

Abstract
Tissue damage increases cancer risk through poorly understood mechanisms1. In the pancreas, pancreatitis associated with tissue injury collaborates with activating mutations in the Kras oncogene to dramatically accelerate the formation of early neoplastic lesions and ultimately pancreatic cancer2,3. By integrating genomics, single-cell chromatin assays and spatiotemporally-controlled functional perturbations in autochthonous mouse models, we show that the combination of Kras mutation and tissue damage promotes a unique chromatin state in the pancreatic epithelium that distinguishes neoplastic transformation from normal regeneration and is selected for throughout malignant evolution. This cancer-associated epigenetic state emerges within 48 hours of pancreatic injury, and involves an acinar-to-neoplasia ‘chromatin switch’ that contributes to the early dysregulation of genes defining human pancreatic cancer. Among the genes most rapidly activated upon tissue damage in the pre-malignant pancreatic epithelium is the alarmin cytokine IL-33, which cooperates with mutant Kras in unleashing the epigenetic remodeling program of early neoplasia and neoplastic transformation in the absence of injury. Collectively, our study demonstrates how gene-environment interactions can rapidly produce gene regulatory programs that dictate early neoplastic commitment and provides a molecular framework for understanding the interplay between genetics and environmental cues in cancer initiation.
Understanding the mechanisms by which tissue damage promotes cancer initiation may expose rational strategies to prevent, detect, and intercept tumors before they evolve to an intractable stage. A paradigm of damage-associated carcinogenesis is pancreatic ductal adenocarcinoma (PDAC), an invariably lethal cancer that lacks effective therapies. Its major oncogene, mutant Kras, is found altered in virtually all patients and is necessary for disease initiation and maintenance4,5. Intriguingly, Kras gene mutations are only weakly oncogenic but potently cooperate with signals emanating from tissue damage and the resulting inflammation (pancreatitis) to initiate the disease2,3,6. Normally, pancreatic injury triggers a rapid cell fate transition characterized by loss of acinar differentiation and concomitant acquisition of a ‘duct-like’ state, a process termed acinar-to-ductal metaplasia (ADM) that resolves via acinar re-differentiation as the tissue regenerates7. However, in the presence of a Kras mutation, metaplasia aberrantly persists and progresses into pancreatic intraepithelial neoplasia (PanIN)8,9. These observations suggest that oncogenic KRAS co-opts otherwise reparative regenerative responses to drive PDAC initiation10. Given that neoplastic lesions emerge rapidly after tissue damage in the setting of mutant Kras but in the apparent absence of additional mutations2,3,6, we hypothesize that uncharacterized epigenetic mechanisms underlie the interplay between cancer-predisposing mutations and environmental insults in cancer pathogenesis.
Injury-induced chromatin states
As a first step towards dissecting how gene–environment interactions reprogram the pancreatic epithelium during tumor development, we generated chromatin accessibility maps of pancreatic epithelial cells freshly isolated from healthy, damaged, early neoplastic or malignant tissues from genetically and pathologically accurate mouse models engineered to enable selective isolation of exocrine pancreatic epithelial cells using the fluorescent reporter mKate2 (Extended Data Fig. 1a–c; Supplementary Fig. 1a, see Methods). Specifically, mKate2+ cells from the following tissue conditions were subjected to ATAC-seq11: (i) normal healthy pancreas (Normal), (ii) normal pancreas undergoing regenerative ADM driven by tissue damage (Injury); (iii) Kras-mutant pancreata undergoing stochastic neoplastic transformation (Kras*), (iv) Kras-mutated pancreata undergoing synchronous neoplastic reprogramming accelerated by tissue damage (Kras*+Injury) and, as reference for advanced disease, (v) PDAC (PDAC) arising in KPflC mice (Ptf1a-Cre;RIK;LSL-KrasG12D;p53fl/+) or upon syngeneic orthotopic transplantation of KrasG12D;p53-null engineered pancreatic organoids (Fig. 1a–b, Extended Data Fig. 1b–d; Supplementary Table 1).
Fig 1. Tissue damage induces cancer-associated chromatin states in pre-malignancy.
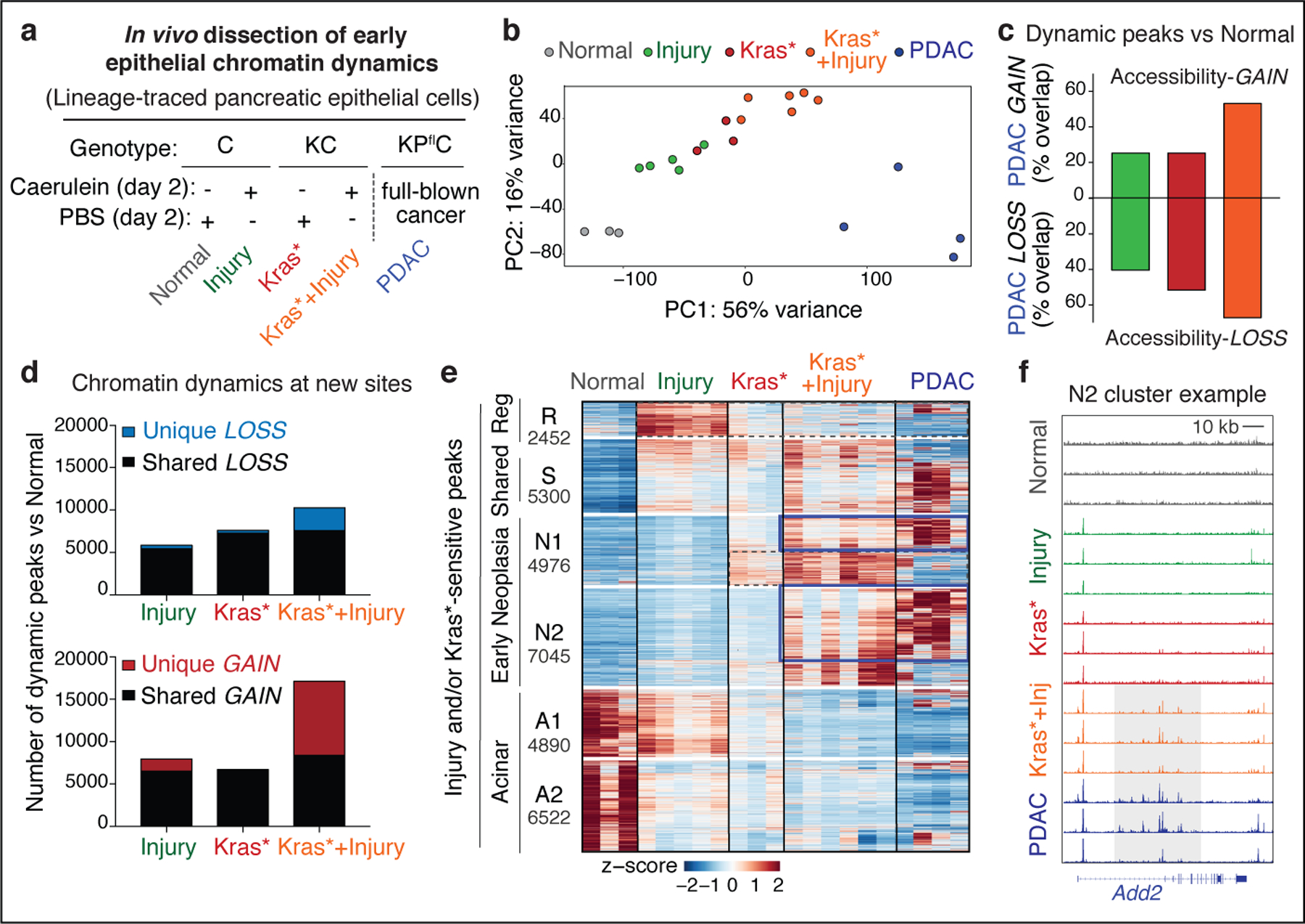
a, Experimental settings to interrogate epithelial neoplastic reprogramming in vivo. Chromatin accessibility (and gene expression, below) analyses were performed on lineage-traced pancreatic epithelial cell populations FACS-isolated from well-defined tissue states (see main text). When applicable, tissue damage was induced by treatment with the synthetic cholecystokinin analogue caerulein6,8. b, Principal Component Analyses of ATAC-seq data from independent biological replicates of pancreatic epithelial cells isolated from tissue states described in (a). c, Proportion of ATAC-peaks significantly gained (top) or lost (bottom) in PDAC compared to Normal pancreas, and found similarly altered in pre-malignant tissues subjected to injury, expressing mutant Kras, or both. Bar color indicates experimental condition, as in (b). d, Number of ATAC-peaks that are significantly lost (top) or gained (bottom) in the indicated conditions vs Normal pancreas, shared or unique to each condition. e, Heatmap representation of peaks gained or lost between Normal, Injury, Kras* and Kras*+Injury conditions. Each column represents an independent mouse. Numbers indicate the number of peaks per cluster. f, ATAC-seq tracks at a N2-cluster locus exhibiting synergistic accessibility-GAINS by combination of injury and mutant Kras (one independent mouse per lane). y-axis scale range per lane [0–60]. In c, d, bar charts summarize the degree of overlap between dynamic ATAC-peaks identified by DESeq2 analyses (log2FC >= 0.58; FDR <=0.1) comparing Injury (n=5 mice), Kras* (n=3 mice), Kras*+Injury (n=6 mice), or PDAC (n=4 mice) conditions versus Normal (n=3 mice).
Differential accessibility analysis was used to identify open chromatin regions (peaks) that were significantly gained or lost in each condition compared to Normal, which we refer to as accessibility-GAIN and accessibility-LOSS regions, respectively (Supplementary Table 2). As illustrated in Fig. 1b, these analyses uncovered large-scale chromatin accessibility changes across conditions, with the majority of changes contributed by cooperative effects of tissue damage and mutant Kras early on in tumorigenesis (PC1: 56%), rather than the later transition from early neoplasia to PDAC (PC2: 16%). Strikingly, cells undergoing synchronous neoplastic reprogramming by the combined effects of tissue damage and oncogenic Kras displayed more than half of the chromatin aberrations that distinguish advanced PDAC from normal pancreas (Fig. 1c), suggesting that PDAC co-opts chromatin regulatory mechanisms from its onset.
Comparison of the dynamic peaks associated with the reversible metaplasia that accompanies physiological regeneration (Injury) to those occurring in persistent, pre-neoplastic metaplasia (Kras* or Kras*+Injury) revealed shared and unique traits. Accessibility-LOSS changes were largely shared and, consistent with the reduction in acinar differentiation that defines both processes10, preferentially affected loci linked to acinar cell functions (Fig. 1d, Extended Data Fig. 1e, Supplementary Table 2). While there was also overlap between accessibility-GAIN regions, the combination of mutant Kras and injury produced a large number (>8,500) of additional chromatin accessibility changes that were not observed in pancreatic epithelial cells expressing mutant Kras or subject to injury alone (Fig. 1d; Extended Data Fig. 1e). Unsupervised hierarchical clustering of all dynamic peaks sensitive to mutant Kras and/or tissue damage identified clusters of open chromatin regions specific to normal healthy (A), regenerative (R) and early neoplastic (N) tissue states as well as shared (S), with a large set of peaks uniquely gained upon co-occurrence of both stimuli (N2) (Extended Data Fig. 2a).
Notably, 67% of the accessibility-GAINS unique to cells undergoing synchronous neoplastic transformation by the combined effects of mutant Kras and injury (cluster N2) were retained in advanced PDAC, whereas those specific to each insult alone (e.g. cluster R) were not (Fig. 1e). These early cancer-associated chromatin configurations arose within 48 hours of tissue damage and were associated with genes linked to PDAC-related pathways, including many cancer-relevant factors (Supplementary Tables 2–3; Extended Data Fig. 2b). The majority of open chromatin regions distinguishing normal (A), regenerating (R), and early neoplastic (N) tissues mapped to non-coding intergenic and intronic regions containing motifs for master transcription factors (TFs) controlling pancreatic cell lineage commitment (e.g. NR5A2, PTF1A) and carcinogenesis (e.g. AP-1, SOX, KLF)12,13 (Extended Data Fig. 2c–d), with TF enrichment and co-occurrence patterns differing across conditions (Extended Data Fig. 2f,g). Of note, Kras mutation and tissue damage cooperatively promoted accessibility-LOSS at loci containing active enhancers and bound by acinar specification TFs in the normal exocrine pancreas14–16 and accessibility-GAIN at loci containing experimentally-validated enhancers of advanced PDAC17 (Extended Data Fig. 3a–c).
Perturbing chromatin output
To functionally relate chromatin changes to cell fate transitions in vivo, we adapted the mouse models described above to incorporate a pancreas-specific and doxycycline (dox)-regulatable GFP-linked short hairpin RNA (shRNA) enabling perturbation of the transcriptional output of active regulatory elements preferentially associated with fate-specifying genes. Specifically, we exploited the activity of a well-characterized chromatin reader, the Bromodomain and Extraterminal (BET) family member Brd4, which binds acetylated (active) chromatin and is particularly important for enhancer-mediated transcription of cell-identity genes18,19. We reasoned that inducible targeting of Brd4 function in Kras-mutant or -wild type pancreatic epithelial cells would perturb and expose, in a TF/context-agnostic manner, transcriptionally-active gene programs defining their states and reveal functional links between chromatin state and phenotypic output in vivo. Moreover, this genetic approach overcomes confounding effects of pharmacological BET inhibition that would simultaneously disrupt epigenetic programs of surrounding stromal cells known to influence pancreatic epithelial cell state/fate20.
To compare pro-neoplastic and regenerative programs, we produced models differing in Kras mutation status and harboring well-validated shRNAs targeting Brd4 (2 independent strains) or Renilla Luciferase (control) (Fig. 2a; Extended Data Fig. 3d; see Methods for details). As expected, dox-treated KCsh (Kras mutant) and Csh (Kras WT) mice expressing Brd4 shRNA (shBrd4), but not the Renilla shRNA (shRen) controls, showed potent suppression of Brd4 protein restricted to mKate2/GFP double-positive cells (Fig. 2b; Extended Data Fig. 3e), and RNA-seq and ATAC-seq data obtained from sorted mKate2/GFP+ cells showed that Brd4 suppression impaired the expression of established enhancer-associated cell-identity genes15,17,21 without decreasing chromatin accessibility at these loci or globally affecting transcription (Fig. 2c, Extended Data Fig. 3f,g; Supplementary Fig. 1b).
Fig. 2. An in vivo approach to perturb chromatin output in regenerating and neoplastic pancreatic epithelia.
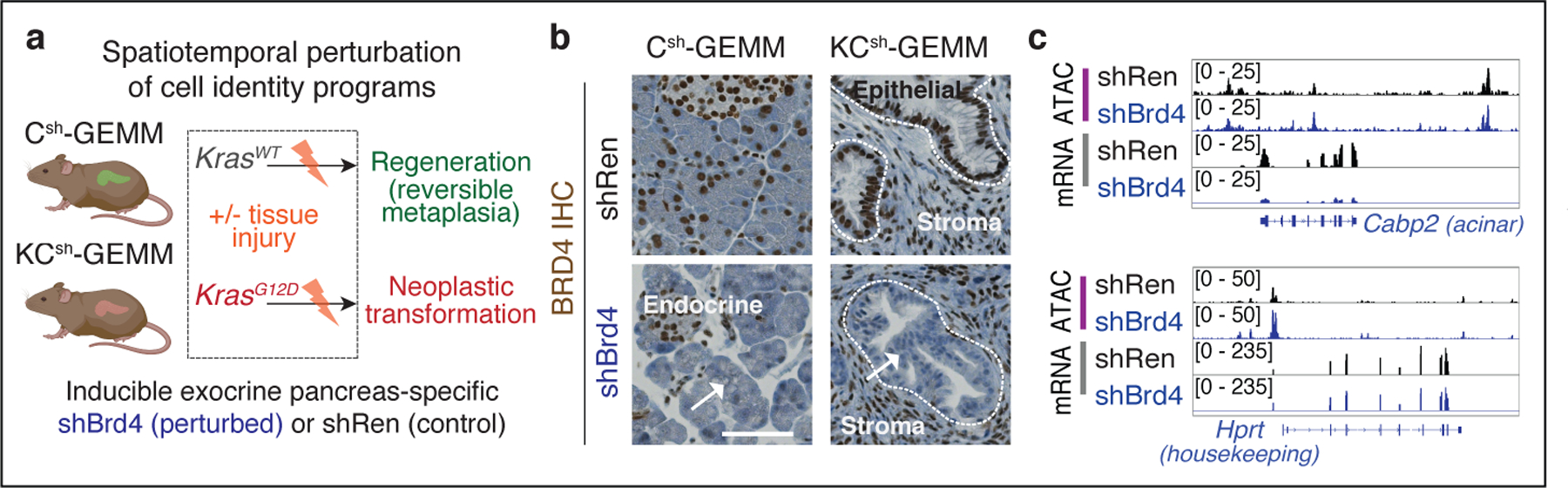
a, Diagram of experimental settings to study regenerative and tumor-initiating epithelial plasticity in response to tissue damage in KC- and C-GEMMs. Illustrations from biorender.com. b, Representative immunohistochemistry (IHC) of Brd4 in Csh (top) or KCsh (bottom) mice (n = 3 mice/group) fed with dox-containing food for 9 days. Surrounded areas represent epithelium; arrows point to Brd4-suppressed exocrine pancreas epithelium in wild-type or mutant Kras mice expressing Brd4.specific (shBrd4.552) but not control (shRen.713) shRNA. Scale bar, 100 μm. c, Representative ATAC-seq and RNA-seq tracks of a known acinar identity gene15 (top) or housekeeping gene locus (bottom) in lineage-traced (mKate2+;GFP+) pancreatic epithelial cells isolated from shRen.713 or shBrd4.552 KCsh mice (n = 3 each), analyzed at the same time point after dox administration as in b.
We next analyzed the phenotypic impact of perturbing Brd4 function on cells undergoing regenerative and pro-neoplastic cell fate transitions profiled above (Extended Data Fig. 4a). Brd4-suppressed cells effectively lost acinar morphology and expression of acinar markers (CPA1, Amylase) while acquiring ductal (SOX9, KRT19) and dedifferentiation (Clusterin) markers in response to tissue damage, mutant Kras, or their combination in both regenerating and neoplastic conditions (Fig. 3a; Extended Data Fig. 4b–g). Therefore, despite the marked chromatin changes detected in damaged tissues (Fig 1), Brd4 is not required for ADM and, in fact, restrains this transition. In contrast, Brd4 suppression impaired both the resolution of ADM during normal regeneration as well as the initiation of neoplasia in the context of mutant Kras. In the regenerative context, Brd4-suppressed cells retained metaplastic features at a time when metaplasia in controls had resolved (Fig. 3a; Extended Data Fig. 4b,e–g). In the neoplastic context, Brd4 suppression prevented the appearance of PanIN lesions (Fig. 3c–d). In both settings, metaplastic cells expressing shBrd4 disappeared over time leading to tissue atrophy (Fig. 3a–c; Extended Data Fig. 5a–c). Of note, epithelial-specific Brd4 suppression did not reduce Myc protein nor recapitulated effects of Myc suppression (Extended Data Fig. 5d–f). These results uncover distinct epigenetic requirements for the induction versus resolution of metaplasia and link Brd4 function to Myc-independent expression programs required for regenerative and neoplastic outcomes of epithelial cell plasticity.
Fig. 3. Neoplastic and regenerative outcomes of injury rely on distinct Brd4-dependent programs.
