

Abstract
SUMOylation has emerged as an important post-translational modification that involves the covalent attachment of the small ubiquitin-like modifier (SUMO) polypeptide to a lysine residue of a target protein. The enzymatic pathway of SUMOylation is very similar to ubiquitinylation and involves an activating enzyme, a conjugating enzyme, ligases and deconjugating enzymes. SUMOylation modulates the function of a number of proteins associated with various pathways, and in fact, dysregulation of the SUMOylation pathway is observed in both cancer and neurological diseases. In many cancers, the SUMO enzymes are upregulated and SUMO levels correlate directly with prognosis and disease progression. As a result, there has been an emphasis on the discovery and development of inhibitors of SUMOylation. In this review, the latest advances in SUMOylation inhibitors are described alongside the methods used to discover small molecule SUMOylation inhibitors, which include natural products, peptidomimetics, as well as synthetic derivatives identified via virtual screens.
Introduction
Post-translational modifications (PTMs) are a critical part of the cellular regulatory process, with over 450 distinct modifications having been identified.1 One important class of PTMs is the ubiquitin-like proteins (UBLs), which share structural similarities to ubiquitin. The small ubiquitin-like modifier (SUMO) was one of the first members of this protein family discovered after ubiquitin.2 Despite belonging to the same protein family, ubiquitin and SUMO have different cellular functions. While ubiquitinylation primarily targets substrate proteins to the proteasome for degradation, SUMOylation has been shown to modulate several cellular processes including cellular localization, transcription regulation, protein-protein interactions, and DNA damage repair.3–5 SUMOylation is a dynamic and reversible process that is controlled by an enzymatic cascade very similar to ubiquitinylation. It requires an ATP-dependent heterodimeric SUMO-activating enzyme dubbed E1, a SUMO conjugating enzyme, E2, and E3 ligases that assist in the recognition of substrates and increase the efficiency of isopeptide formation.6 Finally, SUMO/sentrin specific peptidases (SENPs) hydrolyze SUMO from the substrate, reversing SUMOylation.7
Like many other pathways, the SUMOylation pathway is often dysregulated in cancer. Generally, the SUMO conjugating enzymes are upregulated in cancer, which leads to higher levels of SUMOylated proteins, and often correlates with poor prognosis.8–11 In several cancers, isoforms of the deSUMOylating SENPs are also upregulated, which suggests that SUMOylation may have a more influential role in cancer progression and that SUMOylation may be accelerated in certain cancers as a consequence of increased protein turnover.12–14 Given the dysregulation of this pathway in a variety of cancer types as well as the fact that SUMOylation has been demonstrated to modulate the activity of pathways associated with the hallmarks of cancer, effort has been devoted toward the identification of small molecules that modulate the SUMOylation pathway.8 This review serves as an overview of those efforts, and highlights current progress towards the development of SUMOylation inhibitors.
Overview of the SUMOylation Enzymatic Pathway
As previously stated, the SUMOylation pathway is analogous to the ubiquitinylation pathway, with the primary difference being the number of isozymes identified for each step. 40 ubiquitin E2 enzymes have been identified in humans, however there is only one human SUMO E2 enzyme, Ubc9.3,15 Similarly, 79 functional deubiquitinating enzymes have been characterized as well as more than 600 ubiquitin E3 ligases, compared to six SENPs and ten SUMO E3 ligases.4,16 The number of enzymes required to regulate these pathways is related to the number of proteins modified by each post-translational process, as more than 10,000 substrates have been shown to be modified by ubiquitin, whereas 3,000–6,000 proteins have been identified as substrates for SUMOylation.17 Another key difference is that multiple isoforms of SUMO have been discovered as compared to the single ubiquitin polypeptide. Three isoforms of SUMO (SUMO1–3) are expressed in humans, and there are key differences among them. SUMO2 and SUMO3 share 97% sequence homology and are often denoted as SUMO2/3 as many detection methods cannot distinguish between them.3 Furthermore, it has been demonstrated that proteins that are SUMOylated with SUMO2/3 are generally associated with cellular stress.18 Whereas, SUMO1 is only 50% similar to SUMO2/3 and primarily modulates the physiological state of proteins.19 While all isoforms use the same enzymatic machinery, the underlying mechanisms that dictate the specificity between the isoforms remain unclear.
The first step in the SUMOylation pathway is maturation of the SUMO protein. Two isoforms of SENPs, SENP1 and SENP2, are responsible for cleaving the tetrapeptide C-terminal cap to reveal a di-glycine motif. The di-glycine motif eventually forms the isopeptide bond with the ε-amino group of a lysine residue on the protein substrate.7 After SUMO maturation has occurred, the E1 enzyme will activate the protein for conjugation (Figure 1). The SUMO activating enzyme (SAE) is a heterodimeric protein complex that consists of Aos1/SAE1 and Uba2/SAE2. Activation of mature SUMO protein is ATP-dependent and involves two discrete reactions. The first step is adenylation of the C-terminus of SUMO, which consumes ATP and releases pyrophosphate as a byproduct.17 The catalytic cysteine in the E1 enzyme attacks the acylated-adenylate to form a thioester intermediate.20 After the E1-SUMO complex is formed, SUMO is then transferred to a cysteine residue on the SUMO-conjugating enzyme, SUMO E2, through a transthioesterification reaction.21 SUMO E2 catalyzes formation of the isopeptide bond between the side chain amine of lysine and the C-terminal glycine residue of SUMO, often with the assistance of an E3 ligase, which can increase the rate of SUMOylation. E3 ligases are also involved in colocalization of the E2-SUMO complex and the target protein. However, it has been demonstrated that SUMOylation can occur without the use of an E3 ligase, which is the case for RanGAP1, one of the first SUMO substrates identified.22,23
Figure 1.
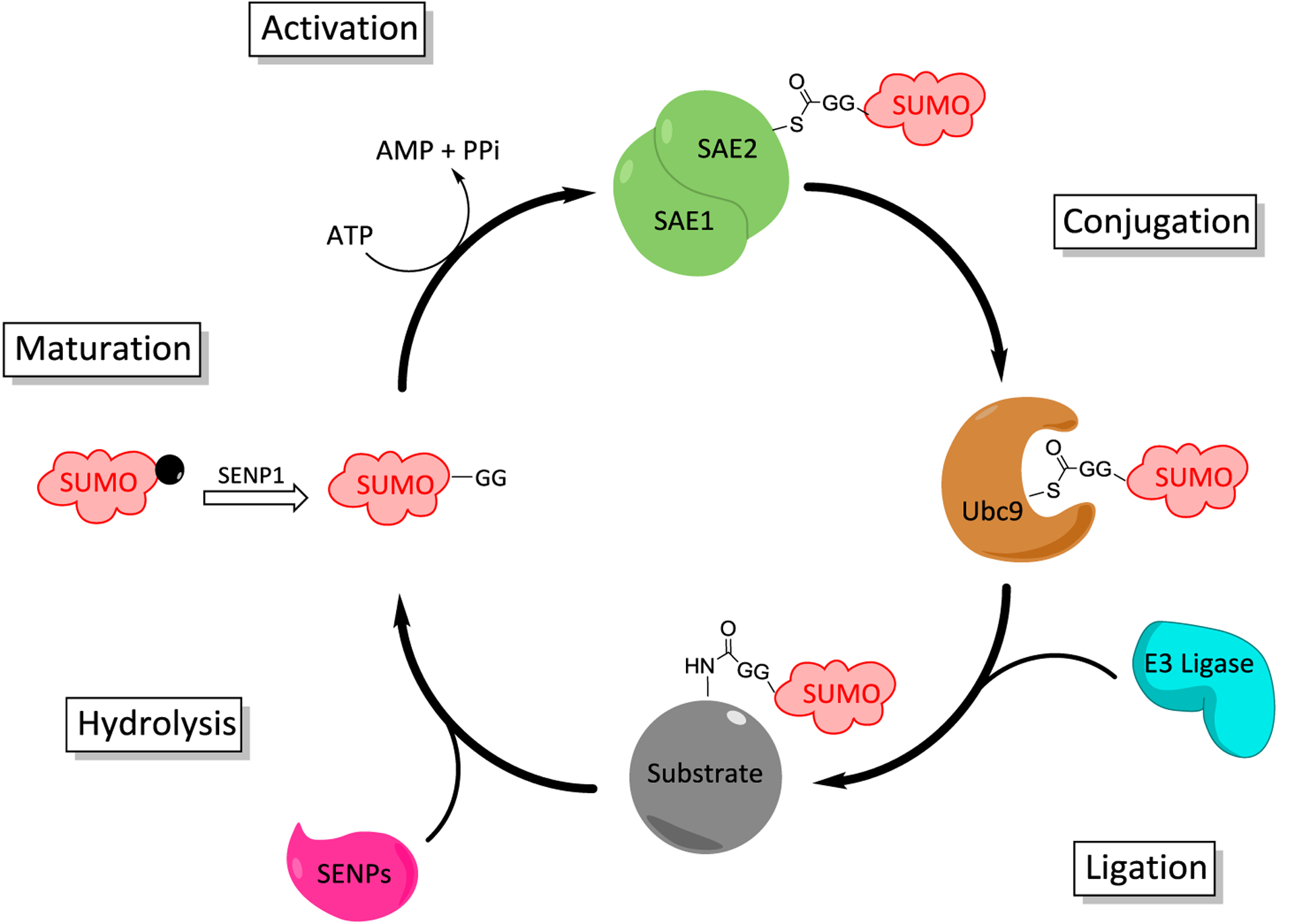
SUMOylation catalytic cycle (Maturation). Immature SUMO has its C-terminal cap cleaved to reveal a diglycine motif by SENP1. (Activation) SUMO is then activated in an ATP-dependent reaction by heterodimeric SAE by forming a thioester between the active site cysteine and the C-terminus of SUMO. (Conjugation) SUMO is then transferred to the active site cysteine residue of the SUMO-conjugating protein, Ubc9. (Ligation) The substrate protein is then SUMOylated on the side chain amine of a lysine residue usually involving an E3 ligase. (De-modification) SUMOylation can be reversed through the action of SENPs.
SUMO-E1 Inhibitors
SUMO activating enzyme (SAE) is responsible for the first step in the SUMOylation enzymatic cascade and is overexpressed in many types of cancer.8 In the case of small cell lung cancer, knockdown of one subunit of SAE was able to decrease the migration and invasiveness of tumors as well as to sensitize them to chemotherapeutics.24 Additionally, upregulation of SAE2 has been implicated in the evolution of multidrug resistance in hepatocellular carcinoma.25 MYC is a regulator of several proto-oncogenes and has been implicated in the progression of many types of cancer. The SUMOylation pathway is critical for the survival of cancer cells that display high levels of MYC activity, as SAE knockdown confers synthetic lethality to such cells.26
Several natural products have been identified as SUMO-E1 inhibitors. The first identified inhibitors of SUMOylation was ginkgolic acid (1), and its analog, anacardic acid (2). These molecules inhibit SAE by blocking formation of the E1-SUMO complex.27 An in vitro assay assessing RanGAP1 SUMOylation measured an IC50 value of 3.0 μM and 2.2 μM for ginkgolic acid and anacardic acid, respectively. In vivo experiments revealed that ginkgolic acid had no effect on protein ubiquitinylation, revealing it to selectively inhibit SUMOylation. A fluorescently tagged ginkgolic acid analog was subsequently prepared, which was used to confirm that SUMO-E1 is the target of ginkgolic acid. Despite its widespread adoption as a SUMOylation inhibitor, ginkgolic acid/anacardic acid also display anti-fungal, anti-bacterial, as well as inhibitory activity toward many different enzymes, confounding the results and the impact of SUMOylation inhibition.28–30 Shortly after they published their study that identified ginkgolic acid as the first SUMOylation inhibitor, the Yoshida group demonstrated kerriamycin B (3) to block formation of the E1-SUMO complex with an in vitro IC50 of 11.7 μM.31 Kerriamycin B was first isolated from Streptomyces violaceolatus and displayed anti-bacterial activity against Gram-positive bacteria. In addition, prior studies had demonstrated kerriamycin B to exhibit antitumor activity as well, however the mechanism remained unclear.32 It is possible that its ability to inhibit SUMOylation is at least partially responsible for this activity, although no studies have conclusively linked the observed activities.
More recently, the Yoshida lab identified another natural product that can inhibit SUMOylation by blocking formation of the E1-SUMO complex. Davidiin (4), isolated from the plant Davidia involucrate, is one of the most potent natural product SUMOylation inhibitors identified, with an in vitro IC50 value of 0.15 μM.33 Davidiin was able to inhibit SUMOylation in vivo and exhibited antiproliferative activity against multiple cancer lines at concentrations that correspond to its SUMOylation inhibitory activity. Prior studies had shown davidiin to downregulate EZH2, an enzyme that methylates histone H3, and is upregulated in hepatocellular carcinoma.34 However, EZH2 is a SUMO substrate, and davidiin was able to inhibit growth of EZH2 overexpressing cancer cells, suggesting that the inhibition of EZH2 modulation is affected by daviidin. A structurally similar compound, tannic acid (5), was identified through a gene expression assay as a nontoxic SUMOylation inhibitor.35 The researchers used a human Liver Receptor Homolog 1 (LRH1) as their SUMO substrate, which has a high level of basal SUMOylation, and determined an in vitro IC50 value of 12.8 μM. Like davidiin and ginkgolic acid, tannic acid blocks the formation of the E1-SUMO complex, however the mechanism through which tannic acid inhibits SUMOylation while remaining nontoxic is unknown. Tannic acid can be a useful tool to study the SUMOylation pathway without affecting cell viability.
