

Abstract
Aims
Pathological cardiac remodelling and subsequent heart failure represents an unmet clinical need. Long non-coding RNAs (lncRNAs) are emerging as crucial molecular orchestrators of disease processes, including that of heart diseases. Here, we report on the powerful therapeutic potential of the conserved lncRNA H19 in the treatment of pathological cardiac hypertrophy.
Method and results
Pressure overload-induced left ventricular cardiac remodelling revealed an up-regulation of H19 in the early phase but strong sustained repression upon reaching the decompensated phase of heart failure. The translational potential of H19 is highlighted by its repression in a large animal (pig) model of left ventricular hypertrophy, in diseased human heart samples, in human stem cell-derived cardiomyocytes and in human engineered heart tissue in response to afterload enhancement. Pressure overload-induced cardiac hypertrophy in H19 knock-out mice was aggravated compared to wild-type mice. In contrast, vector-based, cardiomyocyte-directed gene therapy using murine and human H19 strongly attenuated heart failure even when cardiac hypertrophy was already established. Mechanistically, using microarray, gene set enrichment analyses and Chromatin ImmunoPrecipitation DNA-Sequencing, we identified a link between H19 and pro-hypertrophic nuclear factor of activated T cells (NFAT) signalling. H19 physically interacts with the polycomb repressive complex 2 to suppress H3K27 tri-methylation of the anti-hypertrophic Tescalcin locus which in turn leads to reduced NFAT expression and activity.
Conclusion
H19 is highly conserved and down-regulated in failing hearts from mice, pigs and humans. H19 gene therapy prevents and reverses experimental pressure-overload-induced heart failure. H19 acts as an anti-hypertrophic lncRNA and represents a promising therapeutic target to combat pathological cardiac remodelling.

Keywords: Cardiovascular disease, Hypertrophy, Long non-coding RNA, Gene therapy, Translational research
Graphical Abstract

See page 3475 for the editorial comment on this article (doi: 10.1093/eurheartj/ehaa663)
Translational perspective
Pathological cardiac hypertrophy is an abnormal increase in cardiomyocyte size, leading to an enlargement of the heart muscle with functional deterioration. Therapeutic options to halt or reverse cardiac hypertrophy are extremely limited. Long non-coding RNAs are emerging as key regulators of pathological processes including heart disease and represent novel, druggable molecules. In a step towards translation of non-coding RNA therapeutics, this study presents the preclinical development of murine and human lncRNA H19 for the treatment of cardiac hypertrophy. First toxicological assessments of tailored cardiomyocyte-directed H19 gene therapy vectors are promising, opening up pathways for clinical development of H19-based cardiac therapeutics.
Introduction
Pathological hypertrophy caused by chronic hypertension, genetic predisposition, or aortic stenosis leads to maladaptive remodelling and is often followed by ventricular dysfunction and subsequently cardiac heart failure and death.1 Current treatments with anti-hypertensive drugs, such as β-blockers, calcium antagonists, or angiotensin-converting enzyme (ACE) inhibitors,2 are rather symptomatic approaches and patients with cardiac hypertrophy face an unfavourable prognosis, emphasizing the need for novel therapeutic options.
During the past few decades, several genes have been identified which are involved in cardiac hypertrophy; however, the precise underlying molecular mechanisms for this disease remain ill-understood. In addition to protein-coding transcripts, in the past years, a few long non-coding RNAs (lncRNAs) have been described that are critically involved in cardiac hypertrophy.3–5 For instance, lncRNA cardiac-hypertrophy-associated transcript (Chast) promotes hypertrophy by disrupting beneficial autophagic processes via down-regulation of Plekhm1, a regulator of autophagy located on the opposite strand of Chast.6 LncRNA cardiac-hypertrophy-associated epigenetic regulator (Chaer), that is also pro-hypertrophic, physically interacts with polycomb repressive complex 2 (PRC2). Chaer functions as a decoy for PRC2, thus preventing spreading of repressive chromatin marks at the promoter regions of genes involved in cardiac hypertrophy.7 Another example is lncRNA maternally expressed gene 3 (Meg3) which promotes cardiac fibrosis, a hallmark of prolonged hypertensive stress.8 Pharmacological inhibition of both Chast and Meg3 in a mouse model of pressure overload-induced cardiac hypertrophy was sufficient to prevent cardiac remodelling and fibrosis, respectively, highlighting lncRNAs as valid targets for therapeutic strategies.
Moreover, lncRNA H19 which is transcribed from a highly conserved and paternally imprinted gene locus9 has been demonstrated to play important roles in foetal and early postnatal growth control in mice.10 A number of studies linked H19 to various types of cancer, where it can sponge different microRNAs, serve as host gene for microRNA-675 (miR-675), or interacts with different chromatin remodelling complexes.11 H19 has been previously associated with cardiac diseases. Nevertheless, the current literature is controversial and it remains unclear whether H19 is cardioprotective or whether it promotes heart diseases. For instance, in a rat model of adriamycin-induced dilated cardiomyopathy H19 was described to promote apoptosis,12 while another report proposes H19 as a negative regulator of left ventricular hypertrophy through a mechanism involving H19-encoded miR-675.13 A potential explanation for this discrepancy is the well-known fact that lncRNAs can be differentially expressed in different cell types to exert distinct cell type-specific functions.5,14
Since H19 expression peaks early after the induction of pressure overload-induced left ventricular heart failure by transverse aortic constriction (TAC) but is strongly repressed in the decompensated phase of hypertrophic cardiac remodelling, we hypothesized that H19 may have cardiomyocyte-specific protective functions and therefore, may be exploited as a therapeutic target.
Methods
Detailed methods are provided in the Supplementary material online.
Human tissue sampling
This study was performed with the approval of the institutional ethics committees of the University of Würzburg, Germany, Medical College of Wisconsin, Milwaukee, USA, the University Medical Center Hamburg-Eppendorf, Germany, and the Hannover Medical School, Germany. For details, see Supplementary material online.