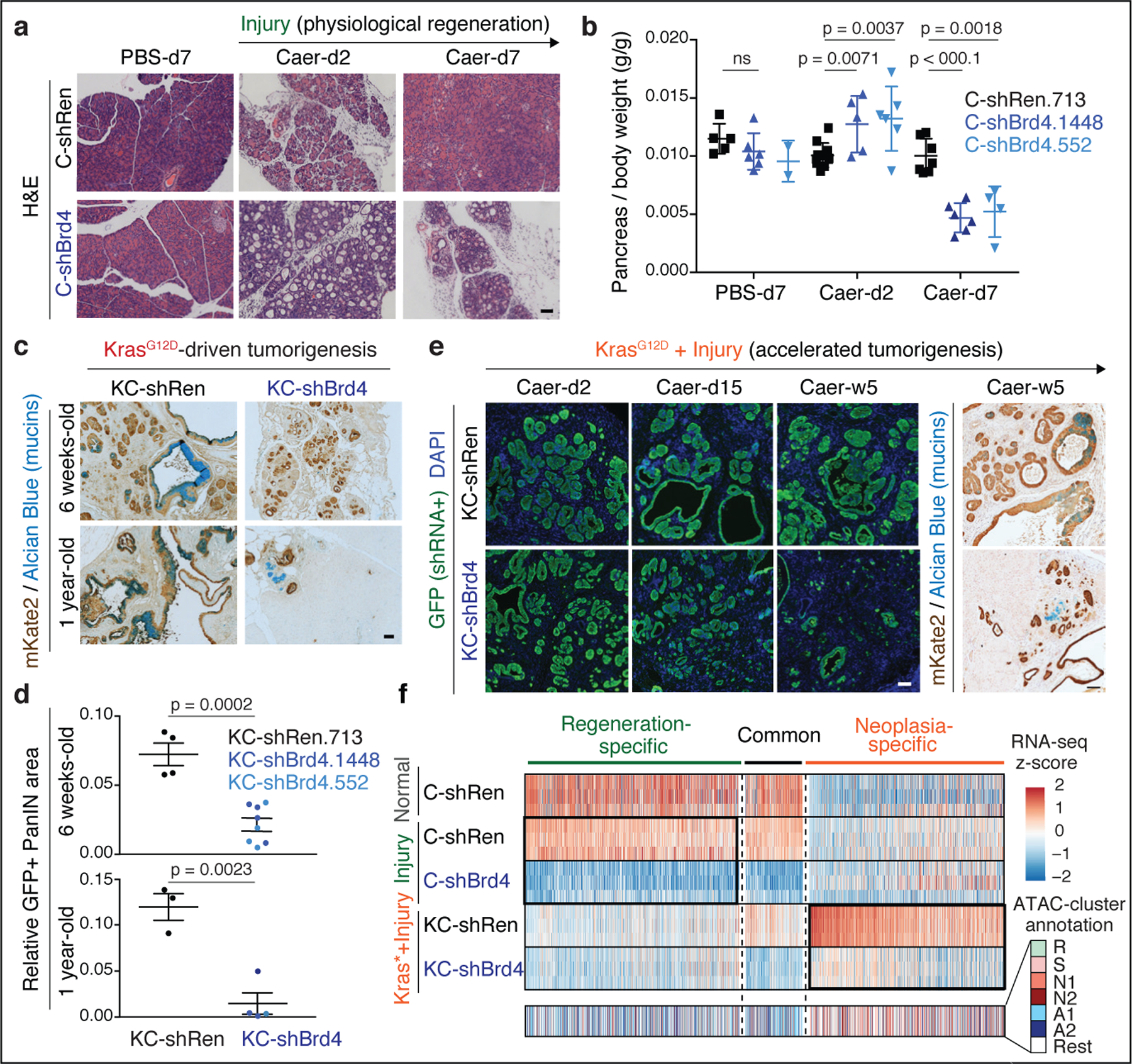
a, Representative H&E of pancreata from Kras wild-type C-shRen (control) or C-shBrd4 (sh552) mice treated with Caerulein (Caer) or PBS harvested at indicated days (d) post-treatment (number of mice/group, as in b). b, Quantification of pancreas-to-body weight ratio of C-shRen or -shBrd4 mice at indicated time-points after caerulein treatment, denoting rapid loss of pancreatic tissue in shBrd4 mice between day-2 and day-7 post-injury. n = 5, 6, 2, 11, 5, 6, 7, 6 or 4 (from left to right) mice/group. c, Representative IHC of mKate2 and Alcian blue in pancreata from KC-GEMM-shRen.713 or -shBrd4.552 mice placed on dox diet since postnatal day 10, analyzed at indicated time points. d, Quantification of PanIN lesion area in pancreata from 6 week-old KC-shRen (n=4) or -shBrd4 (n=8) mice, or 1-year-old KC-shRen (n=3) or KC-shBrd4 (n=4) mice. e, Representative immunofluorescence (GFP) and IHC (mKate2, Alcian blue) to visualize the progression of Kras-mutant cells expressing Ren or Brd4 shRNAs (mKate2+;GFP+) upon injury-accelerated pancreatic neoplasia, analyzed at indicated days (d) or weeks (w) post-Caer (n=3 mice/group). f, (Top) Heatmap of downregulated DEGs upon Brd4 suppression in regenerative metaplasia (Csh:Injury) or neoplastic transformation (KCsh:Kras*+Injury) settings across indicated conditions (as in Extended Data Fig. 4a). n=3 shRen.713 or shBrd4.1448 mice (rows) per condition. Normal C-shRen samples show expression levels of DEGs in healthy pancreas. Black squares delineate genes uniquely sensitive to Brd4 suppression in cells undergoing injury-driven regenerative (left) vs neoplastic (right) transitions. See Supplementary Table 4 for list of shBrd4-sensitive genes in each context. (Bottom) Chromatin accessibility dynamics at regulatory loci of shBrd4-sensitive genes, indicated by ATAC-cluster annotation. In b, d, data presented as means±s.e.m and significance assessed by unpaired two-tailed Student’s t-test (ns, non-significant). Scale bar, 100 μm.
To identify these programs, the chromatin accessibility landscapes from Figure 1 were compared to RNA-seq and ATAC-seq profiles of pancreatic epithelial cells (mKate2/GFP+) triggered to undergo pro-neoplastic or regenerative fate transitions in the presence and absence of Brd4 (Extended Data Fig. 4a, 6a). Consistent with the differential chromatin states observed during regenerative versus pro-neoplastic metaplasia, the Brd4-sensitive gene expression programs in each condition were also distinct (Fig. 3f; Extended Data Fig. 6b). In the regenerative context (Csh: Injury), Brd4 suppression blunted the expression of genes linked to acinar-specific (A) clusters identified through ATAC-seq (Fig. 3f; Extended Data Fig. 6b–d; Supplementary Table 4). Among these genes were master TFs (Ptf1a22) known to stabilize acinar cell identity (Extended Data Fig. 6b,d), providing an explanation for the exacerbated ADM and impairment of regeneration of Brd4-suppressed pancreata after injury.
In the neoplastic setting (KCsh: Kras*+Injury), Brd4 suppression also reduced acinar gene expression (Extended Data Fig. 6b–d-bottom) but, additionally, impaired the activation of a larger set of genes that otherwise are selectively opened and induced in this context (clusters N1/N2) (Fig. 3f; Extended Data Fig. 6c). These neoplasia-specific Brd4 targets included effectors of oncogenic Kras, targets of cancer-associated transcriptional networks, and genes characteristic of advanced human PDAC23 (Extended Data Fig. 6e–k). Consistent with a direct effect on chromatin transcriptional activity, Brd4 perturbation did not prevent the acquisition of injury-driven chromatin states observed in controls (Extended Data. Fig. 6l,m). These results functionally connect the different injury-induced chromatin states of normal and Kras-mutant cells with distinct Brd4-dependent transcriptional programs required for regeneration or neoplastic transformation, respectively.
Epigenetic reprogramming instructs neoplastic commitment
To reveal potential mechanisms whereby chromatin dysregulation redirects regenerative injury responses to neoplasia, we performed RNA-seq analyses across the full spectrum of epithelial states described above (Fig. 1a). Integration of transcriptional, chromatin accessibility, and shBrd4 sensitivity profiles identified two major classes of differentially expressed genes (DEGs) distinguishing pancreatic epithelial (mKate2+) cells from regenerating (Injury), early neoplastic (Kras*; Kras*+Injury) and cancer (PDAC) tissues from healthy normal (Normal): DEGs with ubiquitously opened regulatory elements across all conditions (chromatin-stable DEGs) and DEGs displaying parallel accessibility-GAINS or LOSSES at one or more associated regulatory element (chromatin-dynamic DEGs) (Fig. 4a; Extended Data Fig. 7a,b; Supplementary Table 5). Interestingly, chromatin-stable DEGs were linked to housekeeping cellular processes, whereas ‘chromatin-dynamic DEGs’ encoded factors regulating traits altered in PDAC, represented an enriched fraction in mutant-Kras pancreata subject to injury (Kras*+Injury) and advanced cancer (PDAC), and were particularly sensitive to Brd4 perturbation (Extended Data Fig. 7c,d; Supplementary Table 6).
Fig. 4. A chromatin switch induced by gene – environment interactions defines the neoplastic transition.

a, Proportion of chromatin-dynamic DEGs (blue, red) vs chromatin-stable DEGs (grey) between Brd4-competent pancreatic epithelial cells (mKate2+) isolated from regenerating (Injury, n=5 mice), early neoplasia (Kras*, n=3; Kras*+Injury, n=4 mice) or invasive cancer (PDAC, n=3 mice) tissues vs from normal pancreas (Normal, n=4 mice). b, Unsupervised clustering of dynamic ATAC-peaks associated with DEGs distinguishing pancreatic epithelial cells (mKate2+) isolated from Injury (I), Kras* (K), Kras*+Injury (K+I), or PDAC tissue conditions vs Normal, and colored depending on whether they contain binding sites for acinar (NR5A2 and/or PTF1A) and/or neoplasia-associated (AP-1) TFs. Bar length represents Log2 fold changes of accessibility signals gained or lost in each condition vs Normal, as assessed by DESeq2 analyses (number of mice/group as in Fig. 1). c-d, GSEA comparing the expression of neoplasia-specific downregulated (left) or upregulated (right) epigenetic programs herein identified between human PDAC specimens and human normal pancreas (Moffitt et al. dataset)23 (c), or between shBrd4.1448- vs shRen.713-expressing pancreatic epithelial cells isolated from KC-GEMM mice (Kras*+Injury condition, n=3 per genotype) (d). e, UMAP visualization of single-cell ATAC-seq (scATAC-seq) profiles of 6369 Brd4-competent Kras-mutant pancreatic epithelial cells (mKate2+) isolated from Kras* and Kras*+Injury tissue conditions (n=1 mice each), and colored by the indicated tissue condition (left). Inferred activity scores for the acinar TF NR5A2 and AP-1 per individual cell are portrayed in color (right), displaying a switch in transcription factor activity in Kras-mutant cells upon tissue injury. f, AP-1 and NR5A2 activity scores of Kras-mutant cells (columns) annotated by tissue condition (as in e-left). g, Top-scoring pathways in GREAT ontology analyses of peaks positively or negatively correlated with the chromatin switch defined by increasing AP-1/NR5A2 activity ratios across the single cell epigenetic profiles of Kras-mutant cells shown in f.
Comparison of dynamic cis-regulatory elements of DEGs altered during regeneration, early neoplasia and cancer revealed a common redistribution of transcriptionally-active open chromatin from loci containing binding sites for acinar lineage-specifying TFs (Fig. 4b, blue/green-colored peaks) to newly accessible regions enriched in motifs for wound healing and Ras/cancer-associated TFs including the AP-1 TF family (Fig. 4b, red-colored peaks; Supplementary Table 7). However, unsupervised clustering analysis identified a gene regulatory program that is uniquely induced by cooperative effects of mutant Kras and tissue damage (Fig. 4b). Accordingly, the relative expression of the TFs predicted to bind differentially-active chromatin domains differed between conditions, even among members of the same TF family (Extended Data Fig. 7e–g). Consistent with a role for this neoplasia-specific epigenetic program in carcinogenesis, the gene regulatory activities and expression outputs altered in Kras-mutant pancreata shortly after injury were largely shared with (and further exacerbated in) advanced disease (Extended Data Fig. 7e–g), strongly correlated with signatures defining human PDAC (Fig. 4c), and were blunted in shBrd4 metaplastic cells unable to progress to neoplasia (Fig. 4d). Thus, while loss of acinar differentiation is sufficient to activate certain AP-1 TFs and other cancer-associated networks during physiological metaplasia24, a distinct epigenetic program facilitated by injury-driven chromatin accessibility changes is required for neoplastic commitment.
To discriminate whether this neoplasia-specific chromatin state reflects bona-fide chromatin remodeling (versus a shift in the proportion of pre-existing diverse cell types comprising the epithelium25), we applied single-cell ATAC-seq (scATAC-seq) on over 6,000 Kras-mutant cells isolated from Kras* or Kras*+Injury conditions. These analyses revealed rapid chromatin accessibility shifts induced by injury within and across epigenetically heterogeneous subpopulations of pre-malignant Kras-mutant cells (Extended Data Fig. 8a–e; Supplementary Table 8) through remodeling of specific loci consistent with those detected in bulk populations (r =0.765–0.882) (Extended Data Fig. 8f–i). Also consistent with chromatin remodeling, mapping of activity scores for acinar differentiation (e.g. NR5A2) and Kras/injury-activated (e.g. AP-1) TFs per individual cell showed a shift in TF activity of Kras-mutant cells upon tissue injury (Fig. 4e) and an anticorrelation of their activity scores across single-cell epigenetic profiles (Fig. 4f; Extended Data Fig. 8j). scATAC-seq analyses also captured depletion of cells with specific chromatin states (e.g. acinar-state) coinciding with the emergence of less differentiated subpopulations defined by widespread chromatin opening at neoplasia-associated loci (Extended Data Fig. 8h,i). In contrast, metaplasia-associated genes were found to pre-exist in an open state across most epithelial subpopulations (including in cells with open chromatin at acinar genes) and, in agreement with bulk analyses, did not experience further accessibility gain upon injury (Extended Data Fig. 8i-right). Thus, while the activity of specific TFs that underlie such early epigenetic heterogeneity and injury-driven plasticity currently relies on correlative observations, these results demonstrate that oncogenic mutations and tissue injury cooperatively remodel chromatin to produce neoplasia-specific transcriptional programs. We refer to this process as the ‘acinar-to-neoplasia’ chromatin switch.
IL-33 is a chromatin-activated effector of early neoplasia
Many of the neoplasia-specific chromatin-activated genes encoded membrane-bound and secreted proteins (Fig 4g; Fig 5a). Among the most robustly activated genes was the alarmin cytokine IL-33, an injury-associated factor that coordinates wound healing and tissue repair responses26. Hence, analysis of both bulk and scATAC-seq chromatin accessibility datasets consistently identified numerous peaks at the Il33 locus that were rapidly and selectively gained in Kras-mutant pancreata undergoing the injury-facilitated chromatin switch (Kras*+Injury; Extended Data Fig. 9a–d) and retained in established PDAC (Extended Data 9e). These changes correlated with a Brd4-dependent increase in Il33 expression within the pancreatic epithelium (Fig. 5a,b, Extended Data Fig. 9f–h) that could be cooperatively induced in cultured cells upon transduction with Ras/injury-sensitive TFs previously validated to bind these dynamic loci27 (Extended Data Fig. 9e,i). Accordingly, multiplexed immunoassay for 40 different cytokines identified IL-33 as the most abundant cytokine in Kras-mutant pancreata after injury (Extended Data Fig. 9j).
Fig. 5. Epigenetic dysregulation of IL-33 promotes neoplastic reprogramming.
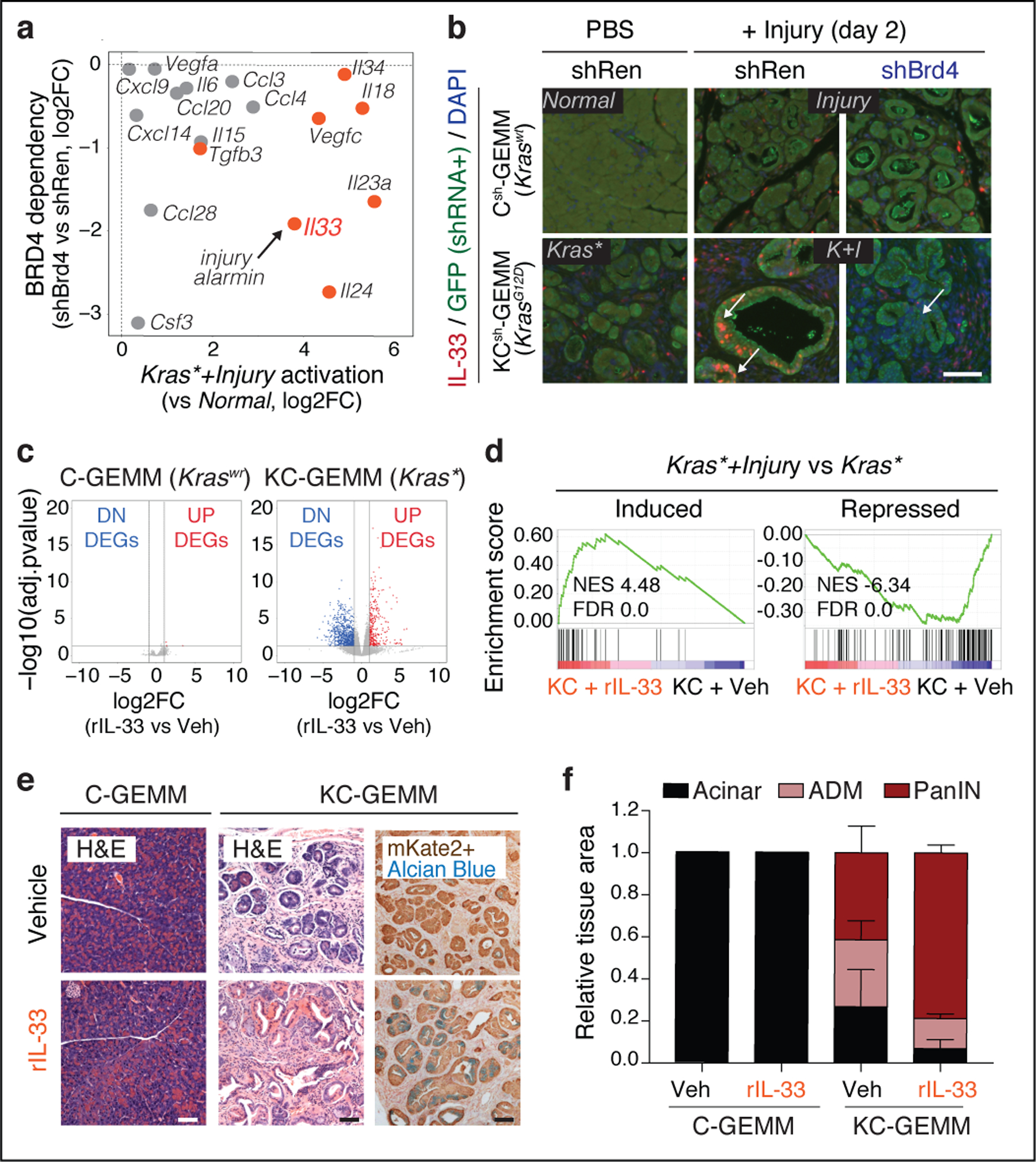
a, Effects of Brd4 suppression (shBrd4.1448 vs shRen.713, y-axis) on the expression of the indicated cytokines or chemokines and their degree of activation in pancreatic epithelial cells during injury-facilitated neoplasia (x-axis). Factors exhibiting neoplasia-specific mRNA upregulation and ATAC-GAIN are marked in orange. b, Representative immunofluorescence of IL-33 (red) and GFP (green) in pancreata from Kras wild-type or Kras-mutant mice 2 days after tissue injury (caerulein) or control (PBS) (n=4 mice/group). Arrows point epithelial-cell (GFP-positive) Brd4-dependent activation of IL-33 in the Kras*+Injury (K+I) condition, which contrasts with the predominantly stromal (GFP-negative) pattern of tissues subject to caerulein (Injury) or Kras gene mutation (Kras*) alone. Nuclei counterstained with DAPI (blue). c,Volcano plots showing the cooperation between rIL-33 and mutant Kras in driving transcriptional reprogramming of the pancreatic epithelium, assessed by RNA-seq of mKate2+ epithelial cells isolated from Kras-wild type (C-GEMM) mice treated with rIL-33 (n=4) or vehicle (PBS; n=5) (left), or Kras-mutant (KC-GEMM) mice treated with rIL-33 (n=4) or vehicle (PBS, n=4) (right), at 21 day. d, GSEA comparing expression of genes induced (top) or repressed (bottom) upon damage (Kras*+Injury vs Kras*) in Kras-mutant epithelial cells isolated from rIL-33-treated (n=4) vs vehicle-treated (n=3) mice (day 0). rIL-33 treatment mimics transcriptional changes of tissue damage in Kras-mutant pancreata. e, Representative H&E or IHC (mKate2, Alcian blue) from Kras-wild type (C-GEMM) or Kras-mutant (KC-GEMM) mice treated with rIL-33 or vehicle (PBS), at 21 days (n=4 mice/group). f, Quantification of normal exocrine tissue (acinar) or metaplastic (ADM) and neoplastic (PanIN) lesion area in Kras-WT (left) or Kras-mutant (right) mice treated with rIL-33 or vehicle control, at 21 days. Pooled data presented as means±s.e.m. n=3, 4, 4 or 5 (from left to right) mice. (p=0.0169 for PanIN area in KC-GEMM rIL-33 vs vehicle, unpaired two-tailed Student’s t-test). Scale bars, 100 μm.
We next examined the extent to which exogenous IL-33 could recapitulate the effects of tissue damage by intraperitoneal administration of recombinant mouse IL-33 (rIL-33) to Kras-mutant- or Kras-wt mice (Extended Data Fig. 10a). Remarkably, rIL-33 mimicked injury in cooperating with mutant Kras to activate the neoplasia-specific, Brd4-dependent gene expression program induced upon tissue damage (Fig. 5c,d; Extended Data Fig. 10b–e), including genes upregulated in human PDAC (Extended Data Fig. 10f). These transcriptional outputs were preceded by accessibility-GAIN and gene expression at cancer-associated loci sensitive to injury in pre-neoplastic Kras-mutant tissues (Extended Data Fig. 10g–i), and associated with an accelerated appearance of PanIN lesions (see ‘KC-GEMM’ panels in Fig. 5e,f; Extended Data Fig. 10j). Notably, rIL-33 had no detectable effects on normal pancreata (Fig. 5e,f; Extended Data Fig. 10j). Of note, rIL-33 did not significantly induce its own mRNA or nuclear protein staining in Kras-mutant epithelial cells (Extended Data Fig. 10k,l), suggesting that its ability to phenocopy injury in the presence of oncogenic Kras is predominantly due to its soluble form. These results identify IL-33 as a target and effector of gene-environment interactions driving early-stage neoplasia and suggest a chromatin-mediated amplification mechanism whereby tissue damage mediators unleash and enforce oncogene-dependent gene expression.
Discussion
Here we document large-scale chromatin accessibility remodeling in damaged tissues that, in the presence of an oncogenic Kras mutation, leads to an epigenetic program – not accessible during physiological regeneration – that contributes to tumor initiation and is selected for during malignant progression. By combining bulk and single-cell profiling with spatiotemporally-controlled in vivo perturbations of mechanisms that regulate cell identity, we show that pancreatic metaplasia involves epigenetic silencing of acinar identity loci that is exacerbated by Brd4 suppression, suggesting that somatic enhancers and/or super-enhancers linked to cell type specification28,29 actively prevent excessive dedifferentiation upon injury and facilitate its resolution. In contrast, progression to neoplasia couples dedifferentiation to a distinctive chromatin remodeling program that diverts DNA accessibility and Brd4-mediated transcription from normal lineage-specifying to cancer-defining loci. Thus, while enhancer remodeling facilitates metastasis in advanced PDAC17,30, these data imply a role for chromatin dysregulation at an early disease stage (see Supplementary Discussion for additional commentary).
