

Abstract
Decoupling the roles of the farnesoid X nuclear receptor and Takeda G-protein-coupled bile acid receptor 5 is essential for the development of novel bile acid therapeutics targeting metabolic and neurodegenerative diseases. Herein, we describe the synthesis of 12β-methyl-18-nor-bile acids which may serve as probes in the search for new bile acid analogues with clinical applicability. A Nametkin-type rearrangement was applied to protected cholic acid derivatives, giving rise to tetra-substituted Δ13,14- and Δ13,17-unsaturated 12β-methyl-18-nor-bile acid intermediates (24a and 25a). Subsequent catalytic hydrogenation and deprotection yielded 12β-methyl-18-nor-chenodeoxycholic acid (27a) and its 17-epi-epimer (28a) as the two major reaction products. Optimization of the synthetic sequence enabled a chromatography-free route to prepare these bile acids at a multi-gram scale. In addition, the first cis-C-D ring-junctured bile acid and a new 14(13 → 12)-abeo-bile acid are described. Furthermore, deuteration experiments were performed to provide mechanistic insights into the formation of the formal anti-hydrogenation product 12β-methyl-18-nor-chenodeoxycholic acid (27a).
Introduction
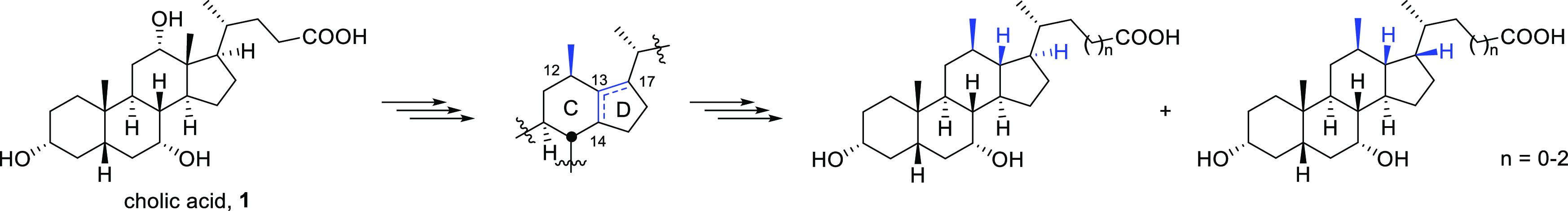
Bile acids are physiologically important molecules that are generated in the liver and constitute the end products of cholesterol catabolism. They are conjugated with either taurine or glycine before being secreted into small bile canaliculi by the bile-salt export pump. From there, they flow through a growing network of bile ducts into the gallbladder for storage. The primary bile acids, cholic acid (CA, 1) and chenodeoxycholic acid (CDCA, 2), are the products of the liver, whereas the secondary bile acids, lithocholic acid (3) and deoxycholic acid (4), originate from bacterial metabolization of primary bile acids in the intestine (Figure 1). The conjugated primary bile acids are the main components of the human bile acid pool. Because of their amphipathic properties, bile acids facilitate the solubilization of lipid-soluble nutrients, enabling their absorption in the digestive tract. Eventually, they are actively reclaimed from the distal small intestine and shuttled back to the liver by a complex system of bile acid-specific transport proteins.1,2
Figure 1.
Primary and secondary bile acids and a selection of some medically important analogues.
Until recently, however, the general knowledge of bile acids did not extend far beyond their role as emulsifiers for lipids in the enterohepatic circulation, rendering this class of compounds rather unappealing for study.3 This paradigm changed when bile acids were found to act as hormones4 by activating the farnesoid X nuclear receptor (FXR) and Takeda G-protein-coupled bile acid receptor 5 (TGR5).3,5,6 FXR is a bile acid sensor that controls cellular bile acid concentrations by mediating their biosynthesis, as well as their cellular uptake and efflux.7,8 Furthermore, FXR is an important metabolic regulator of lipid9,10 and glucose metabolism,10−12 and its activation can also reduce inflammation13 and fibrosis.14 Activation of TGR5 signaling promotes various metabolic actions including an increase in energy expenditure and oxygen consumption in the brown adipose tissue and muscle,15 immunomodulatory effects,6b,16 and stimulation of glucagon-like peptide-1 secretion in enteroendocrine cells, which in turn ameliorate insulin sensitivity and glucose homeostasis, in general.10,17,18
Both of these receptors have important physiological functions due to which they are pursued as potent therapeutic targets for the treatment of various liver and metabolic disorders.19−27 Since their discovery, various bile acid analogues have emerged as potent agonists (i.e., 5–11; Figure 1), and many of these are being evaluated in clinical and pre-clinical trials.8,28−30 The 6α-ethyl-substituted analogue of CDCA (5),30 also known as obeticholic acid or INT-747, is of particular note as it was approved as a first-in-class FXR agonist by the US Food and Drug Administration (FDA) for the treatment of primary biliary cholangitis in 2016. Currently, the compound is also being evaluated in advanced clinical trials for the treatment of primary sclerosing cholangitis and non-alcoholic steatohepatitis.8,28,31 These findings sparked renewed interest in bile acid research and has stimulated the development of new bile-acid-inspired therapeutics.
In this paper, we report on our efforts to synthesize a new class of 12β-methyl-18-nor-bile acids.32 Our inspiration for this project stemmed from publications by Brieskorn and Mosandl (1981) and Shimizu, T. et al. (1999).33,34 In both studies, the authors described the preparation of Δ13,14- and Δ13,17-12β-methyl-18-nor-bile acid derivatives (13/14 and 16/17) that we deemed to be a suitable entry into the synthesis of a new range of bile acids (Scheme 1). In both cases, a 12α-hydroxy-activated bile acid derivative (12 or 15b) was subjected to reaction conditions that enabled migration of the angular C18 methyl group to the neighboring C12 carbon atom in a Nametkin rearrangement, leading to the formation of double bonds between the C13 and C14, or C13 and C17 carbon centers. Shimizu and co-workers also explored mesylate (15c) and triflate (15d) as alternative leaving groups for their reaction, but in these cases the alkene reaction products 16 and 17 were only generated in poor yields.34
Scheme 1. Previously Reported Syntheses of Δ13,14- and Δ13,17-12β-Methyl-18-nor-bile Acid Derivatives.
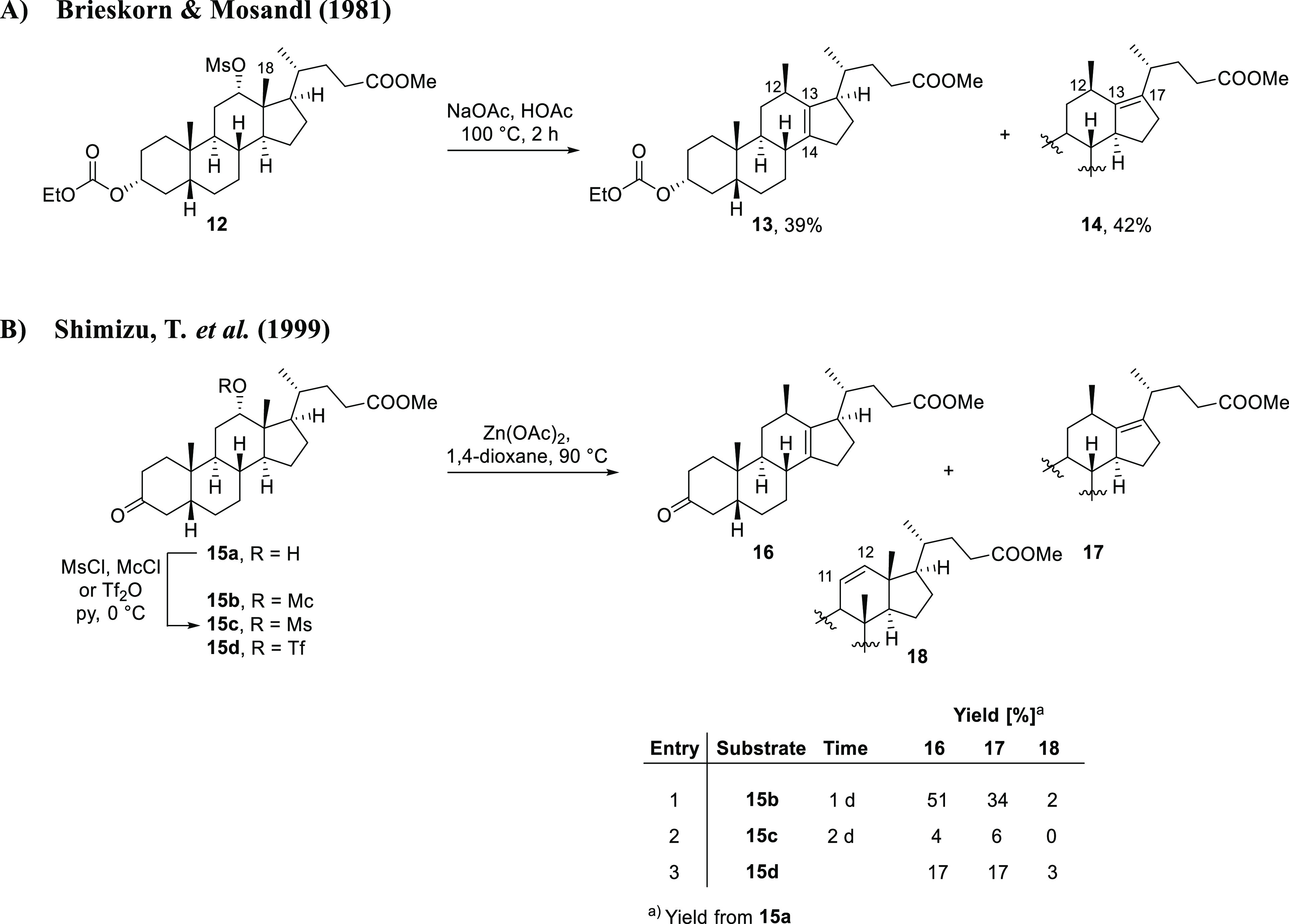
Abbreviations: Tf2O: trifluoromethanesulfonic anhydride; Mc: chloromethanesulfonate (monochlate); Ms: methanesulfonate; py: pyridine.
To the best of our knowledge, there have been no subsequent reports on the utility of such unsaturated 12β-methyl-18-nor-bile acid derivatives or further investigations into the rearrangement, leaving the potential of these substrates unrealized. Moreover, the isolation of rearrangement products has not been reported on a practical scale as either preparative high-performance liquid chromatography (HPLC) or silver chromatography was employed for separation.33,34 Renewed interest in the development of selective bile acid therapeutics prompted us to investigate this rearrangement as a source of new bile acid analogues. Herein, we describe the development of a chromatography-free synthesis and characterization of unnatural 12β-methyl-18-nor-bile acid derivatives formed from an 18 → 12β Nametkin methyl migration of 12α-hydroxy-activated CA derivatives.
Results and Discussion
To access the envisaged alkenes 24a and 25a (Scheme 2), we sought to prepare mesylate 23a as a substrate for the Nametkin rearrangement previously described by Brieskorn and Mosandl.33 Following established methods, our synthetic efforts began by converting CA (1) into its corresponding methyl ester 21a. This was followed by selective acetylation of the 3- and 7-hydroxy groups and the subsequent reaction of 22a with methanesulfonyl chloride to furnish the desired precursor 23a.35,36
Scheme 2. Preparation of Δ13,14- and Δ13,17-12β-Methyl-18-nor-bile Acid Intermediates 24a–c and 25a–c.
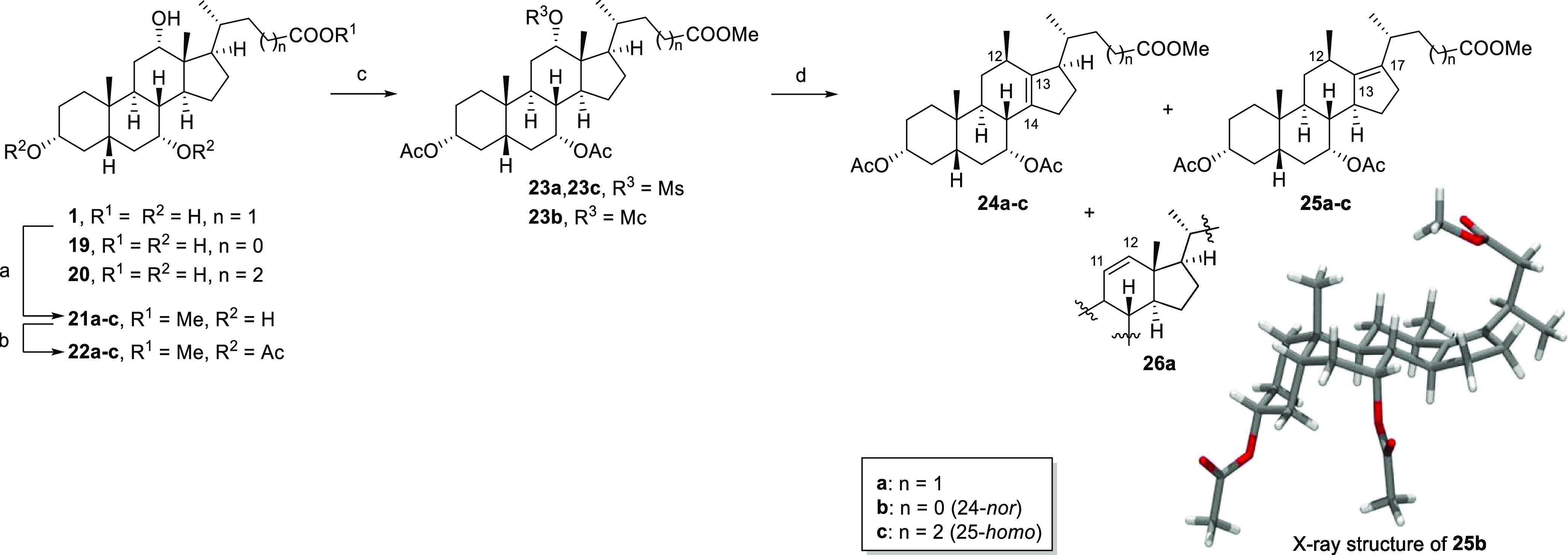
Reagents and conditions: (a) MeOH, p-TsOH·H2O, Δ or rt; 21b–c: 83–84%; (b) for 22a: Ac2O, DMAP, py (2.5 equiv), toluene;36 90%; for 22b–c: DCM, DMAP, NEt3, Ac2O, 0 °C–rt; 2–6 h; 76–86%; (c) for 23a(36)/23c: MsCl, py, 0 °C–rt, o/n; 83–97%; for 23b: McCl, py, 0 °C, 45 min; (d) 24a/c//25a/c: HOAc, NaOAc, 100 °C, 2–3 h; 83–86%; 24b/25b: HOAc, Zn(OAc)2, 100 °C, 1.5 h; 84%. Abbreviations: DCM: dichloromethane; DMAP: N,N-dimethylaminopyridine; o/n: overnight; rt: room temperature; Ts: para-toluenesulfonate.
When mesylate 23a was subjected to the reported reaction conditions (NaOAc, HOAc, and 100 °C), a mixture of unsaturated 12β-methyl-18-nor-bile acid products was obtained in good yield after chromatographic purification. Spectroscopic analysis and HPLC analysis revealed that the Δ13,14-alkene 24a and the Δ13,17-isomer 25a were formed, on average, in a 1:2 ratio, and that the mixture also contained minor amounts of other alkene side products including the Δ11,12-isomer 26a. After isolating the alkene product mixture, as a single fraction by normal-phase chromatography, we employed chromatography on silver nitrate-impregnated silica gel as a means to separate the individual components.37 In this manner, small amounts of the two major rearrangement products 24a and 25a (Scheme 2) were recovered whose structures were assigned by 2D NMR analysis. These assignments were later confirmed by X-ray crystallographic analysis (vide infra). However, silver chromatography37 was limited by the requirement of repeated runs to obtain larger quantities of individual products for further synthesis. In addition, the prohibitive cost of silver nitrate needed to process large quantities of product led us to seek alternatives.
With the knowledge that alkenes 24a and 25a were major components of the rearrangement mixture, we next omitted the purification step on silver-impregnated silica gel before committing the partially purified mixture of alkenes to the final two steps of the synthesis (Scheme 3). Accordingly, catalytic hydrogenation over 10% palladium on charcoal in ethanol yielded a mixture of saturated 12β-methyl bile acid products that again proved inseparable by standard chromatography techniques. Therefore, this saturated product mixture was directly subjected to saponification to deprotect the acetate and methyl ester-protecting groups.
Scheme 3. Preparation and Characterization of New 12β-Methyl-18-nor-bile Acids.

Following global deprotection, HPLC analysis of the mixture revealed that, among other minor products, two major components were present in a 1:2.6 ratio (27a/28a). Both compounds were isolated by a combination of reversed- and normal-phase column chromatography, as well as recrystallization. X-ray diffraction of suitable single crystals of each of these isolated products elucidated their structures as two new 12β-methyl-18-nor-bile acids 27a and 28a. Bile acid 27a is structurally analogous to CDCA (2) except for the relocation of the angular C18 methyl group to the 12β-position. Diastereomer 28a, on the other hand, has an unnatural epimerized C17 carbon center, positioning the side chain on the α-face of the steroid scaffold; a feature that is not found in naturally occurring steroids or bile acids. During efforts to purify 27a and 28a, we also isolated small amounts of two minor byproducts 29 and 30, whose structures were also solved by X-ray diffraction. In addition to the rearranged methyl group, now at the equatorial 12β-position, compound 29 was also found to have an epimerized C14 carbon center, giving rise to an unprecedented diastereomer with a C/D-cis ring junction. Compound 30 featured a C-nor-D-homo ring system which resulted from the saturation of C-nor-D-homo alkenes formed from migration of the C13–14 bond to the C12 position in a Wagner–Meerwein-type rearrangement of mesylate 23a. This C-nor-D-homo motif has been reported previously to be a characteristic structural feature of cyclopamine and related steroidal alkaloids.38
The reaction mixture also contained a minor amount (7–9%) of 12β-methyl-18-nor-lithocholic acid isomers 31 and 32, of which the structure of 31 was confirmed by X-ray crystallographic analysis. As isomer 32 was an oil, confirmation of its stereochemistry was not possible by crystallography, and its analysis by NMR spectroscopy remained inconclusive. Hydrogenation experiments with purified samples of either the Δ13,14- or the Δ13,17-alkene (24a or 25a) demonstrated that 12β-methyl-18-nor-lithocholic acid side products (31 and 32) were only generated from hydrogenation of the Δ13,14-alkene (24a). In contrast, the two major products, 27a and 28a, were the only two products formed from the Δ13,17-alkene (25a).
With the aim of building a drug discovery library of structurally modified 12β-methyl-18-nor-bile acids, we also pursued the synthesis of 24-nor- and 25-homo-analogues (27b/c and 28b/c), as depicted in Scheme 4. For the synthesis of 24-nor-bile acids (27b and 28b, Scheme 5), we investigated the methodology described by Shimizu and co-workers34 (Scheme 1B), by employing monochlate 23b to effect the rearrangement to Δ13,14- and Δ13,17-alkenes (24b and 25b) upon treatment with zinc acetate in dioxane. As per the synthesis of mesylate 23a (Scheme 2), the required monochlate (23b) was prepared from 24-nor-cholic acid (19)39 in three standard transformations, which involved methyl esterification, followed by selective acetylation of the 3- and 7-hydroxy groups, and activation of the 12-hydroxy group with chloromethanesulfonyl chloride. However, when Shimizu’s reported conditions for the rearrangement were applied to 23b (Scheme 1B), the starting material was consumed exceedingly slowly. By switching the solvent from dioxane to acetic acid and repeating the reaction with zinc acetate at 100 °C, the rearrangement was complete within 1 h.
Scheme 4. Synthesis of 24-nor and 25-homo 12β-Methyl-18-nor-bile Acids 27b,c and 28b,c.
