

Abstract
The development of C-terminal heat shock protein (Hsp90) inhibitors has emerged as a potential treatment for cancer. Small molecules that target the mitochondria have proven to be efficacious towards cancer, as the reprogramming of mitochondrial function is often associated with oncogenesis. Herein, we developed triphenylphosphonium (TPP)-conjugated Hsp90 C-terminal inhibitors and evaluated their anticancer activity and their accumulation in the mitochondria. TPP-conjugated Hsp90 C-terminal inhibitors manifested increased activity against various cancer cell lines as compared to the parent compounds.
Keywords: Hsp90 C-terminal inhibitors, triphenylphosphonium, mitochondria, anticancer
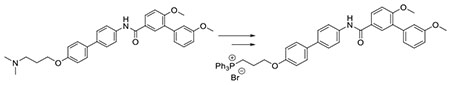
Graphical Abstract
As a member of the molecular chaperone family, heat shock protein 90 (Hsp90) is essential for the conformational maturation of nascent polypeptides and the rematuration of denatured proteins.1 Specifically, Hsp90 regulates the folding, stability, and function of more than 300 client protein subsstrates, many of which are critical for cancer cell survival and malignant transformation.2–7 In fact, Hsp90-dependent client proteins (e.g., Her2, CDK6, Akt) are directly associated with all ten hallmarks of cancer. Consequently, Hsp90 inhibitors may be beneficial for the treatment of cancer as they target multiple oncogenic pathways simultaneously.
Numerous investigations of the Hsp90 N-terminal ATP-binding site have been pursued, which ultimately produced 17 small molecules derived from natural products (eg., Figure 1) that underwent clinical evaluation.8–9 Unfortunately, the pro-survival heat shock response (HSR) that is induced upon the administration of N-terminal inhibitors is detrimental, as increased levels of Hsp90 are produced.10 In contrast, Hsp90 C-terminal inhibitors can segregate the HSR from the degradation of client proteins, which provides an opportunity to develop small molecules that overcome this undesired effect.11–19
Figure 1.
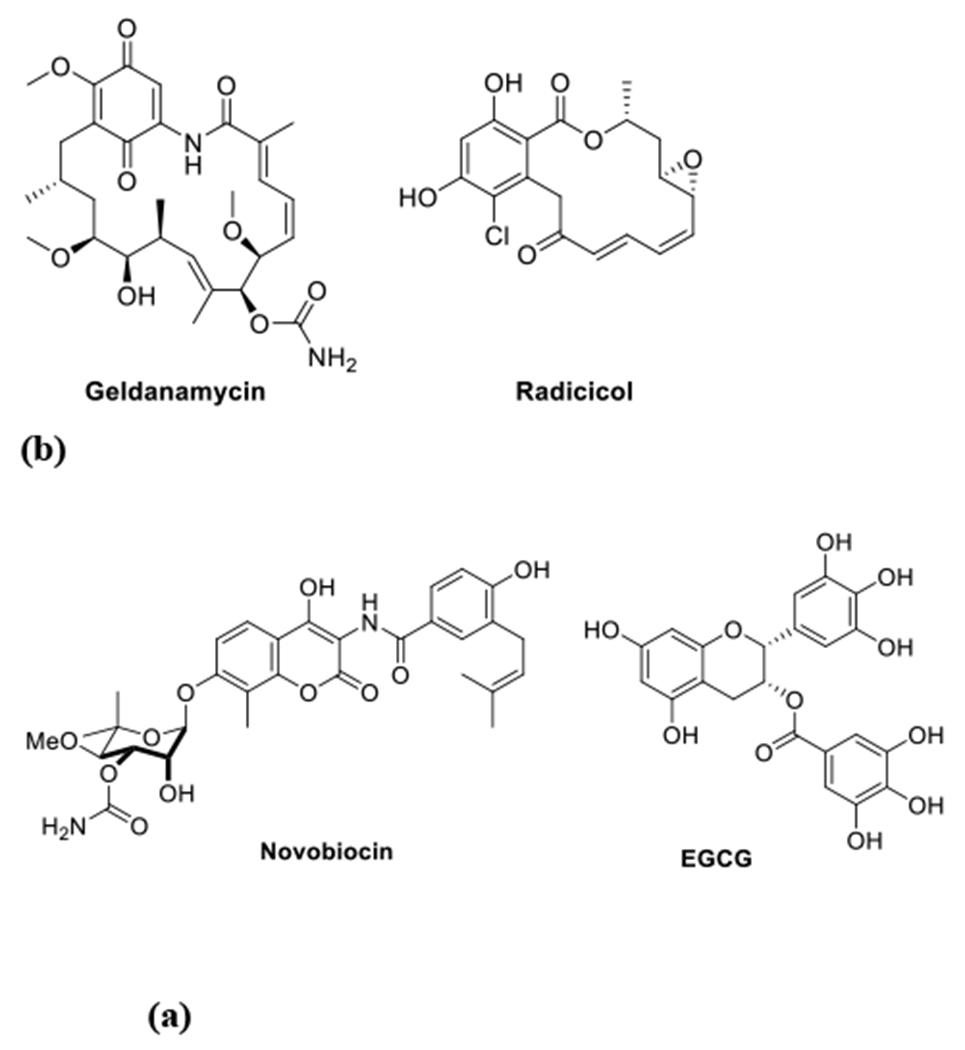
(a) Natural product-derived Hsp90 N-terminal inhibitors
(b) Natural product-derived Hsp90 C-terminal inhibitors
Novobiocin was the first Hsp90 C-terminal inhibitor identified and shown to degrade client protein substrates without induction of the HSR.20–21 Compounds based on the coumarin core of novobiocin and structurally related analogs, have been synthesized to improve their anticancer activity (eg., ~700 μM against SKBr3), which ultimately, led to molecules that manifest ~1000-fold greater activity than novobiocin.22–26 Unfortunately, no co-crystal structure of an inhibitor bound to the Hsp90 C-terminus exists, which makes the development of improved analogs challenging.