Cancer initiation is facilitated by interactions between genetic and environmental insults. Our studies identify an epigenomic mechanism that contributes to this effect, involving an ‘acinar-to-neoplasia’ chromatin switch that can arise in Kras-mutant cells within 48 hours of tissue damage. One key target of this program is IL-33, whose cytokine activity can replace the requirement for injury in accelerating the formation of early neoplastic (PanIN) lesions. While IL-33 can both restrain31 and amplify anti-tumor immunity32 in advanced cancers, it connects tissue damage responses with oncogene-dependent epithelial plasticity during early neoplasia. Further study of these and other epigenetically-dysregulated programs may provide new opportunities for the rational design of early detection and treatment strategies to intercept inflammation- and RAS-driven malignancies such as PDAC at an earlier stage.
Methods
Generation and authentication of KCshBrd4 ESC clones
KC-shBrd4 ESCs (Ptf1a-Cre;LSL-KrasG12D;RIK;CHC33) were targeted with 2 independent GFP-linked Brd4-shRNAs (shBrd4.552 and shBrd4.1448)34,35 cloned into mir30-based targeting constructs36, as previously described33,36. Targeted ESCs were selected and functionally tested for single intregation of the GFP-linked shRNA element into the CHC locus as previously described37. The KC-shRen ESC control clone used in this study has been previously described33,37. Before injection, ESCs were cultured briefly for expansion in KOSR+2i medium38. The identity and genotype of the ESC, resulting chimeric mice and their progeny was authenticated by genomic PCR using a common Col1a1 primer CACCCTGAAAACTTTGCCCC paired with a transgene specific primer: shRen.713: GTATAGATAAGCATTATAATTCCTA; shBrd4.552: TATTGTTCCCATATCCAT; shBrd4.1448: CTAGTTTAGACTTGATTGTG, yielding an ∼250-bp product. ESC were confirmed to be negative for mycoplasma and other microorganisms before injection.
Animal models
All animal experiments in this study were performed in accordance with a protocol approved by the Memorial Sloan-Kettering Institutional Animal Care and Use Committee. Mice were maintained under specific pathogen-free conditions, and food and water were provided ad libitum. All mice strains have been previously described. Ptf1a-Cre39, LSL-KrasG12D40, CHC41, CAGs-LSL-RIK42, and TRE-GFP-shRen43 strains were interbred and maintained on mixed Bl6/129J backgrounds.
To enable selective isolation of epithelial cells from pancreatic tissues we employed the pancreas-specific Cre driver Ptf1a-Cre39,44 and the lineage-tracing allele LSL-rtTA3-IRES-mKate2 (RIK)41,42 that, by themselves or in combination with a Cre-activatable KrasG12D allele40, enable tagging of pancreatic epithelial cells harboring wild-type (WT) or mutant Kras by the fluorescent reporter mKate2. To compare the effects of tissue injury in the transcriptional and chromatin accessibility landscapes of mutant Kras-expressing or Kras wild-type pancreatic epithelial cells, KC-GEMM (Ptf1a-Cre;RIK;LSL-KrasG12D) or C-GEMM (Ptf1a-Cre;RIK) 5-week old male mice were treated with 8 hourly intraperitoneal injections of 80 μg/kg caerulein (Bachem) or PBS for 2 consecutive days, using littermates when possible. To characterize invasive disease, pancreatic ductal adenocarcinoma (PDAC) cells were isolated from cancer lesions arising in autochthonous transgenic models (KPflC-GEMM; Ptf1a-Cre;RIK;LSL-KrasG12D;p53fl/+)5,45 that were macro-dissected away from pre-malignant tissue. As an orthogonal approach, 8–10 weeks old C57Bl/6 female mice (Harlan) were subjected to for orthotopic transplantations with syngeneic ductal organoids harbouring mutant Kras and inactivated Trp53 gene (see below). Prior to transplantation, organoid cultures were dissociated with TrypLE (Gibco) after mechanical dissociation by pipetting and 1–2×105 cells in serum-free advanced DMEM/F12 (Life Technologies) supplemented with 2 mM glutamine and pen-strep were mixed 1:1 with growth factor reduced matrigel (Corning) and injected into the exposed pancreas using a Hamilton syringe fitted with a 26 gauge needle. In all experiments with orthotopic or transgenic PDAC models, tumors did not exceed a maximum volume corresponding to 10 % of the animal’s body weight (typically 12 mm diameter). Mice were evaluated daily for signs of distress or endpoint criteria. Specifically, mice were immediately euthanized if they presented signs of cachexia, weight loss >20% of initial weight, breathing difficulties, or developed tumours 12 mm in diameter. No tumours exceeded this limit.
For studying the effects of epithelial-specific suppression of Brd4 or Myc in a mutant Kras background, chimeric cohorts of male KCshBrd4 mice derived from the ESCs above described were generated by the Center for Pancreatic Cancer Research (CPCR) at MSKCC or the Rodent Genetic Engineering Core at NYU as previously described33. ESC-derived KCshMyc mice have been previously described33. Only KC-shRNA mice with a coat color chimerism of >95 % were included for experiments. Kras wild-type Csh counterparts were generated by strain intercrossing (to remove LSL-KrasG12D allele). For induction of shRNA expression, mice were switched to a doxycycline diet (, at either 4 weeks of age (for injury-driven regeneration or accelerated neoplasia experiments) or at post-natal day (stochastic Kras-driven-neoplasia) to induce shRNA expression. In the case of C-shRNA mice, both female and male mice were used, allocated at randome to sex-matched treatment groups. Mice were switched to a doxycycline diet 625 mg/kg, Harlan Teklad) that was changed twice weekly at either 4 weeks of age (for injury-driven regeneration or accelerated neoplasia settings) or postnatal day 10 (stochastic Kras-driven-neoplasia) to induce shRNA expression.
For treatment with recombinant IL-33, 5 weeks-old C or KC mice were injected intraperitoneally once daily doses with 1 µg of murine recombinant IL-33 (#580504, R&D Systems) or vehicle (PBS) for 5 consecutive days, using sex-matched experimental groups.
Pancreatic epithelial cell isolation
For RNA-seq, ATAC-seq and scATAC-seq analyses in lineage-traced epithelial cells were freshly-isolated isolated from pancreatic tissues from KC, KPflC, or KC-shRNA mice by FACS-sorting. Specifically, pancreata were finely chopped with scissors and incubated with digestion buffer containing 1 mg/ml Collagenase V (C9263, Sigma-Aldrich), 2 U/mL Dispase (17105041, Life Technologies) dissolved in HBSS with Mg2+ and Ca2+ (14025076, Thermo Fisher Scientific) supplemented with 0.1 mg/ml DNase I (Sigma, DN25–100MG) and 0.1 mg/ml Soybean Trypsin Inhibitor (STI) (T9003, Sigma), in gentleMACS C Tubes (Miltenyi Biotec) for 42 min at 37°C using the gentleMACS Octo Dissociator. Normal (non-fibrotic) pancreas samples were dissociated as above, except that the digestion buffer contained 1mg/mL Collagenase D (11088858001, Sigma-Aldrich). After enzymatic dissociation, samples were washed with PBS and further digested with a 0.05% solution of Trypsin-EDTA (15400054, Thermo Fisher Scientific) diluted in PBS for 5 min at 37°C. Trypsin digestion was neutralized with FACS buffer (10 mM EGTA and 2% FBS in PBS) containing STI. Samples were then washed in FACS buffer containing DNase I and STI, filtered through a 100 μm strainer. Cell suspensions were blocked for 5 min at room temperature with rat anti-mouse CD16/CD32 with Fcblock (Clone 2.4G2, BD Biosciences) in FACS buffer containing DNase I and STI, and an APC-conjugated CD45 antibody (Clone 30-F11, Biolegend, 1:200) or APC-Cy7 CD45 antibody (Clone 30-F11, Biolegend, 1:200) was then added and incubated for 10 min at 4°C. Cells were then washed once with in FACS buffer containing DNase I and STI, filtered through a 40 μm strainer, and resuspended in FACS buffer containing DNase I and STI and 300 nM DAPI as live-cell marker. Sorts were performed on a BD FACSAria III cell sorter (Becton Dickinson) for mKate2 (co-expressing GFP for on dox-shRNA mice), excluding CD45+ cells. Cells were sorted directly into Trizol LS (Thermo Fisher Scientific) for RNA-seq or collected in 2% FBS in PBS for ATAC-seq.
Immunofluorescence, immunohistochemistry and histological analyses
Tissues were fixed overnight in 10% neutral buffered formalin (Richard-Allan Scientific), embedded in paraffin and cut into 5 µm sections. Slides were heated for 30 min at 55°C, deparaffinized, rehydrated with an alcohol series and subjected to antigen retrieval with citrate buffer (Vector Laboratories Unmasking Solution, H-3300) for 25 min in a pressure cooker set on high. Sections were treated with 3% H2O2 for 10 min followed by a wash in deionized water (for immunohistochemistry only), washed in PBS, then blocked in PBS/0.1% Triton X-100 containing 1% BSA. Primary antibodies were incubated overnight at 4°C in blocking buffer. The following primary antibodies were used: mKate2 (Evrogen, AB233, 1:1000), GFP (ab13970, Abcam, 1:500; and 2956S, Cell Signaling Technology, 1:200), Brd4 (HPA015055, Sigma-Aldrich, 1:100), Myc (ab32072, Abcam, 1:100), CPA1 (AF2765, R&D, 1:400), Clusterin (sc-6419, SCBT, 1:200), SOX9 (AB5535, Millipore, 1:1000), Amylase (sc-31869, SCBT, 1:1000), KRT19 (Troma III, Developmental Studies Hybridoma Bank, 1:500), FOSL1 (sc-376148, SCBT, 1:100), JUNB (sc-8051, SCBT, 1:100), AGR2 (NBP2–27393, Novus Biologicals, 1:200), DCLK1 (ab109029, Abcam, 1:200), Ki67 (BD Biosciences 550609, 1:200) and IL-33 (AF3626, R&D, 1:150). For mKate2, GFP and cMyc immunohistochemistry, Vector ImmPress HRP kits and ImmPact DAB (Vector Laboratories) were used for secondary detection. Tissues were then counterstained with Haematoxylin or when indicated Alcian blue (pH 2.5) and 0.1% Nuclear Fast Red Solution, dehydrated and mounted with Permount (Fisher). The immunohistochemistry detection of Brd4 was performed at the Molecular Cytology Core Facility of Memorial Sloan Kettering Cancer Center using Discovery XT processor (Ventana Medical Systems-Roche). The tissue sections were blocked for 30 min in 10% normal goat serum, 2% BSA in PBS. A rabbit polyclonal anti-Brd4 antibody (HPA015055, Sigma-Aldrich) was used in 1 ug/ml (1:100) concentrations. The incubation with the primary antibody was done for 6 hours, followed by 60 minutes incubation with biotinylated goat anti-rabbit IgG (PK610, Vector labs) in 5.75ug/mL concentration. Blocker D, Streptavidin- HRP and DAB detection kit (760–124, Ventana Medical Systems-Roche) were used according to the manufacturer instructions. Slides were counterstained with Hematoxylin (760–2021, Ventana), Bluing Reagent (760–2037, Ventana) and coverslipped with Permount (Fisher Scientific).
For immunofluorescence, the following secondary antibodies were used: goat anti-chicken AF488 (A11039, Invitrogen, 1:500), donkey anti-chicken IgY H&L (FITC) (ab63507, Abcam, 1:500), donkey anti-rabbit AF594 (A21207, Invitrogen, 1:500), goat anti-rabbit AF594 (A11037, Thermo Fisher Scientific, 1:500), donkey anti-goat AF488 (A11055, Invitrogen, 1:500) and donkey anti-goat AF594 (A11058, Thermo Fisher Scientific, 1:500). Slides were counterstained with DAPI and mounted in ProLong Gold (Life Technologies). Hematoxylin and eosin (H&E) was performed using standard protocols. Images were acquired on a Zeiss AxioImager microscope using using a 10 × (Zeiss NA 0.3) or 20 × (Zeiss NA 0.17) objective, an ORCA/ER CCD camera (Hamamatsu Photonics, Hamamatsu, Japan), and Axiovision or Zeiss (ZEN 2.3) software. Bright-field and fluorescence images of pancreata gross morphology were acquired using Nikon SMZ1500 microscope and NIS-Element imaging software.
Histological classification and grading of pancreatic lesions into ADM or PanIN PanIN lesions was performed by a veterinary pathologist blinded to genotype and treatment condition in H&E stained-slides using established criteria46. When applicable GFP+ area marking shRNA-expressing epithelial cells from KCsh mice was quantified using “SpotR software”. All lesions in at least 3 representative 20X fields per section were measured and counted. The results were averaged and normalized to total tissue area analyzed. Statistical analyses were performed using unpaired two-tailed Student’s t-test in GraphPad Prism (v7 and v8). Graphs display means ± s.e.m of independent biological replicates (mice).
Multiplexed immunoassays in tissue lysates
0.1% SDS; 1mM EDTA; 1% NP-40, supplemented with fresh protease inhibitors, Complete™ Mini Protease Inhibitor Cocktail, Sigma) using PowerBead Tubes, Ceramic 2.8mm and PowerLyzer 24 Homogenizer (110/220 V, 2 × 30-sec cycles; S 3500) (Qiagen). After homogenization, RIPA lysates were incubated for 30 mins at 4°C and with continued vortexing, and clarified by centrifugation. Same amounts of total tissue protein (25 µg) diluted in RIPA buffer were subjected to quantitative multiplexed ELISA (Discovery Assays) performed by Eve Technologies (Canada).
Cell culture and retroviral infection
Cells were maintained in a humidified incubator at 37 °C with 5% CO2. The 266–6 pancreatic acinar cell tumor cell line was purchased from ATCC (CRL-2151) and was grown in complete DMEM (DMEM, 10% FBS (Gibco), pen-strep) on non-coated, tissue-culture-treated plates. KPflC cells were derived from PDAC arising in a Ptf1a-Cre;LSL-KrasG12D;p53flox/+;RIK mouse generated by blastocyst injection, as previously described47, and propagated in propagated in complete DMEM on collagen-coated plates (PurCol, Advanced Biomatrix, 0.1 mg/ml). Frozen stocks generated within 2 to 3 passages from date of purchase (266–6) or generation (KPflC) and were used for the in vitro experiments. Cell lines were not externally authenticated. All cell lines used were negative for mycoplasma.
For stable transduction of 266–6 and KPflC cells with FOSL1 and/or cJUN or both, VSV-G pseudotyped retroviral supernatants were generated from transduced Phoenix-gp packaging cells and infections were performed as described elsewhere34. The following plasmids were used: p6599 MSCV-IP N-HAonly FOSL1 (Addgene, #34897), p6600 MSCV-IP N-HAonly JUN (Addgene, #34898), pMIEG3-cJun (Addgene, #40348), and their corresponding empty vectors (MSCV-IP N-HAonly-EV or pMIEG3-EV). Empty vectors were generated by removing FOSL1 (from MSCV-IP N-HAonly FOSL1 plasmid) or cJun (from pMIEG3-cJun plasmid) cDNAs by digestion with Xho/EcoRI-HF or XhoI/BamHI-HF restriction enzymes (New England Biolabs), respectively followed by Klenow step (M0210M, New England Biolabs), gel extraction purification of the digested back-bone fragment (QIAquick Gel Extraction Kit, Qiagen), and ligation through T4 DNA Ligase (New England Biolabs), following manufacturer’ instructions. All plasmids were authenticated by test-digestion and sanger sequencing. Infected cells were selected with 2 μg/mL (for 266–6 cells) or 8 μg/mL (for KPflC;RIK cells) puromycin (Sigma) and/or sorted based on GFP-positivity using Sony MA900 Cell Sorter (Sony) (GFP-vectors), depending on whether they were transduced with MSCV-IP-N-HA and/or pMIEG3 vectors, respectively, and were harvested for expression analyses at day 12 (KPflC;RIK) or day 28 (266–6) post-infection.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) analysis
Total RNA was isolated from mKate2+,CD45-DAPI- sorted primary pancreatic epithelial cells using the Trizol LS (Thermo Fisher Scientific), and cDNA was obtained from 500 ng of RNA using the Transcriptor First Strand cDNA Synthesis Kit (Roche) after treatment with DNAse I (Invitrogen) following manufacturer’s instructions. The following primer sets for mouse sequences were used: Il33_F GCTGCGTCTGTTGACACATT, Il33_R GACTTGCAGGACAGGGAGAC, Agr2_F ACAACTGACAAGCACCTTTCTC, Agr2_R GTTTGAGTATCGTCCAGTGATGT, Muc6_F AGCCCACATTCCCTATCAGC, Muc6_R CACAGTGGAAGATTGCGAGAG, Cpa1_F CAGTCTTCGGCAATGAGAACT, Cpa1_R GGGAAGGGCACTCGAACATC, Sox9_F CGTGCAGCACAAGAAAGACCA, Sox9_R GCAGCGCCTTGAAGATAGCAT, Hprt_F TCAGTCAACGGGGGACATAAA, Hprt_R GGGGCTGTACTGCTTAACCAG, Rplp0_F GCTCCAAGCAGATGCAGCA, Rplp0_R CCGGATGTGAGGCAGCAG, Actb_F GGCTGTATTCCCCTCCATCG and Actb_R CCAGTTGGTAACAATGCCATGT. qRT-PCR was carried out in triplicate (5 cDNA ng/reaction) using SYBR Green PCR Master Mix (Applied Biosystems) on the ViiA 7 Real-Time PCR System (Life technologies). Hprt, Rplp0 (aka 36b4) or ActB served as endogenous normalization controls.
Isolation, culture and genetic manipulation of pancreatic organoids
To isolate untransformed ductal organoids for the transplantable PDAC tumorigenesis model, normal pancreas from LSL-KrasG12D mice (pure Bl/6N background) minced and digested with 0.01 2% collagenase XI (C9407, Sigma-Aldrich) and 0.012 % Dispase (17105041, Life Technologies) in in HBSS with Mg2+ and Ca2+ (14025076, Thermo Fisher Scientific) at 37 °C for a maximum of 30 mins. The material was further digested with TrypLE (GIBCO) for 5 min at 37°C, washed twice with DMEM/F12 (Life Technologies) supplemented with 2 mM glutamine and pen-strep, embedded in growth factor reduced matrigel (Corning), and cultured in complete medium, as described in Boj et al 2014. For activation of mutant Kras, organoids harbouring the LSL-KrasG12D allele were transduced with Ad-mCherry-Cre (Vector Biolabs), and Cherry+ cells were sorted from single cell organoid suspension by flow cytometry 36h thereafter. Resulting clones were assessed for LSL-KrasG12D recombination by genotyping PCR in genomic DNA using the following primers: GTCTTTCCCCAGCACAGTGC, CTCTTGCCTACGCCACCAGCTC, and AGCTAGCCACCATGGCTTGAGTAAGTCTGCA. Validated Cre-recombined clones were then subjected to CRISPR-based inactivation of Trp53 using the PX458 vector (Addgene #48138) and gRNA AGTGAAGCCCTCCGAGTGTC sequence. PX458-sgTrp53 was transduced into organoids by transient transfection using the spinoculation method previously described48, with the modification of using the Effectene transfection reagent (Qiagen). PX458-sgTrp53 introduced cells were sorted by GFP positivity with flow cytometry 36 h post-transfection. p53 null status of targeted clones was validated by western blot, using anti-p53 antibody (CM5, Leica Microsystem) and anti-β-actin−peroxidase antibody (Sigma-Aldrich) as normalization control.
Bulk ATAC-seq analysis
Cell preparation, transposition reaction, ATAC-seq library construction and sequencing:
65,000 mKate2+ cells isolated by FACS, washed once with 50 µL of cold PBS, and resuspended in 50ul cold lysis buffer11. Cells were then centrifuged immediately for 10 min at 500 g, 4°C and nuclei pellet was subjected to transposition with Nextera Tn5 transposase (FC-121–1030, Illumina) for 30 min at 37°C, according to manufacturer’s instructions. DNA was eluted using MinElute PCR Purification Kit in 11.5 µl elution buffer (Qiagen). ATAC-seq libraries were prepared using the NEBNext High-Fidelity 2x PCR Master Mix (NEB M0541) as previously described37. Purified libraries were assessed using a Bioanalyzer High-Sensitivity DNA Analysis kit (Agilent). Approximately 200 million paired-end 50 bp reads were sequenced per replicate on a HiSeq 2500 (High Output) at the New York Genome Center.
Mapping, peak calling and dynamic peak calling:
Fastq files were trimmed with trimGalore and cutadapt49, and the filtered, pair-ended reads were aligned to mm9 with bowtie250. Peaks were called over input using MACS2 51, and only peaks with a p-value of <=0.001 and outside the ENCODE blacklist region were kept. All peaks from all samples were merged by combining peaks within 500bp of each. featureCount52 was used to count the mapped reads for each sample. The resulting peak atlas was normalized using DESeq253. For comparison to DepthNorm, samples were normalized to 10 million mapped reads. Normalized bigwig files were created using the normalization factors from DESeq2 as previously described54 and bedtools genomeCoverageBed55. Dynamic ATAC-peaks were called if they had an absolute log2FC >= 0.58 and a FDR <=0.1.
