

Abstract
Tuberous sclerosis complex (TSC) is a multisystem developmental disorder characterized by hamartomas in various organs, such as the brain, lungs, and kidneys. Epilepsy, along with autism and intellectual disability, is one of the neurologic impairments associated with TSC that has an intimate relationship with developmental outcomes and quality of life. Sustained activation of the mammalian target of rapamycin (mTOR) via TSC1 or TSC2 mutations is known to be involved in the onset of epilepsy in TSC. However, the mechanism by which mTOR causes seizures remains unknown. In this study, we showed that, human induced pluripotent stem cell-derived TSC2-deficient (TSC2−/−) neurons exhibited elevated neuronal activity with highly synchronized Ca2+ spikes. Notably, TSC2−/− neurons presented enhanced Ca2+ influx via L-type Ca2+ channels (LTCCs), which contributed to the abnormal neurite extension and sustained activation of cAMP response element binding protein (CREB), a critical mediator of synaptic plasticity. Expression of Cav1.3, a subtype of LTCCs, was increased in TSC2−/− neurons, but long-term rapamycin treatment suppressed this increase and reversed the altered neuronal activity and neurite extensions. Thus, we identified Cav1.3 LTCC as a critical downstream component of TSC-mTOR signaling that would trigger enhanced neuronal network activity of TSC2−/− neurons. We suggest that LTCCs could be potential novel targets for the treatment of epilepsy in TSC.
SIGNIFICANCE STATEMENT There is a close relationship between elevated mammalian target of rapamycin (mTOR) activity and epilepsy in tuberous sclerosis complex (TSC). However, the underlying mechanism by which mTOR causes epilepsy remains unknown. In this study, using human TSC2−/− neurons, we identified elevated Ca2+ influx via L-type Ca2+ channels as a critical downstream component of TSC-mTOR signaling and a potential cause of both elevated neuronal activity and neurite extension in TSC2−/− neurons. Our findings demonstrate a previously unrecognized connection between sustained mTOR activation and elevated Ca2+ signaling via L-type Ca2+ channels in human TSC neurons, which could cause epilepsy in TSC.
Keywords: calcium, epilepsy, LTCC, mTOR, rapamycin, TSC
Introduction
Tuberous sclerosis complex (TSC) is a multisystem developmental disorder characterized by benign tumors called hamartomas in the brain, lungs, and kidneys (Crino et al., 2006). The causal genes for TSC are TSC1 and TSC2, which encode hamartin and tuberin, respectively (Sparagana and Roach, 2000; Montagne et al., 2001). Hamartin and tuberin form a protein complex that acts as a GTPase-activating protein for the small G-protein, Rheb (Yamagata et al., 1994; Garami et al., 2003; Inoki et al., 2003; Zhang et al., 2003). Heterozygous loss-of-function mutations in either TSC1 or TSC2 increase the GTP-bound form of Rheb, resulting in the hyperactivation of the mammalian target of rapamycin (mTOR) and the subsequent development of hamartomas in various tissues (Crino, 2013).
Some of the major clinical symptoms of TSC are neurologic and psychiatric impairments, including epilepsy, autism, and intellectual disabilities. Epilepsy, especially, is the most common neurologic symptom and is a critical issue affecting the quality of life of patients with TSC (Chu-Shore et al., 2010; Thiele, 2010). Up to 90% of patients with TSC have epilepsy that begins in infancy and early childhood, and over half of the cases are refractory to regular antiepileptic drugs (Overwater et al., 2015).
Using rodent models of TSC, previous studies have shown that intractable epilepsy in TSC is likely caused by constitutively activated mTOR. Brain-specific conditional Tsc1 and Tsc2 KO mice develop epilepsy as well as histologic abnormalities in the brain, including megalencephaly, somatic hypertrophy, neural disorganization, and ectopic axonal growth (Uhlmann et al., 2002; Meikle et al., 2007; Choi et al., 2008; Zeng et al., 2008, 2011; Magri et al., 2011; Abs et al., 2013; Koene et al., 2019). mTOR activity levels are correlated with the severity of epilepsy and neuropathology in the mouse brain (Zeng et al., 2011; Nguyen et al., 2019). Importantly, the mTOR inhibitor rapamycin or its derivatives prevent epilepsy, prolong survival, and ameliorate histologic brain abnormalities in Tsc model mice (Zeng et al., 2008, 2011; Carson et al., 2012; Lasarge and Danzer, 2014; Hsieh et al., 2016; Koene et al., 2019). Studies using two- or three-dimensional cultures of human induced pluripotent stem cells (iPSCs) or human embryonic stem cells also support the effect of rapamycin on the altered functional and morphologic changes in human neurons and glial cells with TSC mutations (Costa et al., 2016; Blair et al., 2018; Sundberg et al., 2018; Nadadhur et al., 2019; Winden et al., 2019). In addition, recent clinical studies reported that the use of mTOR inhibitors is a promising treatment for intractable epilepsy (Cardamone et al., 2014; French et al., 2016; Krueger et al., 2016; Mizuguchi et al., 2019). However, the mechanisms underlying seizure development in TSC remain unknown (Lasarge and Danzer, 2014). Why does excess mTOR activity trigger epilepsy in TSC? As mTOR inhibitors have important side effects, such as stomatitis and respiratory infections, new treatments that are more beneficial for patients with TSC are needed (Krueger et al., 2013; Fogarasi et al., 2016; French et al., 2016). However, the precise signaling pathways downstream of mTOR, which cause seizures in TSC, need to be understood in more detail to develop new treatments.
In this study, we generated TSC2-mutant neurons from human iPSCs and showed that TSC2−/− neurons had abnormally extended neurites (Costa et al., 2016; Blair et al., 2018). In addition, TSC2−/− neurons exhibited highly synchronized spontaneous neuronal activity with Ca2+ spikes. We further characterized the physiological properties of TSC2−/− neurons and identified enhanced Ca2+ influx via voltage-dependent L-type Ca2+ channels (LTCCs), possibly Cav1.3, as a downstream effect of sustained TSC-mTOR signaling. Increased Ca2+ influx via LTCCs partially contributed to the abnormally extended neurite elongation of TSC2−/− neurons and triggered sustained activation of the transcription factor, cAMP-responsive element binding (CREB). Our findings provide an important clue for understanding the signaling crosslink between TSC-mTOR and Ca2+ signaling via LTCC, which may be associated with epilepsy onset in TSC.
Materials and Methods
Establishment of human iPSCs with TSC2 mutations
Human iPSCs (HPS0002: 409B2) (Okita et al., 2011) were provided by RIKEN BRC through the National BioResource Project of the MEXT/AMED. The iPSCs were transfected with plasmids encoding sgRNA (ACAATCGCATCCGGATGATAGGG) and Cas9 by electroporation (NEPA21, NepaGene). After 4 d, genomic DNA was extracted from the transfected cells, and the fragment containing exon 3 of the TSC2 gene was amplified by PCR using the following primers: 5′-GATTCTCCTGCCTCACTCTCCC-3′ and 5′-AGTCAGCTGTCAACCATGTTCCT-3′. The fragments were subsequently digested with T7 endonuclease I (New England Biolabs) to verify the presence of mutations. After confirming the mutations, single iPSC clones were isolated, and TSC2 mutations were further confirmed by DNA sequencing. The established iPSCs were maintained in StemMACS iPS-Brew XF human (Miltenyi Biotec) medium in Matrigel matrix (Corning)-coated plastic dishes. This study was approved by the Tokyo Metropolitan Institute of Medical Science Committee.
Differentiation of iPSCs to neural progenitor cells (NPCs) and neurons
Embryoid body (EB) formation was performed as previously described (Brennand et al., 2011) with some modifications. Briefly, human iPSCs were incubated with EDTA dissociation solution (Beers et al., 2012) for 3 min and suspended in StemMACS iPS-Brew XF medium containing 10 μm Y27632. Cell suspensions were spotted on the lids of 10 cm culture dishes, and cell aggregates were formed by hanging drops. The next day, the cell aggregates were transferred into N2 medium (DMEM/F12 [Wako], 1× N2 supplement [Invitrogen], 1× B27 minus vitamin A supplement [Invitrogen], 2 mm GlutaMAX supplement [Invitrogen], 10 μm SB431542 [Selleck], 10 μm dorsomorphin [Abcam]) and maintained in suspension in noncoated plastic dishes for 7 d to form EBs. Thereafter, the EBs were plated onto polyornithine (Sigma)/laminin (Sigma)-coated plastic plates and maintained for 1 week in N2 medium containing 1.0 µg/ml laminin. The rosettes were subsequently detached using the STEMdiff Neural Rosette Selection Reagent (STEMCELL Technologies) and cultured in NPC medium (DMEM/F12, 1xN2 supplement, 1× B27 minus vitamin A supplement, 2 mm GlutaMAX supplement, 1.0 µg/ml laminin, and 20 ng/ml FGF2 [Wako]). NPCs were maintained at a high density and diluted by one-third every week with Accutase (Nacalai).
Neural differentiation was performed as previously described (Shi et al., 2012) with modifications. NPCs were treated with Accutase and suspended in neural culture medium (0.5× DMEM/F12, 0.5× Neurobasal medium, 0.5× N2 supplement, 1× B27 supplement, 0.05 mm mercaptoethanol [Sigma], 2 mm GlutaMAX supplement, 2.5 µg/ml insulin [Sigma], 0.5 mm sodium pyruvate [(Sigma], 0.5× nonessential amino acid [Sigma], 1× CultureOne supplement [Thermo Fisher Scientific], and 1× penicillin-streptomycin-amphotericin B suspension [Wako]). The cell suspensions were filtered using a pluriStrainer-Mini 40 μm (PluriSelect) and plated on polyornithine/polyethyleneimine (Sigma)/laminin (Sigma)-coated 12 mm coverslips (Thermo Fisher Scientific) in 24-well dishes. The cell densities were 0.5 to 1.0 × 105 cells per well. Half volume of the medium was exchanged twice a week.
Immunoblotting
The cells were lysed using lysis buffer (10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1.0% Triton, and 1 mm EDTA) containing 0.1% SDS, PhosSTOP (Sigma-Aldrich), and Complete Protease Inhibitor (Sigma-Aldrich). The lysates were centrifuged at 15,000 × g for 10 min at 4°C, and the protein concentrations of the supernatants were measured using a BCA protein assay (Thermo Fisher Scientific). For Cav1.3 detection, the cells were directly lysed with 1× SDS-PAGE loading buffer, and the protein concentration was determined by XL-Bradford (KY-1030, Integrale). The proteins were separated by 7.5% SDS-PAGE and transferred to a PVDF transfer membrane. The membrane was blocked with PBS/0.05% Tween 20 (PBST) containing 3% skim milk and was then probed with appropriate antibodies for 2 h at room temperature. After washing with PBST for 30 min, the membrane was probed with HRP-conjugated secondary antibodies for 1 h. Next, the membrane was washed with PBST for 30 min, and the protein bands were visualized using Immobilon Western Chemiluminescent HRP Substrate (Millipore) on an LAS3000 (Fujifilm).
Antibodies
The antibodies used in this study include the chicken anti-GFP antibody (Millipore, 06-896), rabbit anti-SOX2 antibody (Millipore, AB5603), anti-Tuberin/TSC2 (D93F12) XP rabbit mAb (Cell Signaling Technology [CST], #4308), anti-Phospho-S6 Ribosomal Protein (Ser235/236) antibody (CST, #2211), anti-S6 Ribosomal Protein (5G10) antibody (CST, #2217), anti-α/β-tubulin antibody (CST, #2148), and anti-microtubule-associated protein 2 (MAP2) antibody (Millipore AB5622). We also used the anti-Phospho-CREB (Ser133) (87G3) antibody (CST #9198), anti-MAP2 monoclonal antibody (AP18) (Invitrogen, #MA5-12 826), anti-β-actin antibody (MBL, M177-3), anti-Nestin clone 10C2 antibody (Millipore, MAB5326), anti-Cav1.3 antibody (Alomone Labs, ACC-005), anti-Cav1.3 antibody (Ab144) (Jenkins et al., 2010), anti-βIII-tubulin antibody (Tuj1, Covance, MMS-435P), and Anti-RFP mAb cocktail antibody (MBL, M208-3). For signal absorption of Cav1.3 (Alomone Labs, ACC-005), the antibodies were preincubated with the Cav1.3/CACNA1D Blocking Peptide (#BLP-CC005).
Construction of the plasmid encoding c.a.Rheb
An expression plasmid expressing mCherry-IRES-Cre under EF1alpha promoter was a gift from Karl Deisseroth (Addgene, plasmid #55632) (Fenno et al., 2014). The vector was digested with AscI and EcoRV, blunted with KOD DNA polymerase (Toyobo), and self-ligated to construct a plasmid encoding mCherry. Human Rheb cDNA was amplified from the cDNA of HeLa cells using PCR with KOD plus (Toyobo) and the following primers: CCGAATTCATGCCGCAGTCCAAGTCCCGGAAG and GGCTCGAGTCACATCACCGAGCATGAAGACTTG, which were then cloned into a cloning vector.
The Q64L and S16H mutations in Rheb were introduced by PCR with a pair of primers, (GTAGACACAGCCGGGCTAGATGAATATTCTATC and GATAGAATATTCATCTAGCCCGGCTGTGTCTAC; GATCCTGGGCTACCGGCATGTGGGGAAATCCTC and GAGGATTTCCCCACATGCCGGTAGCCCAGGATC, respectively). Mutations in S16H and Q64L of Rheb were confirmed by DNA sequencing. mCherry-T2A-c.a.Rheb (Q64L and S16H double mutant) plasmids were constructed using the In-Fusion HD Cloning Kit (Clontech) in the mCherry-expressing plasmid with the following primers: T2A-S, GAGGGCAGAGGCAGTCTGCTGACATGCGGTGACGTGGAAGAGAATCCCGGCCCT; T2A-AS, AGGGCCGGGATTCTCTTCCACGTCACCGCATGTCAGCAGACTGCCTCTGCCCTC; T2A-mCherry-AS, GCAGACTGCCTCTGCCCTCCTTGTACAGCTCGTCCATGC; T2A-Rheb-S, GGAAGAGAATCCCGGCCCTATGCCGCAGTCCAAGTCC; Rheb-WPRE-S, TGCTCGGTGATGTGAGATATCAAGCTTATCGATAATC; and WPRE-Rheb-AS, ATCGATAAGCTTGATATCTCACATCACCGAGCATGAAG.