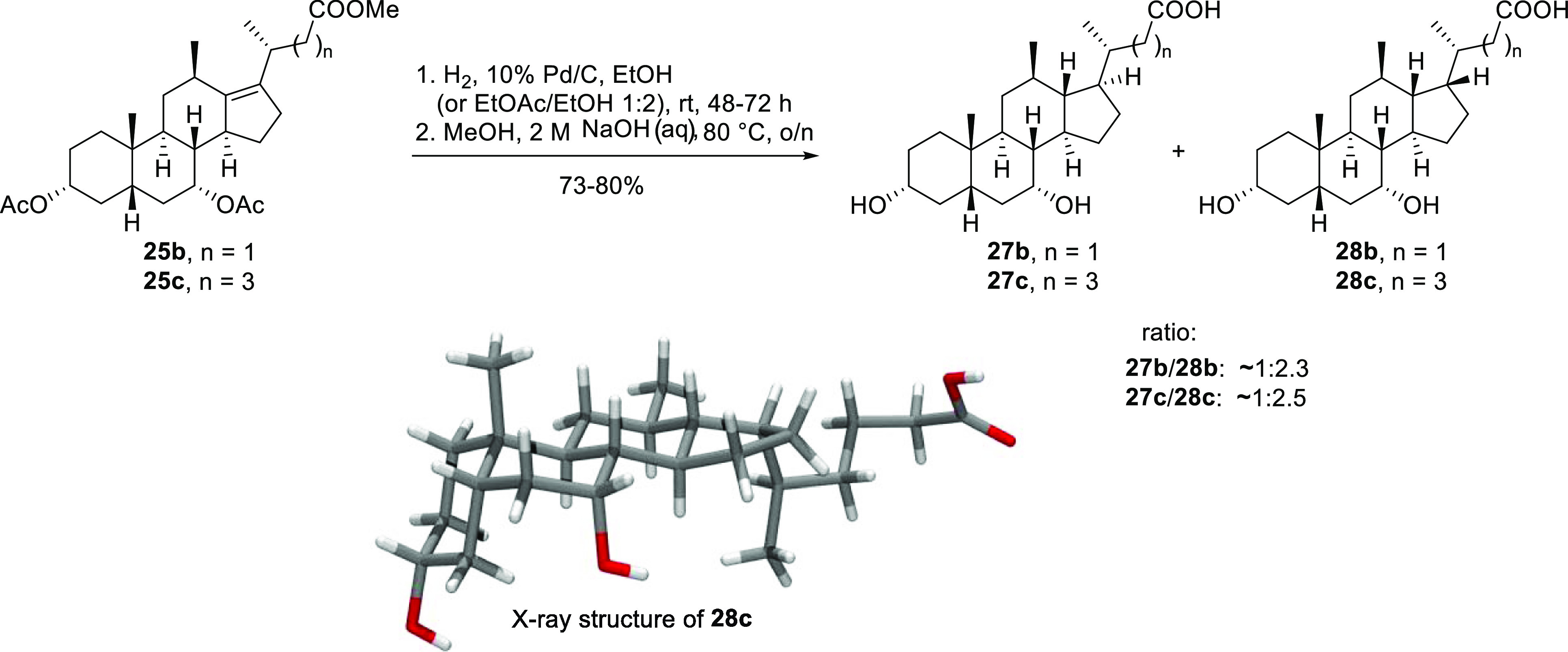
Scheme 5. Chromatography-Free, Large-Scale Synthesis of 12β-Methyl-18-nor-alkene Bile Acids 33 and 34 and 12β-Methyl-18-nor-bile Acids 27a and 28a.
Reagents and conditions: (a) MeOH, p-TsOH·H2O, Δ, 2 h, then rt, 3 d; (b) py, DMAP, EtOAc, Ac2O, 0 °C–rt, o/n; 51% (2 steps, unoptimized); (c) MsCl, py, 0 °C–rt, o/n; (d) NaOAc, HOAc, 100 °C, o/n; (e) 10 M NaOHaq, MeOH, 80 °C, 72 h; 33: analytical sample: 92.5% pure after fractional crystallization; >99% pure after chromatography; 34: ca. 60 g; 95% pure, following fractional crystallization (HPLC analysis); (f) fractional crystallization; (g) H2 (5 bar), 10% Pd/C (0.1 equiv), MeOH/H2O (10:1), rt, 20 h; 27a: 98.8% pure after fractional crystallization; 28a: >99% pure after fractional crystallization (HPLC analysis).
Analysis of this partially purified mixture by HPLC showed that the desired Δ13,14- and Δ13,17-alkenes 24b and 25b were present in a ratio of 1:1.4 (see the Supporting Information). As before, separation of alkenes 24b and 25b was achieved by column chromatography on 20% silver nitrate-impregnated silica gel. Δ13,17-alkene 25b was obtained as a clean fraction, and suitable crystals were grown enabling its structural determination by X-ray crystallographic analysis. On the other hand, only a small amount of analytically pure Δ13,14-isomer 24b was recovered for spectroscopic analysis. Therefore, we subjected the Δ13,17-alkene 25b alone to palladium-catalyzed hydrogenation. As expected, the product mixture from this hydrogenation reaction contained only two products (in an anti/syn ratio of ∼1:2.3, as deduced from the 13C NMR spectrum of the chromatographed mixture), but yet again these products were inseparable by chromatography. Therefore, the mixture of saturated reaction products was saponified to produce the two 12β-methyl-18,24-bisnor-bile acids 27b and 28b, which were easily separated by normal-phase chromatography.
Since the application of the reaction conditions described by Shimizu et al.(34) to our substrates, namely, the use of a monochlate-activated substrate in combination with Zn(OAc)2, afforded no particular benefit in effecting the desired 1,2-methyl shift, we elected to continue with the combination of mesylate/NaOAc/AcOH as the preferred rearrangement conditions. Accordingly, we then prepared the analogous 25-homo-bile acids 27c and 28c (Scheme 4). Following the preparation of the mesyl-activated 25-homo-bile acid precursor (23c) from 25-homo-cholic acid 20(35) and subsequent rearrangement, the corresponding mixture of protected alkenes (24c and 25c) was fractionated by column chromatography on 20% silver nitrate-impregnated silica gel. With the expectation that the Δ13,17-alkene would form only two products in the hydrogenation step, and to avoid the formation of other byproducts, only Δ13,17-alkene 25c was subjected to hydrogenation and saponification. Thus, the two 12β-methyl-25-homo-18-nor-bile acids 27c and 28c were isolated by normal-phase chromatography, and the structure of 28c was determined by X-ray crystallographic analysis (Scheme 4).
Despite developing a general method that enabled the synthesis of 12β-methyl-18-nor-bile acids 27/28(a–c), this parallel approach was still limited to the preparation of small amounts of target materials due to the required use of expensive and laborious silver-impregnated chromatography. To access a diverse drug discovery library would require the production of larger quantities of materials. We therefore turned our attention to upscaling the synthesis of 27a and 28a to access 27b/28b and 27c/28c by side-chain reduction and elongation reactions, respectively.39,40
Following the same reaction sequence outlined in Scheme 2, we set out from 250 g of CA. With only minor modifications and a single chromatographic step to remove an impurity that otherwise inhibited the palladium-catalyzed hydrogenation reaction, products 27a and 28a could be isolated by iterative fractional precipitation procedures (see the Experimental Section). A key feature of the fractional precipitation process takes advantage of the relative insolubility of 28a in methanol/ethyl acetate mixtures when compared with 27a. Therefore, from 250 g of CA, this six-step procedure was amenable to scale-up with 30.4 g (12% yield) of 27a and 32.4 g (13% yield) of 28a being produced, with only a single passage of column chromatography required to remove the impurity that inhibited the catalytic hydrogenation step.
Surprisingly, during the fractional precipitation process to isolate the individual products 27a and 28a, a small amount (3.5 g) of pure deprotected Δ13,14-alkene 34 (Scheme 5) was isolated, pertaining to incomplete hydrogenation of the protected alkene mixture. The compound’s high crystallinity, as a hydrate form, not only enabled its isolation and characterization by single-crystal X-ray diffraction but also, most importantly, indicated that it was stable to the basic hydrolysis conditions employed in the final reaction step. These observations prompted us to investigate reversing the order of the hydrogenation and hydrolysis steps, to attempt purification of the unsaturated 12β-methyl-18-nor-bile acid products by precipitation, prior to hydrogenation. In the envisaged process, we also anticipated removing the impurity that inhibited catalytic hydrogenation.
With this objective in mind, we increased the scale to 500 g of CA for our second campaign and prepared ca. 330 g of the mixture of protected 12β-methyl-18-nor-alkenes (24a, 25a, and others) over four steps (a–d; Scheme 5). Prior to committing the bulk of this material to hydrolysis, we trailed the hydrolysis on 12 g of this mixture. Following workup, it was possible to isolate a mixture (6 g) containing the unsaturated, deprotected 12β-methyl-18-nor-bile acid products from the crude by filtering a powder that precipitated between ethyl acetate and the aqueous layers upon vigorous shaking and scratching. Further chromatography of a sample (2 g) of this powder enabled the isolation and characterization of the two major reaction products 33 and 34. A test hydrogenation of the crude material demonstrated that the saturated bile acids 27a and 28a were indeed formed in an approximate 2.5:1 ratio (as deduced from the 13C NMR spectrum of the crude, by comparison with the samples prepared previously), confirming that the impurities that inhibited the catalytic hydrogenation had been removed by the precipitation process. Interestingly, this is a reversal of the anti/syn selectivity observed when the protected alkene mixture containing 24a and 25a was hydrogenated. The only further optimization required for the hydrogenation was the addition of water to the reaction solvent (methanol) to prevent esterification of the carboxylic acids, which was found to be a significant side reaction when neat methanol was employed as the reaction solvent.
Following the satisfactory outcome of the test hydrolysis and hydrogenation reactions, we next subjected the remainder of the bulk-protected 12β-methyl-18-nor-alkene mixture (318 g) to basic hydrolysis (step e; Scheme 5). The reaction proceeded in an almost identical fashion at scale, and ∼300 g of the crude-deprotected 12β-methyl-18-nor-bile acid alkene mixture was furnished as a powder upon precipitation. Evaluation of the 13C NMR spectrum of the mixture confirmed that alkenes 33 and 34 represented the major reaction products in a 1.5:1 ratio. Further purification, by fractional precipitation of the bile acid alkene mixture, removed many of the side products and yielded ca. 60 g of Δ13,14-alkene 34 (>95% pure) and 144 g of a mixture that was enriched with Δ13,17-alkene 33, but also contained alkene 34 and other minor impurities. For this fractional precipitation, we were able to take advantage of the relative insolubility of Δ13,14-alkene 34 in methanol/ethyl acetate mixtures, when compared with Δ13,17-alkene 33. However, when alkene 33 was the major component of a mixture of the two, it was possible to purify small amounts of it by precipitating from hot, dry ethyl acetate.
For the final hydrogenation step, however, we recombined 46 g of pure Δ13,14-alkene 34 with the Δ13,17-alkene-enriched mixture (144 g). The hydrogenation was conducted (in a calorimeter-equipped 3.4 L stainless-steel pressure vessel) at 25 °C and 5 bar and was complete after 20 h. In contrast to our initial work (vide supra), on hydrogenation of the protected alkene mixture, we found that hydrogenation of the deprotected 12β-methyl-18-nor-alkene bile acids favored the formation of the saturated bile acid 27a in a ratio of 2.5:1 (as deduced from the 13C NMR spectrum of the crude). Following our procedures developed for the fractional precipitation, bile acids 27a and 28a were eventually isolated in 16 and 5% yields, respectively, with purity of greater than 95%, over six steps from CA.
With the availability of gram quantities of the bile acids 27a and 28a, the syntheses of the corresponding 12β-methyl-25-homo-18-nor-bile acids 27c and 28c were accomplished in 36 and 44% yields, respectively, over five steps employing an Arndt–Eistert homologation strategy (Scheme S2), whereas the 24-nor-bile acid analogue 27b was prepared in 22% yield (unoptimized) over three steps using a second-order Beckmann rearrangement as a key transformation following literature procedures (Scheme S3).39,40
To investigate what impact the position of the double bond in the Δ13,14- and Δ13,17-12β-methyl-18-nor-alkene bile acids 33 and 34 would have on the profile of hydrogenation products, we opted to study the hydrogenation of the individual alkene bile acids using an H-Cube apparatus. Once reaction conditions such as the concentration of reactant (2.5 mg/mL), pressure (80 bar), and flow rate (0.3 mL/min) were optimized, alkenes 33 or 34 were hydrogenated under continuous flow in a single pass (Table 1). Thus, hydrogenation of Δ13,17-alkene 33 generated 12β-methyl-18-nor-bile acids 27a and 28a as the only two reaction products, which was consistent with the hydrogenation of the protected congeners. The product ratio was concluded to be 1.3:1 from analysis of the 13C NMR spectrum of the product mixture.
Table 1. Comparison of Product Ratios for H-Cube Hydrogenation of Substrates 33 and 34.
| entry | substratea | product mixture | product ratiob |
|---|---|---|---|
| 1 | 33 (Δ13,17) | 27a/28a | 1.3/1 |
| 2 | 34 (Δ13,14) | 27a/28a/29/31/32 | 3/0.2/1/0.4/0.8 |
50 mg in 20 mL of methanol (2.5 mg/mL; 0.3 mL/min; 80 bar; 25 °C).
Approximate ratio: determined by integration of the 13C NMR spectrum; quantitative conversion of the substrate.
In the case of Δ13,14-alkene 34, however, five hydrogenation products were observed in the 13C NMR spectrum of the crude reaction mixture (Table 1), all of which could be assigned by comparison with authentic samples. Interestingly, under the reaction conditions employed, considerable amounts of CD-cis bile acid 29 and 12β-methyl-18-nor-lithocholic acid derivatives 31 and 32 were formed alongside the major product 27a, whereas the 17-epi-isomer 28a represented only a minor constituent of the product mixture. These results not only corroborate that the 12β-methyl-18-nor-lithocholic acid derivatives (31 and 32) and other side products (29) originate from hydrogenation of Δ13,14-alkene bile acid 34 but also offer insight into the mechanism of the palladium-catalyzed hydrogenation of these alkenes.
To better understand the formation of 12β-methyl-18-nor-bile acid 27a—the product of a formal anti-hydrogenation—we performed a deuteration of Δ13,17-alkene bile acid 33 on a preparative scale in an H-Cube by exchanging water for deuterium oxide (as the source of deuterium gas) and using deuterated methanol as the solvent (Scheme 6). The two deuterated bile acid products 27a–d3 and its C17-epimer (analogous to hydrogenated products 27a and 28a) were then separated by chromatography for analysis.
Scheme 6. Deuteration of Δ13,17-Alkene Bile Acid 33 under Continuous Flow.
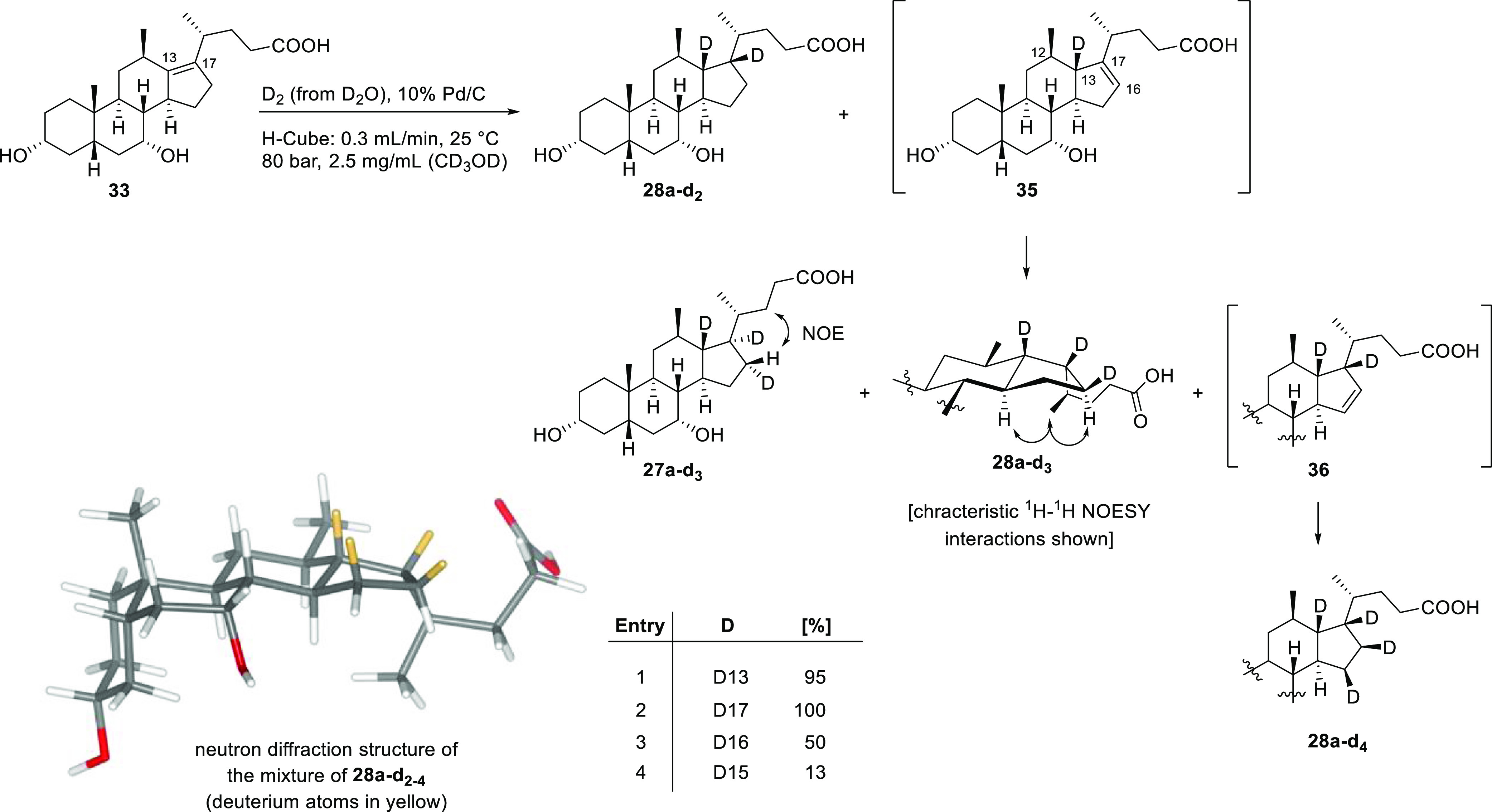
Spectroscopic analysis of 27a–d3 revealed that the compound carried three deuterium atoms (Figure 2). The presence of these newly introduced deuterium atoms was most obvious in the carbon NMR spectrum, as carbon–deuterium coupling caused splitting of the carbon signals giving rise to an apparent loss of signal intensity. This effect was observed for the C13 and C17 signals, but in addition a third 2H atom was bound to C16. The presence of a NOESY interaction between the remaining H16 proton and the side chain demonstrated that 2H16 was oriented toward the α-face.
Figure 2.

Carbon NMR analysis of compound 27a–d3: (A) 13C NMR spectrum of 27a (with power-gated 1H decoupling); (B) 13C NMR spectrum of 27a–d3 (with power-gated 1H decoupling); and (C) semi-quantitative 13C NMR spectrum of 27a–d3 (inverse-gated 1H- and 2H-decoupled). 13C NMR spectra were recorded at 125 MHz in CD3OD.
Less straightforward proved the interpretation of the spectral data for the C17-epimer. As before, the carbon NMR spectrum showed that deuterium atoms were present in positions 13 and 17, which satisfied our expectation. However, additional isotopic effects were observed for the C16 and C15 carbon resonances in the proton- and deuterium-decoupled 13C NMR spectrum, both of which appeared now as a cluster of three signals with different intensities and up-field shifts (Figure 3). The major resonance for C16 at 23.55 ppm was found to be shifted up-field by 2Δ(C) = 0.1 ppm, when compared to the same signal of the non-deuterated compound 28a. This effect is characteristic for a two-bond isotope shift, as caused by the neighboring 2H17. On the other hand, the major signal for C15 at 30.36 ppm is in accord with the shift of the C15 signal of the non-deuterated analogue 28a, which indicated that no deuterium atom was nearby, including C16. These observations allowed us to conclude that these signals originated from a compound that carried only two deuterium atoms, in positions 13 and 17 (28a–d2). A subsequent examination of the second largest signals at 23.22 (C16) and 30.25 ppm (C15) revealed that the C16 signal had undergone a one-bond isotope shift of 1ΔC(D) = 0.43 ppm due to a deuterium atom being attached to C16, whereas the C15 resonance was shifted up-field by 2ΔC(D) = 0.1 ppm as a secondary effect. This suggested that the NMR sample contained a second constituent with three deuterium atoms in positions 13, 17, and 16 (28a–d3). The observation of a NOESY cross-peak between the proton signals of H16 and H21 showed that the 2H16 was facing toward the β-side. Finally, the upfield shift of 1ΔC = 0.5 ppm of both of the least intense signals at 29.83 and 23.11 ppm, relative to their respective non-deuterated 13C signals, pointed to the presence of a third isomer with four deuterium atoms being attached to C13, C17, C16, and C15. Although the orientation of the C15-deuterium could not be elucidated by NMR spectroscopy, due to a lack of characteristic NOESY interactions, we assigned a β-orientation based on the neutron diffraction structure of 28a–d2-4, as depicted in Scheme 6. Moreover, the neutron structure confirmed the presence and β-orientation of each of the four deuterium atoms at C13, C17, C16, and C15. The notion that the analyzed sample was in fact composed of a mixture of three isomers with different deuteration patterns was furthermore supported by the recorded isotope distribution in the mass spectrum, which differed significantly from that expected for a single compound (see the Supporting Information). By comparison of the relative intensities of the respective carbon peaks of each cluster in the semi-quantitative 13C NMR spectrum, the ratio of the deuterated bile acid mixture was estimated to be 4:2:1.
