

Abstract
Background:
In bacteria, PriA protein, a conserved DEXH-type DNA helicase, plays a central role in replication restart at stalled replication forks. Its unique DNA binding property allows it to recognize and stabilize stalled forks and the structures derived from them. PriA plays a very critical role in replication fork stabilization and DNA repair in E. coli and N. gonorrhoeae. In our in vivo expression technology screen, priA gene was induced in vivo when Helicobacter pylori infects mouse stomach.
Materials and Methods:
We decided to elucidate the role of H. pylori PriA protein in survival in mouse stomach, survival in gastric epithelial cells and macrophage cells, DNA repair, acid stress and oxidative stress.
Results:
The priA null mutant strain was unable to colonize mice stomach mucosa after long-term infections. Mouse colonization was observed after one week of infection but the levels were much lower than the wild type HpSS1 strain. PriA protein was found to be important for intracellular survival of epithelial cell / macrophage cell ingested H. pylori. Also, a priA null mutant was more sensitive to DNA damaging agents and was much more sensitive to acid and oxidative stress as compared to the wild type strain.
Conclusions:
These data suggest that the PriA protein is needed for survival and persistence of H. pylori in mice stomach mucosa.
Keywords: Helicobacter pylori, DNA helicase, replication fork stabilization
Introduction
Helicobacter pylori (H. pylori) is a highly successful human pathogen that colonizes roughly one half of the world’s population. It is typically transmitted orally within families during early childhood and can persist for decades in its preferred niche, the gastric mucosa, despite triggering vigorous innate and adaptive immune responses. H. pylori infection causes chronic gastritis, which is asymptomatic in the majority of carriers but is considered a major risk factor for the development of gastric and duodenal ulcers and the two gastric malignancies, mucosa-associated lymphoid tissue lymphoma and gastric adenocarcinoma (1). In addition to its tight association with cancer, H. pylori stands out among other gram negative bacterial pathogens in its ability to persist and establish chronic infection. The capacity of H. pylori to colonize the human stomach can be attributed to the production of specific bacterial products. Numerous H. pylori components have been designated colonization factors based on the demonstration that null mutant strains defective in the production of these factors are impaired in the ability to colonize the stomach in animal models. For example, H. pylori null mutant strains defective in production of urease or flagella are unable to colonize animal models (2, 3). Urease hydrolyzes urea to yield ammonium ions and thereby contributes to the acid resistance of H. pylori (4). Flagella confer the property of motility and enable H. pylori to penetrate the gastric mucus layer. In a signature- tagged mutagenesis analysis, 47 H. pylori genes were found to be essential for colonization of the Mongolian gerbil stomach but not essential for growth of H. pylori in vitro (5). Probably many other H. pylori factors are also required for colonization of the stomach.
In vivo expression technology (IVET) was designed to identify genes of pathogens that are preferentially expressed during infection and has been used extensively (6, 7). IVET is a promoter-trapping technique that selects microbial promoters active in a specified niche during the interaction of a microorganism with its host. Using IVET we were able to identify several H. pylori genes which were induced in vivo during infection of mice stomachs. Thirty one genes were identified. These include genes responsible for a broad and varied group of cellular structures and functions: virulence, cell envelope structures, motility, oxidative stress, nucleic acid and sugar metabolism, translation, protein synthesis, type IV secretion system and a few conserved and hypothetical proteins. Our IVET screening revealed the host-induced expression of several genes involved in nucleic acid metabolism, including hsdM/R, hsdM, recG, priA and tnpB. This class of host-induced genes is involved in DNA synthesis and modification.
PriA is a single-stranded DNA-dependent ATPase, and a 3’ to 5’ DNA translocase/ helicase that was discovered originally because of its in vitro requirement for the conversion of bacteriophage phiX174 viral DNA to the duplex replicative form (8, 9). In Escherichia coli this protein, at the crossroads of DNA replication and recombination, plays a central role in origin- independent, replication restart of collapsed or arrested DNA replication forks and is also involved in DNA recombination (10–12). In Neisseria gonorrhoeae, priA plays a critical role in DNA repair and is important for resisting killing by oxidative damaging agents (13). These activities rely on the ability of PriA to load replication forks at a D loop, an intermediate that forms during homologous recombination, double-strand break-repair, and stable DNA replication. Since the priA gene was induced in our IVET screen (14), and it plays a very critical role in replication fork stabilization and DNA repair in E. coli and N. gonorrhoeae, we decided to elucidate its role in H. pylori virulence.
Materials and Methods
Bacterial strains and growth media.
The H. pylori strain used in this study was Sydney Strain 1, SS1 (15). The strains were grown for 16 to 18 hours at 37°C in a microaerophilic atmosphere in bisulfiteless Brucella broth (BLBB) (16) containing 5% fetal bovine serum (Hyclone, Logan, UT). For BLBB solid medium, 1.7% agar was added. Unless stated otherwise, the antibiotics used in BLBB solid or liquid medium were: kanamycin (kan) 15 μg/ml, chloramphenicol (chl) 12.5 μg/ml, Glaxo Selective Supplement A or GSSA (5 μg /ml of Amphotericin-B, 20 μg /ml of Bacitracin, 1.07 μg /ml of Nalidixic acid, 0.33 μg /ml of Polymyxin-B, and 10 μg /ml of Vancomycin) (17). E. coli strain DH5 α was grown in Luria broth (LB) medium (18).
Isolation of DNA from bacterial strains.
Plasmid DNA was isolated from E. coli DH5 α using the QIAprep Miniprep or QIAfilter plasmid maxi kit (QIAGEN, Valencia, CA) in accordance with manufacturer’s recommended protocols. Genomic DNA was extracted from H. pylori strain SS1 using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI) as described by the manufacturer.
Construction of priA gene knockout strain:
The full length priA gene (1,860 bp) was cloned in pUC19. A priA gene null mutation was generated by insertion mutagenesis. A Kanamycin resistance gene along with its own promoter was inserted at the unique Nsi restriction enzyme site (nucleotide 435) of priA. The resulting plasmid, ppriA::Kan, was used to transform H. pylori strain SS1 strain by electroporation. Kanamycin-resistant transformants for priA disruption in H. pylori SS1 were verified by PCR analysis. The strain was named priAKO.
Generation of a priA gene complemented strain:
The priA gene along with its own promoter (3,300bp) was cloned between KpnI and the BglII sites of the pHel2 shuttle vector (19). This plasmid named ppriA-prom was used to transform strain priAKO. Kanamycin and chloramphenicol resistant transformants were verified for the presence of the ppriA-prom plasmid. This strain was named priAKOComp. Also in an independent experiment, the priA promoter sequence was cloned upstream of a promoterless chloramphenicol (cat) gene to check if it can drive the expression of the cat gene. The promoter sequence was found to be capable of driving the expression of the promoter less cat gene.
Animal housing and diet.
Mice were maintained in a National Institutes of Health (NIH) animal facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (Rockville, MD). They were maintained in a specific-pathogen-free animal care holding room and were confirmed to be free of the following microorganisms: cilium-associated respiratory bacillus, ectromelia, mouse rotavirus, mouse encephalomyelitis virus, lymphocytic choriomeningitis virus, murine cytomegalovirus, mouse hepatitis virus, mouse adenovirus, minute virus of mice, Mycoplasma pulmonis, parvovirus, polyomavirus, pneumonia virus of mice, reovirus, and Sendai virus. Mice were housed in 7.5- by 11.5- by 5-in. sterilized ventilated Thoren cages (Thoren Caging System, Inc., Hazleton, PA) on Tek Fresh bedding (Harlan Teklad, Madison, WI). Cages were changed weekly. The animal holding room was maintained under environmental conditions of 20°C, 40 to 70% relative humidity, 15 air changes/h and a 12-h–12-h light-dark cycle. Mice were fed an autoclaved pelleted rodent diet (rodent NIH-31 autoclavable NA; Zeigler Brothers,Inc., Gardners, PA) ad libitum and provided sterilized individual water bottles for an ad libitum water source. Upon arrival, the mice were acclimated for a minimum of 7 days prior to being used in the experiments. This study was reviewed and approved by the NIH Institutional Animal Care and Use Committee. All procedures and use of animals were in compliance with the Public Health Service Guide for the Care and Use of Laboratory Animals (20).