A 2014 study utilized phage display to probe the sequence variability of the C-terminal of SUMO region by SAE. Researchers synthesized full-length SUMO proteins, but randomized the last seven positions, however, the terminal glycine was left as this residue is necessary for activity. From this initial library of full-length SUMO variants, three SUMO mutants were identified that exhibit faster enzyme kinetics than wt-SUMO. Researchers then synthesized heptamers based on the novel C-terminal sequences from the most active variants. Two of these heptamers, S50 and S90, were able to block wt SUMO from forming a thiosester with SAE.36 Researchers at Sloan-Kettering employed a similar strategy, and developed semisynthetic SUMO analogs with nonhydrolyzable 5′-sulfonyladenosine moieties to mimic the acyl-adenylate intermediate that results from the first half reaction catalyzed by SAE.37 Employing native chemical ligation, CGG-AMSN and CGG-AVN were appended to truncated SUMO that was lacking the C-terminal diglycine residues to produce SUMO-AMSN (6) and SUMO-AVSN (7). Each ligand was designed to specifically inhibit the two half reactions catalyzed by SAE, as the vinyl sulfonamide can act as an electrophilic trap for the catalytic cysteine residues. These adducts were shown to selectively inhibit SAE over the analogous ubiquitin-activating enzyme and block formation of the E1-SUMO complex. These probes were subsequently employed to elucidate structural information about the active site of SAE, and to gain insight into the remodeling that occurs during catalysis.20
The Zhang lab used computational studies to discover novel and synthetic SAE inhibitors. These studies used the ATP-binding pocket as the location for designing new inhibitors. Initially, phenyl urea compounds (8 and 9) were identified as potential SUMOylation inhibitors and in vitro results confirmed that 8 and 9 manifested IC50 values of 11.1 ± 3.1 μM and 11.7 ± 5.3 μM, respectively.38 Follow-up studies revealed these compounds to inhibit SAE by preventing formation of the E1-SUMO complex. While these compounds were the first synthetic SAE inhibitors described, their symmetrical nature presented potential issues with solubility. Consequently, their next study addressed symmetry concerns and identified a series of quinazolinyloxy biaryl urea compounds that served as SAE inhibitors. From this study, compound 10 emerged as the most potent in vitro SUMOylation inhibitor and manifested an IC50 value of 13.4 μM.39 Compound 10 exhibits the same mode of activity as 8 and 9 and prevents formation of the E1-SUMO complex. A third study was used to discover scaffolds distinct from compounds 8-10. This study identified pyrazole urea (11) and thiazole urea (12) as the most potent molecules capable of inhibiting SUMOylation in vitro and manifested IC50 values of 13.8 μM and 33.2 μM, respectively.40 None of these compounds have been evaluated in vivo, and despite the discovery of novel SUMOylation inhibitory scaffolds, there have been no further medicinal chemistry efforts to optimize these scaffolds.
A high throughput screen discovered a SUMO-E1 inhibitor with a mechanism distinct from other modulators of the SUMOylation process. While other inhibitors target the ATP-binding pocket as competitive inhibitors, the Chen lab discovered CID9549553 (13), which covalently targets an allosteric binding pocket of SUMO E1.41 In an in vitro SUMOylation assay, compound 13 manifested an IC50 of 0.54 μM. Subsequent medicinal chemistry efforts led to a more potent SUMOylation inhibitor, COH000 (14), which exhibited an IC50 value of 0.2 μM. To establish that 14 was a selective SAE inhibitor, they evaluated the compound’s activity on ubiquitinylation and it was inactive at the highest concentration tested, 100 μM. Further studies showed that compound 14 was able to induce a conformational change in SUMO-E1, which prevents both SUMO or ATP binding and leads to increased SAE2 ubiquitinylation and subsequent degradation. Compound 14 was active in cells and induced apoptosis, consistent with previous SUMOylation inhibitors. Attempts to generate a stable mutant by converting the proposed target cysteine into a serine residue were unsuccessful. However, the co-crystal structure of 14 bound to SAE confirmed binding of the compound to a previously unknown allosteric binding pocket.42
The Chen lab also identified a class of tricyclic quinoxaline-based SAE inhibitors wherein compound 15 was the most potent and exhibited an in vitro IC50 value of 0.5 μM.43 NMR based experiments demonstrated 15 to target the ATP binding pocket, as it was able to displace ATP from the active-site. In vivo experiments showed 15 to be a selective SUMOylation inhibitor, as it had no effect on the ubiquitinylation pathway. Due to the role that SUMOylation plays in DNA damage repair, inhibition of SUMOylation has been hypothesized to increase the sensitivity of cancer cells to radiotherapy, as increased SENP expression leads to increased sensitivity to radiotherapy.44 Compound 15 increased the sensitivity of multiple cancer cell lines to radiotherapy. In addition, SUMOylation has been shown to modulate proteins associated with viral replication, and therefore, inhibition of SUMOylation has been proposed as a novel strategy towards the treatment of HIV.45 Consistent with this observation, 15 was able to reduce the viral replication of HIV in an infection model, which highlights the potential therapeutic utility of SUMOylation inhibitors beyond cancer.
Pevonedistat is a NEDD8-activating enzyme (NAE) inhibitor that is currently in Phase 2 clinical trials as a combination therapy for the treatment of both acute myeloid leukemia and non-small cell lung cancer.46 Pevonedistat is an AMP mimic that inhibits NAE by forming an adduct with NEDD8, another UBL, in an ATP-dependent manner catalyzed by NAE.47 Using pevonedistat as a starting point, researchers at Takeda Pharmaceuticals developed a selective SAE inhibitor, ML-792 (16). ML-792 manifests a similar mechanism of action as pevonedistat and forms a covalent adduct with SUMO that is catalyzed by SAE. In fact, 16 is the most potent SAE inhibitor identified to date and exhibits an in vitro IC50 value of 3 nM for SUMO1 and 11 nM for SUMO2.48 Against the analogous NAE and ubiquitin activating enzyme, 16 manifested IC50 values of 32 μM and >100 μM, respectively, highlighting compound 16 as both a selective and potent SAE inhibitor. Importantly, 16 is active in cells and allowed for detailed investigation of the SUMOylation process. Although DNA repair pathway proteins are SUMO substrates, 16 did not induce DNA-damage nor did it display synergism with the known DNA damage agents, cisplatin or hydroxyurea. However, compound 16 induced apoptosis in cancer cells and cell lines that expressed high levels of MYC, which were more sensitive to compound 16. An analog of 16, TAK-981 (17) is currently in Phase I clinical trials for the treatment of lymphomas and metastatic solid tumors.49
SUMO-E2 Inhibitors
After SUMO has been activated by the SAE, it is transferred to SUMO-E2, which catalyzes formation of the isopeptide bond between the C-terminal glycine residue of SUMO and the ε-amino group of a lysine residue within the target protein. As previously mentioned, 40 enzymes catalyze the ligation of ubiquitin with a target protein in the ubiquitinylation pathway, whereas there is only one E2 enzyme, Ubc9, that is responsible for conjugation within the SUMOylation pathway. As a result, there is an opportunity to inhibit the SUMOylation pathway by targeting one enzyme, as compared to the many enzymes required for ubiquitinylation. BRCA1 mutation is associated with an increased risk of aggressive breast and ovarian cancer in women. Silencing of Ubc9 in BRCA1 mutant derived ovarian and breast cancer lines decreased proliferation and migration, indicating the role Ubc9 plays in the progression of these cancers.50 Ubc9 expression is also a predictor of chemoresistance in breast cancer.9 Additionally, knockdown of Ubc9 can increase sensitivity to chemotherapeutics in both advanced-stage melanomas and hepatocellular carcinomas.51,52 These studies illustrate the therapeutic potential for a selective inhibitor of Ubc9.
Some of the first identified inhibitors of SUMO-E2 were the natural products spectomycin B1 (18) and structurally related compounds, chaetochromin A (19) and viomellein (20), which were discovered by the same lab that was responsible for reporting the SUMOylation inhibitory activities of ginkgolic acid and kerriamycin B.53 Spectomycin B1 is a Gram-positive antibiotic produced by Streptomyces spectabilis, whereas chaetochromin A is a metabolite of the fungi Chaetomium gracile and displays a diverse array of documented activities, and viomellein is an antibiotic that can be isolated from multiple fungal species.54–58 An in vitro SUMOylation inhibition assay revealed that spectomycin B1, chaetochromin A, and viomellein produce IC50 values of 4.4 μM, 3.7 μM and 10.2 μM respectively. The monomeric analogs of spectomycin B1, spectomycins A1 and A2, have also been evaluated for SUMOylation inhibitory activity and confirmed that the dimeric species is critical for activity, as both monomers were 50-fold less potent.59 Antagonizing Ubc9 function in breast cancer lines is known to inhibit estrogen receptor gene expression. In agreement, treatment of MCF7 cells with spectomycin B reduced pS2 mRNA levels, a primary estrogen-response gene. In addition, spectomycin B also suppressed β-estradiol-dependent proliferation in MCF7 cells, which could be reversed by Ubc9 overexpression. In a study looking at treating liver fibrosis through Farnesoid X receptor (FXR) agonism, researchers used spectomycin to prevent FXR SUMOylation, which also inhibited its transactivation. Spectomycin was demonstrated to rescue FXR activity and synergize with FXR agonists for the treatment of liver fibrosis.60
In their efforts to develop an electrophoretic mobility shift assay to detect SUMOylation inhibitors, the Schneekloth lab discovered flavone 2-D08 (21), which inhibits Ubc9 by preventing the transfer of SUMO from Ubc9 to the substrate.61 Using their assay, they found 21 to exhibit an IC50 value of 6.0 μM, and inhibited IκBα SUMOylation by all three SUMO isoforms. In addition, 21 did not affect global ubiquitinylation, suggesting that 21 exhibits selectivity. Compound 21 was able to inhibit SUMOylation in the presence of varying concentrations of the antioxidant, dithiothreitol (DTT), providing evidence that 21 does not exhibit its activity through formation of reactive oxygen species (ROS). In addition, the study evaluated several structurally related flavonoids and found a very rigid SAR, as none of the other derivatives were as potent as 21. In another study, 2-D08 was used to investigate the role of SUMOylation in the viral replication cycle of Zika and related flaviviruses, as viral proteins have been shown to be post-translationally modified by SUMO.62,63 Another study demonstrated that 21 also exhibits anti-aggregatory and neuroprotective activity against amyloid β.64 However, this activity is likely unrelated to the inhibition of SUMOylation, as other flavonoids used in the study also display similar neuroprotective effects but do not exhibit SUMOylation inhibitory activity.
The Schneekloth lab developed a small molecule microarray assay to elucidate Ubc9 inhibitors, after the discovery of 2-D08 in a generalized SUMOylation assay. Using fluorescently labeled Ubc9 and UbcH5b, an analogous ubiquitin E2, they identified 133 potential Ubc9 inhibitors from their initial screen of 19,200 compounds.65 The hits were evaluated in an in vitro assay with RanGAP1 or a fluorescent peptide as a substrate for SUMOylation. Compound, 22 emerged with an IC50 of 74 ± 5 μM. Its activity was also assessed against two ubiquitin E2s, UBE2B and UbcH5b, and manifested IC50 values of 120 μM and 118 μM respectively, demonstrating that 22 has modest selectivity for the SUMOylation pathway. Compound 22 was unable to block formation of the E1-SUMO complex, confirming that it manifests its activity via the inhibition of Ubc9. Furthermore, compound 21 was still active in the presence of detergents, suggesting that the compound is not acting through aggregate formation.
Scientists at GlaxoSmithKline developed a similar high throughput screen to identify SUMOylation inhibitors, specifically looking at TRPRS1 as a SUMO substrate.66 TRPS1 is a transcriptional repressor of genes responsible for bone and cartilage production and SUMOylation is required for TRPS1 activity.67 In order to develop potential therapeutics for bone and cartilage diseases, they developed a high throughput screen to discover Ubc9 inhibitors. After optimization of the assay, a 2.2 million compound library was screened, which led to the identification of 728 potential hits. Compounds that inhibited E1 and compounds that were thiol reactive were eliminated, and eventually, GSK145A (23) was identified as a lead compound that manifested an IC50 value of 12.5 μM. Subsequent studies revealed 23 to act as a substrate for Ubc9 and to compete with TRPRS1 binding and SUMOylation. Furthermore, LCMS analysis revealed formation of a SUMO-GSK145A covalent species.
In addition to their work to develop assays for the detection of SUMOylation inhibitors, the Schneekloth lab employed a strategy to develop synthetic derivatives of the SUMO consensus region.68 Initially, they investigated the minimum peptide sequence necessary to be a substrate for Ubc9 and determined that the tetrapeptide consensus sequence from the androgen receptor (Ac-IKLE-NH2) could efficiently be SUMOylated by Ubc9. Six analogs of the sequence were made in which each residue or termini was replaced with a hydroxyl amine and then reacted with 100 commercially available aldehydes to generate a library of 600 derivatives. Each compound was screened for its ability to act as a substrate for Ubc9 and to compete against SUMO1. This study identified 24 and 25 as the most active peptide derivatives and manifested of IC50 values 12 ± 0.8 μM and 15 ± 1 μM, respectively. Docking studies suggest the heterocyclic side chains project into a hydrophobic pocket, which highlighted that shorter peptide sequences may mimic key interactions of the larger protein.
The Zhang lab, which discovered synthetic SUMO E1 inhibitors 8-12 via virtual screening applied the same screening method, but utilized the binding pocket on Ubc9 where substrates like RanGAP1 bind and contains a catalytic cysteine residue.69 They discovered two compounds with modest SUMOylation inhibitory activity in an in vitro assay using RanGAP1 as a substrate. The lead compounds, 26 and 27, exhibited IC50 values of 46.3 μM and 68.8 μM, respectively, and inhibited formation of the SUMO-E2 complex, but not the SUMO-E1 complex, confirming their compounds bind and selectively inhibit Ubc9.
Using a fragment-based approach, the Schneekloth lab identified an allosteric binding pocket within Ubc9 by solving its co-crystal structures.70 In addition, they used HSQC to confirm fragment binding to the allosteric region of Ubc9. Using in vitro SUMOylation assays, they demonstrated that the fragments prevented formation of the SUMO-E2 complex, without affecting formation of the SUMO-E1 complex. They also generated Ubc9 mutants by altering residues in the allosteric pocket, which prevented inhibition by these small molecules. Since the fragments are active at the millimolar concentration range, work is required to fully exploit the allosteric binding pocket.