Figure 3.

Carbon NMR analysis of the mixture of 28a–d2–4: (A) 13C NMR spectrum of 28a (with power-gated 1H decoupling); (B) 13C NMR spectrum (with power-gated 1H decoupling) of 28a–d2–4; and (C) semi-quantitative 13C NMR spectrum of the mixture of 28a–d2–4 (inverse gated, 1H- and 2H-decoupled; NS 9600). 13C NMR spectra were recorded at 125 MHz in CD3OD. See the Supporting Information for full spectra.
From these findings, we deduced that during the course of the deuteration the tetrasubstituted Δ13,17-double bond had undergone migration over two bonds, thus accounting for the formation of the anti-deuteration product 27a–d3 and the introduction of β-facing deuterium atoms in positions 16 and 15. These considerations can be rationalized by the half-hydrogenated state mechanism that was introduced by Horiuti and Polanyi in 1934.41,42 In accordance with this mechanism, we can postulate the following sequence of events: Initially, the Δ13,17-alkene reactant (33) is adsorbed onto the palladium surface by its double bond from the convex side of the steroid scaffold, presumably by a combination of π- and σ-bonding. Since no deuterium atom was detected in position 12, we can conclude that the first deuterium–carbon bond was established at C13, whereas the adjacent C17 maintains bonding to the palladium surface giving rise to a half-hydrogenated form. This intermediate can now either react irreversibly with another deuterium to give the 17-epi-bile acid 28a–d2 or, because of the reversible nature of the reaction, dissociate again to either the starting alkene 33 or the isomeric alkene 35. In the event that the isomeric alkene (35) is formed, syn-addition of deuterium from the α-face will yield the anti-deuteration product 27a–d3, while deuteration from the β-face will yield the trideuterium bile acid 28a–d3. Another iteration of this sequence will lead to the intermediate alkene 36 which, upon selective deuteration from the β-face, gives the tetradeuterated analogue 28a–d4.
Conclusions
In summary, a Nametkin-type rearrangement was applied to the CA scaffold to generate a mixture of Δ13,14- and Δ13,17-unsaturated 12β-methyl-18-nor-bile acids. These rearrangement products were hydrogenated, yielding a new series of saturated bile acids with unprecedented structures. The synthetic sequence was optimized, enabling the preparation and isolation of alkene intermediates (33 and 34), and saturated analogues (27–30) at a multi-gram scale by a chromatography-free route. Together, these new chemical entities constitute a new bile acid drug discovery library that is suitable for further derivatization; several projects exploring their synthetic utility have been undertaken in our laboratories and will be reported in due course.
Experimental Section
General Experimental Procedures
Melting points were determined by differential scanning calorimetry (DSC) on a Mettler Toledo DSC1 instrument at a heating rate of 10 K·min–1. Proton (1H) and carbon (13C) NMR spectra were recorded on a three-channel Bruker AVANCE III 500 MHz spectrometer with a Bruker 5 mm BBO probe. Chemical shifts are reported in ppm relative to Me4Si (TMS, δ 0.0), or residual solvent peaks as an internal standard set to δ 7.26 and 77.00 (CDCl3), or δ 3.34 and 49.05 (MeOD) for 1H and 13C, respectively. The 13C spectra were recorded using either power-gated 1H decoupling (standard pulse sequence zgpg30) or inverse-gated simultaneous 1H and 2H decoupling (see the Supporting Information for details). NMR data are reported as follows: chemical shift in ppm, multiplicity (ap = apparent, s = singlet, d = doublet, t = triplet, q = quartet, sext = sextet, sp = septet, br = broad, dd = doublet of doublets, td = triplet of doublets, dt = doublet of triplets, and m = multiplet), coupling constant in Hz, integration.
Electrospray ionization (ESI) mass spectrometry (MS) experiments were performed on a QTOF Premier mass spectrometer (Micromass, UK) under normal conditions. Sodium formate solution was used as a calibrant for high-resolution MS (HRMS) measurements. Specific optical rotations were acquired on a Rudolph Autopol IV automatic polarimeter at ambient temperature (20 °C), unless otherwise stated, λ = 589 nm, and concentration (g/100 mL) in the solvent indicated, using a cell of path length of 100 mm.
All reactions were monitored by thin layer chromatography (TLC) using 0.2 μm silica gel (Merck Kieselgel 60 F254)-precoated aluminum plates, using UV light and an ammonium molybdate or a potassium permanganate staining solution to visualize. Silver nitrate-impregnated TLC plates were prepared by dipping silica gel-precoated aluminium plates in a 20% aqueous silver nitrate solution and drying them in an oven at 120 °C overnight.43 Silver nitrate-impregnated silica gel was prepared by dispersing silica gel in a solution of the corresponding amount of silver nitrate in acetonitrile and concentrating this mixture to dryness. Flash column chromatography was performed on Davisil silica gel (60; particle size, 0.040–0.063 mm), or using Reveleris silica or C-18 reversed phase flash cartridges on a Grace Reveleris automated flash system with continuous gradient facility. Solvents for reactions and chromatography were of analytical grade and were used as supplied unless otherwise stated. Unless otherwise stated, petroleum ether refers to the fraction boiling at 60–80 °C.
Hydrogenation and deuteration reactions in an H-Cube Pro apparatus (ThalesNano) were performed at 100% gas production capacity, using Milli-Q H2O or 99.9% D2O, respectively. Chiral and achiral HPLC analyses were performed on an Agilent 1100 (Quaternary pump) HPLC system with a diode array detector (200–400 nm), employing columns, as indicated in the Supporting Information Injection volumes were typically 10 μL (1–2 mg·mL–1), and data were processed with Agilent Cerity System software. Compounds 21a–23a were initially synthesized by employing the procedures reported in ref (35a)(35b), and (36a) with minor modifications. nor-Cholic acid (19) was prepared by the method of Bhattarai et al. with minor modifications.39a
Experimental Procedures
Synthesis of 12β-Methyl-18-nor-bile Acids
Methyl 3α,7α-Diacetyloxy-12β-methyl-18-nor-5β-chol-13(14)-en-24-oate (24a) and Methyl 3α,7α-Diacetyloxy-12β-methyl-18-nor-5β-chol-13(17)-en-24-oate (25a)
A mixture of mesylate 23a (17.1 g, 29.2 mmol) and anhydrous sodium acetate (37.1 g, 452 mmol) in glacial acetic acid (680 mL) was heated at 100 °C for 3 h. After completion of the reaction (TLC analysis), most of the acetic acid was evaporated, and the remainder was diluted with water and extracted with ethyl acetate (3×). The combined organic phases were washed acid free with a saturated bicarbonate solution and brine, dried over MgSO4, and concentrated. The crude product was partially purified by automated column chromatography (silica gel, ethyl acetate/petroleum ether: 1–20%) to afford 12.3 g (86%) of a mixture of rearrangement products as a heavy light-yellow oil [Rf = 0.48 (ethyl acetate/petroleum ether = 3:7)]. An analytical sample of each of the two major rearrangement products, Δ13,14-alkene 24a and Δ13,17-alkene 25a, was obtained by further purification of the partially pure mixture of rearrangement products on 20% silver nitrate-impregnated silica gel by eluting with a 3:7 mixture of ethyl acetate and petroleum ether under gravity. Both compounds were obtained as colorless oils that foamed. In addition, a minor amount of Δ11,12-alkene 26a(36b) was recovered as a colorless oil that solidified on standing [Rf = 0.35 (20% AgNO3-silica gel, ethyl acetate/petroleum ether = 3:7) and Rf = 0.20 (20% AgNO3-silica gel, ethyl acetate/toluene = 1:9)].
Methyl 3α,7α-Diacetyloxy-12β-methyl-18-nor-5β-chol-13(14)-en-24-oate (24a)
Rf = 0.45 (20% AgNO3-silica gel, ethyl acetate/petroleum ether = 3:7), Rf = 0.48 (20% AgNO3-silica gel, ethyl acetate/toluene = 1:9), [α]D20 = +52.6 (c 0.815, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.08 (dd, 5.7, 2.8 Hz, 1H), 4.60 (tt, 11.3, 4.5 Hz, 1H), 3.65 (s, 3H), 2.59–2.54 (m, 1H), 2.45–2.39 (m, 1H), 2.39–2.30 (m, 2H), 2.24–2.11 (m, 2H), 2.10–1.95 (overlapping signals: m, 4H; 2.010, s, 3H and 2.006, s, 3H), 1.95–1.81 (m, 2H), 1.80–1.62 (m, 6H), 1.62–1.56 (m, 1H), 1.55–1.41 (m, 2H), 1.28–1.20 (m, 1H), 1.13 (td, 14.4, 3.4 Hz, 1H), 1.07–0.97 (overlapping signals: m, 1H and 1.05, d, 7.2 Hz, 3H), 0.93 (d, 6.8 Hz, 3H), 0.92 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 174.48, 170.57 (2C), 140.89, 137.74, 74.08, 70.96, 53.47, 51.39, 40.50, 39.44, 35.63, 34.70 (2C), 34.61, 33.51, 33.29, 32.68, 32.60, 31.66, 31.08, 26.89, 25.28, 25.08, 22.27, 21.58, 21.41, 21.13, 18.88; HRMS (ESI) m/z: calcd for C29H44O6Na+, 511.3030; found, 511.3034.
Methyl 3α,7α-Diacetyloxy-12β-methyl-18-nor-5β-chol-13(17)-en-24-oate (25a)
Rf = 0.41 (20% AgNO3-silica gel, ethyl acetate/petroleum ether = 3:7), Rf = 0.34 (20% AgNO3-silica gel, ethyl acetate/toluene = 1:9), [α]D20 = −24.4 (c 0.66, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.08 (dd, 5.9, 3.2 Hz, 1H), 4.61 (tt, 11.4, 4.5 Hz, 1H), 3.66 (s, 3H), 3.05–2.96 (m, 1H), 2.41–2.34 (m, 1H), 2.25–2.13 (m, 5H), 2.08 (dt, 11.6, 13.0 Hz, 1H), 2.05 (s, 3H), 2.04 (s, 3H), 2.05–1.89 (m, 3H), 1.88–1.80 (m, 1H), 1.78–1.71 (m, 1H), 1.69–1.57 (m, 5H), 1.55–1.44 (m, 2H), 1.29–1.18 (overlapping signals: m, 2H and 1.26, d, 6.9 Hz, 3H), 1.10 (td, 14.4, 3.5 Hz, 1H), 0.98 (d, 6.8 Hz, 3H), 0.90–0.81 (overlapping signals: m, 1H and 0.86, s, 3H); 13C NMR (125 MHz, CDCl3): δ 174.27, 170.60, 170.23, 139.21, 135.25, 74.24, 69.71, 51.44, 49.46, 47.38, 40.81, 36.31, 35.81, 34.76, 34.72, 34.61, 34.02, 32.64, 31.42, 31.16, 30.38, 30.15, 26.96, 26.37, 22.82, 21.45 (2C), 20.75, 19.64; HRMS (ESI) m/z: calcd for C29H44O6Na+, 511.3030; found, 511.3028.
3α,7α-Dihydroxy-12β-methyl-18-nor-5β-cholan-24-oic Acid (27a) and 3α,7α-Dihydroxy-12β-methyl-17-epi-18-nor-5β-cholan-24-oic Acid (28a)
To a solution of the partially purified mixture of alkene rearrangement products (1.11 g, 2.27 mmol) in ethanol (15 mL) was added 10% palladium on charcoal (190 mg), and the atmosphere was exchanged for hydrogen. After being stirred under atmospheric pressure and at room temperature overnight (TLC and/or MS analysis), the reaction mixture was filtered through a pad of Celite, and the filtrate was concentrated to give 1.10 g (99%) of a crude mixture of hydrogenation products as a colorless oil.
The crude product was re-dissolved in methanol (25 mL) and 10 M aqueous sodium hydroxide solution (3 mL) was added. The resulting reaction mixture was heated at 80 °C overnight. After being cooled to room temperature, the reaction was concentrated and the residue re-dissolved in water and acidified to pH 1 by the dropwise addition of a 1 M aqueous hydrochloric acid solution. The formed colorless precipitate was extracted with ethyl acetate (3×), and the combined organic phases were washed with brine, dried over MgSO4, and concentrated to give a light-yellow foam. The crude product was adsorbed onto C-18 silica gel and purified by automated flash column chromatography in two separate batches (C18 silica gel, water/methanol = 2–85%) to yield A: 73.7 mg (8.7%) of a mixture of 12β-methyl-18-nor-lithocholic acid derivatives [31/32, Rf = 0.64/0.53 (acetone/dichloromethane/acetic acid = 3:7:0.1), Rf = 0.10 (C18-silica gel, water/methanol = 1:4)], B: 186 mg (21%) of a 3.4:1 mixture of 27a and 30, as determined by 1H NMR analysis, and C: 380 mg (43%) of 28a containing 6% of CDCA (as determined by 1H NMR analysis) and a minor amount of 29. The presence of CDCA results from hydrogenation/saponification of the Δ11,12-unsaturated intermediate 26a, which was formed as a minor side product in the Nametkin rearrangement.
The latter mixture was recrystallized from ethyl acetate/methanol to afford 241 mg (27%) of pure 28a.
Additional purification of mixture B by automated flash column chromatography on normal phase silica gel [acetone (+1% acetic acid)/dichloromethane (+1% acetic acid) 2–20%] gave rise to 111 mg (12%) of pure 27a and 33.4 mg (4%) of 30 with 96% purity, as determined by HPLC analysis.
Employing the same latter chromatographic method for purifying the mother liquor from recrystallization of mixture C, 35.0 mg (4%) of 29 was recovered with 95% purity, as determined by HPLC analysis.
Separation of the mixture of the 12β-methyl-18-nor-lithocholic acid derivatives (31/32, mixture A) was achieved by flash column chromatography on silica gel [acetone/dichloromethane (+1% acetic acid) = 1:49, 1:9]. Compound 31 was obtained as a solid which could be recrystallized from ethyl acetate/petroleum ether to yield colorless needles. Compound 32 was a colorless foam.
3α,7α-Dihydroxy-12β-methyl-18-nor-5β-cholan-24-oic Acid (27a)
Rf = 0.34 (acetone/dichloromethane/acetic acid = 3:7:0.1), Rf = 0.38 (C18-silica gel, water/methanol = 1:4); mp: 182–184 °C (DSC); [α]D20 = +24.2 (c 1.0, MeOH); 1H NMR (500 MHz, CD3OD): δ 3.91 (dd, 5.8, 3.0 Hz, 1H), 3.38 (tt, 11.2, 4.4 Hz, 1H), 2.37 (ddd, 15.3, 8.8, 5.5 Hz, 1H), 2.26 (dt, 11.6, 13.1 Hz, 1H), 2.22–2.14 (m, 1H), 2.03–1.94 (m, 2H), 1.91–1.80 (m, 3H), 1.79–1.71 (m, 2H), 1.67–1.48 (m, 6H), 1.46–1.21 (m, 6H), 1.03–0.91 (overlapping signals: m, 2H; 0.97, d, 6.6 Hz, 3H and 0.93, d, 6.7 Hz, 3H), 0.89 (s, 3H), 0.88–0.78 (m, 2H); 13C NMR (125 MHz, CD3OD): δ 178.06, 72.97, 69.01, 54.48, 50.46, 48.99, 46.96, 43.23, 40.48, 40.00, 37.22, 36.63, 36.49, 36.15, 35.71, 34.32, 33.72, 31.45, 30.00, 26.44, 25.32, 23.58, 21.81, 19.94; HRMS (ESI) m/z: calcd for C24H40O4Na+, 415.2819; found, 415.2822.
3α,7α-Dihydroxy-12β-methyl-17-epi-18-nor-5β-cholan-24-oic Acid (28a)
Rf = 0.25 (acetone/dichloromethane/acetic acid = 3:7:0.1), Rf = 0.25 (C18-silica gel, water/methanol = 1:4); mp: 225–228 °C (DSC); [α]D20 = −36.5 (c 0.625, MeOH); 1H NMR (500 MHz, CD3OD): δ 3.88 (dd, 5.7, 2.9 Hz, 1H), 3.37 (tt, 11.1, 4.5 Hz, 1H), 2.34–2.19 (m, 4H), 1.96 (ddd, 14.7, 5.5, 3.6 Hz, 1H), 1.96–1.87 (m, 3H), 1.84 (sext d, 6.8, 2.5 Hz, 1H), 1.75 (dt, 12.9, 3.6 Hz, 1H), 1.74–1.59 (m, 4H), 1.57–1.31 (m, 7H), 1.10–1.00 (m, 2H), 1.00–0.90 (overlapping signals: m, 2H and 0.97, d, 6.3 Hz, 3H), 0.89 (d, 6.8 Hz, 3H), 0.86 (s, 3H), 0.69 (dt, 11.5, 12.5 Hz, 1H); 13C NMR (125 MHz, CD3OD): δ 177.99, 72.98, 68.78, 57.70, 48.54, 44.55, 43.32, 42.97, 40.40, 37.06, 36.62, 36.09, 35.70, 34.23, 33.57, 33.37, 33.23, 32.99, 31.49, 30.38, 23.69, 23.65, 21.77, 17.21; HRMS (ESI) m/z: calcd for C24H40O4Na+, 415.2819; found, 415.2824.
3α,7α-Dihydroxy-12β-methyl-14-epi-18-nor-5β-cholan-24-oic Acid (29)
Rf = 0.29 (acetone/dichloromethane/acetic acid = 3:7:0.1); 1H NMR (500 MHz, CD3OD): δ 3.89 (dd, 5.7, 2.9 Hz, 1H), 3.36 (tt, 11.2, 4.3 Hz, 1H), 2.35 (ddd, 15.3, 9.4, 5.2 Hz, 1H), 2.29 (dt, 11.6, 13.4 Hz, 1H), 2.24–2.18 (m, 1H), 2.08 (td, 12.1, 3.2 Hz, 1H), 2.04–1.97 (m, 2H), 1.97–1.74 (m, 5H), 1.70–1.53 (m, 5H), 1.49 (dt, 14.4, 2.2 Hz, 1H), 1.46–1.25 (m, 7H), 0.99 (td, 14.2, 3.4 Hz, 1H), 0.90 (d, 6.4 Hz, 3H), 0.88–0.84 (overlapping signals: d, 3H and 0.86, s, 3H), 0.72 (ap q, 12.2 Hz, 1H); 13C NMR (125 MHz, CD3OD): δ 178.11, 73.12, 71.94, 53.11, 47.89, 46.23, 43.53, 42.99, 40.37, 38.87, 36.76, 36.70, 36.25, 36.11, 34.05, 33.14, 31.79, 30.84, 29.91, 29.52, 28.63, 24.09, 21.21, 18.22; HRMS (ESI) m/z: calcd for C24H40O4Na+, 415.2819; found, 415.2821.
3α,7α-Dihydroxy-14(13 → 12)abeo-13-epi-5β-cholan-24-oic Acid (30)
mp: 181–183 °C (DSC); [α]D20 = −3.4 (c 0.475, MeOH); 1H NMR (500 MHz, CD3OD): δ 4.00 (dd, 5.6, 2.8 Hz, 1H), 3.35 (tt, 11.3, 4.2 Hz, 1H), 2.36 (ddd, 15.5, 8.9, 5.4 Hz, 1H), 2.25 (dt, 12.0, 10.0 Hz, 1H), 2.21–2.10 (m, 2H), 1.94–1.35 (m, 16H), 1.21–1.04 (m, 4H), 1.00–0.93 (m, 2H), 0.92 (d, 6.8 Hz, 3H), 0.87 (s, 3H), 0.85 (d, 6.2 Hz, 3H); 13C NMR (125 MHz, CD3OD): δ 178.16, 72.74, 67.47, 50.69, 45.57, 45.02, 43.04, 40.59, 39.80, 39.13, 37.65, 36.98, 36.71, 35.96, 33.73, 33.40, 31.75, 30.96, 26.52, 26.10, 22.45, 21.92, 18.57, 17.71; HRMS (ESI) m/z: calcd for C24H40O4Na+, 415.2819; found, 415.2818.