Transfection of neurons with Lipofectamine 2000
TSC2+/+ NPCs were differentiated into neurons and transfected with mCherry or mCherry-T2A-c.a.Rheb-expressing plasmids by Lipofectamine 2000 (Thermo Fisher Scientific) on days 8-9 following neural differentiation. Ca2+ imaging was performed on days 9-11 after the transfection. Activation of mTOR in mCherry-T2A-c.a.Rheb-expressing neurons was confirmed by immunostaining with an anti-Phospho-S6 antibody.
Immunocytochemistry
Neurons were fixed in 4.0% PFA/PBS for 10 min at room temperature. The fixed cells were washed once with PBS, permeabilized with 0.2% Triton X-100/PBS for 5 min, and blocked with 3% skim milk/PBS for 1 h. After blocking, the coverslips were incubated with the appropriate primary antibodies overnight at 4°C. Thereafter, the coverslips were washed 3 times with PBS for 30 min in total. The cells were then probed with Alexa-488-, -586-, or -647-conjugated secondary antibodies (Invitrogen) for 1 h at room temperature. After washing 3 times with PBS for 30 min, the coverslips were mounted with Vectashield (Vector Laboratories) and observed under a confocal fluorescence microscope (FV3000 [Olympus] or BZ-X800 [Keyence]).
For CREB phosphorylation (pCREB) immunostaining, neurons were pretreated with 0.5 μm TTX (Nacalai) for 2 h, stimulated with 60 mm KCl for 3 min, and fixed with 4% PFA/PBS at various time points after stimulation. The fixed cells were then sequentially treated with −20°C methanol for 5 min and 0.2% Triton X-100/PBS for 5 min and were then subjected to immunostaining as described above.
Electroporation of NPCs
NPCs (3 × 106 cells) were electroporated with 2.5 µg pmax-GFP using a Mouse Neural Stem Cell Nucleofector TM Kit (Amaxa, VPG-1004) according to the manufacturer's instructions. The electroporated cells (∼7500 cells/well) were mixed with nontransfected cells (1 × 105 cells/well) and plated into 24-well plates. The cells were fixed with 4% PFA/PBS on day 7 of neuronal differentiation, stained with an anti-GFP antibody, and subjected to analysis.
Ca2+ imaging
Cultured neurons on coverslips were loaded with 5 μm Fluo-8 AM (Quest Fluo-8, AAT Bioquest) and 5 μm fura-2 AM (Dojindo) in the recording solution (115 mm NaCl, 5.4 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 20 mm HEPES, 10 mm glucose, pH, 7.42) for 30 min at room temperature. Fluo-8 signals were observed under an inverted microscope (IX80, Olympus) with a U-MNIBA3 filter cube unit (Olympus). The fluorescent images of spontaneous neuronal activity at 37°C were recorded for 3 min at 1 Hz. For measuring Ca2+ influx into neurons on membrane depolarization, TTX (0.5 μm, Nacalai) was added to the recording solution, and the cells were stimulated with 60 mm KCl for 20 s. fura-2 fluorescent images were obtained every 5 s using 340 ± 10 and 380 ± 10 nm excitation filters, a 400 nm dichroic beam splitter, and a bandpass emission filter at 510-550 nm. The mCherry signals were taken with a U-MWIY2 filter cube unit (Olympus).
To measure the Ca2+ store size, we removed CaCl2 from the recording buffer and added 10 μm of cyclopiazonic acid (CPA) (Tocris Bioscience) to inhibit Ca2+ pump activity. Additional drugs used were nifedipine (Wako), ω-conotoxin MVIIC (Peptide Institute, 4283-v), and ω-conotoxin GVIA (Peptide Institute, 4161-v). To make a raster plot of Ca2+ spikes, imaging data analysis was performed using a self-made ImageJ plugin. This plugin detects the local maximum and minimum over a five-length sliding window across neighboring elements of the Ca2+ imaging data. When the local maximum value was higher than 5 times the SD of the resting value, the Ca2+ transient of the local maximum was recognized as the Ca2+ peak.
RNA purification, cDNA synthesis, and qPCR
RNA was purified from neurons using the RNeasy Mini Kit (QIAGEN) according to the manufacturer's instructions. cDNA was synthesized using the ReverTra Ace qPCR RT Master Mix (Toyobo), and qPCR was performed using the StepOne Real-Time PCR system (Applied Biosystems) and the Thunderbird SYBR qPCR Mix (Toyobo) with the appropriate primers. The primers used were as follows: GRIA1-S, 5′-GGGAGGTGATTCCAAGGACAAG-3′; GRIA1-AS, 5′-AGCATGGCTAGTCCAAGTCC-3′; GRIN1-S, 5′-AGGCCGTGAGAGACAACAAG-3′; GRIN1-AS, AACAGCTCTCCAGTCGTCAC-3′; CACNA1C-S, 5′-AGAAGGACTGGGGCAGTTTG-3′; CACNA1C-AS, 5′-TCTATGGTCATGTCGCAGGC-3′, CACNA1D-S, 5′-AGCCAGCACCCTGCTTAATG-3′; CACNA1D-AS, 5′-AGGACCAAAGTCCTGTAGCTC-3′; ADARB1-S, 5′-AGCCCAACGTGTACCATGAG-3′; ADARB1-AS, 5′-GTGAGTGAGAACTGGTCCTG-3′; r18S-S, 5′-TACCTGAGGCTGTTGGTCAAG-3′; and r18S-AS, 5′-TTGGTGCGACTCATAAACAACC-3′. Target gene levels were normalized to those of r18S RNA.
Quantification of the RNA editing of the CACNA1D gene
The 305 bp fragments containing the IQ domain of the CACNA1D gene were amplified by PCR using the 5′-TGAAGAACTTCGGGCTGTGATAAAG-3′ and 5′-GTTTTGTTTCCTCAGGCTCGTCATC-3′ primers, with cDNA as the template. Fragments were purified using a gel extraction kit (QIAGEN); thereafter, direct DNA sequencing was performed using ∼30 ng of fragments as the template. The AG editing ratio was calculated by measuring the electropherogram peak heights of the nucleotides and was expressed as guanosine/(guanosine + adenosine) × 100.
Karyotype analysis
Chromosomal analysis was commercially done by Chromocenter. Briefly, iPSCs were treated with a Colcemid solution (0.2 µg/ml Demecolcine [Sigma]) for 2 h at 37°C. The medium was collected in a 15 ml tube, and the cells were washed with PBS. The cells were treated with 1 ml of Accutase (Nacalai) at 37°C for 5 min and collected in a 15 ml tube with the preserved cultured medium and PBS. The cells were centrifuged at 1500 rpm for 3 min, and the resultant pellet was suspended and treated with 5 ml of hypotonic solution (0.075 m KCl) for 20 min. Then, 8 ml of Carnoy's solution (methanol:acetic acid = 3:1) was added, and the cells were suspended with a Pasteur pipette and centrifuged at 1500 rpm for 3 min. The pellet was suspended in 10 ml of Carnoy's solution and centrifuged at 1500 rpm for 3 min. After repeating the process twice, the cells were suspended with Carnoy's solution and stored at −30°C until use.
Specimen slides were prepared by the HANABI PVI Metaphase Spreader (Adstec). The slides were washed in Milli-Q water and incubated at 80°C in an oven for 42-45 h. After trypsinized for 10-40 s on ice, the slides were stained with 6% Giemsa's Azur eosin methylene blue solution (Merck) in PBS for 6 min. Metaphase chromosome images were captured by an Axio Imager Z2 fluorescence microscope (Carl Zeiss) and analyzed by an Ikaros software program (Metasystems). Karyotype designations were described according to the 2016 edition of An International System for Human Cytogenomic Nomenclature (ISCN 2016).
Experimental design and statistical analysis
The experiments were performed at least 3 times for every figure. Total number of cells and/or cultures used per group were provided in the figure legends. All data were expressed as mean ± SD, and individual data were also provided as scatter plots in the figures. Statistical analysis was performed using EZR software (Kanda, 2013). Normality was analyzed using the Shapiro–Wilk test. For comparisons between multiple groups with normally distributed data, one-way ANOVA followed by post hoc Bonferroni's multiple comparisons tests or Dunnett's multiple comparisons tests were used for statistical analysis. In cases with non-normal distributions, the analyses were performed by the Kruskal–Wallis test followed by post hoc Steel–Dwass or Steel tests. Mann–Whitney U test (two-sided) was used for comparisons between two groups with non-normal distributions. The statistical values and the particular statistical tests used for all analyses were described in each figure legend. Statistical significance was set at p < 0.05.
Results
Establishment and characterization of human iPSC-derived TSC2-deficient neurons
We established human iPSCs harboring TSC2 mutations by using the gene-editing tool CRISPR/Cas9 to target exon 3 of the TSC2 gene. After sequencing, we confirmed frameshifts in the target sites of the human iPSCs and obtained heterozygous and homozygous TSC2 mutants (TSC2+/− and TSC2−/−) having a distinct mutation (Fig. 1A). We also analyzed karyotypes of TSC2+/+ and TSC2-modified iPSCs (Fig. 1B). The karyotypes were TSC2+/+: 47,XX,+12; TSC2+/−: 47,XX,+12,add(17)(q25); TSC2−/−: 47,XX,+12,t(12;12)(p10;p10), which indicated that all TSC2+/+, TSC2+/−, and TSC2−/− iPSCs had trisomy of chromosome 12. In addition, an unidentified small chromosomal fragment was added to the end of the long-arm of chromosome 17 in TSC2+/− iPSCs (Fig. 1B, middle, arrowhead), and TSC2−/− iPSCs had a whole-arm translocation between two copies of chromosome 12 (right, arrowheads). We would like to mention the impact of this issue on our findings and conclusions later in the discussion section.
Figure 1.
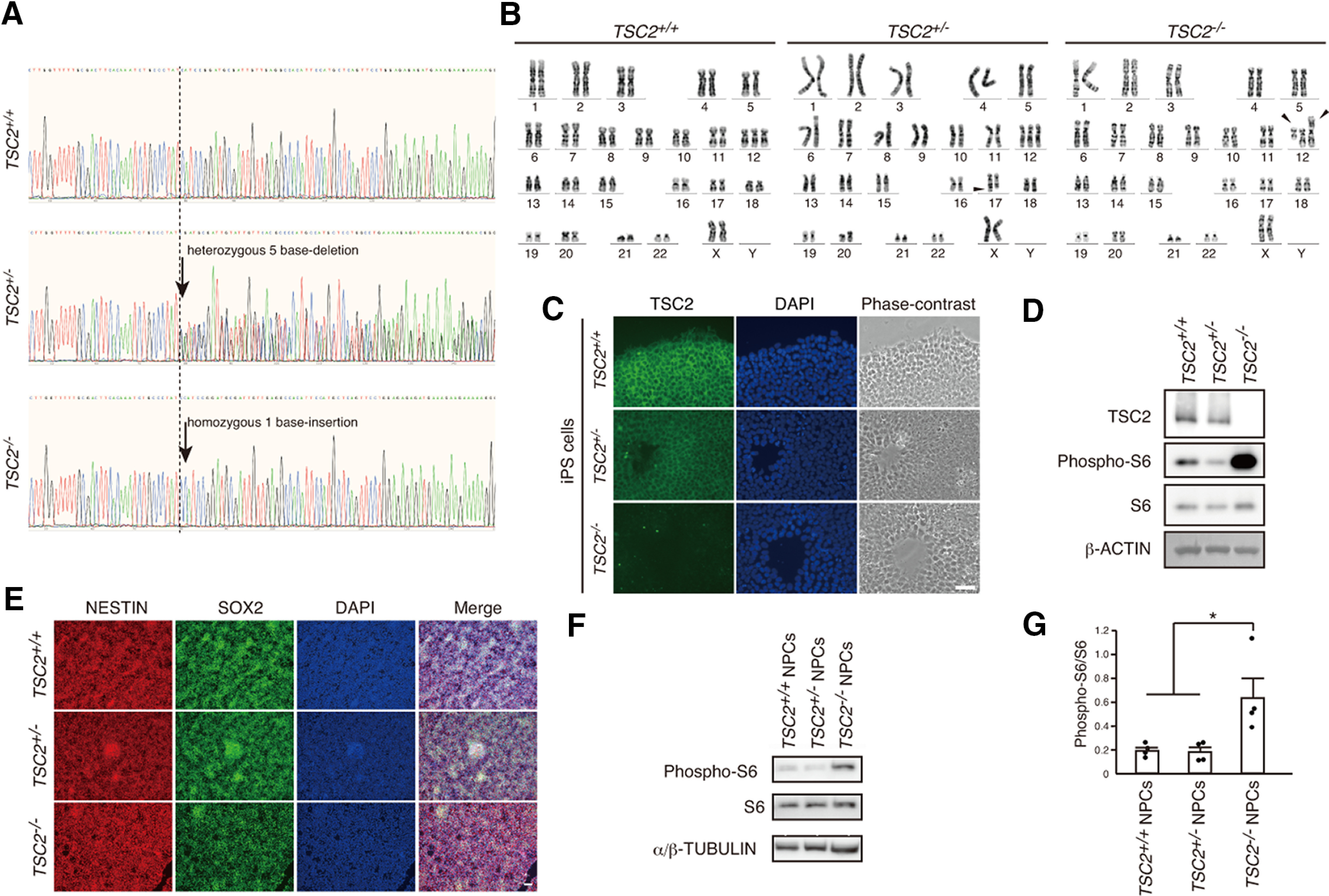
Characterization of TSC2-modified iPSCs and NPCs. A, DNA sequences of exon 3 in the TSC2 gene of TSC2+/+, TSC2+/−, and TSC2−/− iPSCs. TSC2+/− and TSC2−/− iPSCs had a heterozygous 5 base deletion and a homozygous 1 base insertion, respectively, as indicated by the arrows. B, Analysis of karyotypes of TSC2+/+, TSC2+/−, and TSC2−/− iPSCs by G-banding. Middle, Arrowhead indicates addition of an unidentified short fragment to chromosome 17 of TSC2+/− iPSCs. Right, Arrowheads indicate whole-arm translocation between the two copies of chromosome 12 in TSC2−/− iPSCs. C, Immunostaining of TSC2 in TSC2+/+, TSC2+/−, and TSC2−/− iPSCs. Green represents TSC2. Blue represents DAPI. Scale bar, 50 µm. D, TSC2/TUBERIN and S6 phosphorylation (phospho-S6) levels in cell lysates of TSC2+/+, TSC2+/−, and TSC2−/− iPSCs. β-actin was used as the loading control. E, Expression of NESTIN and SOX2 in TSC2+/+, TSC2+/−, and TSC2−/− NPCs. Scale bar, 50 µm. F, Increased phospho-S6 levels in TSC2−/− NPCs. G, Relative phospho-S6 intensity in TSC2+/+, TSC2+/−, and TSC2−/− NPCs. Data are mean ± SD. Experiments were performed 4 times. One-way ANOVA: F(2,9) = 6.845, p = 0.0156. Bonferroni's multiple comparisons test, *p = 0.035, TSC2+/+ versus TSC2−/−; *p = 0.03, TSC2+/− versus TSC2−/−.