Animal procedures and analysis of cardiac function
All procedures involving animals have been reviewed and approved by local animal welfare bodies of the Hannover Medical School. For details, see Supplementary material online. At indicated endpoints post-TAC surgery, echocardiographic measurements were performed with a Vevo 2100 (VisualSonics Inc., Toronto, Canada) and analysed using the Vevo LAB software (VisualSonics Inc., Toronto, Canada).
Cell culture, treatments, and cellular assays
HL-1 cardiomyocyte-like cells were cultured according to standard protocols. Directed differentiation of human-induced pluripotent stem cells (hiPSC) towards cardiomyocytes was achieved according to published protocols. Cell transfection and treatments, as well as subsequent cellular assays, were performed as indicated the Supplementary material online.
Gene and protein expression analysis
Total RNA was isolated applying TriFast method (Peqlab, Erlangen, Germany) or miRNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Between 100 to 1000 ng was reverse transcribed into complementary cDNA and assessed by quantitative real-time PCR analysis (qRT-PCRs) or used for transcriptome profiling. Proteins were analysed by standard immunoblotting using specific primary and secondary antibodies (Supplementary material online).
RNA immunoprecipitation
RNA immunoprecipitation (RIP) was performed in nuclear lysates according to previous established protocols (Supplementary material online).
ChiPSeq and ChiP-qRT-PCR
ChiPSeq sampling, analysis, and validation using qRT-PCRs were performed according to standard laboratory protocols as indicated the Supplementary material online.
Statistics
Data are displayed as means ± SD. Statistical analysis was carried out using GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA). Comparison among two groups was made by two-tailed unpaired Student’s t-test. For comparison of three or more than two groups, one-way analysis of variance (ANOVA) corrected by the Bonferroni post-test was applied. In all cases, P < 0.05 was considered as statistically significant.
Results
Muscle-enriched H19 is dysregulated in experimental cardiac hypertrophy in mice, pigs, and in human diseased heart samples
We first demonstrated in a wide range of different tissues of adult mice that H19 is muscle specific with the highest expression in skeletal muscle and a ∼10-fold enrichment of cardiac H19 over all other tissues (Figure 1A). Within the major cardiac cell types, the expression in cardiomyocytes is lower compared to endothelial cells, but higher than in cardiac fibroblasts (Figure 1B). H19 is strongly down-regulated during cardiac development in postnatal mice and further repressed in aged mice (>2 years) (Figure 1C). Since the activation of a foetal gene program is a hallmark of heart failure,15 we reasoned that H19 may play a role in the development of cardiac hypertrophy and subsequent heart failure. We monitored the cardiac expression of H19 in mice for 13 weeks after the initiation of pressure overload-induced left ventricular hypertrophy by TAC. Consistent with previous reports, H19 expression increased in the initial phase (2 weeks post-TAC).13,16 Nonetheless, during the progression from the compensated stage to the decompensated stage of heart failure (4–6 weeks after TAC) H19 expression was significantly down-regulated and remained low until the experimental endpoint 13 weeks after TAC (Figure 1D and Supplementary material online, Table S1 for full echocardiographic evaluation during HF development). During the decompensated phase, H19 is regulated on the transcriptional level, as shown by a significant down-regulation of the H19 precursor RNA, 6 and 13 weeks post-TAC (Supplementary material online, Figure S1A). Importantly, miR-675, which was suggested to play a role in cardiac hypertrophy,13 did not follow the expression pattern of H19 and remained relatively constant throughout the TAC-time course (Supplementary material online, Figure S1B). MiR-675 was far less abundant compared to the expression of the H19 host gene (Supplementary material online, Figure S1C). Additionally, Camk2d which was the suggested downstream mediator of miR-675 in hypertrophy was also not regulated until 13 weeks post-TAC (Supplementary material online, Figure S1D). In contrast to most lncRNAs, H19 is not only locus but also highly sequence-conserved among mammals (Supplementary material online, Figure S2A,B) implying an important functional role and also making H19 a potential therapeutic target in human cardiovascular disease. To elucidate this possibility, we first measured the expression of H19 in human iPSC-derived cardiomyocytes. Disease modelling with pro-hypertrophic agents such as isoproterenol led to rapid and sustained repression of H19 (Figure 1E). Next, we tested H19 in human engineered heart tissue (hEHT), which was exposed to increased afterload to model pathological hypertrophy.17 Afterload enhancement led to significant contractile force reduction compared to control hEHTs, paralleled by a strong reduction of H19 expression after 7 days of afterload enhancement (Figure 1F).
Figure 1.

Cardiac hypertrophy-associated repression of H19 is conserved among mice, humans, and pigs. (A) Abundance of H19 in various mouse organs (n = 3). (B) Distribution of H19 in a cardiac fraction of murine hearts (CM, cardiomyocytes; CF, cardiac fibroblasts; EC, endothelial cells; n = 8). (C) H19 in stages of murine heart development (n = 4–5). (D) H19 expression levels, heart weight-to-tibia-length ratio (HW/TL), and ejection fraction (EF) over the time course of heart failure progression (TAC, transverse aortic constriction; n = 5–8). (E) H19 gene expression in human induced pluripotent stem cell (iPSC)-derived cardiomyocytes after pro-hypertrophic stimulation (Iso, isoproterenol; n = 3). (F) Contractile force measurements and human H19 levels in human engineered heart tissue with and without afterload enhancement (AE; n = 6–8). (G) Expression of human H19 in patient-derived cardiac material compared to corresponding control tissues (indicated as dashed line) (HOCM, hypertrophic obstructive cardiomyopathy; LVAD, left ventricular assist device). (H) H19 Expression in different heart segments of hypertrophic pigs and Sham animals. Data are means ± SD. P-values were determined by two-tailed unpaired Student’s t-test. *P < 0.05; **P < 0.01; ***P < 0.001.
Of high translational importance, H19 was found to be repressed in human heart samples from patients with different diseases including aortic stenosis, hypertrophic cardiomyopathy, and failing hearts compared to cardiac tissue from healthy individuals (Figure 1G, Supplementary material online, Table S2). In line with high expression levels of H19 during murine heart development, H19 was also highly expressed in human foetal heart tissue (Figure 1G, Supplementary material online, Table S2). Moreover, since H19 is also well conserved in pigs (Supplementary material online, Figure S2B), we tested the regulation of H19 in hypertrophic pig hearts representing a clinically relevant large animal model. Cardiac hypertrophy in pigs was induced by percutaneous implantation of undersized bare-metal stents in the descending aorta of growing pigs resulting in a gradual increase in afterload during the growth of the animals (Supplementary material online, Figure S2C).18 Importantly, H19 levels were strongly decreased in various sections of the hypertrophic left ventricle compared with healthy (Sham) hearts (Figure 1H).