SUMO-E3 Inhibitors
At present, there are no inhibitors of the SUMO-E3 ligases, however they have been implicated in the growth and chemoresistance of several cancers.71–73 The predominant family members of the SUMO-E3 ligases are the protein inhibitors of activated STAT (PIAS), which were originally discovered as transcriptional regulators of the JAK-STAT pathway. These proteins have been shown to regulate 60 different proteins in addition to their role as SUMO-E3 ligases.74–77 A 2014 study demonstrated that PIAS1 upregulation corresponds to increased resistance to docetaxel in prostate cancer, whereas knockdown of PIAS1 resulted in decreased cell proliferation and the induction of apoptosis, highlighting the potential therapeutic potential of targeting PIASs.78
SENP Inhibitors
Sentrin specific peptidases (SENPs) are responsible for the removal of SUMO from the target protein, with SENP1 and SENP2 responsible for the maturation of SUMO. Like other parts of the SUMOylation pathway, dysregulation of SENPs have been implicated in many cancers. SENP1 increases activity of the androgen receptor (AR) and is a driver of prostate cancer progression.79 Increased levels of SENP1 and SENP3 have also been observed in colon, prostate, ovary, and lung cancers.8 Additionally, there is a growing body of literature regarding the role that SENPs play in the radiosensitivity of cancer.44 In fact, deletion of SENP6 in HeLa cells promoted a more robust DNA damage response and led to a decrease in sensitivity to both radiotherapy and chemotherapy.80 The bulk of research to develop inhibitors of specific enzymes in the SUMOylation pathway has focused upon SENP inhibition.
Several natural products have been identified that inhibit one or more of the SENP isoforms. Momordin Ic (28) was identified from a screen of natural products and found to exhibit an in vitro IC50 value of 15.37 μM against SENP1.81 Momordin Ic is isolated from the fruit of Kochia scoparia and has been shown to decrease invasiveness of liver cancer through modulation of the p38 and JNK pathways.82 Researchers demonstrated in vivo activity with transfected HEK293 cells, wherein HIF-1α SUMOylation levels were decreased by momordin Ic treatment. HIF-1α is a known SUMO substrate, and SUMOylated HIF-1α is targeted to the proteasome for degradation. Furthermore, HIF-1α is an important oncogene in prostate cancer. SENP1 is necessary for HIF-1α stability, as SENP1 knockdown leads to HIF-1α degradation and the induction of apoptosis.83,84 Similarly, treatment with momordin Ic induced apoptosis in PC3 cells in vivo as well as in a mouse tumor xenograft model. In addition, they found that sensitivity to treatment correlated with SENP1 expression levels, with higher expression leading to increased sensitivity to SENP1 inhibition.
Streptonigrin (29) was identified as the most potent compound from an iterative screen of the National Cancer Institute’s Developmental Therapeutics Program compound repository.85 Streptonigrin is a Gram-positive antibiotic isolated from Streptomyces flocculus that also possesses anti-tumor activity. Streptonigrin was in clinical trials against multiple types of cancers in the 1960s, but clinical trials did not lead to an FDA-approved drug.86,87 In addition to SENP inhibitory activity, streptonigrin is a potent protein arginine deaminase inhibitor.88 Researchers tested the selectivity of streptonigrin against three of the six SENP isoforms, and found that streptonigrin is selective for SENP1 with an in vitro IC50 value of 0.518 ± 0.100 μM for SENP1 as compared to 6.919 ± 0.676 μM and 5.205 ± 0.853 μM for SENP2 and SENP6, respectively. Streptonigrin treatment also led to a dose-dependent decrease in HIF-1α levels in vivo. Structurally related compounds were also tested, and two analogs that exhibited inhibitory activity, NSC76919 (30) and NSC45384 (31) were identified. Unfortunately, neither compound was as potent as the natural product.
Vialinin A (32) is a potent anti-inflammatory agent that was isolated from the mushroom, Thelephora vialis. It inhibits the production and release of tumor necrosis factor alpha (TNF-α) with an IC50 against murine bone-derived mast cells of 0.04 nM.88 Previous studies suggested that vialinin A is capable of inhibiting the deubiquitinating enzyme, UP5, which is at least partially responsible for its anti-inflammatory activity.90,91 Vialinin A and the structurally related natural product, atromentin (33), were evaluated for their ability to inhibit SENP1 in vitro and found that they inhibited SENP1 with in vitro IC50 values of 1.52 ± 0.06 μM for vialinin A and 6.10 ± 0.11 μM for atromentin, respectively.92 Researchers also demonstrated that vialinin A and atromentin inhibited SENP1 in vivo. They proposed that inhibition of SENP1 could be responsible for the potent anti-inflammatory activity manifested by vialinin A. Synaptotagmins are SUMO substrates that are involved in the vesicular traffic of TNF-α, therefore it’s possible that abhorrent synaptotagmin activity results from SENP1 inhibition and could affect TNF-α secretion.
The first set of synthetic SENP inhibitors developed were peptide-based inhibitors that used the C-terminal amino acid sequence of SUMO to drive selectivity.93 Biotin conjugated to a 5-mer, 9-mer and 13-mer peptide sequence of the SUMO C-terminus was synthesized to contain a vinyl sulfone at the C-terminus that acts as an electrophilic trap. Studies confirmed that all three peptide sequences were recognized by SENP1 and none of them exhibited any cross reactivity with other ubiquitin or ubiquitin-like specific proteases. A similar approach was employed by the Funeriu lab, wherein they synthesized a biotin labeled 7-mer peptide sequence of the SUMO2 C-terminus. In this example, the final residue was a glycine fluoromethylketone that can act as an electrophile.94 In vitro and in vivo experiments confirmed that their peptide derivatives could efficiently and selectively bind to SENP1 and SENP2.
JCP-666 (34) was identified from a screen of cysteine protease inhibitors as an inhibitor of the lone SENP isoform present in Plasmodium falciparum (pfSENP1).95 However, the aza-epoxide was too unstable and a more stable synthetic analog with similar potency, VEA-260 (35), was used in subsequent studies. As the catalytic domain of pfSENP1 is similar to human SENPs, 34 and 35 were screened for selectivity against human SENPs as well as other ubiquitin and ubiquitin-like specific proteases. 34 and 35 exhibited 17.9 ± 1.0 μM and 16.2 ± 1.5 μM IC50 values against pfSENP1, respectively, and were more potent against human SENPs. Against SENP1, 34 and 35 exhibited IC50 values of 9.0 ± 0.5 μM and 7.1 ± 0.6 μM, respectively. Against SENP2, 34 and 35 manifested IC50 values of 4.7 ± 0.5 μM and 3.7 ± 0.2 μM. 35 also inhibited SENP8, which is a NEDD8 specific protease, but neither compound exhibited activity against the ubiquitin specific proteases. In a follow up study, the Bogyo lab developed analogs of 35 and discovered the more potent and selective SENP inhibitors, VAE-499 (36), VAE-500 (37) and VAE-561 (38).96 VAE-499 and VAE-500 manifested 0.25 ± 0.03 μM and 0.86 ± 0.1 μM IC50 values against SENP2, and VEA-500 was inactive against SENPs 1, 6 and 7. VAE-561 was their most potent compound against SENP6 and SENP7, with IC50 values of 4.2 ± 0.6 μM and 4.3 ± 0.7 μM, respectively. Upon screening the library for in vivo activity against HEK293 cells, the aza-epoxide compounds were highly reactive and showed little to no selectivity for SENPs, whereas these second-generation compounds showed no cross reactivity with other proteins in vivo.
The first non-peptide based SENP inhibitors were developed by the Zhou and Nakamura labs. A series of benzodiazepine-based compounds was synthesized based on initial computational studies. The lead compound obtained, 39, exhibited an IC50 value of 9.2 μM against SENP1 in vitro. Compound 39 was also evaluated in vivo, and inhibited the growth of prostate cancer cells with an EC50 value of 13.0 μM.97 The Nakamura lab reported GN6767 (40) as an HIF-1α inhibitor, and a biotinylated analog of 40 was able to bind SENP1 in a pull down assay. Subsequent in vitro evaluation confirmed that 40 also inhibited SENP1. Therefore, a small library of analogs was prepared and eventually led to the discovery of GN6958 (41), which exhibited an IC50 value of 29.6 ± 0.5 μM against SENP1. Compound 41 also displayed selectivity towards SENP1 and did not inhibit SENP2 at concentrations up to 100 μM, nor did it inhibit other proteases including trypsin, chymotrypsin or thermolysin at 100 μM. Consistent with SENP1 inhibition, 41 inhibited HIF-1α accumulation in HeLa cells.98
Virtual screens have been used to discover several classes of SENP inhibitors. In the first such study, researchers screened a library of 180,000 compounds from SPECS.99 The screen identified the lead compound 42, which exhibited an in vitro IC50 value of 2.385 ± 0.059 μM. A small library of derivatives was then synthesized and led to the identification of 43, which had a modest improvement in activity. In their in vitro assay, 43 displayed an IC50 value of 1.080 ± 0.010 μM against SENP1. However, none of the compounds were evaluated for in vivo activity. In another study, researchers from the Chen Lab discovered a novel class of SENP inhibitors from a virtual screen of the Developmental Therapeutics Program’s compound library from the National Cancer Institute.100 The hits were evaluated for their ability to inhibit SENP1 and SENP2 by measuring SUMO1 and SUMO2 maturation. The lead compounds, SPI-01 (44) and SPI-02 (45), were more active at inhibiting SUMO2 maturation, as well as more active against SENP2. In addition to evaluating their compounds against the maturation of SUMO isoforms, they also evaluated their compounds against SENP1, SENP2 and SENP7 using a fluorescent SUMO mimic. In this in vivo assay, 44 manifested IC50 values of 5.9 ± 1.4 μM, 2.9 ± 1.6 μM, and 3.5 ± 1.5 μM against SENP1, SENP2 and SENP7, respectively, whereas 45 exhibited IC50 values of 2.1 ± 1.9 μM, 2.0 ± 2.0 μM, and 2.7 ± 1.9 μM against SENP1, SENP2 and SENP7, respectively. In addition, compound 44 was active in vivo, as they observed a dose-dependent accumulation of SUMOylated proteins within HeLa cells after treatment with 44. In order to determine the mechanism of action, they used chemical shift perturbation (CSP) analysis and kinetic experiments to establish that their compounds were noncompetitive and noncovalent inhibitors of SENPs.
The Zhang lab used virtual screens to discover inhibitors of other enzymes involved in the SUMOylation pathway, which led to the discovery of a novel class of SENP inhibitors.101 Their screen of the Namiki-Shoji compound library (+4 million compounds) and subsequent biological assessment revealed 1,2,5-oxadiazoles as a novel pharmacophore for SENP inhibition. After a secondary screen that focused primarily on the 1,2,5-oxadiazole pharmacophore, they evaluated a total of 80 compounds using an in vitro assay to establish structure-activity relationship trends. From this study, they identified 46 and 47 as lead compounds with IC50 values of 5.9 ± 1.3 μM and 3.7 ± 0.5 μM against SENP2, respectively. Compounds were also evaluated against SENP1 and determined that 46 manifested an IC50 value of 9.7 ± 3.0 μM while 47 manifested no activity at the highest concentration tested, 30 μM. Neither compound was shown to inhibit other proteases, including papain and trypsin. In another study, researchers screened the SPEC database, and evaluated 117 compounds in vitro, which led to the identification of SI2 (48) which exhibited an IC50 value of 1.29 μM against SENP1.102 Compound 48 was tested against other SENP isoforms and displayed activity against SENPs 2 and 3, but not SENP5. Furthermore, 48 was unable to inhibit several classes of proteases. Researchers then used 48 to enrich the population of SUMOylated proteins within PC3 cells to perform proteomic studies and to identify novel SUMO substrates. Using 48, they identified several proteins within the spliceosome that are regulated by SUMOylation.
A study from the Zhou lab screened roughly 200,000 compounds in an effort to identify novel scaffolds that served as SENP inhibitors. They screened their library of potential hits at a single concentration of 50 μM, and 11 of the molecules were shown to exhibit greater than 50% inhibition.103 The two lead compounds, 49 and 50, manifested IC50 values of 15.0 μM and 17.6 μM, respectively against SENP1. They synthesized a small library of analogs based on these structures and ultimately produced compound 51, which exhibits an IC50 value of 3.5 μM, however, they did not evaluate their compound in vivo.
The most recent effort to use virtual screening to discover novel SENP inhibitors was conducted by the Riedl lab, who sought to develop a SENP1 inhibitor with a competitive mechanism of action.104 After in vitro testing of their virtual hits, compound 52 emerged as a lead compound and manifested an IC50 value of 3.7 μM. They synthesized a library of 25 derivatives and selected two additional compounds for further evaluation, 53 and 54. Compound 53 produced an IC50 value of 0.99 μM, however it exhibited a poor selectivity profile against other deubiquitinating enzymes, whereas compound 54, which produced an IC50 value of 7.5 μM had greater selectivity towards SENP1 than 52. Compounds 52 and 54 displayed 10- and 3-fold selectivity for SUMO1 over SUMO2/3 respectively, whereas compound 53 displayed no selectivity against the SUMO isoforms. Preliminary ADME studies were performed, which included 43 for comparison. The ester functionalities present in 43 and 52 represent metabolic liabilities that led to short half-lives of <1 min and 41 min, respectively. When the ester was replaced with an amide as with 54, the half-life increased to >240 min. Kinetic studies were performed with compound 54 and indicated that 54 was a noncompetitive inhibitor, which is interesting given that their virtual screen was designed specifically to identify competitive inhibitors. Finally, they evaluated the in vivo activity of their compounds against prostate cancer cells. Compounds 52 and 53 affected cell viability with EC50 values of 186 μM and 58 μM, respectively, however, 54 did not affect cell viability at the highest concentration tested (200 μM).
Other SUMOylation Inhibitors
Several molecules have been discovered that modulate the SUMOylation pathway wherein the mode of action is either undefined or outside the scope of this review. The first molecule is topotecan (55), a semi-synthetic camptothecin derivative currently approved by the FDA for treatment against cervical, ovarian and small cell lung cancer.105,106 Topotecan is a DNA topoisomerase I inhibitor and has demonstrated off-target activity, including inhibiting HIF-1α.107 As both are known SUMOylation substrates, researchers sought to determine whether topotecan could also affect SUMOylation within glioblastoma multiforme (GBM), wherein elevated levels of SUMOylated proteins have been observed.108,109 Across three GBM cell lines, topotecan was able to decrease global SUMOylation levels in a dose-dependent manner. Interestingly, the previously reported SUMOylation inhibitors ginkgolic acid (1) and anacardic acid (2) were unable to inhibit SUMOylation in these cell lines at concentrations up to 100 μM. Topotecan was also evaluated against other ubiquitin-like protein pathways and was shown to inhibit NEDDylation in two of three GBM cell lines. Other UBLs were unaffected by topotecan. They evaluated expression levels of SUMO-associated enzymes and found they were unaffected by topotecan treatment, however no other attempts to elucidate the mechanism were reported.
