comprehensive bidirectional analysis: differentially expressed genes (DEGs), both upregulated and downregulated genes (Wang et al., 2006) in BE vs. normal esophagus (Figure 1D) were used to rank order the control vs. SPT6-depleted samples, and conversely, DEGs in control vs. SPT6-depleted samples were analyzed in all BE datasets publicly available on NCBI as of February 1, 2021 (Figure 1D). We found that the combined DEGs (upregulated and downregulated genes in BE (Wang et al., 2006); Figure 1E; Table S1) as well as the individual upregulated and downregulated genes (Figures 1F and 1G) were able to independently classify the control and SPT6-depleted samples with perfection (area under the Receiver Operator Characteristic (ROC) curve; ROC AUC: 1.00). The converse was also true, i.e., the combined DEGs (upregulated and downregulated genes in SPT6-depleted samples; Figure 1H, Data S1) as well as the individual upregulated/downregulated signatures (Figures 1I and 1J) were independently able to classify the normal esophageal and BE samples across several independent human datasets. Downregulated genes consistently performed better (ROC AUC ranges from 0.56–1.00 in UP-genes, I, and 0.84–1.00 in DOWN-genes, J). The DEGs from the SPT6-depleted samples also perfectly classified the BE samples derived from mice lacking p63 (Figures 1H–1J); p63−/− mice are the only genetic model of BE known to date (Wang et al., 2011). Finally, a Pearson correlation matrix revealed that SPT6-depleted samples clustered much closer to BE tissues than to the colon (correlation coefficient 0.71–0.73 to BE vs. 0.5–0.51 to colon; Figure 1K and Table S2). These findings demonstrate that the transcriptional profile of the SPT6-depleted keratinocyte is more like BE than the colon or any other tissue type tested.
SPT6 loss resembles intestinal metaplasia, not healthy intestinal differentiation
Prior studies have linked loss of p63, the master regulator of keratinocyte proliferation and differentiation into a stratified lining (Truong et al., 2006; Yang et al., 1999; Senoo et al., 2007, Crum and Mckeon, 2010), as a state that is permissive to the transdifferentiation of stratified squamous epithelium into “intestine-like” metaplasia. In p63−/− mice, the stratified lining of both trachea and esophagus is replaced by a highly ordered, columnar ciliated epithelium that is deficient in basal cells (Daniely et al., 2004). In the same mice, under conditions of programmed damage to the esophageal lining, progenitor cells at the gastroesophageal junction serve as precursors of Barrett’s metaplasia (Wang et al., 2011). SPT6 loss in keratinocytes was also associated with a functional loss of p63 (Li et al., 2021); without SPT6, levels of p63 protein were diminished, and p63-binding sites on the genome were closed, as determined using ATAC seq (Li et al., 2021). Thus, both the SPT6-depleted primary human keratinocyte model and the p63−/− mouse model rely upon a final common pathway that escapes an epidermal fate; both lack functional p63. We noted that in the p63−/− mouse model of BE (Wang et al., 2011), the authors had further delineated that despite the overall similarities, BE segment and intestine tissues have key differences: a set of metaplasia-specific genes is enriched in BE, whereas a set of intestine-specific genes is enriched in the intestine (Wang et al., 2011) (Figure 2A; Table S3). Gene set enrichment analyses (GSEA-preranked) (Subramanian et al., 2005; Mootha et al., 2003) found these differences also in human BE vs. small intestine tissues (GSE13083) (Stairs et al., 2008); (Figure 2B) and in the SPT6-depleted keratinocyte organoid models (Li et al., 2021) vs. small intestine-derived organoids (Figure 2C). Visualization of the same analyses as heatmaps confirm that although the metaplasia-specific signature was induced in both BE tissue (Figure 2D) and in SPT6-depleted (SPT6i) organoids (Figure 2E) compared to their respective small intestine-derived samples, there were subtle differences. For example, the fraction of genes induced within each signature was higher in the organoids (Figure 2E) compared to the tissues (Figure 2D). Because the signatures are derived from mouse tissues (Wang et al., 2011), it is possible that their greater representation in human SPT6i organoids than in human BE tissues [which capture the gene expression patterns contributed by non-epithelial (stromal and immune) cells] reflects the species-specific differences in gene expression in the non-epithelial cells. These findings further support our argument that SPT6 depletion does not merely trigger intestinal transdifferentiation; it induces metaplasia-specific genes in the setting of de-enrichment of intestine-specific genes.
Figure 2.

Downregulation of SPT6 enriches metaplasia-specific genes
(A–E) Schematic in (A) summarizing the workflow for distinguishing BE from intestine. Using TP63−/− as a strategy to induce BE in mice, a prior study showed that compared to intestinal tissues, BE tissue was enriched in a 16-gene metaplasia-specific signature and de-enriched in a 16-gene intestine-specific signature (see Table S3 for the list of genes). These gene sets were analyzed for enrichment (Gene Set Enrichment Analysis – GSEA pre-ranked analysis) in human BE vs. small intestine tissues (GSE13083; B) and SPT6-depleted (SPT6i) vs. small intestine-derived organoids (C; scale bar = 100 μm). Heatmaps (D and E) display the levels of expression of the individual metaplasia-specific and intestine-specific genes in the BE vs. small intestine tissues (D) and organoids SPT6-depleted keratinocyte (SPT6i) vs. small intestine-derived organoids (E).
(F and G) Differentially expressed genes (DEGs; see Data S1) between control (CTLi) and SPT6-depleted (SPT6i) samples were used to rank order normal squamous esophagus (NE) from non-dysplastic Barrett’s esophagus (BE-noD), dysplastic BE (BE-D), and esophageal adenocarcinoma (EAC; n = 12) samples in an RNA seq dataset [E-MTAB-4054] (Maag et al., 2017), either using UP genes alone (G; left), DOWN-genes alone (G; right), or both UP and DOWN signatures together (F). Numbers in parenthesis indicate the number of samples. ROC-AUC in each case is annotated below the bar plots. Welch’s two sample unpaired t test is performed on the composite gene signature score to compute the p values. In multi-group setting, each group is compared to the NE control group and only significant p values are displayed.