Lack of H19 augments cardiac hypertrophy in vivo
We next investigated whether H19 is functionally involved in the development of cardiac hypertrophy in vivo using H19 knock-out (KO) mice,19 which underwent TAC surgery and were monitored for 6 weeks (Figure 2A). Indeed, cardiac mass was significantly increased in H19-KO mice after TAC compared to wild-type mice concomitant with a trend towards increased cardiac dimensions (Supplementary material online, Table S3). The increase in cardiac hypertrophy in H19 KO mice was paralleled by increased cardiomyocyte size and higher Mcip1.4 expression in comparison to wild-type mice after TAC (Figure 2B–D). Interestingly, under basal conditions (Sham-operated mice), a trend towards a higher expression of pro-hypertrophic marker genes Nppb and Mcip1.4 in H19 KO compared to wild-type littermates was already observed (Figure 2D). The moderately increased HW/TL ratio in Sham KO mice can be explained by the known overgrowth phenotype of these mice10 as suggested by higher body weight but similar heart to body weight ratios compared to wild-type controls (Supplementary material online, Figure S3A,B).
Figure 2.

H19 knock-out exacerbates cardiac hypertrophy in vivo. (A) Schematic representation of the experimental design. (B) Heart weight-to-tibia-length ratio (HW/TL) of H19 KO and wild-type littermates (Wt) and (C) cardiomyocyte size (n = 6; DAPI = 4′,6-diamidino-2-phenylindole; TAC, transverse aortic constriction; WGA, wheat germ agglutinin coupled to Alexa Flour 488). (D) Expression of hypertrophic marker genes Nppb (natriuretic peptide B) and Mcip1.4 (myocyte-enriched calcineurin-interacting protein 1.4). (E) Microarray analysis of fractionated cardiomyocytes from H19 KO and Wt animals (n = 3) 6 weeks after TAC represented as heatmaps of indicated gene sets. Data are means ± SD. P-values were determined by one-way ANOVA with Bonferroni correction.
To study cardiomyocyte-specific molecular changes underlying the observed effects of H19, we performed mRNA expression profiling on purified cardiomyocytes from wild-type and H19 KO hearts 6 weeks after TAC surgery. To identify coordinated changes in the expression of functionally related genes, we performed gene set enrichment analysis.20,21 A total of 347 gene sets were enriched in H19 KO cardiomyocytes. Amongst them, we found several pathways associated with cardiac disease and hypertrophy such as the NFATc3 and transforming growth factor (TGF)-beta signalling pathways as well as cardiac hypertrophy-related genes (Figure 2E). In contrast, only four gene sets, all related to inflammatory response pathways, were enriched in wild-type hearts. (Supplementary material online, Table S4 provides a complete list of interrogated gene sets.) Supplementary material online, Figure S4A,B shows a volcano plot of all genes and the 50 most up- or down-regulated genes in single-gene comparison between H19 KO and wild-type cardiomyocytes.
H19 associates with PRC2 to control NFAT signalling
To gain mechanistic insight into the molecular role of H19, we first performed subcellular fractionation of murine cardiomyocytes. H19 was localized in the cytoplasmic as well as in the nuclear fraction, with the majority of nuclear H19 being bound to chromatin (Figure 3A), suggesting a potential involvement in epigenetic regulation of cardiomyocyte gene expression. The cytoplasmic/nuclear distribution was not affected upon hypertrophic stimulation (Supplementary material online, Figure S5). A common function of lncRNAs is their association with regulatory proteins (transcription factors, chromatin remodellers) to tether them as a ribonucleoprotein complex to their target sites.5 In this context, H19 was previously shown to interact with Enhancer Of Zeste 2 (EZH2) in cancer cells, a component of the histone methyltransferase polycomb repressive complex 2 (PRC2) that primarily tri-methylates lysine residue 27 on histone H3 (H3K27me3)22 establishing suppressive histone marks. Indeed, our in silico analysis revealed a high interaction propensity between the central region of H19 and EZH2 (Figure 3B). RNA immunoprecipitation (RIP) with specific antibodies against EZH2 confirmed this interaction in vitro as indicated by 200-fold enrichment of H19 over the IgG control and a stronger enrichment compared to lncRNA Neat1, a known EZH2 interactor.23 Knock-down of H19 by siRNA abolished the enrichment suggesting that the interaction between H19 and EZH2 is specific (Figure 3C). In addition, RIP with antibodies against the other main subunits of PRC2, SUZ12, and embryonic ectoderm development (EED), also pulled down H19 but not the lncRNA Nron which served as a negative control (Figure 3D).24 To gain further insight into the potential regulation of PRC2 by H19, we performed spike-in controlled H3K27me3 ChIP-Seq experiments in HL-1 cells after silencing (siRNA) or overexpression (lentivirus) of H19 (Supplementary material online, Figure S6A,B). Repression of H19 led to a global increase of H3K27me3 as indicated by the emergence of 6386 ChIP-Seq peaks that are unique to the siRNA condition. In contrast, H3K27me3 levels upon H19 overexpression were comparable to controls suggesting that high levels of H19 have no supra-dose effects on PRC2 (Figure 4A–C). Surprisingly, when interrogating the H3K27me3 signatures in the individual loci, we found no changes in the Nfatc3 locus, which was confirmed by ChIP-Seq and ChIP-PCR (Supplementary material online, Figure S6C–E). This suggests a non-direct regulation of the Nfatc3 locus, by H19-mediated PRC2 regulation. Indeed, among the most differentially affected loci we identified the anti-hypertrophic NFAT mediator Tescalcin (Tesc)25,26 locus to be heavily methylated upon H19 siRNA treatment (Figure 4D). This was confirmed in independent ChIP-qPCR experiments (Figure 4E). In line with increased H3K27 tri-methylation, H19 siRNA treatment resulted in lower Tesc mRNA levels which were partially rescued by knock-down of EZH2 (Figure 4E, Supplementary material online, Figure S6F). Overexpression of H19 under basal condition had no effect on Tesc H3K27 tri-methylation or mRNA expression (Figure 4F). Tescalcin is known to act on NFAT signalling by inhibiting calcineurin26 and by keeping GSK3 in its active form to phosphorylate NFAT for nuclear exclusion.25 We therefore tested Nfatc3 expression and activity and found increased NFATc3 protein levels and increased NFATc3 activity on its target promoters of Nppb and Mcip1.4 after H19 knock-down as demonstrated in western blot and ChIP-PCR using specific NFATc3 antibodies (Figure 4G,H). In support of the in vitro data, we found Tesc to be increasingly suppressed during the course of TAC in mice while Nfatc3 mRNA expression was increased (Supplementary material online, Figure S6G,H).
