

Abstract
Flow cytometers have been utilized for the analysis of submicron-sized particles since the late 1970s. Initially, virus analyses preceded extracellular vesicle (EV), which began in the 1990s. Despite decades of documented use, the lack of standardization in data reporting has resulted in a growing body of literature that cannot be easily interpreted, validated, or reproduced. This has made it difficult for objective assessments of both assays and instruments, in-turn leading to significant hindrances in scientific progress, specifically in the study of EVs, where the phenotypic analysis of these submicron-sized vesicles is becoming common-place in every biomedical field. Methods for fluorescence and light scatter standardization are well established and the reagents to perform these analyses are commercially available. However, fluorescence and light scatter calibration are not widely adopted by the small particle community as methods to standardize flow cytometry (FCM) data. In this proof-of-concept study carried out as a resource for use at the CYTO2019 workshop, we demonstrate for the first-time simultaneous fluorescence and light scatter calibration of small particle data to show the ease and feasibility of this method for standardized FCM data reporting. This data was acquired using standard configuration commercial flow cytometers, with commercially available materials, published methods, and freely available software tools. We show that application of light scatter, fluorescence, and concentration calibration can result in highly concordant data between FCM platforms independent of instrument collection angle, gain/voltage settings, and flow rate; thus, providing a means of cross comparison in standard units.
Keywords: calibration, fluorescence, light scatter, EVs, reference materials, small particles, virus
Efforts to standardize flow cytometry (FCM) data began several decades ago, following the production of multiple commercial FCM platforms. Fluorescence standardization methods have been established since the late 1990’s, yet these have not been widely adopted by the FCM community for a variety of reasons. Nonetheless, extensive literature and commercially available materials have been developed to facilitate fluorescence calibration in cellular analysis (1–7). These methods allow for the conversion of fluorescence intensities, which are arbitrary units, into standardized units of fluorescence recognized by the National Institutes of Standards and Technology (NIST), such as molecules of equivalent soluble fluorophore (MESF) or equivalent number of reference fluorophore (ERF) (1–7). These protocols can be readily applied to the analysis of small particles, as demonstrated in this current study. Although it can be assumed that all commercial flow cytometers can fully resolve cellular populations from noise, the same is not true for small particles such as EVs. Therefore, supplementary methods need to be employed to validate small particle FCM analysis.
The detection and characterization of small particles, in the form of viruses, using light scatter triggering was published over 40 years ago (8). Calibration of light scatter from a flow cytometer was demonstrated for small particles in 2009 by Fattacioli et al., and specifically for EVs by van der Pol et al. in 2012 (9, 10). Despite having been established for a decade, the use of light scatter calibration in small particle FCM has been limited, partly owing to the complexity of Mie Theory-based scatter modeling required for light scatter signal normalization. As an answer to this, in 2015, a commercial light scatter calibration assay (Rosetta Calibration by Exometry) was released to facilitate this process and used in an FCM standardization study for the International Society on Thrombosis and Haemostasis (ISTH) (11). In 2019, FCMPASS, a free alternative small particle flow cytometer calibration software package for light scatter and fluorescence became available (12). While there is now both software and materials available for light scatter calibration, further support in the form of education and resource materials is required for the correct implementation and assessment in accuracy of calculated models.
Currently, both fluorescence and light scatter calibration is underutilized in the field of small particle FCM. Calibration for small particle analysis is, however, critical due to the majority of commercial FCM instrumentation working at their detection limits when analyzing EVs and other biological particles <200 nm in diameter. Since flow cytometers have a wide range of optical configurations, methods are required for standardized data reporting such that meaningful biological conclusions can be made. There is currently no consensus method for this. This study was carried out for a CYTO2019 Workshop where the feasibility of combining scatter and fluorescence calibration for small particle FCM was presented (13). Calibrations were performed using FCMPASS software package with commercially available reference materials to convert fluorescence intensity to MESF and light scatter to diameter. A biological reference particle in the form of a fluorescently-tagged virus was used to validate this method (12). Conversion of fluorescence and light scatter intensities from the antibody-labeled virus into PE MESF and nanometers allowed for direct comparison of the data from the same virus sample collected on two different FCM platforms. This is the seminal report of the combined application of fluorescence and light scatter calibration as a method toward standardized data reporting for small particle FCM (12).
Materials and Methods
Sample Preparation
MV-M-sfGFP (ViroFlow Technologies, Canada, Lot#S1003A), murine leukemia virus tagged with super-folder GFP (sfGFP), was reconstituted and diluted as per manufacturer’s instructions according to the particle concentration provided. The virus particles were stained at a concentration of 5 × 108 particles ml−1 and labeled with anti-GFP PE antibody (Clone FM264G, Bio-Legend) at 0.4 μg ml−1 in a 100 μl staining volume for a minimum of 30 min at room temperature, protected from light. To achieve this staining concentration, the lyophilized pellet of MV-M-sfGFP containing 1.8 × 109 particles was resuspended in the manufacturer recommended 200 μl volume of H2O and diluted to 109 particles ml−1 by adding 1.6 ml of PBS. 50 μl of MV-M-sfGFP at 109 particlesml−1 was then added to 50 μl of 0.8 μgml−1 anti-GFP antibody (1 μl of 0.2 mgml−1 antibody stock into 250 μl PBS). Upon completion of incubation, 2 μl of the labeled virus was diluted into 1 ml of PBS (~106 particlesml−1) immediately prior to analysis by FCM. Control samples such as antibody alone, MV-zero (virus expressing no GFP, Lot#Z1003A), and unstained MV-M-sfGFP samples were similarly prepared. All dilutions were made using 0.1 μm filtered PBS (PBS ×1, no Ca2+, noMg2+, Wisent).
Cytometer Configuration
BD LSR Fortessa 50 mW 488 nm laser, 50 mW 561 nm, 488/10 (SSC), 561–586/15 (PE), 488–530/30 (GFP). Beckman Coulter CytoFLEX S, 80 mW 405 nm, 50 mW 561 nm, 405/8 (SSC), 561–585/42 (PE), 488–525/40 (GFP). Further details on cytometer settings and acquisition can be found in attached MIFlowCyt (Supplementary Table 1) and MIFlowCyt-EV (Supplementary Table 2) documents (14, 15).