Inoculation of mice with H. pylori strains:
We did a comparative analysis of Wild type HpSS1 (WT), priA null mutant (priAKO) and priA gene complemented (priAKOComp) strains by inducing mice infections with all three strains. Six week old C57BL/6 mice were inoculated intragastrically with 0.1 ml of 16–18 hours grown cultures of H. pylori once a day every other day for a total of three inoculations. Mouse stomachs were harvested at 4 weeks and 8 weeks of infection to determine the colonization levels. Stomach homogenates were plated on BLBB plates at 100–103 dilutions.
Invasion and intracellular survival assays with gastric epithelial cells:
The day before the assay was performed, GSM06 gastric epithelial cells were seeded in 6-well tissue culture plates to 2 × 105 cells per well in DMEM/Ham F-12 medium (21, 22). An invasion assay and an intracellular bacterial viability assay were performed as described previously (23). GSMO6 cells were infected with 16–18 hours grown cultures of WT, priA KO and priA priAKOComp strains at a multiplicity of infection (MOI) of 100 for 4 and 24 hrs. The monolayers were centrifuged for 5 min at 600 g to synchronize bacterial contact with the monolayer. After 2 hours of incubation at 33°C in a humidified incubator, the monolayers were washed thrice with PBS and then incubated with 2 ml of DMEM/Ham F-12 containing 100 μg/ml gentamicin for 2 hours to kill extracellular bacteria. This was followed by washing the monolayers thrice with PBS and lysis in 1.0 ml sterile water per well (4hours time point) or overnight incubation in DMEM/Ham F-12 containing 1 μg gentamicin/ml (24 hours time point). Next day, the monolayers were washed, lysed and the lysates were plated on BLBB plates at 100–103 dilutions.
Invasion and intracellular survival assays with macrophage cells:
The day before the assay was performed, RAW 264.7 cells were seeded in 6-well tissue culture plates to 2 × 105 cells per well in DMEM medium (24). The protocol was similar to the invasion and intracellular survival assays with gastric epithelial cells (23).
UV Survival Assay:
WT, priA KO and priAKOComp strains were grown on BLBB plates for 18–36 hours. All the strains were grown in liquid culture and were resuspended to an OD600 of 0·2 in BLBB medium. Dilutions (100–105) were performed in PBS and plated on BLBB plates. The plates were exposed to UV fluence ranging from 10, 15, 20, 30 × 100 mj/cm2 in a UV Stratalinker. After UV exposure, the plates were incubated under microaerophilic conditions.
Ciprofloxacin Sensitivity Assay:
WT, priA KO and priAKOComp strains were grown on BLBB plates for 18–36 hours. All the strains were grown in liquid culture containing no ciprofloxacin, 0.0032, 0.0064 and 0.0128 μg/ml of ciprofloxacin for 16–18 hrs. Samples were taken and the absorbance was measured at OD600.
Acid Survival Assay:
WT, priA KO and priAKOComp strains were grown on BLBB plates for 18–36 hours. All the strains were grown from the plates in liquid culture for 16–18 hrs. pH 4 acidic medium supplemented with 0.5 mM urea, which promotes H. pylori survival in acid (25, 26), was prepared by adding 4.8 M HCl to standard growth media and filter sterilized. H. pylori strains were resuspended to an OD600 of 0.35 in pH 4 acidic medium. Incubation was performed at 37°C under microaerophilic conditions and samples were removed for analysis at 0, 2, 4 and 24 hours. Dilutions (100–104) were performed in PBS and plated on BLBB plates.
Oxidative Stress Assay:
WT, priA KO and priAKOComp strains were resuspended to an OD600 of 0.35 in PBS containing fresh unopened 50 mM H2O2 (27). Incubation was performed at 37°C under microaerophilic conditions and samples were removed for analysis at 0, 4 and 24 hours. Dilutions (100–104) were performed in PBS and plated on BLBB plates.
Statistical analysis:
Data are presented as mean ± SE from three independent experiments. The Student’s T-test was used for statistical analysis between values from WT and priA KO strains. The threshold significance level for the statistically significant mean differences between these groups was P < 0.05.
Results
H. pylori priA mutant is impaired in its ability to colonize mice stomach mucosa.
Using pathogen-free C57BL/6 mice we performed a comparative analysis of WT, priAKO and priAKOComp strain colonization at 4 weeks and 8 weeks after infection. Unlike the WT and priAKO Comp strains, the HpSS1 priAKO strain did not colonize the mouse stomachs at either time point (Figure 1). In addition, the restored colonization ability of the HpSS1 priAKOComp strain was significantly lower than the WT HpSS1 strain colonization of mouse stomach at both 4 and 8 weeks.
Figure 1: H.pylori priA mutant is impaired in colonizing mice stomach mucosa.
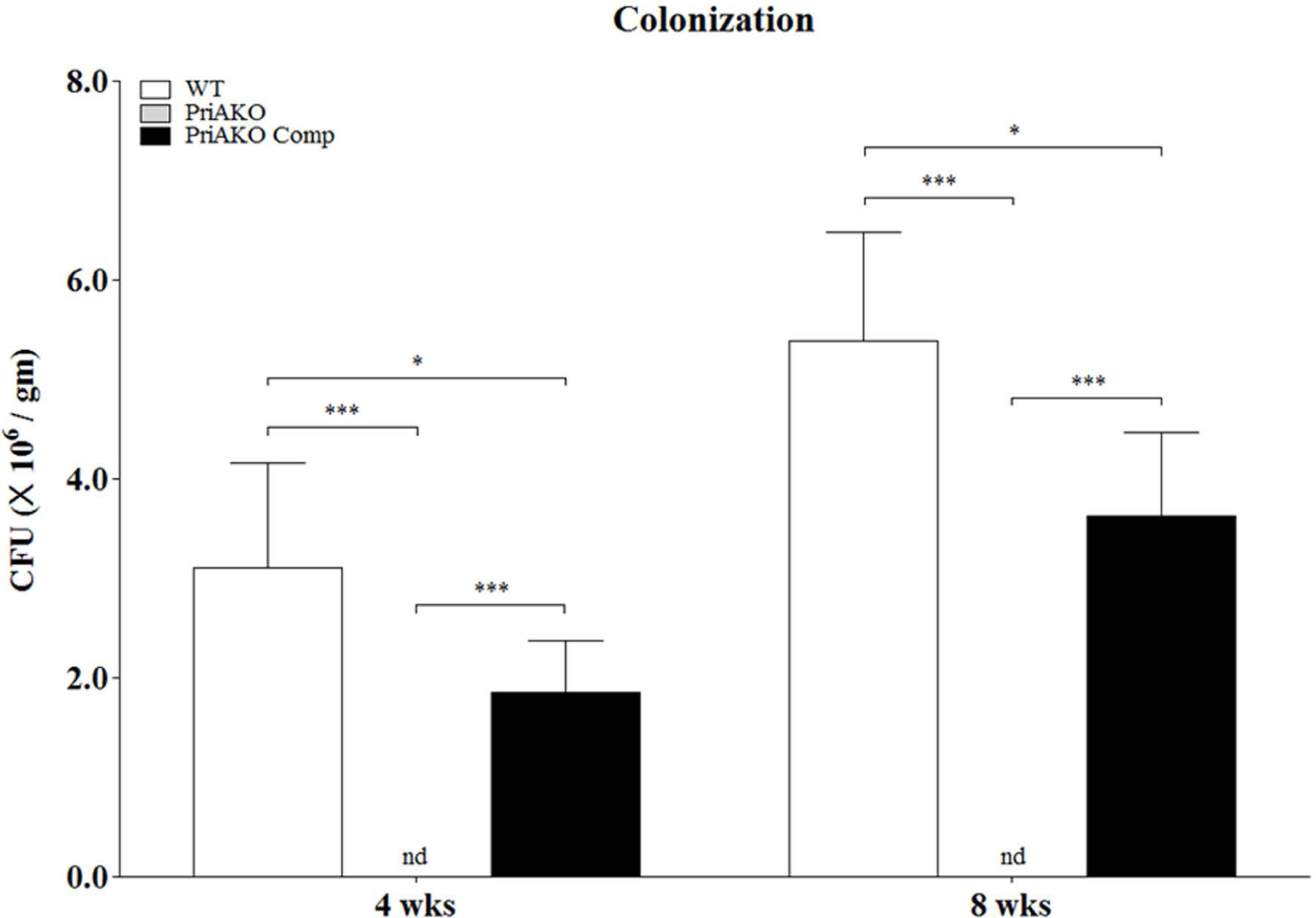
Mice infections were done with WT, priAKO and priAKOComp strains and the stomachs (n=5 for each group) were harvested at 4 weeks and 8 weeks of infection. Colony forming units (CFU) were determined. Error bars represent the standard error for each experimental group. Statistically significant differences determined by the Student’s T-test are indicated by * for a p-value of <0.05 and *** for a p-value of <0.001. nd represents no detection of bacteria.