Since mitochondrial bioenergetics are required for cancer cell survival, anticancer agents that accumulate in the mitochondria may overcome some of the undesired effects resulting from inhibition of the cytosolic Hsp90 isoforms (HSP90α and HSP90β). The mitochondrial Hsp90 homolog, TRAP1, can induce cell death in cancer cells if inhibited.27 Therefore, the development of mitochondrial-targeted Hsp90 inhibitors represents a promising strategy for the development of new anti-cancer agents. Several N-terminal Hsp90 inhibitors have been modified to contain the triphenylphosphonium (TPP) salt, which is commonly used to direct compounds towards the mitochondria.28–29 The TPP containing N-terminal inhibitors demonstrated enhanced anti-proliferative activity as compared to their non-TPP conjugated analogs. Giorgio et al. also reported a TPP-conjugated molecule that manifested ~10 fold greater activity.30 Therefore, the TPP moiety was introduced onto novobiocin-based Hsp90 C-terminal inhibitors and they were evaluated for anticancer activity to determine whether such modifications enhanced Hsp90 C-terminal inhibitory activity.
Three Hsp90 C-terminal inhibitors (1, 2, and 3) were chosen for modification, based on prior evaluations and confirmation of Hsp90 inhibitory activity. Compound 1 contains the coumarin core as found in novobiocin, whereas compounds 2 and 3 were identified as more potent Hsp90 C-terminal inhibitory scaffolds in recent studies (Figure 2). 22, 31–32
Figure 2.

Hsp90 C-terminal inhibitors
Since the benzamide side chain of these molecules is required for anti-cancer activity, the TPP moiety was not introduced onto the benzamide. Instead, studies have shown that the incorporation of an ionizable N, N-dimethylaminopropyl moiety in lieu of the sugar on novobiocin can enhance activity against various cancer cell lines. Furthermore, extensive structure-activity relationship (SAR) studies have shown that modifications to the amine can produce compounds that manifest similar activity. When combined, these data supported modification to the sugar region of novobiocin is solvent-accesible and represents a location at which replacement of the amine can occur along with TPP to give a mitochondrial-targeted Hsp90 C-terminal inhibitor (Scheme 1).
Scheme 1.