Small Particle Sample Acquisition
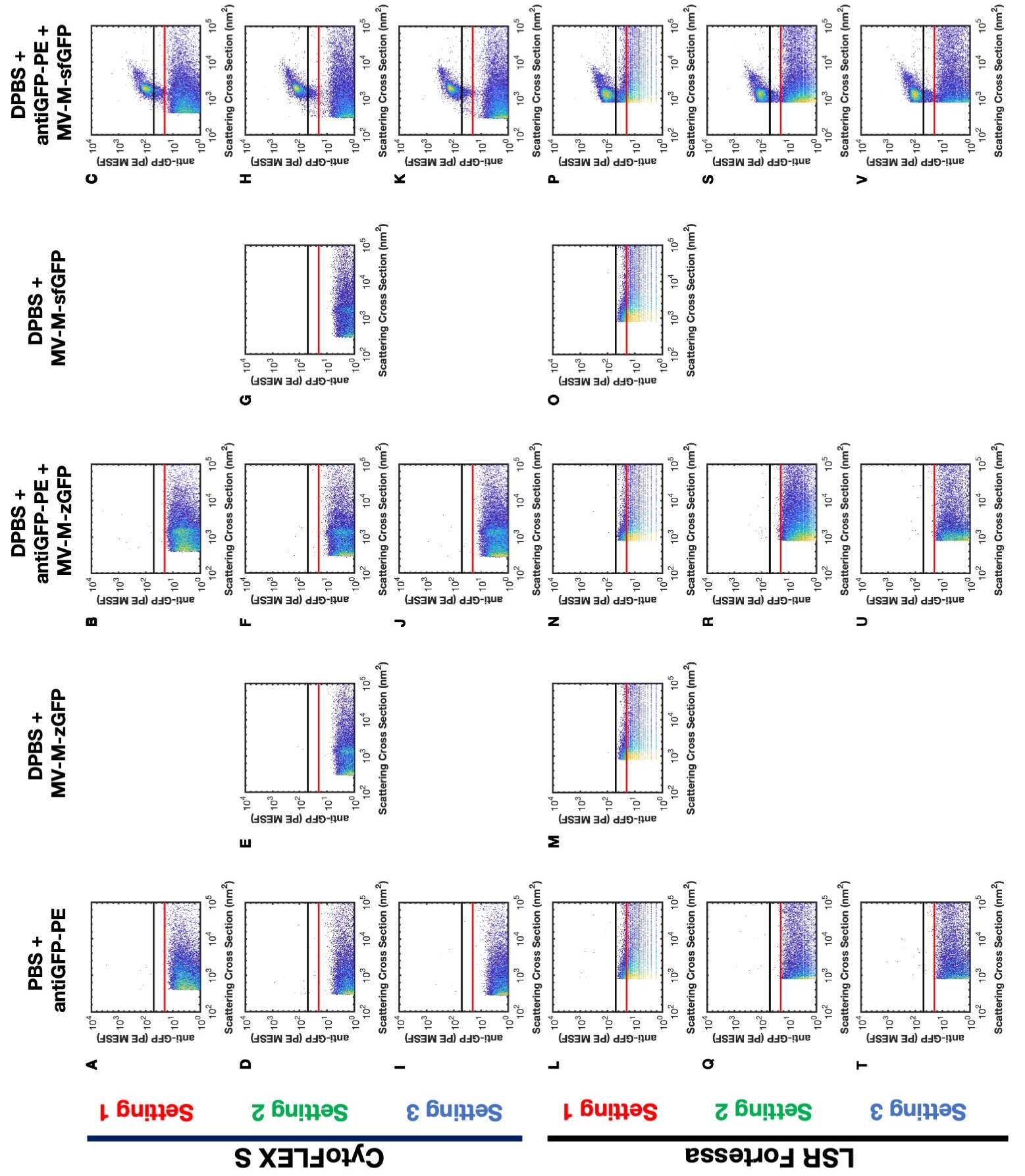
The same bead and antibody-labeled virus samples were acquired on an LSR Fortessa (BD Biosciences) and CytoFLEX S (Beckman Coulter). Samples were acquired using three different detection settings (for SSC and PE) to demonstrate that calibration can normalize data independently of acquisition settings and FCM platform. The settings and acquisition plots are summarized in Table 1 and Supplementary Figure 1, respectively. The detector settings for fluorescence detection on the CytoFLEX S and LSR Fortessa were determined using the PE MESF beads (QuantiBrite, BD Biosciences). On the Fortessa, setting 1 being the lowest voltage where the dimmest bead population was resolved from background and setting 3 being the highest voltage where the brightest bead population was within maximum detection limit. Setting 2 was approximately halfway between settings 1 and 3. Gain settings for fluorescence detection on the CytoFLEX S were similarly determined. For scatter calibration, three separate gains were used on the CytoFLEX S. For setting 1, side scatter (SSC) gain was chosen where the 600 nm NIST-traceable polystyrene bead population was within the range of detection. Settings 2 and 3 SSC voltages were the same for all three settings acquired on the LSR Fortessa. This was due to the LSR Fortessa being unable to fully resolve the virus above the trigger threshold. Since the concentration and comparison of fluorescent data would be heavily influenced by the SSC triggering threshold, a single voltage and trigger threshold was maintained. This provided a consistently detected population to compare across different fluorescent channel settings. The difference between the three scatter data sets on the CytoFLEX S are based on gating of MV-M-sfGFP at 20 and 50 PE MESF as outlined in the respective figures. All samples were acquired on the lowest preset instrument flow rates for 1 min. For the CytoFLEX S this was 10 μl min−1, verified using the built in calibration software and weighing samples of deionized water before and after acquisition. For the LSR Fortessa the flow rate was calculated to be approximately 18.2 μl min−1 by measuring spike-in beads whose concentration was determined using the CytoFLEX S.
Table 1.