3α-Hydroxy-12β-methyl-18-nor-5β-cholan-24-oic Acid (31)
mp: 158–161 °C (DSC); [α]D20 = +50.5 (c 0.645, MeOH); 1H NMR (500 MHz, CD3OD): δ 3.57 (tt, 11.1, 4.7 Hz, 1H), 2.40 (ddd, 15.2, 8.9, 5.5, 1H), 2.25–2.16 (m, 1H), 1.98–1.72 (m, 7H), 1.70–1.47 (m, 7H), 1.45–1.17 (m, 7H), 1.08–0.98 (overlapping signals: m, 2H and 1.00, d, 6.6 Hz, 3H), 0.98–0.92 (overlapping signals: m, 1H; 0.95, s, 3H and d, 3H), 0.91–0.80 (m, 2H); 13C NMR (125 MHz, CD3OD): δ 178.00, 72.53, 55.24, 54.58, 50.56, 43.62, 43.37, 41.30, 40.38, 37.22, 37.21, 36.57, 36.44, 35.67, 33.67, 31.32, 30.47, 28.26, 27.81, 26.38, 25.35, 24.03, 21.86, 19.91; HRMS (ESI) m/z: calcd for C24H40O3Na+, 399.2870; found, 399.2875.
Isomer of 3α-Hydroxy-12β-methyl-18-nor-5β-cholan-24-oic Acid (32)
[α]D20 = +42.6 (c 0.62, MeOH); 1H NMR (500 MHz, CD3OD): δ 3.57 (tt, 11.0, 4.6 Hz, 1H), 2.39 (ddd, 15.4, 9.3, 5.0 Hz, 1H), 2.30–2.20 (m, 1H), 2.00–1.16 (m, 24H), 1.03 (td, 14.2, 3.6 Hz, 1H), 0.93 (d, 6.2 Hz, 3H), 0.92–0.89 (overlapping signals: 0.91, s, 3H and d, 3H), 0.69 (dt, 11.8, 12.3 Hz, 1H); 13C NMR (125 MHz, CD3OD): δ 178.08, 72.58, 51.46, 49.76, 47.11, 43.76, 39.05, 38.70, 37.16, 36.21, 36.12, 35.61, 35.54, 34.16, 33.17, 31.58, 30.69, 29.11, 28.58, 27.92, 26.04, 23.82, 21.26, 18.17; HRMS (ESI) m/z: calcd for C24H40O3Na+, 399.2870; found, 399.2878.
Synthesis of 12β-Methyl-18,24-bisnor-chenodeoxycholic Acids (27b and 28b)
Methyl 3α,7α,12α-Trihydroxy-24-nor-5β-cholan-23-oate (21b)
24-nor-Cholic acid 19 (3.48 g, 8.82 mmol) was dissolved in dry methanol (120 mL) and a catalytic amount of para-toluenesulfonic acid monohydrate (0.15 g, 0.83 mmol) was added. The reaction was stirred under an atmosphere of argon for 3 d. Solid sodium carbonate was added to the reaction mixture before concentrating to dryness. The crude was purified by automated column chromatography on silica gel eluting with a gradient of chloroform/methanol (0–50%) to yield 3.60 g (83%) of the corresponding methyl ester 21b. The recorded spectral data of 21b were consistent with those reported in the literature.39a
Methyl 3α,7α-Diacetyloxy-12α-hydroxy-24-nor-5β-cholan-23-oate (22b)
To a solution of methyl 24-nor-cholate 21b (5.00 g, 12.2 mmol) in dichloromethane (50 mL) was added 4-dimethylaminopyridine (DMAP; 0.15 g, 1.2 mmol) and triethylamine (6.86 mL, 49.2 mmol). The solution was cooled to 0 °C before acetic anhydride (2.39 mL, 0.25 mmol) was added dropwise. After 2 h, at 0 °C, water (10 mL) was added to the reaction mixture followed by 1 M aqueous hydrochloric acid (100 mL). The organic phase was separated, washed with 1 M aqueous hydrochloric acid (50 mL) and brine (50 mL), dried over MgSO4, and concentrated. The crude product was purified by automated flash column chromatography (silica gel, ethyl acetate/toluene = 10–35%) to give 5.21 g (86%) of 22b as a colorless foam. [α]D20 = +35.0 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 4.91–4.88 (m, 1H), 4.58 (tt, 11.4, 4.5 Hz, 1H), 4.02–3.99 (m, 1H), 3.66 (s, 3H), 2.43 (dd, 14.8, 3.5 Hz, 1H), 2.23 (td, 11.8, 5.4 Hz, 1H), 2.13–2.01 (overlapping signals: m, 2H), 2.06 (s, 3H), 2.02 (s, 3H), 1.98–1.68 (m, 7H), 1.66–1.36 (m, 9H), 1.33–1.23 (m, 1H), 1.19–1.01 (overlapping signals: m, 2H), 1.05 (d, 6.4 Hz, 3H), 0.93 (s, 3H), 0.73 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 173.92, 170.70, 170.54, 74.16, 72.60, 70.92, 51.46, 47.27, 46.75, 42.22, 41.27, 41.04, 38.19, 34.86, 34.62, 34.45, 33.49, 31.39, 28.74, 28.23, 27.48, 26.75, 23.05, 22.62, 21.70, 21.52, 18.66, 12.61; HRMS (ESI) m/z: calcd for C28H44O7Na+, 515.2979; found, 515.2980.
Methyl 3α,7α-Diacetyloxy-12α-chloromethylsulfonyl-24-nor-5β-cholan-23-oate (23b)
To a solution of 22b (6.72 g, 11.1 mmol) in dry pyridine (110 mL) was dropwise added chloromethylsulfonyl chloride (1.51 mL) at 0 °C. After 45 min, water (100 mL) and diethyl ether (100 mL) were added before the mixture was transferred to a separating funnel. More water and diethyl ether were added until there were two phases. The aqueous phase was separated and extracted with diethyl ether (3 × 50 mL). The combined organics were washed sequentially with water (2 × 50 mL), 1 M aqueous hydrochloric acid (2 × 50 mL), and brine, dried over MgSO4, and concentrated. The crude product was used in the next step without further purification. 1H NMR (500 MHz, CDCl3): δ 5.24–5.22 (m, 1H), 4.93–4.90 (m, 1H), 4.69 (d, 12.6 Hz, 1H), 4.64 (d, 12.6 Hz, 1H), 4.57 (tt, 11.3, 4.3 Hz, 1H), 3.66 (s, 3H), 2.40 (dd, 15.0, 3.1 Hz, 1H), 2.30 (td, 12.4, 4.3 Hz, 1H), 2.14 (dt, 15.3, 3.8 Hz, 1H), 2.11–2.00 (overlapping signals: m, 2H; 2.07, s, 3H and 2.03, s, 3H), 1.99–1.77 (m, 6H), 1.75–1.43 (m, 8H), 1.36–1.27 (m, 1H), 1.16–1.06 (overlapping signals: m, 2H), 1.09 (d, 6.4 Hz, 3H), 0.94 (s, 3H), 0.82 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 173.64, 170.78, 170.37, 86.84, 74.00, 70.55, 54.78, 51.52, 46.62, 46.44, 42.79, 40.99 (2C), 37.94, 34.90 (2C), 34.61, 33.91, 31.39, 28.48, 27.69, 27.03, 26.74, 23.03, 22.62, 21.64, 21.56, 18.80, 12.32; HRMS (ESI) m/z: calcd for C29H45ClO9SNa+, 627.2365; found, 627.2359.
Methyl 3α,7α-Diacetyloxy-12β-methyl-18,24-bisnor-5β-chol-13(14)-en-23-oate (24b) and Methyl 3α,7α-Diacetyloxy-12β-methyl-18,24-bisnor-5β-chol-13(17)-en-23-oate (25b)
To a solution of crude monochlate 23b (11.1 mmol) in acetic acid (74 mL) was added zinc acetate (10 equiv, 20.4 g) before the mixture was heated to 100 °C. After 1.5 h, the reaction was cooled to room temperature, filtered, and the filtrate concentrated in vacuo. The crude reaction product was purified by automated column chromatography eluting with a gradient of 5–30% ethyl acetate in petroleum ether to afford a partially pure mixture of alkene products which was one spot by silica gel TLC analysis (Rf = 0.32, eluent: 30% ethyl acetate in petroleum ether). Subsequently, the partially pure alkene mixture was separated by column chromatography on 20% silver-impregnated silica gel, eluting with 8% ethyl acetate in toluene to afford three main fractions—in the order of elution: (i) a mixture of Δ13,14 alkene 24b and an unknown reaction product with the same mass (2.76 g, 52%), (ii) pure Δ13,14 alkene 24b (260 mg, 5%), and (iii) Δ13,17 alkene 25b (1.44 g, 27%).
Methyl 3α,7α-Diacetyloxy-12β-methyl-18,24-bisnor-5β-chol-13(14)-en-23-oate (24b)
1H NMR (500 MHz, CDCl3): δ 5.08 (dd, 5.6, 2.8 Hz, 1H), 4.60 (tt, 11.3, 4.6 Hz, 1H), 3.65 (s, 3H), 2.63–2.58 (m, 1H), 2.57–2.48 (m, 1H), 2.41–2.34 (m, 2H), 2.29 (dd, 14.8, 3.2 Hz, 1H), 2.24–2.17 (m, 1H), 2.09–1.84 (overlapping signals: m, 6H; 2.012, s, 3H; 2.007, s, 3H), 1.78–1.56 (m, 6H), 1.54–1.41 (m, 2H), 1.17–1.09 (overlapping signals: m, 1H and 1.13, d, 6.9 Hz, 3H), 1.05–0.95 (overlapping signals: m, 1H and 0.98, d, 6.8 Hz, 3H), 0.91 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 174.49, 170.54 (2C), 141.06, 138.02, 74.04, 70.87, 52.47, 51.36, 40.48, 39.47, 35.40, 34.71 (2C), 34.63, 33.46 (2C), 33.19, 32.59, 31.51, 31.10, 26.89, 24.87, 22.19, 21.57, 21.41, 21.01, 19.57; HRMS (ESI) m/z: calcd for C28H42O6Na+, 497.2874; found, 497.2870.
Methyl 3α,7α-Diacetyloxy-12β-methyl-18,24-bisnor-5β-chol-13(17)-en-23-oate (25b)
[α]D20 = −45.3 (c 0.64, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.10–5.06 (m, 1H), 4.61 (tt, 11.4, 4.5 Hz, 1H), 3.63 (s, 3H), 3.60–3.54 (m, 1H), 2.40–2.22 (m, 5H), 2.20–2.13 (m, 1H), 2.12–1.89 (overlapping signals: m, 4H; 2.044, s, 3H and 2.035, s, 3H), 1.88–1.80 (m, 1H), 1.78–1.71 (m, 1H), 1.66–1.57 (m, 3H), 1.55–1.43 (m, 2H), 1.35 (d, 6.9 Hz, 3H), 1.30–1.22 (m, 1H), 1.19 (td, 11.2, 3.3 Hz, 1H), 1.09 (td, 14.4, 3.4 Hz, 1H), 1.02 (d, 6.9 Hz, 3H), 0.89–0.80 (overlapping signals: m, 1H and 0.84, s, 3H); 13C NMR (125 MHz, CDCl3): δ 173.04, 170.46, 170.09, 138.91, 134.40, 74.17, 69.62, 51.25, 49.35, 47.22, 40.83, 40.78, 36.16, 35.81, 34.75, 34.69, 34.57, 33.96, 31.38, 30.55, 29.34, 26.92, 26.24, 22.78, 21.40 (2C), 20.29, 19.48; HRMS (ESI) m/z: calcd for C28H42O6Na+, 497.2874; found, 497.2885.
3α,7α-Dihydroxy-12β-methyl-18,24-bisnor-5β-cholan-23-oic Acid (27b) and 3α,7α-Dihydroxy-12β-methyl-17-epi-18,24-bisnor-5β-cholan-23-oic Acid (28b)
To a solution of Δ13,17 alkene 25b (671 mg, 1.41 mmol) in ethyl acetate (5 mL) and ethanol (10 mL) was added 10% palladium on charcoal (150 mg), and the mixture was placed under an atmosphere of hydrogen. After 24 h, the catalyst loading was increased by 80 mg and the hydrogenation continued for another 16 h. The mixture was filtered through Celite, and the filtrate was concentrated and purified by automated column chromatography on silica gel, eluting with a gradient of 5–20% ethyl acetate in toluene, to afford 600 mg (89%) of a mixture of saturated products (Rf = 0.58, eluent: 10% ethyl acetate in toluene). This mixture was carried to the next step without separation of isomers.
Employing the same procedure as for the preparation of 27a/28a, the mixture of hydrogenation products (600 mg, 1.26 mmol) in methanol (7 mL) was saponified in the presence of sodium hydroxide solution to yield a residue of unprotected 12β-methyl-18,24-bisnor bile acids. Subsequent purification of this mixture by automated column chromatography (silica gel; acetone in dichloromethane, 0–90%) gave 87 mg of 27b (Rf = 0.33, eluent: 30% acetone in dichloromethane), 141 mg of 28b (Rf = 0.10, eluent: 30% acetone in dichloromethane), and a mixture of the two products (153 mg). Combined yield over two steps: 80%.
3α,7α-Dihydroxy-12β-methyl-18,24-bisnor-5β-cholan-23-oic Acid (27b)
[α]D20 = +9.4 (c 0.5, MeOH); 1H NMR (500 MHz, CD3OD): δ 3.92–3.88 (m, 1H), 3.38 (tt, 11.2, 4.5 Hz, 1H), 2.40 (dd, 14.5, 3.1 Hz, 1H), 2.37–2.30 (m, 1H), 2.26 (dt, 11.6, 13.2 Hz, 1H), 2.03–1.76 (m, 6H), 1.69–1.56 (m, 4H), 1.53 (dt, 14.7, 1.9 Hz, 1H), 1.49–1.25 (m, 6H), 1.02 (d, 6.6 Hz, 3H), 1.00–0.90 (overlapping signals: m, 2H and 0.98, d, 6.8 Hz, 3H), 0.89 (s, 3H), 0.82 (ap q, 12.3 Hz, 1H), 0.72 (ap q, 10.2 Hz, 1H); 13C NMR (125 MHz, CD3OD): δ 178.14, 72.95, 68.94, 54.96, 49.71, 48.90, 46.93, 43.20, 40.47, 39.83, 37.20, 36.62, 36.51, 36.14, 35.70, 34.29 (2C), 31.44, 29.80, 25.42, 23.58, 21.76, 20.85; HRMS (ESI) m/z: calcd for C23H38O4Na+, 401.2662; found, 401.2666.
3α,7α-Dihydroxy-12β-methyl-17-epi-18,24-bisnor-5β-cholan-23-oic Acid (28b)
[α]D20 = −41.1 (c 1.005, MeOH); 1H NMR (500 MHz, CD3OD): δ 3.88 (dd, 5.9, 3.2 Hz, 1H), 3.37 (tt, 11.2, 4.4 Hz, 1H), 2.40 (sext d, 7.0, 2.4 Hz, 1H), 2.30–2.18 (m, 2H), 2.15 (dd, 14.6, 7.6 Hz, 1H), 2.08 (dd, 14.6, 7.4 Hz, 1H), 1.99–1.86 (m, 4H), 1.80–1.60 (m, 5H), 1.59–1.48 (m, 3H), 1.43–1.27 (m, 2H), 1.10–0.90 (overlapping signals: m, 4H), 1.02 (d, 6.2 Hz, 3H), 0.93 (d, 6.8 Hz, 3H), 0.86 (s, 3H), 0.69 (q, 12.1 Hz, 1H); 13C NMR (125 MHz, CD3OD): δ 177.16, 72.89, 68.68, 57.55, 48.42, 44.35, 43.23, 43.06, 42.96, 40.31, 36.99, 36.57, 36.03, 35.63, 34.17, 33.35, 31.43, 31.09, 30.25, 23.69, 23.63, 21.65, 17.36; HRMS (ESI) m/z: calcd for C23H38O4Na+, 401.2662; found, 401.2665.
Synthesis of 12β-Methyl-25-homo-18-nor-chenodeoxycholic Acids (27c and 28c)
Methyl 3α,7α,12α-Trihydroxy-25-homo-5β-cholan-25-oate (21c)40b
A solution of 25-homo-cholic acid (20; 6.39 g, 15.1 mmol) and a catalytic amount of p-toluenesulfonic acid monohydrate (200 mg, 1.05 mmol) in dry methanol (90 mL) were heated under reflux overnight. After being cooled to room temperature, the reaction was diluted with ethyl acetate before being washed with saturated sodium bicarbonate solution and brine. The organic phase was separated and dried over MgSO4 to yield a colorless solid, which was subsequently triturated with a 20% mixture of ethyl acetate in hexane and recovered by filtration to afford 5.52 g (84%) of 21c as a colorless amorphous solid. [α]D20 = +23.9 (c 0.585, CHCl3); 1H NMR (500 MHz, CD3OD): δ 3.97–3.93 (m, 1H), 3.81–3.77 (m, 1H), 3.65 (s, 3H), 3.37 (tt, 11.1, 4.4 Hz, 1H), 2.34–2.21 (m, 4H), 2.01–1.92 (m, 2H), 1.91–1.84 (m, 2H), 1.81 (dt, 14.3, 3.2 Hz, 1H), 1.77–1.68 (m, 2H), 1.68–1.62 (m, 1H), 1.62–1.47 (m, 6H), 1.47–1.34 (m, 4H), 1.30–1.20 (m, 1H), 1.16–1.06 (m, 2H), 1.04–0.94 overlapping signals (m, 1H and 1.02, d, 6.6 Hz, 3H), 0.92 (s, 3H), 0.71 (s, 3H); 13C NMR (125 MHz, CD3OD): δ 176.13, 74.11, 72.95, 69.13, 51.97, 48.22, 47.51, 43.27, 43.04, 41.10, 40.54, 37.02, 36.60, 36.56, 35.96, 35.91, 35.29, 31.25, 29.65, 28.79, 27.95, 24.27, 23.21, 22.82, 17.96, 13.02; HRMS (ESI) m/z: calcd for C26H44O5Na+, 459.3081; found, 459.3059.
Methyl 3α,7α-Diacetyloxy-12α-hydroxy-25-homo-5β-cholan-25-oate (22c)44
To a solution of methyl 25-homo-cholate (21c, 5.52 g, 12.6 mmol), dry triethylamine (5.3 mL, 38.0 mmol), and a catalytic amount of 4-dimethylaminopyridine (DMAP; 154 mg, 1.26 mmol) in dry dichloromethane (60 mL) was dropwise added acetic anhydride (2.6 mL, 27.5 mmol) at room temperature over the course of 2 h. After being stirred for 4 h, the reaction was diluted with water and extracted. The organic phases were combined, dried over MgSO4, and concentrated. The crude product was triturated with a 20% mixture of ethyl acetate in hexane to yield 5.00 g (76%) of 22c as a colorless, amorphous solid. [α]D20 = +32.7 (c 0.575, CHCl3); 1H NMR (500 MHz, CDCl3): δ 4.91–4.87 (m, 1H), 4.58 (tt, 11.3, 4.5 Hz, 1H), 4.01 (t, 2.7 Hz, 1H), 3.67 (s, 3H), 2.34–2.16 (m, 3H), 2.11–2.05 (m, 1H), 2.06 (s, 3H), 2.02 (s, 3H), 1.94 (ddd, 15.5, 5.4, 3.7 Hz, 1H), 1.90–1.79 (m, 3H), 1.77–1.66 (m, 4H), 1.65–1.54 (m, 7H), 1.54–1.45 (m, 4H), 1.28–1.19 (m, 1H), 1.16–1.03 (m, 3H), 0.99 (d, 6.6 Hz, 3H), 0.92 (s, 3H), 0.68 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 174.24, 170.65, 170.50, 74.10, 72.79, 70.88, 51.44, 47.32, 46.56, 42.10, 40.97, 38.12, 35.25, 35.14, 34.78, 34.55, 34.44, 34.38, 31.31, 28.49, 28.16, 27.33, 26.68, 22.96, 22.53, 21.63, 21.51, 21.45, 17.67, 12.50; HRMS (ESI) m/z: calcd for C30H48O7Na+, 543.3292; found, 543.3288.