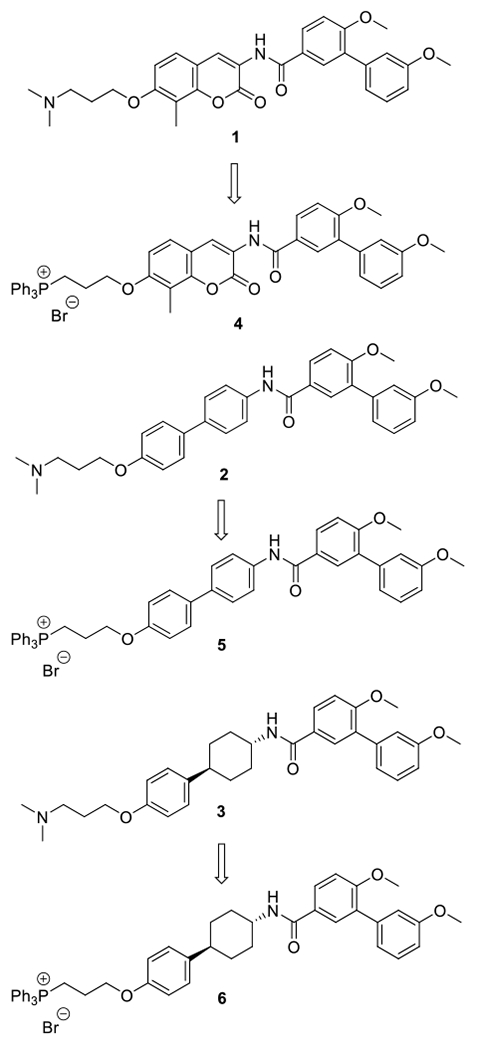
Design of TPP conjugated Hsp90 C-terminal inhibitors
Syntheses of the proposed analogs began via their corresponding phenols, 7, 9 and 11, which have been previously reported.22, 31–32 In general, 1-bromoethanol was attached to the phenol via a Mitsunobu coupling reaction. The resulting alkyl bromides were then heated at reflux in the presence of triphenyl phosphine in acetonitrile until the corresponding TPP salts precipitated from solution to give 4, 5 and 6, respectively. (Scheme 2).
Scheme 2. Synthesis of TPP conjugated Hsp90 C-terminal inhibitors 4, 5, 6.
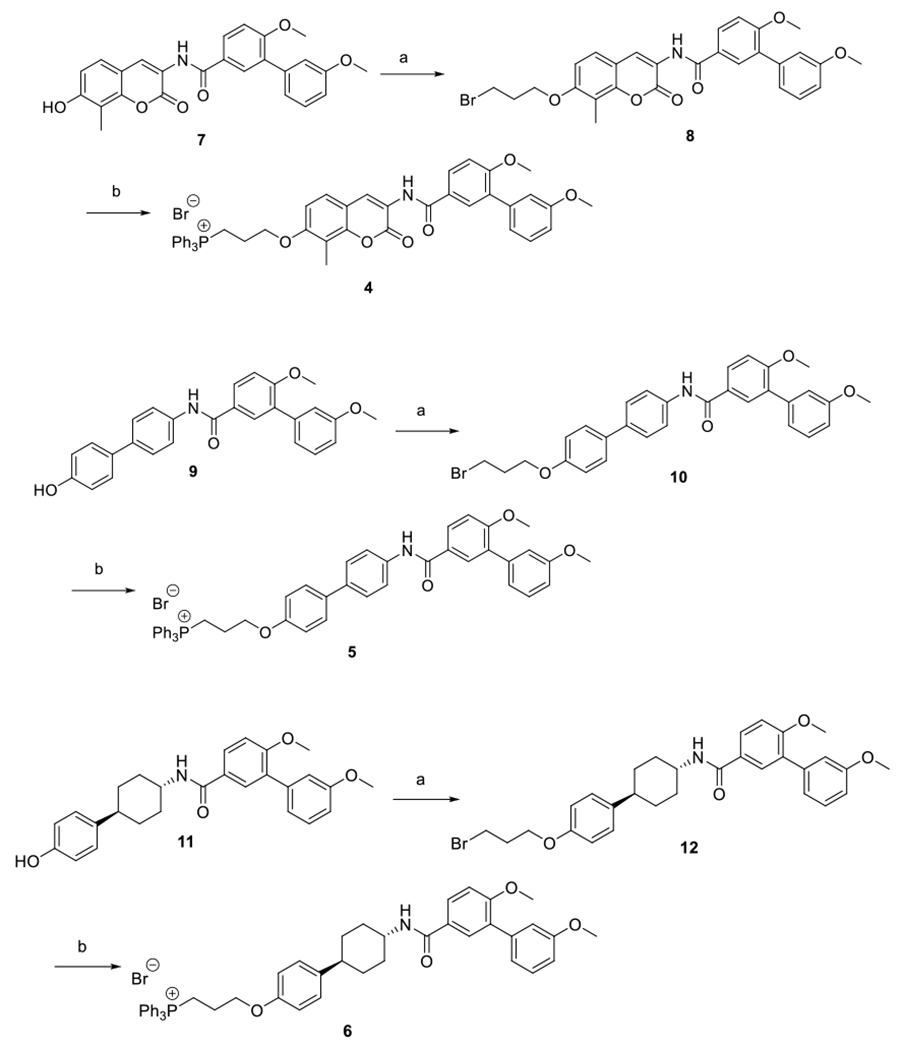
(a) 1-bromoethanol, DIAD, PPh3, THF, 0 °C to r.t., overnight, 30-50% (b) PPh3, acetonitrile, reflux, overnight 25-45%
An MTT assay was conducted to evaluate the anticancer activities of 4, 5 and 6, and all of the compounds were found to exhibit efficacious activity against various cancer cell lines (SKBr3, MCF-7, PC3 and HCT-116) as shown in Table 1. A two-fold increase in anti-proliferative activity was observed for 4, 5 and 6, against the MCF-7 cell line when compared to the amino-counterparts 1, 2 and 3, respectively. Compound 5 manifested ~ 10-fold increased activity against the SKBr3 cell line when compared to amine 2 (Table 1).
Table 1.
Cytotoxicity of 4, 5 and 6 in comparison of their parent compounds
| MCF-7 (μM) | SKBr3 (μM) | PC3 (μM) | HCT-116 (μM) | |
|---|---|---|---|---|
| 4 | 0.80 ±0.07 | 0.31 ± 0.19 | 1.94 ± 0.84 | 0.64 ±0.21 |
| 1 | 1.80 ± 1.48 | 0.85 ± 0.10 | 1.01 ± 0.06 | 0.83 ±0.20 |
| 5 | 0.93 ±0.25 | 0.21 ±0.05 | 2.94 ± 1.32 | 0.45 ± 0.15 |
| 2 | 2.14 ± 0.75 | 2.03 ± 0.23 | 3.53 ± 0.91 | 1.49 ± 0.47 |
| 6 | 0.17 | 0.23 ±0.04 | 1.56 ±0.19 | 0.38 ±0.06 |
| 3 | 0.34 ±0.02 | 0.25 ±0.05 | N.D. | N.D. |