priA is important for intracellular survival of H. pylori.
WT, priAKO and priAKOComp strains were used to infect GSM06 gastric epithelial cells as described in the materials and methods section. To determine whether the priAKO mutant strain was invasion defective, which could contribute to the observed reduced stomach colonization, we determined the numbers of CFU/ml in infected GSM06 cells at several time points. We did not observe any difference in the CFU/ml after 4 hours of infection for the priAKO strain and the WT strain, which indicates that the priAKO stain was fully capable of invading the gastric epithelial cells. However, the level of survival of the priAKO strain was significantly lower than the levels of survival of the WT and priAKO Comp strains at 24 hours after infection (Figure 2). Similarly, as shown in figure 3, priAKO strain was fully capable of invading RAW 264.7 macrophages after 4 hours of infection. However, the level of survival of the priAKO strain was significantly lower than both WT and priAKOComp strains at 24 hours after infection in RAW 264.7 cells (Figure 3).
Figure 2: priA is important for intracellular survival of H. pylori.
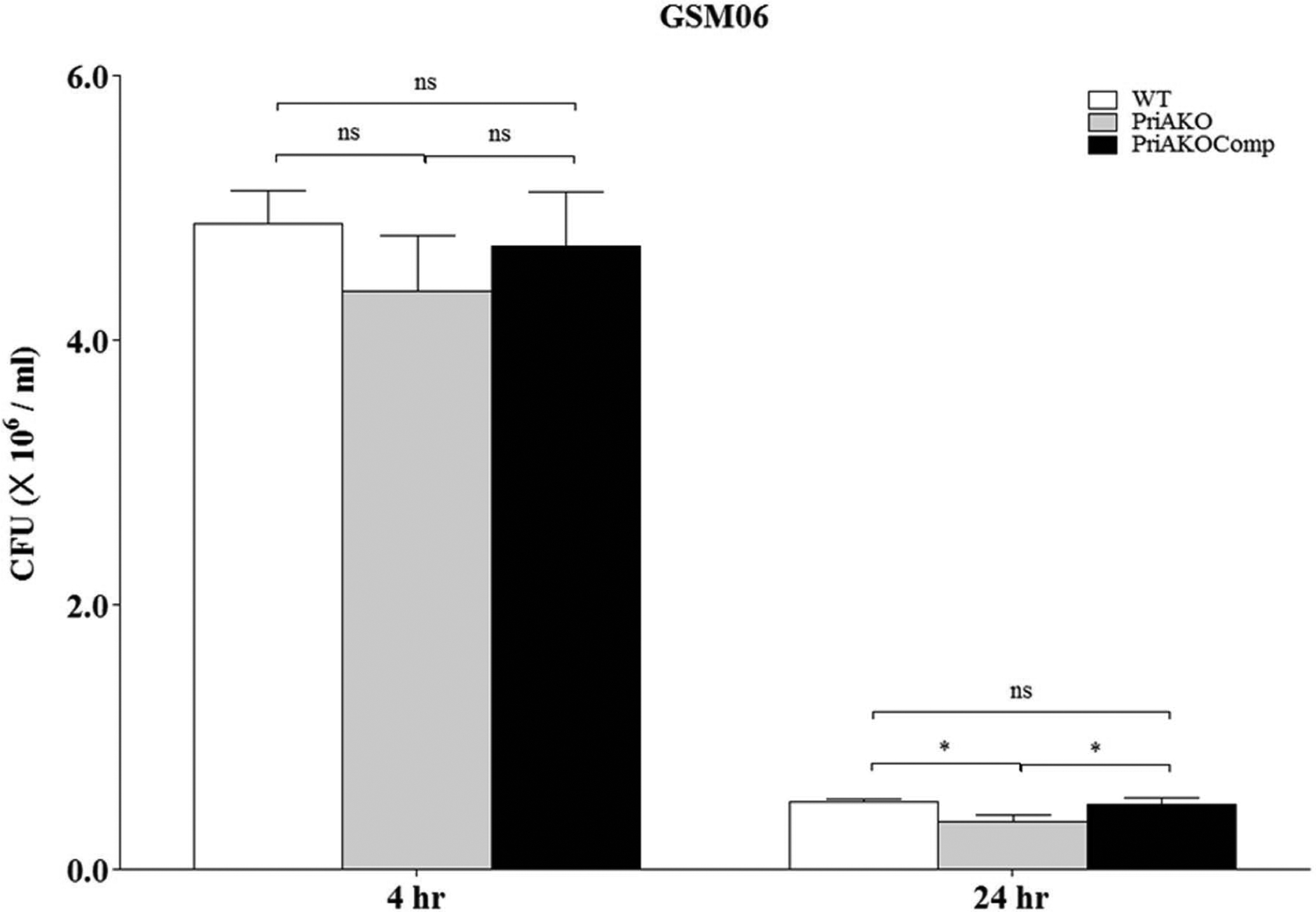
GSMO6 cells (2 × 105/well) were infected with WT, priAKO and priAKOComp strains at an MOI of 100, treated with gentamicin and incubated for the indicated times. After lysis, colony forming units (CFU) from the intracellular bacteria were scored. Error bars represent the standard error of the mean of three experiments performed in duplicate. Statistically significant differences determined by the Student’s T-test are indicated by * for a p-value of <0.05 and ns represents no significant difference.
Figure 3: priA is important for intracellular survival of H. pylori.