Methyl 3α,7α-Diacetyloxy-12α-mesyloxy-25-homo-5β-cholan-25-oate (23c)
To a solution of diacetate methyl ester 22c (6.45 g, 12.4 mmol) in dry pyridine (120 mL) was added methanesulfonyl chloride (1.34 mL, 17.3 mmol) at 0 °C, and the resulting mixture was stirred at room temperature overnight. The reaction was quenched in ice and extracted with ethyl acetate (3×). The combined organic phases were washed with brine, dried over MgSO4, and concentrated. The crude product was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 2:3) to yield 7.17 g (97%) of the corresponding mesylate 23c as a colorless oil that solidified on standing. An analytical sample was re-crystallized from ethyl acetate/petroleum ether to yield colorless prisms. mp: 157–160 °C; [α]D20 = +58.2 (c 0.655, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.12–5.09 (m, 1H), 4.92–4.89 (m, 1H), 4.57 (tt, 11.4, 4.4 Hz, 1H), 3.66 (s, 3H), 3.08 (s, 3H), 2.34–2.21 (m, 3H), 2.12–2.04 (m, 1H), 2.09 (s, 3H), 2.05–2.00 (m, 1H), 2.03 (s, 3H), 1.95 (ddd, 15.5, 5.4, 3.6 Hz, 1H), 1.91–1.79 (m, 3H), 1.77–1.67 (m, 3H), 1.67–1.57 (m, 4H), 1.57–1.47 (m, 3H), 1.47–1.35 (m, 3H), 1.31–1.22 (m, 1H), 1.16–1.05 (m, 3H), 1.01 (d, 6.6 Hz, 3H), 0.93 (s, 3H), 0.76 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 174.15, 170.68, 170.37, 84.61, 74.02, 70.53, 51.45, 46.98, 46.01, 42.97, 40.96, 39.67, 37.80, 35.32, 35.19, 34.81, 34.77, 34.40, 34.34, 31.27, 28.53, 27.34, 27.07, 26.67, 22.82, 22.62, 21.62, 21.48, 21.37, 18.08, 12.25; HRMS (ESI) m/z: calcd for C31H50O9SNa+, 621.3068; found, 621.3065.
Methyl 3α,7α-Diacetyloxy-12β-methyl-25-homo-18-nor-5β-chol-13(14)-en-25-oate (24c) and Methyl 3α,7α-Diacetyloxy-12β-methyl-25-homo-18-nor-5β-chol-13(17)-en-25-oate (25c)
Using the same procedure as described for the preparation of 24a/25a, a mixture of mesylate 23c (4.57 g, 7.63 mmol) and anhydrous sodium acetate (4.57 g, 55.7 mmol) in glacial acetic acid (180 mL) was heated at 100 °C for 2.5 h. The crude mixture of alkenes (light tan foam) was partially purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 5–10%) to afford 3.80 g (99%) of a crude mixture of alkenes. This mixture was then separated by gravity column chromatography on 20% silver nitrate-impregnated silica gel eluting with a mixture of 5% ethyl acetate in toluene to yield 1.33 g (35%) of Δ13,14-alkene 24c as a colorless oil, 1.27 g (33%) of Δ13,17-alkene 25c as a colorless oil that solidified on standing, and 561 mg (15%) of mixed fractions.
Methyl 3α,7α-Diacetyloxy-12β-methyl-25-homo-18-nor-5β-chol-13(14)-en-25-oate (24c)
[α]D20 = +43.6 (c 1.49, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.08 (dd, 5.7, 2.8 Hz, 1H), 4.60 (tt, 11.3, 4.5 Hz, 1H), 3.65 (s, 3H), 2.58–2.53 (m, 1H), 2.42–2.16 (m, 5H), 2.10–1.95 (overlapping signals: m, 4H; 2.010, s, 3H and 2.005, s, 3H), 1.95–1.89 (m, 1H), 1.89–1.80 (m, 1H), 1.78–1.56 (m, 7H), 1.55–1.36 (m, 3H), 1.32–1.23 (m, 1H), 1.13 (td, 14.4, 3.5 Hz, 1H), 1.06 (d, 7.1 Hz, 3H), 1.03–0.89 (overlapping signals: 0.99, dt, 11.5, 12.2 Hz, 1H; m, 1H; 0.93, d, 6.8 Hz, 3H and 0.92, s, 3H); 13C NMR (125 MHz, CDCl3): δ 174.17, 170.58, 170.56, 141.12, 137.45, 74.07, 70.93, 53.61, 51.36, 40.50, 39.47, 35.84, 34.69 (2C), 34.60, 34.52, 33.48, 33.40, 32.56, 31.67, 31.09, 29.35, 26.88, 25.04, 23.54, 22.21, 21.57, 21.40, 21.32, 19.12; HRMS (ESI) m/z: calcd for C30H46O6Na+, 525.3187; found, 525.3188.
Methyl 3α,7α-Diacetyloxy-12β-methyl-25-homo-18-nor-5β-chol-13(17)-en-25-oate (25c)
[α]D20 = −25.2 (c 1.055, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.11–5.06 (m, 1H), 4.61 (tt, 11.2, 4.4 Hz, 1H), 3.65 (s, 3H), 3.02–2.93 (m, 1H), 2.41–2.32 (m, 1H), 2.29 (t, 7.6 Hz, 2H), 2.24–2.13 (m, 3H), 2.13–1.89 (overlapping signals: m, 4H; 2.04, s, 3H and 2.03, s, 3H), 1.87–1.78 (m, 1H), 1.78–1.71 (m, 1H), 1.66–1.43 (m, 7H), 1.38–1.19 (overlapping signals: m, 4H and 1.30, d, 6.8 Hz, 3H), 1.09 (td, 14.3, 3.2 Hz, 1H), 0.95 (d, 6.8 Hz, 3H), 0.92–0.81 (overlapping signals: m, 1H and 0.85, s, 3H); 13C NMR (125 MHz, CDCl3): δ 174.14, 170.59, 170.23, 138.28, 136.09, 74.26, 69.76, 51.40, 49.47, 47.19, 40.82, 36.36, 35.81, 35.07, 34.76, 34.72, 34.60, 34.26, 34.02, 31.50, 31.42, 30.24, 26.96, 26.27, 23.53, 22.79, 21.45 (2C), 20.86, 19.79; HRMS (ESI) m/z: calcd for C30H46O6Na+, 525.3187; found, 525.3184.
3α,7α-Dihydroxy-12β-methyl-25-homo-18-nor-5β-cholan-25-oic Acid (27c) and 3α,7α-Dihydroxy-12β-methyl-17-epi-25-homo-18-nor-5β-cholan-25-oic Acid (28c)
Employing the same procedure as outlined for the preparation of 27a/28a, a solution of Δ13,17-olefin 27b (881 mg, 1.75 mmol) in ethanol (60 mL) was hydrogenated in the presence of 10% palladium on charcoal (290 mg) at room temperature for 72 h. The crude residue was re-dissolved in methanol (45 mL) and treated with a 10 M aqueous sodium hydroxide solution (8 mL) under gentle reflux overnight. The crude product was purified by automated flash column chromatography in two separate batches (C18 silica gel, water/methanol = 2–85%) to yield 147 mg (21%) of 27c and 373 mg (52%) of 17-epi-isomer 28c, both as colorless solids, which were re-crystallized from ethyl acetate/petroleum ether.
3α,7α-Dihydroxy-12β-methyl-25-homo-18-nor-5β-cholan-25-oic Acid (27c)
[α]D20 = +25.7 (c 0.435, MeOH); [α]D = +29.6 (c 0.365, EtOH); 1H NMR (500 MHz, CD3OD): δ 3.92–3.88 (m, 1H), 3.38 (tt, 11.1, 4.5 Hz, 1H), 2.34–2.21 (m, 3H), 2.02–1.93 (m, 2H), 1.88 (dt, 14.2, 3.1 Hz, 1H), 1.86–1.79 (m, 1H), 1.79–1.69 (m, 3H), 1.68–1.59 (m, 3H), 1.59–1.44 (m, 5H), 1.44–1.24 (m, 5H), 1.09–0.95 (overlapping signals: m, 2H and 0.98, d, 6.6 Hz, 3H), 0.95–0.86 (overlapping signals: m, 1H; 0.93, d, 6.7 Hz, 3H and 0.88, s, 3H), 0.86–0.75 (m, 2H); 13C NMR (125 MHz, CD3OD): δ 177.90, 72.97, 68.99, 54.55, 50.73, 48.96, 46.98, 43.23, 40.48, 39.97, 37.28, 36.63 (2C), 36.14, 35.71, 35.48, 34.31, 31.45, 30.40, 29.89, 25.37, 24.83, 23.59, 21.96, 20.31; HRMS (ESI) m/z: calcd for C25H42O4Na+, 429.2975; found, 429.2979.
3α,7α-Dihydroxy-12β-methyl-17-epi-25-homo-18-nor-5β-cholan-25-oic Acid (28c)
mp: 174–176 °C; [α]D20 = −40.6 (c 0.45, MeOH); 1H NMR (500 MHz, CD3OD): δ 3.91 (dd, 5.8, 3.0 Hz, 1H), 3.41 (tt, 11.2, 4.5 Hz, 1H), 2.33–2.22 (overlapping signals: m, 2H and 2.29, t, 7.4 Hz, 2H), 2.02–1.90 (m, 4H), 1.86 (sext d, 6.9, 2.6 Hz, 1H), 1.82–1.72 (m, 2H), 1.72–1.51 (m, 8H), 1.46–1.35 (m, 2H), 1.31–1.14 (m, 2H), 1.12–0.93 (overlapping signals: m, 4H and 1.02, d, 6.2 Hz, 3H), 0.91 (d, 6.8 Hz, 3H), 0.90 (s, 3H), 0.72 (dt, 11.5, 12.5 Hz, 1H); 13C NMR (125 MHz, CD3OD): δ 177.69, 72.98, 68.79, 57.75, 48.54, 44.56, 43.33, 43.13, 40.40, 37.98, 37.07, 36.63, 36.09, 35.71, 35.28, 34.24, 33.39, 33.18, 31.50, 30.38, 24.21, 23.71 (2C), 21.83, 17.47; HRMS (ESI) m/z: calcd for C25H42O4Na+, 429.2975; found, 429.2980.
Procedures for Obtaining 12β-Methyl-18-nor-bile Acids 27a and 28a following the Rearrangement Reaction (Step d; Scheme 2 or 5); Beginning from 250 g of CA
Partial Purification and Hydrogenation of a Mixture of Protected Alkenes (24a/25a/Other)
Prior to hydrogenation, the crude mixture of protected alkenes containing 24a and 25a, obtained from the rearrangement reaction (step d; Scheme 2 or 5), was partially purified by chromatography on silica gel (eluting with 5 to 25% ethyl acetate/petroleum ether mixtures) to remove head and tail impurities containing an impurity that inhibits direct hydrogenation of the crude mixture. The thus-obtained mixture (173 g) was dissolved in ethanol (750 mL) before 100 mL of ethanol was evaporated under reduced pressure using a rotary evaporator (water bath at 50 °C). The solution was transferred into a 3.4 L stainless-steel pressure vessel fitted to an HEL Simular reaction calorimeter to allow purging and gassing of the system. The vessel was fitted with a 45 mm diameter gassing impeller and stirred at 600 rpm and held at 20 °C through control of the external jacket temperature. After being degassed, 10% palladium on charcoal (5 g) was added under an inert atmosphere. The solution was degassed before hydrogen (g) was introduced at 5 bar. After 16 h, the mixture was carefully degassed, purged with nitrogen, and further 10% palladium on charcoal (7 g) was added. The solution was degassed before hydrogen (g) was introduced at 5 bar. The reaction mixture was left stirring for an additional 48 h before being filtered through a pad of Celite and concentrated.
Hydrolysis of the Saturated Alkene Mixture
The crude thus obtained was dissolved in methanol (300 mL) before a solution of sodium hydroxide (123 g, 3 mol) in water (300 mL) was added. The mixture was heated at 80 °C for 72 h. Methanol was removed by concentration in vacuo. Water (1 L) was added to the crude, which was then cooled in an ice bath. Concentrated hydrochloric acid (270 mL) was added dropwise to the crude, forming a gum. To the suspension containing the gum was added an excess of ethyl acetate, and the gum was vigorously agitated until a white powder precipitated. The suspension was transferred to a separating funnel and after standing the acidic aqueous layer was removed. The suspension in ethyl acetate was washed with water and the aqueous layer was removed. The suspension in ethyl acetate was filtered to yield a colorless powder (∼100 g; fraction A), and the filtrate was concentrated to give a brown oil (∼50 g; fraction B).
Fractional Crystallization
Fraction B (∼50 g) could be fractionally crystallized as follows: methanol (ca. 300 mL) was added, and near complete dissolution was achieved (a fine white precipitate remained). This suspension was concentrated to approximately half the volume before ethyl acetate (500 mL) was added. This suspension was heated to reflux for 1 h before cooling it to r.t. and ageing overnight. The precipitate was filtered, affording compound 28a (8 g, >99% pure).
Briefly, fraction A (∼100 g) could be fractionally crystallized by refluxing as a suspension in ethyl acetate (for 1 h), cooling, ageing, and filtering. The concentration of the filtrate gave fraction C (ca. 13.5 g), while the precipitate—fraction D (ca. 85 g)—was collected separately.
Ethyl acetate and water were added to fraction C before the aqueous phase was discarded. The concentration of the resultant ethyl acetate suspension to dryness (at 60 °C, in vacuo), followed by the addition of fresh ethyl acetate, heating to reflux (1 h), and cooling, led to the precipitation of pure compound 27a (5.7 g).
Fraction D (ca. 85 g) was boiled in ethyl acetate (1 h) before the suspension was concentrated to dryness (at 60 °C, in vacuo). The resulting precipitate (63 g) was dissolved in hot ethanol (750 mL total), filtered, and concentrated. Ethyl acetate and methanol were used to dissolve this material, which was then slowly concentrated (at 60 °C, in vacuo) to diminish the amount of methanol in the mixture. By this process and by seeding with powdered compound 28a that had been isolated from fraction B (above), it was possible to precipitate pure compound 28a (24.7 g). This process was repeated until the amount of compound 28a in the remaining mixture had diminished to the point that the next material to precipitate preferentially (under the same conditions) was alkene 34 (3.5 g of a pure sample was isolated). Continuing this precipitation process eventually cleanly delivered compound 27a (24.7 g from various crops).
Overall Yields
Over four steps from 0.415 mol of recrystallized diacetate 22a:27a (30.4 g, 0.077 mol, 19%) and 28a (32.7 g, 0.083 mol, 20%). The combined yields of 27a and 28a over six steps from 250 g of CA (1) from this sequence was 28%.
Chromatography-Free Synthesis of 12β-Methyl-18-nor-bile Acids 27a and 28a; Beginning from 500 g of CA (Scheme 5)
Scheme 5, Step a. Methyl 3α,7α,12α-Trihydroxy-5β-cholan-24-oate (21a)41
CA (1, 501.6 g, 1.23 mol) was suspended in dry methanol (1.5 L) and p-toluenesulfonic acid monohydrate (4.0 g, 0.02 mol) was added. The mixture was heated to reflux for 2 h until dissolution, then the reaction was left to stir at room temperature for 3 d. The precipitate was filtered and washed with ice cold methanol (500 mL). The filtrate was concentrated, and further precipitate was collected by filtration. Ethyl acetate (1 L) was added to the combined solid portions and the suspension was concentrated to dryness to afford CA methyl ester 21a (524 g, quant).
Scheme 5, Step b. Methyl 3α,7α-Diacetyloxy-12α-hydroxy-5β-cholan-24-oate (22a)
Methyl ester 21a (524 g, 1.23 mol) was stirred in a refluxing mixture of ethyl acetate (1.9 L), pyridine (300 mL), and N,N-dimethylaminopyridine (DMAP; 7.0 g, 0.056 mol) until dissolution. The solution was cooled to 3 °C before acetic anhydride (261 mL, 2.74 mol) was added dropwise over 2 h. The solution was allowed to warm to 20 °C and stirred for 16 h. Water (3 L) was added and the mixture stirred vigorously for 30 min before transferring to a separating funnel. The aqueous phase was removed before 1 M aqueous hydrochloric acid (1.5 L) was added, shaken vigorously, and separated. The organic phase was washed with brine (2.4 L), dried over MgSO4, and concentrated. Ethyl acetate (1 L) was added and the mixture was concentrated to dryness. The crude was dissolved in twice the volume (per weight) of boiling ethyl acetate before petroleum ether (3 to 6 volumes) was added to the solution. The mixture was allowed to cool slowly to effect crystallization. After aging, the amorphous precipitate was collected by filtration. The mother liquor was concentrated, and additional crops of pure product were obtained from mixtures of ethyl acetate/petroleum ether. Combined yield of 22a: 318 g, 51% over two steps from 1 [Rf = 0.63; (ethyl acetate/petroleum ether, 1:1)].
Scheme 5, Step c. Methyl 3α,7α-Diacetyloxy-12α-mesyloxy-5β-cholan-24-oate (23a)
CA 3,7-diacetate methyl ester 22a (318 g, 0.62 mol) was dissolved in pyridine (1.5 L) before concentrating it to half volume. More pyridine was added (1 L), and the total volume was concentrated to about 1.6 L. This mixture was cooled in an ice bath to below 3 °C under an atmosphere of argon. Methanesulfonyl chloride (100 mL, 1.28 mol) was added dropwise and the temperature was maintained below 8 °C. After completion of the addition, the mixture was allowed to warm to room temperature and was left to stir overnight. The mixture was cooled in an ice bath before water (100 mL) was added dropwise. The reaction mixture was concentrated before ethyl acetate (1 L) was added and the mixture re-concentrated. Ethyl acetate (1 L) and water (1 L) were added. The organic phase was separated; washed with water (500 mL), 1 M hydrochloric acid (500 mL), and brine (800 mL); dried over MgSO4; and concentrated. The crude product was used in the next step without further purification. Rf = 0.61 (23a); 0.34 (22a) (acetone/dichloromethane, 1:19).
Scheme 5, Step d. Methyl 3α,7α-Diacetyloxy-12β-methyl-18-nor-5β-chol-13(14)-en-24-oate (24a) and Methyl 3α,7α-Diacetyloxy-12β-methyl-18-nor-5β-chol-13(17)-en-24-oate (25a)
The crude mesylate 23a (∼405 g) was dissolved in acetic acid (500 mL) at 100 °C under an atmosphere of argon before sodium acetate (170 g, 2.1 mol) was added in one portion. After being stirred at 100 °C overnight, the reaction was allowed to cool to room temperature. Toluene (800 mL) was added and the mixture was concentrated. Toluene (1 L) was added and the mixture was re-concentrated. Ethyl acetate (1 L) was added and the mixture was concentrated. The residue was partitioned between ethyl acetate and water, and the organic phase was separated. The aqueous phase was washed with ethyl acetate, and the combined organic phases were washed with brine, dried over MgSO4, and concentrated to dryness to afford 330 g of the crude alkene mixture (24a/25a/others).
Scheme 5, Step e. 3α,7α-Dihydroxy-12β-methyl-18-nor-5β-chol-13(17)-en-24-oic Acid (33) and 3α,7α-Dihydroxy-12β-methyl-18-nor-5β-chol-13(14)-en-24-oic Acid (34)
The crude alkene mixture (318 g) (24a/25a/others) was dissolved in hot methanol (650 mL) before a 10 M aqueous sodium hydroxide solution (265 g of sodium hydroxide in 650 mL of water) was added. This mixture was heated at 90 °C overnight. Methanol was evaporated before fresh methanol was added to obtain the total volume to 1.8 L. Separately, 800 mL of concentrated hydrochloric acid was added to 5.2 L of water to generate a diluted hydrochloric acid solution. This aqueous hydrochloric acid solution was cooled in an ice bath before the methanolic, basic, and crude reaction mixture was added dropwise with vigorous stirring over 48 h, forming a white precipitate. After the addition was complete, the acidic suspension was filtered, and the precipitate was washed with water.
Scheme 5, Step f. Fractional Precipitation of Deprotected Alkene Mixture (33/34/Others)
The precipitate (from step e) was dissolved in methanol and concentrated to a point just before precipitation occurred and then an equivolumetric amount of ethyl acetate was added. The solution was then concentrated to half the volume (rotary evaporator, 45 °C). An equivolumetric amount of ethyl acetate was added and the solution was concentrated to half volume once again. This process was repeated until a precipitate formed. The precipitate, which was pure Δ13,14-alkene 34 (60 g), was collected by filtration. Further fractional crystallization by this method yielded mixtures containing mainly alkenes 33 (major component) and 34 (minor component) among other minor alkene side-products (144 g).