Compounds 4, 5 and 6 were also evaluated via western blot analyses of SKBr3 cell lysates following incubation for 24h in an effort to confirm that TPP conjugated compounds localized in the mitochondria. Since GDA is a known Hsp90 inhibitor and resides in the cytoplasm, client protein degradation (such as Her2 and AKT) was observed. In contrast, 4, 5 and 6 did not degrade related proteins at concentrations that mirrored the cellular IC50 value. Previous results showed that parent compounds 1, 2, and 3 function as Hsp90 C-terminal inhibitors and induce degradation of these client proteins.22, 31–32 Consequently, we proposed that installation of TPP group to these parent compounds will redirect them into the mitochondria. As a consequence, cytoplasmic client proteins will not be degraded by the mitochondrial-targeted Hps90 inhibitors. The TPP conjugated compounds continued to produce steady levels of Hsp90 and Hsp70, which is a hallmark of Hsp90 C-terminal inhibition.
A mitochondria-targeted accumulation assay was also performed to quantify the relative concentrations of 4, 5 and 6. These compounds all showed significant accumulation in the mitochondria as compared to the cytoplasm. Surprising, the parent compounds also showed similar levels of accumulation in the mitochondria. It appears that the N,N-dimethyl amino moiety present in the parent compound is ionized under physiological conditions and appears to facilitate their transport into the mitochondria, much like the TPP moiety. Nonetheless, compounds 4 and 6 showed increased concentrations of mitochondria accumulation as compared to their corresponding parent compounds. The TPP moiety not only can drive these compounds into the mitochondria, but is capable of having similar effects as other ionizable functional groups.