Summary of scatter and fluorescence gain/voltage settings used for detection of anti-GFP PE labeled MV-M-sfGFP
| CYTOFLEX S (GAIN) | LSR FORTESSA (VOLTAGE) | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| SETTING | 1 | 2 | 3 | 1 | 2 | 3 |
|
| ||||||
| SSC channel | 125 | 180 | 250 | 410 | 410 | 410 |
| GFP channel | 3,000 | 3,000 | 3,000 | 540 | 540 | 540 |
| PE channel | 300 | 1,000 | 2,000 | 463 | 605 | 650 |
| Trigger | VSSC | VSSC | VSSC | SSC | SSC | SSC |
| Threshold | 1,400 | 1,500 | 2,000 | 200 | 200 | 200 |
Light Scatter Calibration
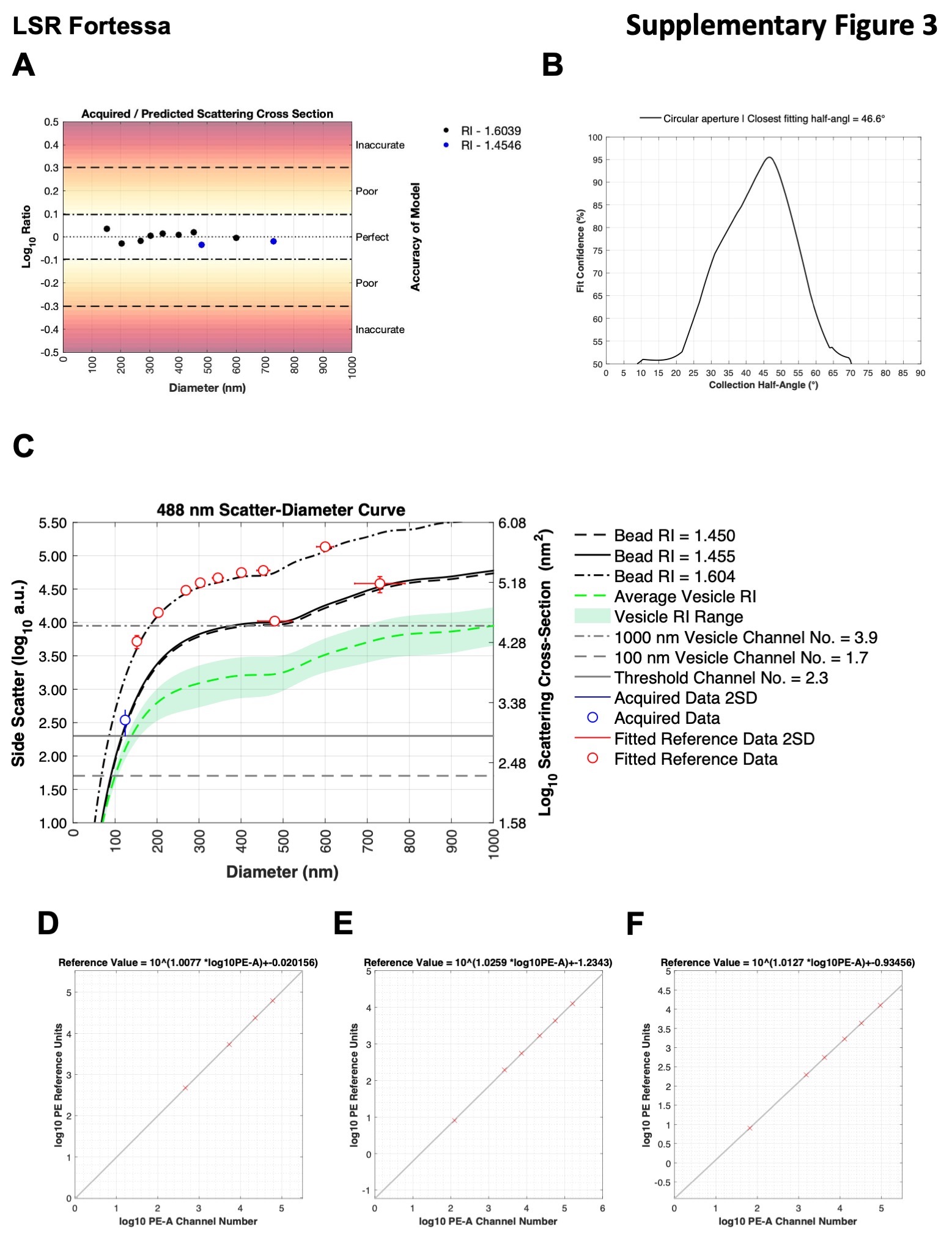
81, 100, 152, 203, 269, 303, 345, 401, 453, 568, and 600 nm polystyrene NIST-traceable beads (ThermoFisher Scientific) and 480 and 730 nm silica NIST-traceable beads (ThermoFisher Scientific) were acquired on each cytometer. Median SSC-H intensity (488 nm SSC on LSR Fortessa, 405 nm SSC-H on CytoFLEX S) were gated using FlowJo (v10.5.3). Mie modeling and subsequent conversion of light scatter intensity to diameter was performed using FCMPASS software (v2.17, http://nanopass.ccr.cancer.gov) (12). The background of how Mie theory and the FCMPASS software work is beyond the scope of this technical note and can be found in previously published literature (16, 17). Model input settings used including refractive indices, bead information, and statistics can be found in Supplementary Tables 3 and 4, with model outputs shown in Supplementary Figures 2 and 3. The effective refractive index (RI) of the MV-M-sfGFP virus was assumed to be 1.45 based on published literature and dispersion was accounted for using Sellmeier equations for water (18), (19). Samples were acquired with one scatter detector setting on the LSR Fortessa while three different scatter detector settings were acquired on the CytoFLEX S. A single bead population (152 nm) was used to cross-calibrate detector settings 2 with settings 1 and 3 on the CytoFLEX S with the full set of light scatter calibration beads acquired using detector setting 1.
Fluorescence Calibration
Linear regression was performed by converting acquired PE channel statistics and PE MESF reference bead values (QuantiBrite PE, Cat# 340495, Lot 73318, BD Biosciences) to logarithmic values before performing regression (Supplementary Figures 2D–F and 3D–F). PE MESF reference beads were acquired at a single voltage/gain on each instrument that allowed for the brightest bead to be within the range of detection of the instrument. To account for differing spectral filters and as a demonstration of utilizing ergonomic calibration methods, 8-peak rainbow beads (Cat# RCP-30–5A, Lot AF01, Spherotech) were cross calibrated to PE MESF values on each cytometer at the same voltage/gain as the PE MESF beads were acquired at. The derived MESF values are summarized in Supplementary Table 5. Median PE signals for each population were gated using FlowJo. The linear regressions for fluorescence calibration and conversion of .fcs files to MESF units was performed using FCMPASS software (v2.17, http://nanopass.ccr.cancer.gov) (12). The cross calibrated 8-peak rainbow beads were used to calibrate the PE intensity scales at detector settings 2 and 3 for both the LSR Fortessa and settings 1 and 3 for the CytoFLEX S.
Concentration Calibration
Virus concentration on the CytoFLEX S and LSR Fortessa was normalized using 200 nm fluorescent polystyrene (Green FluoSpheres, Cat# F8848, ThermoFisher Scientific) using the mean concentration of beads acquired using the CytoFLEX S postcalibration of the fluidic system at a low flow rate. Due to composition of the spike-in beads, aggregates were observed. Populations appearing as doublets, triplets, and so forth, were counted as a single bead.