We next asked if the metaplastic BE-like signature that is induced upon SPT6 depletion stays induced during the progression of BE to esophageal adenocarcinoma (EAC). To this end, we analyzed the DEGs in SPT6-depleted keratinocytes (SPT6i) in an RNA seq dataset comprising 51 tissue samples, which included normal squamous esophagus, BE metaplasia without dysplasia, BE with low-grade dysplasia (LGD), and EAC [E-MTAB-4054] (Maag et al., 2017); these were collected at endoscopy from 44 patients. We found that the combined DEGs (upregulated and downregulated genes) in SPT6-depleted samples classified normal esophagus nearly perfectly (ROC AUC: 0.96) from BE samples and perfectly (ROC AUC: 1.00) from both dysplastic BE and EAC samples (Figure 3F). When we analyzed the upregulated genes (Figure 3G; left) or the downregulated genes (Figure 3G; right) separately, we found that the individual upregulated/downregulated signatures were independently able to classify the normal esophageal samples from all other esophageal tissues representative of progression through the metaplasia-dysplasia-neoplasia cascade. These findings suggest that the BE-like gene expression pattern we observe in SPT6-depleted organoids is conserved during subsequent progression of BE to EAC.
Figure 3.

Downregulation of SPT6 can be triggered by exposure to acid
(A) Publicly available microarray dataset from esophageal epithelial cells (immortalized with hTERT) treated with pH 4.5 for 2 and 6 h were analyzed for SPT6 expression.
(B) Graph displays the abundance of SPT6 transcripts; the x axis represents the log2(X + 1) transformation of unexposed (0), 2 and 6 h after exposure to pH 4.5, and the y axis represents log2 normalized SPT6 expression. Significance was determined by linear regression, where Y = −0.4828∗X + 5.1847 and p = 0.028.
(C–E) Schematic in (C) shows the workflow for the analyses we did here on human primary keratinocytes in 3D culture, after transiently exposing them to acid. Bar graphs in (D) display the relative expression of SPT6 (top) and TP63 (bottom) in primary keratinocytes exposed to pH 7.5 or 4.5, as determined by qPCR. Error bars represent S.E.M. Significance as determined by t test, n = 3. Immunoblots (IBs) in (E) display the abundance of SPT6 and TP63 proteins in equal aliquots (50 μg) of whole-cell lysates of the keratinocytes. O.D = optical density, as determined by band densitometry. Representative immunoblots from 3 independent repeats are shown.
(F) Schematic summarizing the evidence we present here, showing the keratinocyte stem cell as the cell of origin of BE; upon chronic acid (low pH) injury, SPT6 is downregulated in the keratinocyte stem cells. SPT6 suppression, either due to acid exposure (physiologic) or transiently with siRNA (experimentally), causes the spontaneous transdifferentiation of epidermal cells into Barrett's metaplasia. Prior work (Li et al., 2021) demonstrated that such transdifferentiation was due to the stalled transcription of the master regulator of epidermal fate p63.
Acid exposure suppresses SPT6 in keratinocytes
Because BE is a consequence of prolonged acid exposure (Mcdonald et al., 2015), we next asked what, if any, might be the impact of low pH on the levels of SPT6 expression in keratinocytes. We found that SPT6 transcripts are downregulated in immortalized esophageal keratinocytes exposed to acid (Figures 3A and 3B). Prior work has also shown that exposure of esophageal keratinocytes to bile and acid causes a reduction in p63 (Roman et al., 2007). When we exposed primary keratinocytes (same cell line used previously in Li et al. (2021)) to pH 4.5 (Figure 3C), we found that both SPT6 and TP63 transcripts were reduced (Figure 3D). Most importantly, reduced SPT6 and TP63 transcripts in these cells translated to reduced SPT6 and, to a lesser extent, TP63 proteins upon acid challenge (Figure 3E). These findings help link the SPT6→p63 mechanism(s) outlined earlier (Li et al., 2021) to one of the most definitive physiologic triggers of BE, i.e., exposure to acid.
Discussion
Our findings augment the impact of the discoveries reported earlier (Li et al., 2021) in three ways: (i) First, they validate transient SPT6-depletion in keratinocytes as an effective strategy for studying origin of BE. Although organoids derived from segments of established BE have been successfully grown in long-term cultures (Sato et al., 2011), attempts to model the initiation of BE had thus far been unsuccessful (Kong et al., 2009). (ii) Second, they weigh in on the long-standing debate surrounding the cell of origin in BE. Controversies exist as to whether BE results from a direct conversion of differentiated cells via a process called transdifferentiation or whether BE develops from niche stem or progenitor cells at the gastroesophageal junction (Dvorak et al., 2011; Chen et al., 2011). Taken together with the evidence presented earlier (Li et al., 2021), our findings argue strongly for keratinocyte transdifferentiation or transcommitment as the mechanism. (iii) Third, our findings in SPT6i keratinocytes suggest a mechanism for initiation of BE while being able to connect the dots between physiological triggers of the disease, i.e., chronic acid reflux (Figure 3F). For example, acid exposure has been shown to promote intestinal differentiation in both BE explants (Fitzgerald et al., 1996) and BE-derived adenocarcinoma cell lines (Souza et al., 2002), and here we show that acid exposure reduces SPT6 mRNA and protein. Another intriguing coincidence is that SPT6 is located on the long arm of Chr 17q11.2, and loss of heterozygosity (LOH) at this locus is frequently encountered in BE-associated adenocarcinomas (Swift et al., 1995; Dunn et al., 2000), representing one of the most frequent LOH in BE (Dunn et al., 1999). Notably, loss of p53, which is located on the short arm of Chr 17p13, signals risk for BE to adenocarcinoma progression (Kastelein et al., 2013). Unlike the timing of loss of tp53, which happens later in BE-to-cancer progression, microdeletions in the distal long arm of Chr 17q has been proposed as early event in BE initiation (Petty et al., 1998). Because microsatellites might be sensitive indicators of disrupted mechanisms and indicate a propensity to mutagenesis, we speculate that microdeletions at Chr17q11.2 could impact SPT6 expression and trigger the transdifferentiation of keratinocytes into BE. It is possible that such microdeletions are a consequence of DNA damage due to repetitive acid injury. Regardless of the exact mechanism by which SPT6 is lost, it appears that its loss is capable of derailing both the expression (this work) and transcriptional activity of TP63 [shown earlier (Li et al., 2021)], a key determinant of keratinocyte fate. While our manuscript was in review, another group (Nowicki-Osuch et al., 2021) has since shown using lineage tracing studies that all EACs arise from BE even if BE is not visible at the time of cancer diagnosis and even if these BE cells had migrated away from the gastroesophageal junction toward the gastric cardia. While here we did not investigate BE to cancer progression, or the precise location of BE cells in the foregut, our conclusions shed light on some of the earliest events that may trigger the initiation of BE from acid-injured esophageal lining. Our studies on human keratinocytes, vetted against numerous human tissue datasets, also lend strong support to the original discoveries made in animal models, wherein the role of TP63+ transitional basal progenitors was reported as the cell of origin for BE in mouse models (Jiang et al., 2017). The BE model described here also upholds the prevailing concept that BE arises from the “transcommitment” or transdifferentiation of esophageal stem cells to produce an intestine-like epithelium (Xian et al., 2019). The importance of our findings are further enhanced in light of recent multiscale computational modeling studies (Curtius et al., 2021) and now, molecular studies (Nowicki-Osuch et al., 2021), both independently verifying that BE is the invariant precursor to EACs, or in other words, all EACs originate from BE.