An analytical sample of Δ13,17-alkene 33 could be obtained by dissolving a portion of the 144 g mixture of alkenes (obtained from the fractional crystallization) in hot ethyl acetate and concentrating to dryness (several times to remove water) followed by recrystallization from ethyl acetate. Alternatively, analytical samples of alkenes 33 and 34 could be obtained by column chromatography over silica gel eluting with 10–30% acetone in dichloromethane + 1% acetic acid.
3α,7α-Dihydroxy-12β-methyl-18-nor-5β-chol-13(17)-en-24-oic Acid (33)
[α]D20 = −36.3 (c 1.0, MeOH); 1H NMR (500 MHz, CD3OD): δ 3.94–3.90 (m, 1H), 3.39 (tt, 11.1, 4.4 Hz, 1H), 3.12–3.04 (m, 1H), 2.65–2.57 (m, 1H), 2.31–2.14 (m, 6H), 2.08 (td, 11.8, 3.4 Hz, 1H), 2.04–1.98 (m, 1H), 1.96 (ddd, 14.7, 5.4, 3.6 Hz, 1H), 1.89 (dt, 14.3, 3.1 Hz, 1H), 1.70–1.55 (m, 6H), 1.44–1.29 (m, 3H), 1.29 (d, 7.0 Hz, 3H), 1.16 (td, 11.3, 3.1 Hz, 1H), 1.05–0.96 (overlapping signals: m, 1H and 0.99, d, 6.9 Hz, 3H), 0.92 (ap q, 12.2 Hz, 1H), 0.85 (s, 3H); 13C NMR (125 MHz, CD3OD): δ 177.74, 141.96, 135.17, 72.94, 67.96, 50.82, 50.55, 43.05, 40.42, 37.73, 37.26, 36.45, 36.21, 35.79, 34.46, 33.52, 32.39, 31.49, 31.45, 31.09, 27.30, 23.62, 21.40, 20.30; HRMS (ESI) m/z: calcd for C24H38O4Na+, 413.2662; found, 413.2664.
3α,7α-Dihydroxy-12β-methyl-18-nor-5β-chol-13(14)-en-24-oic Acid (34)
[α]D20 = +60.6 (c 1.0, MeOH); 1H NMR (500 MHz, CDCl3): δ 4.09–4.04 (m, 1H), 3.38 (tt, 11.2, 4.4 Hz, 1H), 2.66–2.60 (m, 1H), 2.40–2.23 (m, 6H), 2.14–2.06 (m, 2H), 2.04–1.93 (m, 3H), 1.86–1.78 (m, 2H), 1.77–1.60 (m, 4H), 1.54 (dt, 14.7, 2.1 Hz, 1H), 1.44–1.31 (m, 2H), 1.30–1.19 (m, 1H), 1.10–1.01 (overlapping signals: m, 2H and 1.08, d, 7.0 Hz, 3H), 0.96 (d, 6.8 Hz, 3H), 0.91 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 178.06, 142.28, 140.23, 72.89, 68.99, 54.96, 42.75, 42.51, 40.73, 37.18, 36.51, 36.13, 35.92, 34.95, 34.52, 33.90, 32.84, 32.74, 31.55, 26.76, 25.55, 22.96, 21.76, 19.33; HRMS (ESI) m/z: calcd for C24H38O4Na+, 413.2662; found, 413.2668.
Scheme 5, Step g. 3α,7α-Dihydroxy-12β-methyl-18-nor-5β-cholan-24-oic Acid (27a) and 3α,7α-Dihydroxy-12β-methyl-17-epi-18-nor-5β-cholan-24-oic Acid (28a)
From step f, Δ13,14-alkene bile acid 34 (46 g) was combined with the mixture of alkenes 33 and 34 (144 g) (ratio not determined) to afford the starting material (190 g) used in this step. This mixture of alkenes (190 g) was dissolved in methanol (1.8 L) before water (200 mL) was added. The solution was transferred into a 3.4 L high-pressure reactor (as described above) and degassed before 10% palladium on charcoal (wet, 18 g) was added under an inert atmosphere. The solution was degassed before hydrogen (g) atmosphere was introduced at 5 bar. After 16 h, the mixture was carefully degassed before filtration through a pad of Celite and concentrated. Products 27a and 28a were isolated from the crude by fractional crystallization (as described above) from mixtures of methanol/ethyl acetate, ethyl acetate/water, ethanol/ethyl acetate, and methanol (with heating, concentration, and ageing) to yield 70.5 g (179.6 mmol, 29%) of 27a and 27.2 g (69.3 mmol, 11%) of 28a over four steps from recrystallized 3,7-diacetate CA methyl ester 22a (0.62 mol).
Obtaining Minor Byproducts
After fractional crystallization of the major products, fractions of the mother liquor were partitioned by column chromatography over silica gel eluting with acetone (+1% acetic acid)/dichloromethane (+1% acetic acid) mixtures (2–50%). Analytical samples of 14(13 → 12)-abeo bile acid 30 (400 mg) and C,D-cis-ring-juncture compound 29 (200 mg) were obtained by further recrystallization and chromatography.
13β,16α,17α-Trideutero-3α,7α-dihydroxy-12β-methyl-18-nor-5β-cholan-24-oic Acid (27a–d3) and a Mixture of 13β,17β-Di-, 13β,16β,17β-Tri- and 13β,15β,16β,17β-Tetradeutero-3α,7α-dihydroxy-12β-methyl-18-nor-5β-cholan-24-oic Acids (28a–d2–4)
A solution of alkene 33 (175 mg; 0.448 mmol) in CD3OD (2.5 mg/mL; the starting material (33) was co-evaporated a few times with CD3OD prior to use) was reacted in an H-Cube apparatus with deuterium oxide as a deuterium source (after thorough pre-purging of the system with D2O) at 25 °C, 80 bar pressure, and a flow rate of 0.3 mL/min. After the deuteration was complete, the solution was concentrated and the crude product purified by automated flash column chromatography [silica gel, acetone (+1% acetic acid)/dichloromethane (+1% acetic acid) = 0–30%] to afford 76.2 mg (41%) of 27a–d3 and 71.3 mg (38%) of 28a–d2–4 as colorless solids along with mixed fractions.
13β,16α,17α-Trideutero-3α,7α-dihydroxy-12β-methyl-18-nor-5β-cholan-24-oic Acid (27a–d3)
[α]D20 +23.5 (c 2.48, MeOH); 1H NMR (500 MHz, CD3OD): δ 3.91 (dd, 5.7, 2.9 Hz, 1H), 3.38 (tt, 11.1, 4.4 Hz, 1H), 2.37 (ddd, 15.3, 8.9, 5.5 Hz, 1H), 2.26 (dt, 11.6, 13.1 Hz, 1H), 2.22–2.14 (m, 1H), 2.03–1.94 (m, 2H), 1.88 (dt, 14.3, 3.2 Hz, 1H), 1.86–1.79 (m, 2H), 1.78–1.70 (m, 1H), 1.68–1.59 (m, 3H), 1.53 (dt, 14.7, 2.0 Hz, 1H), 1.49 (d, 8.9 Hz, 1H) 1.44–1.21 (m, 6H), 1.03–0.87 overlapping signals (0.99, td, 14.2, 3.4 Hz, 1H; 0.97, d, 6.6 Hz, 3H; m, 1H and 0.93, d, 6.8 Hz, 3H), 0.89 (s, 3H), 0.82 (dt, 12.0, 12.3 Hz, 1H); 13C NMR (125 MHz, CD3OD): δ 178.03, 72.97, 69.02, 53.83 (t, 18.3 Hz), 48.91, 46.94, 43.23, 40.49, 39.87, 37.22, 36.64, 36.36, 36.16, 35.72, 34.32, 33.68, 31.45, 29.93, 26.44, 24.87 (t, 18.6 Hz), 23.58, 21.80, 19.90; HRMS (ESI) m/z: calcd for C24H37D3O4Na+, 418.3007; found, 418.3008.
Mixture of 13β,17β-Di-, 13β,16β,17β-Tri-, and 13β,15β,16β,17β-Tetradeutero-3α,7α-dihydroxy-12β-methyl-18-nor-5β-cholan-24-oic Acids (28a–d2–4)
[α]D20 −37.1 (c 0.44, MeOH); 1H NMR (500 MHz, CD3OD): δ 3.88 (dd, 5.7, 2.9 Hz, 1H), 3.37 (tt, 11.2, 4.4 Hz, 1H), 2.34–2.20 (m, 3H), 1.96 (ddd, 14.8, 5.6, 3.7 Hz, 1H), 1.93–1.80 (m, 4H), 1.78–1.68 overlapping signals (1.75, dt, 12.9, 3.6 Hz, 1H and m, 1H), 1.68–1.59 (m, 2.6H), 1.56–1.40 (m, 5H), 1.40–1.31 (m, 2H), 1.09–1.00 (m, 2H), 1.00–0.94 overlapping signals (m, 1H and 0.97, d, 6.3 Hz, 3H), 0.89 (d, 6.8 Hz, 3H), 0.86 (s, 3H), 0.68 (dt, 11.6, 12.4 Hz, 1H); 13C NMR (125 MHz, CD3OD): δ 177.98, 72.98, 68.78, 56.98 (t, 17.8 Hz), 48.51, 44.47, 43.32, 42.32 (m), 40.40, 37.05, 36.63, 36.09, 35.70, 34.23, 33.52, 33.28, 33.22, 32.87, 31.49, 30.37/30.26/29.96 (m), 23.70, 23.53/23.21 (t, 17.6 Hz), 21.75, 17.20; HRMS (ESI) m/z: calcd for C24H38D2O4Na+, 417.2944; found, 417.2946; m/z: calcd for C24H37D3O4Na+, 418.3007; found, 418.3003; m/z: calcd for C24H36D4O4Na+, 419.3070; found, 419.3060.
Synthesis of 25-homo-Cholic Acid (20; Scheme S1)
25-Diazo-3α,7α,12α-triformyloxy-24-oxo-25-homo-5β-cholane (S2)40b,40c,46
To a solution of tris(formyloxy)-cholic acid S1(45) (21.0 g, 42.6 mmol) in dry benzene (200 mL) was added oxalyl chloride (8.00 mL, 94.5 mmol) at 5 °C. After being stirred for 5 h at room temperature, the reaction mixture was concentrated, and the residue was dried under high vacuum (light-yellow foam). The acid chloride was re-dissolved in a mixture of dry toluene and diethyl ether before freshly prepared diazomethane was distilled into the reaction flask under ice cooling. [Diazomethane was prepared by adding a solution of Diazald (27.9 g, 0.13 mol) in diethyl ether (160 mL) dropwise to a solution of potassium hydroxide (12 g, 0.21 mol) in water (60 mL) and ethanol (70 mL) at 70 °C using a diazomethane kit and a blast shield]. The reaction was allowed to warm to room temperature and was stirred overnight. Then, the yellow solution was concentrated, and the crude was used in the next reaction step without further purification. An analytical sample of diazoketone S2 was obtained as a light-yellow foam by automated flash column chromatography (silica gel, ethyl acetate/petroleum ether = 5–40%). [α]D20 = +65.2 (c 0.675, CHCl3); 1H NMR (500 MHz, CDCl3): δ 8.16 (s, 1H), 8.10 (s, 1H), 8.02 (s, 1H), 5.28–5.25 (m, 1H), 5.20 (sbr, 1H), 5.09–5.05 (m, 1H), 4.72 (tt, 11.4, 4.6 Hz, 1H), 2.39–2.28 (mbr, 1H), 2.27–2.09 (m, 3H), 2.06–1.99 (m, 1H), 1.98–1.84 (m, 2H), 1.83–1.62 (m, 8H), 1.61–1.51 (m, 2H), 1.50–1.24 (m, 5H), 1.17–1.05 (m, 2H), 0.95 (s, 3H), 0.84 (d, 6.6 Hz, 3H), 0.76 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 195.12, 160.53 (2C), 160.48, 75.31, 73.74, 70.69, 54.24, 47.37, 45.07, 42.99, 40.84, 37.76 (2C), 34.84, 34.54, 34.48, 34.30, 31.36, 30.81, 28.59, 27.19, 26.60, 25.58, 22.80, 22.34, 17.63, 12.18; HRMS (ESI) m/z: calcd for C28H40N2O7Na+, 539.2728; found, 539.2739.
Methyl 7α,12α-Diformyloxy-3α-hydroxy-25-homo-5β-cholan-25-oate (S3)
To a solution of crude diazoketone S2 in methanol (780 mL) was added a solution of silver nitrate (38.0 g, 224 mol) in a 1:2 mixture of water (120 mL) and methanol (260 mL) [prepared by dissolving the silver salt in water first and then diluting this with methanol], and the resulting reaction mixture was heated under reflux for 4 h. After being cooled to room temperature, the reaction was concentrated to half volume, diluted with a mixture water and ethyl acetate, and subsequently filtered through Celite. The aqueous phase was separated and extracted with ethyl acetate (3×). The organic fractions were combined, washed with water and brine, dried over MgSO4, and concentrated. The crude product was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 20–60%) to yield 13.0 g (62%) of S3 as a colorless foam over three steps. [α]D20 = +64.1 (c 0.64, CHCl3); 1H NMR (500 MHz, CDCl3): δ 8.14 (s, 1H), 8.10 (s, 1H), 5.28–5.24 (m, 1H), 5.08–5.04 (m, 1H), 3.66 (s, 3H), 3.50 (tt, 11.0, 4.4 Hz, 1H), 2.32–2.19 (m, 2H), 2.13 (td, 12.4, 4.4 Hz, 1H), 2.07–1.97 (m, 2H), 1.96–1.89 (m, 1H), 1.86 (tt, 9.5, 3.6 Hz, 1H), 1.82–1.72 (m, 2H), 1.72–1.62 (m, 5H), 1.62–1.51 (m, 2H), 1.51–1.41 (m, 4H), 1.40–1.32 (m, 2H), 1.30–1.18 (m, 2H), 1.14–0.97 overlapping signals (m, 2H and 1.01, td, 14.3, 3.3 Hz, 1H), 0.93 (s, 3H), 0.85 (d, 6.6 Hz, 3H), 0.75 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 174.17, 162.72, 160.57, 75.39, 71.55, 70.87, 51.44, 47.22, 44.97, 42.96, 41.03, 38.60, 37.81, 35.13, 34.95, 34.90, 34.39, 34.28, 31.51, 30.40, 28.60, 27.22, 25.52, 22.81, 22.38, 21.43, 17.78, 12.11; HRMS (ESI) m/z: calcd for C28H44O7Na+, 515.2979; found, 515.2986.
3α,7α,12α-Trihydroxy-25-homo-5β-cholan-25-oic Acid (20)46
To a solution of bis(formyloxy)-methyl ester S3 (8.57 g, 17.4 mmol) in methanol (160 mL) was added a 2 M aqueous solution of sodium hydroxide (80 mL, 160 mmol). After being refluxed for 3 h, the reaction was cooled to room temperature, the solvent evaporated, and the residue diluted with water and added slowly to a 0.5 M aqueous solution of hydrochloric acid. The resulting mixture was extracted with ethyl acetate (3×), and the combined organic fractions were washed with brine, dried over MgSO4, and concentrated. The crude product was triturated with a 50% mixture of ethyl acetate in hexane and recovered by filtration to give 6.39 g (87%) of 20 as a colorless amorphous solid. [α]D20 = +49.7 (c 0.38, MeOH); 1H NMR (500 MHz, CD3OD): δ 3.95 (t, 2.7 Hz, 1H), 3.81–3.77 (m, 1H), 3.37 (tt, 11.1, 4.4 Hz, 1H), 2.32–2.18 (m, 4H), 2.02–1.92 (m, 2H), 1.91–1.83 (m, 2H), 1.81 (dt, 14.3, 3.2 Hz, 1H), 1.77–1.68 (m, 2H), 1.68–1.62 (m, 1H), 1.62–1.34 (m, 10H), 1.31–1.21 (m, 1H), 1.17–1.05 (m, 2H), 1.04–0.94 overlapping signals (1.02, d, 6.6 Hz, 3H and 0.98, td, 14.2, 3.5 Hz, 1H), 0.92 (s, 3H), 0.72 (s, 3H); 13C NMR (125 MHz, CD3OD): δ 177.91, 74.13, 72.95, 69.15, 48.28, 47.52, 43.28, 43.04, 41.10, 40.55, 37.06, 36.67, 36.56, 35.96, 35.91, 35.55, 31.25, 29.65, 28.80, 27.95, 24.28, 23.21, 22.96, 17.99, 13.03; HRMS (ESI) m/z: calcd for C25H42O5Na+, 445.2924; found, 445.2924.
Synthesis of 12β-Methyl-25-homo-18-nor-chenodeoxycholic Acids 27c and 28c from 12β-Methyl-18-nor-chenodeoxycholic Acids 27a and 28a (Scheme S2)
3α,7α-Diformyloxy-12β-methyl-18-nor-5β-cholan-24-oic Acid (S4)
To a solution of compound 27a (7.50 g, 19.1 mmol) in 90% formic acid (113 mL) was added a catalytic amount of 70% perchloric acid (0.26 mL), and the reaction mixture was then stirred at 50 °C for 2.5 h. After being cooled to 40 °C, acetic anhydride (90 mL) was introduced dropwise over a period of 30 min ensuring that the temperature did not exceed 60 °C (effervescence was observed toward the end of the addition). The reaction was stirred at room temperature for 70 min and subsequently quenched in ice water. The cream-colored precipitate was filtered off and re-dissolved in ethyl acetate. The organic solution was washed with brine, dried over MgSO4, and concentrated. The crude product was purified by automated flash column chromatography (silica gel, ethyl acetate/petroleum ether = 5–35%) to give 8.64 g (quant.) of S4 as a colorless foam. 1H NMR (500 MHz, CDCl3): δ 8.07 (s, 1H), 8.03 (d, 0.8 Hz, 1H), 5.18–5.14 (m, 1H), 4.73 (tt, 11.3, 4.4 Hz, 1H), 2.45 (ddd, 15.7, 9.8, 5.3 Hz, 1H), 2.24 (ddd, 15.9, 9.1, 6.8 Hz, 1H), 2.14 (dt, 11.9, 13.0 Hz, 1H), 2.05–1.99 (m, 1H), 1.99–1.90 (m, 2H), 1.85–1.69 (m, 4H), 1.68–1.44 (m, 8H), 1.42 (td, 10.9, 3.2 Hz, 1H), 1.36–1.23 (m, 3H), 1.10 (td, 14.4, 3.4 Hz, 1H), 0.96 (d, 6.6 Hz, 3H), 0.94–0.88 (overlapping signals: m, 1H; 0.92, s, 3H and 0.91, d, 6.7 Hz, 3H), 0.86–0.77 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 178.14, 160.70, 160.68, 74.10, 70.98, 52.91, 48.92, 47.41, 43.69, 40.94, 38.31, 35.72, 34.92, 34.77, 34.72, 34.55, 33.98, 32.23, 31.35, 28.56, 26.81, 24.76, 24.08, 22.75, 21.12, 19.40; HRMS (ESI) m/z: calcd for C26H40O6Na+, 471.2717; found, 471.2714.
25-Diazo-3α,7α-diformyloxy-12β-methyl-24-oxo-25-homo-18-nor-5β-cholane (S6)
To a solution of S4 (8.64 g, 19.3 mmol) in dry toluene (80 mL) was added oxalyl chloride (6.00 mL, 70.9 mmol) at room temperature. After being stirred for 2.5 h, the reaction mixture was concentrated, and the residue was dried under high vacuum. The acid chloride was re-dissolved in dry toluene (20 mL) and subsequently added dropwise to a pre-prepared diazomethane solution at 0 °C [prepared from N-nitroso-N-methylurea (MNU; 20 g, 194 mmol), 50% aqueous potassium hydroxide solution (60 mL), and diethyl ether (80 mL) at −5 to −10 °C; the diazomethane solution was dried over potassium hydroxide before use]. The reaction was allowed to warm to room temperature, and after being stirred overnight the yellow solution was concentrated and the residue was purified by automated flash column chromatography (silica gel, ethyl acetate/petroleum ether = 5–40%) to afford 6.80 g (75%) of the desired diazoketone S6. [α]D20 = +21.4 (c 0.935, CHCl3); 1H NMR (500 MHz, CDCl3): δ 8.07 (s, 1H), 8.03 (d, 0.8 Hz, 1H), 5.24 (sbr, 1H), 5.18–5.13 (m, 1H), 4.73 (tt, 11.3, 4.5 Hz, 1H), 2.48–2.33 (mbr, 1H), 2.26–2.09 (overlapping signals: m, 1H and 2.14, dt, 11.9, 13.1 Hz, 1H), 2.02 (ddd, 15.4, 5.1, 4.1 Hz, 1H), 1.99–1.90 (m, 2H), 1.84–1.68 (m, 4H), 1.68–1.59 (m, 3H), 1.59–1.38 (m, 6H), 1.36–1.20 (m, 3H), 1.10 (td, 14.4, 3.5 Hz, 1H), 0.96 (d, 6.6 Hz, 3H), 0.94–0.87 (overlapping signals: m, 1H; 0.91, s, 3H and 0.89, d, 6.7 Hz, 3H), 0.87–0.78 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 195.33, 160.64 (2C), 74.08, 70.96, 54.21 (br), 52.84, 48.97, 47.38, 43.64, 40.91, 39.57 (br), 38.29, 35.68, 35.03, 34.74, 34.70, 34.53, 33.95, 31.33, 28.49, 26.79, 25.15 (br), 24.05, 22.73, 21.12, 19.54; HRMS (ESI) m/z: calcd for C27H40N2O5Na+, 495.2829; found, 495.2838.