Figure 3.

Nuclear H19 interacts with EZH2 to regulate NFAT signalling. (A) Cytoplasmatic, nuclear soluble, and chromatin-associated abundance of H19 and controls in subcellular fractions of HL-1 cardiomyocytes (ActB, actin beta; Gapdh, glyceraldehyde-3-phosphate dehydrogenase; Neat1, nuclear paraspeckle assembly transcript 1; Xist, X inactive specific transcript) (n = 3). (B) Interaction score and binding propensities for EZH2 interaction with H19 predicted by CatRAPID. The heatmap indicates the interaction score (ranging from −3 to +3) of individual amino acid and nucleotide pairs. (C) Validation of the direct binding between H19 and EZH2 by RNA immunoprecipitation (RIP) in nuclear lysates of HL-1 cardiomyocytes in the presence and absence (si-H19) of H19 compared to controls. (D) Validation of direct interaction of H19 with components of the PRC2 complex (EED, Embryonic Ectoderm Development, SUZ12, SUZ12 Polycomb Repressive Complex 2 Subunit). Data are means ± SD.
Figure 4.

Modulation of H19 expression alters H3K27me3 patterns in HL-1 cells. (A) Venn diagram analysis of H3K27me3 ChIP-Seq data. (B) Peak correlation scatterplot for pairwise comparison. (C) Cluster heatmaps showing peak intensities of H3K27me3. (D) H3K27me3 peak profile in the Tescalcin (Tesc) locus depicted with Integrated Genome Viewer (IGV). (E) ChIP-qPCR validation for H3K27me3 in the Tesc locus (pTesc: −716 to −596 nt) and Tesc expression after H19 and Ezh2 knockdown. (F) ChIP-qPCR validation for H3K27me3 in the Tesc locus (pTesc: −716 to −596 nt) and Tesc expression after lentiviral-based H19 overexpression. (G) Immunoblotting of NFATc3 upon H19 knockdown. (H) ChIP-qPCR for NFATc3 binding to the Nppb and Mcip1.4 promoter after H19 knockdown. Data are means ± SD. P-values were determined by two-tailed unpaired Student’s t-test.
Altogether, this suggests H19 to function as a key suppressor of NFAT signalling through preventing PRC2-mediated epigenetic repression of the Tesc locus.
H19 is anti-hypertrophic in vitro
To test whether in contrast to the loss of H19, its overexpression may ameliorate hypertrophic responses, we stably overexpressed H19 in HL-1 cells. Similar to hiPSC-derived cardiomyocytes, induction of cellular hypertrophy after treatment with phenylephrine and isoproterenol (P + I) led to a reduction of H19 expression in these cells (Supplementary material online, Figure S7), while overexpression of H19 blunted the hypertrophic response (Figure 5A). This correlated with NFATc3 protein levels and, importantly, also with the levels of nuclear translocation of NFATc3 which were both increased after P + I stimulation, but rescued by H19 overexpression (Figure 5B,C). This was paralleled by lower NFATc3 activity as evidenced by lower expression of the molecular markers of hypertrophy (Nppa) and direct NFAT targets (Nppb and Mcip1.4) (P = 0.061 for Nppb) upon H19 overexpression (Figure 5D). To further confirm the H19-NFAT-Tesc-axis, we designed Tesc-specific siRNAs. P + I stimulation under simultaneous overexpression of H19 and repression of Tesc significantly increased the direct NFAT targets Nppb and Mcip1.4 (Figure 5E). Collectively, these data suggest that H19 acts upstream of Tesc. Ectopic induction of H19 expression is sufficient to suppress cardiomyocyte hypertrophy in vitro highlighting a potential for therapeutic approaches in vivo.
Figure 5.

Lentiviral overexpression of H19 blocks cardiac hypertrophy in vitro. (A) Size of H19 overexpressing (pLV+H19) cardiomyocytes after hypertrophic stimulation (48 h; DAPI, 4′,6-diamidino-2-phenylindole; I, isoproterenol; Phalloidin, Phalloidin–Tetramethylrhodamine B isothiocyanate; P, phenylephrine). (B) Immunohistochemical analysis of NFATc3 localization. (C) Immunoblotting of NFATc3 in cell lysates. (D) Expression levels of cardiac stress markers Nppa (natriuretic peptide A), Nppb (natriuretic peptide B), and Mcip1.4 (myocyte-enriched calcineurin-interacting protein 1.4; n = 3 independent experiments). (E) Validation of Tesc-specific siRNA (si-Tesc) and expression levels of Nppb and Mcip1.4 in H19-overexpressing cells (pLV+H19) after hypertrophic stimulation in presence and absence of Tesc (P + I, 48 h). Data are means ± SD. P-values were determined by one-way ANOVA with Bonferroni correction.