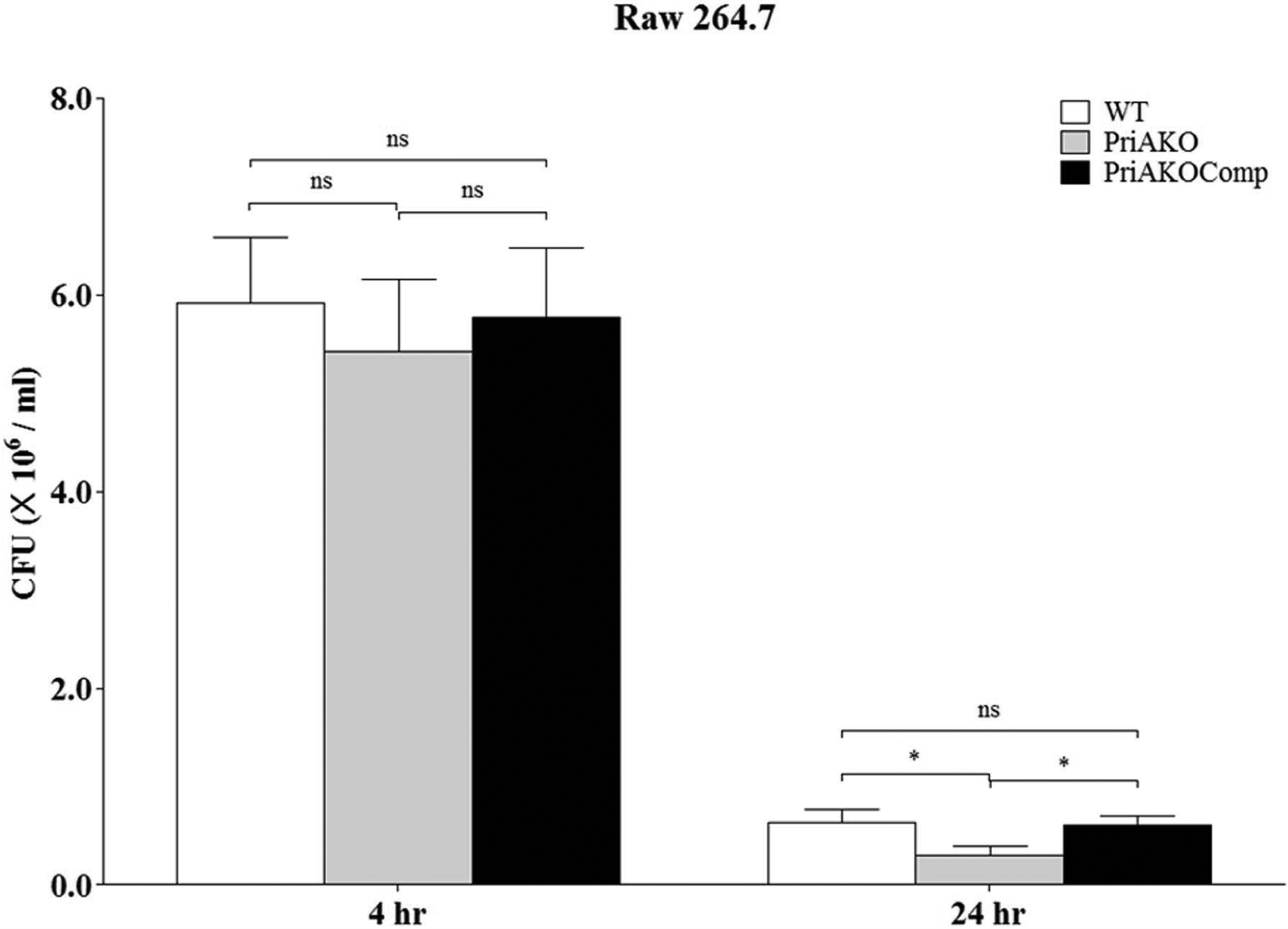
RAW264.7 cells (2 × 105/well) were infected with WT, priAKO and priAKOComp strains at an MOI of 100, treated with gentamicin and incubated for the indicated times. After lysis, colony forming units (CFU) from the intracellular bacteria were scored. Error bars represent the standard error of the mean of three experiments performed in duplicate. Statistically significant differences determined by the Student’s T-test are indicated by * for a p-value of <0.05 and ns represents no significant difference.
priA is involved in DNA repair in H. pylori.
In E. coli and in N. gonorrhoeae, priA mutants display increased sensitivities to UV light (13, 28). To test whether H. pylori priA plays a role in DNA repair, H. pylori strains were exposed to increasing doses of UV light. We observed that the priAKO strain was more sensitive to UV light than the WT strain at all levels of UV exposure (Figure 4A). This repair defect was restored in the priAKOComp strain. We also tested whether inactivation of the priA gene in H. pylori increased the sensitivity of the bacteria to the DNA gyrase inhibitor ciprofloxacin. Ciprofloxacin induces double strand (ds) breaks in the DNA (29, 30). We found that the priA KO strain displayed a reduction in the ability to repair ds breaks as compared to the WT strain (Figure 4B). Survival after ciprofloxacin exposure was restored in priAKOComp strain.
Figure 4: priA is involved in DNA repair in H. pylori.
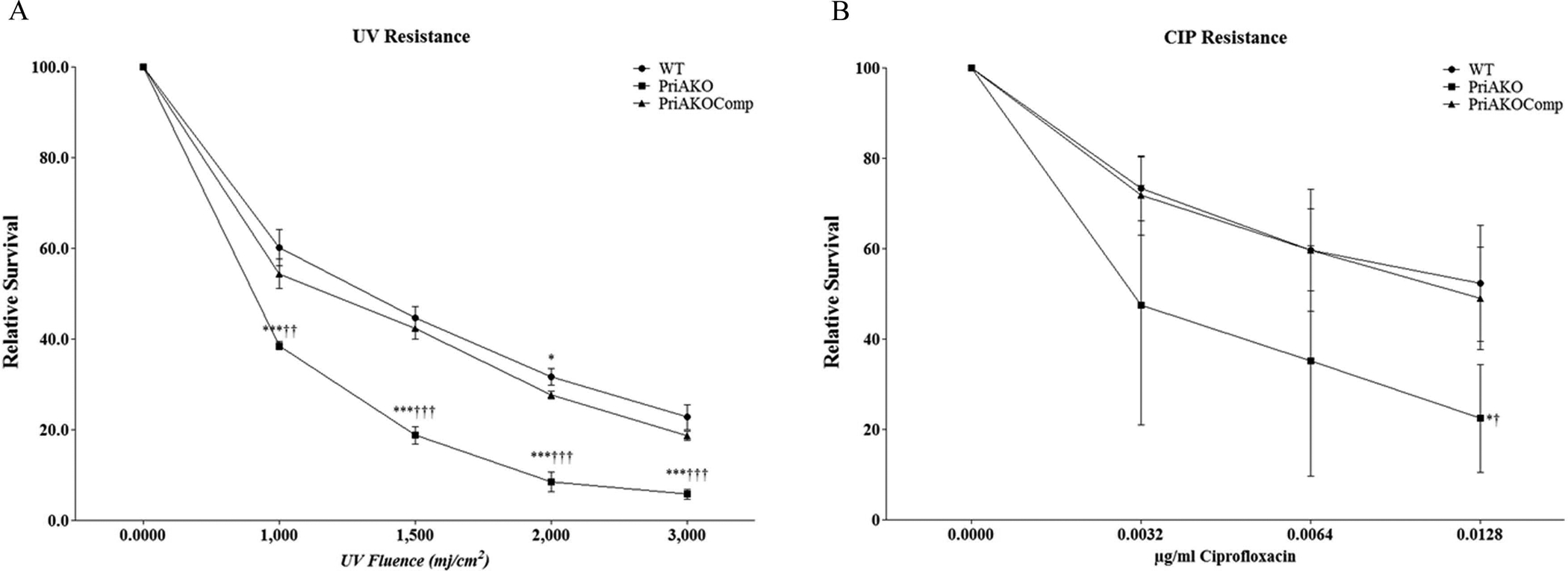
Relative survival of WT, priAKO and priAKOComp strains after irradiation with UV light (A) and double strand breaks induced by ciprofloxacin (B) were determined. Error bars represent the standard error of the mean of three experiments performed in duplicate. * denotes a comparison between WT and priAKO and † denotes a comparison between priAKOComp and priAKO. Statistically significant differences determined by the Student’s T-test are indicated by */† for a p-value of <0.05, **/†† for a p-value of <0.01 and ***/††† for a p-value of <0.0001.
H. pylori priA mutant is more sensitive to acid challenge.
WT, priAKO and priAKOComp strains were incubated in pH 4 acidic medium and samples were taken at 0, 2, 4 and 24 hours and plated on BLBB plates. These results show that the priAKO strain was capable of surviving the acid challenge at 4 and 24 hours of incubation; however it was somewhat more susceptible to acid than the WT strain. (Figure 5). At each time point, the priAKOComp strain was capable of surviving the acid challenge comparable to the WT strain.
Figure 5: H. pylori priA mutant is more susceptible to acid challenge.