Limitations of the study
While the strength of our study lies in the rigorous computational validation of one disease model (SPT6-depleted keratinocyte) against diseased tissues from diverse cohorts, there are some notable limitations. For example, although we showed that the impact of suppressing the SPT6→TP63 axis on the gene expression pattern is widely reflected in most if not all BE data sets, whether SPT6 suppression itself is a major and widely prevalent trigger event in BE initiation remains to be established. Similarly, acid exposure reduced both SPT6 mRNA and protein levels, but head-to-head comparisons of the genetic (SPT6 depletion) and physiologic (repetitive acid challenge) triggers need to be analyzed systematically using gene and pathway overlap assessments to fully understand which model is closest to BE and what role SPT6 depletion may plan in its initiation. Finally, prolonged acid injury was not attempted; such studies in conjunction with genomic and epigenomic studies will be insightful to understand how acid injury may lead to SPT6 loss and/or suppression.
STAR★ Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-SPT6 | Thermo Fisher Scientific | A300-801A |
| Anti-TP63 | Abcam | ab53039 |
| Rabbit polyclonal anti-β-tubulin | Santa Cruz Biotechnology | sc-9104 |
| Mouse monoclonal anti-GAPDH | Santa Cruz Biotechnology | sc-365062 |
| IRDye 800CW Goat anti-Mouse IgG Secondary | LI-COR Biosciences | 926-32210 |
| IRDye 680RD Goat anti-Rabbit IgG Secondary | LI-COR Biosciences | 926-68071 |
| Experimental Models: Cells | ||
| Human Epidermal Keratinocyte Culture (from neonatal foreskin) | UC San Diego HUMANOID Center of Research Excellence | Li et al.(2021) |
| Human intestinal organoids (terminal ileum), adult, male and female | UC San Diego HUMANOID Center of Research Excellence | Sharma et al.(2021) |
| L-WRN cells | ATCC | CRL-3276 (Miyoshi and Stappenbeck, 2013) |
| Chemicals, peptides, and recombinant proteins | ||
| PVDF Transfer Membrane, 0.45μM (for blotting) | Thermo Scientific | 88518 |
| PowerUp™ SYBR™ Green Master Mix (for qPCR) | Applied Biosciences | A25741 |
| qScript™ cDNA SuperMix (for qPCR) | QuantaBio | 101414 |
| Ethanol | Koptec | UN1170 |
| Protease inhibitor cocktail (for cell lysis) | Roche | 11 873 580 001 |
| Tyr phosphatase inhibitor cocktail (for cell lysis) | Sigma-Aldrich | P5726 |
| Ser/Thr phosphatase inhibitor cocktail (for cell lysis) | Sigma-Aldrich | P0044 |
| 100% Methanol (for priming PVDF membrane) | Supelco | MX0485 |
| Glycine | Fisher Scientific | BP381-5 |
| Bovine Serum Albumin | Sigma-Aldrich | A9647-100G |
| Triton-X 100 (for cell lysis) | Sigma-Aldrich | X100-500ML |
| TrypLE Select | Thermo Scientific | 12563-011 |
| Advanced DMEM/F-12 | Thermo Scientific | 12634-010 |
| HEPES Buffer | Life Technologies | 15630080 |
| Glutamax | Thermo Scientific | 35050-061 |
| Penicillin-Streptomycin | Thermo Scientific | 15140-122 |
| Collagenase Type I | Thermo Scientific | 17100-017 |
| Matrigel | Corning | 354234 |
| B-27 | Thermo Scientific | 17504044 |
| N-acetyl-L-cysteine | Sigma-Aldrich | A9165 |
| Nicotinamide | Sigma-Aldrich | N0636 |
| FGF-7 (KGF) | PeproTech | 100-19-50ug |
| FGF10 | PeproTech | 100-26-50ug |
| A-83-01 | Bio-Techne Sales Corp. | 2939/50 |
| SB202190 | Sigma-Aldrich | S7067-25MG |
| Y-27632 | R&D Systems | 1254/50 |
| DPBS | Thermo Scientific | 14190-144 |
| Ultrapure Water | Invitrogen | 10977-015 |
| EDTA | Thermo Scientific | AM9260G |
| Hydrocortisone | STEMCELL Technologies | 7925 |
| Heparin | Sigma Aldrich | H3149 |
| Fetal Bovine Serum | Sigma-Aldrich | F2442-500ML |
| EpiVita Media | Cell Applications | 141-500a |
| Animal Component-Free Cell Dissociation Kit | STEMCELL Technologies | 5426 |
| Red Blood Cell Lysis Buffer | Invitrogen | 00-4333-57 |
| Cell Recovery Solution | Corning | 354253 |
| Sodium Azide (for antibody dilutions) | Fisher Scientific | S227I-100 |
| Quick-RNA MicroPrep Kit | Zymo Research | R1051 |
| Quick-RNA MiniPrep Kit | Zymo Research | R1054 |
| Ethyl alcohol, pure | Sigma-Aldrich | E7023 |
| TRI Reagent | Zymo Research | R2050-1-200 |
| 2x SYBR Green qPCR Master Mix | Bimake | B21203 |
| qScript cDNA SuperMix | Quanta Biosciences | 95048 |
| Applied Biosystems TaqMan Fast Advanced Master Mix | Thermo Scientific | 4444557 |
| 18S, Hs99999901_s1 | Thermo Scientific | 4331182 |
| Oligonucleotides and Primer Sequences | ||
| Human SPT6; Forward: CCGTGTCCACCCTGAGAC Human SPT6; Reverse: CATAGCCCTGCCTCTCCA |
This paper | n/a |
| Human TP63; Forward: GACAGGAAGGCGGATGAAGATAG Human TP63; Reverse: TGTTTCTGAAGTAAGTGCTGGTGC |
This paper | n/a |
| Human 18S; Forward: GTAACCCGTTGAACCCCATT Human 18S; Reverse: CCATCCAATCGGTAGTAGCG |
Thermo Scientific | 4331182 |
| Software and algorithms | ||