Methyl 7α-Formyloxy-3α-hydroxy-12β-methyl-25-homo-18-nor-5β-cholan-25-oate (S8)
To a solution of diazoketone S6 (6.80 g, 14.4 mmol) in methanol (20 mL) was added a solution of silver nitrate (10.5 g, 61.8 mmol) in a 1:9 mixture of water (35 mL) and methanol (315 mL) [prepared by dissolving the silver salt in water first and then diluting it with methanol], and the resulting reaction mixture was heated under reflux for 3–4 h. After being cooled to room temperature, the reaction mixture was filtered through Celite and methanol was evaporated. The aqueous phase was extracted with ethyl acetate (3×), washed with water (2×) and brine, dried over MgSO4, and concentrated. The crude product was purified by automated flash column chromatography (silica gel, ethyl acetate/petroleum ether = 0–50%) to yield 3.90 g (60%) of S8 as a colorless foam. 1H NMR (500 MHz, CDCl3): δ 8.06 (s, 1H), 5.17–5.12 (m, 1H), 3.67 (s, 3H), 3.51 (tt, 11.0, 4.3 Hz, 1H), 2.37–2.24 (m, 2H), 2.04–1.92 (m, 3H), 1.89 (dt, 14.4, 3.3 Hz, 1H), 1.80–1.33 (m, 16H), 1.33–1.20 (m, 2H), 1.08–0.93 (overlapping signals: m, 1H; 1.03, td, 14.2, 3.4 Hz, 1H and 0.96, d, 6.6 Hz, 3H), 0.93–0.84 (overlapping signals: m, 1H; 0.89, d, 3H and 0.90, s, 3H), 0.82–0.70 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 174.21, 160.79, 71.76, 71.09, 53.04, 51.42, 49.15, 47.39, 43.79, 41.14, 38.81, 38.30, 35.83, 35.14, 34.99, 34.65, 34.45, 33.99, 31.48, 30.72, 29.12, 28.49, 24.09, 23.51, 22.79, 21.27, 19.74; HRMS (ESI) m/z: calcd for C27H44O5Na+, 471.3081; found, 471.3086.
3α,7α-Dihydroxy-12β-methyl-25-homo-18-nor-5β-cholan-25-oic Acid (27c)
To a solution of S8 (4.90 g, 10.9 mmol) in methanol (65 mL) was added 2 M aqueous sodium hydroxide solution (35 mL), and the resulting reaction mixture was heated under reflux for 3 h. After complete conversion, the reaction was allowed to cool to room temperature and subsequently concentrated. The residue was re-dissolved in water and then acidified to pH 1 by adding dropwise a 1 M aqueous hydrochloric acid solution. The resulting mixture was extracted with ethyl acetate (3×), and the combined organic layers were dried over MgSO4 and concentrated. The crude product was purified by automated flash column chromatography [silica gel, acetone (+1% acetic acid)/dichloromethane (+1% acetic acid) = 5–40%] to afford 3.55 g (80%) of 27c as a colorless solid. The spectroscopic data of this compound are consistent with those reported earlier for the same compound 27c.
3α,7α-Diformyloxy-12β-methyl-17-epi-18-nor-5β-cholan-24-oic Acid (S5)
Using the same protocol as outlined for the preparation of compound S4, a catalytic amount of 72% perchloric acid (0.33 mL) was added to a solution of 28a (9.50 g, 24.2 mmol) in 90% formic acid (142 mL) at 50 °C, and the resulting reaction mixture was stirred for another 3 h at this temperature. Subsequently, the reaction temperature was lowered to 40 °C and acetic anhydride (115 mL) was added dropwise so that the reaction temperature was maintained between 40 and 50 °C. The reaction was stirred for another 60 min while cooling to room temperature. After work-up and purification, 9.90 g (91%) of the desired bisformyl protected acid S5 was recovered. [α]D20 = −14.9 (c 0.67, CHCl3); 1H NMR (500 MHz, CDCl3): δ 8.07 (s, 1H), 8.03 (d, 0.9 Hz, 1H), 5.19–5.15 (m, 1H), 4.74 (tt, 11.3, 4.5 Hz, 1H), 2.41–2.30 (m, 2H), 2.22–2.16 (m, 1H), 2.12 (dt, 11.9, 13.1 Hz, 1H), 2.05–1.98 (m, 1H), 1.95 (dt, 14.5, 3.3 Hz, 1H), 1.87 (td, 11.8, 3.3 Hz, 1H), 1.84–1.70 (m, 3H), 1.70–1.59 (m, 4H), 1.59–1.39 (m, 7H), 1.19 (td, 10.7, 3.4 Hz, 1H), 1.15–0.99 (m, 2H), 0.99–0.92 (overlapping signals: m, 1H and 0.95, d, 6.3 Hz, 3H), 0.89 (s, 3H), 0.84 (d, 6.8 Hz, 3H), 0.69 (dt, 11.8, 12.4 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 179.41, 160.68, 160.59, 74.12, 70.82, 56.06, 45.03, 43.08, 41.44, 41.00, 35.44, 34.76, 34.66, 34.50, 33.83, 32.07, 31.94, 31.85, 31.61, 31.28, 29.11, 26.82, 22.83, 22.51, 21.03, 16.68; HRMS (ESI) m/z: calcd for C26H40O6Na+, 471.2717; found, 471.2715.
25-Diazo-3α,7α-diformyloxy-12β-methyl-24-oxo-17-epi-25-homo-18-nor-5β-cholane (S7)
Employing the same procedure as described for the preparation of S6, oxalyl chloride (6.10 mL, 72.1 mmol) was added dropwise to a solution of compound S5 (9.90 g, 22.1 mmol) in dry toluene (80 mL) at 10 °C. After being stirred for 2.5 h and concentrated, the crude acid chloride was re-dissolved in dry toluene (40 mL) and subsequently introduced dropwise into a pre-prepared solution of diazomethane in diethyl ether [prepared from N-nitroso-N-methylurea (21.0 g, 204 mmol), 50% aqueous potassium hydroxide solution (60 mL), and diethyl ether (120 mL) at −5 to −10 °C; the diazomethane solution was dried over potassium hydroxide before use] to obtain 8.70 g (83%) of S7 as a light yellow, crystalline solid. mp/decomp.: 142–145 °C; [α]D20 = −11.1 (c 0.75, CHCl3); 1H NMR (500 MHz, CDCl3): δ 8.06 (s, 1H), 8.03 (d, 0.5 Hz, 1H), 5.22 (sbr, 1H), 5.19–5.15 (m, 1H), 4.74 (tt, 11.3, 4.5 Hz, 1H), 2.37–2.26 (mbr, 2H), 2.21–2.09 (overlapping signals: m, 1H and 2.14, dt, 11.9, 13.0 Hz, 1H), 2.05–1.98 (m, 1H), 1.95 (dt, 14.5, 3.3 Hz, 1H), 1.86 (td, 11.8, 3.4 Hz, 1H), 1.83–1.70 (m, 3H), 1.69–1.59 (m, 4H), 1.58–1.39 (m, 7H), 1.19 (td, 10.7, 3.3 Hz, 1H), 1.14–0.99 (overlapping signals: 1.09, td, 14.4, 3.4 Hz, 1H and m, 1H), 0.99–0.91 (overlapping signals: m, 1H and 0.95, d, 6.3 Hz, 3H), 0.89 (s, 3H), 0.83 (d, 6.8 Hz, 3H), 0.69 (dt, 11.7, 12.4 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 195.19, 160.63, 160.54, 74.10, 70.80, 56.08, 54.14 (br), 45.03, 43.08, 41.54, 41.00, 39.20 (br), 35.43, 34.75, 34.66, 34.50, 33.83, 32.32 (br), 31.94, 31.79, 31.29, 29.11, 26.83, 22.83, 22.55, 21.09, 16.80; HRMS (ESI) m/z: calcd for C27H40N2O5Na+, 495.2829; found, 495.2830.
Methyl 7α-Formyloxy-3α-hydroxy-12β-methyl-17-epi-25-homo-18-nor-5β-cholan-25-oate (S9)
Employing the same procedure as described for the preparation of S8, a solution of silver nitrate (13.0 g, 76.5 mmol) in a mixture of water (43 mL) and methanol (90 mL) was added to a solution of diazoketone S7 (8.40 g, 17.8 mmol) in methanol (300 mL), and the resulting reaction mixture was refluxed for 2.5 h to give rise to 5.15 g (65%) of S9 as a colorless foam. [α]D20 = −40.4 (c 0.96, MeOH), [α]D = −32.6 (c 0.57, CHCl3); 1H NMR (500 MHz, CDCl3): δ 8.06 (s, 1H), 5.17–5.13 (m, 1H), 3.67 (s, 3H), 3.51 (tt, 11.0, 4.4 Hz, 1H), 2.28 (t, 7.6 Hz, 2H), 2.18 (tt, 8.6, 2.9 Hz, 1H), 2.06–1.96 (m, 2H), 1.94–1.84 (m, 2H), 1.81–1.68 (m, 3H), 1.67–1.55 (m, 7H), 1.55–1.46 (m, 2H), 1.46–1.35 (m, 3H), 1.22–1.07 (m, 3H), 1.06–0.98 (m, 2H), 0.96 (d, 6.3 Hz, 3H), 0.93 (dt, 8.4, 11.8 Hz, 1H), 0.87 (s, 3H), 0.80 (d, 6.8 Hz, 3H), 0.67 (dt, 11.8, 12.3 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 174.20, 160.71, 71.74, 70.97, 56.10, 51.40, 45.04, 43.08, 41.47, 41.17, 38.71, 36.65, 35.45, 35.11, 34.58, 34.33, 33.85, 31.94, 31.73, 31.40, 30.68, 29.10, 22.98, 22.85, 22.49, 21.15, 16.89; HRMS (ESI) m/z: calcd for C27H44O5Na+, 471.3081; found, 471.3084.
3α,7α-Dihydroxy-12β-methyl-17-epi-25-homo-18-nor-5β-cholan-25-oic Acid (28c)
Using the same procedure as outlined for the preparation of 27c, a 2 M aqueous sodium hydroxide solution (35 mL) was added to a solution of compound S9 (5.14 g, 11.5 mmol) in methanol (65 mL), and the resulting reaction mixture was refluxed for 3 h to yield 4.17 g (90%) of 28c as a colorless, crystalline solid. The spectroscopic data of this sample were consistent with those reported earlier for the same compound 28c.
Synthesis of 12β-Methyl-18,24-bisnor-chenodeoxycholic Acid 27b from 12β-Methyl-18-nor-chenodeoxycholic Acid 27a (Scheme S3)
3α,7α-Diformyloxy-12β-methyl-18,24-bisnor-5β-cholane-23-nitrile (S10)
To a solution of 3α,7α-diformyloxy-12β-methyl-18-nor-5β-cholan-24-oic acid S4 (542 mg, 1.21 mmol) in trifluoroacetic acid (1.91 mL) was added dropwise trifluoroacetic anhydride (0.51 mL, 3.63 mmol) at room temperature. The solution was cooled to 0 °C and with a slow flow of dry argon gas purging the reaction mixture, solid sodium nitrite (91 mg, 1.32 mmol) was added portionwise. The mixture was stirred for 30 min and allowed to warm slowly to room temperature before heating to 40 °C for 1.5 h. The reaction mixture was concentrated to dryness and stripped twice from dry toluene before ethyl acetate (20 mL) and water (20 mL) were added. The aqueous phase was separated and extracted with ethyl acetate (2 × 20 mL). The combined organics were washed sequentially with saturated aqueous bicarbonate solution (2 × 30 mL) and brine, dried over MgSO4, and concentrated. The crude product was purified by automated flash column chromatography (silica gel, ethyl acetate/petroleum ether = 10–35%) to give 280 mg (56%) of S10 as a colorless foam. 1H NMR (500 MHz, CDCl3): δ 8.07 (s, 1H), 8.02 (d, 0.7 Hz, 1H), 5.17–5.14 (1H, m), 4.73 (tt, 11.3, 4.5 Hz, 1H), 2.39 (dd, 16.5, 4.0 Hz, 1H), 2.30–2.21 (m, 1H), 2.20–1.89 (m, 5H), 1.87–1.74 (m, 2H), 1.68–1.56 (m, 5H), 1.56–1.47 (m, 2H), 1.44–1.20 (m, 4H), 1.15–1.06 (overlapping signals: m, 1H), 1.12 (d, 6.9 Hz, 3H), 1.01 (d, 6.6 Hz, 3H), 0.95–0.87 (overlapping signals: m, 1H), 0.91 (s, 3H), 0.82 (q, 12.3 Hz, 1H), 0.68 (q, 10.3 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 160.64, 160.54, 119.93, 73.97, 70.67, 53.58, 47.47, 47.30, 43.67, 40.84, 38.03, 35.62, 34.72, 34.68, 34.52, 33.90, 33.39, 31.28, 28.38, 26.79, 23.77, 22.67, 21.15, 20.07, 18.58; HRMS (ESI) m/z: calcd for C25H37NO4Na+, 438.2615; found, 438.2612.
3α,7α-Dihydroxy-12β-methyl-18,24-bisnor-5β-cholan-23-oic Acid (27b) and 3α,7α-Dihydroxy-12β-methyl-18,24-bisnor-5β-cholan-23-amide (S11)
A 20% (w/v) aqueous potassium hydroxide solution (4 mL) was added to a solution of nitrile S10 (280 mg, 0.67 mmol) in ethanol (4 mL). The mixture was heated at 90 °C for 72 h before cooling to room temperature and pouring slowly into an ice-cooled 10% aqueous hydrochloric acid solution. Ethyl acetate was added (20 mL) before the aqueous phase was separated and extracted with ethyl acetate (3 × 20 mL). The combined organics were washed with brine, dried over MgSO4, and concentrated. The crude product was purified by automated flash column chromatography [silica gel, acetone (+1% acetic acid)/dichloromethane (+1% acetic acid) = 5–100%] to afford 100 mg (39%) of 27b and 100 mg (39%) of amide S11 as white solids. The spectral data for 27b were identical to those obtained earlier for the same compound.
3α,7α-Dihydroxy-12β-methyl-18,24-bisnor-5β-cholan-23-amide (S11)
mp: 198–199 °C (DSC); [α]D20 +13.3 (c 0.3, MeOH); 1H NMR (500 MHz, CD3OD): δ 3.92–3.89 (m, 1H), 3.42–3.34 (m, 1H), 2.35–2.21 (m, 3H), 2.04–1.77 (m, 6H), 1.69–1.27 (m, 11H), 1.07–0.82 (overlapping signals: m, 3H), 1.04 (d, 6.6 Hz, 3H), 0.97 (d, 6.7 Hz, 3H), 0.88 (s, 3H), 0.77 (q, 10.1 Hz, 1H); 13C NMR (125 MHz, CD3OD): δ 179.39, 72.89, 68.93, 54.76, 49.64, 48.98, 46.87, 43.15, 40.41, 39.93, 37.44, 37.07, 36.58, 36.11, 35.66, 34.47, 34.26, 31.40, 29.89, 25.54, 23.53, 21.69, 20.50; HRMS (ESI) m/z: calcd for C23H39NO3Na+, 400.2822; found, 400.2827.
Acknowledgments
We thank the New Zealand Ministry of Business, Innovation and Employment (MBIE; 29336-HVMSTR-IRL) and New Zealand Pharmaceuticals Limited (NZP) for financial support. We also thank Dr Graham Caygill and GlycoSyn Ltd. for assistance with, and use of, a high-pressure hydrogenation calorimeter and Bruce Hamilton for invaluable assistance with HPLC analysis. We are also grateful for the beamtime we obtained on the KOALA beamline at the Australian Centre for Neutron Scattering, a facility of the Australian Nuclear Science and Technology Organisation (ANSTO).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c04199.
Additional reaction schemes, 1H and 13C NMR spectra and HPLC chromatograms for compounds as well as crystallographic data and structure refinement tables for compounds 25b, 27a, 28a, 28c, 29, 30, 31, 33, and 34 and neutron diffraction data for 28a–d2–4. CCDC 2094561–2094569 and 2094996 contain the supplementary crystallographic data for this paper (PDF)
Author Contributions
# A.L. and L.D.H. contributed equally.
The authors declare no competing financial interest.
Author Status
¶ Deceased.