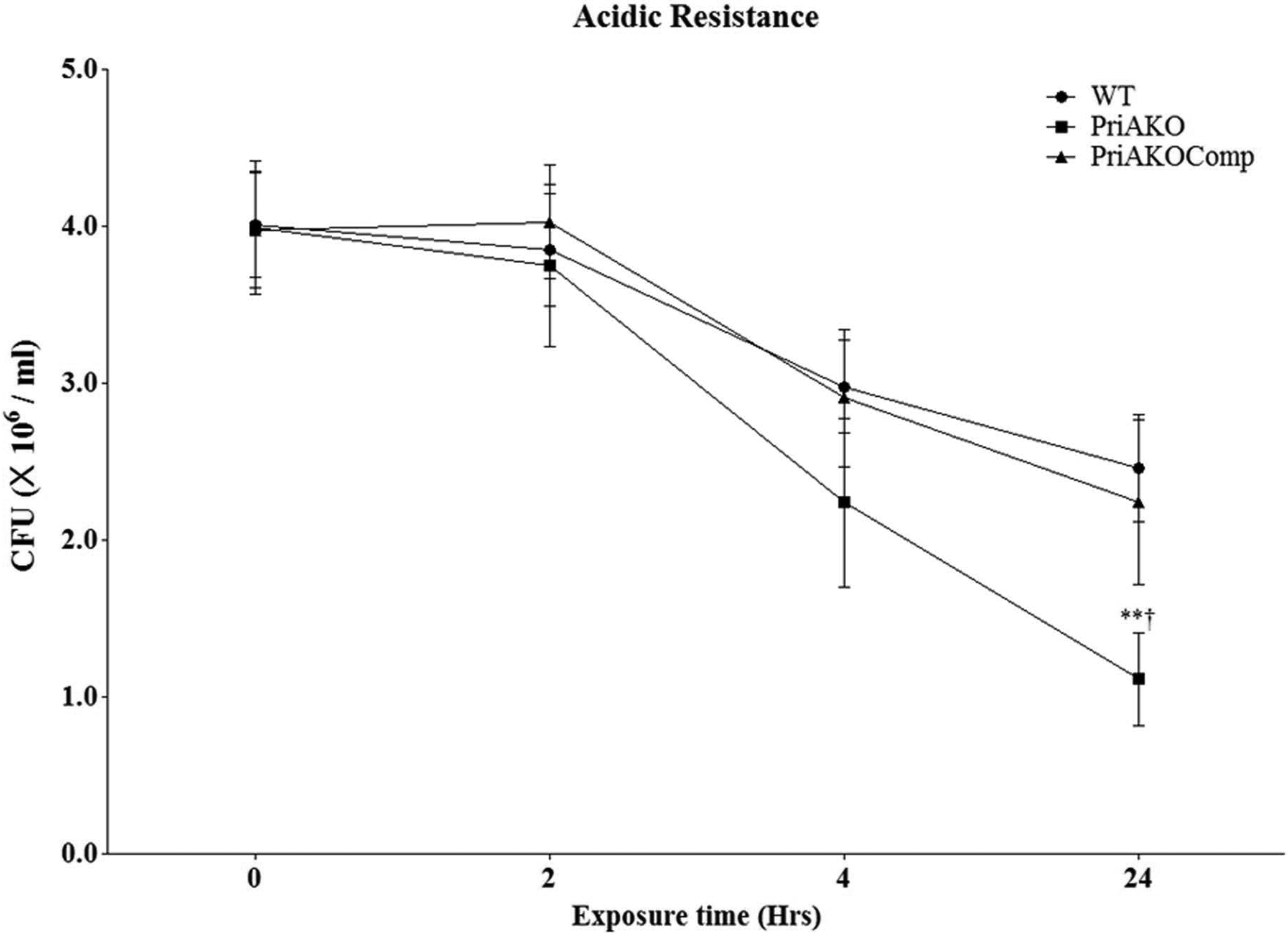
WT, priAKO and priAKOComp strains were grown in pH 4.0 acidic medium. Samples were collected at the indicated time points and the colony forming units (CFU) were determined. Error bars represent the standard error of the mean of three experiments. * denotes a comparison between WT and priAKO and † denotes a comparison between priAKOComp and priAKO. Statistically significant differences determined by the Student’s T-test are indicated by ** for a p-value of <0.001 and † for a p-value of <0.05.
H. pylori priA mutant is more sensitive to oxidative damage.
WT, priAKO and priAKOComp strains were incubated in PBS containing 50mM H2O2. Assessment of viability after exposure to hydrogen peroxide demonstrated that the priAKO strain was much more sensitive to oxidative stress when compared to the WT strain (Figure 6). The phenotype could be complemented back to WT levels in the priAKOComp strain.
Figure 6: H. pylori priA mutant is more sensitive to oxidative damage.
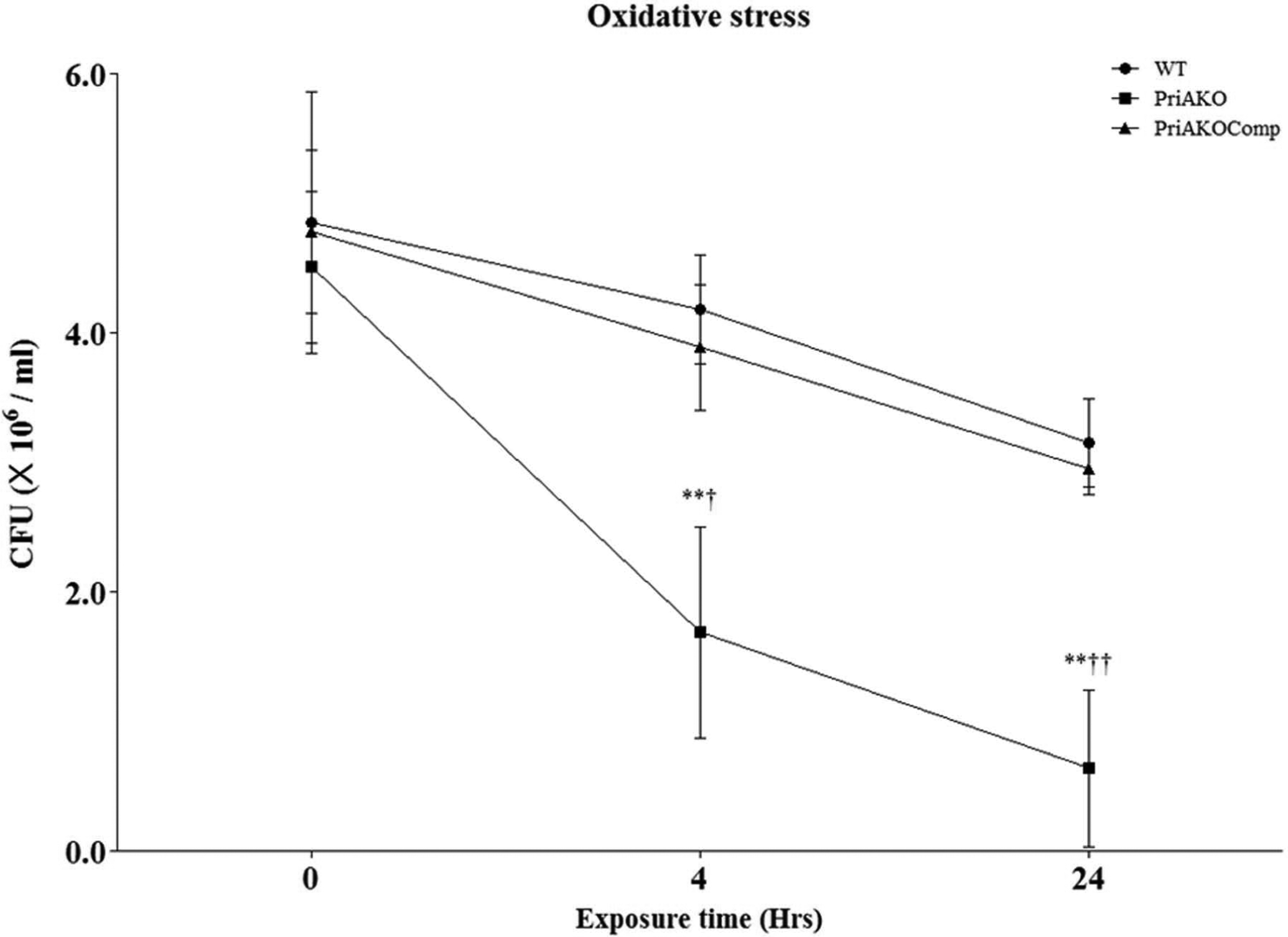
WT, priAKO and priAKOComp strains were exposed to 50 mM H2O2. Samples were collected at the indicated time points and the colony forming units (CFU) were determined. Error bars represent the standard error of the mean of three experiments. * denotes a comparison between WT and priAKO and † denotes a comparison between priAKOComp and priAKO. Statistically significant differences determined by the Student’s T-test are indicated by **/†† for a p-value of <0.001 and † for a p-value of <0.05.