Data and Statistical Analysis
All data analyses and figures were carried out using MATLAB (R2019b, Mathworks) unless otherwise stated. Samples were gated as shown in representative example (Fig. 1) and exhaustively in Supplementary Figure 1 with control samples shown in Supplementary Figure 4.
Fig 1.

Representative data demonstrating the comparability of uncalibrated and calibrated data across flow cytometers. (A and B) the side scatter versus anti-GFP-PE fluorescence of MV-MsfGFP are shown on the LSR Fortessa and CytoFLEX S, respectively. A comparison gate is drawn from 4 × 103 to 6 × 104 SSC-H and 1 × 103 to 3 × 104 PE-A. (C and D) the scatter cross section versus anti-GFP-PE (MESF) of MV-M-sfGFP are shown on the LSR Fortessa and CytoFLEX S, respectively. A comparison gate is drawn from 1 × 103 to 1 × 104 nm2 and 20 to 400 PE MESF. (E and F) the diameter (nm) versus anti-GFP-PE (MESF) of MV-M-sfGFP are shown on the LSR Fortessa and CytoFLEX S, respectively. A comparison gate is drawn from 100 to 180 nm to and 20 to 400 PE MESF (black). A second comparison gate is drawn from 118 to 180 nm to and 50 to 400 PE MESF (red dotted). (G and H) the diameter (nm) versus anti-GFP-PE (MESF nm−2) of MV-M-sfGFP are shown on the LSR Fortessa and CytoFLEX S, respectively. A comparison gate is drawn from 100 to 180 nm to and 5 × 10−4 to 8 × 10−3 PE MESF nm−2 (black).
RESULTS
Fluorescence Calibration
The median PE intensity of anti-GFP-PE labeled MV-MsfGFP from the LSR Fortessa and CytoFLEX S (gated from 50–400 PE MESF) ranged in arbitrary units of intensity from 111 to 17,604 (a 159-fold difference) (Table 2), across the three different detectors settings per cytometer (Table 1). The result of this was that a single gate could not be used irrespective of instruments or detector settings (Figs. 1A,B and 2B and Supplementary Fig. 1A–F). Upon calibration of fluorescence intensity to PE MESF units, median values ranged from 99 to 128 PE MESF (29% difference) across flow cytometer platforms. Within cytometry platforms this variation was seen to be 103–110 (7% difference) and 99–128 (29% difference) PE MESF for the LSR Fortessa and CytoFLEX S, respectively (Table 2). Variation within and between FCM platforms irrespective of detector settings was therefore considerably more consistent upon fluorescence calibration. Furthermore, a single gate from 20 to 400 PE MESF could be used to gate labeled MV-M-sfGFP irrespective of instrument or detector settings (Fig. 2E).
Table 2.
Summary of statistics obtained for detection of anti-GFP PE labeled MV-M-sfGFP. Light scatter and fluorescence statistics were calculated using a gate from 50 to 400 PE MESF units, with the 50th percentile (5th, 95th percentile) shown. Count and concentration statistics were calculated using a 50–400 PE MESF gate with a comparison gate at 50–400 PE MESF and 118–180 nm to account for the trigger threshold of the LSR Fortessa, which appears to cut of a small portion of the MV-M-sfGFP population
| CYTOFLEX S | LSR FORTESSA | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| SETTING | 1 | 2 | 3 | 1 | 2 | 3 |
|
| ||||||
| Light scatter | 6,542 | 9,349 | 13,121 | 348 | 348 | 347 |
| (a.u.) | (4,790, 10,264) | (6,722, 13,430) | (9,425, 19,751) | (240, 620) | (240, 620) | (240, 632) |
| Scatter cross section | 1,894 | 1,889 | 1,903 | 1,326 | 1,326 | 1,323 |
| (nm2) | (1,387, 2,981) | (1,358, 2,723) | (1,367, 2,889) | (915, 2,358) | (915, 2,349) | (915, 2,409) |
| Diameter | 120.7 | 120.6 | 120.8 | 130.3 | 130.3 | 130.2 (121, 146) |
| (nm) | (113, 133) | (113, 130) | (113, 132) | (121, 146) | (121, 146) | |
| PE | 2,117 | 8,925 | 17,604 | 111 | 847 | 1,454 |
| (a.u.) | (1,232, 3,368) | (4,824, 12,432) | (9,630, 25,227) | (64, 175) | (508, 1,283) | (894, 2,204) |
| PE | 99 | 128 | 128 | 110 | 108 (64, 164) | 103 |
| (MESF) | (58, 156) | (70, 178) | (70, 182) | (63, 173) | (62, 157) | |
| PE | 2.16 | 2.80 | 2.78 | 2.06 | 2.02 | 1.93 |
| (×10−3 MESF nm−2) | (0.133, 0.310) | (0.160, 0.362) | (0.162, 0.366) | (0.120, 0.300) | (0.122, 0.285) | (0.119, 0.273) |
| Count ×104 (50–400 PE MESF) | 1.38 | 1.85 | 1.68 | 2.55 | 2.05 | 2.38 |
| Count ×104 (50–400 PE MESF, 118–180 nm) | 1.12 | 1.49 | 1.37 | 2.55 | 2.05 | 2.38 |
| Concentration ×106 ml−1 (50–400 PE MESF) | 1.46 | 1.75 | 1.69 | 1.25 | 1.19 | 1.24 |
| Concentration ×106 ml−1 (50–400 PE MESF, 118–180 nm) | 1.18 | 1.41 | 1.38 | 1.25 | 1.19 | 1.24 |
Fig 2.

Overlays of uncalibrated versus calibrated data from anti-GFP-PE labeled MV-M-sfGFP on flow cytometers at different detector settings. (A) Uncalibrated light scatter intensity of anti-GFP-PE labeled MV-M-sfGFP is shown on the LSR Fortessa (black) and CytoFLEX S (blue). A comparison gate is drawn from 4 × 103 to 6 × 104 SSC-H. (B) Uncalibrated fluorescence intensity of anti-GFP-PE labeled MV-MsfGFP is shown on the LSR Fortessa (black) and CytoFLEX S (blue). A comparison gate is drawn from 1 × 103 to 3 × 104 PE-A. (C) Calibrated light scatter intensity in scatter cross section units of anti-GFP-PE labeled MV-M-sfGFP is shown on the LSR Fortessa (black) and CytoFLEX S (blue). A comparison gate is drawn from 1 × 103 to 1 × 104 nm2. (D) Calibrated light scatter intensity in diameter units of anti-GFP-PE labeled MV-M-sfGFP is shown on the LSR Fortessa (black) and CytoFLEX S (blue). A comparison gate is drawn from 100 to 180 nm (gray) and a second comparison gate from 118 to 180 nm (red) is drawn to allow for comparisons above the LSR Fortessa SSC-H trigger threshold. (E) Calibrated fluorescence intensity in PE MESF units of anti-GFP-PE labeled MV-M-sfGFP is shown on the LSR Fortessa (black) and CytoFLEX S (blue). A comparison gate is drawn from 20 to 400 PE MESF (gray) and a second comparison gate from 50 to 400 nm (red) is drawn to allow for comparisons above the background signal from the LSR Fortessa at low voltages. (F) Calibrated fluorescence intensity in PE MESF nm−2 units of anti-GFP-PE labeled MV-M-sfGFP is shown on the LSR Fortessa (black) and CytoFLEX S (blue). A comparison gate is drawn from 5 × 10−4 to 8 × 10−3 PE MESF nm−2 (gray).