We immunostained the iPSCs using an anti-TSC2 antibody and confirmed decreased TSC2 levels in TSC2+/− iPSCs and the complete loss of TSC2 in TSC2−/− iPSCs (Fig. 1C). Western blotting could not detect TSC2 in TSC2−/− cells (Fig. 1D).
It is well known that mutations in either TSC1 or TSC2 activate the mTOR pathway. Therefore, we examined the phosphorylation state of S6, a downstream component of the mTOR pathway. As in previously published studies (Blair et al., 2018), we observed a clear increase of phospho-S6 signals in TSC2−/− iPSCs but not in TSC2+/− cells compared with TSC2+/+ cells (Fig. 1D). Thus, at least under our present culture conditions, homozygous deletion of TSC2 was necessary to activate the mTOR pathway in human iPSCs.
We isolated EBs from these iPSCs and differentiated them into excitatory cortical neurons (Brennand et al., 2011; Shi et al., 2012). All NPCs were positive for NESTIN and SOX2 (Fig. 1E). As in previous studies (Costa et al., 2016; Blair et al., 2018), a significant increase of S6 phosphorylation was only detected in TSC2−/− NPCs (Fig. 1F,G).
We then differentiated the NPCs into cortical neurons. The cultured neurons were positive for VGLUT1, an excitatory neural marker, but were negative for GAD65, an inhibitory neural marker (Fig. 2A,B). Multiple neurons were positive for BRN2 (TSC2+/+: 72.49 ± 7.9%; TSC2+/−: 57.2 ± 16.29%; TSC2−/−: 60.82 ± 11.45%, mean ± SD, n = 5 or 6; Kruskal–Wallis test, p = 0.11) and SATB2 (TSC2+/+: 80.03 ± 13.44%; TSC2+/−: 66.77 ± 22.75%; TSC2−/−: 65.63 ± 24.74%, mean ± SD, n = 3 experiments; Kruskal–Wallis test, p = 0.59), but the percentage of CTIP2-positive neurons was small, indicating that most of the cultured neurons were differentiated into cortical upper-layer neurons (Fig. 2C,D).
Figure 2.
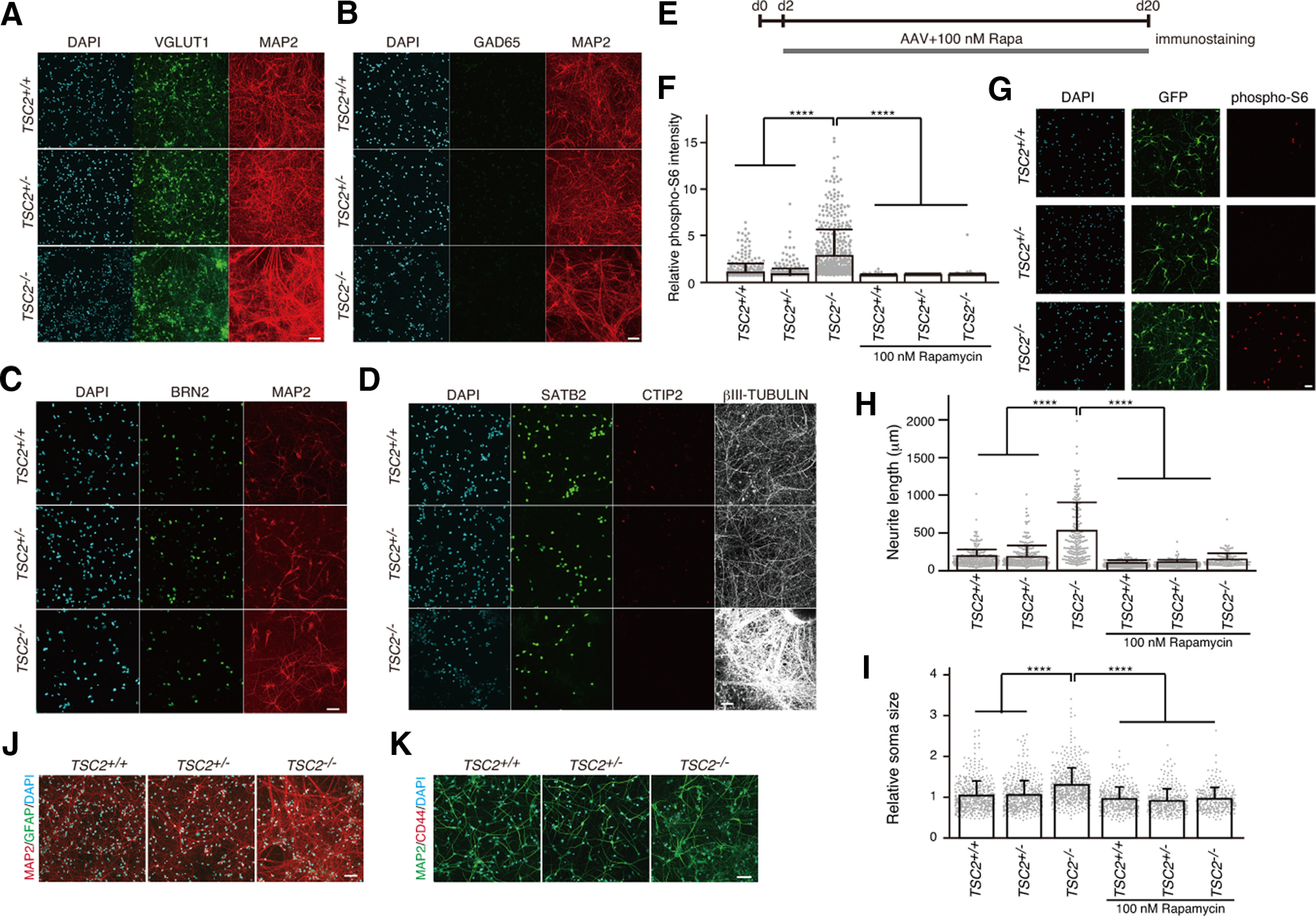
mTOR-dependent abnormal morphology of human iPSC-derived TSC2−/− neurons. A, B, Immunostaining of iPSC-derived neurons with anti-vGLUT1, anti-GAD65, and anti-MAP2 antibodies at 23 d after neural differentiation from NPCs. C, D, Immunostaining of iPSC-derived neurons with anti-BRN2, anti-SATB2, anti-CTIP2, and anti-βIII-tubulin antibodies at ∼30 d after neural differentiation from NPCs. E, Schedule of application of AAV encoding EGFP and treatment with rapamycin after plating NPCs for neural differentiation at day 0 (d0). F, Relative phospho-S6 intensity of TSC2+/+, TSC2+/−, and TSC2−/− neurons at 23 d of neuronal differentiation. Kruskal–Wallis test, p < 0.0001. Steel test compared with TSC2−/− DMSO, ****p < 0.00001. The experiments were performed 3 times, and the number of neurons used for analysis was TSC2+/+ DMSO, 552; TSC2+/− DMSO, 522; TSC2−/− DMSO, 567; TSC2+/+ Rapa, 393; TSC2+/− Rapa, 349; and TSC2−/− Rapa, 289. G, Phospho-S6 signals in iPSC-derived TSC2+/+, TSC2+/−, and TSC2−/− neurons after 23 d. The cells were sparsely infected with AAVs encoding EGFP and stained with anti-GFP and anti-phospho S6 antibodies. Scale bar, 50 µm. H, Neurite length of TSC2+/+, TSC2+/−, and TSC2−/− neurons with or without 100 nm rapamycin treatment for 22 d from day 1 of neuronal differentiation. We measured the length of the longest and thin neurites of individual GFP-expressing neurons and defined them as the neurite length. Experiments were performed 4 times. Data are mean ± SD. Kruskal–Wallis test, p < 0.0001. Steel test compared with TSC2−/− DMSO, ****p < 0.00001. The number of cells used for analysis was TSC2+/+ DMSO, 298; TSC2+/− DMSO, 286; TSC2−/− DMSO, 196; TSC2+/+ Rapa, 396; TSC2+/− Rapa, 222; and TSC2−/− Rapa, 129. I, Relative soma size of TSC2+/+, TSC2+/−, and TSC2−/− neurons with or without 100 nm rapamycin treatment for 22 d from day 1 of neuronal differentiation. The experiments were performed 3 times. Data are mean ± SD. Kruskal–Wallis test, p < 0.0001. Steel test compared with TSC2−/− DMSO, ****p < 0.00001. The number of cells used for analysis was TSC2+/+ DMSO, 552; TSC2+/− DMSO, 522; TSC2−/− DMSO, 555; TSC2+/+ Rapa, 393; TSC2+/− Rapa, 349; and TSC2−/− Rapa, 289. J, K, Immunofluorescent staining of cultured cells with anti-GFAP, anti-CD44, and anti-MAP2 antibodies at ∼30 d after neural differentiation from NPCs. Scale bar, 50 µm.
We sparsely infected these neurons with adeno-associated virus (AAV, serotype 2)-encoding EGFP under the synapsin I promoter to characterize the morphology of the differentiated neurons and treated them with or without rapamycin (Fig. 2E). Immunostaining showed that the S6 phosphorylation level was increased in TSC2−/− neurons, but not in TSC2+/− neurons (Fig. 2F,G). In addition, TSC2−/− neurons had longer neurites and larger somas than TSC2+/− and TSC2+/+ neurons (Fig. 2H,I). These morphologic changes in TSC2−/− neurons were diminished when they were chronically treated with rapamycin (Fig. 2H,I), as was the case with phospho-S6 signals (Fig. 2F). Since our cultured cells were almost negative for the astrocyte marker, GFAP or CD44, (Fig. 2J,K), it was unlikely that glial cells caused the morphologic differences in TSC2−/− neurons.
TSC2−/− neurons exhibit abnormal neuronal activity with highly synchronous Ca2+ spikes
To examine the functional differences in developing TSC2-mutated neurons, we subsequently analyzed the network activity of a large population of neurons by Ca2+ imaging. We loaded the cells with Fluo-8 AM, a Ca2+ indicator, and analyzed the intracellular Ca2+ signals for >3 successive months. At 8 d of neuronal differentiation, neurons of all genotypes sparsely exhibited spontaneous Ca2+ spikes without differences in neuronal activity between TSC2+/+ neurons and TSC2-mutated neurons. However, after 20 d of neuronal differentiation, TSC2−/− neurons, but not the other neurons, often showed synchronized neuronal firing, in which >50% of neurons in a recording field showed Ca2+ spikes at the same time (Fig. 3A; Movies 1, 2, 3). The raster plots of Ca2+ spikes clearly revealed the synchronous firing of a large number of TSC2−/− neurons (Fig. 3B). The number of observations of highly synchronized Ca2+ spikes in TSC2−/− neurons gradually increased up to 80 d of neuronal differentiation (100%, 12 of 12 experiments), but slightly decreased at 100 d, possibly owing to the difficulty in maintaining neuronal networks in culture (Fig. 3C). Although the percentage of neurons presenting Ca2+ spikes did not significantly differ between the genotypes except at 80 d of neuronal differentiation (Fig. 3D), the frequency of Ca2+ spikes of TSC2−/− neurons was higher than that of TSC2+/− or TSC2+/+ neurons (Fig. 3E). When we divided the TSC2−/− neurons into two groups, namely, the synchronically or the sporadically Ca2+ spiking neuronal groups, the synchronized TSC2−/− neurons exhibited a higher frequency of Ca2+ spikes than the nonsynchronized ones (Fig. 3F), suggesting the effect of enhanced neural connections on higher neural activities in synchronized TSC2−/− neurons. Because the density of cultured neurons, which was measured by counting the Fluo-8-stained neurons in the optical field, was similar among the three genotypes of the neurons, neural density would not explain the typical Ca2+ spike patterns of TSC2−/− neurons compared with other genotypes of neurons (TSC2+/+: 62.0 ± 18.45 [n = 16]; TSC2+/−: 68.44 ± 33.14 [n = 18]; TSC2−/−: 57.0 ± 22.98 [n = 18] (cells/FOV); mean ± SD, Kruskal–Wallis test, χ2 = 0.58 021, df = 2, p = 0.748, not significant).
Figure 3.