H19 therapy attenuates cardiac hypertrophy in vivo
We next tested whether H19 gene therapy may aid in preventing hypertrophy in vivo. We generated cardiotropic adeno-associated viruses (AAV9) expressing H19 under the control of the cardiomyocyte selective cardiac troponin T promoter (see Supplementary material online, Figure S8A for vector map). We administered 1.75+E12 viral particles per mouse via a single tail vein injection which led to a ∼80-fold induction of H19 expression in the heart (Supplementary material online, Figure S8B). Directly after TAC surgery, mice were injected with AAV9 to ensure full exogenous expression of H19 once the endogenous gene starts to get silenced (Figure 6A). Cardiomyocyte-specific H19 gene therapy significantly ameliorated the development of hypertrophy as demonstrated by reduced heart weight to tibia length ratios and smaller cardiomyocyte sizes compared to mice injected with AAV9-empty control virus (Figure 6B,C). This was in line with significantly improved cardiac function parameters in H19-treated mice (left ventricular volume and ejection fraction) as determined by echocardiography (Figure 6D, Supplementary material online, Table S5). We assessed potential adverse effects in liver and kidney as primary off-targets of the viral-based H19 gene therapy. H19 expression in the kidney was not affected, whereas the expression in the liver showed a slight but not significant increase (Supplementary material online, Figure S8C). Detailed histopathological analysis of liver and kidney tissue did not reveal any abnormalities in response to AAV-H19 treatment (Supplementary material online, Table S6). In addition, pre-clinical laboratory chemistry analyses of standard kidney, liver, and inflammation markers revealed no detrimental effects in response to H19 gene therapy (Supplementary material online, Figure S8D), collectively suggesting that at least for the tested period of 6 weeks this therapy is safe.
Figure 6.

H19 gene therapy prevents cardiac remodelling. (A) Schematic representation of the experimental design with cardiomyocyte-specific AAV9-H19 or control virus (AAV9-empty). (B) Heart-weight-to-Tibia-length ratios (HW/TL) of Sham- or TAC-operated mice and (C) cardiomyocyte size (n = 6; DAPI = 4′,6-diamidino-2-phenylindole; WGA, wheat germ agglutinin coupled to Alexa Flour 488; TAC, transverse aortic constriction). (D) Echocardiographic analysis of cardiac dimensions and function. Data are means ± SD. P-values were determined by one-way ANOVA with Bonferroni correction.
To better mimic clinical situations, we next tested whether H19 therapy is also effective when cardiac hypertrophy is already established. Importantly, to further investigate the translational potential, we included an additional group which was treated with the human (in place of murine) H19 gene (AAV-hH19, Supplementary material online, Figure S9A). Mice were injected with murine or human AAV9-H19 4 weeks after the induction of cardiac hypertrophy by TAC (Figure 7A). Strikingly, 4 weeks later AAV9-H19 as well as AAV-hH19-treated mice presented with significantly smaller hearts concomitant with smaller cardiomyocytes sizes compared to AAV-empty-treated controls (Figure 7B,C). The capillary density determined by CD31 immunostaining revealed no differences between groups suggesting cardiomyocyte-specific therapeutic effects (Supplementary material online, Figure S9B). The number of immune cells (CD45+) was lower in H19-treated mice, presumably reflecting on lower clearing demands of stressed/dying cells (Supplementary material online, Figure S9C). Cardiac functional parameters as determined by echocardiography were improved in mice receiving either murine or human H19 (Figure 7D, Supplementary material online, Table S7). This observation was paralleled by significantly increased Tesc mRNA expression in H19-treated mice (Figure 7E) and further correlated with lower mRNA expression of the H19-PRC2-Tesc target Nfatc3 and the hypertrophy markers Nppb and Mcip1.4 in cardiac tissue (Figure 7F). The gene therapy also provoked a slight increase in miR-675 (relative to the induction of H19). Nevertheless, no change in the expression of Camk2d, the putative downstream target of miR-675, was observed, suggesting that the gene therapy effects are H19-specific (Supplementary material online, Figure S9D,E). Finally, we infected human iPSC-derived cardiomyocytes with AAV6-hH19 and prepared three-dimensional hEHTs. We observed a significantly increased force development over time in comparison to control hEHTs (AAV6-empty) (Figure 8A–C), again highlighting H19’s translational potential.
Figure 7.

H19 therapy is effective in hypertrophic hearts. (A) Schematic representation of the experimental design with murine and human H19 (AAV9-H19, AAV9-hH19 compared to empty vector AAV9-empty). (B) Heart-weight-to-Tibia-length ratios (HW/TL) of Sham- or TAC-operated mice and (c) cardiomyocyte size (n = 6; DAPI = 4′,6-diamidino-2-phenylindole; TAC, transverse aortic constriction; WGA, wheat germ agglutinin coupled to Alexa Flour 488). (D) Echocardiographic analysis. (E) Tesc expression levels. (F) Expression levels of Nfatc3, Nppb, and Mcip1.4. Data are means ± SD. P-values were determined by one-way ANOVA with Bonferroni correction.
Figure 8.
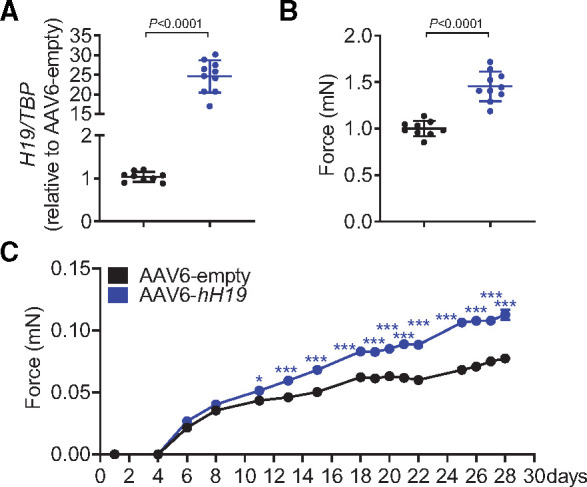
Administration of human H19 (hH19) improves contractile force of human engineered heart tissues. (A) AAV6-mediated overexpression of H19 in human engineered heart tissue (EHT) compared to control (AAV6-empty). (B) Increased contractile force 28 days after EHT preparation. (C). Force development over time. Data are means ± SD. P-values were determined by two-tailed unpaired Student’s t-test or one-way with Bonferroni correction.