| ImageJ | https://imagej.nih.gov/ij/index.html | n/a |
| GraphPad Prism | https://www.graphpad.com/scientific-software/prism/ | n/a |
| QuantStudio Design & Analysis Software | https://www.thermofisher.com/us/en/home/global/forms/life-science/quantstudio-3-5-software.html | n/a |
| Illustrator (ADOBE) | https://www.adobe.com/products/illustrator.html | n/a |
| ImageStudio Lite (LI-COR Sciences) | https://www.licor.com/bio/image-studio-lite/ | n/a |
| The source code is available at https://github.com/sahoo00/BoNE. | This work | n/a |
| Other | ||
| 6-well Tissue Culture Plate | Genesee Scientific | 25-105 |
| 12-well Tissue Culture Plate | CytoOne | CC7682-7512 |
| Cell Scraper | Millipore Sigma | C5981-100EA |
| Countess Cell Counting Chamber Slides | Invitrogen | C10312 |
| Trypan Blue Stain | Invitrogen | T10282 |
| 70 um Cell Strainer | Thermo Fisher Scientific | 22-363-548 |
| 100 um Cell Strainer | Corning | 352360 |
| RNase Away | Thermo Fisher Scientific | 14-375-35 |
| Countess II Automated Cell Counter | Thermo Fisher Scientific | AMQAX1000 |
| Canon Rebel XS DLSR | Canon | n/a |
| MiniAmp Plus Thermal Cycler | Applied Biosystems | A37835 |
| QuantStudio5 | Applied Biosystems | A28140 |
| Light Microscope (brightfield images) | Carl Zeiss LLC | Axio Observer, Inverted; 491917-0001-000 |
| Deposited Data | ||
| RNA Seq dataset for control vs. SPT6i keratinocytes | Publicly released with prior publication (Li et al., 2021). | GEO: GSE153129 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Pradipta Ghosh (prghosh@ucsd.edu).
Materials availability
-
•
This study did not generate new unique reagents.
-
•
The RNA Seq dataset for control vs. SPT6i keratinocytes was publicly released with prior publication (Li et al., 2021), and can be accessed here: GSE153129.
Experimental model and subject details
Human epidermal keratinocyte culture
SPT6 knock-down and wild-type human epidermal keratinocytes were cultured under 3D spheroid conditions (Ghosh et al., 2020) for colon-derived organoids. Briefly, primary human epidermal keratinocytes derived from human neonatal foreskin were used for all cell culture studies. Cells were seeded in Matrigel (Corning, 354234) domes at 5E4 cells per well in a 24 well plate. To allow for complete polymerization of the Matrigel, the plate was inverted and incubated at 37°C for 10 minutes before proliferation media (50% conditioned media prepared from L-WRN cells (ATCC, CRL-3276(Miyoshi and Stappenbeck, 2013)) containing Wnt3a, R-spondin, and noggin) or differentiation media (5% conditioned media) was added to each well. Cells were maintained at 37°C/5% CO2 humidified conditions and media was changed every 2-3 days until spheroids were fully formed, and crypt budding was visible.
Method details
Computational methods
Gene expression databases
Publicly available microarray and gene expression databases were downloaded from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus website (GEO) (Edgar et al., 2002; Barrett et al., 2005, 2013). If the dataset is not normalized, RMA (Robust Multichip Average) (Irizarry et al., 2003a, 2003b) is used for microarrays and CPM (Counts Per Millions) (Li and Dewey, 2011; Pachter, 2011) is used for RNASeq data for normalization. We used log2(CPM+1) to compute the final log-reduced expression values for RNASeq data. Accession numbers for these crowdsourced datasets are GSE153129, GSE100843, GSE65013, GSE64894, GSE39491, GSE49292, GSE26886, GSE34619, GSE13083, GSE96831, GSE120795, GSE129153, GSE148818, GSE58963, GSE157059, GSE9768 and GSE70051 and are provided in the figures and manuscript (Dalerba et al., 2011, 2016; Volkmer et al., 2012).
Boolean analysis
Boolean logic is a simple mathematic relationship of two values, i.e., high/low, 1/0, or positive/negative. The Boolean analysis of gene expression data requires first the conversion of expression levels into two possible values. The StepMiner algorithm is used to perform Boolean analysis of gene expression data (Sahoo et al., 2008). Boolean analysis is a statistical approach that creates binary logical inferences that explain the relationships between phenomena. Boolean analysis is performed to determine the relationship between the expression levels of pairs of genes. The StepMiner algorithm is applied to gene expression levels to convert them into Boolean values (high and low). In this algorithm, first the expression values are sorted from low to high and a rising step function is fitted to the series to identify the threshold. Middle of the step is used as the StepMiner threshold. This threshold is used to convert gene expression values into Boolean values. A noise margin of 2-fold change is applied around the threshold to determine intermediate values, and these values are ignored during Boolean analysis.