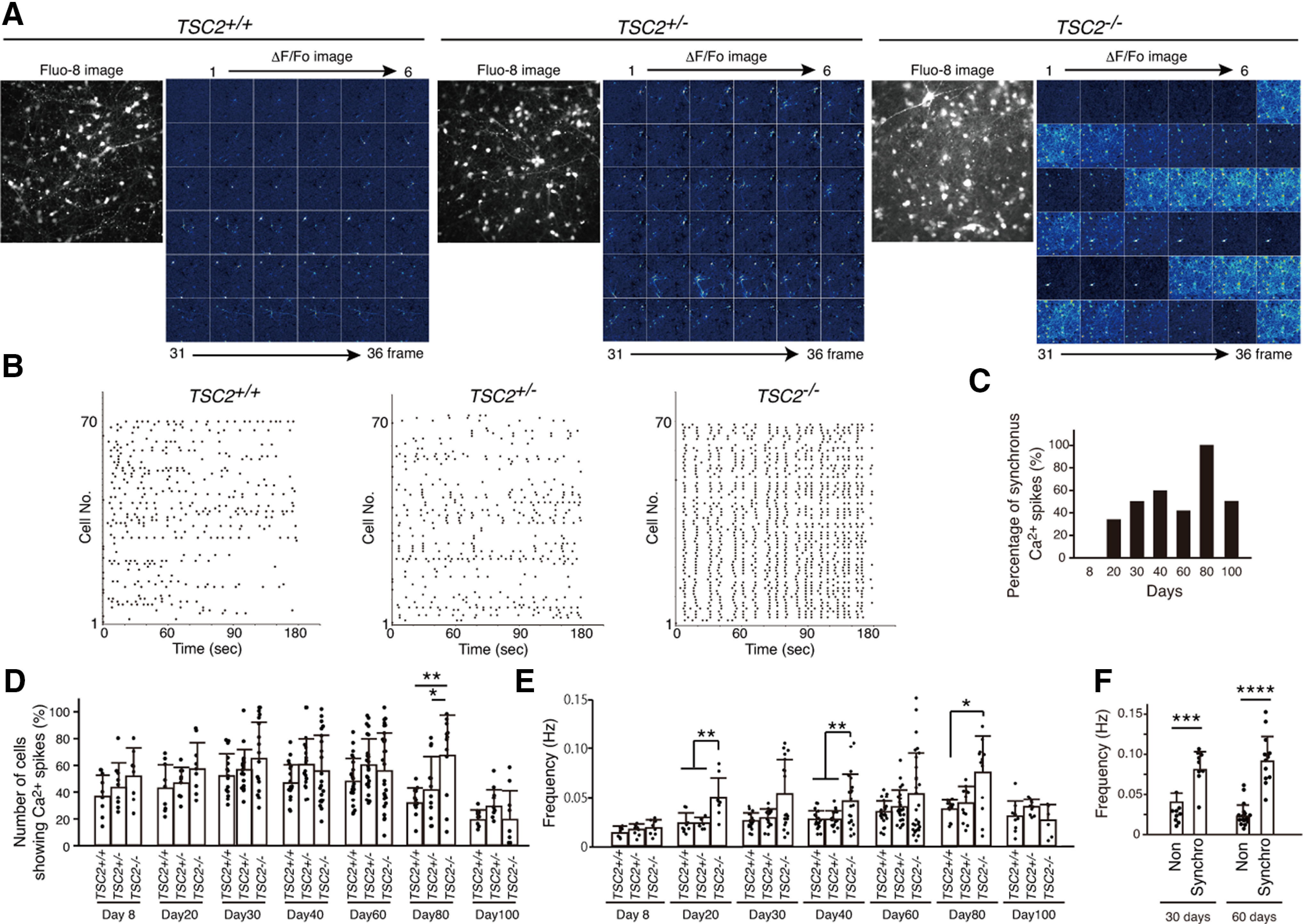
Cultured TSC2−/− neurons exhibited synchronous neuronal activity. A, Ca2+ dynamics of iPS-derived TSC2+/+, TSC2+/−, and TSC2−/− neurons at 33 d. Fluorescence images of Fluo-8 and the ΔF/F0 changes in Fluo-8 signals are shown. Images were taken at 1 Hz. Thirty-six frames of ΔF/F0 changes in Fluo-8 are presented. B, Raster plots of Ca2+ spikes in TSC2+/+, TSC2+/−, and TSC2−/− neurons. Vertical axis represents the individual cells analyzed (∼1 in 70 cells). C, Developmental change in the percentage of experiments in which synchronous Ca2+ spikes were observed in TSC2−/− neurons. Each value was obtained from 6 to 29 independent experiments. D, Percentages of neurons exhibiting spontaneous Ca2+ spikes in TSC2+/+, TSC2+/−, and TSC2−/− neurons on various days of culture. Data are mean ± SD. The experimental numbers were 9-29 for each group. Day 8: one-way ANOVA, F(2,23) = 1.258, p = 0.296. Day 20: one-way ANOVA, F(2,24) = 2.042, p = 0.152. Day 30: one-way ANOVA, F(2,49) = 2.029, p = 0.142. Day 40: one-way ANOVA, F(2,57) = 2.031, p = 0.141. Day 60: one-way ANOVA, F(2,75) = 2.034, p = 0.138. Day 80: one-way ANOVA, F(2,34) = 7.907, p = 0.00151. Bonferroni's multiple comparisons test, **p = 0.0013, TSC2+/+ (n = 13) versus TSC2−/− (n = 12); *p = 0.0324, TSC2+/− (n = 12) versus TSC2−/− (n = 12). Day 100: one-way ANOVA, F(2,26) = 1.98, p = 0.158. E, Frequencies of Ca2+ spikes in TSC2+/+, TSC2+/−, and TSC2−/− neurons on various culture days of neuronal differentiation. The experimental numbers were 9-29 for each group. Day 8: one-way ANOVA, F(2,23) = 1.517, p= 0.24. Day 20: Kruskal–Wallis test, p = 0.001084, Steel–Dwass test, **p= 0.004896, TSC2+/+ (n = 9) versus TSC2−/− (n = 9); **p = 0.004896, TSC2+/− (n = 9) versus TSC2−/− (n = 9). Day 30: Kruskal–Wallis test, p = 0.0968. Day 40: one-way ANOVA, F(2,57) = 7.323, p = 0.00148, Bonferroni's multiple comparisons test, **p =0.0036, TSC2+/+ (n = 19) versus TSC2−/− (n = 19); **p = 0.0086, TSC2+/− (n = 19) versus TSC2−/− (n = 19). Day 60: Kruskal–Wallis test, p = 0.6337. Day 80: Kruskal–Wallis test, p = 0.0299, Steel test compared with TSC2−/−, *p = 0.03611, TSC2+/+ (n = 13) versus TSC2−/− (n = 12). Day 100: one-way ANOVA, F(2,23) = 2.644, p = 0.0926. F, Difference in Ca2+ spike frequencies between synchronized (synchro) and nonsynchronized (non) TSC2−/− neurons at 30 and 60 d. Data are mean ± SD. Day 30: Mann–Whitney U test, ***p = 0.00057. Day 60: Mann–Whitney U test, ****p = 0.0000105. The experimental number was synchronized (n = 9) and nonsynchronized (n = 9) at 30 d. The experimental number was synchronized (n = 12) and nonsynchronized (n = 17) at 60 d.
Spontaneous Ca2+ signals of TSC2+/+ neurons at 33 d.
Spontaneous Ca2+ signals of TSC2+/– neurons at 33 d.
Spontaneous Ca2+ signals of TSC2−/− neurons at 33 d (accelerated ninefold).
TSC2−/− neurons exhibited enhanced Ca2+ influx via LTCCs
Since the intracellular Ca2+ level is one of the critical factors regulating neuronal activity, we further examined the intracellular Ca2+ dynamics of TSC2 mutant neurons. As Fluo-8 AM is a nonratiometric Ca2+ indicator, and its fluorescence is affected by the loading conditions, such as the cell thickness and the amount of dye in the loaded cells, we subsequently used fura-2 AM, a ratiometric Ca2+ indicator, to precisely analyze the Ca2+ dynamics of TSC2 mutant neurons. TTX was added to the recording solution to suppress the spontaneous neuronal activity. Under these conditions, the synchronous Ca2+ spikes of TSC2−/− neurons were diminished, suggesting the requirement of Na+-dependent electrical activity for generating Ca2+ spikes.
We depolarized 30-d-old neurons using 60 mm KCl in the presence of TTX. Surprisingly, we found that TSC2−/− neurons exhibited much higher increases in Ca2+ levels than TSC2+/− or TSC2+/+ neurons on membrane depolarization (Fig. 4A,B). This difference was likely not caused by the developmental delay of the TSC2+/+ and TSC2+/− neurons since the increment was also observed in 150-d-old TSC2−/− neurons (Fig. 4B). In contrast, resting Ca2+ levels were not significantly different between TSC2−/−, TSC2+/−, and TSC2+/+ neurons (Fig. 4B). Thus, TSC2−/− neurons exhibited enhanced Ca2+ influx on membrane depolarization.
Figure 4.

TSC2−/− neurons showed enhanced Ca2+ influx via LTCCs on membrane depolarization. A, Ca2+ response on membrane depolarization in iPSC-derived neurons with TSC2 mutations. The fluorescence ratio (340/380 nm) of fura-2 is shown. Bars represent 60 mm KCl stimulation for 20 s. B, Resting cytoplasmic Ca2+ levels and peak amplitude of Ca2+ influx into neurons on 60 mm KCl stimulation of 30-d-old (left) and 150-d-old (right) cultures. Data are mean ± SD. Resting Ca2+ level: day 30, one-way ANOVA, F(2,132) = 2.932, p = 0.0568; day 150, one-way ANOVA, F(2,83) = 2.824, p = 0.0651. The number of cells analyzed was 32-58 (30-d-old) and 23-35 (150-d-old). KCl response: day 30, Kruskal–Wallis test, p < 0.0001, Steel–Dwass test, ***p < 0.0001, TSC2+/+ (n = 58) versus TSC2−/− (n = 45); ***p < 0.0001, TSC2+/− (n = 32) versus TSC2−/− (n = 45). Day 150, Kruskal–Wallis test, p < 0.0001, Steel–Dwass test, ***p < 0.0001, TSC2+/+ (n = 35) versus TSC2−/− (n = 23); ***p < 0.0001, TSC2+/− (n = 29) versus TSC2−/− (n = 23). C, The effect of L-, P/Q-, and N-type VGCC blockers on Ca2+ signals of TSC2−/− neurons on membrane depolarization. Nif: 5 μm nifedipine; MVIIC: 1 μm ω-conotoxin MVIIC; GVIA: 1 μm ω-conotoxin GVIA. D, Ca2+ increases in neurons treated with Ca2+ channel blockers. Data are mean ± SD. Kruskal–Wallis test, p < 0.0001, Steel test compared with TSC2−/−, ***p < 0.0001, TSC2−/− (n = 156) versus TSC2−/− + Nif (n = 99); p = 0.705, TSC2−/− (n = 156) versus TSC2−/− + GVIA (n = 222); p = 0.9949, TSC2−/− (n = 156) versus TSC2−/− + MVIIC (n = 212). The number of cells used for analysis was 201, 82, 102, 102, 156, 99, 222, and 212 from the left bar to the right bar in the figure.
Several types of voltage-gated calcium channels (VGCCs) are expressed in the brain (Simms and Zamponi, 2014). To further clarify the nature of enhanced Ca2+ influx into TSC2−/− neurons, we depolarized neurons with 60 mm KCl in the presence of inhibitors for L-, N-, or P/Q-type VGCCs. As shown in Figure 4C, we found that nifedipine, an LTCC inhibitor, significantly decreased Ca2+ influx into TSC2−/− neurons on membrane depolarization. In contrast, neither ω-conotoxin MVIIC, a P/Q-type VGCC blocker, nor ω-conotoxin GVIA (GVIA), an N-type VGCC blocker, had this effect (Fig. 4C,D). These results suggest that increased Ca2+ influx through LTCCs underlies the altered Ca2+ dynamics in TSC2−/− neurons on membrane depolarization.
Cav1.2 and Cav1.3, encoded by the CACNA1C and CACNA1D genes, respectively, are two critical members of the LTCC family that play important roles in normal brain development and plasticity. Therefore, we quantified their expression in TSC2−/− neurons using qPCR. We found that, compared with TSC2+/+ neurons, CACNA1D expression, but not CACNA1C expression, was significantly increased in TSC2−/− neurons at day 30 of neuronal differentiation (Fig. 5A). In contrast, we did not observe a significant difference in the gene expression of NMDARs (GRIN1) or AMPARs (GRIA1). Increased expression of CACNA1D was also observed at day 60 of neuronal differentiation (Fig. 5B), indicating that the difference among TSC2+/+, TSC2+/−, and TSC2−/− neurons was not because of the developmental time course.
Figure 5.

CACNA1D expression was increased in TSC2−/− neurons. A, Relative gene expression in iPSC-derived neurons with TSC2 mutations at 30 d of neuronal differentiation. Data are mean ± SD. CACNA1C: Kruskal–Wallis test, p = 0.01387, Steel–Dwass test, p = 0.167, TSC2+/+ (n = 12) versus TSC2−/− (n = 12); p = 0.3740, TSC2+/+ (n = 12) versus TSC2+/− (n = 12); *p = 0.0154, TSC2+/− (n = 12) versus TSC2−/− (n = 12). CACNA1D: Kruskal–Wallis test, p < 0.0001, Steel–Dwass test, ****p = 0.000083, TSC2+/+ (n = 12) versus TSC2−/− (n = 12); **p = 0.00190, TSC2+/− (n = 12) versus TSC2−/− (n = 12). GRIA1: one-way ANOVA, F(2,32) = 0.2749, p = 0.761. GRIN1: one-way ANOVA, F(2,21) = 0.6872, p = 0.514. The experiments were performed using the cDNA from 4 to 6 independent cultures. B, Relative CACNA1D expression on day 60. Data are mean ± SD. Kruskal–Wallis test, p = 0.000899, Steel–Dwass test, **p = 0.00194, TSC2+/+ (n = 10) versus TSC2−/− (n = 10), **p = 0.00897, TSC2+/− (n = 10) versus TSC2−/− (n = 10). The data were obtained using cDNA from five independent cultures. C, Expression levels of Cav1.3 in TSC2+/+, TSC2+/−, and TSC2−/− neurons. The same amounts of protein lysates (∼10 µg) were immunoblotted with an anti-Cav1.3 antibody (Ab144). β-Actin was used as the loading control. Right, Fold change in Cav1.3 expression normalized to β-actin. One-way ANOVA, F(2,10) = 8.205, **p = 0.00778, Bonferroni's multiple comparisons test, **p = 0.007, TSC2+/+ (n = 4) versus TSC2−/− (n = 5); p = 0.234, TSC2+/+ (n = 4) versus TSC2+/− (n = 4), p = 0.229, TSC2+/− (n = 4) versus TSC2−/− (n = 5). D, Immunostaining of Cav1.3 (green, Alomone Labs, ACC-005) and MAP2 (red) in TSC2+/+, TSC2+/−, and TSC2−/− neurons. Scale bar, 20 µm. Inset, Magnified image of the white box area. E, Relative immune intensity of Cav1.3 in neurons at the soma. Kruskal–Wallis χ2 = 106.08, df = 2, ***p < 2.2 × 10−16. Steel test, ****p < 1 × 10−9, TSC2−/− (n = 112) versus TSC2+/+ (n = 141), ****p < 1 × 10−9, TSC2−/− (n = 112) versus TSC2+/− (n = 104). F, A-to-I editing of the IQ domain of CACNA1D. The electropherograms of direct sequencing of the CACNA1D gene from TSC2+/+, TSC2+/−, and TSC2−/− neurons are shown. Arrows indicate the adenine-to-guanine conversion signals. G, Percentage of A-to-I editing of CACNA1D cDNA on day 30 (left). Right, Expression levels of ADARB1 mRNA in the neurons. Data are mean ± SD. A-to-I editing: one-way ANOVA, F(2,12) = 20.70, p = 0.000129, Bonferroni's multiple comparisons test, ***p = 0.00011, TSC2+/+ (n = 5) versus TSC2−/− (n = 5); **p = 0.00383, TSC2+/− (n = 5) versus TSC2−/− (n = 5). ADARB1: one-way ANOVA, F(2,27) = 67.80, p < 0.0001, Bonferroni's multiple comparisons test, ***p < 0.0001, TSC2+/+ (n = 10) versus TSC2−/− (n = 10); ***p < 0.0001, TSC2+/− (n = 10) versus TSC2−/− (n = 10). All data were obtained from five independent cultures. H, A-to-I editing (%) and relative ADARB1 expression in 60-d-old neurons. Data are mean ± SD. A-to-I editing: one-way ANOVA, F(2,12) = 1.117, p = 0.359, n = 5 experiments. ADARB1: one-way ANOVA, F(2,21) = 20.66, p = 0.000011, Bonferroni's multiple comparisons test, ****p = 0.00002, TSC2+/+ (n = 8) versus TSC2−/− (n = 8); ***p = 0.00015, TSC2+/− (n = 8) versus TSC2−/− (n = 8). Data were obtained from five independent cultures.