Four ortho-quinone natural products have also been identified as potent SUMOylation inhibitors, nocardione A (56), 33-DINOR-dunnione (57), 33-DINOR-dehydrodunnione (58), and β-lapachone (59).110 These compounds were first isolated from the bark of Handroanthus impetiginosus, and 56 has previously demonstrated tyrosine phosphatase inhibitory activity as well as antifungal activity. Antiproliferative activity against cancer cells has been demonstrated with compounds 56 and 59.110,111 All four compounds inhibited SUMOylation in a dose-dependent manner and manifested IC50 values of 3.9, 1.1, 1.2, and 0.4 μM, for 56-59, respectively, in an in vitro assay. All compounds also inhibited SUMOylation in vivo. As quinones are redox active, molecules that generate reactive oxygen species (ROS) are likely responsible for the compound’s activity. Treatment with 56 resulted in a dose-dependent increase in ROS. Hydrogen peroxide treatment inhibits SUMOylation by inducing formation of a disulfide crosslink between the E1 and E2 enzymes, and similarly, treatment with 56-59 induced formation of the same disulfide crosslink.112,113 Interestingly, when the catalytic cysteine residue of SUMO E1 was mutated to a serine and used in the same assay, disulfide bond formation still occurred, suggesting that the crosslink does not necessarily rely upon the catalytic cysteine residue. The inhibitory effects of 56-59 could be blocked by treatment with the antioxidant N-acetyl-L-cysteine, providing further evidence for the proposed mechanism of action.
A pyrroloiminoquinone, macrophilone A (60), isolated from Macrorhynchia philippina, also demonstrated SUMOylation inhibitory activity through formation of ROS.114 In order to verify the structure, the Schneekloth lab synthesized the natural product. The methoxy derivative (61) was also synthesized and evaluated and similar to quinones 56-59, iminoquinones 60 and 61 also generated ROS and subsequent E1 and E2 crosslinking. The crosslinking could be prevented by treatment with N-acetyl-L-cysteine. In their in vitro assay, the synthetic analog 61 was more active and exhibited an IC50 value of 2.5 μM, whereas the natural product, 60, manifested an IC50 value of 8.0 μM. In vivo evaluation of 61 against lung cancer cells revealed it to be incredibly cytotoxic, with an EC50 value of 145 nM. Neither N-acetyl-L-cysteine nor N-acetyl-L-cysteine amide were able to mitigate the cytotoxicity, suggesting the cytotoxicity of 61 goes beyond its ability to generate ROS. A follow-up study of other members of the macrophilone family revealed many members of this family are capable of inhibiting SUMOylation in vitro, although none were as potent as 60.116 In addition, compound 60 also inhibited the ERK cascade through reduction of Raf and MEK protein levels.
Triptolide (62) is a diterpene natural product isolated from the herb Tripterygium wilfordii and exhibits immunosuppressive activity as well as antiproliferative activity against melanoma, breast cancer, and prostate cancer.117,118 Triptolide was tested against two prostate cancer cell lines, PC3 (androgen-independent) and LNCaP (androgen-dependent), where it manifested EC50 values of 20.3 nM and 9.75 nM, respectively. Real-time PCR revealed that SENP1 mRNA levels were decreased in a dose and time-dependent manner by triptolide, with an IC50 value of 70.7 nM against PC3 cells. Consistent with decreased mRNA levels, SENP1 protein levels were also suppressed by triptolide. Similarly, triptolide downregulated the expression of c-Jun and the androgen receptor, both of which are SUMO-substrates. The overexpression or knockdown of these three proteins reduced the efficacy of triptolide, suggesting the importance of these proteins for triptolide activity. Triptolide has also been shown to induce the proteasome-mediated degradation of the largest subunit of RNA polymerase II, Rpb1, which in turn leads to global inhibition of transcription.119,120 Several triptolide derivatives have been synthesized, and multiple derivatives remain in clinical trials for the treatment of cancer or rheumatoid arthritis.121,122
While most approaches to increase the level of SUMOylated proteins have focused on the inhibition of SENPs, a screen of over 100,000 compounds revealed compound N106 (63) to be capable of increasing SUMOylation through the activation of SUMO-E1.123 Researchers were looking for compounds that could increase SUMOylation of SERCA2a, as a potential treatment for heart failure. In their dose-response studies, researchers determined that 63 produced an EC50 value of 2.45 ± 0.13 μM, but 63 was no longer efficacious at concentrations above 50 μM. In order to determine the mechanism of action, they performed computational studies and identified a potential binding pocket for 63 at the interface between the SAE1 and SAE2 subunits of SUMO-E1. They mutated residues within this proposed binding pocket and observed that 63 was inactive against these mutants, providing additional evidence for the binding mode. Additionally, 63 was evaluated in the same manner against the ubiquitinylation pathway, where it had no effect, thus confirming that 63 is selective for the SUMOylation pathway. Compound 63 is the first molecule capable of increasing SUMOylation through E1 activation.
Conclusion
The role played by SUMOylation during cancer progression is rapidly emerging and as a result, efforts to develop selective inhibitors have increased significantly. This review attempts to summarize modern inhibitors that target various enzymes involved in the SUMOylation process, which when combined, has helped to validate the SUMOylation pathway as a good target for the development of cancer chemotherapeutics. While a considerable amount of effort has been devoted toward the development of SUMO-E1 and SENP inhibitors, (one SUMO-E1 inhibitor is currently in clinical trials), little effort to develop SUMO-E2 inhibitors has been sought. Ubc9 represents an underinvestigated target within the SUMOylation pathway, and is the only enzyme responsible for conjugation, which is in contrast to the ~40 enzymes associated with the ubiquitinylation process. In addition, Ubc9 can SUMOylate substrates without E3 ligases, which in contrast to ubiquitinylation. Due to the existence of multiple isoforms of SUMO, the exact mechanism through which Ubc9 can modify substrates with different SUMO isoforms remains unclear, however, the process likely involves specific SUMO E3-ligases. SUMO-E3 ligases have also been implicated in cancer progression, suggesting that E3 may represent an excellent target for the discovery of novel anti-cancer agents. Several scaffolds have been developed that inhibit SENP activity, but many of them suffer from low potency or poor isoform selectivity, and some have not been validated in vivo, which represents an additional opportunity to optimize more potent and selective inhibitors. While much work has been done, there are significant opportunities to develop more selective and potent SUMOylation inhibitors for the treatment of cancer as well as neurological disorders as highlighted in this review.
Figure 2.
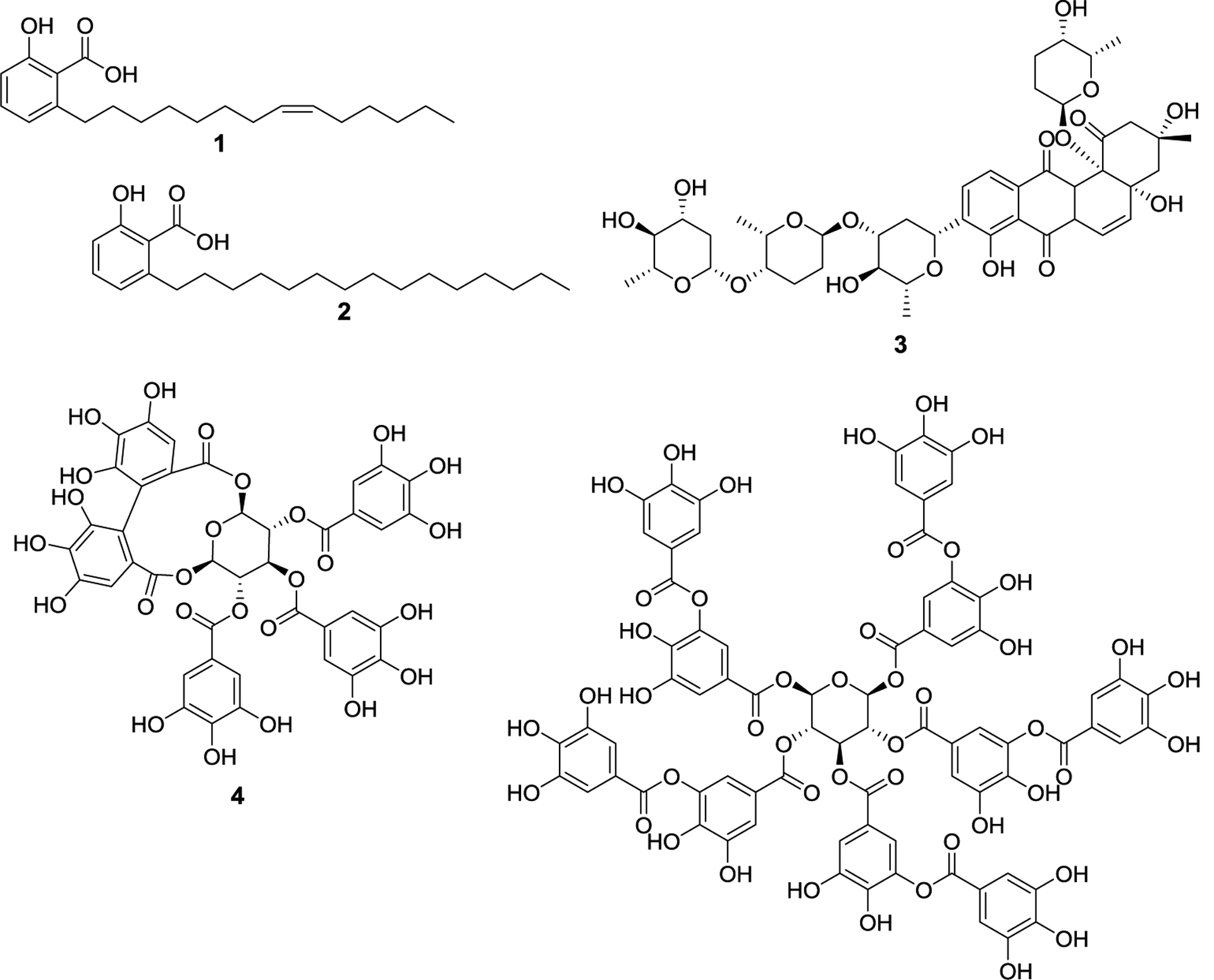
Natural product E1 inhibitors. First identified SUMOylation inhibitors, ginkgolic acid (1), anacardic acid (2), and kerriamycin B (3), and polyphenolic SUMOylation inhibitors, davidiin (4), and tannic acid (5).
Figure 3.
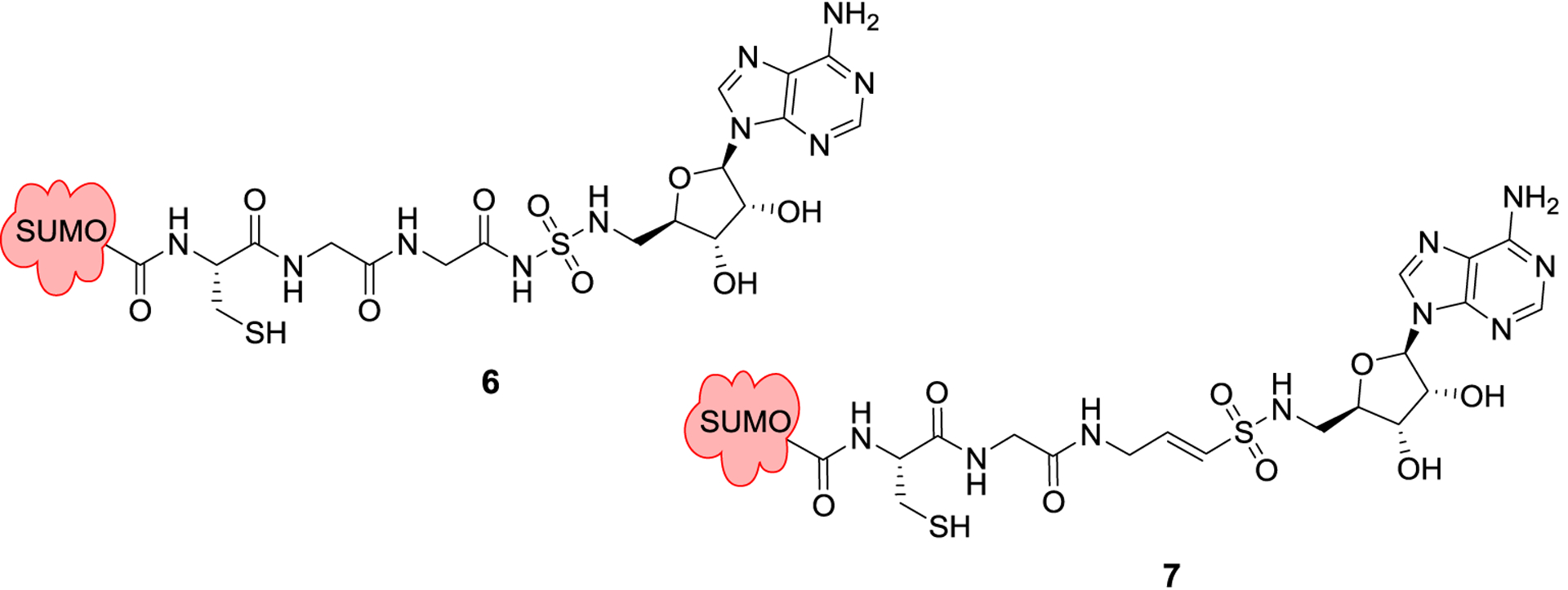
Semisynthetic SUMO adducts mimicking the acyl-adenylate intermediate, 6-7.
Figure 4.