In this study, we developed three TPP conjugated Hsp90 C-terminal inhibitors and evaluated their antiproliferative activity, as well as their mitochondria accumulation. Experimental results clearly showed these modifications produced compounds that manifested increased anti-proliferative activity. A mitochondria accumulation assay confirmed that TPP conjugation with Hsp90 C-terminal inhibitors produced compounds that target the mitochondria. Future research will be conducted to generate additional compounds that contain TPP in an effort to enhance activity and to study the consequences mechanism of mitochondrial inhibition.
Figure 3. Western bolt for TPP conjugated Hsp90 C-terminal inhibitors 4, 5, 6.
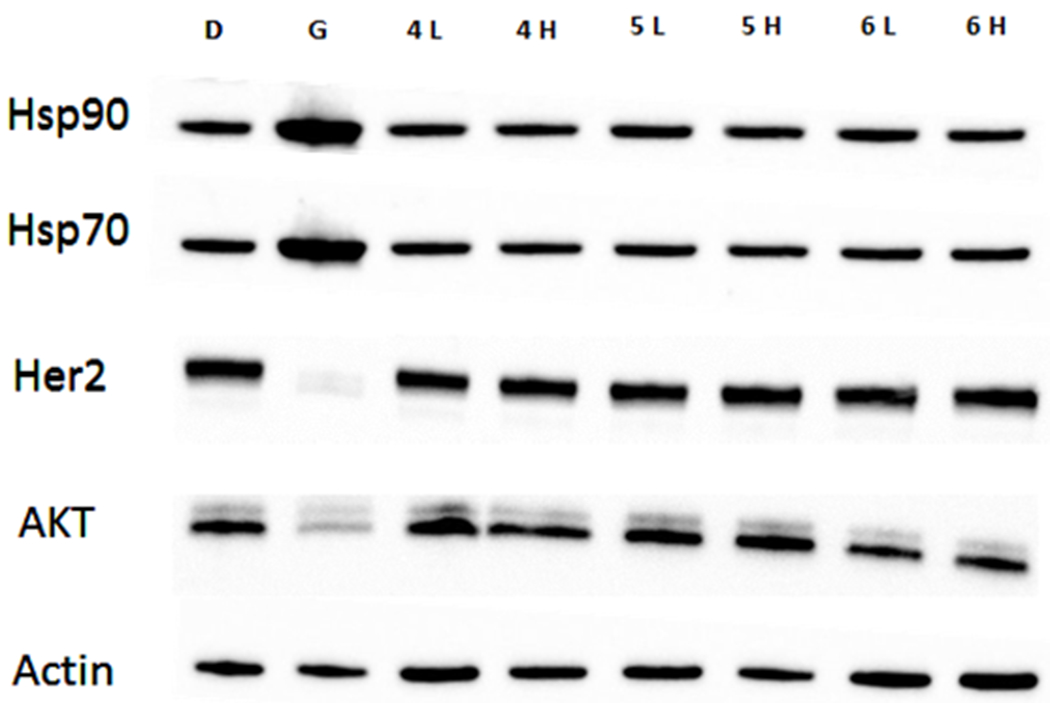
D: DMSO, G: geldanamycin (GDA), 4, 5, 6 showed as low & high concentrations, L: ½ of IC50, H: 5 × IC50.
Figure 4.