Light Scatter Calibration
Side-scatter intensity of MV-M-sfGFP was compared between the LSR Fortessa and CytoFLEX S. Three scatter settings were tested on the CytoFLEX S and one setting on the LSR Fortessa (defined in Methods section and Table 1). The SSC signal of the MV-M-sfGFP virus between instruments ranged from 347 to 13,121 arbitrary units (a 37.8-fold difference) prior to calibration (Table 2). The result of this was that a single gate could not be used irrespective of instruments or detector settings (Figs. 1A,B and 2A and Supplementary Fig. 1A–F).
While scattering cross section is a valid metric to report calibrated light scatter measurements, it describes the statistical quantity of light that will reach the detector. This metric is influenced by the collection optics of the instrument, namely that larger collection angles will collect more light. The light scatter cross section can, therefore, normalize results from instruments with the same collection angle irrespective of detector settings, as seen in Figure 2C. Upon calibration of CytoFLEX S SSC signal from arbitrary units to scatter cross section the range reduced from a 37.8-fold difference to <1%. Scatter cross section as a unit cannot, however, normalize data between instruments with different collection angles. If the same population is acquired on two instruments with differing collection angles, the instrument with the larger collection angle will most likely result in a larger scattering cross section. This is observed when the CytoFLEX S and LSR Fortessa data are compared (Figs. 1C,D and 2C). The scatter cross section of the MV-M-sfGFP virus on the CytoFLEX S (collection half angle of 53.2°) was 1903 nm2, while the LSR Fortessa (collection half angle of 46.6°) had a scattering cross section of 1,323 nm2 (a difference of 44%) (Table 2).
To normalized light scatter signals irrespective of collection angle a scatter-diameter curve must be generated (Supplementary Figs. 2C and 3C) which takes into account the collection angle of the system. In order for the approximated diameter to be accurate, knowledge and incorporation of a suitable refractive index (RI) of the particles being normalized is required. It can be seen that when the MV-MsfGFP data is converted to units of diameter, the range between the instruments is reduced to 121–130 nm (a 10.7% difference) (Table 2 and Figs. 1E,F and 2D). Within the CytoFLEX S platform the variation in derived diameter irrespective of detector settings was seen to be <0.2% with derived diameter varying from 120.6 to 120.8 nm (Table 2). The comparability of the data between uncalibrated and calibrated values across the two CytoFLEX S and LSR Fortessa can be seen in representative examples (Figs. 1 and 2), and for every setting (Supplementary Fig. 1). Furthermore, a single gate from 100 to 180 nm can be used irrespective of instrument and detector settings to identify the virus population (Fig. 2D).
Epitope-Density/MESF-Density Calibration
Epitope-density describes the number of the epitopes over a unit of surface area, for example, epitope number nm−2. An alternative, in cases where there is more than one fluorophore per antibody, could be the number of fluorophores over unit of surface area, for example, MESF nm−2. The epitope number can be considered equivalent to the MESF values obtained from fluorescence calibration when the following criteria are met: (a) fluorophore to protein (F:P) ratio is 1:1 (20). Due to the large size of PE, this can generally be assumed when conjugated IgG, whereas small dyes such as fluorescein can vary considerably. (b) All antibodies have the same dye. Tandem dyes for example can result in variability of fluorescence due to the tandem not being present on all antibodies.(c) Antibody-labeling is optimized, that is, all epitopes are labeled. (d) One antibody is bound per one epitope, and (e) no steric hindrance impeding the antibody from binding to all expressed epitopes.
When compared to MV-M-sfGFP, EVs have a very wide diameter distribution (Fig. 3A) which is proportional to their surface area distribution (Fig. 3B). If the surface area of EVs scale with their epitope number, this too will be highly variable (Fig. 3C,D) (21–23). EVs have a lower RI and the majority are likely smaller than MV-M-sfGFP, resulting in decreased light scatter detectability. While high abundance epitopes can exist on EVs, many epitopes are likely much lower than GFP-expression on MV-M-sfGFP (22,23). Statistics, such as a median or mean, for reporting fluorescence and diameter are heavily dependent on how much of a population is being resolved. Therefore, if a population is not fully resolved the reporting of any statistic to describe the population in terms of light scatter, fluorescence, or concentration parameters requires it to accompanied by the limit of detection to be reproducible, for example, 3 × 106 particles ml−1 (30–300 PE MESF).
Fig 3.

Comparisons of uncalibrated versus calibrated abundance of detected anti-GFP-PE labeled MV-M-sfGFP on flow cytometers at different detector settings. (A) Comparison of acquired counts in 1 min on LSR Fortessa (black outline) and CytoFLEX S (blue outline) gated from 50 to 400 PE MESF units (gray symbol) and 50–400 PE MESF and >118 nm (red symbol). (B) Comparison of acquired concentration per ml in 1 min on LSR Fortessa (black outline) and CytoFLEX S (blue outline) gated from 50 to 400 PE MESF units (gray symbol) and 50–400 PE MESF and >118 nm (red symbol).
Upon calibration of fluorescence intensity to PE MESF nm−2 units, median values ranged from 2.02 × 10−3 to 2.78 × 10−3 PE MESF nm−2 (38% difference) across flow cytometer platforms. Within cytometry platforms this variation was seen to be 1.93 × 10−3 to 2.06 × 10−3 (7% difference) and 2.16 × 10−3 to 2.80 × 10−3 (30% difference) PE MESF for the LSR Fortessa and CytoFLEX S, respectively (Table 2). Variation within and between FCM platforms irrespective of detector settings was therefore considerably more consistent using PE MESF nm−2 than no calibration, with variation similar to that of MESF calibration (Figs. 1G,H and 2F).
