

Abstract
Rationale: Data on the molecular mechanisms that regulate platelet–pulmonary endothelial adhesion under conditions of hypoxia are lacking, but may have important therapeutic implications.
Objectives: To identify a hypoxia-sensitive, modifiable mediator of platelet–pulmonary artery endothelial cell adhesion and thrombotic remodeling.
Methods: Network medicine was used to profile protein–protein interactions in hypoxia-treated human pulmonary artery endothelial cells. Data from liquid chromatography–mass spectrometry and microscale thermophoresis informed the development of a novel antibody (Ab) to inhibit platelet–endothelial adhesion, which was tested in cells from patients with chronic thromboembolic pulmonary hypertension (CTEPH) and three animal models in vivo.
Measurements and Main Results: The protein NEDD9 was identified in the hypoxia thrombosome network in silico. Compared with normoxia, hypoxia (0.2% O2) for 24 hours increased HIF-1α (hypoxia-inducible factor-1α)–dependent NEDD9 upregulation in vitro. Increased NEDD9 was localized to the plasma-membrane surface of cells from control donors and patients with CTEPH. In endarterectomy specimens, NEDD9 colocalized with the platelet surface adhesion molecule P-selectin. Our custom-made anti-NEDD9 Ab targeted the NEDD9–P-selectin interaction and inhibited the adhesion of activated platelets to pulmonary artery endothelial cells from control donors in vitro and from patients with CTEPH ex vivo. Compared with control mice, platelet–pulmonary endothelial aggregates and pulmonary hypertension induced by ADP were decreased in NEDD9−/− mice or wild-type mice treated with the anti-NEDD9 Ab, which also decreased chronic pulmonary thromboembolic remodeling in vivo.
Conclusions: The NEDD9–P-selectin protein–protein interaction is a modifiable target with which to inhibit platelet–pulmonary endothelial adhesion and thromboembolic vascular remodeling, with potential therapeutic implications for patients with disorders of increased hypoxia signaling pathways, including CTEPH.
Keywords: thrombosis, platelets, hypoxia
At a Glance Commentary
Scientific Knowledge on the Subject
Hypoxia signaling pathways are upregulated in the pulmonary endothelium in clinical pathophenotypes characterized by pathogenic platelet–endothelial interactions and thromboembolic remodeling. However, the molecular mechanisms that regulate platelet-pulmonary endothelial biology under hypoxic conditions in pulmonary thromboembolic disorders are poorly understood.
What This Study Adds to the Field
We show hypoxia inducible factor-1-dependent upregulation of NEDD9 in pulmonary artery endothelial cells. We focused on a functional NEDD9 peptide expressed on the extracellular surface of pulmonary artery endothelial cells, which participates in a previously unrecognized protein-protein interaction with platelet P-Selectin. By developing a novel anti-NEDD9 antibody to antagonize NEDD9-P-Selectin complex formation, we inhibited platelet–endothelial cell adhesion mediated by hypoxia or inflammation in vitro, prevented acute platelet–endothelial aggregate formation and pulmonary hypertension in vivo, and decreased in situ chronic thromboembolic remodeling in rats. Our anti-NEDD9 antibody also inhibited adhesion of platelets to endothelial cells isolated from patients with chronic thromboembolic pulmonary hypertension, and we observed that NEDD9 is increased in endarterectomy specimens from patients. Overall, this study identifies NEDD9 as a modifiable mediator of platelet–endothelial interactions with potential therapeutic relevance to diseases characterized by hypoxia, activated platelets, and pulmonary thromboembolic remodeling.
Interaction between activated platelets and the endothelium is a key pathogenetic event underlying numerous pulmonary vascular diseases (1). Upregulation of hypoxia signaling pathways in human pulmonary artery endothelial cells (HPAECs) is also common to clinical disorders characterized by pulmonary thromboembolic arterial remodeling (2). The ramifications of hypoxic vascular injury on endothelial dysfunction, including proliferation and fibrosis, have been reported in the lung circulation (3). However, few data are available on the molecular mechanisms that regulate platelet–pulmonary endothelial interactions under hypoxic conditions.
In carcinoma cell lines, hypoxia increases the hypoxia-inducible factor-1α– dependent transcriptional regulation of NEDD9 (4), which is a scaffolding protein that controls numerous protein–protein interactions involved in metastasis. In turn, metastasis is associated with abnormal platelet–endothelial adhesion (5), thereby suggesting NEDD9 may be unrecognized in the pathogenesis of prothrombotic pathophenotypes (6). Increased vascular hypoxia–inducible factor-1α expression is observed after luminal pulmonary embolism (7), often the antecedent event to chronic thromboembolic pulmonary hypertension (CTEPH) (8), and in cells from CTEPH pulmonary endarterectomy specimens (9). Furthermore, accumulation of pulmonary endothelial NEDD9 promotes fibrotic vascular remodeling, endothelial dysfunction, and pulmonary hypertension (10). On the basis of these converging observations, we hypothesized that NEDD9 upregulation by hypoxia is an unrecognized mechanism that regulates pathogenic platelet–pulmonary endothelial adhesion.