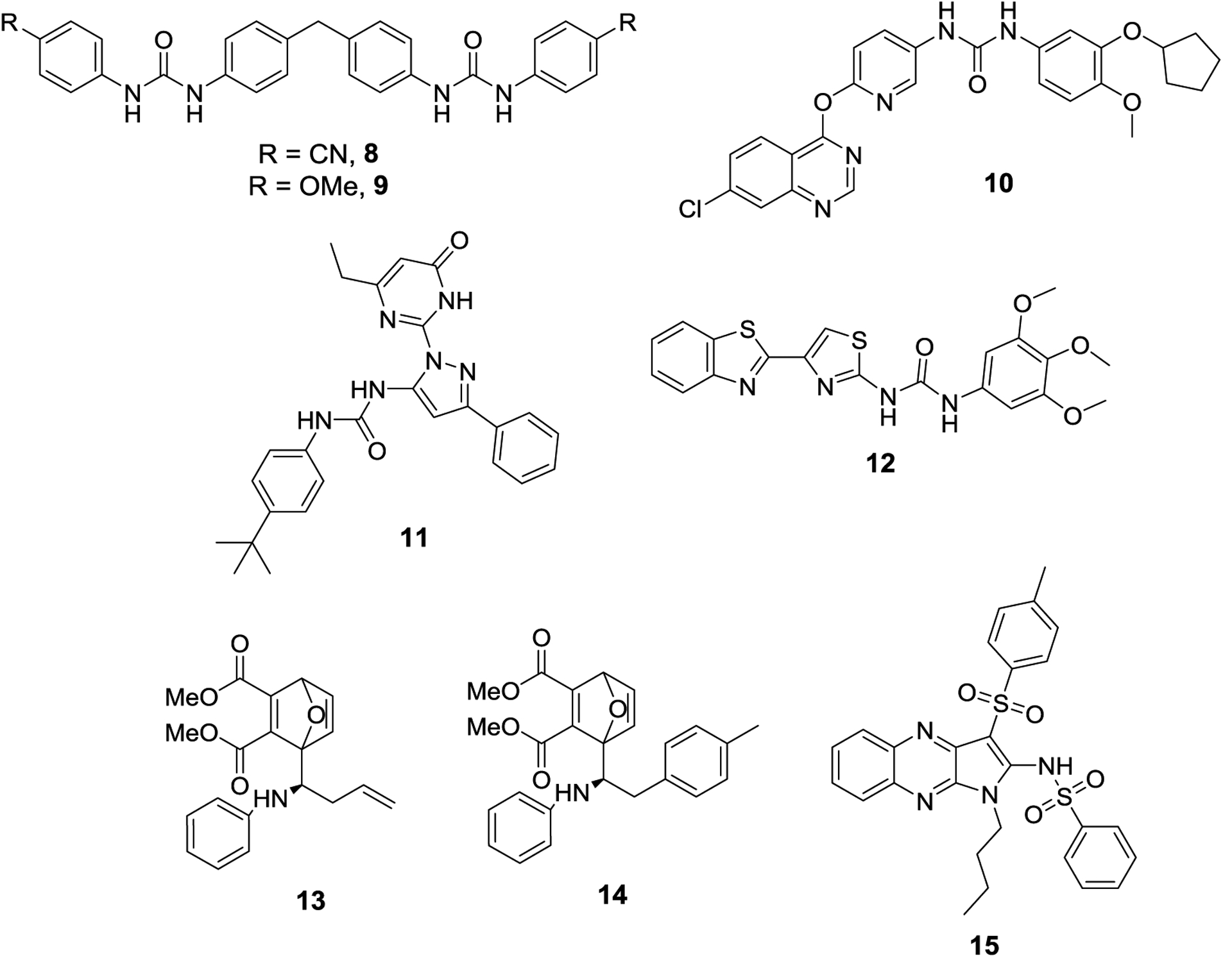
Synthetic SUMOylation inhibitors discovered from virtual screens, 8-12. Initial hit compound CID9549553 (13) and lead compound COH000 (14), and quinoxaline SAE inhibitor (15).
Figure 5.
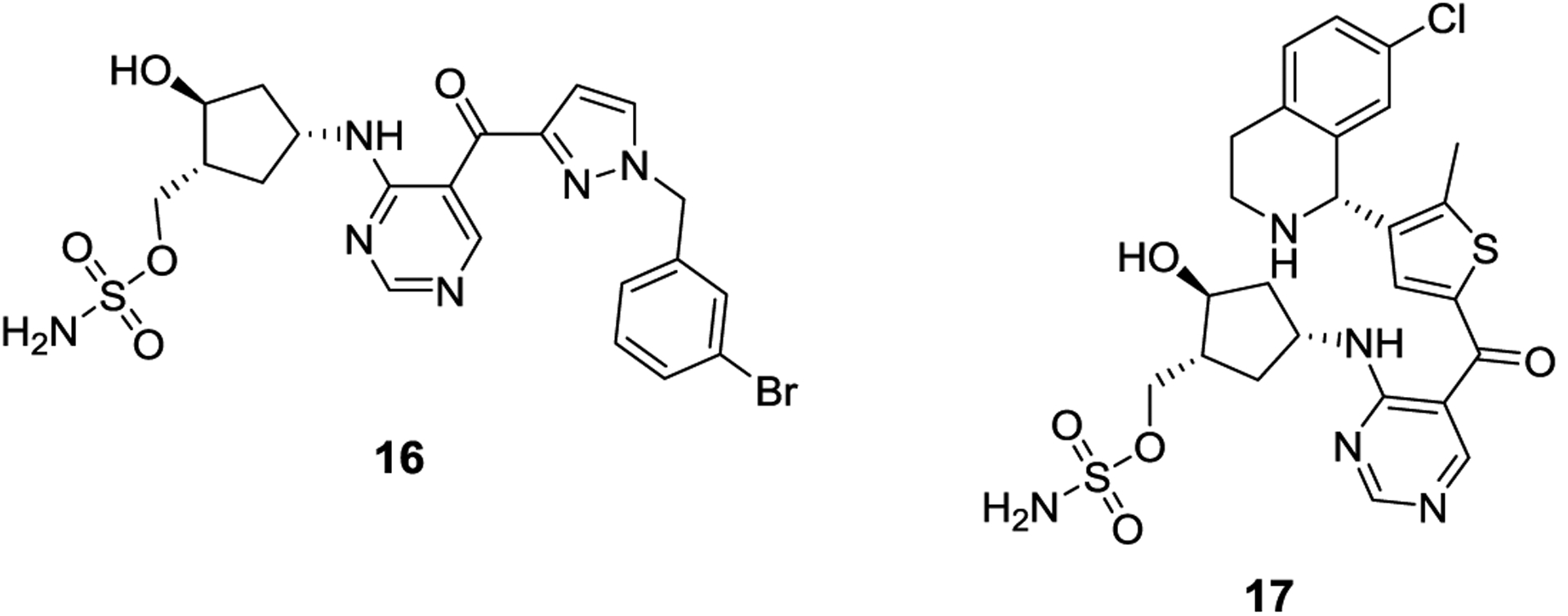
AMP mimic SAE inhibitors developed by Takeda, ML-792 (16) and TAK-981 (17).
Figure 6.
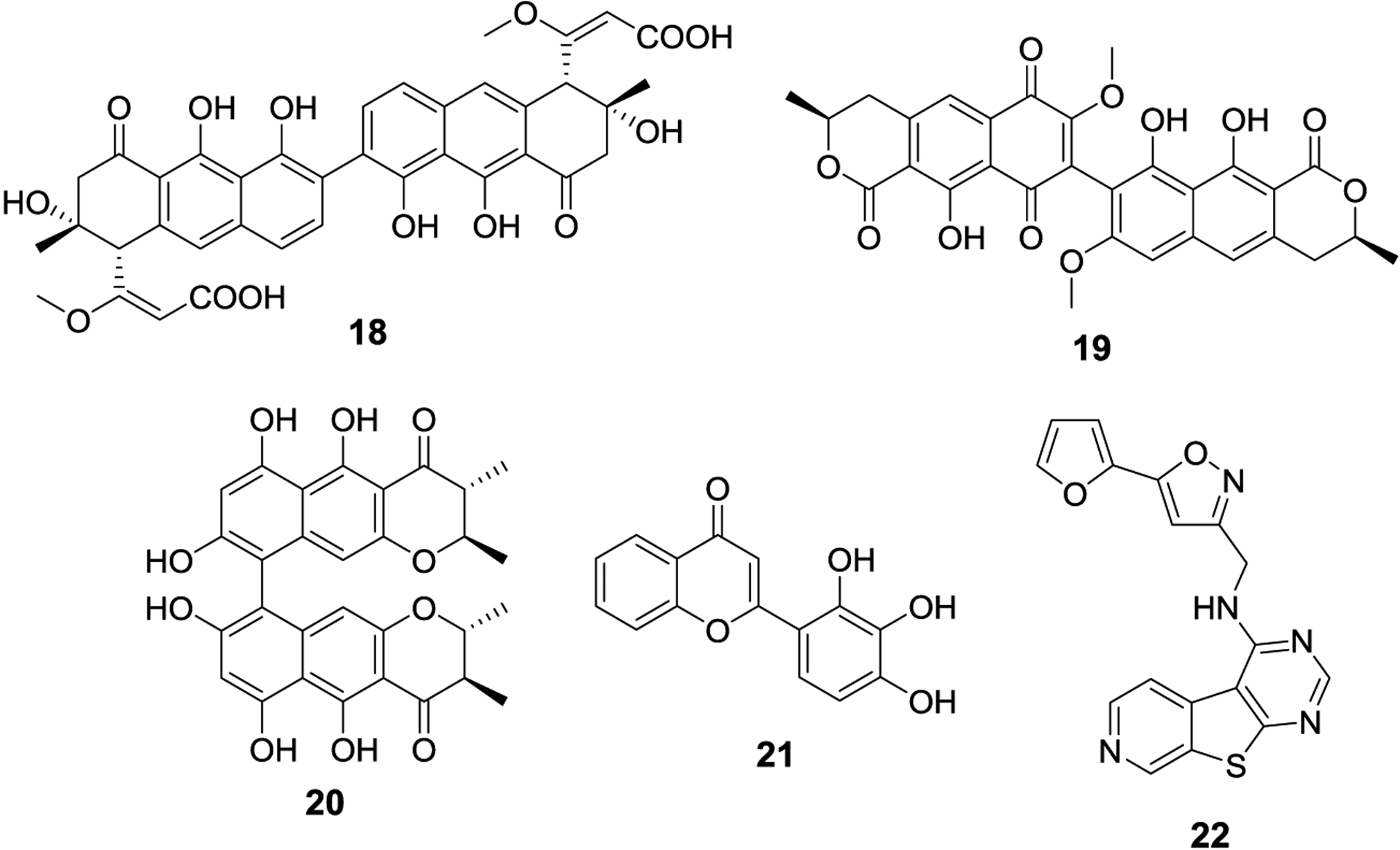
First identified natural product inhibitors of SUMO-E2, spectomycin B (18), viomellein (19), and chaetochromin A (20). Compounds discovered from developed high throughput assays, 2-D08 (21) and 22.
Figure 7.
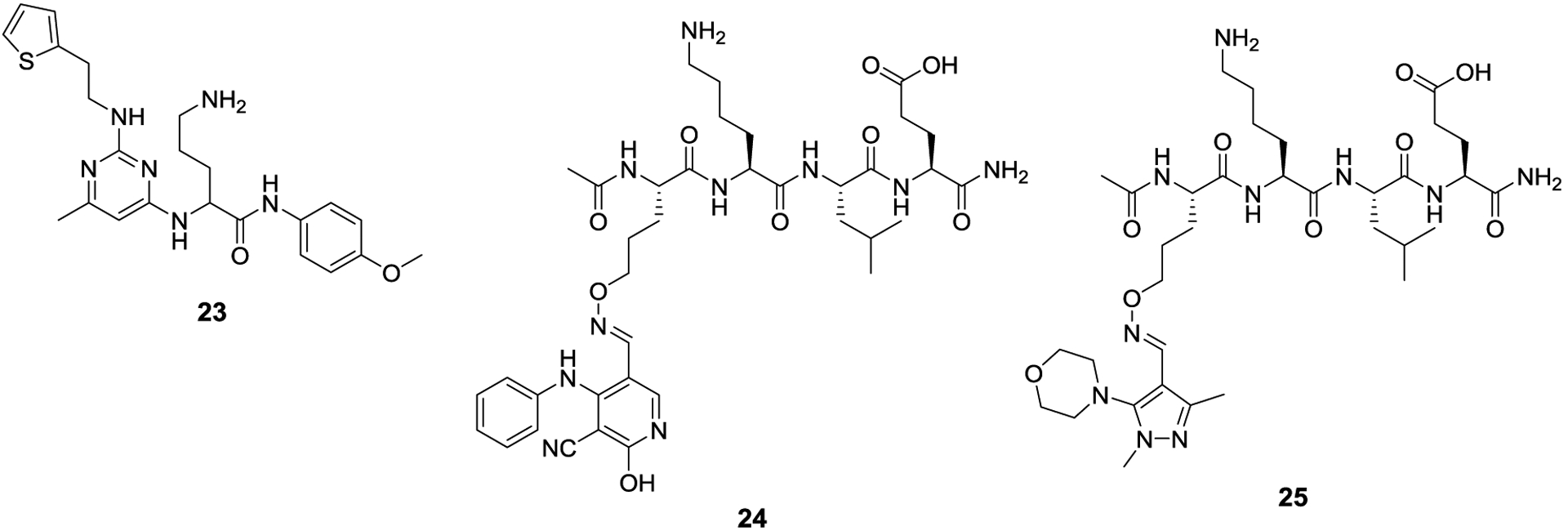
Ubc9 inhibitors that can effectively compete against target proteins as substrates for SUMOylation.
Figure 8.