To analyze the protein expression level of Cav1.3 in neurons, we performed Western blotting and confirmed that TSC2−/− neurons had more Cav.1.3 proteins (∼250 kDa) than TSC2+/+ neurons at 30 d (Fig. 5C).
We also immunostained the cortical neurons with an anti-Cav1.3 antibody and detected punctuate immunosignals of Cav1.3 at the dendrites and soma of human neurons (Fig. 5D). Cav1.3 immunosignals of TSC2−/− neurons at the soma were stronger than those of TSC2+/+ and TSC2+/− neurons (Fig. 5E). Preincubation of the anti-Cav1.3 antibody with an antigen peptide diminished the Cav1.3 immunosignals, suggesting the specificity of the immunosignals (Fig. 5D, bottom).
Since adenosine-to-inosine (A-to-I) RNA editing within the IQ domain of Cav1.3 diminishes the Ca2+-dependent inactivation (CDI) by Ca2+/calmodulin and consequently increases channel activity (Huang et al., 2012; Bazzazi et al., 2013), we also examined A-to-I RNA editing of CACNA1D along with the expression of an adenosine deaminase enzyme (ADARB1), which catalyzes the A-to-I conversion. Although the editing ratio was quite low (∼7.5%-18%), compared with TSC2+/+ or TSC2+/− neurons, we detected an ∼2-fold increase in CACNA1D RNA editing in 30-d-old TSC2−/− neurons (Fig. 5F,G). ADARB1 expression in TSC2−/− neurons was also increased by 2.5-fold at 30 d (Fig. 5G). However, we could not detect a significant difference in CACNA1D RNA editing among 60-d-old TSC2+/+, TSC2+/−, and TSC2−/− neurons regardless of the higher expression of ADARB1 in TSC2−/− neurons than in TSC2+/+ and TSC2+/− neurons (Fig. 5H). Thus, increased Cav1.3 levels predominantly contributed to enhanced Ca2+ influx into TSC2−/− neurons.
c.a.Rheb increased Ca2+ influx and Cav1.3 expression in TSC2+/+ neurons
To further confirm the mTOR-dependent increase in Ca2+ influx in human cortical neurons, we transfected TSC2+/+ neurons with a plasmid encoding mCherry-T2A-c.a.Rheb to activate mTOR and examined Ca2+ influx on KCl stimulation. Activation of the mTOR signaling pathway in c.a.Rheb-transfected neurons was checked by immunostaining the transfected cells with an anti-phospho-S6 antibody (Fig. 6A). There was a positive correlation between mCherry and phospho-S6 immunosignal intensity in the mCherry-T2A-c.a.Rheb-expressing TSC2+/+ neurons, but not in mCherry-expressing TSC2+/+ neurons (Fig. 6B), and the mean intensity of phospho-S6 immunosignals was significantly higher in c.a.Rheb-transfected neurons than in mCherry-transfected cells (Fig. 6C). The relative soma size of c.a.Rheb-transfected neurons was also larger than that of mCherry-transfected cells (Fig. 6D). These observations supported the activation of mTOR signaling pathways in mCherry-T2A-c.a.Rheb-transfected TSC2+/+ neurons.
Figure 6.
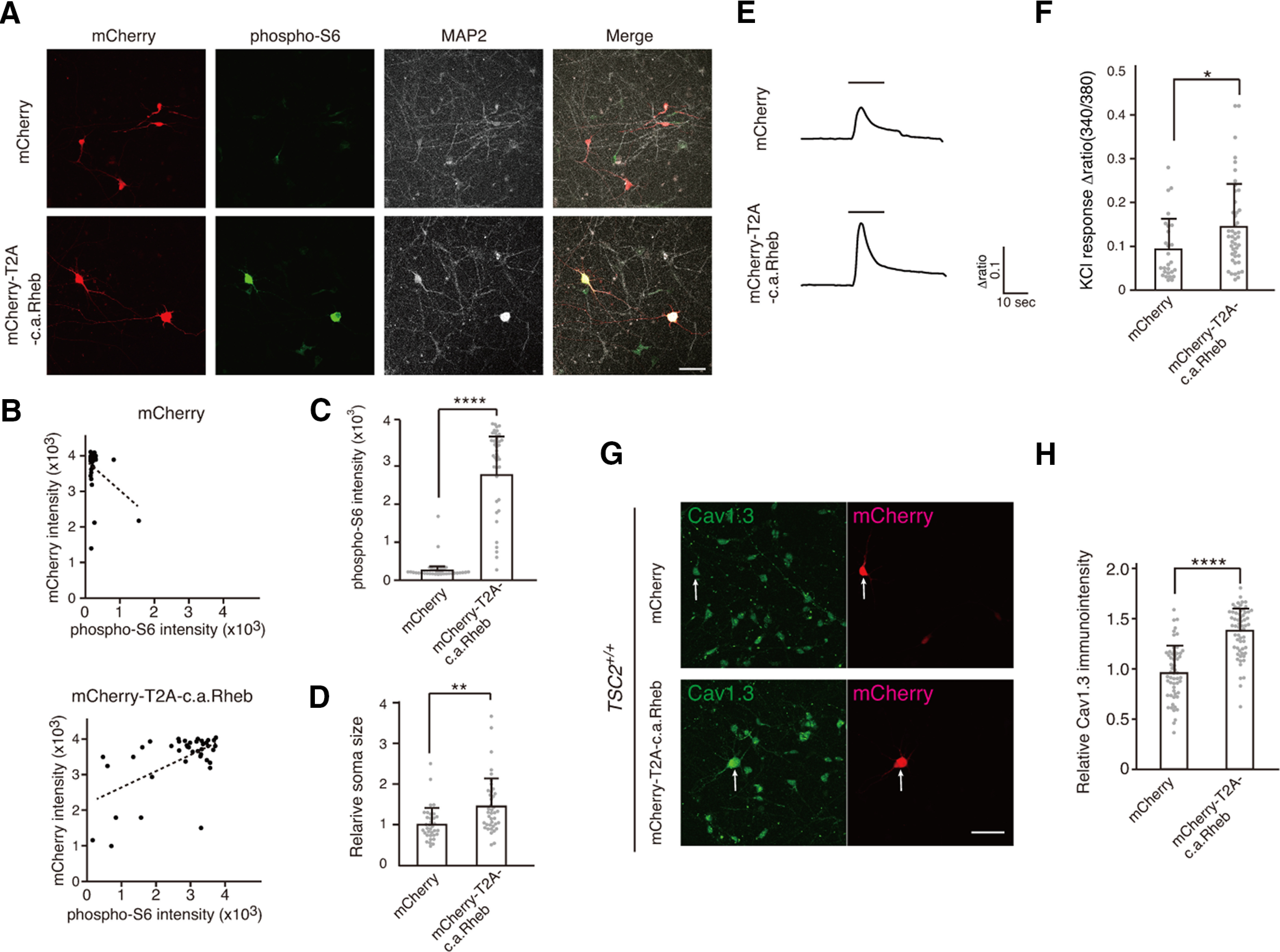
c.a.Rheb increased Ca2+ influx and Cav1.3 expression in TSC2+/+ neurons. A, c.a.Rheb increases phospho-S6 intensity in TSC2+/+ neurons. TSC2+/+ neurons were transfected with plasmids encoding mCherry (top panels) or mCherry-T2A-c.a.Rheb (bottom panels) and stained with anti-phospho-S6 and MAP2 antibodies. Scale bar, 50 µm. B, Scatter plots for mCherry versus phospho-S6 signals in mCherry (top) or mCherry-T2A-c.a.Rheb (bottom)-transfected neurons. C, Mean signal intensity of phospho-S6 in mCherry (n = 35) and mCherry-T2A-c.a.Rheb (n = 40) transfected neurons. Mann–Whitney U test, ****p = 1.09 × 10−18. D, Soma size of mCherry (n = 35) and mCherry-T2A-c.a.Rheb (n = 40) transfected neurons. Welch two-sample t test, **p = 0.00104. E, Ca2+ signals in mCherry or mCherry-T2A-c.a.Rheb-expressing neurons on KCl stimulation. TSC2+/+ neurons were transfected with mCherry or mCherry-T2A-c.a.Rheb-expressing plasmids on days 8-9, and Ca2+ signals were measured with fura 2-AM on days 9-11 after transfection. Bars represent 60 mm KCl application. F, Quantification of peak height of Ca2+ transient on KCl stimulation in mCherry (n = 27) or mCherry-T2A-c.a.Rheb (n = 41) expressing neurons. Mann–Whitney U test, *p = 0.016. Data were obtained from 9 dishes of 3 independent cultures for each plasmid. G, Immunostaining of Cav1.3 (Alomone Labs, ACC-005) in mCherry or mCherry-T2A-c.a.Rheb-expressing TSC2+/+ neurons. Arrows indicate mCherry-positive neurons. Scale bar, 50 µm. H, Relative immunoactivity of Cav1.3 in mCherry (n = 56) or mCherry-T2A-c.a.Rheb (n = 61) expressing neurons. Immunosignals were normalized to Cav1.3 immunosignals of mCherry-negative cells in each well. Kolmogorov–Smirnov test, mCherry: p = 0.9148; mCherry-T2A-c.a.Rheb: p = 0.2991; F test= 0.76. p = 0.3068. Student t test, ****p = 4.835 × 10−14.
As we expected, we observed a much larger Ca2+ response on depolarization in mCherry-T2A-c.a.Rheb-expressing neurons compared with those of mCherry-expressing neurons (Fig. 6E,F). We also found stronger immunosignals of Cav1.3 in mCherry-T2A-c.a.Rheb-expressing neurons at the soma than in mCherry-expressing neurons (Fig. 6G,H). Together, these results suggest that mTOR activation augments Ca2+ influx on depolarization in human cortical excitatory neurons by enhancing Cav1.3 expression.
Long-term, but not short-term, rapamycin treatment rescued the abnormal neural activity of TSC2−/− neurons
Since rapamycin has been reported to prevent epilepsy in TSC (Cardamone et al., 2014; Lasarge and Danzer, 2014; French et al., 2016; Krueger et al., 2016; Mizuguchi et al., 2019) and suppress abnormal axon extension of TSC2−/− neurons (Choi et al., 2008; Nadadhur et al., 2019), we treated neurons with 10 nm rapamycin for 18 d, starting from 2 d of neuronal differentiation and performed Ca2+ imaging to examine whether rapamycin suppressed the synchronous neuronal activity of a large population of TSC2−/− neurons (Fig. 7A). We found that the rapamycin treatment changed the synchronous Ca2+ spike patterns of TSC2−/− neurons into sporadic patterns similar to those found in TSC2+/+ and TSC2+/− neurons (Fig. 7B). We could not detect the synchronous firing of a large number of TSC2−/− neurons. The effect of rapamycin on Ca2+ spike patterns was clearly shown by raster plots (Fig. 7C). The Ca2+ spike frequencies of TSC2−/− neurons were decreased to levels similar to those in the control neurons (Fig. 7D). This effect was not likely because of rapid protein modifications, such as phosphorylation, since short-term application of rapamycin for up to 30 min did not significantly affect synchronous Ca2+ spikes and spike frequencies in TSC2−/− neurons (Fig. 7E).
Figure 7.

Long-term treatment of TSC2−/− neurons with rapamycin changed Ca2+ dynamics from synchronous to sporadic patterns. A, Schematic illustration of rapamycin treatment and Ca2+ imaging after plating NPCs for differentiation. B, Ca2+ dynamics of DMSO-treated (left) or rapamycin-treated (right) TSC2−/− neurons. The ΔF/F0 changes of the Fluo-8 signals for 36 frames are shown. Neurons that were treated with DMSO or 10 nm rapamycin for >18 d from day 2 of neuronal differentiation were used for the analysis. C, Raster plots of Ca2+ spikes from B. D, Ca2+ spike frequencies of TSC2+/+, TSC2+/−, and TSC2−/− neurons with or without rapamycin treatment. Data are mean ± SD. One-way ANOVA, F(5,29) = 26.88, p < 0.0001, Dunnett's multiple comparisons test compared with TSC2−/−, ****p < 0.00001. The number of experiments was 6 for each group except for TSC2−/− + Rapa (n = 5). E, Ca2+ spike frequencies of TSC2−/− neurons after treatment with 50 nm rapamycin for 5, 10, or 30 min. We used 50 nm rapamycin to completely and rapidly suppress the mTOR activity. The experiments were performed 3 times. One-way ANOVA, F(3,8) = 0.009871, p = 0.999.
We also measured Ca2+ influx on membrane depolarization in TSC2−/− neurons after long-term rapamycin treatment (Fig. 8A). We found that chronic rapamycin treatment significantly decreased Ca2+ influx into TSC2−/− neurons on membrane depolarization to levels similar to those in TSC2+/+ and TSC2+/− neurons (Fig. 8B). The average values of the peak amplitudes, the amount of Ca2+ (area under the curve), and the Ca2+ decay from the Ca2+ peak (tau) were high in TSC2−/− neurons, but decreased to levels similar to those of TSC2+/+ neurons after rapamycin treatment (Fig. 8C). In addition, we also confirmed that rapamycin treatment reduced CACNA1D expression in TSC2−/− neurons to the levels of TSC2+/+ and TSC2+/− neurons (Fig. 8D). The intracellular Ca2+ store size of TSC2−/− neurons, which was measured by inhibiting Ca2+ pump activity with CPA in the absence of extracellular Ca2+, was larger than that of neurons of the other genotypes (Fig. 8E). In addition, the amplitude of Ca2+ release was quite small compared with the ratio change in Ca2+ levels on KCl stimulation. These data ruled out the possibility that decreased Ca2+ pump activity underlies the increased Ca2+ elevation in TSC2−/− neurons on depolarization.
Figure 8.