ATAC-seq heatmap clustering:
The dynamic peaks of regeneration and early neoplasia determined by comparing Normal, Injury, Kras*, and Kras*+Injury conditions (as defined in Fig. 1a and Extended Data Fig. 1a) were clustered using z-score and a kmeans of 6 and plotted using ComplexHeatmap56.
ATAC-seq peak annotation and pathway enrichment analysis.
Peaks were associated with genes based on UCSC.mm9.knownGene57 using ChIPseeker package58. The peaks were further analyzed for genic location (annotatePeaks). The distance of a peak to the nearest TSS was identified and annotated the peak to that gene. For pathway enrichment analysis of genes associated with regeneration and early neoplasia ATAC-seq clusters, genes uniquely associated with peaks belonging to each cluster were subjected to pathway analyses using enrichr59. Genes associated with > 1 ATAC cluster were excluded such that each gene is uniquely associated to one module.
TF motif enrichment and co-occurrence analyses:
Motif enrichment analysis was performed individually on each of the 6 ATAC-peak clusters using the HOMER de novo motif discovery tool60 using findMotifsGenome command with size = given and length = 8 parameters. Motif enrichment scores of the de novo predicted motifs identified from the above analyses were calculated for all 6 ATAC-clusters or accessibility-GAIN or -LOSS regions between Injury, Kras*, Kras*+Injury and PDAC vs Normal conditions were calculated by applying the findMotifsGenome command with the size = given and length = 8 in each peak-set, and results were visualized as heatmaps (Fig. 1g) or bubble plots (Extended Data Fig. 2f) plotted with R package ggplot. For each of the 6 ATAC-cluster peaks, TF motif occurrence in a given peak was defined as previously described14. In brief, Pearson correlation coefficient method was used to determine the similarity between TF pairs based on the whether a specific motif was present (1) or absent (0), and the results were visualized using the R package ggcorrplot with hierarchical clustering.
Metagene plots:
Metagene plots were created with the deepTools package using ATAC-seq peak centers and regions extended to +/− 3,000 bp with 10 bp bins. To evaluate differential enrichment from ATAC-seq meta-profiles between experimental conditions, average signals for each experimental condition were calculated (200 bins) for +/−1 Kb around the peak center, and compared. p-value was determined by Kolmogorov–Smirnov test.
Intersection of ATAC-seq and publicly available ChIP-seq data:
To analyze accessibility dynamics at H3K27ac-enriched regions defining PDAC metastasis, we first extracted 849 peaks significantly enriched in PDAC metastasis vs normal pancreas-derived organoids17 (based on enrichment cut-off of more than 10-fold increase of H3K27ac signal in any of the metastasis-derived organoids compared to the average of normal pancreas previously used17). We then matched the mm9 coordinates of H3K27Ac-GAIN ChIP-seq regions with positive ATAC-seq peak calls from the MACS2 output for PDAC samples over input and used genome_left_join function from R package fuzzyjoin to identify coordinates that were overlapping between PDAC positive peaks and H3K27Ac ChIP-seq of organoid-cultured vs freshly-isolated PDAC cells, respectively. This found positive ATAC-seq match for 53% of metastasis-associated H3K27Ac. In vivo accessibility dynamics at these PDAC opened-loci was evaluated by calculating the proportion of metastasis-associated ATAC-seq loci overlapping with ATAC-GAIN regions between Injury, Kras*, Kras*+Injury and PDAC conditions vs Normal, as well as by measuring differential enrichment of ATAC-seq signals centered on the middle of this metastasis-associated peakset across these same conditions using the metagene plot analyses above described. In addition, the Gene Transcription Regulation Database (GTRD, v20.06) was used to extract publicly available ChIP-seq experiment information on TFs and mine for those validated to bind regulatory regions of the Il33 locus displaying ATAC-GAIN between Injury, Kras* or Kras*+Injury vs Normal conditions. Specifically, dynamic ATAC-seq peaks associated with Il33 were converted from mm9 to mm10 coordinates using the UCSC liftover tool61 and were then intersected with GTRD’s ChIP-Seq datasets. Selected TFs whose binding sites are enriched in these differentially accessible Il33 ATAC-peaks identified in our study and experimentally validated to bind these regions in other contexts are indicated in Extended Data Fig. 9e.
Integration of TF-associated RNA-seq and ATAC-seq data:
To assign motifs enriched at differentially-accessible loci (identified by HOMER de novo analyses) to specific TFs most likely to bind these sequences in each experimental condition, we applied a similar workflow as previously described62 that integrates the motif enrichment and mRNA fold change data between 2 conditions. In brief, we first identified motifs significantly enriched in the 6 ATAC-seq clusters defined from dynamic peaks of regeneration and early neoplasia as described above, to expose potential regulatory nodes linked to accessibility-GAINS or -LOSSES driven by Injury (Injury), mutant Kras (Kras*) or their combination (Kras*+Injury). For each of these enriched motifs, we extracted HOMER’s best matches to known motifs TFs to generate a list of putative TF factors. For each of these TFs, we calculated RNA-seq and ATAC-seq absolute scores reflecting the degree of enrichment of their assigned motif (- log10 p-value, defined by HOMER) or magnitude of their mRNA expression fold change (log2FC) between Injury, Kras*, Kras*+Injury and PDAC conditions vs Normal, respectively. To avoid having results from one of the comparisons dominate the entire analysis, a weight γ was calculated for each TF and comparison by dividing the TF’s absolute score in that specific comparison by the sum of the absolute scores across all:
The combined ATAC-RNA scores for each TF and comparison was calculated by multiplying these weighted RNA- and ATAC- scores:
To reflect directionality of the TF expression change in each tissue state compared to Normal, resulting scores were multiplied by −1 or +1 depending on whether the TF was downregulated or upregulated in each comparison, and motif enrichment values for either ATAC-LOSS or -GAIN peaks of the same comparison were used, respectively. The combined ATAC-RNA scores top 12 TFs with highest combined-scores in the Kras*+Injury and PDAC (vs Normal) are displayed as heatmaps in Extended Data Fig. 7e.
Circos visualization (Fig. 4b):
Dynamic peaks linked to genes displaying consistent gene expression changes (FC>=2, p-adj < 0.05, see below) across Injury, Kras*, Kras*+Injury and PDAC conditions (vs Normal) were clustered across the indicated conditions using log2FC values with single clustering method. Resulting clusters were plotted using ‘circos’ (v0.69–8) and annotated for AP1-motifs (annotatePeaks with AP-1.GSE21512.motif) or NR5A2- or PTF1A-bound loci in normal pancreas (extracted from GSE3429516 or GSE8626215, respectively). In addition, this same class of dynamic peaks were investigated for motif enrichment using HOMER findMotifsGenome.
Single-cell ATAC-seq analysis
Cell preparation, transposition reaction, scATAC-seq library construction and sequencing:
Approximately 50,000 mKate2+ cells (mKate2+;CD45-;DAPI-) were isolated by FACS and subjected to scATAC-Seq protocol (10X Genomics, CG000168 RevA)63. Briefly, FACS-sorted cells were lysed in cold-lysis buffer (0.1% NP-40, 0.1% Tween 20, 0.01% Digitonin, 10 mM NaCl, 3 mM MgCl2 and 10 mM Tris-HCl [pH 7.4]), washed and processed according to ‘Nuclei Isolation for Single-Cell ATAC Sequencing’ protocol (CG000169 RevD). Resulting nuclei suspension was subjected to transposition reaction for 60 min at 37°C and then encapsulated in microfluidic droplets using 10X Chromium instrument following manufacturer’s instructions with a targeted nuclei recovery ~ 5,000. Barcoded DNA material was cleaned and prepared for sequencing according to the Chromium Single Cell ATAC Reagent Kits User Guide (10x Genomics; CG000168 RevA). Purified libraries were assessed using a Bioanalyzer High-Sensitivity DNA Analysis kit (Agilent) and sequenced on a Illumina HiSeq 2500 (High Output) platform at approx. 150M reads (R1 50bp, R2 50 bp, i7 8bp, i5 16bp) per 1 sample (~ 5000 nuclei) at MSKCC’s Integrated Genomics Operation Core.
Pre-processing of scATAC-seq data:
Fastq files for each sample were pre-processed to a cell-by-peak count matrix through the CellRanger ATAC pipeline63 with several modifications as follows: Reads were aligned to the mm10 reference, barcodes were counted, and peaks were called using CellRanger’s default peak-caller. However, since CellRanger frequently calls unusually large peaks, a custom modification of the CellRanger pipeline allowed all peaks within 10 bps of one another to be merged (as opposed to the 500 bps window in the default pipeline), creating an initial peak atlas with sufficiently narrow peaks. This modification increased the resolution of called peaks, and thus allowed us to distinguish nearby peaks that are variable across sub-populations of cells.
To reduce the large number of peaks in this atlas, we filtered out low-coverage peaks, unless these characterized a subpopulation of cells using the following strategy: First, an unbiased clustering of cells was performed using Phenograph64 on these initial peak features to define major cellular compartments. The peaks in the initial atlas were then retained in the final count matrix only if either (1) their coverage (reads per peak) normalized by peak width was above a certain threshold, hence they are confident peak calls, and/or (2) the peak was determined to be enriched in any cluster using a Fisher’s exact test (adjusted p-value<.01), hence they are differentially accessible across clusters of cells. For this analysis, we automatically determined the coverage threshold for filter (1) based on the distribution of per peak coverage for each sample. Specifically, peaks with coverage less than the sample median coverage (across all peaks) failed this filter (1) and were then passed to filter (2) to test for differential accessibility. All downstream CellRanger steps for cell filtering were applied after the above steps as usual. CellRanger’s aggregation function was then used to combine cells across samples to a unified peak atlas, using the depth normalization function to normalize cells across the two samples. A window of 10bp was again used to merge nearby peaks, as opposed to the default 500 bp. The above procedure resulted in a dataset of 11712 putative cells and 152991 peaks from both samples.
scATAC-seq filtering, normalization and visualization:
Low coverage cells were filtered by inspecting the per cell coverage (sum of peak counts per cell) and removing all cells in the lower mode of this distribution, resulting in a final count of 6369 cells. A binary matrix was then produced from the filtered count matrix by setting all nonzero values to 1. This allowed each peak feature to be represented as either open or closed, avoiding biases from wide peaks where counts may be extraordinarily high.
The data was then normalized by regressing out the sum of total counts per cell from this binarized matrix using ordinary linear regression. This normalization step is performed to partially correct for sampling biases across cells. A similar approach has been used in previous scATAC-seq studies65. Specifically, a linear model was fit that explains the counts for each peak , for peaks , with coverage as the sole regressor:
After obtaining estimates for model parameters , corrected values for each peak were given as follows:
Extraordinarily wide (>2000bp) or narrow (<2bp) peaks were removed from the count matrix following normalization, so as to filter non-specific peaks containing multiple transcription factor bindings and/or nucleosome-occupied regions within one called element. Principal Components Analysis (PCA) was then performed for dimensionality reduction, and the top 20 components--which were chosen by inspecting the cumulative variance explained across PCs using the knee point method--were then used as features for a Uniform Manifold Approximation and Projection (UMAP)66 visualization of the cells in two dimensions (Figure 5A). Global structure of the visualization was robust to choice of thresholds on peak width, principal components, and number of UMAP neighbors.
scATAC-seq clustering and differential peak analysis:
Phenograph clustering of the top 20 principal components was used with number of nearest neighbors K=25 to determine highly granular subsets of cells. Clusters were then merged to larger, coherent subsets based on accessibility patterns of peaks nearby known cell type markers. In particular, a Fisher’s exact test was performed on the binarized data for each peak, testing in each case for enrichment of peak accessibility in a Phenograph cluster versus all other cells. Significant peaks were then mapped to nearest target genes based on distance to the transcription start site (using a maximum allowable distance of 10kb), and enriched accessibility patterns were compared across clusters. Clusters were merged based on degree of overlap among cluster-defining peaks and shared opened chromatin at known pancreas cell state markers. To then identify differential peaks across these large compartments, each major subpopulation was compared to the rest by Fisher’s exact test (Fisher’s exact test, adjusted p-value < 0.05) to obtain a final set of significant, compartment-specific enrichments (listed Supplementary Table 8).
Visualization of per-peak accessibility:
To inspect accessibility dynamics across populations, we developed a visualization strategy relying on the binarized matrix to help overcome its sparsity. For each peak, a Gaussian kernel density estimate was fitted on UMAP embedding coordinates for cells in which the peak was open. The density was then estimated at each cell’s location (regardless of accessibility status) in the embedding, producing a continuous-valued metric corresponding to the density of cells harboring the open peak in a particular region of the visualization. These estimates are valid for visualization of cells with open chromatin at a specific locus on the UMAP itself, but do not provide a general estimate of peak-accessibility density within a region of high-dimensional phenotypic space.
Comparison to bulk ATAC-seq dynamics:
To first study reproducibility of bulk accessibility patterns in single cells, signals from corresponding bulk samples were compared to each single cell library where cells were aggregated to produce a ‘pseudo-bulk’ sample. First, bulk ATAC-seq peaks were converted from mm9 to mm10 coordinates using the UCSC liftover tool61. scATAC-seq BAM files (combining reads from all cells) were then converted to normalized bigwig files using the bamCoverage function from the deeptools package67, and accessibility signals were collected per lifted-over peak. Global reproducibility was assessed by correlation between bulk signals per peak (derived from DESeq-corrected signal tracks) and the single cell pseudo-bulk signals. To then assess whether dynamics in accessibility were consistent across bulk and single cell datasets, volcano plots were generated using results from DESeq differential accessibility analysis of bulk data. Fold changes in each peak region based on single cell pseudo-bulk signals were directly compared to inferred dynamics in lifted-over peaks from DESeq results of bulk datasets.
Comparison to bulk ATAC-seq peak modules:
A major goal of the scATAC-seq analysis is to deconvolve the accessibility differences identified at the bulk level, and therefore we sought to investigate the accessibility patterns of bulk peak modules across single cells. To achieve this, coordinates of peaks identified in bulk were converted from mm9 to mm10 coordinates using the UCSC liftover tool. To directly compare these to de novo identified peaks in scATAC-seq, bedtools intersect tool55 was used to find overlapping regions between bulk peak sets and those included in the single cell count matrix. All peaks with any non-zero overlap were retained for downstream analysis. These subsets of peaks were visualized by computing the proportion of cells in each compartment and each biological condition (independent animal) harboring an open peak, which were then z-scored across clusters and conditions for visualization in heatmaps. To ensure coverage differences between conditions did not impact these values systematically, the analysis was performed on down-sampled data, where all cells’ counts were randomly sampled to a total peak count 5000 counts per cell. Global shifts in peak accessibility across clusters and between conditions were robust to various thresholds on total peak count, and were globally consistent in the original dataset without downsampling.
Compartment-specific signal tracks:
To further evaluate accessibility differences across major subpopulations and between conditions, signal tracks were generated per sample per cellular compartment for visualization in the Integrative Genomics Viewer68. First, BAM files for each sample were separated into compartment-specific bins using information about the CellRanger –corrected cell barcode for each read, thus creating a new BAM file per compartment per condition. A signal track bigwig file was then produced from each BAM file using two normalization strategies to ensure biases due to cluster size and sample-specific sequencing depths did not impact visualizations. The first strategy utilized the bamCoverage function from the deeptools package for normalization accounting for total read counts of the sample-specific, cluster-specific BAM file. The second strategy corrected for these differences by randomly sub-sampling each BAM file to the total read count of the smallest, leaving each file with approximately 5,529,294 total reads. In the latter case, the smallest two clusters were excluded from the analysis, as their coverage was too poor due to low cell count. To correct for cell count and sequencing depth disparities among the different conditions compared, display of accessibility signals between conditions are downsampled to same coverage across all tracks shown.
Transcription factor activity scores:
To evaluate the activity of acinar- (NR5A2) and injury/neoplasia-activated (e.g. AP-1) transcription factors in individual cells, we first identified genomic regions with differential chromatin accessibility and linked to genes displaying consistent changes in gene expression between early neoplasia (Kras*+Injury) and normal pancreas (Normal) through integrative analyses of bulk-ATAC and bulk-RNA-seq data (mm9, see below). These regions were annotated for binding sites for NR5A216 or AP-1 (AP-1.GSE21512.motif), respectively, and were lifted over to mm10 coordinates for comparison with scATAC-seq profiles, as above. In this case, lifted over coordinates were then associated with scATAC-seq peaks only when coordinates were fully overlapping using bedtools ‘intersect minimum overlap’ function, to ensure significant overlap between TF binding sites and scATAC-seq peaks. To then quantify binding activity per cell, the proportion of all open peaks per cell overlapping with a binding site was computed. These values were then visualized on UMAP and in heatmaps (values in heatmaps are logged with a .0001 pseudocount). We quantified the relative accessibility of AP1 and NR5A2 binding sites per cell by computing a ratio of logged binding activity scores, which scales with increasing AP1 activity and decreasing NR5A2 activity.
Identification of peaks correlating with AP-1/NR5A2 ratio across individual cells.
To identify individual peaks whose dynamics are associated increasing AP-1/NR5A2 TF activity ratios across individual Kras-mutant cells, we computed Pearson correlation of each normalized peak with AP-1/ NR5A2 ratio across cells. Peaks with a relatively strong correlation magnitude (|r|>.1) were selected for further analysis, and visualized with normalized accessibility trends over cells ranked by increasing AP-1/ NR5A2 ratio. Gene annotations per peak were derived by mapping each peak coordinate to its nearest target gene within a 50 kb window. To confirm the association of positively or negatively correlated-peaks with the AP-1/NR5A2 TF activity switch (e.g. Il33 or Cpa1-associated peaks) we performed a unpaired two-tailed Student’s t-test comparing AP-1/NR5A2 TF activity score ratios of all cells where the identified correlated-peak is open (i.e. at least one scATAC-seq count in that peak) versus those where the peak is closed. GREAT tools69 was used to compare the ontology of genes associated with positively or negatively -correlated peaks, using single nearest gene’ within 1000 kb as input parameters, and with comparable results obtained using the ‘two nearest genes’ option. Top pathways from ‘GO Biological Process’ and ‘GO Molecular Function’ categories are displayed in Fig. 4g.
RNA-seq analysis
RNA extraction, RNA-seq library preparation and sequencing:
Total RNA was isolated from primary mKate2+,CD45-DAPI- pancreatic epithelial cells isolated from normal, regenerating (Injury), early neoplastic (Kras*, Kras*+Injury) and cancer (PDAC) tissues into TRIzolLS and assessed using a Agilent 2100 Bioanalyzer. Sequencing and library preparation was performed at the Integrated Genomics Operation (IGO) at MSKCC. RNA-seq libraries were prepared from total RNA. After RiboGreen quantification and quality control by Agilent BioAnalyzer, 100–500ng of total RNA underwent polyA selection and TruSeq library preparation according to instructions provided by Illumina (TruSeq Stranded mRNA LT Kit, RS-122–2102), with 8 cycles of PCR. Samples were barcoded and run on a HiSeq 4000 or HiSeq 2500 in a 50bp/50bp paired end run, using the HiSeq 3000/4000 SBS Kit or TruSeq SBS Kit v4 (Illumina) at MSKCC’s Integrated Genomics Operation Core. An average of 41 million paired-end reads was generated per sample. At the most the ribosomal reads represented 0.01% of the total reads generated and the percent of mRNA bases averaged 53%.
RNA-seq read mapping, differential expression analysis and heatmap visualization:
Resulting RNA-Seq data was analyzed by removing adaptor sequences using Trimmomatic70. RNA-Seq reads were then aligned to GRCm38.91 (mm10) with STAR71 and transcript count was quantified using featureCounts 52 to generate raw count matrix. Differential gene expression analysis was performed using DESeq2 package (Love et al., 2014) between experimental conditions, using 3–5 independent biological replicates (individual mouse) per condition, implemented in R (http://cran.r-project.org/). Principal component analysis (PCA) was performed using the DESeq2 package in R. Differentially expressed genes (DEGs) were determined by > 2-fold change in gene expression with adjusted P-value < 0.05. For heatmap visualization of DEGs, samples were z-score normalized and plotted using ‘pheatmap’ package in R.
Intersection of gene expression (RNA-seq) and chromatin accessibility (bulk ATAC-seq) data:
To define ‘chromatin-dymamic’ vs ‘chromatin-stable’ DEGs, upregulated and downregulated DEGs for the indicated comparisons (Injury, Kras*, Kras*+Injury or PDAC vs Normal; FC>2, padj<0.05) were classified into the following chromatin-categories based on the accessibility change at associated peaks in the same tissue state vs Normal: DEG with accessibility-GAIN (at least one associated peak showing significant accessibility-GAIN), DEG with accessibility-LOSS (at least one associated peak showing significant accessibility-LOSS) or ‘chromatin stable’-DEG (none of the associated peaks showing significant changes in chromatin accessibility). In the instance that different peaks associated with the same gene showed opposing dynamic accessibility patterns (which as noted for < 4% of DEGs in average), the gene was only classified into accessibility -GAIN or -LOSS categories if the contribution of GAIN or -LOSS peaks were at least 3 times more represented within the dynamic peaks, respectively. DEGs with no associated ATAC-peaks detected in that same tissue state were classified as ND. The chromatin accessibility status of the DEGs in regenerating, early-stage neoplasia and malignant PDAC tissue states in GEMMs is summarized in Supplementary Table 5. Expression dynamics of gene sets associated with bulk ATAC-seq clusters across Normal, Injury, Kras*, Kras*+Injury and PDAC sample were visualized as heatmap of normalized median expression values plotted with seaborn in python. To define these gene sets, peaks from each of the 6 ATAC-seq clusters were associated with genes based on UCSC.mm9.knownGene57 using ChIPseeker package58, as above, and ATAC-cluster-defining genes were defined as those uniquely associated with one cluster. For each RNA-seq sample, the median expression across genes associated to each ATAC-cluster was computed, and z-scored for visualization.