Methods
Additional methods are available in the online supplement.
Cell Culture and Treatments
Details for all cell types and biological reagents used in this study are provided in Tables E1 and E2 in the online supplement, respectively. Primary HPAECs, other endothelial cell types, and human pulmonary artery smooth muscle cells were grown to confluence using Endothelial Basal Medium-2 (Lonza) and Smooth Muscle Growth Medium-2 (Lonza), respectively, unless otherwise specified. Human brain microvascular endothelial cells were selected as a control owing to the unique consequences of hypoxia on cerebral perfusion, the importance of endothelial dysfunction in cerebral bleeding, and intracranial hemorrhage that is reported in some patients prescribed anticoagulant therapies to treat thromboembolic pulmonary vascular diseases (11, 12). All media were supplemented with a cell type–specific Bulletkit (Lonza). C57BL/6 mouse primary pulmonary artery endothelial cells and human brain microvascular endothelial cells were grown to confluence using Cell Biologics Endothelial Cell Medium with Kit (Cell Biologics). Cells (passages 3–8) were incubated at 37°C in 5.0% CO2 and dissociated using 0.5% trypsin/ethylenediaminetetraacetic acid. In selected experiments, cells were treated with hypoxia (10%, 2%, 1%, or 0.2% O2) using a tightly sealed modular hypoxia chamber incubated at 37°C for 24 hours, as reported previously (10).
Platelet–Endothelial Cell Adhesion Assay
Endothelial cells were seeded on a 96-well opaque-bottom plate (Thermo Fisher Scientific) and grown to 100% confluence at 37°C and at 5.0% CO2. Human platelets from healthy volunteers were isolated (Partners Institutional Review Board [IRB] #2016P001640) and fluorescently labeled with 5-chloromethylfluorescein diacetate before activation with 10 μM of TRAP (thrombin receptor agonist peptide) (Sigma), as described previously (13, 14). Platelet isolation methods are provided in the online supplement. In some experiments, HPAECs were stimulated with the inflammatory cytokine IL-6 (25 ng/ml) (Peprotech) for 24 hours, followed by treatment with an anti–P-selectin antibody (Ab) (10 μg/ml) (Sigma), anti–P-selectin glycoprotein ligand-1 Ab (clone KPL-1) (15 μg/ml) (15) (Sigma), or purified t-PA (tissue plasminogen activator) (15 ng/ml) (16) (Abcam) at 37°C in a water bath for 15 minutes before incubating with HPAECs for 45 minutes. The platelet number was counted by fluorescence-activated cell sorting and adjusted to 2 × 108/ml and then incubated with cell monolayers for 45 minutes at 37°C and at 5.0% CO2. The total fluorescence (485/535 nm) was measured using a multilabel counter plate reader (Molecular Devices) before and after three serial washes with phosphate-buffered saline. The percentage of platelet adhesion was calculated as follows: (remaining fluorescence – blank) ÷ (total fluorescence – blank) × 100.
Human CTEPH Samples
Demographic and hemodynamic data for patients with CTEPH undergoing pulmonary endarterectomy were collected prospectively and are provided in Tables 1–3 (G.A.A., G.E., R.N.C.) (IRB #2016P001640). In the operating room, specimens were divided into proximal and distal sections, and snap-frozen in liquid nitrogen or preserved in 10% formalin. The samples were acquired on the basis of availability. The CTEPH HPAECs were isolated at the time of surgery (S.R., J.C.H., Y.-R.Y.) (n = 3) (IRB #00082338) using an aseptic technique in a tissue culture hood according to published methods (17) and as detailed in the online supplement.
Table 1.
Demographic and Cardiopulmonary Hemodynamic Profile of Patients Treated with Thrombectomy for Luminal PE or DVT or with PEA for CTEPH
| Patient Group | Age (yr) | Sex (F) [n (%)] | mPAP (mm Hg) | PVR (WU) | CO (L/min) | CI (L/min/m2) |
|---|---|---|---|---|---|---|
| Acute PE or DVT, n = 6 | 56 ± 5.1 | 3 (50) | — | — | — | — |
| CTEPH, n = 7 | 55 ± 6.2 | 4 (57) | 46 ± 4.8 | 8.0 ± 1.3 | 4.4 ± 0.6 | 2.1 ± 0.2 |
Definition of abbreviations: CI = cardiac index; CO = cardiac output; CTEPH = chronic thromboembolic pulmonary hypertension; DVT = deep vein thrombosis; mPAP = mean pulmonary artery pressure; PE = pulmonary embolism; PEA = pulmonary endarterectomy; PVR = pulmonary vascular resistance; WU = Wood units.