Long-term rapamycin treatment ameliorated enhanced Ca2+ influx into TSC2−/− neurons on membrane depolarization. A, Schematic illustration of rapamycin treatment and Ca2+ imaging after plating NPCs for differentiation. We applied rapamycin from nine day onward following NPC differentiation, since the Ca2+ response seen on depolarization did not likely depend on neural networks. B, Ca2+ dynamics of TSC2+/+, TSC2+/−, TSC2−/− and rapamycin-treated TSC2−/− neurons on stimulation with 60 mm KCl. The fura-2 ratio (380/340 nm) is shown. Bar represents the 60 mm KCl stimulation for 20 s. C, Peak amplitude (left), area under the curve (AUC, middle), and tau (right) of Ca2+ transients of TSC2+/+, TSC2+/−, TSC2−/− and rapamycin-treated TSC2−/− neurons for >11 d. A total of 104-149 neurons were analyzed for each group. Peak amplitude: Kruskal–Wallis test, p < 0.0001, Steel–Dwass test, ***p < 0.0001, TSC2+/+ (n = 126) versus TSC2−/− (n = 104); ***p < 0.0001, TSC2+/− (n = 105) versus TSC2−/− (n = 104); ***p < 0.0001, TSC2−/− (n = 104) versus TSC2−/− + Rapa (n = 149). AUC: Kruskal–Wallis test, p < 0.0001, Steel–Dwass test, ***p < 0.0001, TSC2+/+ (n = 126) versus TSC2−/− (n = 104); ***p < 0.0001, TSC2+/− (n = 105) versus TSC2−/− (n = 104); ***p < 0.0001, TSC2−/− (n = 104) versus TSC2−/− + Rapa (n = 149). Tau: Kruskal–Wallis test, p = 0.000109, Steel–Dwass test, **p < 0.001, TSC2+/+ (n = 126) versus TSC2−/− (n = 104); **p < 0.001, TSC2−/− (n = 104) versus TSC2−/− + Rapa (n = 149). D, Rapamycin treatment decreased CACNA1D expression. Data are mean ± SD. The number of experiments was 12 for each group. Kruskal–Wallis test, p < 0.0001, Steel test compared with TSC2−/−, *p = 0.01144, TSC2−/− (n = 12) versus TSC2+/+ (n = 12); *p = 0.01204, TSC2−/− (n = 12) versus TSC2+/− (n = 12); ***p = 0.000159, TSC2−/− (n = 12) versus TSC2+/+ + Rapa (n = 12); **p = 0.001035, TSC2−/− (n = 12) versus TSC2+/− + Rapa (n = 12); ***p = 0.000158, TSC2−/− (n = 12) versus TSC2−/− + Rapa (n = 12). E, Ca2+ store sizes of TSC2+/+, TSC2+/−, and TSC2−/− neurons. The peak amplitudes of intracellular Ca2+ levels on CPA treatment in the absence of extracellular Ca2+ were evaluated. Data are mean ± SD. The experiments were performed 3 times. For each experiment, 26-58 cells were used in the analysis. One-way ANOVA, F(2,6) = 49.67, p = 0.000185, Bonferroni's multiple comparisons test, ***p = 0.00077, TSC2+/+ (n = 3) versus TSC2−/− (n = 3); ***p = 0.00026, TSC2+/− (n = 3) versus TSC2−/− (n = 3).
Increased Ca2+ influx through LTCCs partially contributed to the abnormally extended neurite extensions of human TSC2−/− neurons
VGCCs play an important role in normal brain function and development (Simms and Zamponi, 2014; Dolphin, 2016). They are the primary mediators of Ca2+ entry into neurons on membrane depolarization and control various Ca2+-dependent processes, including gene expression, neurite outgrowth, migration, and neuronal firing (Heyes et al., 2015). Recent studies have suggested that spontaneous Ca2+ fluctuations caused by LTCCs play critical roles in neurite extension in developing cortical neurons in rodents (Tang et al., 2003; Kamijo et al., 2018). Since long-term rapamycin treatment ameliorated both the abnormal neurite extension (Fig. 2H) and the enhanced Ca2+ influx via LTCCs in human TSC2−/− neurons (Fig. 8), we speculated that the increased Ca2+ influx through LTCCs could underlie the abnormal neurite extension in human TSC2−/− neurons. We therefore transfected NPCs with a GFP-encoding plasmid and cultured them in vitro with various concentrations of nifedipine (1, 3, and 5 μm) or DMSO (0 μm) for 5 d from day 2 and examined their neurite extension. We found that the reduction in neurite lengths of TSC2−/− neurons by nifedipine was larger than those of TSC2+/+ or TSC2+/− neurons: 5 μm nifedipine treatment reduced neurite extension in TSC2−/− neurons by ∼ 230 µm, whereas neurite lengths of TSC2+/+ and TSC2+/− were reduced by ∼70 and ∼−17 µm, respectively (Fig. 9). Nevertheless, TSC2−/− neurons treated with 5 μm nifedipine still had longer neurites than DMSO-treated TSC2+/+ and TSC2+/− neurons (Kruskal–Wallis test, p < 0.00001, Steel–Dwass test, ****p < 0.00001, DMSO-treated TSC2+/+ [n = 197] vs 5 μm nifedipine-treated TSC2−/− [n = 280]; ****p < 0.00001, DMSO-treated TSC2+/− [n = 312] vs 5 μm nifedipine-treated TSC2−/− [n = 280]), indicating that the abnormal neurite extension of TSC2−/− neurons was not entirely because of Ca2+ influx through LTCCs. Together with the finding that TSC2−/− neurons exhibited enhanced Ca2+ influx via LTCCs in an mTOR-dependent manner, these results suggest that enhanced TSC-mTOR signaling caused by homozygous TSC2 deletion partially contributed to the abnormal neurite extension of human TSC2−/− neurons through enhanced Ca2+ influx via LTCCs.
Figure 9.

Suppression of LTCCs partially ameliorated the aberrant neurite extensions in TSC2−/− neurons. Neurite length of DMSO- or nifedipine-treated TSC2+/+, TSC2+/−, and TSC2−/− neurons is shown. After transfection with pMax-GFP (Lonza), NPCs were differentiated into neurons, and neurite lengths were measured on day 7. For the nifedipine (Nif) treatment, neurons were incubated with three concentrations of nifedipine (1, 3, or 5 μm) or DMSO for 5 d from day 2 of neuronal differentiation. Experiments were performed 4 times. TSC2+/+, DMSO: 409.62 ± 244.4 (n = 197); 1 μm Nif: 395.58 ± 244.58 (n = 254); 3 μm Nif: 390.47 ± 268.61 (n = 276); 5 μm Nif: 341.54 ± 219.4 (n = 255). TSC2+/−, DMSO: 465.34 ± 297.23 (n = 312); 1 μm Nif: 477.92 ± 319.56 (n = 225); 3 μm Nif: 494.76 ± 337.62 (n = 313); 5 μm Nif: 482.75 ± 296.32 (n = 282). TSC2−/−, DMSO: 845.75 ± 472.27 (n = 179); 1 μm Nif: 617.02 ± 319.59 (n = 248); 3 μm Nif: 642.92 ± 364.58 (n = 256); 5 μm Nif: 615.16 ± 332.45 (n = 280). Data are mean ± SD. p < 0.00001 (Kruskal–Wallis test). Steel–Dwass test: ****p < 0.00001, DMSO-treated TSC2−/− (n = 179) versus 1 μm nifedipine-treated TSC2−/− (n = 248); ****p < 0.00001, DMSO-treated TSC2−/− (n = 179) versus 3 μm nifedipine-treated TSC2−/− (n = 256); ****p < 0.00001, DMSO-treated TSC2−/− (n = 179) versus 5 μm nifedipine-treated TSC2−/− (n = 280); ****p < 0.00001, DMSO-treated TSC2−/− (n = 179) versus DMSO-treated TSC2+/+ (n = 197); ****p < 0.00001, DMSO-treated TSC2−/− (n = 179) versus 1 μm nifedipine-treated TSC2+/+ (n = 254); ****p < 0.00001, DMSO-treated TSC2−/− (n = 179) versus 3 μm nifedipine-treated TSC2+/+ (n = 276); ****p < 0.00001, DMSO-treated TSC2−/− (n = 179) versus 5 μm nifedipine-treated TSC2+/+ (n = 255); ****p < 0.00001, DMSO-treated TSC2−/− (n = 179) versus DMSO-treated TSC2+/− (n = 312); ****p < 0.00001, DMSO-treated TSC2−/− (n = 179) versus 1 μm nifedipine-treated TSC2+/− (n = 225); ****p < 0.00001, DMSO-treated TSC2−/− (n = 179) versus 3 μm nifedipine-treated TSC2+/− (n = 313); ****p < 0.00001, DMSO-treated TSC2−/− (n = 179) versus 5 μm nifedipine-treated TSC2+/+ (n = 282); p = 0.0785, DMSO-treated TSC2+/+ (n = 197) versus 5 μm nifedipine-treated TSC2+/+ (n = 225); p = 0.9997, DMSO-treated TSC2+/− (n = 312) versus 5 μm nifedipine-treated TSC2+/− (n = 282). All statistical data are presented in Extended Data Figure 9.
All statistical data are shown. Download Figure 9-1, XLSX file (25.2KB, xlsx) .
Enhanced Ca2+ influx triggers temporally distinct and sustained CREB activation in TSC2−/− neurons
In addition to brain development, Ca2+ influx via LTCCs contributes to excitation-transcription coupling and plasticity through various signal transduction cascades (Dolmetsch et al., 2001; Deisseroth et al., 2003; Ch'ng and Martin, 2011). CREB is a primary mediator that is activated by the phosphorylation of Ser-133 (Gonzalez and Montminy, 1989). Previous studies demonstrated that Ser-133 phosphorylation is regulated by two temporally distinct signaling cascades: one is the rapid CaM kinase-dependent pathway (∼10 min) and the other is the slower Ras/MAPK-dependent pathway (∼ 60 min) (Wu et al., 2001). Although both pathways rely on cytosolic Ca2+ level elevation via LTCCs, the Ras/MAPK-dependent pathway requires larger intracellular Ca2+ transients than the CaM kinase-dependent pathway (Wu et al., 2001).
Since TSC2−/− neurons showed much higher Ca2+ influx than TSC2+/+ neurons (Fig. 4A), we examined whether TSC2−/− neurons had different pCREB kinetics after membrane depolarization. We depolarized cultured neurons with 60 mm KCl for 3 min and tracked the dynamics of pCREB by immunostaining with anti-pCREB antibodies (Fig. 10A). TSC2+/+ neurons showed a prominent increase in pCREB signals in the nuclei at 3 min after KCl stimulation (Fig. 10B). The pCREB levels gradually decreased as the fixation time was delayed by 10 and 60 min. The intensity histogram clearly showed the time-dependent change in the pCREB signal intensities in the neurons (Fig. 10C). The percentage of TSC2+/+ neurons that showed relatively high pCREB intensities with pCREB (arbitrary fluorescence intensity >3000) was 40.0% (3 min), 24.2% (10 min), and 16.74% (60 min) (Fig. 10D). The change in pCREB intensity in TSC2+/− neurons was similar to that in TSC2+/+ neurons. The percentage of TSC2+/− neurons with a pCREB intensity >3000 was 45.5% (3 min), 23.66% (10 min), and 19.27% (60 min). On the other hand, although TSC2−/− neurons showed a transient increase and subsequent decrease in pCREB intensity at 10 min, similar to TSC2+/+ neurons, the pCREB levels at 10 min were rather sustained at 60 min (Fig. 10D). The percentage of TSC2−/− neurons with a relatively high pCREB intensity was 43.9% (3 min), 27.54% (10 min), and 34.87% (60 min). The sustained activation of pCREB at 60 min was abolished in rapamycin-treated TSC2−/− neurons (Fig. 10B–D, bottom panels). Together with the strong Ca2+ influx into TSC2−/− neurons on membrane depolarization (Fig. 4), these results suggest that enhanced Ca2+ influx via LTCCs in TSC2−/− neurons contributed to the sustained activation of the CREB transcription factor on membrane depolarization.
Figure 10.
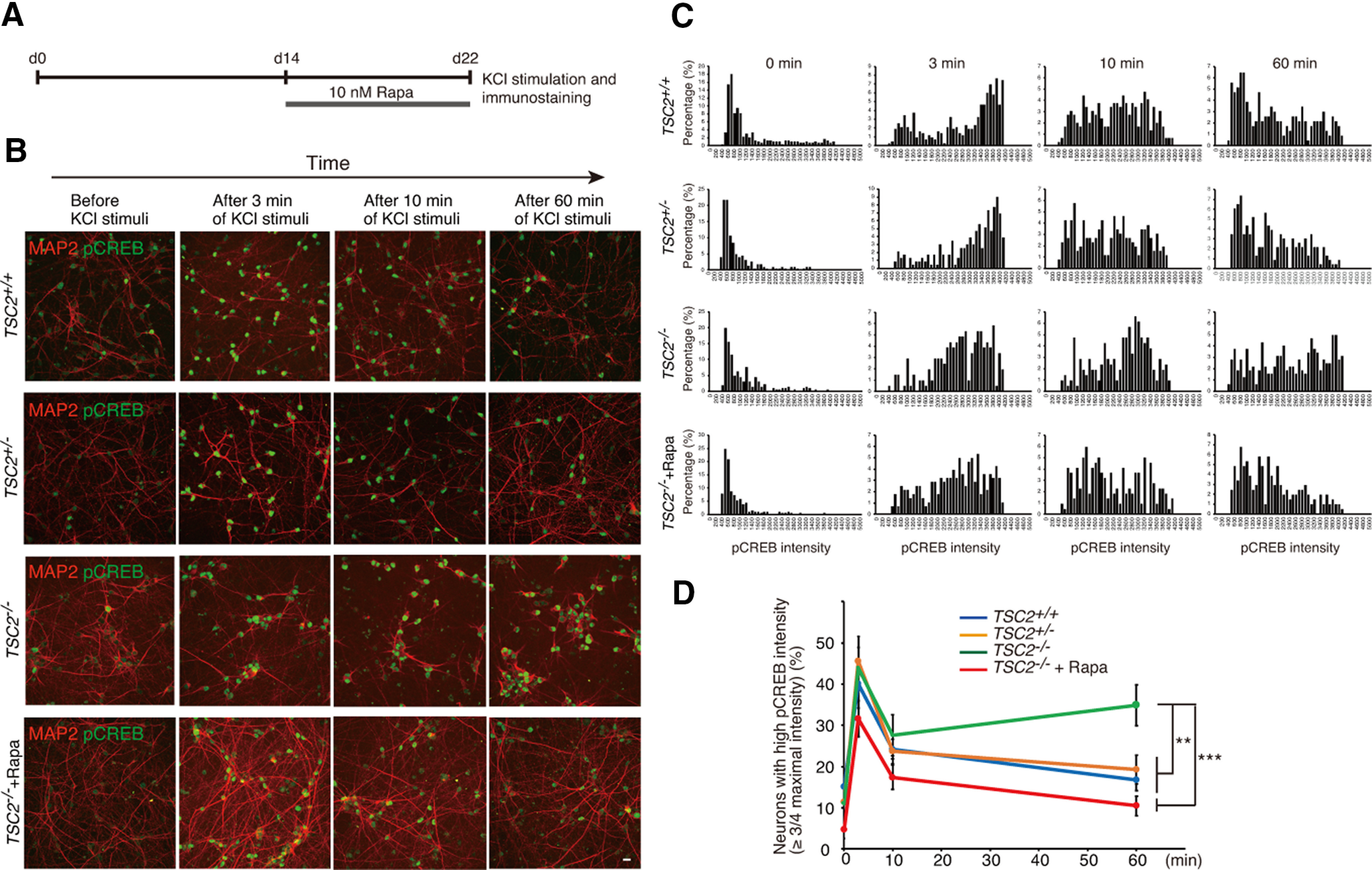
Sustained activation of CREB in TSC2−/− neurons on membrane depolarization. A, Schematic illustration of rapamycin treatment, KCl stimulation, and immunostaining after plating NPCs for neuronal differentiation. B, Immunostaining of phospho-CREB (green) in TSC2+/+, TSC2+/−, TSC2−/−, and rapamycin-treated TSC2−/− neurons after membrane depolarization. Red represents MAP2. Scale bar, 50 µm. C, The intensity histograms of pCREB (arbitrary units) are shown. Experiments were performed at least 3 times with independent cultures. Representative data are shown. D, Percentage of neurons with high pCREB immunoreactivity (intensity> 3000). Data are mean ± SD; n = 4-8 experiments. For each experiment, 200-750 neurons were analyzed. Two-way repeated-measures ANOVA (genotype, F(3,56) = 3.6403), *p = 0.03038. Tukey HSD test, **p = 0.0051, TSC2+/− (n = 7) versus TSC2−/− (n = 6); ***p = 0.00073, TSC2−/− (n = 6) versus TSC2−/− + Rapa (n = 4); **p = 0.0013, TSC2+/+ (n = 8) versus TSC2−/− (n = 6).