Supplementary Material
References
- a Fiorucci S.; Distrutti E.. Handbook of Experimental Pharmacology 256: Bile Acids and Their Receptors. 1st Ed.; Springer International Publishing, 2019. [DOI] [PubMed] [Google Scholar]; b Tazuma S.; Takikawa H.. Bile Acids in Gastroenterology: Basic and Clinical. 1st Ed.; Springer Japan, 2017. [Google Scholar]; c Jenkins G. J.; Hardie L. J.. Bile Acids: Toxicology and Bioactivity; Royal Society of Chemistry: Cambridge, 2008. [Google Scholar]
- a Dawson P. A.; Karpen S. J. Intestinal transport and metabolism of bile acids. J. Lipid Res. 2015, 56, 1085–1099. 10.1194/jlr.r054114. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hofmann A. F. The enterohepatic circulation of bile acids in mammals: form and functions. Front. Biosci. 2009, 14, 2584–2598. 10.2741/3399. [DOI] [PubMed] [Google Scholar]
- a Jansen P. L. M. A new life for bile acids. J. Hepatol. 2010, 52, 937–938. 10.1016/j.jhep.2010.02.003. [DOI] [PubMed] [Google Scholar]; b Duane W. C. Bile acids: developments new and very old. J. Lipid Res. 2009, 50, 1507–1508. 10.1194/jlr.e900004-jlr200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer S. A.; Mangelsdorf D. J. Bile Acids as Hormones: The FXR-FGF15/19 Pathway. Dig. Dis. 2015, 33, 327–331. 10.1159/000371670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Makishima M.; Okamoto A. Y.; Repa J. J.; Tu H.; Learned R. M.; Luk A.; Hull M. V.; Lustig K. D.; Mangelsdorf D. J.; Shan B. Identification of a Nuclear Receptor for Bile Acids. Science 1999, 284, 1362–1365. 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]; b Parks D. J.; Blanchard S. G.; Bledsoe R. K.; Chandra G.; Consler T. G.; Kliewer S. A.; Stimmel J. B.; Willson T. M.; Zavacki A. M.; Moore D. D.; Lehmann J. M. Bile Acids: Natural Ligands for an Orphan Nuclear Receptor. Science 1999, 284, 1365–1368. 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]; c Wang H.; Chen J.; Hollister K.; Sowers L. C.; Forman B. M. Endogenous Bile Acids Are Ligands for the Nuclear Receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- a Maruyama T.; Miyamoto Y.; Nakamura T.; Tamai Y.; Okada H.; Sugiyama E.; Nakamura T.; Itadani H.; Tanaka K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. 10.1016/s0006-291x(02)02550-0. [DOI] [PubMed] [Google Scholar]; b Kawamata Y.; Fujii R.; Hosoya M.; Harada M.; Yoshida H.; Miwa M.; Fukusumi S.; Habata Y.; Itoh T.; Shintani Y.; Hinuma S.; Fujisawa Y.; Fujino M. A G Protein-coupled Receptor Responsive to Bile Acids. J. Biol. Chem. 2003, 278, 9435–9440. 10.1074/jbc.m209706200. [DOI] [PubMed] [Google Scholar]
- Kim I.; Ahn S.-H.; Inagaki T.; Choi M.; Ito S.; Guo G. L.; Kliewer S. A.; Gonzalez F. J. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 2007, 48, 2664–2672. 10.1194/jlr.m700330-jlr200. [DOI] [PubMed] [Google Scholar]
- Chiang J. Y. L.; Ferrell J. M. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G554–G573. 10.1152/ajpgi.00223.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Lee F. Y.; Barrera G.; Lee H.; Vales C.; Gonzalez F. J.; Willson T. M.; Edwards P. A. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 1006–1011. 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y.; Lu Y.; Li X.-Y. Farnesoid X receptor: a master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol. Sin. 2015, 36, 44–50. 10.1038/aps.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad T. R.; Haeusler R. A. Bile acids in glucose metabolism and insulin signalling—mechanisms and research needs. Nat. Rev. Endocrinol. 2019, 15, 701–712. 10.1038/s41574-019-0266-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Renga B.; Mencarelli A.; D’Amore C.; Cipriani S.; Baldelli F.; Zampella A.; Distrutti E.; Fiorucci S. Glucocorticoid receptor mediates the gluconeogenic activity of the farnesoid X receptor in the fasting condition. FASEB J. 2012, 26, 3021–3031. 10.1096/fj.11-195701. [DOI] [PubMed] [Google Scholar]; b Ma K.; Saha P. K.; Chan L.; Moore D. D. Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Invest. 2006, 116, 1102–1109. 10.1172/jci25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.-D.; Chen W.-D.; Wang M.; Yu D.; Forman B. M.; Huang W. Farnesoid X receptor antagonizes nuclear factor κB in hepatic inflammatory response. Hepatology 2008, 48, 1632–1643. 10.1002/hep.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorucci S.; Rizzo G.; Antonelli E.; Renga B.; Mencarelli A.; Riccardi L.; Orlandi S.; Pruzanski M.; Morelli A.; Pellicciari R. A Farnesoid X Receptor-Small Heterodimer Partner Regulatory Cascade Modulates Tissue Metalloproteinase Inhibitor-1 and Matrix Metalloprotease Expression in Hepatic Stellate Cells and Promotes Resolution of Liver Fibrosis. J. Pharmacol. Exp. Ther. 2005, 314, 584–595. 10.1124/jpet.105.084905. [DOI] [PubMed] [Google Scholar]
- Watanabe M.; Houten S. M.; Mataki C.; Christoffolete M. A.; Kim B. W.; Sato H.; Messaddeq N.; Harney J. W.; Ezaki O.; Kodama T.; Schoonjans K.; Bianco A. C.; Auwerx J. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006, 439, 484–489. 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- a Keitel V.; Reinehr R.; Gatsios P.; Rupprecht C.; Görg B.; Selbach O.; Häussinger D.; Kubitz R. The G-Protein Coupled Bile Salt Receptor TGR5 Is Expressed in Liver Sinusoidal Endothelial Cells. Hepatology 2007, 45, 695–704. 10.1002/hep.21458. [DOI] [PubMed] [Google Scholar]; b Pols T. W. H.; Nomura M.; Harach T.; Lo Sasso G.; Oosterveer M. H.; Thomas C.; Rizzo G.; Gioiello A.; Adorini L.; Pellicciari R.; Auwerx J.; Schoonjans K. TGR5 Activation Inhibits Atherosclerosis by Reducing Macrophage Inflammation and Lipid Loading. Cell Metab. 2011, 14, 747–757. 10.1016/j.cmet.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Thomas C.; Gioiello A.; Noriega L.; Strehle A.; Oury J.; Rizzo G.; Macchiarulo A.; Yamamoto H.; Mataki C.; Pruzanski M.; Pellicciari R.; Auwerx J.; Schoonjans K. TGR5-Mediated Bile Acid Sensing Controls Glucose Homeostasis. Cell Metab. 2009, 10, 167–177. 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Katsuma S.; Hirasawa A.; Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 2005, 329, 386–390. 10.1016/j.bbrc.2005.01.139. [DOI] [PubMed] [Google Scholar]
- a Perino A.; Pols T. W. H.; Nomura M.; Stein S.; Pellicciari R.; Schoonjans K. TGR5 reduces macrophage migration through mTOR-induced C/EBPβ differential translation. J. Clin. Invest. 2014, 124, 5424–5436. 10.1172/jci76289. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sonne D. P.; Hansen M.; Knop F. K. MECHANISMS IN ENDOCRINOLOGY: Bile acid sequestrants in type 2 diabetes: potential effects on GLP1 secretion. Eur. J. Endocrinol. 2014, 171, R47–R65. 10.1530/eje-14-0154. [DOI] [PubMed] [Google Scholar]
- a Massafra V.; Pellicciari R.; Gioiello A.; van Mil S. W. C. Progress and challenges of selective Farnesoid X Receptor modulation. Pharmacol. Therapeut. 2018, 191, 162–177. 10.1016/j.pharmthera.2018.06.009. [DOI] [PubMed] [Google Scholar]; b Han C. Update on FXR Biology: Promising Therapeutic Target?. Int. J. Mol. Sci. 2018, 19, 2069. 10.3390/ijms19072069. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Guo C.; Chen W.-D.; Wang Y.-D. TGR5, Not Only a Metabolic Regulator. Front. Physiol. 2016, 7, 646. 10.3389/fphys.2016.00646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows V.; Kennedy L.; Kundu D.; Alpini G.; Francis H. Bile Acid Receptor Therapeutics Effects on Chronic Liver Diseases. Front. Med. 2020, 7, 15. 10.3389/fmed.2020.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Đanić M.; Stanimirov B.; Pavlović N.; Goločorbin-Kon S.; Al-Salami H.; Stankov K.; Mikov M. Pharmacological Applications of Bile Acids and Their Derivatives in the Treatment of Metabolic Syndrome. Front. Pharmacol. 2018, 9, 1382. 10.3389/fphar.2018.01382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Li T.; Chiang J. Y. L. Bile acid-based therapies for non-alcoholic steatohepatitis and alcoholic liver disease. Hepatobiliary Surg. Nutr. 2020, 9, 152–169. 10.21037/hbsn.2019.09.03. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gottlieb A.; Canbay A. Why Bile Acids Are So Important in Non-Alcoholic Fatty Liver Disease (NAFLD) Progression. Cells 2019, 8, 1358. 10.3390/cells8111358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ferrell J. M.; Chiang J. Y. L. Understanding Bile Acid Signaling in Diabetes: From Pathophysiology to Therapeutic Targets. Diabetes Metab. J. 2019, 43, 257–272. 10.4093/dmj.2019.0043. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhong M. TGR5 as a Therapeutic Target for Treating Obesity. Curr. Top. Med. Chem. 2010, 10, 386–396. 10.2174/156802610790980576. [DOI] [PubMed] [Google Scholar]
- Tiratterra E.; Franco P.; Porru E.; Katsanos K. H.; Christodoulou D. K.; Roda G. Role of bile acids in inflammatory bowel disease. Ann. Gastroenterol. 2018, 31, 266–272. 10.20524/aog.2018.0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park R.; Ji J. D. The Role of Bile Acid Receptors in Chronic Inflammatory Diseases. J. Rheum. Dis. 2017, 24, 253–260. 10.4078/jrd.2017.24.5.253. [DOI] [Google Scholar]
- Miyazaki-Anzai S.; Masuda M.; Kohno S.; Levi M.; Shiozaki Y.; Keenan A. L.; Miyazaki M. Simultaneous inhibition of FXR and TGR5 exacerbates atherosclerotic formation. J. Lipid Res. 2018, 59, 1709–1713. 10.1194/jlr.m087239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicciari R.; Fiorucci S.; Camaioni E.; Clerici C.; Costantino G.; Maloney P. R.; Morelli A.; Parks D. J.; Willson T. M. 6α-Ethyl-Chenodeoxycholic Acid (6-ECDCA), a Potent and Selective FXR Agonist Endowed with Anticholestatic Activity. J. Med. Chem. 2002, 45, 3569–3572. 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- Pellicciari R.; Pruzanski M.; Gioiello A.. The Discovery of Obeticholic Acid (Ocaliva): First-in-Class FXR Agonist. In Successful Drug Discovery; Fischer J., Klein C., Childers W. E., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA, 2018; Vol. 3, chapter 8, pp 197–244. [Google Scholar]
- a Festa C.; Renga B.; D’Amore C.; Sepe V.; Finamore C.; De Marino S.; Carino A.; Cipriani S.; Monti M. C.; Zampella A.; Fiorucci S. Exploitation of Cholane Scaffold for the Discovery of Potent and Selective Farnesoid X Receptor (FXR) and G-Protein Coupled Bile Acid Receptor 1 (GP-BAR1) Ligands. J. Med. Chem. 2014, 57, 8477–8495. 10.1021/jm501273r. [DOI] [PubMed] [Google Scholar]; b Pellicciari R.; Passeri D.; De Franco F.; Mostarda S.; Filipponi P.; Colliva C.; Gadaleta R. M.; Franco P.; Carotti A.; Macchiarulo A.; Roda A.; Moschetta A.; Gioiello A. Discovery of 3α,7α,11β-Trihydroxy-6α-ethyl-5β-cholan-24-oic Acid (TC-100), a Novel Bile Acid as Potent and Highly Selective FXR Agonist for Enterohepatic Disorders. J. Med. Chem. 2016, 59, 9201–9214. 10.1021/acs.jmedchem.6b01126. [DOI] [PubMed] [Google Scholar]; c Pellicciari R.; Gioiello A.; Sabbatini P.; Venturoni F.; Nuti R.; Colliva C.; Rizzo G.; Adorini L.; Pruzanski M.; Roda A.; Macchiarulo A. Avicholic Acid: A Lead Compound from Birds on the Route to Potent TGR5 Modulators. ACS Med. Chem. Lett. 2012, 3, 273–277. 10.1021/ml200256d. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Rizzo G.; Passeri D.; De Franco F.; Ciaccioli G.; Donadio L.; Rizzo G.; Orlandi S.; Sadeghpour B.; Wang X. X.; Jiang T.; Levi M.; Pruzanski M.; Adorini L. Functional Characterization of the Semisynthetic Bile Acid Derivative INT-767, a Dual Farnesoid X Receptor and TGR5 Agonist. Mol. Pharmacol. 2010, 78, 617–630. 10.1124/mol.110.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Pellicciari R.; Fiorucci S.; Pruzanski M.. Dérivés de l’acide biliaire en tant que des ligands de fxr pour la prévention ou le traitement de maladies ou conditions médiées par le fxr. WO 2008002573 A2, 03, 01, 2008.; f D’Amore C.; Di Leva F. S.; Sepe V.; Renga B.; Del Gaudio C.; D’Auria M. V.; Zampella A.; Fiorucci S.; Limongelli V. Design, Synthesis, and Biological Evaluation of Potent Dual Agonists of Nuclear and Membrane Bile Acid Receptors. J. Med. Chem. 2014, 57, 937–954. 10.1021/jm401873d. [DOI] [PubMed] [Google Scholar]
- Pellicciari R.; Gioiello A.; Macchiarulo A.; Thomas C.; Rosatelli E.; Natalini B.; Sardella R.; Pruzanski M.; Roda A.; Pastorini E.; Schoonjans K.; Auwerx J. Discovery of 6α-Ethyl-23(S)-methylcholic Acid (S-EMCA, INT-777) as a Potent and Selective Agonist for the TGR5 Receptor, a Novel Target for Diabesity. J. Med. Chem. 2009, 52, 7958–7961. 10.1021/jm901390p. [DOI] [PubMed] [Google Scholar]
- Anfuso B.; Tiribelli C.; Adorini L.; Rosso N. Obeticholic acid and INT-767 modulate collagen deposition in a NASH in vitro model. Sci. Rep. 2020, 10, 1699. 10.1038/s41598-020-58562-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- During this work, a patent application by Pellicciari et al. described the conversion methyl 3α-acetyloxy-6α-ethyl-12α-hydroxy-7-oxo-5β-cholan-24-oate to 12β-methyl-13-epi-18-nor-obeti cholic acid in a four-step procedure involving a triflic anhydride-promoted rearrangement, palladium-catalyzed hydrogenation, ester hydrolysis and reduction of the 7-oxo group with sodium borohydride. To the best of our knowledge, this is the only mention of a 12β-methyl-18-nor-bile acid in the literature. However, the analytical data provided for the 12β-methyl-13-epi-18-nor-obeti cholic acid in the patent application is inconsistent with the given structure and compounds with a C13-epi-configuration have not been observed in course of the study presented in this paper. Pellicciari R.; Gioiello A.; Gavin G.; Lewis R. D.; Yanik M.; Kim M. G.; Leusink F. R.; Koning B.; Hensel T.. Methods and intermediates for the preparation of bile acid derivatives. WO 2018237350 A1, 27, 12, 2018.
- Brieskorn C. H.; Mosandl H. Olefine der Cholanreihe aus Desoxycholsäure und Cholsäure. Arch. Pharm. 1981, 314, 118–127. 10.1002/ardp.19813140205. [DOI] [Google Scholar]
- Shimizu T.; Ohzeki T.; Hiramoto K.; Hori N.; Nakata T. Chloromethanesulfonate as an Efficient Leaving Group: Rearrangement of the Carbon-Carbon Bond and Conversion of Alcohols into Azides and Nitriles. Synthesis 1999, 1999, 1373–1385. 10.1055/s-1999-3541. [DOI] [Google Scholar]
- a Fengyun Y.Method for producing ursodesoxycholic acid from 98.0% of cholic acid of cattle and sheep. CN 2008101215310 A, 2008.; b Salunke D. B.; Ravi D. S.; Pore V. S.; Mitra D.; Hazra B. G. Amino Functionalized Novel Cholic Acid Derivatives Induce HIV-1 Replication and Syncytia Formation in T Cells. J. Med. Chem. 2006, 49, 2652–2655. 10.1021/jm051114u. [DOI] [PubMed] [Google Scholar]; c Fieser L. F.; Rajagopalan S. Oxidation of Steroids. III. Selective Oxidations and Acylations in the Bile Acid Series. J. Am. Chem. Soc. 1950, 72, 5530–5536. 10.1021/ja01168a046. [DOI] [Google Scholar]
- a Harburn J. J.; Loftus G. C.; Marples B. A. Synthesis of Novel Steroidal Inhibitors of HIV-I Protease. Tetrahedron 1998, 54, 11907–11924. 10.1016/s0040-4020(98)83048-2. [DOI] [Google Scholar]; b Namegawa K.; Iida K.; Omura K.; Ogawa S.; Hofmann A. F.; Iida T. Chemical Synthesis of Rare Natural Bile Acids: 11α-Hydroxy Derivatives of Lithocholic and Chenodeoxycholic Acids. Lipids 2018, 53, 403–411. 10.1002/lipd.12013. [DOI] [PubMed] [Google Scholar]
- a Mander L. N.; Williams C. M. Chromatography with silver nitrate: part 2. Tetrahedron 2016, 72, 1133–1150. 10.1016/j.tet.2016.01.004. [DOI] [Google Scholar]; b Williams C. M.; Mander L. N. Chromatography with silver nitrate. Tetrahedron 2001, 57, 425–447. 10.1016/s0040-4020(00)00927-3. [DOI] [Google Scholar]
- a Heretsch P.; Giannis A.. The Veratrum and Solanum Alkaloids. In Alkaloids: Chemistry and Biology; Knölker H.-J., Ed.; Elsevier Inc, 2015; Vol. 74, chapter 4, pp 201–232. [DOI] [PubMed] [Google Scholar]; b Heretsch P.; Tzagkaroulaki L.; Giannis A. Cyclopamine and Hedgehog Signaling: Chemistry, Biology, Medical Perspectives. Angew. Chem., Int. Ed. 2010, 49, 3418–3427. 10.1002/anie.200906967. [DOI] [PubMed] [Google Scholar]
- a Bhattarai K. M.; Davis A. P.; Perry J. J.; Walter C. J.; Menzer S.; Williams D. J. A New Generation of “Cholaphanes”: Steroid-Derived Macrocyclic Hosts with Enhanced Solubility and Controlled Flexibility. J. Org. Chem. 1997, 62, 8463–8473. 10.1021/jo971272w. [DOI] [PubMed] [Google Scholar]; b Schteingart C. D.; Hofmann A. F. Synthesis of 24-nor-5 beta-cholan-23-oic acid derivatives: a convenient and efficient one-carbon degradation of the side chain of natural bile acids. J. Lipid Res. 1988, 29, 1387–1395. 10.1016/s0022-2275(20)38445-5. [DOI] [PubMed] [Google Scholar]
- a Dayal B.; Ertel N. H.; Padia J.; Rapole K. R.; Salen G. 7β-hydroxy bile alcohols: Facile synthesis and 2D 1H NMR studies of 5β-cholestane-3α,7β,12α,25-tetrol. Steroids 1997, 62, 409–414. 10.1016/s0039-128x(97)00007-x. [DOI] [PubMed] [Google Scholar]; b Dayal B.; Shefer S.; Tint G. S.; Salen G.; Mosbach E. H. Synthesis of 5β-cholestane-3α, 7α, 12α, 25-tetrol and 5β-cholestane-3α, 7α, 12α, 24ξ, 25-pentol. J. Lipid Res. 1976, 17, 74–77. 10.1016/s0022-2275(20)37019-x. [DOI] [PubMed] [Google Scholar]; c Dayal B.; Bagan E.; SpeckSalen J. G. A facile synthesis of 5β-cholestane-3α,7α,12α,25-tetrol. Steroids 1980, 35, 439–444. 10.1016/0039-128x(80)90144-0. [DOI] [PubMed] [Google Scholar]
- Horiuti I.; Polanyi M. Exchange reactions of hydrogen on metallic catalysts. Trans. Faraday Soc. 1934, 30, 1164–1172. 10.1039/tf9343001164. [DOI] [Google Scholar]
- a Zaera F. Key unanswered questions about the mechanism of olefin hydrogenation catalysis by transition-metal surfaces: a surface-science perspective. Phys. Chem. Chem. Phys. 2013, 15, 11988–12003. 10.1039/c3cp50402f. [DOI] [PubMed] [Google Scholar]; b Mattson B.; Foster W.; Greimann J.; Hoette T.; Le N.; Mirich A.; Wankum S.; Cabri A.; Reichenbacher C.; Schwanke E. Heterogeneous Catalysis: The Horiuti-Polanyi Mechanism and Alkene Hydrogenation. J. Chem. Educ. 2013, 90, 613–619. 10.1021/ed300437k. [DOI] [Google Scholar]
- Iqbal M.; Black R. J. G.; Winn J.; Reeder A. T.; Blake A. J.; Clarke P. A. Studies on transannulation reactions across a nine-membered ring: the synthesis of natural product-like structures. Org. Biomol. Chem. 2011, 9, 5062–5078. 10.1039/c1ob05448a. [DOI] [PubMed] [Google Scholar]
- Kuramoto T.; Kawamoto K.; Moriwaki S.; Hoshita T. Synthesis of homoursodeoxycholic acid and [11,12-3H]homoursodeoxycholic acid. Steroids 1984, 44, 549–559. 10.1016/s0039-128x(84)80036-7. [DOI] [PubMed] [Google Scholar]
- a Sepe V.; Ummarino R.; D’Auria M. V.; Renga B.; Fiorucci S.; Zampella A. The First Total Synthesis of Solomonsterol B, a Marine Pregnane X Receptor Agonist. Eur. J. Org. Chem. 2012, 5187–5194. 10.1002/ejoc.201200619. [DOI] [Google Scholar]; b Adhikari R.; Cundy D. J.; Francis C. L.; Gebara-Coghlan M.; Krywult B.; Lubin C.; Simpson G. W.; Yang Q. A Scalable Stereoselective Synthesis of Scymnol. Aust. J. Chem. 2005, 58, 34–38. 10.1071/ch04175. [DOI] [Google Scholar]
- a Kazuno T.; Moori A.; Sasaki K.; Mizuguchi M. The Synthesis of Trihydroxybisnorsterocholane and Homocholic Acid. Proc. Jpn. Acad. 1952, 28, 421–424. 10.2183/pjab1945.28.421. [DOI] [Google Scholar]; b Marples B. A.; Stretton R. J.. Anti-fungal pharmaceutical compositions comprising cholane derivatives. GB 2243550 A, 06, 11, 1991.; c Pearlman W. H. The Preparation of C-27 Steroids from Bile Acids. I. Coprostanetetrol-3(α), 7(α),12(α),25. J. Am. Chem. Soc. 1947, 69, 1475–1476. 10.1021/ja01198a066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.