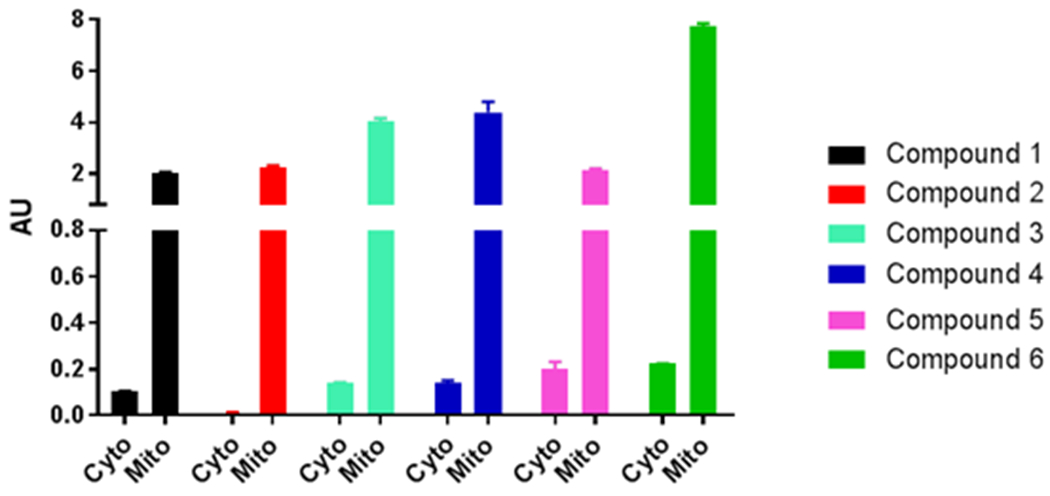
Mitochondrial targeting of compounds. LC-MS quantification of compound 1 (black), compound 2 (red), compound 3 (cyan), compound 4 (blue), compound 5 (magenta), compound 6 (green) in isolated fractions of 24 h treated PC3 cells. Cyto, cytosol; mito, mitochondria and AU is the ratio of the compound and the internal control.
Acknowledgments
Funding Sources
This work was supported by grants from The National Institutes of Health to B.S.J.B [CA120458].
Footnotes
The authors declare no competing financial interest
REFERENCES
- 1.Khandelwal A; Crowley VM; Blagg BSJ Natural product inspired N-Terminal HSP90 Inhibitors: From Bench to Beside? Med. Res. Rev 2016, 36, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hall JA; Forsberg LK; Blagg BSJ Alternative approaches to HSP90 modulation for the treatment of cancer. Fut. Med. Chem 2014, 6, 1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao H; Mary LM; Blagg BSJ HSP90 modulation for the treatment of Alzheimer’s disease. Adv. Pharmacol 2012, 64, 1–25. [DOI] [PubMed] [Google Scholar]
- 4.Karagoz GE; Rudiger SG; Hsp90 interaction with clients. Trends Biochem Sci 2015, 40, 117. [DOI] [PubMed] [Google Scholar]
- 5.Yoshihiko M; Hitoshi N; Len N Current Pharmaceutical Design 2013, 19, 347.22920906 [Google Scholar]
- 6.Pratt WB; Morishma Y; Gestwicki JE; Lieberman AP; Osawa Y Experimental biology and medicine 2014, 239, 1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trepel J; Mollapour M; Giaccone G; Neckers L Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neckers L; Trepel JB Clinical cancer research: an official journal of the American Association for Cancer Research 2014, 239, 1405. [DOI] [PubMed] [Google Scholar]
- 9.Neckers L; Workman P Clinical cancer research: an official journal of the American Association for Cancer Research 2012, 18, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong Banerji, U; Tavana B; George GC; Aaron J; Kurzock R Cancer Treatment Reviews 2013, 39, 375–387 [DOI] [PubMed] [Google Scholar]
- 11.Shelton SN; Shawgo ME; Comer SB; Lu Y; Donnelly AC; Szabla K; Tanol M; Vielhauer GA; Rajewski RA; Matts RL; Blagg BSJ; Robertson JD KU135, a novel novobiocin-derived C-terminal inhibitor of HSP90, exerts potent antiproliferative effects in human leukemic cells. Mol. Pharmacol 2009, 76, 1314–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conde R; Belak ZR; Nair M; O’Carroll RF; Ovsenek N Modulation of Hsf1 activity by novobiocin and geldanamycin. Biochem. Cell Biol 2009, 87, 845–851 [DOI] [PubMed] [Google Scholar]
- 13.Zhao H; Blagg BSJ In: Inhibitors of Molecular Chaperones as Therapeutic Agents. Timothy D, Machajewski GZ, editors. London: RSC Publishing; 2014. p. 259 [Google Scholar]
- 14.Cohen MS; Mukerji R; Samadi AK; Zhang X; Zhao H; Blagg BSJ Novel C-terminal Hsp90 inhibitor for head and neck squamous cell cancer (HNSCC) with in vivo efficacy and improved toxicity profiles compared with standard agents. Ann. Surg. Oncol 2012, 19, S483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burlison JA; Avila C; Vielhauer G; Lubbers DJ; Holzbeierlein J; Blagg BSJ Development of novobiocin analouges that manifest anti-proliferative activity against several cancer cell lines. J. Org. Chem 2008, 73, 2130–2137. [DOI] [PubMed] [Google Scholar]