Discussion
In this study, we generated human iPSC-derived cortical neurons with TSC2 mutations and characterized their developmental and physiological properties. We found that TSC2−/− iPSCs showed a robust increase in the level of S6 phosphorylation. In contrast, we did not observe any difference in phospho-S6 levels between TSC2+/+ and TSC2+/− cells. This tendency was also observed during their development into neurons, as we detected increased phospho-S6 signals only in TSC2−/− NPCs and mature TSC2−/− neurons. Consistent with these observations, TSC2−/− but not TSC2+/− neurons had abnormal morphology, such as larger soma sizes and longer neurites, compared with TSC2+/+ neurons. Analysis of neuronal population activity by using Ca2+ imaging revealed that TSC2−/− neurons displayed high-frequency firing with synchronized Ca2+ spikes in our culture conditions. Our findings are also consistent with those of a recent study in which multielectrode array analysis was used to show increased spontaneous activity of human TSC2−/− neurons (Nadadhur et al., 2019). Importantly, the described abnormalities in TSC2−/− neurons were ameliorated by long-term rapamycin treatment. We also found the potential molecular mechanism underlying these abnormalities: increased Ca2+ entry via LTCCs, perhaps Cav1.3, in TSC2−/− neurons. TSC2−/− neurons showed an mTOR-dependent increase in CACNA1D expression and enhanced Ca2+ influx. In addition, activation of mTOR by c.a.Rheb overexpression in TSC2+/+ neurons enhanced Cav.1.3 expression and augmented Ca2+ influx on depolarization. Thus, our findings suggest that LTCCs, possibly Cav1.3, are an important downstream component of the mTOR pathway and that enhanced Ca2+ influx via LTCCs may be a critical factor underlying epilepsy onset in TSC. LTCCs could be novel molecular targets for the treatment of epilepsy in patients with TSC.
Although many studies have proposed the possibility of an intimate relationship between mTOR hyperactivation and seizures in TSC, the molecular mechanisms by which mTOR causes epilepsy are not completely understood. We found that TSC2−/− neurons had significantly enhanced Ca2+ influx through LTCCs as well as increased Cav1.3 levels. It is known that Cav1.3 has a relatively negative activation threshold in contrast to Cav1.2 and that it enables neurons to exhibit a slowly inactivating Ca2+ influx in response to rather weak depolarization (Koschak et al., 2001; Xu and Lipscombe, 2001). This property of Cav1.3 appears to induce tonic firing in neurons by shifting the membrane potential to a slightly depolarized state (Olson et al., 2005; Liu et al., 2014). It would be interesting for future experiments to examine whether increased Cav1.3 expression affects membrane potential in TSC2−/− neurons and causes high-frequency neuronal firing with synchronized Ca2+ spikes.
LTCCs contribute to neurite growth in developing mouse cortical neurons (Tang et al., 2003; Kamijo et al., 2018). Both Cav1.2 and Cav1.3 are expressed in developing cortical neurons and generate spontaneous regenerative Ca2+ transients at the axonal tips, which contribute to neurite extension (Kamijo et al., 2018). Since we showed that inhibition of LTCCs, albeit in part, ameliorated the excessive neurite extensions of human TSC2−/− neurons, it would be interesting to examine whether human TSC2−/− neurons have more spontaneous regenerative Ca2+ transient events in the axonal tips than TSC2+/+ neurons. In addition, since spontaneous Ca2+ transients by LTCCs also affect the radial migration of cortical neurons (Kamijo et al., 2018), enhanced Ca2+ influx via LTCCs in human cortical TSC2−/− neurons might also lead to aberrant neuronal positioning as well as axonal development in the TSC.
We also showed that TSC2−/− neurons exhibited prolonged activation of CREB after KCl application compared with TSC2+/+ neurons. CREB is a critical transcription factor that regulates the expression of many genes involved in intrinsic excitability and synaptic plasticity (Benito and Barco, 2010). Thus, sustained CREB activation of TSC2−/− neurons on membrane depolarization, which we observed in this study, may also contribute to the increased activity of TSC2−/− neurons by modifying the efficacy of neuronal transmission as well as intrinsic excitability. Our findings may be an underlying cause of the lower threshold of late-phase LTP in the Schaffer collateral pathway and increased excitability of hippocampal CA1 neurons in Tsc1-deficient mice (Abs et al., 2013). Gain-of function mutations of Cav1.3 have also been reported in patients with ASD (Azizan et al., 2013; Scholl et al., 2013; De Rubeis et al., 2014; Pinggera et al., 2015; Limpitikul et al., 2016). Therefore, enhanced Ca2+ influx via Cav1.3 LTCCs in TSC2−/− neurons might be, at least in part, an underlying cause for the psychiatric phenotypes of patients with TSC in addition to epilepsy.
Cav1.3, which can undergo several forms of RNA editing, gives rise to functional diversity (Huang et al., 2012; Bazzazi et al., 2013). A-to-I editing within the IQ motif especially decreases CDI and induces high-frequency firing with Ca2+ spikes in suprachiasmatic nucleus neurons (Huang et al., 2012). Therefore, we examined RNA editing within the IQ motif in Cav1.3 and found a very small increase in A-to-I editing in TSC2−/− neurons (∼16%) compared with TSC2+/+ cells (∼9%) in 30-d-old cultures. However, this increase was not observed at 60 d of neuronal differentiation, regardless of the higher ADARB1 expression in TSC2−/− neurons than that in TSC2+/+ neurons. Therefore, A-to-I editing within the IQ motif in Cav1.3 could contribute to the enhanced Ca2+ influx in the early developmental stage but is negligible in more mature TSC2−/− neurons.
It should be stressed that abnormal neuronal phenotypes were only seen in TSC2−/− cells but not in TSC2+/− cells. Since TSC is an autosomal dominant disease, additional mechanisms, such as the second hit model (Blair et al., 2018), are needed to cause mTOR hyperactivation in the brain of patients with TSC. Alternatively, the phenotypes of TSC2+/− cells may be affected by experimental cell culture conditions, as other studies have also reported certain differences in phospho-S6 signal levels in TSC2 heterozygous cells (Costa et al., 2016; Li et al., 2017). Distinct neuronal environments between in vitro culture and the human brain may affect TSC expression from the residual normal allele and/or cause altered TSC-Rheb protein complex regulation. The lack of phenotypes in the TSC2+/− cells could also be caused by a distinct mutation in TSC2−/− cells, which may have a different outcome on TSC2 function/expression.
Considerable genetic drift and instability can occur over the course of human iPSC culture and after single-cell passaging following gene editing. During the review process of our present study, we found trisomy of chromosome 12 in our TSC2+/+, TSC2+/−, and TSC2−/− iPSCs. The karyotypes were TSC2+/+: 47,XX,+12; TSC2+/−: 47,XX,+12,add(17)(q25); TSC2−/−: 47,XX,+12,t(12;12)(p10;p10). Owing to these chromosome abnormalities, our results should be treated with caution. Moreover, we did not absolutely exclude the possibility that the abnormal chromosome triggered our TSC2−/− phenotypes. Nevertheless, we think that it is reasonable to conclude that the TSC2−/− phenotypes we found in this study were independent of the abnormal chromosome, for the following reasons: (1) Enhanced Ca2+ influx and Cav1.3 expression in TSC2−/− neurons are dependent on the activity of mTOR, which is localized on chromosome 1 in humans. (2) CACNA1D is localized on chromosome 3 in humans, and amplification of CACNA1D is not likely to be involved in the increase in CACNA1D expression in TSC2−/− neurons. (3) Overexpression of the constitutively active form of Rheb in TSC2+/+ neurons increased Cav1.3 protein levels and Ca2+ influx in the transfected cells. (4) Since all genotypes of our iPSCs had trisomy 12, it is difficult to explain that an increase in the number of chromosome 12 affects only TSC2−/− phenotypes.
In conclusion, we demonstrated enhanced Ca2+ influx via LTCCs, likely Cav1.3, which would underlie the aberrant neuronal firing of TSC2−/− neurons. The augmentation of Ca2+ influx via LTCCs in TSC2−/− neurons depends on mTOR activity. To the best of our knowledge, this is the first study highlighting the signal crosslink between the TSC-mTOR pathway and Ca2+ signaling via LTCCs. Thus, our findings provide critical evidence for the understanding of the molecular mechanisms underlying the developmental and psychiatric phenotypes of TSC. Since altered mTOR signaling is associated with several other genetic syndromes in addition to TSC, such as phosphatase and tensin homolog deleted on chromosome 10-related syndrome, neurofibromatosis Type 1, and fragile X syndrome (Switon et al., 2017), our findings could be a fundamental molecular mechanism triggering epilepsy in these diseases. This idea would be supported by a recent study showing the increased Ca2+ influx via LTCC in human NPCs lacking fragile X mental retardation protein (Danesi et al., 2018). It is important for future studies to examine whether the neurons of patients with TSC have enhanced Ca2+ influx via LTCCs and altered Cav1.3 levels in the brain.
Footnotes
This work was supported by Grant-in-Aid for Scientific Research (B), Grant JP25293239/18H02536 to K.Y.; Japan Agency for Medical Research and Development Grant JP18ek0109311h0001/19ek0109311h0002/20ek0109311h0003 to K.Y.; SENSHIN Medical Research Foundation to K.Y.; National Institutes of Health Grant R01EY026817 to A.L.; and Grant-in-Aid for Scientific Research (C), Grant 19K08334 to C.H. We thank RIKEN BRC for providing the human iPS cells (HPS0002: 409B2). pAAV-hSyn-EGFP was a gift from Dr. Bryan Roth (Addgene plasmid #50465). We thank Ms. Tomoko Kawano for helping with the maintenance of the cell cultures; Dr. Frederick J. Livesey (University of Cambridge) and Dr. Julia Tcw (Icahn School of Medicine at Mount Sinai) for kind and fruitful advice regarding cell culturing; Dr. Hiroyuki Miyamoto for valuable comments on the manuscript; and Editage (www.editage.com) for English language editing.
The authors declare no competing financial interests.