Definition of neoplasia-specific epigenetic programs.
ATAC-seq and RNA-seq datasets were overlapped to identify genes exhibiting significant chromatin accessibility and expression changes in cells undergoing injury-accelerated neoplasia (Kras*+Injury) and advanced PDAC (PDAC) but not in regenerative metaplasia (Injury alone) when compared to normal healthy pancreas (Normal).
Functional annotations of gene sets:
Pathway enrichment analysis was performed in the indicated gene sets with the Reactome and KEGG database using enrichR59. Significance of the tests was assessed using combined score defined by enrichR, described as c = log(p) * z, where c is the combined score, p is Fisher exact test p-value, and z is z-score for deviation from expected rank.
Gene set enrichment analysis (GSEA):
GSEA72 was performed using the GSEA-Preranked tool for conducting gene set enrichment analysis of data derived from RNA-seq experiments (version 2.07) against signatures in the MSigDB database (http://software.broadinstitute.org/gsea/msigdb), signatures derived herein, and published expression signatures derived from human23,73 or organoid samples17.
Overlap with human gene expression datasets:
2 independent public datasets of microarray data from human PDAC and normal pancreas samples (GSE7172923 and GSE6245273) were used. Differential expression analysis was then applied using limma package74 to define differentially expressed genes (DEGs) between PDAC vs normal samples, using > 2-fold change and adjusted p-value < 0.05 cut-off.
Statistics and Reproducibility
Statistical analyses were performed with GraphPad Prism (v7/8) and R (v3.5.1 and v1.26.0), and Python programming language (python version 3.6.4). Pooled data are presented as mean values ± s.e.m. Sample size, error bars and statistical methods are reported in the figure legends. P-values are shown in figures or associated legends. Statistical significance of differences between two experimental groups were assessed by unpaired two-tailed Student’s t-test. In RNA-seq data, significance for differential gene expression between groups was based on adjusted p-value < 0.05. For pathway enrichment analysis of RNA-seq gene clusters, the significance of gene lists was assessed by adjusted p-value and z-score59. Significance of gene sets from GSEA was based on the normalized enrichment score (NES) and the false discovery rate q-value (FDR q-val). In bulk ATAC-seq data, dynamic peaks were called if they had an absolute log2FC >= 0.58 and a FDR <=0.1. Motif enrichment scores were evaluated by p-values scores defined by HOMER60. Kolmogorov–Smirnov test was used to obtain p value to assess differential enrichment from bulk ATAC-seq meta-profiles. Correlation between scATAC-seq data and bulk ATAC-seq data was evaluated using Pearson correlation analysis. The correlation of individual peaks with increasing TF activity ratios across individual cells was evaluated using Pearson correlation analyses, and the significance of biological functions linked to identified positively or negatively correlated peak sets was evaluated by GREAT69 FDR q-value.
No statistical methods were used to pre-determine sample size in the mouse studies, and mice with matched sex and age were randomized into different treatment groups (eg. PBS control, caerulein). The investigators were not blinded to allocation during experiments and outcome assessment, except for the histological assessment of pancreatic lesions, which were classified and graded by a veterinary pathologist blinded to genotype and treatment conditions. All experiments were reliably reproduced. Specifically all in vivo experiments, except for omics data (i.e. RNA-seq, ATAC-seq and scATAC-seq), were performed independently at least two times, with the total number of biological replicates (independent animals) indicated in the corresponding figure legends. Caerulein and rIL-33 treatments (and their respective phenotypic/molecular readouts) yielded similar results irrespective of the experimental cohort (eg. mouse litter). The in vitro TF overexpression experiment was repeated twice (with datapoints representing two independent wells each) with similar results. ATAC/RNA-seq data from freshly isolated cells harvested at different dates are also reliably reproduced, with biological replicates (independent mice) from the same experimental groups clustering together in PCA and hierarchical clustering methods irrespective of experimental cohort and sample processing dates. Mouse illustrations were created with ©BioRender - biorender.com. Figures were prepared using Illustrator CC 2019/2020 (Adobe).
Extended Data
Extended Data Fig. 1. Chromatin accessibility dynamics during pancreatic regeneration and early neoplasia.
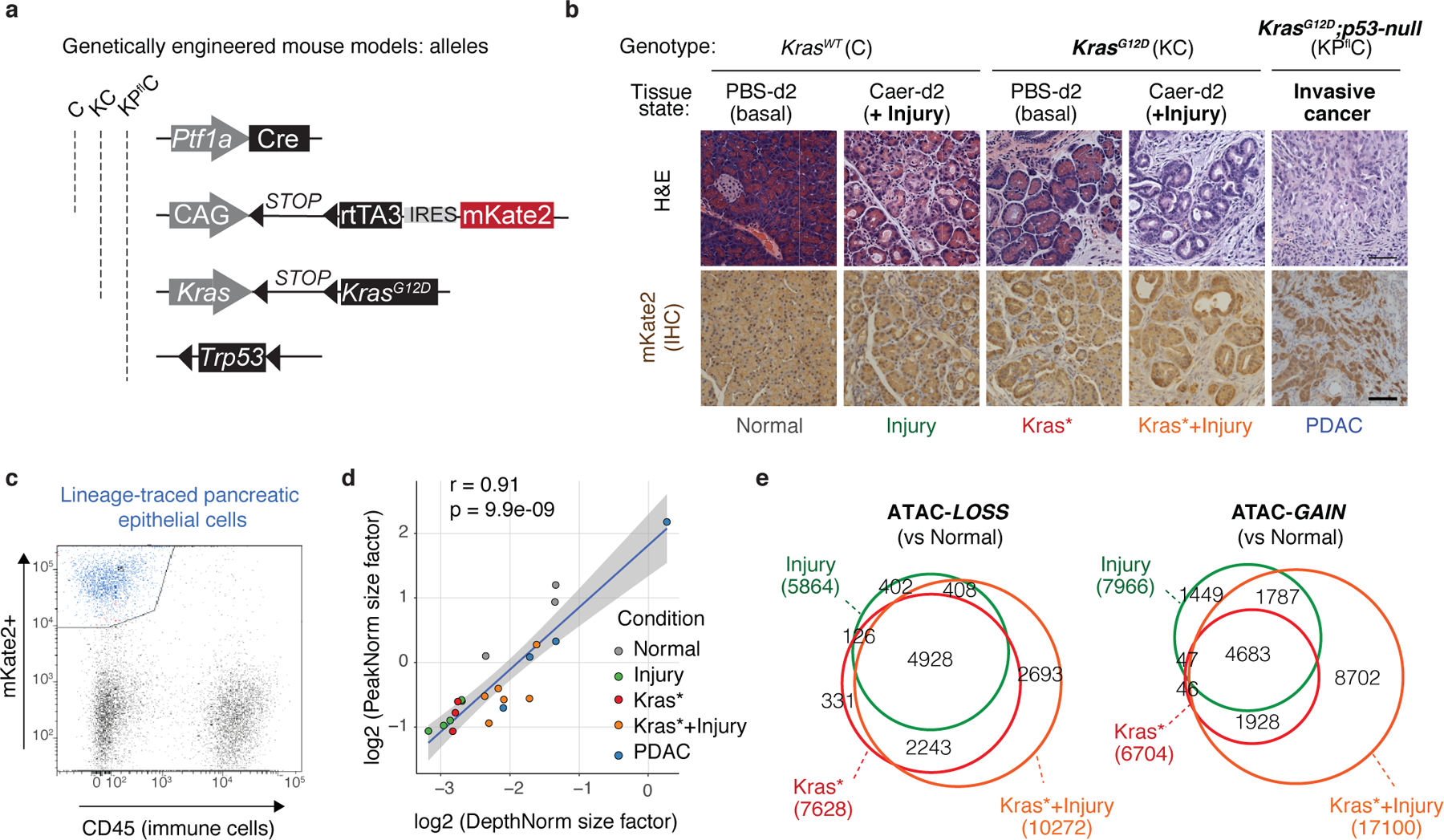
a, Schematic representation of the allele configurations used to trace Cre-recombined wild-type, Kras-mutant or Kras-mutant;p53-null pancreatic epithelial cells in transgenic mice. b, Representative H&E (top) or mKate2 IHC (bottom) of pancreata from the indicated mouse models and treatment conditions (n=3 mice per group), illustrating the defined tissue states, spanning normal healthy (Normal), regenerating (reversible metaplasia, Injury), early neoplastic (Kras*; Kras*+Injury) and malignant (PDAC) tissues, used for in vivo profiling of chromatin and transcriptional dynamics underlying physiological or pathological exocrine pancreas plasticity. Mouse model genotype abbreviations are as follows: C = Ptf1a-Cre / RIK; KC = Ptf1a-Cre / RIK / LSL-KrasG12D; KPflC = Ptf1a-Cre / RIK / LSL-KrasG12D / p53fl/+. The RIK allele enables tracing of Cre-recombined pancreatic epithelial cells through the reporter mKate2. Scale bar, 100 μm. c, Example of gating strategy to isolate pancreatic epithelial cells expressing the lineage-tracing marker mKate2. Live mKate2+;CD45-;DAPI- cells were isolated from single-cell suspensions of pancreata from the autochthonous models of PDAC tumorigenesis (KC or KPflC) or normal pancreas counterparts (C) described in a, b (see also Supplementary Fig. 1). d, Correlation plot showing ATAC-seq size factors used for data normalization of the indicated experimental conditions with two different methods (n=3, 5, 3, 6 or 4 mice per group, as labeled from top to bottom). PeakNorm uses the in-built DESeq2 normalization for all filtered reads mapped to the peak atlas, whereas DepthNorm uses the number of filtered mapped reads irrespective of if reads are within or outside the peak atlas. The shaded region represents the 95% confidence interval for the regression between normalization types. e, Overlap between the dynamic ATAC-peaks lost (left) or gained (right) in the indicated tissue conditions vs Normal. Numbers reflect peaks in each category.
Extended Data Fig. 2. Shared and distinctive features of accessibility-GAIN and -LOSS regions induced by mutant Kras, tissue damage or their combination.

a, Heatmap representation of chromatin accessibility at ATAC-peaks significantly gained or lost between Normal, Injury, Kras* and Kras*+Injury conditions, as assessed by DESeq2 analyses of ATAC-seq data. Unsupervised clustering identified 6 major modules of peaks, that are either shared (S, A2) or specifically altered during physiological regenerative metaplasia (R) versus neoplastic transformation (N1, N2, A1). Each column represents one independent biological replicate (animal). Fig. 1e shows these same clusters plotted with the PDAC condition to illustrate their accessibility status in advanced disease. b, Metagene representation of the mean ATAC-seq signal for 6 ATAC-cluster regions identified in the above analyses in the indicated epithelial states, with the number of mice analyzed per condition indicated in the brackets. c, Genomic annotations of dynamic peaks comprising each ATAC-seq cluster. Note accessibility dynamics predominantly occur at intronic and distal intergenic cis-regulatory elements, with an enriched contribution of promoter or transcriptional start site (TSS) regions in regeneration-associated gained (R) or lost (A2) clusters. d, Top-scoring transcription factor motifs identified by HOMER de novo motif analyses per ATAC-seq cluster. The number in the brackets indicates enrichment p-values. e, Heatmap representing the relative motif enrichment for the top-scoring motifs across the same clusters of peaks sensitive to effects of injury and/or mutant Kras shown in d. Injury and mutant Kras cooperatively produce gain (e.g. AP-1, KLF, ETS, RUNX, SOX, MAF), loss (e.g. HNF), and redistribution (e.g. FOX, GATA) of accessible putative TF binding sites for a multi-pronged network of TF families, including TFs known to control pro-oncogenic (e.g. AP-113, RUNX375, KLF4/576,77, SOX9/179,78) or tumor-suppressive programs (e.g. HNF1A79, KLF1480, NR5A281, PTF1A22) in PDAC. f, Correlation matrices showing the differential degree of co-occurrence of motifs from different classes of TFs at the peaks comprising each ATAC-seq cluster (defined in a), revealing TF modules (marked with black rectangles). Note that AP-1-motif positive peaks gained uniquely during regenerative metaplasia (R cluster) show co-occurrence with pancreas lineage TF (GATA, FOX) motifs, whereas those gained during pro-neoplastic (Kras-mutant) metaplasia (S, N1, N2) do not. g, Bubble plots showing the relative enrichment of the indicated motifs (identified as top-scoring by HOMER analyses described in d) in the ATAC-peaks that are significantly gained (left, right) or lost (blue, right) in the Injury (I, n=5 mice), Kras* (K, n=3 mice), Kras*+Injury (K+I, n=6 mice) or advanced cancer (PDAC, n=4 mice) conditions vs normal healthy pancreas (Normal, n=3), as defined in Fig. 1a.
Extended Data Fig. 3. Early chromatin accessibility changes impact cell-identity genes associated with experimentally-validated enhancers of normal and malignant pancreatic epithelial cells.
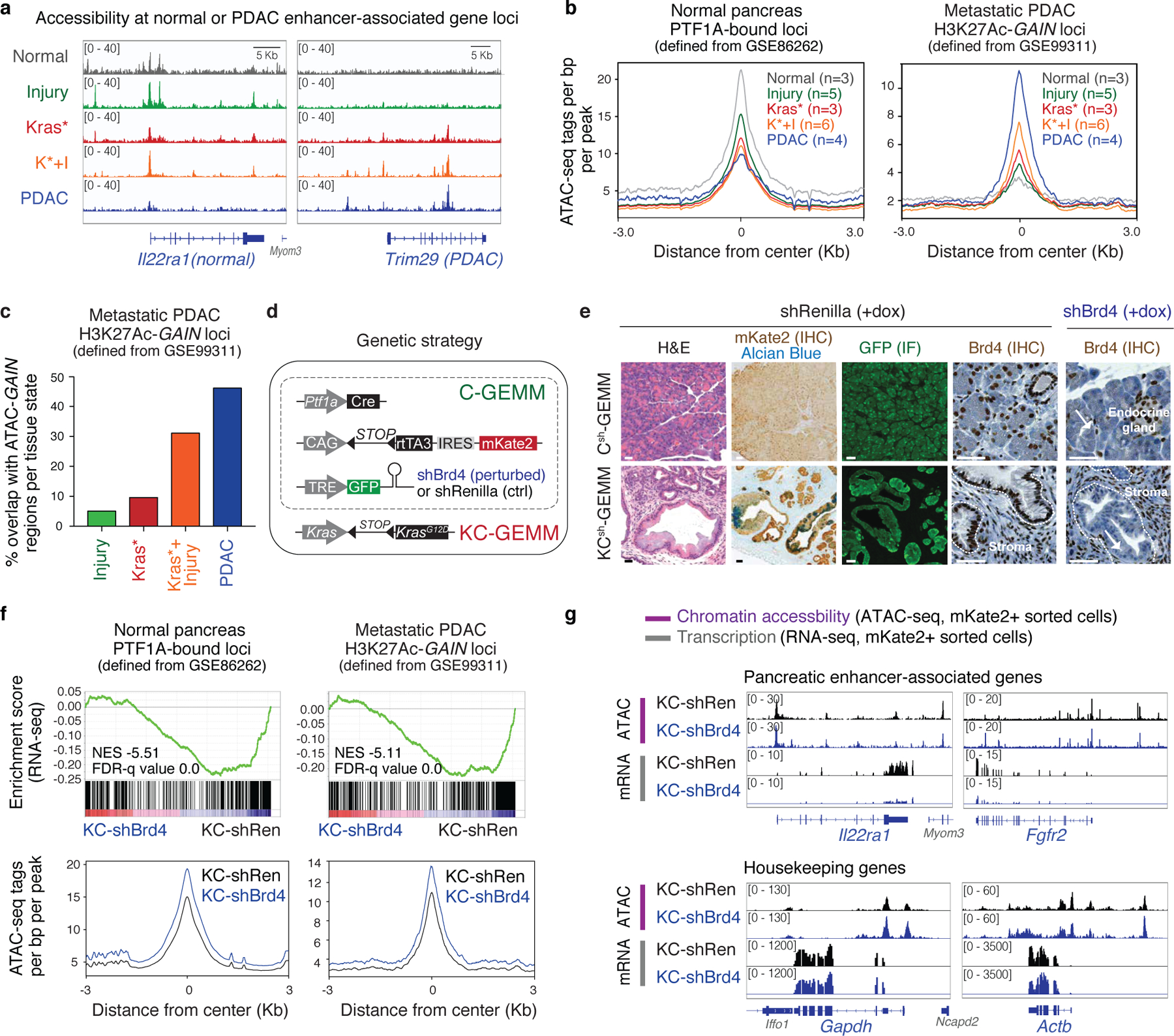
a, Representative ATAC-seq tracks showing dynamic accessibility at gene loci previously described to harbour active enhancers (top) in normal acinar (left, Il22ra82) or advanced PDAC (right, Trim2917) cells across mKate2+ sorted cells freshly-isolated from the indicated tissue states as defined in Fig. 1. n=3, 5, 3, 6 or 4 (from top to bottom) mice per group. b, Metagene representation of the mean ATAC-seq signal for regions bound by the acinar lineage-determining TF PTF1A in normal pancreas (left) (defined from GSE8626215) or H3K27ac-GAIN regions of metastasis-derived PDAC organoids vs normal pancreas counterparts (defined from GSE9931117) in mKate2+ sorted cells freshly-isolated from the indicated tissue states. The number of mice analyzed per condition is indicated in the brackets. c, Proportion of genomic regions showing a significant gain of H3K72Ac signal in metastasis-derived cultured organoids compared to their normal counterparts (H3K27Ac ChIP-seq data from GSE99311) that gain accessibility in pancreatic epithelial (mKate2+) cell populations freshly-isolated from Injury (n=5 mice), Kras* (n=3 mice), Kras*+Injury (n=6 mice) or PDAC (n=4 mice) tissue states as compared to Normal pancreas counterparts (n=3 mice), as defined by overlapping ChIP-seq and ATAC-seq datasets. d, Schematic representation of the genetic configuration used to induce exocrine pancreas-specific suppression of the chromatin reader Brd4. We generated mice (KCsh-GEMM) harboring the following alleles: (i) a pancreas-specific Cre driver (Ptf1a-Cre), (ii) a Cre-activatable LSL-KrasG12D allele and (iii) two additional alleles [LSL-rtTA3-IRES-mKate (RIK) and the collagen homing cassette, (CHC)] that allow for inducible expression of a GFP-linked shRNA targeting Brd4 (shBrd4) or a neutral shRNA (shRenilla) in Cre-recombined cells labeled by the fluorescent reporter mKate2. Upon receiving a doxycycline (dox)-containing diet, a GFP-linked shRNA targeting Brd4 (or Renilla, control) is induced selectively in mKate2-labeled pancreatic epithelial cells. Analogous models harboring the dox-inducible shRNAs without the LSL-KrasG12D allele (referred to as Csh-GEMM) were generated to compare and contrast epigenetic requirements of pro-neoplastic vs regenerative pancreas plasticity. e, Representative H&E, immunohistochemistry (IHC) or immunofluorescence (IF) analyses of the indicated proteins in pancreata from Csh (top) or KCsh (top) mice (n=3 per group) placed on dox fed at 5 weeks old and analyzed 9 days later. mKate2 staining marks Kras-wild-type (bottom) or Kras-mutant (bottom) pancreatic exocrine cells where Ptf1a-Cre has been expressed. GFP staining corresponds to shRNA expression and is coupled with Brd4 suppression in that same compartment (but not in surrounding stroma) in mice harbouring shRNAs targeting Brd4 (shown for the shBrd4.552 strain) but not Renilla (control). Dashed lines demark boundaries between epithelium and stroma, and arrows point to Brd4-suppressed exocrine pancreas compartment of shBr4 mice. The same Brd4 IHC panels are shown in Fig. 2b. Scale bar, 50 μm. f, GSEA and metagene plots showing the relative expression (top) and accessibility (bottom) status, respectively, of the same loci defining normal acinar state (left, with top-500 PTF1A-bound peaks) or harbouring activated enhancers in metastatic PDAC cells (right) shown in (b) in shBrd4 vs shRen mKate2+ metaplastic epithelial cells isolated from KCsh-GEMM mice (Kras*+Injury) described above (n=3 mice per genotype). Brd4 suppression selectively impairs transcription of lineage-specific and PDAC enhancer-associated genes in the Kras-mutant metaplastic epithelium without impairing chromatin accessibility at those loci. Genome-wide profiles of these same conditions are shown in Extended Data Fig. 6m. g, Representative ATAC-seq and RNA-seq tracks of genes known to be associated with active lineage-specific enhancers (top) of acinar (e.g. Cabp282) or pancreatic progenitor cells (e.g. Fgfr221) in shRen.713 (black) or shBrd4.552 (blue) mKate2+ epithelial cells freshly-isolated from metaplastic pancreata from KCsh-GEMM mice (n=3 per genotype) at the 48h post-caerulein treatment time-point (thus matching the Kras*+Injury condition). Brd4 suppression impairs transcription of pancreatic enhancer-associated genes without altering chromatin accessibility at that same loci. Tracks of housekeeping genes (bottom) are shown as specificity controls. Mice were placed on dox 6 days prior to the caerulein treatment, to induce ADM in the presence or absence of Brd4 (as summarized in Extended Data Fig. 4a below). See also Extended Data Fig. 6l,m.
Extended Data Fig. 4. Brd4 suppression is dispensable for both regenerative and neoplasia-associated ADM.