Generation of gene signature scores
Gene expression values were normalized according to a modified Z-score approach centered around StepMiner threshold (formula = (expr - SThr)/3∗stddev). The samples were ordered according to average of the normalized gene expression values in the given gene list. Gene signature score is computed as a linear combination of the normalized gene expression values (the modified Z-score as described above). Samples are ordered using the gene signature score and the strength of the association between gene expression and disease annotation is computed using ROC-AUC measurement. A barplot is used to visualize the sample ordering with different color codes for the sample annotation. Additionally, a set of violin plots is used just below the barplot to demonstrate the distribution of the gene signature score across different sample annotations.
Measurement of classification strength or prediction accuracy
Receivers operating characteristic (ROC) curves were computed by simulating a score based on the ordering of samples that illustrates the diagnostic ability of binary classifier system as its discrimination threshold is varied along the sample order. The ROC curves were created by plotting the true positive rate (TPR) against the false positive rate (FPR) at various threshold settings. The area under the curve (often referred to as simply the AUC) is equal to the probability that a classifier will rank a randomly chosen IBD samples higher than a randomly chosen healthy samples. In addition to ROC AUC, other classification metrics such as accuracy ((TP + TN)/N; TP: True Positive; TN: True Negative; N: Total Number), precision (TP/(TP+FP); FP: False Positive), recall (TP/(TP+FN); FN: False Negative) and f1 (2 ∗ (precision ∗ recall)/(precision + recall)) scores were computed. Precision score represents how many selected items are relevant and recall score represents how many relevant items are selected. Fisher exact test is used to examine the significance of the association (contingency) between two different classification systems (one of them can be ground truth as a reference).
Test and validation of Barrett’s esophagus datasets
A Boolean Network Explorer (BoNE) computational tool was introduced recently (Sahoo et al., 2021) to model natural progressive time-series changes in major cellular compartments that initiate, propagate and perpetuate inflammation in IBD and are likely to be important for disease progression. BoNE provides an integrated platform for the construction, visualization and querying of a network of progressive changes much like a disease map. A published gene signature for Barrett’s Esophagus UP genes and DOWN genes (Wang et al., 2006) were used in BoNE to train Boolean models that distinguish SPT6-depleted and Control samples in GSE153129. Another gene signature for SPT6-depleted versus Control (GSE153129) was found using the differentially expressed gene file provided earlier (Li et al., 2021) (Table S2), where genes were considered significant if they had a log fold change of greater than 10 or less than -10 and p-value of less than 0.1. The gene signature for SPT6-depleted versus Control samples (GSE153129) was used train Boolean models that distinguish normal esophagus and Barrett’s metaplasia in humans (GSE100843, GSE65013, GSE64894, GSE39491, GSE49292, GSE26886, GSE34619, GSE13083, E-MTAB-4054), or WT (wildtype) versus p63-/- in mice (GSE96831). In this model, a path score is computed as mentioned in section “Generation of gene signature scores” that is used to order the samples. The sample ordering is evaluated using the sample annotation (Normal, Barrett’s Esophagus) using ROC-AUC.
Correlation analysis
Correlation analysis was performed using Python pandas.DataFrame.corr (version 1.1.5). Normalized counts (that are not log-reduced) for up- and down- regulated genes (filtered by Fold-change (SPT6i vs. CTLi) > 10) were used to recreate the correlation matrix published earlier (Li et al., 2021), with the addition of Barrett’s metaplasia samples. The analysis includes a selection of samples from the following GEO datasets: GSE153129 (SPT6-depleted and Control), GSE120795 (colon, brain, skeletal muscle), GSE129153 (adipocyte), GSE148818 (trachea) and GSE58963 (BE) Specific sample numbers used to generate the correlation matrix are provided in Table S1.
GeneSet enrichment analysis (GSEA)
GeneSet Enrichment Analysis (GSEA) was performed using Python gseapy (0.10.2 package). Difference in average expression values of two groups is used to compute gene rank file. Metaplasia-specific and intestine-specific mouse genes (Wang et al., 2011) were converted to human genes using BoNE. Intestine-specific genes only, metaplasia-specific genes only, and combined metaplasia-specific and intestine-specific genes correspond to the three genesets tested for Barrett’s metaplasia versus small intestine samples (GSE13083), and SPT6-depleted (GSE153129) versus small intestine samples (GSE157059). GSEA pre-ranked analysis is performed on the precomputed rank file to check the significance of geneset enrichment score and generate the enrichment plot. GSEA computes four key statistics for the gene set enrichment analysis report: Enrichment Score (ES), Normalized Enrichment Score (NES), False Discovery Rate (FDR), Nominal P Value.
Isolation and culture of human small intestine organoids
Intestinal organoids were isolated and cultured following methods optimized previously (Sharma et al., 2021) and explained here briefly. Human ileum biopsies were collected from healthy adult male and female subjects undergoing routine colonoscopy for colon cancer screening using the protocol approved by the Human Research Protection Program Institutional Review Board (Project ID# 190105). To isolate organoids, human ileal tissue specimens were digested in collagenase type I at 37°C. Vigorous pipetting was performed every 10 minutes until tissue fragments dissociated into single epithelial units. Subsequently, wash media (DMEM/F12, 1X glutamax, 10% FBS) was added to neutralize the collagenase digestion, and the cell suspension was passed through a 70 μm filter. The cells were seeded in Matrigel domes and cultured in proliferation media in a 37°C/5% CO2 humidified incubator. Media changes were performed every 2 days until organoids formed and reached confluency.
For all the deidentified human subjects, information including age, ethnicity, gender, previous history of the disease, and the use of medications was collected from the chart following the rules of HIPAA. Each human participant was recruited to the study following an approved human research protocol and signed an informed consent form approved by the Human Research Protection Program at the University of California, San Diego. Each donor agrees that their gastro-intestinal specimens will be used to generate an enteroid line at UC San Diego’s HUMANOIDTM Center of Research Excellence (CoRE) for functional studies.
Exposure to acid injury
Human epidermal keratinocytes were cultured under 2-D and 3-D conditions in Human EpiVita Media (Cell Applications, 141-500a). Keratinocytes were seeded in monolayers at 2E4 cells per well and in Matrigel at 1E5 cells per well in a 12 well plate. Media changes were performed every 2-3 days. Cultures were maintained at 37°C/5% CO2 humidified conditions for 5-6 days until the monolayers reached confluency, and spheroids formed in the 3D culture. Subsequently, cells were cultured in acidic media with a pH range of 4.5-7.5. The media was prepared by adding 1N HCl drop-wise to EpiVita media until the desired pH was achieved. After 2 hours, the acidic media was removed from the keratinocytes and replaced with fresh EpiVita media. Cells were cultured for an additional 6 hours.