References
- Abs E, Goorden SM, Schreiber J, Overwater IE, Hoogeveen-Westerveld M, Bruinsma CF, Aganovic E, Borgesius NZ, Nellist M, Elgersma Y (2013) TORC1-dependent epilepsy caused by acute biallelic Tsc1 deletion in adult mice. Ann Neurol 74:569–579. 10.1002/ana.23943 [DOI] [PubMed] [Google Scholar]
- Azizan EA, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, Maniero C, Garg S, Bochukova EG, Zhao W, Shaikh LH, Brighton CA, Teo AE, Davenport AP, Dekkers T, Tops B, Küsters B, Ceral J, Yeo GS, Neogi SG, et al. (2013) Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet 45:1055–1060. 10.1038/ng.2716 [DOI] [PubMed] [Google Scholar]
- Bazzazi H, Ben Johny M, Adams PJ, Soong TW, Yue DT (2013) Continuously tunable Ca2+ regulation of RNA-edited CaV1.3 channels. Cell Rep 5:367–377. 10.1016/j.celrep.2013.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers J, Gulbranson DR, George N, Siniscalchi LI, Jones J, Thomson JA, Chen G (2012) Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat Protoc 7:2029–2040. 10.1038/nprot.2012.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito E, Barco A (2010) CREB's control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci 33:230–240. 10.1016/j.tins.2010.02.001 [DOI] [PubMed] [Google Scholar]
- Blair JD, Hockemeyer D, Bateup HS (2018) Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat Med 24:1568–1578. 10.1038/s41591-018-0139-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, McCarthy S, Sebat J, Gage FH (2011) Modelling schizophrenia using human induced pluripotent stem cells. Nature 473:221–225. 10.1038/nature09915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardamone M, Flanagan D, Mowat D, Kennedy SE, Chopra M, Lawson JA (2014) Mammalian target of rapamycin inhibitors for intractable epilepsy and subependymal giant cell astrocytomas in tuberous sclerosis complex. J Pediatr 164:1195–1200. 10.1016/j.jpeds.2013.12.053 [DOI] [PubMed] [Google Scholar]
- Carson RP, Van Nielen DL, Winzenburger PA, Ess KC (2012) Neuronal and glia abnormalities in Tsc1-deficient forebrain and partial rescue by rapamycin. Neurobiol Dis 45:369–380. 10.1016/j.nbd.2011.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch'ng TH, Martin KC (2011) Synapse-to-nucleus signaling. Curr Opin Neurobiol 21:345–352. 10.1016/j.conb.2011.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Di Nardo A, Kramvis I, Meikle L, Kwiatkowski DJ, Sahin M, He X (2008) Tuberous sclerosis complex proteins control axon formation. Genes Dev 22:2485–2495. 10.1101/gad.1685008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA (2010) The natural history of epilepsy in tuberous sclerosis complex. Epilepsia 51:1236–1241. 10.1111/j.1528-1167.2009.02474.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa V, Aigner S, Vukcevic M, Sauter E, Behr K, Ebeling M, Dunkley T, Friedlein A, Zoffmann S, Meyer CA, Knoflach F, Lugert S, Patsch C, Fjeldskaar F, Chicha-Gaudimier L, Kiialainen A, Piraino P, Bedoucha M, Graf M, Jessberger S, et al. (2016) mTORC1 inhibition corrects neurodevelopmental and synaptic alterations in a human stem cell model of tuberous sclerosis. Cell Rep 15:86–95. 10.1016/j.celrep.2016.02.090 [DOI] [PubMed] [Google Scholar]
- Crino PB (2013) Evolving neurobiology of tuberous sclerosis complex. Acta Neuropathol 125:317–332. 10.1007/s00401-013-1085-x [DOI] [PubMed] [Google Scholar]
- Crino PB, Nathanson KL, Henske EP (2006) The tuberous sclerosis complex. N Engl J Med 355:1345–1356. 10.1056/NEJMra055323 [DOI] [PubMed] [Google Scholar]
- Danesi C, Achuta VS, Corcoran P, Peteri UK, Turconi G, Matsui N, Albayrak I, Rezov V, Isaksson A, Castren ML (2018) Increased calcium influx through L-type calcium channels in human and mouse neural progenitors lacking fragile X mental retardation protein. Stem Cell Rep 11:1449–1461. 10.1016/j.stemcr.2018.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, Fu SC, Aleksic B, Biscaldi M, Bolton PF, Brownfeld JM, Cai J, Campbell NG, et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515:209–215. 10.1038/nature13772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K, Mermelstein PG, Xia H, Tsien RW (2003) Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol 13:354–365. 10.1016/s0959-4388(03)00076-x [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME (2001) Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294:333–339. 10.1126/science.1063395 [DOI] [PubMed] [Google Scholar]
- Dolphin AC (2016) Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol 594:5369–5390. 10.1113/JP272262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenno LE, Mattis J, Ramakrishnan C, Hyun M, Lee SY, He M, Tucciarone J, Selimbeyoglu A, Berndt A, Grosenick L, Zalocusky KA, Bernstein H, Swanson H, Perry C, Diester I, Boyce FM, Bass CE, Neve R, Huang ZJ, Deisseroth K (2014) Targeting cells with single vectors using multiple-feature Boolean logic. Nat Methods 11:763–772. 10.1038/nmeth.2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarasi A, De Waele L, Bartalini G, Jozwiak S, Laforgia N, Verhelst H, Petrak B, Pedespan JM, Witt O, Castellana R, Crippa S, Gislimberti G, Gyorsok Z (2016) EFFECTS: an expanded access program of everolimus for patients with subependymal giant cell astrocytoma associated with tuberous sclerosis complex. BMC Neurol 16:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, Curatolo P, de Vries PJ, Dlugos DJ, Berkowitz N, Voi M, Peyrard S, Pelov D, Franz DN (2016) Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet 388:2153–2163. 10.1016/S0140-6736(16)31419-2 [DOI] [PubMed] [Google Scholar]
- Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G (2003) Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell 11:1457–1466. 10.1016/s1097-2765(03)00220-x [DOI] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR (1989) Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 59:675–680. 10.1016/0092-8674(89)90013-5 [DOI] [PubMed] [Google Scholar]
- Heyes S, Pratt WS, Rees E, Dahimene S, Ferron L, Owen MJ, Dolphin AC (2015) Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. Prog Neurobiol 134:36–54. 10.1016/j.pneurobio.2015.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh LS, Wen JH, Claycomb K, Huang Y, Harrsch FA, Naegele JR, Hyder F, Buchanan GF, Bordey A (2016) Convulsive seizures from experimental focal cortical dysplasia occur independently of cell misplacement. Nat Commun 7:11753. 10.1038/ncomms11753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Tan BZ, Shen Y, Tao J, Jiang F, Sung YY, Ng CK, Raida M, Kohr G, Higuchi M, Fatemi-Shariatpanahi H, Harden B, Yue DT, Soong TW (2012) RNA editing of the IQ domain in Cav1.3 channels modulates their Ca2+-dependent inactivation. Neuron 73:304–316. 10.1016/j.neuron.2011.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Xu T, Guan KL (2003) Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 17:1829–1834. 10.1101/gad.1110003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MA, Christel CJ, Jiao Y, Abiria S, Kim KY, Usachev YM, Obermair GJ, Colbran RJ, Lee A (2010) Ca2+-dependent facilitation of Cav1.3 Ca2+ channels by densin and Ca2+/calmodulin-dependent protein kinase II. J Neurosci 30:5125–5135. 10.1523/JNEUROSCI.4367-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo S, Ishii Y, Horigane SI, Suzuki K, Ohkura M, Nakai J, Fujii H, Takemoto-Kimura S, Bito H (2018) A critical neurodevelopmental role for L-type voltage-gated calcium channels in neurite extension and radial migration. J Neurosci 38:5551–5566. 10.1523/JNEUROSCI.2357-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda Y (2013) Investigation of the freely available easy-to-use software 'EZR' for medical statistics. Bone Marrow Transplant 48:452–458. 10.1038/bmt.2012.244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koene LM, Grondelle SE, Proietti Onori M, Wallaard I, Kooijman NH, Oort A, Schreiber J, Elgersma Y (2019) Effects of antiepileptic drugs in a new TSC/mTOR-dependent epilepsy mouse model. Ann Clin Transl Neurol 6:1273–1291. 10.1002/acn3.50829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J (2001) alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J Biol Chem 276:22100–22106. 10.1074/jbc.M101469200 [DOI] [PubMed] [Google Scholar]
- Krueger DA, Care MM, Agricola K, Tudor C, Mays M, Franz DN (2013) Everolimus long-term safety and efficacy in subependymal giant cell astrocytoma. Neurology 80:574–580. 10.1212/WNL.0b013e3182815428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DA, Wilfong AA, Mays M, Talley CM, Agricola K, Tudor C, Capal J, Holland-Bouley K, Franz DN (2016) Long-term treatment of epilepsy with everolimus in tuberous sclerosis. Neurology 87:2408–2415. 10.1212/WNL.0000000000003400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasarge CL, Danzer SC (2014) Mechanisms regulating neuronal excitability and seizure development following mTOR pathway hyperactivation. Front Mol Neurosci 7:18. 10.3389/fnmol.2014.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Cao J, Chen M, Li J, Sun Y, Zhang Y, Zhu Y, Wang L, Zhang C (2017) Abnormal neural progenitor cells differentiated from induced pluripotent stem cells partially mimicked development of TSC2 neurological abnormalities. Stem Cell Rep 8:883–893. 10.1016/j.stemcr.2017.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limpitikul WB, Dick IE, Ben-Johny M, Yue DT (2016) An autism-associated mutation in CaV1.3 channels has opposing effects on voltage- and Ca2+-dependent regulation. Sci Rep 6:27235. 10.1038/srep27235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Harding M, Pittman A, Dore J, Striessnig J, Rajadhyaksha A, Chen X (2014) Cav1.2 and Cav1.3 L-type calcium channels regulate dopaminergic firing activity in the mouse ventral tegmental area. J Neurophysiol 112:1119–1130. 10.1152/jn.00757.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magri L, Cambiaghi M, Cominelli M, Alfaro-Cervello C, Cursi M, Pala M, Bulfone A, Garcìa-Verdugo JM, Leocani L, Minicucci F, Poliani PL, Galli R (2011) Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell 9:447–462. 10.1016/j.stem.2011.09.008 [DOI] [PubMed] [Google Scholar]
- Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, Jensen FE, Kwiatkowski DJ (2007) A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci 27:5546–5558. 10.1523/JNEUROSCI.5540-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi M, Ikeda H, Kagitani-Shimono K, Yoshinaga H, Suzuki Y, Aoki M, Endo M, Yonemura M, Kubota M (2019) Everolimus for epilepsy and autism spectrum disorder in tuberous sclerosis complex: EXIST-3 substudy in Japan. Brain Dev 41:1–10. 10.1016/j.braindev.2018.07.003 [DOI] [PubMed] [Google Scholar]
- Montagne J, Radimerski T, Thomas G (2001) Insulin signaling: lessons from the Drosophila tuberous sclerosis complex, a tumor suppressor. Sci STKE 2001:pe36. 10.1126/stke.2001.105.pe36 [DOI] [PubMed] [Google Scholar]
- Nadadhur AG, Alsaqati M, Gasparotto L, Cornelissen-Steijger P, van Hugte E, Dooves S, Harwood AJ, Heine VM (2019) Neuron-glia interactions increase neuronal phenotypes in tuberous sclerosis complex patient iPSC-derived models. Stem Cell Rep 12:42–56. 10.1016/j.stemcr.2018.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LH, Mahadeo T, Bordey A (2019) mTOR hyperactivity levels influence the severity of epilepsy and associated neuropathology in an experimental model of tuberous sclerosis complex and focal cortical dysplasia. J Neurosci 39:2762–2773. 10.1523/JNEUROSCI.2260-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, Hong H, Nakagawa M, Tanabe K, Tezuka K, Shibata T, Kunisada T, Takahashi M, Takahashi J, Saji H, Yamanaka S (2011) A more efficient method to generate integration-free human iPS cells. Nat Methods 8:409–412. 10.1038/nmeth.1591 [DOI] [PubMed] [Google Scholar]
- Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I, Surmeier DJ (2005) G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J Neurosci 25:1050–1062. 10.1523/JNEUROSCI.3327-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwater IE, Bindels-De Heus K, Rietman AB, Ten Hoopen LW, Vergouwe Y, Moll HA, De Wit MC (2015) Epilepsy in children with tuberous sclerosis complex: chance of remission and response to antiepileptic drugs. Epilepsia 56:1239–1245. 10.1111/epi.13050 [DOI] [PubMed] [Google Scholar]
- Pinggera A, Lieb A, Benedetti B, Lampert M, Monteleone S, Liedl KR, Tuluc P, Striessnig J (2015) CACNA1D de novo mutations in autism spectrum disorders activate Cav1.3 L-type calcium channels. Biol Psychiatry 77:816–822. 10.1016/j.biopsych.2014.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl UI, Goh G, Stölting G, de Oliveira RC, Choi M, Overton JD, Fonseca AL, Korah R, Starker LF, Kunstman JW, Prasad ML, Hartung EA, Mauras N, Benson MR, Brady T, Shapiro JR, Loring E, Nelson-Williams C, Libutti SK, Mane S, et al. (2013) Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet 45:1050–1054. 10.1038/ng.2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Kirwan P, Livesey FJ (2012) Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat Protoc 7:1836–1846. 10.1038/nprot.2012.116 [DOI] [PubMed] [Google Scholar]
- Simms BA, Zamponi GW (2014) Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron 82:24–45. 10.1016/j.neuron.2014.03.016 [DOI] [PubMed] [Google Scholar]
- Sparagana SP, Roach ES (2000) Tuberous sclerosis complex. Curr Opin Neurol 13:115–119. 10.1097/00019052-200004000-00001 [DOI] [PubMed] [Google Scholar]
- Sundberg M, Tochitsky I, Buchholz DE, Winden K, Kujala V, Kapur K, Cataltepe D, Turner D, Han MJ, Woolf CJ, Hatten ME, Sahin M (2018) Purkinje cells derived from TSC patients display hypoexcitability and synaptic deficits associated with reduced FMRP levels and reversed by rapamycin. Mol Psychiatry 23:2167–2183. 10.1038/s41380-018-0018-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Switon K, Kotulska K, Janusz-Kaminska A, Zmorzynska J, Jaworski J (2017) Molecular neurobiology of mTOR. Neuroscience 341:112–153. 10.1016/j.neuroscience.2016.11.017 [DOI] [PubMed] [Google Scholar]
- Tang F, Dent EW, Kalil K (2003) Spontaneous calcium transients in developing cortical neurons regulate axon outgrowth. J Neurosci 23:927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele EA (2010) Managing and understanding epilepsy in tuberous sclerosis complex. Epilepsia 51 Suppl 1:90–91. 10.1111/j.1528-1167.2009.02458.x [DOI] [PubMed] [Google Scholar]
- Uhlmann EJ, Wong M, Baldwin RL, Bajenaru ML, Onda H, Kwiatkowski DJ, Yamada K, Gutmann DH (2002) Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol 52:285–296. 10.1002/ana.10283 [DOI] [PubMed] [Google Scholar]
- Winden KD, Sundberg M, Yang C, Wafa SM, Dwyer S, Chen PF, Buttermore ED, Sahin M (2019) Biallelic mutations in TSC2 lead to abnormalities associated with cortical tubers in human iPSC-derived neurons. J Neurosci 39:9294–9305. 10.1523/JNEUROSCI.0642-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW (2001) Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA 98:2808–2813. 10.1073/pnas.051634198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Lipscombe D (2001) Neuronal CaV1.3α1L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci 21:5944–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Sanders LK, Kaufmann WE, Yee W, Barnes CA, Nathans D, Worley PF (1994) rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J Biol Chem 269:16333–16339. 10.1016/S0021-9258(17)34012-7 [DOI] [PubMed] [Google Scholar]
- Zeng LH, Xu L, Gutmann DH, Wong M (2008) Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol 63:444–453. 10.1002/ana.21331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Rensing NR, Zhang B, Gutmann DH, Gambello MJ, Wong M (2011) Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of Tuberous Sclerosis Complex. Hum Mol Genet 20:445–454. 10.1093/hmg/ddq491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D (2003) Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol 5:578–581. 10.1038/ncb999 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spontaneous Ca2+ signals of TSC2+/+ neurons at 33 d.
Spontaneous Ca2+ signals of TSC2+/– neurons at 33 d.
Spontaneous Ca2+ signals of TSC2−/− neurons at 33 d (accelerated ninefold).
All statistical data are shown. Download Figure 9-1, XLSX file (25.2KB, xlsx) .