In summary, these data suggest that not the initial peak of H19 expression, but its subsequent repression is detrimental in the development of cardiac hypertrophy in various species. Murine and human H19 gene therapy to prevent this repression during the decompensated stage is not only sufficient to suppress the development of cardiac hypertrophy but also its progression into heart failure when H19 is applied in a therapeutic approach.
Discussion
LncRNA H19 suppresses pro-hypertrophic signalling
Non-coding RNAs including microRNAs and lncRNAs are emerging as key mediators of developmental and pathological cues. In particular, lncRNAs have a wide spectrum of functions and can interact with regulatory proteins to orchestrate gene expression at any level to regulate these processes.4,5 Here, we report on the highly conserved and muscle-enriched lncRNA H19 which suppresses pro-hypertrophic signalling in mice after pressure overload-induced left ventricular cardiac failure. Experimental pressure overload of the left ventricle induced a transient increase of H19, which was followed by sustained repression of upon the transition to the decompensated stage of heart failure, suggesting that loss of H19 is detrimental. As hypothesized, the hypertrophy phenotype 6 weeks post-TAC in H19 KO mice was significantly worsened in comparison to wild-type controls as demonstrated on the histopathological level as well as on the molecular level using microarray analysis on isolated cardiomyocytes.
H19 interacts with PRC2 to control NFAT signalling via epigenetic control of the Tescalcin locus
Mechanistically, different lncRNAs were shown to interact with the epigenetic remodelling complex PRC2 to favour or to prevent binding and methylation of chromatin at specific subsets of genes.7,27,28 H19 was demonstrated to associate with the PRC2 subunit EZH2 in the setting of cancer cells.22,29 Since it was unknown whether this link exists in cardiac cells, we performed in silico and in vitro binding assays that demonstrated that H19 physically interacts with all major subunits of PRC2. Moreover, we found that H19 cooperates with EZH2/PRC2 to suppress pro-hypertrophic NFAT signalling. This is in contrast to previous studies suggesting that inhibition of EZH2 leads to cardiac hypertrophy.30,31 However, in our study PRC2 remains intact as a complex and our ChIP-seq experiments revealed that the H19-PRC2 complex controls only a subset of genes. As highly relevant for cardiac hypertrophy, we identified that in the absence of H19, PRC2 binds and hyper-methylates the anti-hypertrophic Tesc locus. Tescalcin is a known negative regulator of NFAT. Accordingly, our data show that loss of H19 leads to repression of Tesc in vitro and in vivo, which in turn increases NFATc3 levels and activity on its hypertrophy-related target genes Nppb and Mcip1.4. In contrast, increased NFAT signalling by hypertrophic stimuli in vitro (P + I) and in vivo (TAC) was abrogated in cells or cardiac tissue overexpressing H19 which further corroborates the suggested H19-PRC2-Tesc axis to suppress hypertrophic signalling (Take home figure). Importantly, we did not find any evidence for the previously described anti-hypertrophic effect of miR-675, which is encoded within the H19 locus.13 MiR-675 and its postulated downstream target Camk2d remain unaffected in the development of pressure overload-induced cardiac hypertrophy. Neither repression nor overexpression of H19 has an influence on the expression levels of Camk2d, reinforcing that the anti-hypertrophic effects described here are H19-specific and independent of miR-675. A possible explanation is the early time point (2 weeks post-TAC) which was analysed by Lantao and co-workers. In line, Camk2d levels increase early after TAC, peak at Day 4, and start to decrease as early as 7 days post-TAC surgery.32 Moreover, in vivo inhibition of miR-675 by miRNA inhibitors, which exacerbated cardiac hypertrophy,13 is not cell-type specific. This is in marked contrast to our cardiomyocyte-specific, therapeutic approach to prevent or reverse left ventricular heart failure, which seems to be independent of miR-675. With regards to H19 manipulation, cell-type specificity is extremely important since H19 also fulfils an important role in the vasculature. For example, while H19 is beneficial for endothelial cell function in a hind limb ischaemia model,33 it drives the progression of abdominal aortic aneurysm via smooth muscle cell-specific functions.34 Importantly, and in support of our findings, in both studies H19 functions independently of miR-675.
Take home figure.
H19 gene therapy blocks PRC2-mediated methylation of the Tescalcin promoter and disrupts NFAT signaling which subsequently prevents and reverses cardiac hypertrophy-mediated heart failure.
H19 gene therapy attenuates cardiac hypertrophy
Importantly, our in vitro rescue experiments indicated that exogenous H19 expression results in restored or gain of function. While this is prerequisite for therapeutic approaches in vivo, ectopic overexpression of the lncRNA transcript does not necessarily replace the endogenous function for reasons that are not fully understood to date. Based on the in vitro data, we designed AAV9 vectors for in vivo cardiomyocyte-specific H19 therapy, which significantly attenuated cardiac hypertrophy after TAC on the pathological and functional level. Importantly, in our therapeutic approach the viral particles were administered 4 weeks post-TAC when hypertrophy was already developed, and heart function declined. To further stress the translational perspective, we also performed gene therapy using the human H19 gene. Comparison of echocardiography assessment at this time point and at the experimental endpoint indicates that both murine and human H19 delivery is sufficient to stall the progression of hypertrophy. These results demonstrate that viral-based gene therapy may indeed be a therapeutic strategy to target lncRNA reconstitution to the heart, and specifically hypertrophic cardiomyocytes. However, based on our results further studies with longer post-TAC periods and more frequent echocardiography assessments are warranted. In contrast to inhibitory strategies, targeted delivery of lncRNAs is technically more challenging and this branch is less advanced. Thus, further research is needed to gain deeper insights into the potentially broad functional involvement of lncRNAs in cardiac homeostasis and disease as well as into their therapeutic exploitation. With regards to H19, future studies should also address the cell type-specific and potentially distinct roles in endothelial cells and cardiac fibroblasts.