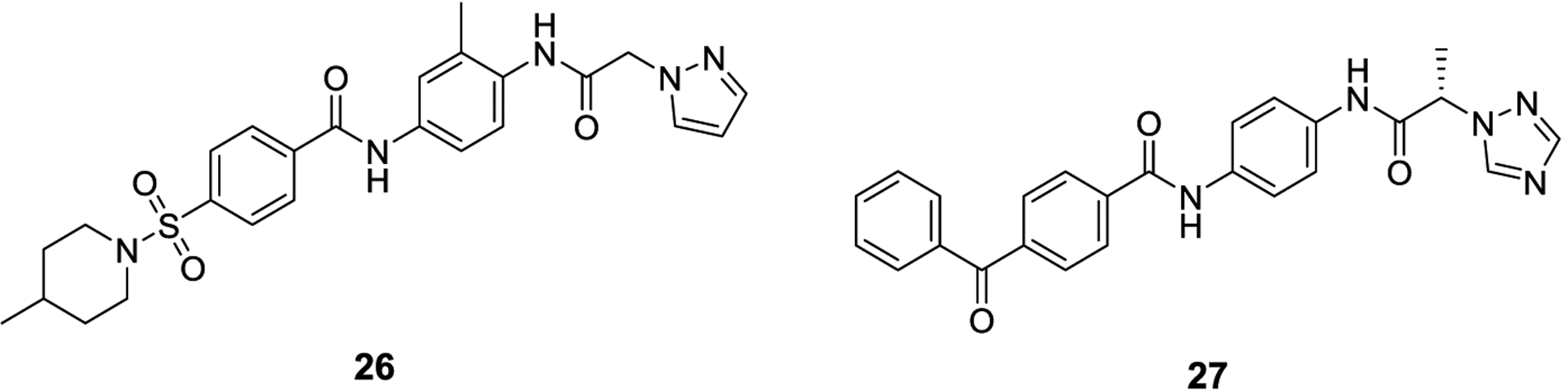
Synthetic inhibitors of Ubc9 discovered from a virtual screen, 26 and 27.
Figure 9.

Natural product SENP inhibitors, momordin Ic (28), streptonigrin (29), NSC76919 (30), NSC45384 (31), vialinin A (32), and atromentin (33).
Figure 10.
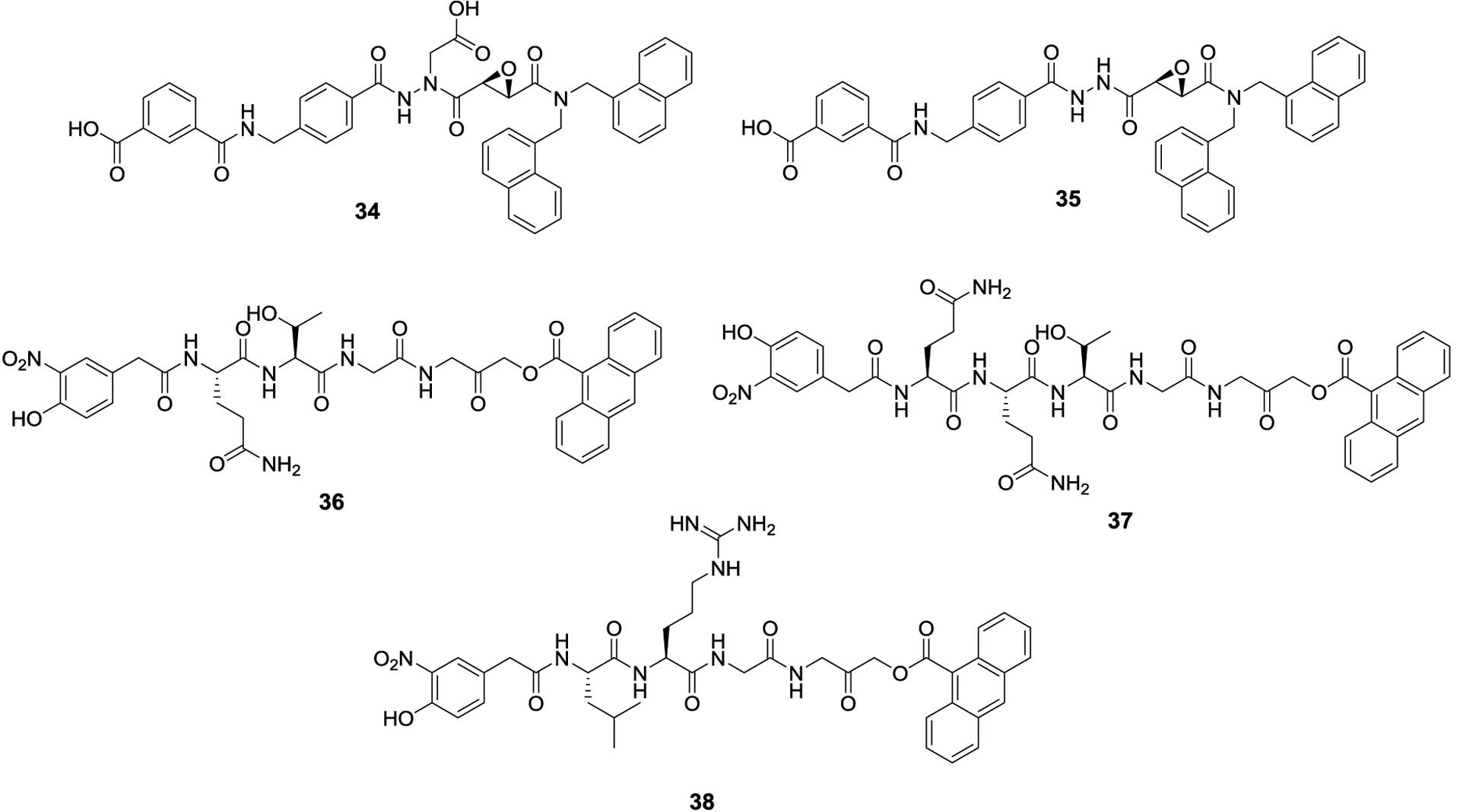
Aza-epoxide SENP inhibitors 34 and 35, and second generation SENP inhibitors 36-38.
Figure 11.
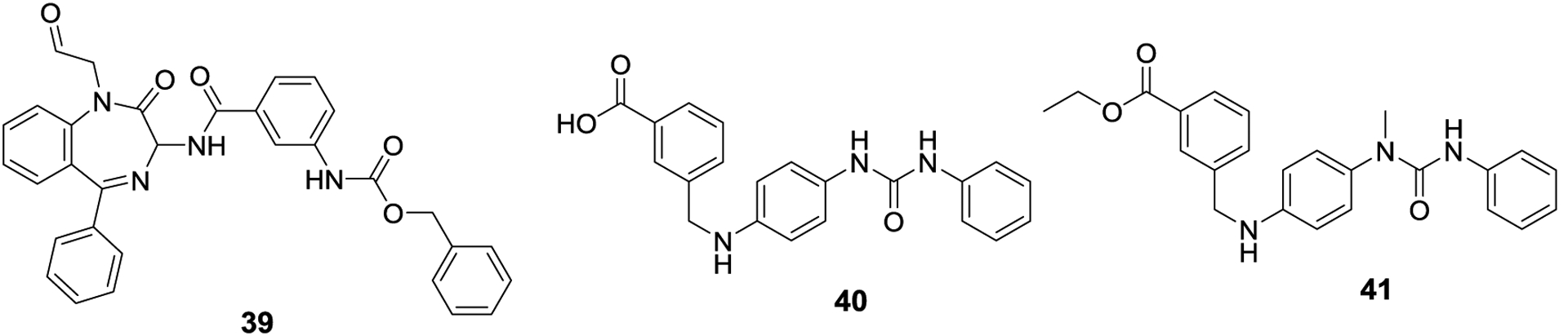
First synthetic small molecule inhibitors of SENPs, 39-41.
Figure 12.
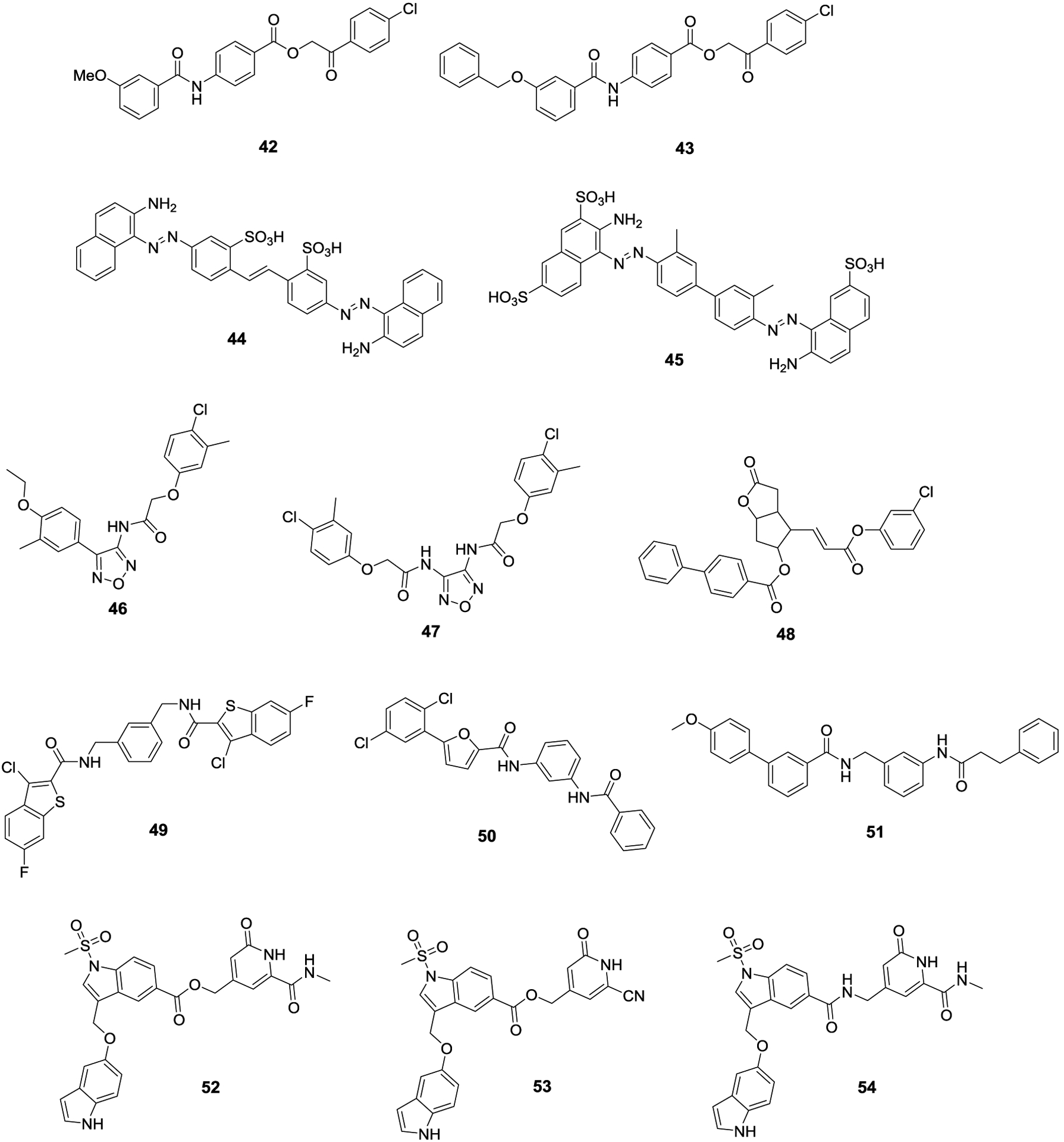
SENP inhibitors discovered through virtual screen studies 42-54.
Figure 13.
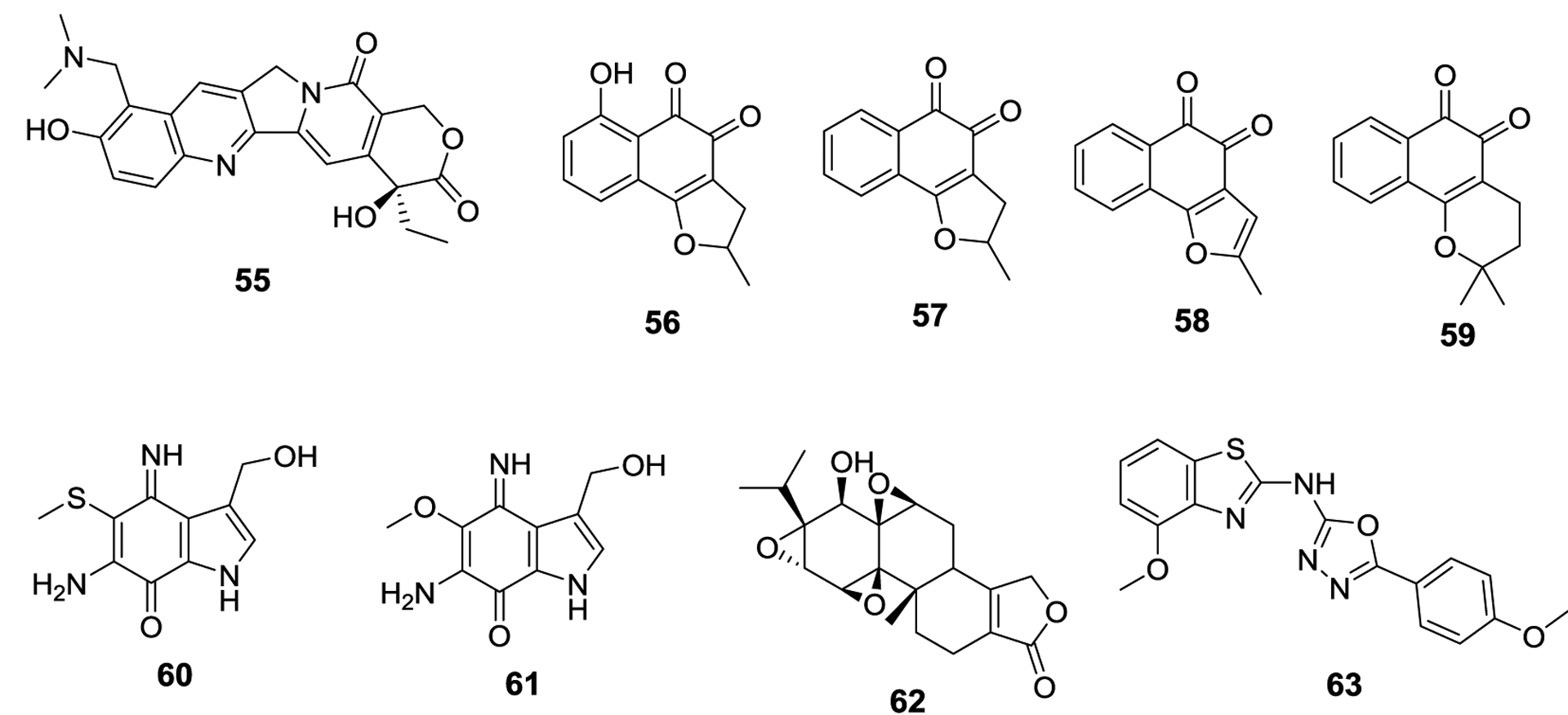
Modulators of the SUMOylation pathway with novel mechanisms of action 55-63.