RNA isolation
The Matrigel domes were scraped from the surface of the 12 well plate, and spheroids were collected in cell recovery solution (Corning, 354253) and incubated for 1 h at 4°C under constant rotation. Monolayers and spheroids were lysed in 200 μl of RNA lysis buffer per well, and RNA Isolation was performed following instructions from the Zymo Research Quick-RNA MicroPrep Kit (R1051).
Quantitative (q)RT-PCR
Gene expression in spheroids and monolayers was measured by qRT-PCR using 2x SYBR Green qPCR Master Mix (Bimake, B21203). cDNA was amplified with gene-specific primer/probe set for SPT6 and Barrett’s Esophagus markers and qScript cDNA SuperMix 5x (Quanta Biosciences, 95048). qRT-PCR was performed with the Applied Biosystems QuantStudio 5 Real-Time PCR System. Cycling parameters were as follows: 95°C for 20 s, followed by 40 cycles of 1 s at 95°C and 20 s at 60°C. All samples were assayed in triplicate and eukaryotic 18S ribosomal RNA was used as a reference. Primer sequences are provided in Table of reagents (above).
Quantitative immunoblotting
For immunoblotting, keratinocytes protein samples were boiled in Laemmli sample buffer, separated by SDS-PAGE and transferred onto 0.4mm PVDF membrane (Millipore) prior to blotting. Post transfer, membranes were blocked using 5% Non-fat milk or 5% BSA dissolved in PBS. Primary antibodies (anti-SPT6; Thermo Fisher, A300-801A; dilution 1:500) were prepared in blocking buffer containing 0.1% Tween-20 and incubated with blots, rocking overnight at 4°C. After incubation, blots were incubated with secondary antibodies for one hour at room temperature, washed, and imaged using a dual-color Li-Cor Odyssey imaging system.
Quantification and statistical analysis
Statistical analyses in computational studies
All statistical tests were performed using R version 3.2.3 (2015-12-10). Standard t-tests were performed using Python scipy.stats.ttest_ind package (version 0.19.0) with Welch’s Two Sample t-test (unpaired, unequal variance (equal_var=False), and unequal sample size) parameters. Linear regression was performed using Python scipy.stats.linregress package (version 1.7.0). Multiple hypothesis correction were performed by adjusting p-values with statsmodels.stats.multitest.multipletests (fdr_bh: Benjamini/Hochberg principles) (Fabregat et al., 2018). Violin, Swarm and Bubble plots are created using Python seaborn package version 0.10.1.
Statistical analyses in experimental studies and replications
All experiments were repeated at least three times, and results were presented either as one representative experiment or as average ± S.E.M. Statistical significance was assessed with unpaired Student’s t test. For all tests, a p-value of 0.05 was used as the cutoff to determine significance. The actual p-values are indicated in each figure. All statistical analysis was performed using GraphPad prism 8.
Acknowledgments
We thank George Sen for transparent communications and for providing access to reagents. This paper was supported by the National Institute of Health (NIH, United States) R01 AI141630 and R01 CA100768 (to P.G), R01 GM138385 (to D.S), T32 GM8806 (to D.V) and R01 AI155696, UG3TR003355 and UG3TR002968 (to D.S, P.G and S.D).
Author contributions
D.V. carried out the computational analysis, with supervision from D.S and P.G. M.F and C.T carried out the studies on 3D organoids under the supervision of S.D and P.G. D.S., S.D., and P.G. provided transdisciplinary expertise, resources, and wrote and edited the manuscript. P.G conceived and directed the project.
Declaration of interests
The authors declare no competing interests.
Published: October 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103121.
Contributor Information
Soumita Das, Email: sodas@ucsd.edu.
Debashis Sahoo, Email: dsahoo@ucsd.edu.
Pradipta Ghosh, Email: prghosh@ucsd.edu.
Supplemental information
Data and code availability
The source code is available at https://github.com/sahoo00/BoNE. A bash script scr-be is provided to download all the datasets from our Hegemon web server using a perl script. A Jupyter notebook BE-Analysis.ipynb is provided to perform the analysis and generate the figures in this manuscript. Software programs (listed in Key resources table) are all publicly accessible through valid licenses.