- 16.Burlison JA; Blagg BSJ Synthesis and Evaluation of Coumermycin A1 Analogues that Inhibit the Hsp90 Protein Folding Machinery. Org. Lett 2006, 8, 4855–4858. [DOI] [PubMed] [Google Scholar]
- 17.Burlison JA; Neckers L; Smith AB; Maxwell A; Blagg BSJ Novobiocin: Redesigning a DNA Gyrase Inhibitor for Selective Inhibitor for Selective Inhibition of Hsp90. J. Am. Chem. Soc 2006, 128, 15529–15536. [DOI] [PubMed] [Google Scholar]
- 18.Donnelly A; Mays JR; Burlison JA; Nelson JT; Vielhauer G; Holzbeierlein J; Blagg BSJ The Design, Synthesis, and Evaluation of Coumarin Ring Derivatives of the Novobiocin Scaffold that exhibit Antiproliferative activity. J. Org, Chem 2008, 73, 8901–8920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu XM; Shen G; Neckers L; Blake H; Holzbeierlein J; Cronk B; Blagg BSJ HSP90 inhibitors identified from a library of novobiocin analogues. J. Am. Chem. Soc 2005, 127, 12778–12779. [DOI] [PubMed] [Google Scholar]
- 20.Burlison JA; Neckers L; Smith AB; Maxwell A; Blagg BSJ Novobiocin: Redesigning a DNA Gyrase Inhibitor for Selective Inhibitor for Selective Inhibition of Hsp90. J. Am. Chem. Soc 2006, 128 (48), 15529–15536. [DOI] [PubMed] [Google Scholar]
- 21.Neckers L Curr. Top Med Chem 2006, 6 (11), 1163–1171. [DOI] [PubMed] [Google Scholar]
- 22.Zhao H; Donnelly AC; Kusuma BR; Brandt GEL; Brown D; Rajewski RA; Vielhauer G; Holzbeierlein J; Cohen MS; Blagg BSJ J. Med. Chem 2011, 54, 3839–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao H; Garg G; Blagg BSJ Eur. J. Med. Chem 2014, 442–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burlison J; Avila C; Blagg BSJ J. Org. Chem 2008, 73, 2130–2137. [DOI] [PubMed] [Google Scholar]
- 25.Zhao H; Moroni E; Colombo G; Blagg BSJ ACS Med. Chem. Lett 2014, 5, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donnelly AC; Mays JR J. Org. Chem 2008, 73, 8901–8920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rondanin R; Lettini G; Oliva P; Baruchello R; Costantini C; Trapella C; Simoni D; Bernardi T; Sisinni L; Pietrafesa M; Ponterini G; Costi MP; Vignudelli T; Luciani R; Matassa DS; Esposito F; Landriscina M Bioorg. Med. Chem. Lett 2018, 28 (13), 2289–2293. [DOI] [PubMed] [Google Scholar]
- 28.Lee C; Park HK; Jeong H; Lim J; Lee AJ; Cheon KY; Kim CS; Thomas AP; Bae B; Kim ND; Kim SH; Suh PG; Ryu JH; Kang BH J. Am. Chem. Soc 2015, 137 (13), 4358–4367. [DOI] [PubMed] [Google Scholar]
- 29.Bryant KG; Chae YC; Martinez RL; Gordon JC; Elokely KM; Kossenkov AV; Grant S; Childers WE; Abou-Gharbia M; Altieri DC Oncotarget 2017, 8 (68), 112184–112198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D’ Annessa L; Sattin S; Tao J; Pennati M; Sanchez-Martin C; Moroni E; Rasola A; Zaffaroni N; Agard DA; Bernardi A; Colombo G Chem. A. Eur. J 2017, 23 (22), 5188–5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garg G; Forsberg LK; Zhao H; Blagg BSJ Chem. Eur. J 2017, 23, 16574–16585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao H; Blagg BSJ PCT int. Appl, 2015070091 [Google Scholar]