Discussion
During the past 20 years, H. pylori has emerged as an important example of a persistent bacterial pathogen. Not only does this bacterium successfully colonize the hostile environment of the human stomach, but the infection regularly persists for the lifetime of the host in the face of a constant, vigorous innate and adaptive immune response. In most infected people, H. pylori infection causes superficial chronic gastritis, which is usually clinically asymptomatic, although histologically apparent. However, significant subsets of infected individuals are at risk of the subsequent development of duodenal and peptic ulcers, and 1% of those that are infected will develop adenocarcinoma or lymphoma of the stomach (31, 32). Most basic research in the Helicobacter field has focused on the study of bacterial virulence determinants particularly in the context of their association with severe gastrointestinal sequelae of infection. Many different techniques have been developed to study bacterial genes that are expressed during growth in specific niches (33–36). A useful tool for identifying genes involved in virulence is in vivo expression technology (IVET) (37, 38). Using IVET we were able to identify several H. pylori genes that were induced in vivo in infected mice stomachs. Some of these genes are known virulence factors. priA gene was one of the genes identified using IVET (14).
PriA was originally discovered as a factor essential for the conversion of single-stranded circular DNA (SS) to the replicative-form DNA (RF, double-stranded circular DNA) of fX174single- stranded phage in vitro. It binds to a specific hairpin DNA structure on the fX174 genome, hydrolyzes ATP and triggers the assembly of a primosome responsible for the primer RNA synthesis on the single-stranded DNA genome (39). PriA is a DEXH-type DNA helicase containing a unique zinc-finger structure inserted among the helicase conserved motifs. It is highly conserved in eubacteria, suggesting its functional conserved roles in bacterial physiology. priA null mutants are viable, but grow very poorly (28, 40). SOS responses (an inducible DNA repair system that allows bacteria to survive sudden increases in DNA damage) are chronically activated in portions of the cell population, resulting in filamentous morphology (28, 40). Most notably, priA null cells are highly sensitive to genotoxic agents, such as UV (28, 41) and mitomycin C (42), which block ongoing replication. In Neisseria gonorrhoeae, priA was shown to play a role in DNA repair and DNA transformation processes and another study on Neisseria meningitidis, provides evidence for a role of priA in preventing both oxidative and nitrosative injury, and in intracellular meningococcal replication (13, 43).
Since the priA gene was induced in our in vivo screen, and it plays a very critical role in replication fork stabilization and DNA repair in E. coli and N. gonorrhoeae, we decided to elucidate its role in H. pylori. In order to understand the role of priA in H. pylori, the priA gene was disrupted, and assessed for its role in the survival of H. pylori in the gastric mucosa. To determine whether a deficiency in PriA activity is important for H. pylori host colonization, the relative abilities of WT, priAKO and priA gene complemented (priAKOComp) strains to colonize the stomach were evaluated in a mouse infection model. The priAKO strain could colonize mice stomach after one week of infection but at much lower levels than the WT strain (data not shown). Four and eight weeks post infections (p.i.), 5 infected mice from each group were sacrificed and H. pylori survival in the gastric tissue was determined. Four weeks p.i., WT infected animals had good colonization whereas the priA KO infected mice had no colonization. The bacterial load in the WT population increased from 0.31 × 106 CFU/gm of stomach tissue to 0.54 × 106 CFU/gm of stomach tissue at eight weeks p.i. However, no H. pylori was detected in priAKO infected mice during the same period. These data suggest that the PriA protein is needed for survival during mouse stomach colonization. To rule out the possibility that cells lacking PriA protein had an intrinsic impaired growth capability, the in vitro growth rate of priAKO strain was measured for 1 week. priAKO strain showed some reduction in the growth rate but it wasn’t significant when compared to the WT strain (data not shown).
H. pylori is able to invade epithelial cells, survive within large, late endosomal vacuoles, modify the molecular make-up of these vacuoles and later egress from this intracellular niche (44). It has been observed that viable H. pylori bacteria in large cytoplasmic vacuoles can repopulate the extracellular environment in parallel with the disappearance of intravacuolar bacteria (44). This intravacuolar niche is a place for the release of the bacteria and the place for evasion of the hostile microenvironment of the host. Another study reported that the plasma membranes are the site for H. pylori to replicate (45). A recent study found that H. pylori can multiply not only in macrophages and bone marrow-derived dendritic cells (46, 47) but also in epithelial cells (48). This finding has several implications for the biological life cycle of H. pylori in the host. H. pylori can be considered as an intracellular microorganism because it can invade cells, undergo replication within cells, and leave the infected cells for further spread. H. pylori invasion and intracellular survival assays of WT, priAKO and priAKOComp strains were performed with the murine gastric epithelial cell line GSM06 (21). The mouse gastric surface cell line GSM06 has been established from a primary culture of gastric mucosal cells of transgenic mice harboring a temperature-sensitive simian virus 40 (tsSV40) large T-antigen gene (22). GSM06 cells have been shown to retain many of the characteristics of normal gastric surface mucus cells (21). It’s an in vitro model that closely resembles in vivo conditions leading to infection by H. pylori. In our assays we observed that the invasion capability of the priAKO strain was very similar to the WT strain after 4 hours of infection. However, intracellular survival of the priAKO strain was significantly reduced after 24 hours of infection. In these assays gentamicin is used after 2 hours to kill extracellular bacteria. Consequently, the CFU of surviving bacteria represent intracellular bacteria that invaded the GSM06 cells. Our data demonstrated that the ability of the priAKO strain to invade epithelial cells appears to be normal. However, the ability of the priAKO strain to survive in the epithelial cells is defective. Consistent with these results, microscopic imaging of live H. pylori in cells showed that while the numbers of intracellular bacteria were comparable for the WT, priAKO and priAKOComp strains at 4 hours after infection, the numbers of priAKO bacteria were reduced after 24 hours compared to the numbers of WT and priAKOComp strains (data not shown). These data suggest a role of PriA protein in the intracellular survival of epithelial cell ingested H. pylori.
H. pylori has developed multiple mechanisms to evade elimination by host innate immune responses, including macrophages, which may account for the persistence of this bacterium in the host (49, 50). Once inside macrophages, H. pylori interferes with phagosome maturation, which leads to the formation of large phagosomes called megasomes (51–53). H. pylori resides in these megasomes during phagocytosis (51–53). The megasomes apparently do not fuse with the lysosomes in macrophages, thus protecting the bacteria from elimination. The number of priAKO strain colonies recovered from H. pylori infected RAW264.7 macrophages was essentially the same as the numbers of WT and priAKOComp strains colonies recovered 4 hours after infection. However, the level of survival of the priAKO strain in RAW264.7 cells was significantly lower than the survival of the WT and priAKOComp strains 24 hours after infection. Fluorescence microscopy experiments yielded similar findings which indicate that the priAKO strain has impaired survival 24 hours after infection in macrophages (data not shown). Collectively, these data indicate a role for PriA protein in the intracellular survival of H. pylori in both epithelial cells and macrophages. Additionally, our results suggest that the PriA protein plays an important role in the mechanisms used by H. pylori for bacterial evasion of elimination in macrophages.