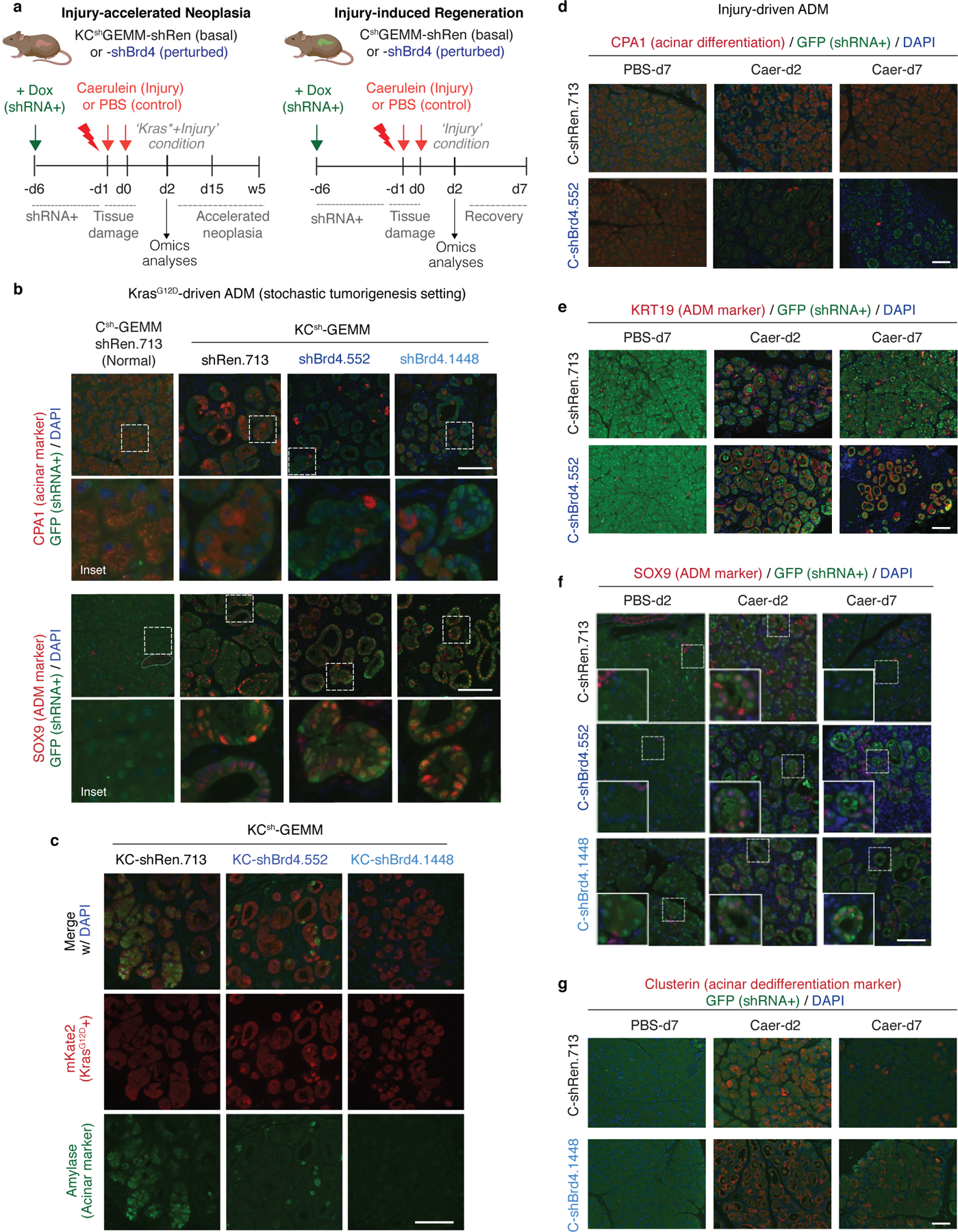
a, Experimental strategy to address the functional impact of spatiotemporally-controlled perturbation of Brd4 during injury-accelerated tumorigenesis or physiological regeneration in KCsh or Csh mice, respectively. 4 weeks old mice were placed on dox diet to induce expression of shRNA targeting Brd4 or Ren (control) in the pancreatic epithelium, and pancreatic injury was induced by caerulein treatment 6 days thereafter to trigger synchronous ADM throughout the organ in the presence or absence of epithelial Brd4 function, respectively. Tissue responses were evaluated at the indicated days (d) or weeks (w) post-caerulein or PBS (control) treatment. Specifically, to match our previous profiling experiments, we examined pancreatic ADM at 48 hours post-caerulein treatment, a time point corresponding to the distinct, genotype-specific chromatin accessibility profiles identified above. Subsequent regeneration (Kras wild-type context) or neoplasia (Kras mutant context) were evaluated 5 days or 2–3 weeks thereafter, respectively. In addition, separate cohorts of dox-treated KCsh mice placed were analyzed at 6 weeks and 1 year of age to track effects the context of stochastic Kras-driven neoplasia. Mouse illustrations were made using ©BioRender - biorender.com. b-c, shBrd4 perturbation does not impair mutant Kras-driven ADM. Representative immunofluorescence stains of the acinar markers (CPA1, Amylase) or the ductal metaplasia marker SOX9 co-stained with lineage-tracer markers (mKate2/GFP) in Kras-mutant pancreata from 6 weeks old KC-shRen or -shBrd4 mice (n=6 per group) in the stochastic tumorigenesis setting. d-g shBrd4 perturbation does not blunt injury-induced ADM but impairs subsequent acinar regeneration. Representative immunofluorescence (IF) staining of pancreata from Kras-wild type Csh mice expressing shRen or shBrd4 treated with Caerulein or PBS control and analyzed at the indicated days (d) post treatment for protein expression of the acinar marker CPA1 (d), metaplasia markers KRT19 (e), SOX9 (f) or clusterin (g) co-stained with GFP (marking shRNA expressing cells) and DAPI (nuclei). n=5 mice per group. Scale bar, 100 μm.
Extended Data Fig. 5. Brd4 suppression impairs regenerative and neoplastic fate outcomes of injury-driven pancreas plasticity.
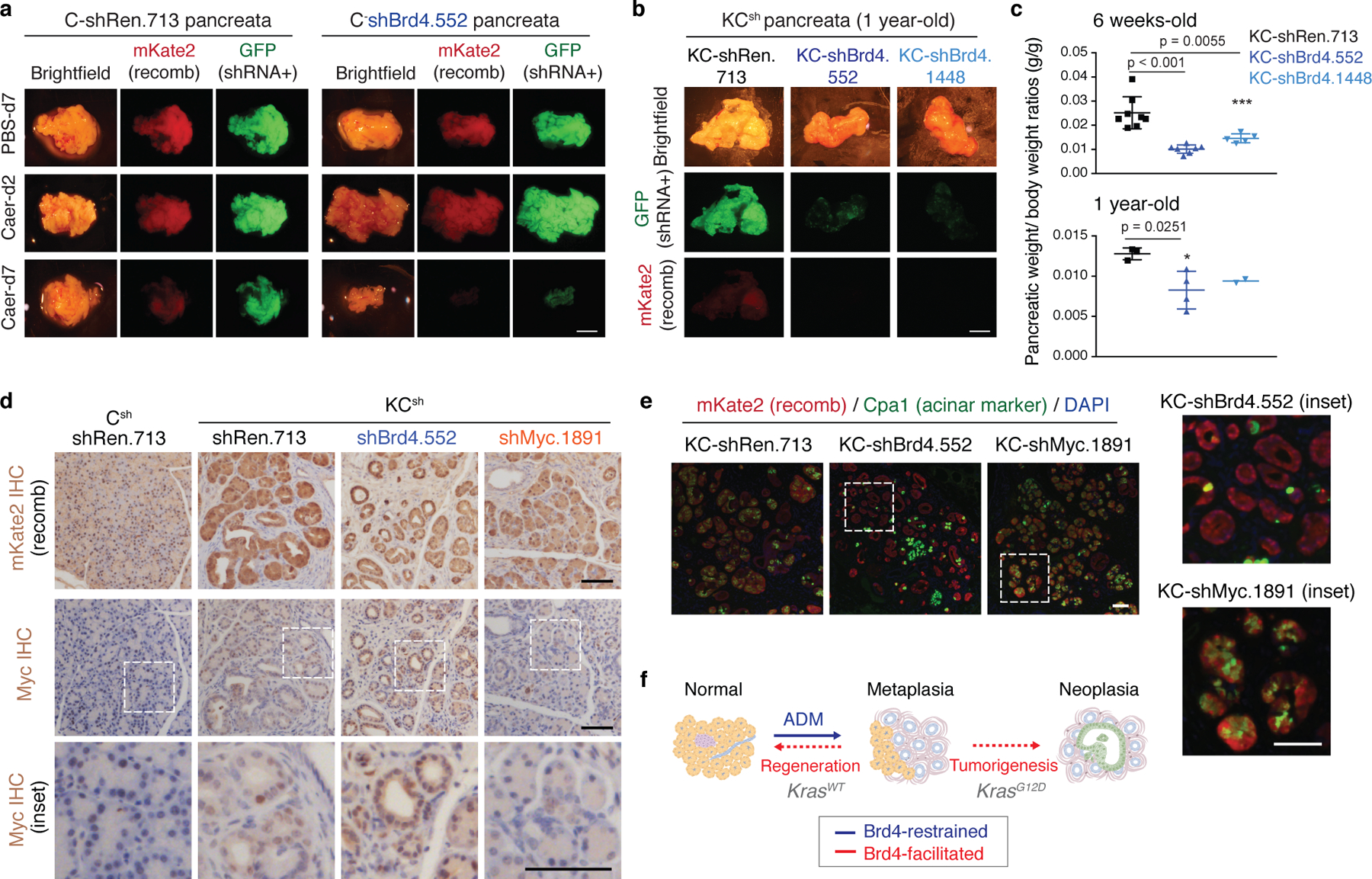
a Representative bright-field and fluorescence images showing gross morphology of pancreata of C-shRen and -shBrd4 mice treated with caerulein or PBS control and analyzed at the indicated time points in days (d). Lineage-traced pancreatic epithelial cells expressing shRNA are marked by the fluorescent reporters mKate2 and GFP. Reduced mKate2 and GFP signals denote loss of pancreatic tissue expressing shBrd4. Scale bar, 5 mm. b, Representative bright-field and fluorescence images showing gross morphology of pancreata of KC-shRen and -shBrd4 mice placed on dox since postnatal day 10 to induce shRNA expression and analyzed at 1-year of age. Reduced mKate2 and GFP signals denote loss of shBrd4-expressing mutant Kras pancreatic epithelial cells. Scale bar, 5 mm. c, Quantification of pancreatic weight normalized to animal body weight by genotype. Data are presented as means ± s.e.m; n=8, 7 or 5 (top, from left to right) mice, or n=3, 4 or 2 (bottom, from left to right) mice; unpaired two-tailed Student’s t-test. d, Representative immunohistochemistry stains of mKate2 (top) and Myc (bottom) in pancreata from 6 weeks old mice KCsh of the indicated genotypes and placed on dox fed at day 10 after birth (stochastic tumorigenesis setting). Lower panels show high magnification images of regions marked with dashed line boxes for visualization of Myc nuclear localization. While oncogenic Myc expression can require Brd4-associated enhancers in some settings35,83,84 and is suppressed by systemic BET inhibition in KC mice85, epithelial-specific Brd4 suppression did not reduce Myc protein in our model. Scale bar, 100 μm. e, Representative co-IF stains of mKate2 (red) and the acinar marker CPA1 (green) in pancreata from 6 weeks old mice KCsh mice of the indicated genotypes and placed on dox fed at day 10 after birth, as above. Right panels show high magnification images of regions marked with dashed line boxes. KCsh mice harboring a validated shRNA targeting Myc (instead of Brd4) exhibited impaired rather than accelerated ADM. The reduction of CPA1 observed in KC-shBrd4 mice is not phenocopied in KC-shMyc mice, which retain Cpa1 expression. Scale bar, 100 μm. f, Schematic representation of the phenotypic output of pancreas-specific suppression of Brd4 during mutant Kras-driven neoplasia and tissue injury-driven regeneration: Brd4 is dispensable for acinar-to-ductal metaplasia induction in both contexts but mediates subsequent neoplastic progression to PanIN or regenerative plasticity, respectively.
Extended Data Fig. 6. Brd4 suppression uncover distinct chromatin-associated transcriptional programs in normal vs Kras-mutant damaged pancreata.
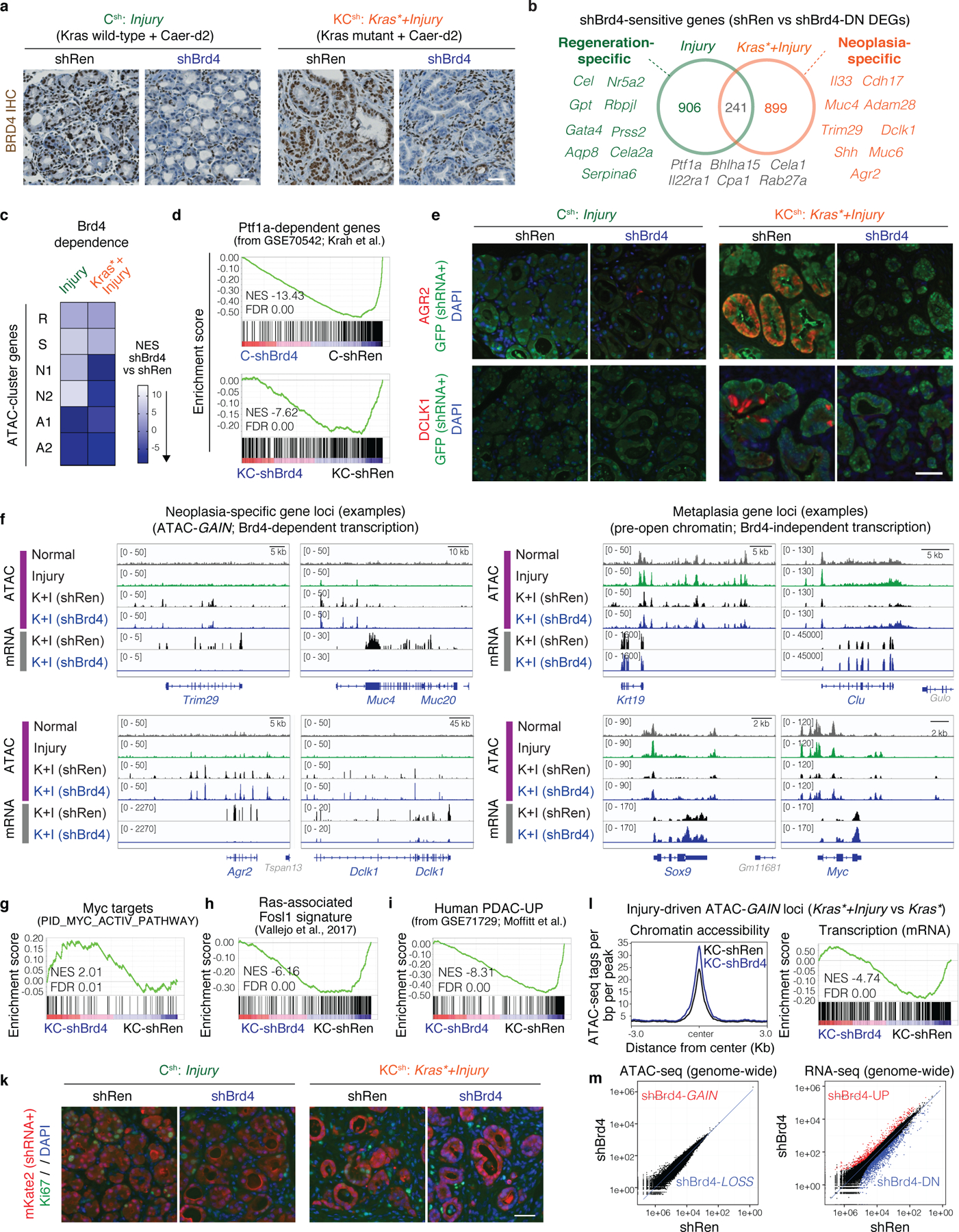
a, Representative immunohistochemical staining (IHC) of Brd4 in pancreata from Csh-GEMM (left) or KCsh-GEMM (right) mice (n=3 per group) harbouring shRen.713 or shBrd4.1448 at 48 hours post-caerulein, placed on dox fed 6 days before caerulein treatment start. b, Overlap of DEGs downregulated upon Brd4 suppression in the Injury (regeneration) or Kras*+Injury (neoplastic transformation) settings. Examples of Brd4-dependent genes, shared or unique to each context are shown. c, Heatmap representation of normalized enrichment scores (NES) comparing the mRNA expression of genes associated with the ATAC-seq clusters identified in Fig. 1 between shBrd4.1448 vs shRen.713 pancreatic epithelial cells (mKate2/GFP+) isolated from Kras wild-type (Injury, left) or Kras-mutant (Kras*+Injury; right) metaplastic tissues, as analyzed by GSEA at the 48 hours post-caerulein time-point. Negative normalized enrichment scores (NES) indicate downregulation of gene set in shBrd4 cells as compared to shRen counterparts. Consistent with the accelerated ADM but blunted neoplastic transformation phenotype (Fig. 3), Brd4 suppression impairs the expression of genes linked to the acinar ATAC-seq clusters (A1/A2) in both WT and Kras-mutant cells and, additionally, of genes linked to the neoplasia-specific ATAC-seq clusters (N1, N2) in Kras-mutant cells. Shared (S) and regeneration-specific (R) and ATAC-seq clusters are not blunted in either context, suggesting these reflect injury-driven ADM states that can be induced in the absence of Brd4 in both WT and Kras-mutant contexts. d, GSEA comparing the expression of known Ptf1a-dependent genes22 between shBrd4 and shRen cells isolated from Kras-WT (Csh; top) or Kras-mutant (KCsh; bottom) mice triggered to undergo regenerative (Injury) or pro-neoplastic (Kras*+Injury) metaplasia, respectively. e-f, Impact of Brd4 suppression on the protein (e) or mRNA (f) levels of known drivers of pancreatic tumorigenesis linked to ATAC-GAIN loci specific to early neoplasia (Kras*+Injury; K+I) that remain in a closed chromatin state in both regenerative metaplasia (Injury) and normal pancreas. Panels in ‘e’ show representative immunofluorescence stains of the indicated neoplasia-activated factors (red) co-stained with GFP (green, marking epithelial cells) in pancreata from wild-type or Kras-mutant shRNA-expressing mice 2 days after tissue injury (caerulein) or control (PBS). Nuclei are counterstained with DAPI (blue). Representative ATAC-seq and RNA-seq tracks of these and other neoplasia-activated genes herein identified to be induced by during pancreatitis-induced neoplasia (Kras*+Injury condition) in a Brd4-independent manner are shown in ‘f’. g, GSEA comparing the expression of a mutant Kras-associated FOSL1 gene signature between shBrd4 and shRen cells isolated from KCsh-GEMM mice (Kras*+Injury condition). h, GSEA comparing the expression of genes upregulated in human PDAC specimens vs human normal pancreas between shBrd4 and shRen cells isolated from KCsh-GEMM mice (Kras*+Injury condition). Similar results were obtained with GSE62452 dataset. i, Representative ATAC-seq and RNA-seq tracks of classic metaplasia-associated genes that, in contrast to the above programs, pre-exist in an opened chromatin in normal pancreas and are induced in a Brd4-dependent manner, consistent with the dispensability of Brd4 for ADM. j, GSEA comparing the expression of Myc activated genes between Kras-mutant shBrd4 and shRen cells (Kras*+Injury condition), showing retained expression in shBrd4 populations. Similar results were obtained with additional Myc signatures86,87 (not shown). k, Representative immunofluorescence stains the proliferation marker Ki67 (green) co-stained with mKate2 (red, marking epithelial cells) in pancreata from wild-type or Kras-mutant shRNA-expressing mice 2 days after tissue injury (caerulein) or control (PBS). Nuclei are counterstained with DAPI (blue). Brd4 suppression induces aberrant activation of Cdkn1a and other stress response p53-activated genes in both WT and Kras-mutant metaplastic cells (see Supplementary Table 4) which, accordingly, showed reduced proliferation. l, Metagene and GSEA plots showing the relative accessibility (left) and expression (right) status, respectively, of ATAC-GAIN regions induced by tissue damage in Kras-mutant pancreata (Kras*+Injury vs Kras*) in Kras-mutant shlBrd4 vs shRen cells isolated from the same Kras*+Injury tissue condition. m, Scatter plot comparing the genome-wide chromatin accessibility (left) and transcriptional (right) landscapes of Kras-mutant shBrd4 vs shRen cells isolated from the same Kras*+Injury tissue condition (n=3 mice per genotype). Each dot represented an ATAC-seq peak (left) or transcript (right; differentially accessible loci (log2FC >= 0.58, FDR <=0.1) or differentially expressed genes (FC>2, p-val <0.05) between genotypes are marked in red (gained) or blue (lost). shBrd4 populations display ATAC-seq profiles indistinguishable from those of shRen controls, ruling out that the observed Brd4-dependent transcriptional changes result from confounding secondary effects of acute Brd4 perturbation on chromatin state or epithelial tissue cell composition. Scale bar, 50 μm.
Extended Data Fig. 7. Early dysregulation of chromatin regulatory features of advanced PDAC.