LncRNA H19 as a translational therapeutic target
Although often locus conserved, lncRNAs generally show a poor degree of sequence conservation even among closely related species, which raised the question about the translation of results from pre-clinical models to humans. However, H19 is remarkably well conserved among higher vertebrates and we found H19 consistently down-regulated in murine and human cardiomyocytes after the hypertrophic stimulus, in hEHTs exposed to afterload enhancement, as well in vivo in diverse human diseased heart tissues, and in hypertrophic murine and pig hearts. Thus, these data are encouraging and further testing of an H19 therapy in large animals is justified. With regard to H19 expression in human heart tissue it should be noted that, due to differences in mean age between diseased and healthy hearts, lower levels of H19 may be partially attributed to an age-related reduction. Nevertheless, consistent repression of H19 in age-matched hypertrophic pig hearts and hEHTs clearly identify hypertrophy as the driving force for H19 repression.
H19 has also been termed as oncofetal lncRNA owing to its elevated expression in the embryo and in different human tumours. Cautions with regards to enforced H19 overexpression is therefore warranted.11 It remains unclear to date whether H19 alone can drive tumour initiation. Our H19 gene therapy approach used cardiotropic AAV9 particles in combination with the cardiomyocyte-specific cTNT promoter to restrict H19 expression to cardiomyocytes. After a maximum of 6 weeks of enforced H19 expression, we did not observe any signs of increased cancer formation or any abnormalities in the histopathological analysis of liver and kidney tissue (the most probable off-target sites of the gene therapy). In addition, toxicological assessments of plasma markers in mice showed no adverse effects. Nevertheless, further Tox studies in additional animal species (e.g. rat and pig) are required to complete preclinical development.
In summary, our data highlight H19 as an anti-hypertrophic lncRNA, which through genetic restoration attenuated cardiac hypertrophy in mice. H19 is highly conserved and dysregulated in hypertrophic hearts of pigs and humans. Thus, our study paves the way for further pre-clinical and clinical development of H19 therapies for the treatment of pathological cardiac hypertrophy.
Data availability
ChiP-seq data (GEO: GSE153375) and Microarray-based transcriptome profiling data (GEO: GSE153344) are available in the Gene Expression Omnibus.
Supplementary Material
Acknowledgements
We thank K. Zimmer, K. Scherf, I. Riedel, A. Just, and J. Bode for excellent technical support. We thank T. Maetzig and A. Schambach for providing the pLV+ lentiviral backbone.
Funding
Deutsche Forschungsgemeinschaft (KFO311/TH903/20-1 to T.T. and BA5631/2-1 to C.B.), the ERC Consolidator Grant LONGHEART (to T.T. under grant agreement No. 648038), and the European Union’s Horizon 2020 research and innovation programme under GA No. 825670 (CARDIOREGENIX). Hypertrophic pig experiments were supported by the European Commission FP7 Programme (FIBROTARGETS project grant HEALTH-2013-6029047, GA No. 602904).
Conflict of interests J.V. and T.T. have filed and licensed patents about the diagnostic and therapeutic use of several cardiovascular lncRNAs including H19. J.V. and S.B. are currently full-time employees of Cardior Pharmaceuticals GmbH. T.T. and S.B. are founders and shareholders of Cardior Pharmaceuticals GmbH. T.E. and M.N.H. are co-founders of EHT Technologies GmbH, which provides technical equipment for generation and video-optical analysis of EHTs.
References
- 1.Hill JA, Olson EN.. Cardiac plasticity. N Engl J Med 2008;358:1370–1380. [DOI] [PubMed] [Google Scholar]
- 2.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O'Flaherty M, Palaniappan LP, Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P; ; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee.. Heart disease and stroke statistics—2018 update: a report from the American Heart Association. Circulation 2018;137:e67–e492. [DOI] [PubMed] [Google Scholar]
- 3.Thum T, Condorelli G.. Long noncoding RNAs and microRNAs in cardiovascular pathophysiology. Circ Res 2015;116:751–762. [DOI] [PubMed] [Google Scholar]
- 4.Beermann J, Piccoli M-T, Viereck J, Thum T.. Non-coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiol Rev 2016;96:1297–1325. [DOI] [PubMed] [Google Scholar]
- 5.Bär C, Chatterjee S, Thum T.. Long noncoding RNAs in cardiovascular pathology, diagnosis, and therapy. Circulation 2016;134:1484–1499. [DOI] [PubMed] [Google Scholar]
- 6.Viereck J, Kumarswamy R, Foinquinos A, Xiao KK, Avramopoulos P, Kunz M, Dittrich M, Maetzig T, Zimmer K, Remke J, Just A, Fendrich J, Scherf K, Bolesani E, Schambach A, Weidemann F, Zweigerdt R, Windt L. D, Engelhardt S, Dandekar T, Batkai S, Thum T.. Long noncoding RNA Chast promotes cardiac remodeling. Sci Transl Med 2016;8:326ra22. [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, Zhang X-J, Ji Y-X, Zhang P, Deng K-Q, Gong J, Ren S, Wang X, Chen I, Wang H, Gao C, Yokota T, Ang YS, Li S, Cass A, Vondriska TM, Li G, Deb A, Srivastava D, Yang H-T, Xiao X, Li H, Wang Y.. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat Med Nat Res 2016;22:1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piccoli M-T, Gupta S, Viereck J, Foinquinos A, Samolovac S, Kramer F, Garg A, Remke J, Zimmer K, Batkai S, Thum T.. Inhibition of the cardiac fibroblast-enriched lncRNA Meg3 prevents cardiac fibrosis and diastolic dysfunction. Circ Res 2017;121:575–583. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Tycko B.. Monoallelic expression of the human H19 gene. Nat Genet 1992;1:40–44. [DOI] [PubMed] [Google Scholar]
- 10.Gabory A, Jammes H, Dandolo L.. The H19 locus: role of an imprinted non-coding RNA in growth and development. BioEssays 2010;32:473. [DOI] [PubMed] [Google Scholar]