References
- Barrett T., Suzek T.O., Troup D.B., Wilhite S.E., Ngau W.C., Ledoux P., Rudnev D., Lash A.E., Fujibuchi W., Edgar R. NCBI GEO: mining millions of expression profiles—database and tools. Nucleic Acids Res. 2005;33:D562–D566. doi: 10.1093/nar/gki022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Fang Y., Tevebaugh W., Orlando R.C., Shaheen N.J., Chen X. Molecular mechanisms of Barrett's esophagus. Dig. Dis. Sci. 2011;56:3405–3420. doi: 10.1007/s10620-011-1885-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crum C.P., Mckeon F.D. p63 in epithelial survival, germ cell surveillance, and neoplasia. Annu. Rev. Pathol. 2010;5:349–371. doi: 10.1146/annurev-pathol-121808-102117. [DOI] [PubMed] [Google Scholar]
- Curtius K., Rubenstein J.H., Chak A., Inadomi J.M. Computational modelling suggests that Barrett’s oesophagus may be the precursor of all oesophageal adenocarcinomas. Gut. 2021;70:1435–1440. doi: 10.1136/gutjnl-2020-321598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalerba P., Kalisky T., Sahoo D., Rajendran P.S., Rothenberg M.E., Leyrat A.A., Sim S., Okamoto J., Johnston D.M., Qian D. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat. Biotechnol. 2011;29:1120–1127. doi: 10.1038/nbt.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalerba P., Sahoo D., Paik S., Guo X., Yothers G., Song N., Wilcox-Fogel N., Forgo E., Rajendran P.S., Miranda S.P. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N. Engl. J. Med. 2016;374:211–222. doi: 10.1056/NEJMoa1506597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniely Y., Liao G., Dixon D., Linnoila R.I., Lori A., Randell S.H., Oren M., Jetten A.M. Critical role of p63 in the development of a normal esophageal and tracheobronchial epithelium. Am. J. Physiol. Cell Physiol. 2004;287:C171–C181. doi: 10.1152/ajpcell.00226.2003. [DOI] [PubMed] [Google Scholar]
- Dunn J., Garde J., Dolan K., Gosney J.R., Sutton R., Meltzer S.J., Field J.K. Multiple target sites of allelic imbalance on chromosome 17 in Barrett's oesophageal cancer. Oncogene. 1999;18:987–993. doi: 10.1038/sj.onc.1202371. [DOI] [PubMed] [Google Scholar]
- Dunn J.R., Garde J., Dolan K., Gosney J.R., Oates B.C., Watson A.J., Fielding P., Field J.K. The evolution of loss of heterozygosity on chromosome 17 during the progression to Barrett's adenocarcinoma involves a unique combination of target sites in individual specimens. Clin. Cancer Res. 2000;6:4033–4042. [PubMed] [Google Scholar]
- Dvorak K., Goldman A., Kong J., Lynch J.P., Hutchinson L., Houghton J.M., Chen H., Chen X., Krishnadath K.K., Westra W.M. Molecular mechanisms of Barrett's esophagus and adenocarcinoma. Ann. N. Y Acad. Sci. 2011;1232:381–391. doi: 10.1111/j.1749-6632.2011.06062.x. [DOI] [PubMed] [Google Scholar]
- Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A., Jupe S., Matthews L., Sidiropoulos K., Gillespie M., Garapati P., Haw R., Jassal B., Korninger F., May B. The reactome pathway knowledgebase. Nucleic Acids Res. 2018;46:D649–D655. doi: 10.1093/nar/gkx1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald R.C. Molecular basis of Barrett's oesophagus and oesophageal adenocarcinoma. Gut. 2006;55:1810–1820. doi: 10.1136/gut.2005.089144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald R.C., Omary M.B., Triadafilopoulos G. Dynamic effects of acid on Barrett's esophagus. An ex vivo proliferation and differentiation model. J. Clin. Invest. 1996;98:2120–2128. doi: 10.1172/JCI119018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P., Swanson L., Sayed I.M., Mittal Y., Lim B.B., Ibeawuchi S.R., Foretz M., Viollet B., Sahoo D., Das S. The stress polarity signaling (SPS) pathway serves as a marker and a target in the leaky gut barrier: implications in aging and cancer. Life Sci Alliance. 2020;3:e201900481. doi: 10.26508/lsa.201900481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gindea C., Birla R., Hoara P., Caragui A., Constantinoiu S. Barrett esophagus: history, definition and etiopathogeny. J. Med. Life. 2014;7:23–30. [PMC free article] [PubMed] [Google Scholar]
- Gonzales K.A.U., Fuchs E. Skin and its regenerative powers: an alliance between stem cells and their niche. Dev. Cell. 2017;43:387–401. doi: 10.1016/j.devcel.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry R.A., Bolstad B.M., Collin F., Cope L.M., Hobbs B., Speed T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry R.A., Hobbs B., Collin F., Beazer-Barclay Y.D., Antonellis K.J., Scherf U., Speed T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Jiang M., Li H., Zhang Y., Yang Y., Lu R., Liu K., Lin S., Lan X., Wang H., Wu H. Transitional basal cells at the squamous-columnar junction generate Barrett's oesophagus. Nature. 2017;550:529–533. doi: 10.1038/nature24269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastelein F., Biermann K., Steyerberg E.W., Verheij J., Kalisvaart M., Looijenga L.H., Stoop H.A., Walter L., Kuipers E.J., Spaander M.C. Aberrant p53 protein expression is associated with an increased risk of neoplastic progression in patients with Barrett's oesophagus. Gut. 2013;62:1676–1683. doi: 10.1136/gutjnl-2012-303594. [DOI] [PubMed] [Google Scholar]
- Kong J., Nakagawa H., Isariyawongse B.K., Funakoshi S., Silberg D.G., Rustgi A.K., Lynch J.P. Induction of intestinalization in human esophageal keratinocytes is a multistep process. Carcinogenesis. 2009;30:122–130. doi: 10.1093/carcin/bgn227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Xu X., Tiwari M., Chen Y., Fuller M., Bansal V., Tamayo P., Das S., Ghosh P., Sen G.L. SPT6 promotes epidermal differentiation and blockade of an intestinal-like phenotype through control of transcriptional elongation. Nat. Commun. 2021;12:784. doi: 10.1038/s41467-021-21067-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maag J.L.V., Fisher O.M., Levert-Mignon A., Kaczorowski D.C., Thomas M.L., Hussey D.J., Watson D.I., Wettstein A., Bobryshev Y.V., Edwards M. Novel aberrations uncovered in Barrett's esophagus and esophageal adenocarcinoma using whole transcriptome sequencing. Mol. Cancer Res. 2017;15:1558–1569. doi: 10.1158/1541-7786.MCR-17-0332. [DOI] [PubMed] [Google Scholar]
- Mcdonald S.A., Lavery D., Wright N.A., Jansen M. Barrett oesophagus: lessons on its origins from the lesion itself. Nat. Rev. Gastroenterol. Hepatol. 2015;12:50–60. doi: 10.1038/nrgastro.2014.181. [DOI] [PubMed] [Google Scholar]
- Miyoshi H., Stappenbeck T.S. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 2013;8:2471–2482. doi: 10.1038/nprot.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Nowicki-Osuch K., Zhuang L., Jammula S., Bleaney C.W., Mahbubani K.T., Devonshire G., Katz-Summercorn A., Eling N., Wilbrey-Clark A., Madissoon E. Molecular phenotyping reveals the identity of Barrett's esophagus and its malignant transition. Science. 2021;373:760–767. doi: 10.1126/science.abd1449. [DOI] [PubMed] [Google Scholar]
- Pachter L. Models for transcript quantification from RNA-Seq. 2011. https://ui.adsabs.harvard.edu/abs/2011arXiv1104.3889P arXiv e-prints.