As observed in E. coli and N. gonorrhoeae, the priAKO strain displayed increased sensitivity to UV irradiation. The sensitivity of the priAKO strain is not as severe as that of a H. pylori recA mutant strain (54); however, it is very similar to that seen in E. coli and N. gonorrhoeae priA mutants (13, 28, 41). It is likely that efficient UV repair systems can repair UV-mediated DNA lesions, and that only a subset of lesions that interfere with replication fork progression require PriA activity after fork collapse. Our investigations also suggest a role for H. pylori priA gene in protection against the DNA gyrase inhibitor ciprofloxacin (29, 30). Ciprofloxacin induces ds breaks in the DNA, a target for AddAB-mediated repair. H. pylori AddAB nuclease and helicase plays a very important role in DNA repair (55). As compared to the WT strain, the priAKO strain displayed a reduction in the ability to repair ds breaks as compared to the WT strain. These data support the importance of PriA protein in maintaining H. pylori viability by facilitating DNA repair following host-inflicted DNA damage.
The ability of H. pylori to survive in an acidic environment is critical to its ability to chronically infect the human stomach. Acid stress is likely encountered by H. pylori during transmission between hosts and transit through the gastric lumen before the bacteria can situate themselves in the neutral pH environment of the gastric mucosa (25). To check acid survival ability in vitro, WT, priAKO and priAKOComp strains were grown in pH 4.0 growth medium supplemented with 0.5 mM urea for 2, 4 and 24 hours. During the first 2 hours of acidic exposure all strains were relatively unaffected by the treatment. After 4 hours, however, the priAKO strain exhibited a decrease in viability relative to the WT strain. At 24 hours of incubation, the priAKO strain was significantly more susceptible to acid relative to the WT and priAKOComp strains, which indicates that the PriA protein is important for survival of H. pylori in an acidic environment.
In its physiological environment, H. pylori frequently suffers both acid and oxidative stress, leading to DNA damage. H. pylori has developed a battery of antioxidant proteins such as superoxide dismutase, catalase, and peroxiredoxins that enable it to avoid oxidative destruction (56). Urease hydrolyzes urea to yield ammonium ions and thereby contributes to the acid resistance of H. pylori (4). In addition to diverse oxidant detoxification enzymes and potent acid avoidance mechanisms, efficient DNA repair systems are required for H. pylori to survive in the host (56, 57). To repair DNA double strand breaks and blocked replication forks, H. pylori is equipped with an efficient system of DNA recombinational repair. To determine whether priA is involved in protection against oxidative stress, WT, priAKO and priAKOComp strains were exposed to hydrogen peroxide in broth culture and viable counts were taken at 4 and 24 hours. The data shows that the WT strain survived significantly better than the priAKO strain under these conditions. The decreased survival of priA mutant to oxidative damaging agents indicates that replication restart serves as an additional defense for countering the effects of toxic oxygen species. These findings provide an evidence for a role of priA and replication restart in resistance to oxidative damage.
In this study, we provide evidence that priA, whose expression increases in mice stomach mucosa plays a key role in the colonization of mice stomach mucosa, is important for intracellular survival of epithelial cell ingested and macrophage cell ingested H. pylori and protects H. pylori from DNA damage, acid stress and oxidative stress. These results show that H. pylori priA provides a bacterial defense against the host response.
Acknowledgements
This research was supported by the intramural research program of the National Institute of Diabetes and Digestive and Kidney Diseases, and the intramural research program of the National Institute on Minority Health and Health Disparities. We are thankful to Dr. Reed B. Wickner, NIDDK, NIH, for critical reading of the manuscript.
References
- 1.Wroblewski LE, Peek RM Jr., Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. 2010October;23(4):713–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eaton KA, Krakowka S. Effect of gastric pH on urease-dependent colonization of gnotobiotic piglets by Helicobacter pylori. Infect Immun. 1994September;62(9):3604–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eaton KA, Suerbaum S, Josenhans C, Krakowka S. Colonization of gnotobiotic piglets by Helicobacter pylori deficient in two flagellin genes. Infect Immun. 1996July;64(7):2445–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marshall BJ, Barrett LJ, Prakash C, McCallum RW, Guerrant RL. Urea protects Helicobacter (Campylobacter) pylori from the bactericidal effect of acid. Gastroenterology. 1990September;99(3):697–702. [DOI] [PubMed] [Google Scholar]
- 5.Kavermann H, Burns BP, Angermuller K, Odenbreit S, Fischer W, Melchers K, et al. Identification and characterization of Helicobacter pylori genes essential for gastric colonization. J Exp Med. 2003April7;197(7):813–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rediers H, Rainey PB, Vanderleyden J, De Mot R. Unraveling the secret lives of bacteria: Use of in vivo expression technology and differential fluorescence induction promoter traps as tools for exploring niche-specific gene expression. Microbiology and Molecular Biology Reviews. 2005June;69(2):217–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Angelichio MJ, Camilli A. In vivo expression technology. Infect Immun. 2002December;70(12):6518–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schekman R, Weiner JH, Weiner A, Kornberg A. Ten proteins required for conversion of phiX174 single-stranded DNA to duplex form in vitro. Resolution and reconstitution. The Journal of biological chemistry. 1975August10;250(15):5859–65. [PubMed] [Google Scholar]
- 9.Wickner S, Hurwitz J. Conversion of phiX174 viral DNA to double-stranded form by purified Escherichia coli proteins. Proceedings of the National Academy of Sciences of the United States of America. 1974October;71(10):4120–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marians KJ. PriA: at the crossroads of DNA replication and recombination. Prog Nucleic Acid Res Mol Biol. 1999;63:39–67. [DOI] [PubMed] [Google Scholar]
- 11.Lovett ST. Connecting replication and recombination. Mol Cell. 2003March;11(3):554–6. [DOI] [PubMed] [Google Scholar]
- 12.Lovett ST. Filling the gaps in replication restart pathways. Mol Cell. 2005March18;17(6):751–2. [DOI] [PubMed] [Google Scholar]
- 13.Kline KA, Seifert HS. Mutation of the priA gene of Neisseria gonorrhoeae affects DNA transformation and DNA repair. Journal of bacteriology. 2005August;187(15):5347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh A, Hodgson N, Yan M, Joo J, Gu L, Sang H, et al. Screening Helicobacter pylori genes induced during infection of mouse stomachs. World journal of gastroenterology : WJG. 2012August28;18(32):4323–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee A, O’Rourke J, De Ungria MC, Robertson B, Daskalopoulos G, Dixon MF. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology. 1997April;112(4):1386–97. [DOI] [PubMed] [Google Scholar]
- 16.Hawrylik SJ, Wasilko DJ, Haskell SL, Gootz TD, Lee SE. Bisulfite or sulfite inhibits growth of Helicobacter pylori. J Clin Microbiol. 1994March;32(3):790–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McColm AA. Nonprimate Animal Models of H. pylori Infection. Methods Mol Med. 1997;8:235–51. [DOI] [PubMed] [Google Scholar]
- 18.Luria SE, Burrous JW. Hybridization between Escherichia coli and Shigella. Journal of bacteriology. 1957October;74(4):461–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heuermann D, Haas R. A stable shuttle vector system for efficient genetic complementation of Helicobacter pylori strains by transformation and conjugation. Mol Gen Genet. 1998March;257(5):519–28. [DOI] [PubMed] [Google Scholar]
- 20.Institute of Laboratory Animal Resources (U.S.). Committee on Care and Use of Laboratory Animals. Guide for the care and use of laboratory animals. In: NIH publication (vol. Bethesda, Md.: U.S. Dept. of Health and Human Services, Public Health Service; p. v. [Google Scholar]