References.
- 1.Venne AS, Kollipara L, Zahedi RP The next level of complexity: crosstalk of posttranslational modifications. Proteomics, 2014. 14, 513–524. [DOI] [PubMed] [Google Scholar]
- 2.Yeh ET, Gong L, Kamitani T Ubiquitin-like proteins: new wines in new bottles. Gene, 2000. 248,1–14. [DOI] [PubMed] [Google Scholar]
- 3.Han ZJ, Feng YH, Gu BH, Li YM, Chen H The post-translational modification, SUMOylation, and cancer (Review). Int J Oncol, 2018. 52, 1081–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gareau JR, Lima CD The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol, 2010. 11, 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.García-Rodríguez N, Wong RP, Ulrich HD Functions of Ubiquitin and SUMO in DNA Replication and Replication Stress. Front Genet, 2016, 7:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gill G SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev, 2004. 18, 2046–2059. [DOI] [PubMed] [Google Scholar]
- 7.Nayak A, Müller S SUMO-specific proteases/isopeptidases: SENPs and beyond. Genome Biol, 2014. 15, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seeler J, Dejean A SUMO and the robustness of cancer. Nat Rev Cancer, 2017. 17, 184–197. [DOI] [PubMed] [Google Scholar]
- 9.Chen SF, Gong C, Luo M, Yao HR, Zeng YJ, Su FX Ubc9 expression predicts chemoresistance in breast cancer. Chin J Cancer, 2011. 30, 638–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang H, Kuai X, Ji Z, Li Z, Shi R Over-expression of small ubiquitin-related modifier-1 and sumoylated p53 in colon cancer. Cell Biochem Biophys, 2013. 67, 1081–1087. [DOI] [PubMed] [Google Scholar]
- 11.Moschos S, Smith A, Mandic M et al. SAGE and antibody array analysis of melanoma-infiltrated lymph nodes: identification of Ubc9 as an important molecule in advanced-stage melanomas. Oncogene, 2007. 26, 4216–4225. [DOI] [PubMed] [Google Scholar]
- 12.Sun Z, Hu S, Luo Q, Ye D, Hu D, Chen F Overexpression of SENP3 in oral squamous cell carcinoma and its association with differentiation. Oncol Rep, 2013. 29, 1701–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding X, Sun J, Wang L, et al. Overexpression of SENP5 in oral squamous cell carcinoma and its association with differentiation. Oncol Rep, 2008. 20, 1041–1045. [PubMed] [Google Scholar]
- 14.Bawa-Khalfe T, Yeh ET SUMO Losing Balance: SUMO Proteases Disrupt SUMO Homeostasis to Facilitate Cancer Development and Progression. Genes Cancer, 2010. 1, 748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stewart MD, Ritterhoff T, Klevit RE, Brzovic PS E2 enzymes: More than Just Middle Men. Cell Res, 2016. 26, 423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nijman SM, Luna-Vargas MP, Velds A, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell, 2005. 123, 773‐786. [DOI] [PubMed] [Google Scholar]
- 17.Cappadocia L, Lima CD Ubiquitin-like Protein Conjugation: Structures, Chemistry, and Mechanism. Chem Rev, 2018. 118, 889–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saitoh H, Hinchey J Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J Biol Chem, 2000. 275, 6252‐6258. [DOI] [PubMed] [Google Scholar]
- 19.Celen AB, Sahin U Sumoylation on its 25th anniversary: mechanisms, pathology, and emerging concepts. FEBS J, 2020. 10, 1111/febs.15319. [DOI] [PubMed] [Google Scholar]
- 20.Olsen SK, Capili AD, Lu X, Tan DS, Lima CD Active site remodeling accompanies thioester bond formation in the SUMO E1. Nature, 2010. 463, 906–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pichler A, Fatouros C, Lee H, Eisenhardt N SUMO conjugation – a mechanistic view. BioMol Concepts, 2017. 8, 13–36. [DOI] [PubMed] [Google Scholar]
- 22.Tozluoğlu M, Karaca E, Nussinov R, Haliloğlu T A mechanistic view of the role of E3 in sumoylation. PLoS Comput Biol, 2010. 6(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahajan R, Delphin C, Guan T, Gerace L, Melchior F A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell, 1997. 88, 97–107. [DOI] [PubMed] [Google Scholar]
- 24.Liu X, Xu Y, Pang Z, et al. Knockdown of SUMO-activating enzyme subunit 2 (SAE2) suppresses cancer malignancy and enhances chemotherapy sensitivity in small cell lung cancer. J Hematol Oncol, 2015. 8, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qin Y, Bao H, Pan Y, et al. SUMOylation alterations are associated with multidrug resistance in hepatocellular carcinoma. Mol Med Rep, 2014. 9, 877–881. [DOI] [PubMed] [Google Scholar]
- 26.Kessler JD, Kahle KT, Sun T, et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science, 2012. 335, 348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fukuda I, Ito A, Hirai G, et al. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem Biol, 2009. 16, 133–140. [DOI] [PubMed] [Google Scholar]
- 28.Prithiviraj B, Manickam M, Singh UP, Ray AB Antifungal activity of anacardic acid, a naturally occurring derivative of salicylic acid. Can. J. Bot, 1997. 75, 207–211. [Google Scholar]
- 29.Muroi H, Kubo I Bactericidal activity of anacardic acids against Streptococcus mutans and their potentiation. J. Agric. Food Chem, 1993. 41, 1780–1783. [Google Scholar]
- 30.Hemshekhar M, Sebastin Santhosh M, Kemparaju K, Girish KS Emerging roles of anacardic acid and its derivatives: a pharmacological overview. Basic Clin Pharmacol Toxicol, 2012. 110, 122–132. [DOI] [PubMed] [Google Scholar]
- 31.Fukuda I, Ito A, Uramoto M, et al. Kerriamycin B inhibits protein SUMOylation. J Antibiot (Tokyo), 2009. 62, 221–224. [DOI] [PubMed] [Google Scholar]
- 32.Hayakawa Y, Adachi K, Iwakiri T, Imamura K, Furihata K, Seto H, Otake N Kerriamycins A B and C, New Isotetracenone Antibiotics. Agric. Biol. Chem, 1987. 51, 1397–1405. [Google Scholar]
- 33.Takemoto M, Kawamura Y, Hirohama M, et al. Inhibition of protein SUMOylation by davidiin, an ellagitannin from Davidia involucrata. 2014, J Antibiot (Tokyo). 67, 335–338. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, Ma J, Chow SC, et al. A potential antitumor ellagitannin, davidiin, inhibited hepatocellular tumor growth by targeting EZH2. Tumour Biol, 2014. 35, 205–212. [DOI] [PubMed] [Google Scholar]
- 35.Suzawa M, Miranda DA, Ramos KA, et al. A gene-expression screen identifies a non-toxic sumoylation inhibitor that mimics SUMO-less human LRH-1 in liver. Elife. 2015, 4:e09003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao B, Villhauer EB, Bhuripanyo K, Kiyokawa H, Schindelin H, Yin J SUMO-mimicking peptides inhibiting protein SUMOylation. Chembiochem, 2014. 15, 2662–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu X, Olsen SK, Capili AD, Cisar JS, Lima CD, Tan DS Designed semisynthetic protein inhibitors of Ub/Ubl E1 activating enzymes. J Am Chem Soc, 2010. 132, 1748–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar A, Ito A, Hirohama M, Yoshida M, Zhang KY Identification of Sumoylation Activating Enzyme 1 Inhibitors by Structure-Based Virtual Screening. J Chem Inf Model, 2013. 53, 809–820. [DOI] [PubMed] [Google Scholar]
- 39.Kumar A, Ito A, Hirohama M, Yoshida M, Zhang KY Identification of quinazolinyloxy biaryl urea as a new class of SUMO activating enzyme 1 inhibitors. Bioorg Med Chem Lett, 2013. 23, 5145–5149. [DOI] [PubMed] [Google Scholar]
- 40.Kumar A, Ito A, Hirohama M, Yoshida M, Zhang KY Identification of new SUMO activating enzyme 1 inhibitors using virtual screening and scaffold hopping. Bioorg Med Chem Lett, 2016. 26, 1218–1223. [DOI] [PubMed] [Google Scholar]
- 41.Li YJ, Du L, Wang J, et al. Allosteric Inhibition of Ubiquitin-like Modifications by a Class of Inhibitor of SUMO-Activating Enzyme. Cell Chem Biol, 2019. 26, 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lv Z, Yuan L, Atkison JH, et al. Molecular mechanism of a covalent allosteric inhibitor of SUMO E1 activating enzyme. Nat Commun, 2018. 9, 5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Y, Li YJ, Divlianska D, Bobkova E, Roth G, Jun PU, Khan P: Bicyclic and Tricyclic Inhibitors of SUMOylation Enzymes and Methods for their use. US Patent 20130245032 A1.Filed May 9, 2013; issuedSeptember19, 2013.
- 44.Hu C, Jiang X. The SUMO-Specific Protease Family Regulates Cancer Cell Radiosensitivity. Biomed Pharmacother, 2019. 109, 66–70. [DOI] [PubMed] [Google Scholar]
- 45.Bosque A, Nilson KA, Macedo AB, et al. Benzotriazoles Reactivate Latent HIV-1 through Inactivation of STAT5 SUMOylation. Cell Rep, 2017. 18, 1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature, 2009. 458, 732–736. [DOI] [PubMed] [Google Scholar]
- 47.Brownell JE, Sintchak MD, Gavin JM, et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell, 2010. 37, 102–111. [DOI] [PubMed] [Google Scholar]
- 48.He X, Riceberg J, Soucy T, et al. Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat Chem Biol, 2017. 13, 1164–1171. [DOI] [PubMed] [Google Scholar]
- 49.Duffey MO, England DB, Hu ZG, Ito M, Langston SP, Mcintyre C, Mizutani H, Xu H Heteroaryl compounds useful as inhibitors of SUMO activating enzyme. WO Patent 2015002994 A2. Filed July 1, 2014; issuedJanuary8, 2015.