- 11.Raveh E, Matouk IJ, Gilon M, Hochberg A.. The H19 long non-coding RNA in cancer initiation, progression and metastasis – a proposed unifying theory. Mol Cancer 2015;14:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Zhang M, Xu W, Chen J, Zhou X.. The long non-coding RNA H19 promotes cardiomyocyte apoptosis in dilated cardiomyopathy. Oncotarget 2017;8:28588–28594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu L, An X, Li Z, Song Y, Li L, Zuo S, Liu N, Yang G, Wang H, Cheng X, Zhang Y, Yang X, Wang J.. The H19 long noncoding RNA is a novel negative regulator of cardiomyocyte hypertrophy. Cardiovasc Res 2016;111:56–65. [DOI] [PubMed] [Google Scholar]
- 14.Chatterjee S, Bär C, Thum T.. Linc-ing the noncoding genome to heart function: beating hypertrophy. Trends Mol Med 2017;23:577–579. [DOI] [PubMed] [Google Scholar]
- 15.Kuwahara K, Nishikimi T, Nakao K.. Transcriptional regulation of the fetal cardiac gene program. J Pharmacol Sci 2012;119:198–203. [DOI] [PubMed] [Google Scholar]
- 16.Greco S, Zaccagnini G, Perfetti A, Fuschi P, Valaperta R, Voellenkle C, Castelvecchio S, Gaetano C, Finato N, Beltrami AP, Menicanti L, Martelli F.. Long noncoding RNA dysregulation in ischemic heart failure. J Transl Med 2016;14:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirt MN, Sörensen NA, Bartholdt LM, Boeddinghaus J, Schaaf S, Eder A, Vollert I, Stöhr A, Schulze T, Witten A, Stoll M, Hansen A, Eschenhagen T.. Increased afterload induces pathological cardiac hypertrophy: a new in vitro model. Basic Res Cardiol 2012;107:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gyöngyösi M, Pavo N, Lukovic D, Zlabinger K, Spannbauer A, Traxler D, Goliasch G, Mandic L, Bergler-Klein J, Gugerell A, Jakab A, Szankai Z, Toth L, Garamvölgyi R, Maurer G, Jaisser F, Zannad F, Thum T, Bátkai S, Winkler J.. Porcine model of progressive cardiac hypertrophy and fibrosis with secondary postcapillary pulmonary hypertension. J Transl Med 2017;15:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ripoche M. A, Kress C, Poirier F, Dandolo L.. Deletion of the H19 transcription unit reveals the existence of a putative imprinting control element. Genes Dev 1997;11:1596–1604. [DOI] [PubMed] [Google Scholar]
- 20.Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC.. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003;34:267–273. [DOI] [PubMed] [Google Scholar]
- 21.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo M, Li Z, Wang W, Zeng Y, Liu Z, Qiu J.. Long non-coding RNA H19 increases bladder cancer metastasis by associating with EZH2 and inhibiting E-cadherin expression. Cancer Lett 2013;333:213–221. [DOI] [PubMed] [Google Scholar]
- 23.Wang S, Zuo H, Jin J, Lv W, Xu Z, Fan Y, Zhang J, Zuo B.. Long noncoding RNA Neat1 modulates myogenesis by recruiting Ezh2. Cell Death Dis 2019;10:505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma S, Findlay GM, Bandukwala HS, Oberdoerffer S, Baust B, Li Z, Schmidt V, Hogan PG, Sacks DB, Rao A.. Dephosphorylation of the nuclear factor of activated T cells (NFAT) transcription factor is regulated by an RNA-protein scaffold complex. Proc Natl Acad Sci USA 2011;108:11381–11386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobayashi S, Nakamura TY, Wakabayashi S.. Calcineurin B homologous protein 3 negatively regulates cardiomyocyte hypertrophy via inhibition of glycogen synthase kinase 3 phosphorylation. J Mol Cell Cardiol 2015;84:133–142. [DOI] [PubMed] [Google Scholar]
- 26.Gutierrez-Ford C, Levay K, Gomes AV, Perera EM, Som T, Kim Y-M, Benovic JL, Berkovitz GD, Slepak VZ.. Characterization of tescalcin, a novel EF-hand protein with a single Ca2+-binding site: metal-binding properties, localization in tissues and cells, and effect on calcineurin. Biochemistry 2003;42:14553–14565. [DOI] [PubMed] [Google Scholar]
- 27.Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding H, Butty VL, Torrey L, Haas S, Abo R, Tabebordbar M, Lee RT, Burge CB, Boyer LA.. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell 2013;152:570–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grote P, Wittler L, Hendrix D, Koch F, Währisch S, Beisaw A, Macura K, Bläss G, Kellis M, Werber M, Herrmann BG.. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell 2013;24:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang D-M, Lin Z-Y, Yang Z-H, Wang Y-Y, Wan D, Zhong J-L, Zhuang P-L, Huang Z-Q, Zhou B, Chen W-L.. IncRNA H19 promotes tongue squamous cell carcinoma progression through β-catenin/GSK3β/EMT signaling via association with EZH2. Am J Transl Res 2017;9:3474–3486. [PMC free article] [PubMed] [Google Scholar]
- 30.Delgado-Olguín P, Huang Y, Li X, Christodoulou D, Seidman CE, Seidman JG, Tarakhovsky A, Bruneau BG.. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat Genet 2012;44:343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mathiyalagan P, Okabe J, Chang L, Su Y, Du X-J, El-Osta A.. The primary microRNA-208b interacts with Polycomb-group protein, Ezh2, to regulate gene expression in the heart. Nucleic Acids Res 2014;42:790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Bers DM, Brown JH.. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res 2003;92:912–919. [DOI] [PubMed] [Google Scholar]
- 33.Hofmann P, Sommer J, Theodorou K, Kirchhof L, Fischer A, Li Y, Perisic L, Hedin U, Maegdefessel L, Dimmeler S, Boon RA.. Long non-coding RNA H19 regulates endothelial cell aging via inhibition of STAT3 signalling. Cardiovasc Res 2019;115:230–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li DY, Busch A, Jin H, Chernogubova E, Pelisek J, Karlsson J, Sennblad B, Liu S, Lao S, Hofmann P, Bäcklund A, Eken SM, Roy J, Eriksson P, Dacken B, Ramanujam D, Dueck A, Engelhardt S, Boon RA, Eckstein H-H, Spin JM, Tsao PS, Maegdefessel L.. H19 induces abdominal aortic aneurysm development and progression. Circulation 2018;138:1551–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChiP-seq data (GEO: GSE153375) and Microarray-based transcriptome profiling data (GEO: GSE153344) are available in the Gene Expression Omnibus.