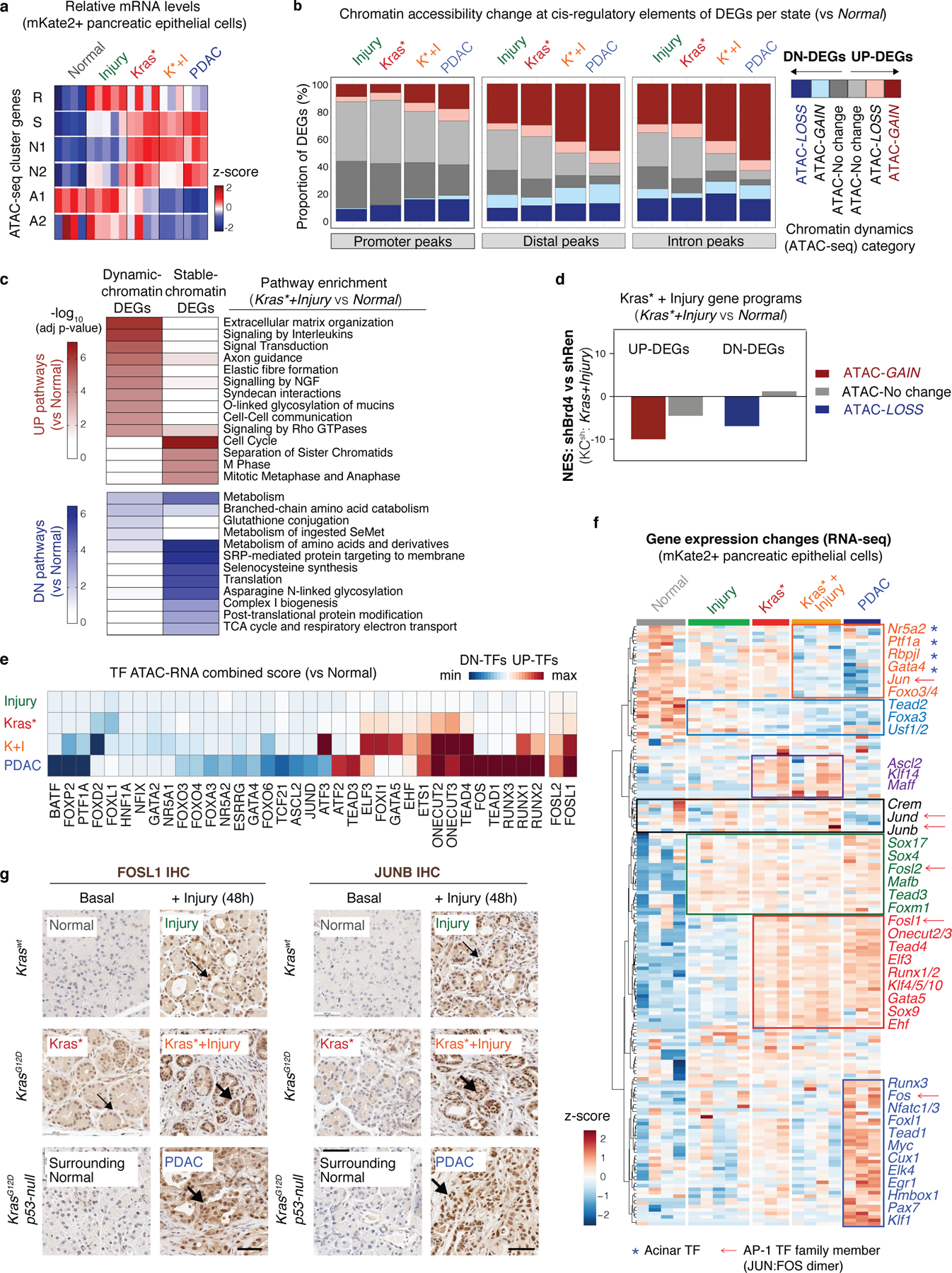
a, Heatmap representation of RNA-seq data showing the relative expression of gene sets associated with ATAC clusters identified in Fig. 1 across mKate2+ pancreatic epithelial cells isolated from Normal, Injury, Kras*, Kras*+Injury and PDAC tissue states (as defined in Fig. 1a). Heatmap color represents median expression of all genes associated with each cluster, z-scored for comparison across conditions. Each colum represents an independent mouse. b, Chromatin dynamics at ATAC-peaks at promoter, distal, exon, or intro regions associated with differentially expressed genes (DEGs; RNA-seq Fold change>2, p-adj<0.05) between mKate2+ pancreatic epithelial cells isolated from the indicated tissue states vs normal pancreas (n = independent mice per condition as in a). DEGs were classified depending on whether they exhibit significant chromatin accessibility change (chromatin-dynamic DEGs) or no accessibility change (chromatin-stable DEGs, in grey) at associated peaks in the respective experimental condition vs normal pancreas. UP-DEGs, upregulated genes; DN-DEGs, downregulated genes. c, Heatmap of RNA-seq data showing top upregulated pathways in the Kras*+Injury condition, separated depending on whether they exhibit ATAC-GAINS at associated peaks (promoter or distal). Upregulated genes associated with accessibility-GAIN (left side) are linked to distinct biological traits commonly acquired in PDAC (e.g. differentiation, inflammation, fibrosis, signaling), whereas those with no ATAC change (i.e. ‘primed’ in normal pancreas) are linked to general cellular processes (e.g. cell proliferation, translation) (right side). See Supplementary Table 6 for additional tissue states and pathways. d, Relative enrichment of the indicated gene sets in shBrd4.1448 vs shRen.713-expressing pancreatic epithelial cells (mKate2/GFP+) isolated from KCsh-GEMM mice (n=3 per shRNA genotype, Kras+Injury) as determined by GSEA. UP-DEGs (left bars) and DN-DEGs (right bars) between the Kras*+Injury vs Normal conditions were classified depending on whether they exhibit or not significant accessibility changes (ATAC-GAIN, or ATAC-LOSS) at associated ATAC-peaks. Negative normalized enrichment scores (NES) indicate downregulation of gene sets in shBrd4 cells as compared to shRen counterparts. e, Heatmap representation of ATAC-RNA combined scores for the indicated TFs and tissue states. The ATAC-RNA combined score infers the probability of differential binding of a specific TF to a motif significantly enriched in the accessibility-GAIN or LOSS regions of each condition vs Normal, based on a consistent gene expression change in the same comparison (see Methods for details). Top TFs scoring for the Kras*+Injury or PDAC conditions vs Normal are shown. f, Heatmap of RNA-seq data from lineage-traced (mKate2+) pancreatic epithelial cells isolated from the indicated tissue states, showing the relative expression of transcription factors (TFs) whose binding motifs are enriched in loci that gain or lose accessibility by effects of tissue damage, mutant Kras or the combination of both (i.e. Fig. 1g-ATAC-clusters) or in the transition to full-blown adenocarcinoma PDAC (PDAC vs Kras*+Injury). Each column represents an individual animal. The boxes highlight modules of TFs that are: (i) similarly expressed in normal regeneration and cancer context (green, black); (ii) selectively induced in early neoplasia and PDAC (red); (iii) selectively overexpressed in late disease (dark blue); (iv) that become increasingly suppressed by effects of injury, mutant Kras (light blue) or both (orange); or (v) that are selectively induced in early-stage but not late disease (purple), with names of TF examples to the right. Injury and mutant Kras differentially induce diverse members of the same TF families, including several AP-1 JUN:FOS complex members (marked with arrows) and other TFs known known to also bind AP-1 motifs. In addition, note the Kras*/injury combination suppresses the expression of master regulators of acinar differentiation (marked with asterisks) more potently than either insult alone. g, Representative immunohistochemical (IHC) stains of two AP-1 family members in Kras-mutant or WT pancreatic tissues in the presence and absence of tissue damage (48 hours post-caerulein) and compared to advanced PDAC (n=4 mice per group). While AP-1 family member FOSL1 is induced in non-injured Kras mutant pancreata, JUNB protein levels increase only after injury with a potent co-activation occurring upon presence of both stimuli, suggesting cooperative gene – environment interactions shape AP-1 TF complex member expression. Scale bar, 100 μm.
Extended Data Fig. 8. Single cell analysis of chromatin dynamics in early-stage neoplasia.
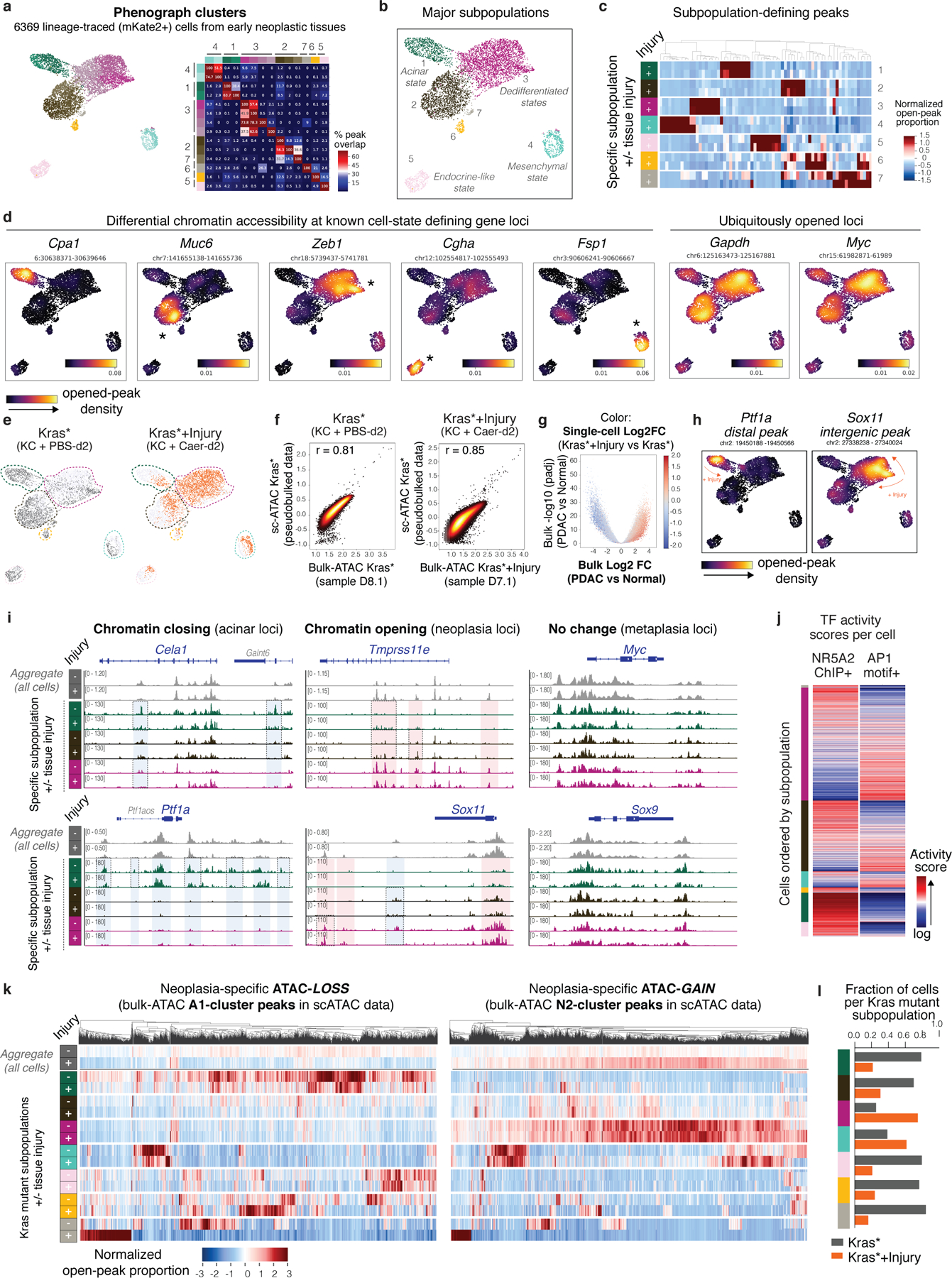
a, UMAP representation of single-cell ATAC-seq (scATAC-seq) profiles of mKate2+ cells isolated from Kras* and Kras*+Injury tissue conditions (n=1 mice each) and co-embedded together, revealing chromatin heterogeneity across Kras-mutant pancreatic epithelial cells from pre-malignant tissue states. Dots represent individual cells (n=6369) and colors indicated cluster identity based on initial phenograph clustering (left). The heatmap shows the degree of intersection of significantly-enriched peaks (Fisher’s exact test, adjusted p-value<0.05) between each pair of phenograph cluster (colored matching UMAP plot), normalized by the total number of enriched peaks in the cluster for that row (left). Rows and columns are ordered according to their grouping into seven larger subpopulations derived from merging of Phenograph clusters based on the overlap of their differentially accessible peak sets (see Methods for details). b, UMAP representation of the same mKate2+ scATAC-seq profiles shown in (a) colored by major subpopulations (see Methods for details). c, Heatmaps showing patterns of accessibility at subpopulation-defining peaks, shown across each of the major subpopulations defined in (b) separated by tissue injury (+/−) condition. Color illustrates the proportion of all cells in each subpopulation and condition with an accessible peak, where values have been z-scored. The complete list of subpopulation-defining peaks are listed in Supplementary Table 8. d, Visualization of differential chromatin opening for the indicated peaks associated with known pancreatic cell-state defining markers or the housekeeping gene Gapdh, illustrated by opened-peak density plots for nearby proximal or distal elements within 50 kb of the transcription start site. Color scale indicates a Gaussian kernel density estimate of cells harboring the open peak in the UMAP visualization, with yellow signal marking increased density of cells with open chromatin at that specific locus. e, UMAP projection of scATAC-seq profiles of Kras-mutant (mKate2+) epithelial cells shown in (a-d) colored by the indicated tissue states. f, Correlation analysis comparing normalized accessibility signals per peak captured in scATAC- and bulk ATAC-seq analyses of in the indicated conditions. For scATAC-seq data, values representing pooling of all individual cells to generate depth-normalized accessibility signals per condition (pseudo-bulk) are shown. For bulk ATAC-seq data, values from a representative sample (independent animal) of a total of n=3 (Kras*) or n=6 (Kras*+Injury) are shown. g, Volcano plot showing dynamic peaks identified between PDAC and Normal conditions in bulk ATAC-seq analyses (Fig. 1), colored according to their relative accessibility fold change detected between Kras*+Injury and Kras* samples in scATAC-seq analyses. Peaks gained or lost in PDAC vs Normal are found differentially represented in scATAC-seq data from early stage neoplasia, correlating with tissue injury status. h, UMAP projection illustrating examples of peaks exhibiting chromatin closing (left) or opening (right) within the same mutant-Kras cell cluster upon tissue injury (+), visualized by opened-peak density plots in which color indicates a Gaussian kernel density estimate of cells harboring the open peak in the UMAP visualization. i, scATAC-seq tracks of the indicated loci showing chromatin accessibility patterns across the indicated subpopulations, marked with color labels matching (b) and separated by experimental condition. The first two rows (aggregate, in grey) show global patterns from pooling all cells from each condition, regardless of subpopulation identity, and population-specific dynamics are shown below. Blue- and red-colored boxes mark ATAC-GAINS or -LOSS detected in aggregate populations, and dashed boxes highlight examples of peaks displaying injury-associated accessibility changes between Kras-mutant cells from the same subpopulation. j, AP-1 and NR5A2 activity scores are anticorrelated across single cell epigenetic profiles, separated by subpopulation. Logged activity scores are plotted as a heatmap, with cells (columns) ordered by ratio of AP-1:NR5A2 activity within each subpopulation. k, Heatmaps showing accessibility signals for the indicated cluster of peaks (columns) identified from bulk ATAC-seq analyses (see Fig. 1e) across each major subpopulation of Kras-mutant cells, separated by experimental condition. The color scale represents the proportion of all cells in each subpopulation and condition with an accessible peak, where values have been z-scored. As above, the first two rows (aggregate) show global accessibility patterns from pooling all individual cells in each condition, regardless of subpopulation; and subpopulation-specific dynamics are shown below. l, Proportion of mKate2+ cells per cluster (marked with color labels matching Extended Data Fig. 8c) derived from Kras* (grey) or Kras*+Injury (orange) tissue conditions.
Extended Data Fig. 9. Epigenetic dysregulation of IL-33 during injury-facilitated neoplastic transformation.
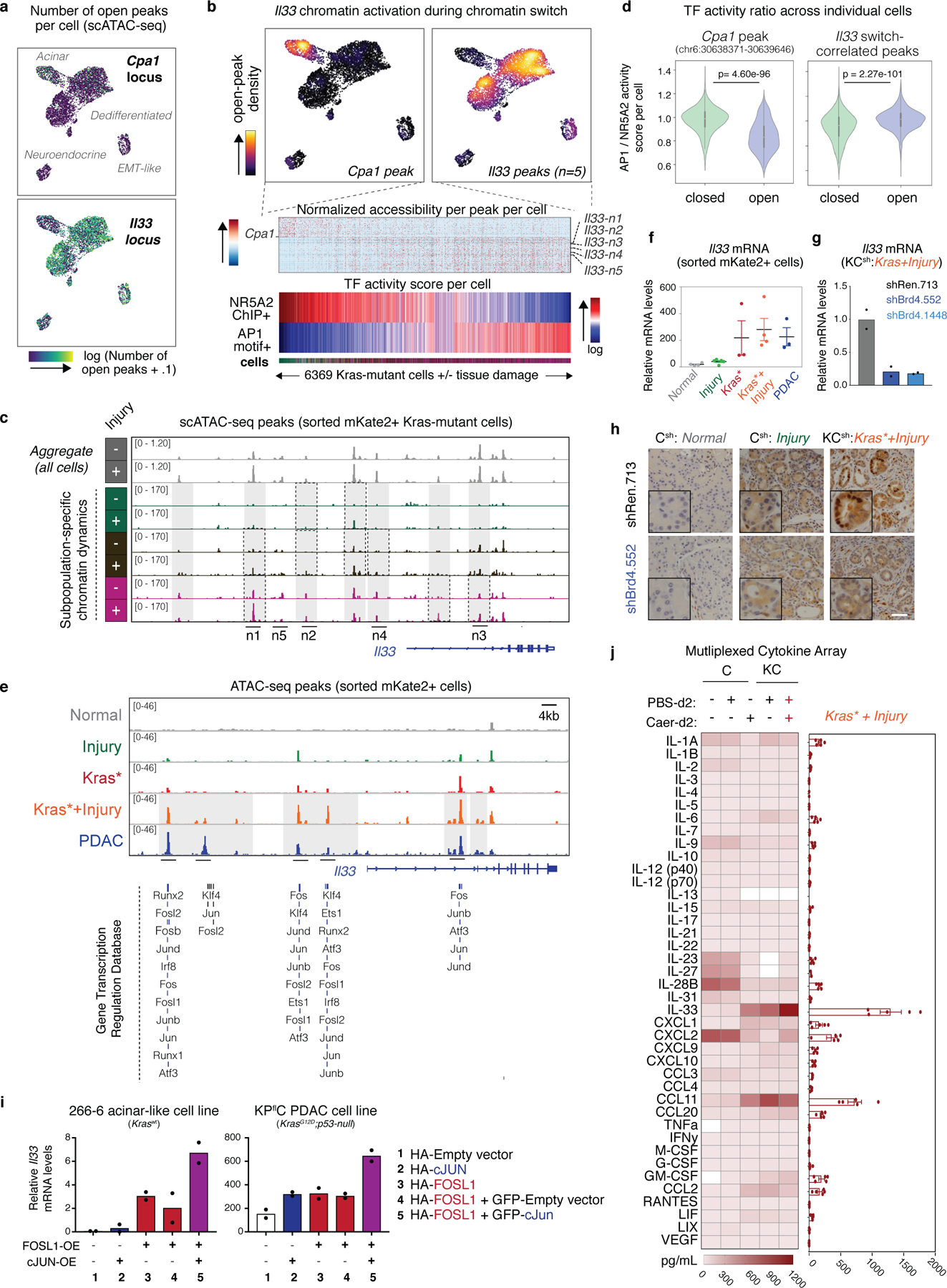
a, UMAP visualization of the number (N) of total open peaks at the Il33 (top) of Cpa1 (locus) per individual Kras-mutant cell in the scATAC-seq analyses applied to 6369 individual cells freshly isolated from Kras* or Kras*+Injury conditions (n=1 mice each) and co-embedded together. Peaks nearby proximal or distal elements within 50 kb of the transcription start site were counted. Color scale indicates log-transformed counts of open peaks in the vicinity of the transcription start site. Note increased accessibility at Il33 gene regulatory loci in dedifferentiated populations, but not in the more differentiated acinar chromatin state or neuroendocrine-like subpopulations. b, scATAC-seq analyses identifies accessibility changes strongly correlated with AP-1/NR5A2 activity ratio across individual Kras-mutant cells isolated from pancreata undergoing early neoplastic cell fate transitions (Kras* and Kras*+Injury conditions). Bottom panels show normalized accessibility values for peaks (rows) displaying a strong (r>.1) positive (e.g. 5 Il33-associated peaks) or negative (r<−.1) (e.g. acinar Cpa1-associated peak) correlation with AP-1/NR5A2 activity scores (as in Fig. 4f) across individual Kras-mutant cells (columns, marked with color labels matching Extended Data Fig. 8b). The identified 5 switch-correlated peaks (n1-n5) at the Il33 locus which overlap with those captured as sensitive to effects of injury and/or mutant Kras in ATAC-seq analyses of bulk populations (see panel e, below). c, Signal tracks of the Il33 loci showing rapid chromatin accessibility gain (in grey boxes) in the mutant Kras epithelium upon tissue injury in single cell populations, separated by cluster (3 clusters shown) and condition (Kras* vs Kras*+Injury). Note accessibility gains are detected upon injury even within a defined cell cluster (examples marked with dashed lines), supporting bona fide chromatin remodeling at these loci. The 5 chromatin switch-correlated peaks identified in ‘b’ are labeled as n1-n5. All tracks show accessibility signals downsampled to same coverage to correct for cell count and sequencing depth disparities across conditions. d, Violin plots showing the AP-1/NR5A2 activity scores of Kras-mutant (mKate2+) pancreatic cells displaying an opened (blue) state for the indicated acinar Cpa1-associated peak, or any of the 5 chromatin switch-correlated Il33 peaks versus those that do not (green). Il33-accessible cell populations exhibit an enhanced AP-1 activity, whereas Cpa1-accessible cells do not. n=288, 6081, 2228 or 4141 (from left to right) individual cells obtained from n=2 mice (Kras*, Kras*+Injury conditions). Significance was assessed by unpaired two-tailed Student’s t-test. e, (Top) Representative ATAC-seq tracks of the Il33 locus in lineage traced (mKate2+) pancreatic epithelial cells isolated from normal (Normal, n=3 mice), regenerating (Injury, n=5 mice), stochastic neoplasia (Kras*, n=3 mice), synchronous injury-accelerated neoplasia (Kras*+Injury, n=6 mice) or cancer (PDAC, n=4) experimental conditions, as described in Fig. 1a. (Bottom) Independent ChIP-seq experiments (lines) from the 2019 GTRD database summarizing experimentally validated binding of certain AP-1 subunits (and other top scoring TFs associated with injury transitions) across different cellular contexts to Kras*/Injury-sensitive Il33 peaks identified in our study. f, Relative mRNA levels (RNA-seq DESeq2-normalized counts) of Il33 in FACS-sorted mKate2+;CD45- cell populations isolated from the indicated tissue states. Data are presented as means ± s.e.m. of n=4, 5, 3, 4 or 3 (from left to right) independent biological replicates (mice) per group. g, qRT-PCR analyses validating downregulation of Il33 mRNA in Brd4-suppressed mutant Kras pancreatic cell populations (mKate2+) isolated from mice (n=2 per genotype) triggered to undergo synchronous pro-neoplastic transitions upon tissue damage in KCsh mice placed on dox-diet 6 days before (as in Extended Data Fig. 4a). Cells were isolated for expression analysis at 48 hours after caerulein treatment, i.e. matching the Kras*+Injury condition of the omics analyses revealing rapid gain in accessibility and expression at the Il33 locus. h, Representative IHC stains of IL-33 protein in normal (left) or metaplastic (middle, right) pancreata expressing shRen or shBrd4 from Kras-WT (Csh) or Kras-mutant (KCsh) mice (n=4 per condition) treated with caerulein-induced pancreatic injury and analyzed 48 hours thereafter, as in Extended Fig. 4a above. Scale bar, 50 μm. i, Relative Il33 mRNA levels in pancreatic acinar 266–6 (left) or KPflC PDAC (right) cultured cells stably transduced with vectors encoding for the indicated proteins, as assessed by qRT-PCR and normalized to β-actin housekeeping control. Representative results of 2 independent experiments performed with n=2 biological replicates (wells) each with individual data points shown. j, Multiplexed immunoassay detecting the indicated cytokines or chemokines in protein lysates from normal or mutant Kras pancreata, 2 days after induction of caerulein (Caer)-induced tissue injury or treatment with PBS (control). n=2, 3, 4, 2 or 5 (from left to right) independent animals per condition. The bar-graphs to the right displays pooled data (means ± s.e.m) from n=5 independent animals (Kras*+Injury condition), revealing IL-33 as a major pancreas injury ‘alarmin’ induced by combined effects of Kras gene mutation and tissue damage.