- 21.Tabuchi Y, Sugiyama N, Horiuchi T, Furuhama K, Furusawa M. Biological characterization of gastric surface mucous cell line GSM06 from transgenic mice harboring temperature-sensitive simian virus 40 large T-antigen gene. Digestion. 1996;57(2):141–8. [DOI] [PubMed] [Google Scholar]
- 22.Sugiyama N, Tabuchi Y, Horiuchi T, Obinata M, Furusawa M. Establishment of gastric surface mucous cell lines from transgenic mice harboring temperature-sensitive simian virus 40 large T-antigen gene. Exp Cell Res. 1993December;209(2):382–7. [DOI] [PubMed] [Google Scholar]
- 23.Elsinghorst EA. Measurement of invasion by gentamicin resistance. Methods in enzymology. 1994;236:405–20. [DOI] [PubMed] [Google Scholar]
- 24.Zhou YN, Coleman WG Jr., Yang Z, Yang Y, Hodgson N, Chen F, et al. Regulation of cell growth during serum starvation and bacterial survival in macrophages by the bifunctional enzyme SpoT in Helicobacter pylori. Journal of bacteriology. 2008December;190(24):8025–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sachs G, Weeks DL, Melchers K, Scott DR. The gastric biology of Helicobacter pylori. Annual review of physiology. 2003;65:349–69. [DOI] [PubMed] [Google Scholar]
- 26.Mouery K, Rader BA, Gaynor EC, Guillemin K. The stringent response is required for Helicobacter pylori survival of stationary phase, exposure to acid, and aerobic shock. Journal of bacteriology. 2006August;188(15):5494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Rourke EJ, Chevalier C, Pinto AV, Thiberge JM, Ielpi L, Labigne A, et al. Pathogen DNA as target for host-generated oxidative stress: role for repair of bacterial DNA damage in Helicobacter pylori colonization. Proceedings of the National Academy of Sciences of the United States of America. 2003March4;100(5):2789–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee EH, Kornberg A. Replication deficiencies in priA mutants of Escherichia coli lacking the primosomal replication n’ protein. Proceedings of the National Academy of Sciences of the United States of America. 1991April15;88(8):3029–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolfson JS, Hooper DC. The fluoroquinolones: structures, mechanisms of action and resistance, and spectra of activity in vitro. Antimicrobial agents and chemotherapy. 1985October;28(4):581–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sioud M, Forterre P. Ciprofloxacin and etoposide (VP16) produce a similar pattern of DNA cleavage in a plasmid of an archaebacterium. Biochemistry. 1989May2;28(9):3638–41. [DOI] [PubMed] [Google Scholar]
- 31.Peek RM Jr., Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002January;2(1):28–37. [DOI] [PubMed] [Google Scholar]
- 32.Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001September13;345(11):784–9. [DOI] [PubMed] [Google Scholar]
- 33.Falkow S Perspectives series: host/pathogen interactions. Invasion and intracellular sorting of bacteria: searching for bacterial genes expressed during host/pathogen interactions. J Clin Invest. 1997July15;100(2):239–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee SH, Butler SM, Camilli A. Selection for in vivo regulators of bacterial virulence. Proceedings of the National Academy of Sciences of the United States of America. 2001June5;98(12):6889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valdivia RH, Falkow S. Fluorescence-based isolation of bacterial genes expressed within host cells. Science. 1997September26;277(5334):2007–11. [DOI] [PubMed] [Google Scholar]
- 36.Scott DR, Marcus EA, Wen Y, Oh J, Sachs G. Gene expression in vivo shows that Helicobacter pylori colonizes an acidic niche on the gastric surface. Proceedings of the National Academy of Sciences of the United States of America. 2007April24;104(17):7235–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mahan MJ, Slauch JM, Mekalanos JJ. Selection of bacterial virulence genes that are specifically induced in host tissues. Science. 1993January29;259(5095):686–8. [DOI] [PubMed] [Google Scholar]
- 38.Rainey PB, Preston GM. In vivo expression technology strategies: valuable tools for biotechnology. Curr Opin Biotechnol. 2000October;11(5):440–4. [DOI] [PubMed] [Google Scholar]
- 39.Shlomai J, Polder L, Arai K, Kornberg A. Replication of phi X174 dna with purified enzymes. I. Conversion of viral DNA to a supercoiled, biologically active duplex. The Journal of biological chemistry. 1981May25;256(10):5233–8. [PubMed] [Google Scholar]
- 40.Nurse P, Zavitz KH, Marians KJ. Inactivation of the Escherichia coli priA DNA replication protein induces the SOS response. Journal of bacteriology. 1991November;173(21):6686–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sandler SJ, Samra HS, Clark AJ. Differential suppression of priA2::kan phenotypes in Escherichia coli K-12 by mutations in priA, lexA, and dnaC. Genetics. 1996May;143(1):5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kogoma T, Cadwell GW, Barnard KG, Asai T. The DNA replication priming protein, PriA, is required for homologous recombination and double-strand break repair. Journal of bacteriology. 1996March;178(5):1258–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tala A, De Stefano M, Bucci C, Alifano P. Reverse transcriptase-PCR differential display analysis of meningococcal transcripts during infection of human cells: up-regulation of priA and its role in intracellular replication. BMC Microbiol. 2008;8:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amieva MR, Salama NR, Tompkins LS, Falkow S. Helicobacter pylori enter and survive within multivesicular vacuoles of epithelial cells. Cell Microbiol. 2002October;4(10):677–90. [DOI] [PubMed] [Google Scholar]
- 45.Tan S, Tompkins LS, Amieva MR. Helicobacter pylori usurps cell polarity to turn the cell surface into a replicative niche. PLoS Pathog. 2009May;5(5):e1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang YH, Wu JJ, Lei HY. The autophagic induction in Helicobacter pylori-infected macrophage. Exp Biol Med (Maywood). 2009February;234(2):171–80. [DOI] [PubMed] [Google Scholar]
- 47.Wang YH, Gorvel JP, Chu YT, Wu JJ, Lei HY. Helicobacter pylori impairs murine dendritic cell responses to infection. PLoS One. 2010;5(5):e10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chu YT, Wang YH, Wu JJ, Lei HY. Invasion and multiplication of Helicobacter pylori in gastric epithelial cells and implications for antibiotic resistance. Infect Immun. 2010October;78(10):4157–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Allen LA. Phagocytosis and persistence of Helicobacter pylori. Cell Microbiol. 2007April;9(4):817–28. [DOI] [PubMed] [Google Scholar]
- 50.Ramarao N, Gray-Owen SD, Backert S, Meyer TF. Helicobacter pylori inhibits phagocytosis by professional phagocytes involving type IV secretion components. Molecular microbiology. 2000September;37(6):1389–404. [DOI] [PubMed] [Google Scholar]
- 51.Allen LA, Allgood JA. Atypical protein kinase C-zeta is essential for delayed phagocytosis of Helicobacter pylori. Curr Biol. 2002October15;12(20):1762–6. [DOI] [PubMed] [Google Scholar]
- 52.Allen LA, Allgood JA, Han X, Wittine LM. Phosphoinositide3-kinase regulates actin polymerization during delayed phagocytosis of Helicobacter pylori. J Leukoc Biol. 2005July;78(1):220–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Allen LA, Schlesinger LS, Kang B. Virulent strains of Helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. J Exp Med. 2000January3;191(1):115–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Amundsen SK, Fero J, Hansen LM, Cromie GA, Solnick JV, Smith GR, et al. Helicobacter pylori AddAB helicase-nuclease and RecA promote recombination-related DNA repair and survival during stomach colonization. Molecular microbiology. 2008August;69(4):994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Amundsen SK, Fero J, Salama NR, Smith GR. Dual nuclease and helicase activities of Helicobacter pylori AddAB are required for DNA repair, recombination, and mouse infectivity. The Journal of biological chemistry. 2009June19;284(25):16759–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang G, Alamuri P, Maier RJ. The diverse antioxidant systems of Helicobacter pylori. Molecular microbiology. 2006August;61(4):847–60. [DOI] [PubMed] [Google Scholar]
- 57.Borlace GN, Keep SJ, Prodoehl MJ, Jones HF, Butler RN, Brooks DA. A role for altered phagosome maturation in the long-term persistence of Helicobacter pylori infection. Am J Physiol Gastrointest Liver Physiol. 2012July15;303(2):G169–79. [DOI] [PubMed] [Google Scholar]