- Petty E.M., Kalikin L.M., Orringer M.B., Beer D.G. Distal chromosome 17q loss in Barrett's esophageal and gastric cardia adenocarcinomas: implications for tumorigenesis. Mol. Carcinog. 1998;22:222–228. doi: 10.1002/(sici)1098-2744(199808)22:4<222::aid-mc3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Que J., Garman K.S., Souza R.F., Spechler S.J. Pathogenesis and cells of origin of Barrett's esophagus. Gastroenterology. 2019;157:349–364 e1. doi: 10.1053/j.gastro.2019.03.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman S., Pétré A., Thépot A., Hautefeuille A., Scoazec J.Y., Mion F., Hainaut P. Downregulation of p63 upon exposure to bile salts and acid in normal and cancer esophageal cells in culture. Am. J. Physiol. Gastrointest. Liver Physiol. 2007;293:G45–G53. doi: 10.1152/ajpgi.00583.2006. [DOI] [PubMed] [Google Scholar]
- Sahoo D., Dill D.L., Gentles A.J., Tibshirani R., Plevritis S.K. Boolean implication networks derived from large scale, whole genome microarray datasets. Genome Biol. 2008;9:R157. doi: 10.1186/gb-2008-9-10-r157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo D., Swanson L., Sayed I.M., Katkar G.D., Ibeawuchi S.R., Mittal Y., Pranadinata R.F., Tindle C., Fuller M., Stec D.L. Artificial intelligence guided discovery of a barrier-protective therapy in inflammatory bowel disease. Nat. Commun. 2021;12:4246. doi: 10.1038/s41467-021-24470-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T., STANGE D.E., Ferrante M., Vries R.G., van Es J.H., van den Brink S., van Houdt W.J., Pronk A., van Gorp J., Siersema P.D., Clevers H. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology. 2011;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- Senoo M., Pinto F., Crum C.P., Mckeon F. p63 Is essential for the proliferative potential of stem cells in stratified epithelia. Cell. 2007;129:523–536. doi: 10.1016/j.cell.2007.02.045. [DOI] [PubMed] [Google Scholar]
- Sharma A., lee J., Fonseca A.G., Moshensky A., Kothari T., Sayed I.M., Ibeawuchi S.R., Pranadinata R.F., Ear J., Sahoo D. E-cigarettes compromise the gut barrier and trigger inflammation. iScience. 2021;24:102035. doi: 10.1016/j.isci.2021.102035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza R.F., Shewmake K., Terada L.S., Spechler S.J. Acid exposure activates the mitogen-activated protein kinase pathways in Barrett's esophagus. Gastroenterology. 2002;122:299–307. doi: 10.1053/gast.2002.30993. [DOI] [PubMed] [Google Scholar]
- Stairs D.B., Nakagawa H., Klein-Szanto A., Mitchell S.D., Silberg D.G., Tobias J.W., Lynch J.P., Rustgi A.K. Cdx1 and c-Myc foster the initiation of transdifferentiation of the normal esophageal squamous epithelium toward Barrett's esophagus. PLoS One. 2008;3:e3534. doi: 10.1371/journal.pone.0003534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift A., Risk J.M., Kingsnorth A.N., Wright T.A., Myskow M., Field J.K. Frequent loss of heterozygosity on chromosome 17 at 17q11.2-q12 in Barrett's adenocarcinoma. Br. J. Cancer. 1995;71:995–998. doi: 10.1038/bjc.1995.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong A.B., Kretz M., Ridky T.W., Kimmel R., Khavari P.A. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev. 2006;20:3185–3197. doi: 10.1101/gad.1463206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmer J.P., Sahoo D., Chin R.K., Ho P.L., Tang C., Kurtova A.V., Willingham S.B., Pazhanisamy S.K., Contreras-Trujillo H., Storm T.A. Three differentiation states risk-stratify bladder cancer into distinct subtypes. Proc. Natl. Acad. Sci. U. S. A. 2012;109:2078–2083. doi: 10.1073/pnas.1120605109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.S., Canto M.I. Predicting neoplastic progression in Barrett's esophagus. Ann. Gastroentol. Hepatol. 2010;1:1–10. [PMC free article] [PubMed] [Google Scholar]
- Wang S., Zhan M., Yin J., Abraham J.M., Mori Y., Sato F., Xu Y., Olaru A., Berki A.T., Li H. Transcriptional profiling suggests that Barrett's metaplasia is an early intermediate stage in esophageal adenocarcinogenesis. Oncogene. 2006;25:3346–3356. doi: 10.1038/sj.onc.1209357. [DOI] [PubMed] [Google Scholar]
- Wang X., Ouyang H., Yamamoto Y., Kumar P.A., Wei T.S., Dagher R., Vincent M., Lu X., Bellizzi A.M., Ho K.Y. Residual embryonic cells as precursors of a Barrett's-like metaplasia. Cell. 2011;145:1023–1035. doi: 10.1016/j.cell.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt F.M. Mammalian skin cell biology: at the interface between laboratory and clinic. Science. 2014;346:937–940. doi: 10.1126/science.1253734. [DOI] [PubMed] [Google Scholar]
- Xian W., Duleba M., Zhang Y., Yamamoto Y., Ho K.Y., Crum C., Mckeon F. The cellular origin of Barrett's esophagus and its stem cells. Adv. Exp. Med. Biol. 2019;1123:55–69. doi: 10.1007/978-3-030-11096-3_5. [DOI] [PubMed] [Google Scholar]
- Xian W., Ho K.Y., Crum C.P., Mckeon F. Cellular origin of Barrett's esophagus: controversy and therapeutic implications. Gastroenterology. 2012;142:1424–1430. doi: 10.1053/j.gastro.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A., Schweitzer R., Sun D., Kaghad M., Walker N., Bronson R.T., Tabin C., Sharpe A., Caput D., Crum C., Mckeon F. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- Zhang W., Wang D.H. Origins of metaplasia in Barrett's esophagus: is this an esophageal stem or progenitor cell disease? Dig. Dis. Sci. 2018;63:2005–2012. doi: 10.1007/s10620-018-5069-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source code is available at https://github.com/sahoo00/BoNE. A bash script scr-be is provided to download all the datasets from our Hegemon web server using a perl script. A Jupyter notebook BE-Analysis.ipynb is provided to perform the analysis and generate the figures in this manuscript. Software programs (listed in Key resources table) are all publicly accessible through valid licenses.