Extended Data Fig. 10. IL-33 cytokine signaling shapes the transcriptional, chromatin accessibility and histological state of the Kras-mutant pancreatic epithelium.

a, Schematic representation of the experimental design to interrogate the impact of recombinant IL-33 (rIL-33) on the transcriptional, chromatin and phenotypic state of the pancreatic epithelium from Kras-mutant (KC-GEMM) or wild-type (C-GEMM) mice. Molecular analyses were performed in lineage-traced (mKate2+) pancreatic epithelial cells purified by FACS-sorting from of rIL-33 or vehicle treated mice at day 0 (ATAC-seq), or day 0 and day 21 days (RNA-seq) after treatment. b, GSEA comparing the expression of the early chromatin activated gene program identified in Fig. 4 analyses (left), or of genes overexpressed in human PDAC specimens compared to normal pancreas (Moffitt et al. dataset)23 (right), in Kras-mutant cells isolated from rIL-33 treated vs PBS-treated mice (day 21 time point). The chromatin activated genes queried are the chromatin-dynamic DEGs identified to be upregulated during injury-accelerated neoplasia (Kras*+Injury) and in advanced disease (PDAC) but not during normal regeneration (Injury alone) and blunted by Brd4 suppression in metaplastic Kras-mutant cells (KCsh: Kras+Injury). c-d, GSEA comparing the expression of genes induced by the combination of mutant Kras + rIL-33 in either shBrd4 vs shRen Kras-mutant pancreatic epithelial cells (mKate2+) isolated from KCsh-GEMM (Kras*+Injury) (c) or in Kras-mutant populations isolated from caeruelin-treated (Kras*+Injury) vs resting (Kras*) KC mice (d). The queried gene sets were identified as significantly upregulated in Kras-mutant pancreatic epithelial cell populations (mKate2+) isolated from rIL-33 (vs PBS) treated mice (KC+rIL-33 vs KC+Veh) at either day 0 (d0) or day 21 (d21) time points. e, qRT-PCR analysis of rIL-33 effects in the mRNA levels of acinar differentiation (Cpa1), metaplasia (Sox9) and Kras-dependent neoplasia (Agr2, Muc6) markers in pancreatic epithelial cell (mKate2+) populations isolated from Kras-WT (C) or Kras-mutant (KC) mice (n=2 each) treated with rIL-33 or Vehicle (PBS) and analyzed 21 days thereafter. f, GSEA comparing the expression of genes induced by the combination of mutant Kras + rIL-33 in human PDAC specimens vs human normal pancreas (Moffitt et al. dataset)23. g, Volcano plots comparing the chromatin accessibility landscape of Kras-mutant pancreatic epithelium of rIL-33-treated vs vehicle-treated mice, as assessed by ATAC-seq performed at the day 0 time-point. h, Top-scoring motifs identified by HOMER de novo analysis in accessibility-GAIN peaks identified in Kras-mutant pancreatic epithelial cells (mKate2+) isolated from rIL-33-treated mice vs from PBS-treated counterparts, assessed by ATAC-seq analyses performed at the day 0 time point. The significance of the enrichment is shown in brackets. i, Metagene representation of the mean ATAC-seq signal (n=3 mice per condition) at accessibility-GAIN regions driven by injury in the Kras-mutant pancreatic epithelium (Kras*+Injury vs Kras*) (top) or at accessibility-GAIN regions linked to the neoplasia-specific gene activation program (identified in Fig. 4b analyses, right) in Kras-mutant pancreatic epithelial cells (mKate2+) from isolated from rIL-33 treated vs PBS-treated mice (n=3 each, day 0 time point). rIL-33 treatment promotes accessibility at injury-sensitive sites. p-values were determined by Kolmogorov–Smirnov test. j, Quantification of the relative number of ADM and PanIN lesions in pancreata from Kras wild-type (C-GEMM) or Kras mutant (KC-GEMM) mice treated with rIL-33 or vehicle (PBS) and analyzed at the indicated time points in days (d) after treatment. Data are presented as means ± s.e.m and significance was assessed by unpaired two-tailed Student’s t-test (ns, not significant). n=3, 4, 4, 5, 3 or 4 (from left to right) independent animals per experimental condition. k, Representative immunofluorescence stains of IL-33 protein (green) co-stained with the lineage-tracer marker mKate2 (red) marking pancreatic epithelial cells from mice (n=3 per group) harbouring wild-type (Normal) or mutant Kras in the indicated tissue states. Scale bar, 100 μm. l, Relative mRNA levels (RNA-seq tpm counts) of Il33 (left) or the indicated mutant Kras effector (Agr288), middle) or acinar TF (Cpa1, right) in FACS-sorted mKate2+ pancreatic epithelial cell populations isolated from rIL-33-treated or PBS-treated mice harbouring WT or mutant Kras. n=3, 4, 4, 4, 5 or 4 (from left to right) biological replicates (independent mice) per group; median and upper/lower quantile values per group are indicated.
Supplementary Material
Acknowledgements
We thank Mayerlin Chalarca, Sarah Ackermann, Janelle Simon, Alex Wuest and MSKCC animal facility for technical support with animal colonies; Sang Yang, So Young, Zhen Zhao and Ambereen Kahn for assistance with the generation of ESC-derived GEMM; Julián Valdés, Surajit Dhara, Timour Baslan, Sha Tian and Alexa Osterhoudt for support with profiling experiments; Ignas Masilionis and Ojavsi Chaduhary for support with scATAC-seq experiments; Vincent Lavallée for his input and discussion on scATAC-seq data analysis; Rui Garner and the MSKCC Core Facility Staff for assistance with cell sorting; and José Reyes and other members of the Lowe laboratory for helpful advice and discussions. We also acknowledge the use of the Integrated Genomics Operation Core, funded by the NCI Cancer Center Support Grant (CCSG, P30 CA08748), Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. D.A-C. was supported by the Spanish Fundación Ramón Areces Postdoctoral Fellowhip; C.B. is supported by an NIH F31 grant (F31CA246901); J.P.M. IV was supported by an American Cancer Society Postdoctoral Fellowship (126337-PF-14–066-01-TBE); R.C. is supported by the Pancreatic Cancer Action Network-AACR Pathway to Leadership Award; H-A.C is supported by a NIH F99 Grant (F99CA245797); K.M.T is supported by the Jane Coffin Childs Memorial Fund for Medical Research; F.M.B is supported by MSKCC’s Translational Research Oncology Training Fellowship (5T32CA160001–08); N.T. is supported by an NIH K99 grant (K99CA237736); G.L. was supported by an NIH F32 grant (1F32CA177072–01) and American Cancer Society Fellowship (PF-13–037-01-DMC); E.A. is supported by a NIH K99 Grant (K99CA23019). D.P. This work was additionally supported by MSKCC’s David Rubenstein Center for Pancreatic Research Pilot Project (to S.W.L) and The Alan and Sandra Gerry Metastasis and Tumor Ecosystems Center (to S.W.L./D.P.); the Lustgarten Foundation Research Investigator Award (to S.W.L.); the Agilent Thought Leader Program (to S.W.L.); and the NIH’s Grants P01CA13106 (to S.W.L.), R01CA204228 and P30CA023108 (to S.D.L.), and U54CA209975 (to D.P.). S.W.L. is an investigator in the Howard Hughes Medical Institute and the Geoffrey Beene Chair for Cancer Biology.
Footnotes
Code availability
Custom codes for processing, filtering, and visualization of single-cell ATAC-seq were performed using Python and are demonstrated in a Jupyter notebook available for download at (https://github.com/dpeerlab/pdac-tumorigenesis-scATAC/).
Competing interest
A patent application (PTC/US2019/041670, internationally filing date 12 July 2019) has been submitted based in part on results presented in this manuscript covering methods for preventing or treating KRAS mutant pancreas cancer with inhibitors of Type 2 cytokine signaling. D.A.C and S.W.L are listed as the inventors. S.W.L. is a founder and scientific advisory board member of Blueprint Medicines, Mirimus Inc., and ORIC pharmaceuticals, and Faeth Therapeutics, and on the scientific advisory board of Constellation Pharmaceuticals and PMV Pharmaceuticals. S.D.L. is on the scientific advisory board of Nybo Therapeutics and Episteme Prognostics.
Additional information
Reprints and permissions information is available at www.nature.com/reprints.
Correspondence and requests for materials should be addressed to S.W.L (lowes@mskcc.org).
Data Availability
All ATAC-seq, RNA-seq, and scATAC-seq data generated in this study have been deposited in the Gene Expression Omnibus (GEO) database under the super-series GSE132330. Publicly available RNA-seq, gene expression microarray and ChIP-Seq data reanalyzed for this study is available under the accession codes GSE86262 (PTF1A ChIP-seq), GSE34295 (NR5A2 ChIP-seq), GSE99311 (H3K27Ac ChIP-seq), GSE62452 (human specimen gene expression microarray), and GSE71729 (human specimen gene expression microarray) and in the Gene Transcription Regulation Database (GTRD https://gtrd.biouml.org). All other data supporting the findings of this study are available from the corresponding authors upon reasonable request.
Main References
- 1.Giroux V & Rustgi AK Metaplasia: tissue injury adaptation and a precursor to the dysplasia-cancer sequence. Nat Rev Cancer 17, 594–604, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guerra C et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11, 291–302, (2007). [DOI] [PubMed] [Google Scholar]
- 3.Habbe N et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A 105, 18913–18918, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collins MA et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 122, 639–653, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hingorani SR et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Carriere C, Young AL, Gunn JR, Longnecker DS & Korc M Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun 382, 561–565, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strobel O et al. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology 133, 1999–2009, (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morris JP, Cano DA, Selkine S, Wang SC & Hebrok M beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. Journal of Clinical Investigation 120, 508–520, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kopp JL et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 22, 737–750, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Storz P Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat Rev Gastroenterol Hepatol 14, 296–304, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–29, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stanger BZ & Hebrok M Control of cell identity in pancreas development and regeneration. Gastroenterology 144, 1170–1179, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vallejo A et al. An integrative approach unveils FOSL1 as an oncogene vulnerability in KRAS-driven lung and pancreatic cancer. Nat Commun 8, 14294, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arda HE et al. A Chromatin Basis for Cell Lineage and Disease Risk in the Human Pancreas. Cell Syst 7, 310–322e314, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoang CQ et al. Transcriptional Maintenance of Pancreatic Acinar Identity, Differentiation, and Homeostasis by PTF1A. Mol Cell Biol 36, 3033–3047, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holmstrom SR et al. LRH-1 and PTF1-L coregulate an exocrine pancreas-specific transcriptional network for digestive function. Genes Dev 25, 1674–1679, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roe JS et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 170, 875–888e820, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loven J et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi J & Vakoc CR The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell 54, 728–736, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sherman MH Stellate Cells in Tissue Repair, Inflammation, and Cancer. Annu Rev Cell Dev Biol 34, 333–355, (2018). [DOI] [PubMed] [Google Scholar]
- 21.Cebola I et al. TEAD and YAP regulate the enhancer network of human embryonic pancreatic progenitors. Nat Cell Biol 17, 615–626, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krah NM et al. The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. Elife 4, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moffitt RA et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 47, 1168–1178, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cobo I et al. Transcriptional regulation by NR5A2 links differentiation and inflammation in the pancreas. Nature 554, 533–537, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wollny D et al. Single-Cell Analysis Uncovers Clonal Acinar Cell Heterogeneity in the Adult Pancreas. Dev Cell 39, 289–301, (2016). [DOI] [PubMed] [Google Scholar]
- 26.Liew FY, Girard JP & Turnquist HR Interleukin-33 in health and disease. Nat Rev Immunol 16, 676–689, (2016). [DOI] [PubMed] [Google Scholar]
- 27.Yevshin I, Sharipov R, Kolmykov S, Kondrakhin Y & Kolpakov F GTRD: a database on gene transcription regulation-2019 update. Nucleic Acids Res 47, D100–D105, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hnisz D et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whyte WA et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McDonald OG et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet 49, 367–376, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li A et al. IL-33 Signaling Alters Regulatory T Cell Diversity in Support of Tumor Development. Cell Rep 29, 2998–3008e2998, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moral JA et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature 579, 130–135, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 33.Saborowski M et al. A modular and flexible ESC-based mouse model of pancreatic cancer. Genes Dev 28, 85–97, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tasdemir N et al. BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov 6, 612–629, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuber J et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dow LE et al. A pipeline for the generation of shRNA transgenic mice. Nat Protoc 7, 374–393, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Livshits G et al. Arid1a restrains Kras-dependent changes in acinar cell identity. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gertsenstein M et al. Efficient generation of germ line transmitting chimeras from C57BL/6N ES cells by aggregation with outbred host embryos. PLoS One 5, e11260, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawaguchi Y et al. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet 32, 128–134, (2002). [DOI] [PubMed] [Google Scholar]
- 40.Jackson EL et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15, 3243–3248, (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beard C, Hochedlinger K, Plath K, Wutz A & Jaenisch R Efficient method to generate single-copy transgenic mice by site-specific integration in embryonic stem cells. Genesis 44, 23–28, (2006). [DOI] [PubMed] [Google Scholar]
- 42.Dow LE et al. Conditional reverse tet-transactivator mouse strains for the efficient induction of TRE-regulated transgenes in mice. PLoS One 9, e95236, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Premsrirut PK et al. A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell 145, 145–158, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heiser PW et al. Stabilization of beta-catenin induces pancreas tumor formation. Gastroenterology 135, 1288–1300, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rhim AD et al. EMT and dissemination precede pancreatic tumor formation. Cell 148, 349–361, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gopinathan A, Morton JP, Jodrell DI & Sansom OJ GEMMs as preclinical models for testing pancreatic cancer therapies. Dis Model Mech 8, 1185–1200, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morris J. P. t. et al. alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 573, 595–599, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Rourke KP et al. Transplantation of engineered organoids enables rapid generation of metastatic mouse models of colorectal cancer. Nat Biotechnol 35, 577–582, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 (2011). [Google Scholar]
- 50.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930, (2014). [DOI] [PubMed] [Google Scholar]
- 53.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pronier E et al. Targeting the CALR interactome in myeloproliferative neoplasms. JCI Insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gu Z, Eils R & Schlesner M Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849, (2016). [DOI] [PubMed] [Google Scholar]
- 57.Carlson M & Maintainer BP TxDb.Dmelanogaster. UCSC.dm3.ensGene: Annotation package for TxDb object(s) 2015, 2015).
- 58.Yu G, Wang LG & He QY ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383, (2015). [DOI] [PubMed] [Google Scholar]
- 59.Chen EY et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kent WJ et al. The human genome browser at UCSC. Genome Res 12, 996–1006, (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Z et al. Loss of CHD1 Promotes Heterogeneous Mechanisms of Resistance to AR-Targeted Therapy via Chromatin Dysregulation. Cancer Cell 37, 584–598e511, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Satpathy AT et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol 37, 925–936, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Levine JH et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 162, 184–197, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Danese A, Richter ML, Fischer DS, Theis FJ & Colomé-Tatché M EpiScanpy: integrated single-cell epigenomic analysis (2019). [DOI] [PMC free article] [PubMed]
- 66.McInnes L, Healy J, Saul N & L., G. UMAP: Uniform Manifold Approximation and Projection (2018).
- 67.Ramirez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robinson JT et al. Integrative genomics viewer. Nat Biotechnol 29, 24–26, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McLean CY et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28, 495–501, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550, (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang S et al. A Novel MIF Signaling Pathway Drives the Malignant Character of Pancreatic Cancer by Targeting NR3C2. Cancer Res 76, 3838–3850, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Whittle MC et al. RUNX3 Controls a Metastatic Switch in Pancreatic Ductal Adenocarcinoma. Cell 161, 1345–1360, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wei D et al. KLF4 Is Essential for Induction of Cellular Identity Change and Acinar-to-Ductal Reprogramming during Early Pancreatic Carcinogenesis. Cancer Cell 29, 324–338, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Diaferia GR et al. Dissection of transcriptional and cis-regulatory control of differentiation in human pancreatic cancer. EMBO J 35, 595–617, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Delgiorno KE et al. Identification and manipulation of biliary metaplasia in pancreatic tumors. Gastroenterology 146, 233–244e235, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kalisz M et al. HNF1A recruits KDM6A to activate differentiated acinar cell programs that suppress pancreatic cancer. EMBO J 39, e102808, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Truty MJ, Lomberk G, Fernandez-Zapico ME & Urrutia R Silencing of the transforming growth factor-beta (TGFbeta) receptor II by Kruppel-like factor 14 underscores the importance of a negative feedback mechanism in TGFbeta signaling. J Biol Chem 284, 6291–6300, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.von Figura G, Morris J. P. t., Wright CV & Hebrok M Nr5a2 maintains acinar cell differentiation and constrains oncogenic Kras-mediated pancreatic neoplastic initiation. Gut 63, 656–664, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiang M et al. MIST1 and PTF1 Collaborate in Feed-forward Regulatory Loops that Maintain the Pancreatic Acinar Phenotype in Adult Mice. Mol Cell Biol, (2016). [DOI] [PMC free article] [PubMed]
- 83.Dawson MA et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478, 529–533, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Delmore JE et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mazur PK et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med 21, 1163–1171, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim J, Lee JH & Iyer VR Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS One 3, e1798, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bian B et al. Gene expression profiling of patient-derived pancreatic cancer xenografts predicts sensitivity to the BET bromodomain inhibitor JQ1: implications for individualized medicine efforts. EMBO Mol Med 9, 482–497, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dumartin L et al. ER stress protein AGR2 precedes and is involved in the regulation of pancreatic cancer initiation. Oncogene 36, 3094–3103, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All ATAC-seq, RNA-seq, and scATAC-seq data generated in this study have been deposited in the Gene Expression Omnibus (GEO) database under the super-series GSE132330. Publicly available RNA-seq, gene expression microarray and ChIP-Seq data reanalyzed for this study is available under the accession codes GSE86262 (PTF1A ChIP-seq), GSE34295 (NR5A2 ChIP-seq), GSE99311 (H3K27Ac ChIP-seq), GSE62452 (human specimen gene expression microarray), and GSE71729 (human specimen gene expression microarray) and in the Gene Transcription Regulation Database (GTRD https://gtrd.biouml.org). All other data supporting the findings of this study are available from the corresponding authors upon reasonable request.