- 50.Xu J, Footman A, Qin Y, et al. BRCA1 Mutation Leads to Deregulated Ubc9 Levels which Triggers Proliferation and Migration of Patient-Derived High Grade Serous Ovarian Cancer and Triple Negative Breast Cancer Cells. Int J Chronic Dis Ther, 2016. 2, 31–38. [PMC free article] [PubMed] [Google Scholar]
- 51.Moschos SJ, Smith AP, Mandic M, et al. SAGE and antibody array analysis of melanoma-infiltrated lymph nodes: identification of Ubc9 as an important molecule in advanced-stage melanomas. Oncogene, 2007. 26, 4216–4225. [DOI] [PubMed] [Google Scholar]
- 52.Fang S, Qiu J, Wu Z, Bai T, Guo W. Down-regulation of UBC9 increases the sensitivity of hepatocellular carcinoma to doxorubicin. Oncotarget, 2017. 8, 49783–49795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirohama M, Kumar A, Fukuda I, et al. Spectomycin B1 as a Novel SUMOylation Inhibitor that Directly Binds to SUMO E2. ACS Chem Biol, 2013. 8, 2635–2642. [DOI] [PubMed] [Google Scholar]
- 54.Staley AL, Rinehart KL. Spectomycins, new antibacterial compounds produced by Streptomyces spectabilis: isolation, structures, and biosynthesis. J Antibiot (Tokyo), 1994. 47, 1425–1433. [DOI] [PubMed] [Google Scholar]
- 55.Koyama K, Natori S. Biosynthesis of chaetochromin A, a bis(naphtho-gamma-pyrone), in Chaetomium spp. Chem Pharm Bull (Tokyo), 1989. 37, 2022–2025. [DOI] [PubMed] [Google Scholar]
- 56.Cardellina JH 2nd, Roxas-Duncan VI, Montgomery V, et al. Fungal bis-Naphthopyrones as Inhibitors of Botulinum Neurotoxin Serotype A. ACS Med Chem Lett, 2012. 3, 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kawai K, Hisada K, Mori S, Nozawa Y, Koyama K, Natori S The impairing effect of chaetochromin A and related mycotoxins on mitochondrial reaction. Proc Jpn Assoc Mycotoxicol, 1991. 33, 31–35. [Google Scholar]
- 58.Stack ME, Mislivec PB Production of xanthomegnin and viomellein by isolates of Aspergillus ochraceus, Penicillium cyclopium, and Penicillium viridicatum. Appl Environ Microbiol, 1978. 36, 552–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nomura Y, Thuaud F, Sekine D, et al. Synthesis of All Stereoisomers of Monomeric Spectomycin A1/A2 and Evaluation of Their Protein SUMOylation-Inhibitory Activity. Chemistry, 2019. 25, 8387–8392. [DOI] [PubMed] [Google Scholar]
- 60.Zhou J, Cui S, He Q, et al. SUMOylation inhibitors synergize with FXR agonists in combating liver fibrosis. Nat Commun, 2020. 11, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim YS, Nagy K, Keyser S, Schneekloth JS Jr. An Electrophoretic Mobility Shift Assay Identifies a Mechanistically Unique Inhibitor of Protein Sumoylation. Chem Biol, 2013. 20, 604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu Z, Chu H, Wen L, et al. Targeting SUMO Modification of the Non-Structural Protein 5 of Zika Virus as a Host-Targeting Antiviral Strategy. Int J Mol Sci, 2019. 20, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Varadaraj A, Mattoscio D, Chiocca S SUMO Ubc9 enzyme as a viral target. IUBMB Life, 2014. 66, 27–33. [DOI] [PubMed] [Google Scholar]
- 64.Marsh DT, Das S, Ridell J, Smid SD Structure-activity relationships for flavone interactions with amyloid β reveal a novel anti-aggregatory and neuroprotective effect of 2’,3’,4’-trihydroxyflavone (2-D08). Bioorg Med Chem, 2017. 25, 3827–3834. [DOI] [PubMed] [Google Scholar]
- 65.Zlotkowski K, Hewitt WM, Sinniah RS, et al. A Small-Molecule Microarray Approach for the Identification of E2 Enzyme Inhibitors in Ubiquitin-Like Conjugation Pathways. SLAS Discov, 2017. 22, 760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brandt M, Szewczuk LM, Zhang H, et al. Development of a high-throughput screen to detect inhibitors of TRPS1 sumoylation. Assay Drug Dev Technol, 2013. 11, 308–325. [DOI] [PubMed] [Google Scholar]
- 67.Kaiser FJ, Ludecke HJ, Weger S SUMOylation modulates transcriptional repression by TRPS1. Biol Chem, 2007. 388, 381–390. [DOI] [PubMed] [Google Scholar]
- 68.Leyva MJ, Kim YS, Peach ML, Schneekloth JS Jr. Synthetic derivatives of the SUMO consensus sequence provide a basis for improved substrate recognition. Bioorg Med Chem Lett, 2015. 25, 2146–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kumar A, Ito A, Hirohama M, Yoshida M, Zhang KY Identification of sumoylation inhibitors targeting a predicted pocket in Ubc9. J Chem Inf Model, 2014. 54, 2784–2793. [DOI] [PubMed] [Google Scholar]
- 70.Hewitt WM, Lountos GT, Zlotkowski K, et al. Insights Into the Allosteric Inhibition of the SUMO E2 Enzyme Ubc9. Angew Chem Int Ed Engl, 2016. 55, 5703–5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hoefer J, Schäfer G, Klocker H, et al. PIAS1 is increased in human prostate cancer and enhances proliferation through inhibition of p21. Am J Pathol, 2012. 180, 2097–2107. [DOI] [PubMed] [Google Scholar]
- 72.Puhr M, Hoefer J, Eigentler A, et al. PIAS1 is a determinant of poor survival and acts as a positive feedback regulator of AR signaling through enhanced AR stabilization in prostate cancer. Oncogene, 2016. 35, 2322–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data [published correction appears in Cancer Discov. 2012 Oct;2(10):960]. Cancer Discov, 2012. 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chung CD, Liao J, Liu B, et al. Specific inhibition of Stat3 signal transduction by PIAS3. Science, 1997. 278, 1803–1805. [DOI] [PubMed] [Google Scholar]
- 75.Shuai K Regulation of cytokine signaling pathways by PIAS proteins. Cell Res, 2006. 16, 196–202. [DOI] [PubMed] [Google Scholar]
- 76.Kotaja N, Karvonen U, Jänne OA, Palvimo JJ PIAS proteins modulate transcription factors by functioning as SUMO-1 ligases. Mol Cell Biol, 2002. 22, 5222–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rabellino A, Andreani C, Scaglioni PP The Role of PIAS SUMO E3-Ligases in Cancer. Cancer Res, 2017. 77, 1542–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Puhr M, Hoefer J, Neuwirt H, et al. PIAS1 is a crucial factor for prostate cancer cell survival and a valid target in docetaxel resistant cells. Oncotarget, 2014. 5, 12043–12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kaikkonen S, Jääskeläinen T, Karvonen U, et al. SUMO-specific protease 1 (SENP1) reverses the hormone-augmented SUMOylation of androgen receptor and modulates gene responses in prostate cancer cells. Mol Endocrinol, 2009. 23, 292–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dou H, Huang C, Singh M, Carpenter PB, Yeh ET Regulation of DNA repair through deSUMOylation and SUMOylation of replication protein a complex. Mol. Cell, 2010. 39, 333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wu J, Lei H, Zhang J, et al. Momordin Ic, a new natural SENP1 inhibitor, inhibits prostate cancer cell proliferation. Oncotarget, 2016. 7, 58995–59005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang J, Liu Q, Xiao H, Luo X, Liu X Suppressive effects of Momordin Ic on HepG2 cell migration and invasion by regulating MMP-9 and adhesion molecules: Involvement of p38 and JNK pathways. Toxicol In Vitro, 2019. 56, 75–83. [DOI] [PubMed] [Google Scholar]
- 83.Zhong H, Semenza GL, Simons JW, De Marzo AM Up-regulation of hypoxia-inducible factor 1alpha is an early event in prostate carcinogenesis. Cancer Detect Prev, 2004. 28, 88–93. [DOI] [PubMed] [Google Scholar]
- 84.Cheng J, Kang X, Zhang S, Yeh ET SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell, 2007. 131, 584–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ambaye N, Chen CH, Khanna S, Li YJ, Chen Y Streptonigrin Inhibits SENP1 and Reduces the Protein Level of Hypoxia-Inducible Factor 1α (HIF1α) in Cells. Biochemistry, 2018. 57, 1807–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Harris MN, Medrek TJ, Golomb FM, Gumport SL, Postel AH, and Wright JC Chemotherapy with Streptonigrin in Advanced Cancer. Cancer, 1965. 18, 49–57. [DOI] [PubMed] [Google Scholar]
- 87.Xu X, Han J, Lin R, Polyak SW, Song F Two New Piperazine-Triones from a Marine-Derived Streptomycetes sp. Strain SMS636. Mar Drugs, 2019. 17, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dreyton CJ, Anderson ED, Subramanian V, Boger DL, Thompson PR Insights into the mechanism of streptonigrin-induced protein arginine deiminase inactivation. Bioorg Med Chem, 2014. 22, 1362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ye YQ, Koshino H, Onose J, Yoshikawa K, Abe N, Takahashi S First total synthesis of vialinin A, a novel and extremely potent inhibitor of TNF-alpha production. Org Lett, 2007. 9, 4131–4134. [DOI] [PubMed] [Google Scholar]
- 90.Okada K, Ye YQ., Taniguchi K, et al. Vialinin A is a ubiquitin-specific peptidase inhibitor. Bioorg Med Chem Lett, 2013. 23, 4328–4331. [DOI] [PubMed] [Google Scholar]
- 91.Yoshioka Y, Ye YQ, Okada K, et al. Ubiquitin-specific peptidase 5, a target molecule of vialinin A, is a key molecule of TNF-α production in RBL-2H3 cells. PLoS One, 2013. 8, e80931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yoshioka Y, Namiki D, Makiuchi M, et al. Vialinin A and thelephantin G, potent inhibitors of tumor necrosis factor-α production, inhibit sentrin/SUMO-specific protease 1 enzymatic activity. Bioorg Med Chem Lett, 2016. 26, 4237–4240. [DOI] [PubMed] [Google Scholar]
- 93.Borodovsky A, Ovaa H, Meester WJ, et al. Small-molecule inhibitors and probes for ubiquitin- and ubiquitin-like-specific proteases. Chembiochem, 2005. 6, 287–291. [DOI] [PubMed] [Google Scholar]
- 94.Dobrotă C, Fasci D, Hădade ND, et al. Glycine fluoromethylketones as SENP-specific activity based probes. Chembiochem, 2012. 13,80–84. [DOI] [PubMed] [Google Scholar]
- 95.Ponder EL, Albrow VE, Leader BA, et al. Functional characterization of a SUMO deconjugating protease of Plasmodium falciparum using newly identified small molecule inhibitors. Chem Biol, 2011. 18, 711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Albrow VE, Ponder EL, Fasci D, et al. Development of small molecule inhibitors and probes of human SUMO deconjugating proteases. Chem Biol, 2011. 18, 722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Qiao Z, Wang W, Wang L, et al. Design, synthesis, and biological evaluation of benzodiazepine-based SUMO-specific protease 1 inhibitors. Bioorg Med Chem Lett, 2011. 21, 6389–6392. [DOI] [PubMed] [Google Scholar]
- 98.Uno M, Koma Y, Ban HS, Nakamura H Discovery of 1-[4-(N-benzylamino)phenyl]-3-phenylurea derivatives as non-peptidic selective SUMO-sentrin specific protease (SENP)1 inhibitors. Bioorg Med Chem Lett, 2012. 22, 5169–5173. [DOI] [PubMed] [Google Scholar]
- 99.Chen Y, Wen D, Huang Z, et al. 2-(4-Chlorophenyl)-2-oxoethyl 4-benzamidobenzoate derivatives, a novel class of SENP1 inhibitors: Virtual screening, synthesis and biological evaluation. Bioorg Med Chem Lett, 2012. 22, 6867–6870. [DOI] [PubMed] [Google Scholar]
- 100.Madu IG, Namanja AT, Su Y, Wong S, Li YJ, Chen Y Identification and characterization of a new chemotype of noncovalent SENP inhibitors. ACS Chem Biol, 2013. 8, 1435–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kumar A, Ito A, Takemoto M, Yoshida M, Zhang KY Identification of 1,2,5-oxadiazoles as a new class of SENP2 inhibitors using structure based virtual screening. J Chem Inf Model, 2014. 54, 870–880. [DOI] [PubMed] [Google Scholar]
- 102.Wen D, Xu Z, Xia L, et al. Important role of SUMOylation of Spliceosome factors in prostate cancer cells. J Proteome Res, 2014.13, 3571–3582. [DOI] [PubMed] [Google Scholar]
- 103.Zhao Y, Wang Z, Zhang J, Zhou H Identification of SENP1 inhibitors through in silico screening and rational drug design. Eur J Med Chem, 2016. 122, 178–184. [DOI] [PubMed] [Google Scholar]
- 104.Lindenmann U, Brand M, Gall F, et al. Discovery of a Class of Potent and Selective Non-competitive Sentrin-Specific Protease 1 Inhibitors. ChemMedChem, 2020. 15, 675–679. [DOI] [PubMed] [Google Scholar]
- 105.Delgado JL, Hsieh CM, Chan NL, Hiasa H Topoisomerases as anticancer targets. Biochem J, 2018. 475, 373–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Brave M, Dagher R, Farrell A, et al. Topotecan in combination with cisplatin for the treatment of stage IVB, recurrent, or persistent cervical cancer. Oncology (Williston Park), 2006. 20, 1401–1416. [PubMed] [Google Scholar]
- 107.Rapisarda A, Uranchimeg B, Scudiero DA, et al. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res, 2002. 62, 4316–4324. [PubMed] [Google Scholar]
- 108.Bernstock JD, Ye D, Gessler FA, et al. Topotecan is a potent inhibitor of SUMOylation in glioblastoma multiforme and alters both cellular replication and metabolic programming. Sci Rep, 2017.7,7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yang W, Wang L, Roehn G, et al. Small ubiquitin-like modifier 1–3 conjugation [corrected] is activated in human astrocytic brain tumors and is required for glioblastoma cell survival. Cancer Sci, 2013. 104, 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fukuda I, Hirohama M, Ito A, et al. Inhibition of protein SUMOylation by natural quinones. J Antibiot (Tokyo), 2016. 69, 776–779. [DOI] [PubMed] [Google Scholar]
- 111.Schaffner-Sabba K, Schmidt-Ruppin KH, Wehrli W, Schuerch AR, Wasley JW beta-Lapachone: synthesis of derivatives and activities in tumor models. J Med Chem, 1984. 27, 990–994. [DOI] [PubMed] [Google Scholar]
- 112.Alapati K, Muvva V Evaluation of bioactive compounds produced by Nocardia levis MK-VL_113 & Streptomyces tendae TK-VL_333 for cytotoxic activity. Indian J Med Res, 2013. 137, 391–393. [PMC free article] [PubMed] [Google Scholar]
- 113.Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ Role of quinones in toxicology. Chem Res Toxicol, 2000. 13, 135–160. [DOI] [PubMed] [Google Scholar]
- 114.Stankovic-Valentin N, Melchior F Control of SUMO and Ubiquitin by ROS: Signaling and disease implications. Mol Aspects Med, 2018. 63, 3–17. [DOI] [PubMed] [Google Scholar]
- 115.Zlotkowski K, Hewitt WM, Yan P, et al. Macrophilone A: Structure Elucidation, Total Synthesis, and Functional Evaluation of a Biologically Active Iminoquinone from the Marine Hydroid Macrorhynchia philippina. Org Lett, 2017. 19, 1726–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yan P, Ritt DA, Zlotkowski K, et al. Macrophilones from the Marine Hydroid Macrorhynchia philippina Can Inhibit ERK Cascade Signaling. J Nat Prod, 2018. 81, 1666–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huang W, He T, Chai C, et al. Triptolide inhibits the proliferation of prostate cancer cells and down-regulates SUMO-specific protease 1 expression. PLoS One, 2012. 7, e37693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Noel P, Von Hoff DD, Saluja AK, Velagapudi M, Borazanci E, Han H Triptolide and Its Derivatives as Cancer Therapies. Trends Pharmacol Sci, 2019. 40, 327–341. [DOI] [PubMed] [Google Scholar]
- 119.Wang Y, Lu JJ, He L, Yu Q Triptolide (TPL) inhibits global transcription by inducing proteasome-dependent degradation of RNA polymerase II (Pol II). PLoS One, 2011. 6, e23993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yi JM, Huan XJ, Song SS, Zhou H, Wang YQ, Miao ZH Triptolide Induces Cell Killing in Multidrug-Resistant Tumor Cells via CDK7/RPB1 Rather than XPB or p44. Mol Cancer Ther, 2016. 15, 1495‐1503. [DOI] [PubMed] [Google Scholar]
- 121.Noel P, Von Hoff DD, Saluja AK, Velagapudi M, Borazanci E, Han H Triptolide and Its Derivatives as Cancer Therapies. Trends Pharmacol Sci, 2019. 40, 327–341. [DOI] [PubMed] [Google Scholar]
- 122.Hou W, Liu B, Xu H Triptolide: Medicinal chemistry, chemical biology and clinical progress. Eur J Med Chem, 2019. 176, 378‐392. [DOI] [PubMed] [Google Scholar]
- 123.Kho C, Lee A, Jeong D, et al. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat Commun, 2015. 6, 7229. [DOI] [PMC free article] [PubMed] [Google Scholar]