The epitope- or MESF-density may potentially provide a useful metric to allow better differentiation and consistent reporting of epitope expression. This statistic removes variability that arises due to heterogeneity of surface area in samples where there is a large size range (e.g., EVs) and available instrumentation will have differing limits of detection. Given the current challenges to fully resolve all EVs in a sample, epitope density offers a means to report on expression level per particle when knowledge of expression within the whole population is not yet feasible. To further illustrate this point, a hypothetical example of surface marker expression on EVs is shown with values reported in epitope number in comparison with epitope density (Fig. 3D,E), assuming the limit of detection is 20 PE MESF (Fig. 3D, red dashed line). There can be significant overlap in expression between the example markers when simply reported as epitope number, while minimal overlap is seen in epitope density populations despite having a coefficient of variation of ±20%. Provided the epitope density scales consistently with the population, it is therefore feasible that the epitope/MESF-density would be consistent even if only a portion of the population is detectable. This would allow a much more consistent method of reporting and comparing data between instruments with differing limits of detection, although further validation would be required. This metric has not previously been demonstrated for small particles and does require care in implementation and interpretation due to potential inaccuracies. The reliability of epitope/MESF-density as a statistic will be dependent upon the precision of the MESF and diameter derivation methods.
Concentration Calibration
The technical specifications for the low flow rate on the LSR Fortessa is 12 μl min−1, while the lowest calibrated flow rate on the CytoFLEX S is 10 μl min−1. Using spike-in beads, the mean flow rate was found to be 18.2 μl min−1 on the LSR Fortessa. The recorded number of MV-M-sfGFP events if fully detected should theoretically have been 20% higher than those of the CytoFLEX S. In practice, the mean total count difference between instruments was 39% (Table 2 and Fig. 4A). This indicates that either the flow rate between the two instruments is not as stated, or the virus was resolved to different extents between the instruments, or a combination of the two. Differences between advertised and observed flow rates has previously been reported in small particle standardization studies (11).
Fig 4.

Reporting epitope abundance. (A) the diameter distribution of extracellular vesicles based on the current literature (dotted), assuming a log-normal distribution with a modal point at 100 nm, versus the diameter distribution of murine leukemia virus, 124 ± 14 (solid) (21). (B) a comparison of surface area distribution of extracellular vesicles (dotted) versus murine leukemia virus based on the diameter distribution. (C) Comparison of epitope number across EVs based on scaling previously published leukocyte (10 μm) epitope abundance for CD14 (110,000 epitopes) (blue), CD11a (55,000) (orange), and CD16 (10,000) (yellow) (22). Each has a variation of ±20%. The calculated GFP number of MV-M-sfGFP assuming one phycoerythrin molecule per antibody, one antibody per GFP, no steric hindrance, and enumeration using PE MESF regression. MV-M-sfGFP epitope data was based on acquired data from the CytoFLEX S at setting 2 (Table 1) is shown at 124 ± 14 nm and 128 ± 50 GFP epitopes. (D) Demonstrates epitope number distribution based on the surface area data (B) and the epitope number versus diameter plot (C). (E) Demonstrates reporting epitope-density distribution of each marker (CD14, CD11a, CD16, GFP) based on the data from plot C.
When spike-in beads were used to normalize the MVM-sfGFP concentration between the two instruments, the calculated mean concentration was shown to be 33% higher on the CytoFLEX S (1.63 × 106 particles ml−1) than on the LSR Fortessa (1.23 × 106 particles ml−1) (Table 2 and Fig. 4B). The inability of the LSR Fortessa to fully resolve MV-MsfGFP is a possible source of the concentration discrepancy between the two cytometers. This can be observed on the SSC parameter’s triggering threshold channel at 200 on Figures 1A and 2D and Supplementary Figure 1A–C. To investigate if this accounts for the difference observed in concentration, a gate above the scatter trigger threshold of the LSR Fortessa was constructed from 50–400 PE MESF and 118–180 nm (Figs. 1E,F and 2D,E). Using this gate, the difference in mean MV-M-sfGFP concentration on the CytoFLEX S was reduced to just 7.8% at 1.32 × 106 particle ml−1 versus 1.23 × 106 particles ml−1 (Table 2 and Figs. 2E,F and 4A,B).
While the spike-in beads used here demonstrate that concentration can provide effective normalization between platforms when flow cytometer sensitivities are accounted for through calibration, the accuracy of the concentration spike-in beads can vary. It was noted in this work that the choice of spike-in beads in the form of 200 nm FluoSpheres was suboptimal due to their tendency to aggregate. Future experiments should utilize more stable spike-in beads for added confidence in concentration enumeration.
Calibration Overview
When uncalibrated MV-M-sfGFP fluorescence and light scattering data is overlaid between the CytoFLEX S and LSR Fortessa, very few comparisons can be made despite this being the same sample acquired on both instruments (Fig. 2A,B). This is illustrative of the current state of much of the small particle literature. The use of any one of the calibration metrics (MESF units, scatter cross section, diameter, MESF nm−2) all considerably increased the comparability of data in contrast to uncalibrated data (Fig. 2C–F). Not only has this work exhibited the utility of fluorescence and light scatter calibration, but the relative ease of these methods have also been demonstrated. Once an instrument has been initially calibrated using MESF reference materials and a set of light scatter reference materials, future instrument measurements and samples can be cross calibrated using just two calibration samples; one for fluorescence (cross calibrated 8-peak beads) and one for light scatter (a single light scatter reference bead). On stable instrument optical models, this data can be transposed using a single standard due to the collection angle being unlikely to change. Once cross calibrated on a single platform, 8-peak rainbow beads should also be sufficient for MESF calibration, assuming collection filter sets are not altered. While calibration can be very ergonomic as outlined and demonstrated by cross calibrating, any time an instrument is modified or altered, for example, from a regular service, MESF and light scatter calibration should be conducted in full again. Furthermore, it is recommended that this calibration be done periodically for good practice and also to monitor instrument performance. Users may not always be aware of modifications made to core instruments and these calibrations would ideally have help from shared resource laboratory managers.
Discussion
Two main factors contributed to the consistency of the results in this report. (a) The same validation sample (PE-labeled MV-M-sfGFP) was used for all of the data acquisitions. This eliminated inconsistencies in staining intensity and highlights the importance of accurate reporting of both antibody concentrations as well as particle concentrations used for antibody-labeling experiments in small particle FCM. Discrepancies in either of these parameters greater than a factor of two can significantly impact staining intensity of small particles (24). (b) The specific types of beads used to perform the calibrations were pivotal for this standardization process. For the light scatter calibration beads, it is critical that beads are accurately sized and have, preferably measured, refractive indices. Light scatter calibration is best done with nonfluorescent beads, as fluorophores can alter the light scatter properties of beads. Light scatter calibration should preferably be performed on samples acquired by SSC-trigger threshold. This allowed for confident determination of the limit of light scatter sensitivity. If a fluorescent trigger is used and the SSC intensities are below those that could be acquired with a SSC-trigger threshold, it is critical to demonstrate that the models retain accuracy in the region they are being extrapolated to. For fluorescence calibration, beads with matching fluorophore to the biological particle were used. This was essential for the fluorescence intensities to be comparable between instruments with differing optical configurations, such as filter sets and laser wavelengths for excitation (24). In summary, the methods employed here compellingly show that standardized data reporting of fluorescence and light scatter is achievable for small particle FCM data when it is converted to standard units of MESF and diameter.
It is important to stress that the primary purpose of light scatter calibration is not to determine the absolute size of biological particles, but to demonstrate a means of comparing data in standardized units with a consistent population. MVM-sfGFP was chosen as a proof-of-principle sample due to it being a well-defined, homogeneous population. The assumed effective RI of the virus, 1.45 (18), significantly impacts the particle diameter predicted by FCMPASS, which in this case is congruent with published EM measurements of the virus. Validation of these methodologies for the extracellular vesicle (EV) field are required, and will be more challenging due to the heterogeneity of EV populations, with the majority of particles likely below the triggering threshold with a log-normal distribution (21). This will mean that normalization of the light scatter and fluorescence data between instruments will heavily rely upon accurate quantitation of the concentration. This is challenging as variations can occur in concentration with identical samples on the same platform, as we have shown in this pilot study. Further testing of fluidic stability and acquisition time for their effect on recorded concentration variation with an easily detectable, homogeneous population should therefore be a priority for laboratories attempting to use concentration measurements and would be the next step in this line of work.
Ultimately, calibration is critical to allow for meaningful biological conclusions and allow reproduction of work. The use of calibration can also aid researchers in identifying optimal assays and equipment for their work. Literature is beginning to emerge where fluorescence calibration is implemented; however, small particle studies and standardization efforts have predominantly used light scatter calibration as a normalization method (11, 25–30). While light scatter calibration has great utility, fluorescence calibration should be seen as a priority in any study utilizing fluorescence staining, whether it is immunophenotyping or fluorescent dyes. This is due to the general convention of reporting only the counts of fluorescent events instead of total detected counts of all events. To this end, a follow up standardization study is required that utilizes both fluorescence and light scatter calibration to compare instrument sensitivities and further validate the findings of this pilot study.
While the need for calibration is essential for the field to progress, its use alone does not ensure quality of research or that instrument settings are optimal. The use of optimal experimental design with controls such as serial dilution to ensure single particle detection, buffer with reagent controls to demonstrate lack of artifacts, and isotype or negative staining controls to show specificity are all required (15). A consensus for reporting of the methods and experimental design needs to be utilized for researchers to make easy comparisons between studies and allow for reproduction of data. This effort is actively being undertaken by the International Society of Extracellular Vesicles, International Society for Advancement of Cytometry, and International Society for Thrombosis and Haemostasis Flow Cytometry Working Group who recently laid out a standard reporting framework (MIFlowCyt-EV) and minimal experiment requirement (15).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
J.A.W. is supported by the Translational Nanobiology Section at the U.S. National Institutes of Health, National Cancer Institute (grant number: 1ZIA-BC011502). V.A.T. acknowledges the University of Ottawa Faculty of Medicine and Flow Cytometry and Virometry Core Facility for providing funds to support this work. The authors thank M.A. Langlois, Ph.D. for the use of antibody and an aliquot of ViroFlow Technologies product.
Grant sponsor: Translational Nanobiology Section at the U.S. National Institutes of Health, National Cancer Institute, Grant number: 1ZIA-BC011502; Grant sponsor: University of Ottawa Faculty of Medicine; Grant sponsor: Flow Cytometry and Virometry Core Facility.
Declaration of Interest Statement
J.A.W. and J.C.J. are coinventors on NIH patents and patent applications related to EV analysis. J.A.W. is a 2019-2023 ISAC Marylou Ingram Scholar. V.A.T. is a coinventor on University of Ottawa patent applications related to use of viruses as reference particles for FCM and is a 2018-2022 ISAC SRL Emerging Leader. V.A.T. was a past CSO of ViroFlow Technologies.
Footnotes
Additional Supporting Information may be found in the online version of this article.
Literature Cited
- 1.Wood JC. Fundamental flow cytometer properties governing sensitivity and resolution. Cytometry 1998;33(2):260–266. [PubMed] [Google Scholar]
- 2.Wood JC, Hoffman RA. Evaluating fluorescence sensitivity on flow cytometers: An overview. Cytometry 1998;33(2):256–259. [PubMed] [Google Scholar]
- 3.Hoffman RA (2005). Standardization, Calibration, and Control in Flow Cytometry. Curr Protoc in Cytom, 32, 1.3.1–1.3.21. 10.1002/0471142956.cy0103s32. [DOI] [PubMed] [Google Scholar]
- 4.Wang L, Hoffman RA. Standardization, calibration, and control in flow cytometry. Curr Protoc Cytom 2017;79:131–1327. [DOI] [PubMed] [Google Scholar]
- 5.Gaigalas AK, Wang L, Schwartz A, Marti GE, Vogt RF Jr. Quantitating fluorescence intensity from fluorophore: Assignment of MESF values. J Res Natl Inst Stand Technol 2005;110(2):101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwartz A, Gaigalas AK, Wang L, Marti GE, Vogt RF, Fernandez-Repollet E. Formalization of the MESF unit of fluorescence intensity. Cytometry Part B 2004;57B (1):1–6. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz A, Wang L, Early E, Gaigalas A, Zhang YZ, Marti GE, Vogt RF. Quantitating fluorescence intensity from fluorophore: The definition of MESF assignment. J Res Natl Inst Stand Technol 2002;107(1):83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hercher M, Mueller W, Shapiro HM. Detection and discrimination of individual viruses by flow cytometry. J Histochem Cytochem 1979;27(1):350–352. [DOI] [PubMed] [Google Scholar]
- 9.Fattaccioli J, Baudry J, Emerard JD, Bertrand E, Goubault C, Henry N, Bibette J. Size and fluorescence measurements of individual droplets by flow cytometry. Soft Matter 2009;5(11):2232–2238. [Google Scholar]
- 10.van der Pol E, van Gemert MJ, Sturk A, Nieuwland R, van Leeuwen TG. Singlevs. swarm detection of microparticles and exosomes by flow cytometry. J Thromb Haemost 2012;10(5):919–930. [DOI] [PubMed] [Google Scholar]
- 11.van der Pol E, Sturk A, van Leeuwen T, Nieuwland R, Coumans F, group, I.-S.-V.W. Standardization of extracellular vesicle measurements by flow cytometry through vesicle diameter approximation. J Thromb Haemost 2018;16(6):1236–1245. [DOI] [PubMed] [Google Scholar]
- 12.Tang VA, Fritzsche AK, Renner TM, Burger D, van der Pol E, Lannigan JA, Brittain GC, Welsh JA, Jones JC, Langlois M-A. Engineered retroviruses as fluorescent biological reference particles for small particle flow cytometry. bioRxiv 2019; 614461. 10.1101/614461. [DOI] [Google Scholar]
- 13.Czechowska K, Lannigan J, Aghaeepour N, Back JB, Begum J, Behbehani G,Bispo C, Bitoun D, Fernández AB, Boova ST, (2019). Cyt-Geist: Current and Future Challenges in Cytometry: Reports of the CYTO 2019 Conference Workshops. Cytometry, 95;(12):1236–1274. 10.1002/cyto.a.23941. [DOI] [PubMed] [Google Scholar]
- 14.Lee JA, Spidlen J, Boyce K, Cai J, Crosbie N, Dalphin M, Furlong J, Gasparetto M,Goldberg MGoralczyk EM (2008). MIFlowCyt: The minimum information about a flow cytometry experiment. Cytometry, 73A;(10):926–930. 10.1002/cyto.a.20623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Welsh JA, Van Der Pol E, Arkesteijn GJA, Bremer M, Brisson A, Coumans F,Dignat-George F, Duggan E, Ghiran I, Giebel B (2020). MIFlowCyt-EV: a framework for standardized reporting of extracellular vesicle flow cytometry experiments. Journal of Extracellular Vesicles, 9, (1), 1713526. 10.1080/20013078.2020.1713526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Welsh JA, Horak P, Wilkinson JS, Ford VJ, Jones JC, Smith D, Holloway JA,Englyst NA (2019). FCMPASS Software Aids Extracellular Vesicle Light Scatter Standardization. Cytometry, 10.1002/cyto.a.23782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Rond Leonie Coumans Frank A. W., Rienk Nieuwland, van Leeuwen Ton G.,van der Pol Edwin (2018). Deriving Extracellular Vesicle Size From Scatter Intensities Measured by Flow Cytometry. Curr Protoc Cytom, 86;(1):e43. 10.1002/cpcy.43. [DOI] [PubMed] [Google Scholar]
- 18.Ma L, Zhu S, Tian Y, Zhang W, Wang S, Chen C, Wu L, Yan X (2016). Label-FreeAnalysis of Single Viruses with a Resolution Comparable to That of Electron Microscopy and the Throughput of Flow Cytometry. Angew Chem Int Ed Engl, 55; (35):10239–10243. 10.1002/anie.201603007. [DOI] [PubMed] [Google Scholar]
- 19.Daimon M, & Masumura A (2007). Measurement of the refractive index of distilled water from the near-infrared region to the ultraviolet region. App Opt, 46(18), 3811–3820. [DOI] [PubMed] [Google Scholar]
- 20.Vira S, Mekhedov E, Humphrey G, & Blank PS (2010). Fluorescent-labeled antibodies: Balancing functionality and degree of labeling. Anal Biochem, 402(2), 146–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van der Pol E (2015). Detection of extracellular vesicles: size does matter, Amsterdam, Netherlands: University of Amsterdam. [Google Scholar]
- 22.Nolan JP (2015). Flow Cytometry of Extracellular Vesicles: Potential, Pitfalls, andProspects. Curr Protoc Cytom, 73;13:14.1–13.14.16. [DOI] [PubMed] [Google Scholar]
- 23.Nolan JP, & Jones JC (2017). Detection of platelet vesicles by flow cytometry. Platelets, 28(3):256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang VA, Fritzsche AK, Renner TM, Burger D, Lannigan JA, Brittain GC,Ouellet CV, van der Pol E, & Langlois M-A (2019). Engineered Retroviruses as Fluorescent Biological Reference Particles for Nanoscale Flow Cytometry. bioRxiv, 614461. 10.1101/614461. [DOI] [Google Scholar]
- 25.Morales-Kastresana A, Musich TA, Welsh JA, Telford W, Demberg T, Wood JCS, Bigos M, Ross CD, Kachynski A, & Dean A (2019). High-fidelity detection and sorting of nanoscale vesicles in viral disease and cancer. J Extracell Vesicles, 8(1), 1597603. 10.1080/20013078.2019.1597603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stoner SA, Duggan E, Condello D, Guerrero A, Turk JR, Narayanan PK, Nolan JP(2016). High sensitivity flow cytometry of membrane vesicles. Cytometry, 89;(2): 196–206. 10.1002/cyto.a.22787. [DOI] [PubMed] [Google Scholar]
- 27.Görgens A, Bremer M, Ferrer-Tur R, Murke F, Tertel T, Horn PA, Thalmann S,Welsh JA, Probst C, Guerin C, (2019). Optimisation of imaging flow cytometry for the analysis of single extracellular vesicles by using fluorescence-tagged vesicles as biological reference material. J Extracell Vesicles, 8;(1):1587567. 10.1080/20013078.2019.1587567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lacroix R, Robert S, Poncelet P, Kasthuri RS, Key NS, Dignat-George F, &Workshop IS (2010). Standardization of platelet-derived microparticle enumeration by flow cytometry with calibrated beads: results of the International Society on Thrombosis and Haemostasis SSC Collaborative workshop. Journal of Thrombosis and Haemostasis, 8;(11):2571–2574. 10.1111/j.1538-7836.2010.04047.x. [DOI] [PubMed] [Google Scholar]
- 29.Welsh JA, Scorletti E, Clough GF, Englyst NA, Byrne CD (2018). Leukocyte extracellular vesicle concentration is inversely associated with liver fibrosis severity in NAFLD. J Leukoc Biol, 104;(3):631–639. 10.1002/jlb.5a1217-501r. [DOI] [PubMed] [Google Scholar]
- 30.Gasecka A, Nieuwland R, Budnik M, Dignat-George F, Eyileten C, Harrison P,Lacroix R, Leroyer A, Opolski G, Pluta K, (2020). Ticagrelor attenuates the increase of extracellular vesicle concentrations in plasma after acute myocardial infarction compared to clopidogrel. J Thromb Haemost, 18;(3):609–623. 10.1111/jth.14689. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.