Data are presented as mean ± SE unless otherwise noted. There was no statistical difference in age between groups (P = 0.94) or sex (P = 0.70) by two-sample Student’s t test and Chi-square analyses, respectively.
Table 2.
Anatomic Locations of the Harvested Thromboembolic Specimens from Patients Treated with Thrombectomy for Luminal PE or DVT or with PEA for CTEPH
| Acute PE/DVT (n = 6) | Anatomic Location of Specimen |
|---|---|
| 1 | Left brachial vein thrombus |
| 2 | Left lung pulmonary artery embolus |
| 3 | Right lung pulmonary artery embolus |
| 4 | Right lung pulmonary artery embolus |
| 5 | Left lung pulmonary artery embolus |
| 6 | Right pulmonary artery embolus |
Definition of abbreviations: CTEPH = chronic thromboembolic pulmonary hypertension; DVT = deep vein thrombosis; PE = pulmonary embolism; PEA = pulmonary endarterectomy.
Table 3.
Individual Characteristics of Patients with CTEPH
| CTEPH (n = 7) | Age (yr) | Sex | mPAP (mm Hg) | PVR (WU) | CO (L/min) | CI (L/min/m2) | Before PEA Therapy |
|---|---|---|---|---|---|---|---|
| 1 | 48 | M | 54 | 10.2 | 4.4 | 2.1 | Riociguat |
| 2 | 64 | M | 39 | 5.3 | 3.2 | 1.7 | None |
| 3 | 47 | M | 63 | 14.5 | 3.1 | 1.5 | None |
| 4 | 74 | F | 39 | 6.6 | 4.7 | 2.6 | None |
| 5 | 46 | F | 31 | 7.6 | 2.9 | 1.4 | None |
| 6 | 32 | F | 61 | 6.3 | 7.5 | 3.1 | None |
| 7 | 76 | F | 37 | 5.2 | 4.8 | 2.6 | None |
Definition of abbreviations: CI = cardiac index; CO = cardiac output; CTEPH = chronic thromboembolic pulmonary hypertension; mPAP = mean pulmonary artery pressure; PEA = pulmonary endarterectomy; PVR = pulmonary vascular resistance; WU = Wood units.
Statistical Analyses
Data are expressed as the mean ± SEM unless otherwise indicated. For continuous data, all comparisons between two groups with n ≤ 6/condition were performed using the Mann-Whitney test. For comparisons between two groups with n > 6/condition, the Mann-Whitney test was used for continuous data that were not distributed normally based on results of the Shapiro-Wilks test. If these data were distributed normally, the Student’s unpaired two-tailed t test was used to compare differences between the two groups. A one-way ANOVA was used to examine differences in response to treatments between groups. A post hoc analysis was performed by the method of Tukey. To avoid overemphasizing false-negative statistical results, an additional adjustment for multiple statistical testing beyond the method of Tukey was not included in the ANOVA comparisons. For categorical variables, the chi-square (χ2) proportion test was used to examine differences between the two groups. The Pearson and Spearman correlation coefficients are presented for linear regression analyses involving normally and nonnormally distributed data, respectively. A P value < 0.05 and a false discovery rate (FDR) < 0.05 were considered significant.
Results
Individual data points for each result are provided in Data File E1 in the online supplement.
Hypoxia Induces HIF-1α–Dependent Upregulation of NEDD9 in HPAECs
Cultured HPAECs were treated with normoxia or hypoxia (10%, 2%, and 0.2% O2) for 24 hours, and NEDD9 expression was analyzed using a commercially available anti-NEDD9 Ab (Ab 1) (Abcam #18056) (10). Compared with normoxia, hypoxia-induced a dose-dependent increase in NEDD9 measured by immunoblot (Figure 1A) and immunofluorescence (Figure 1B). Maximal hypoxia (0.2% O2) for 24 hours also increased NEDD9 in saphenous vein endothelial cells (Figure E1A). By contrast, hypoxia decreased NEDD9 protein expression in human brain microvascular endothelial cells (Figure 1C) and did not affect NEDD9 significantly in other vascular cell types (Figure E1A).
Figure 1.
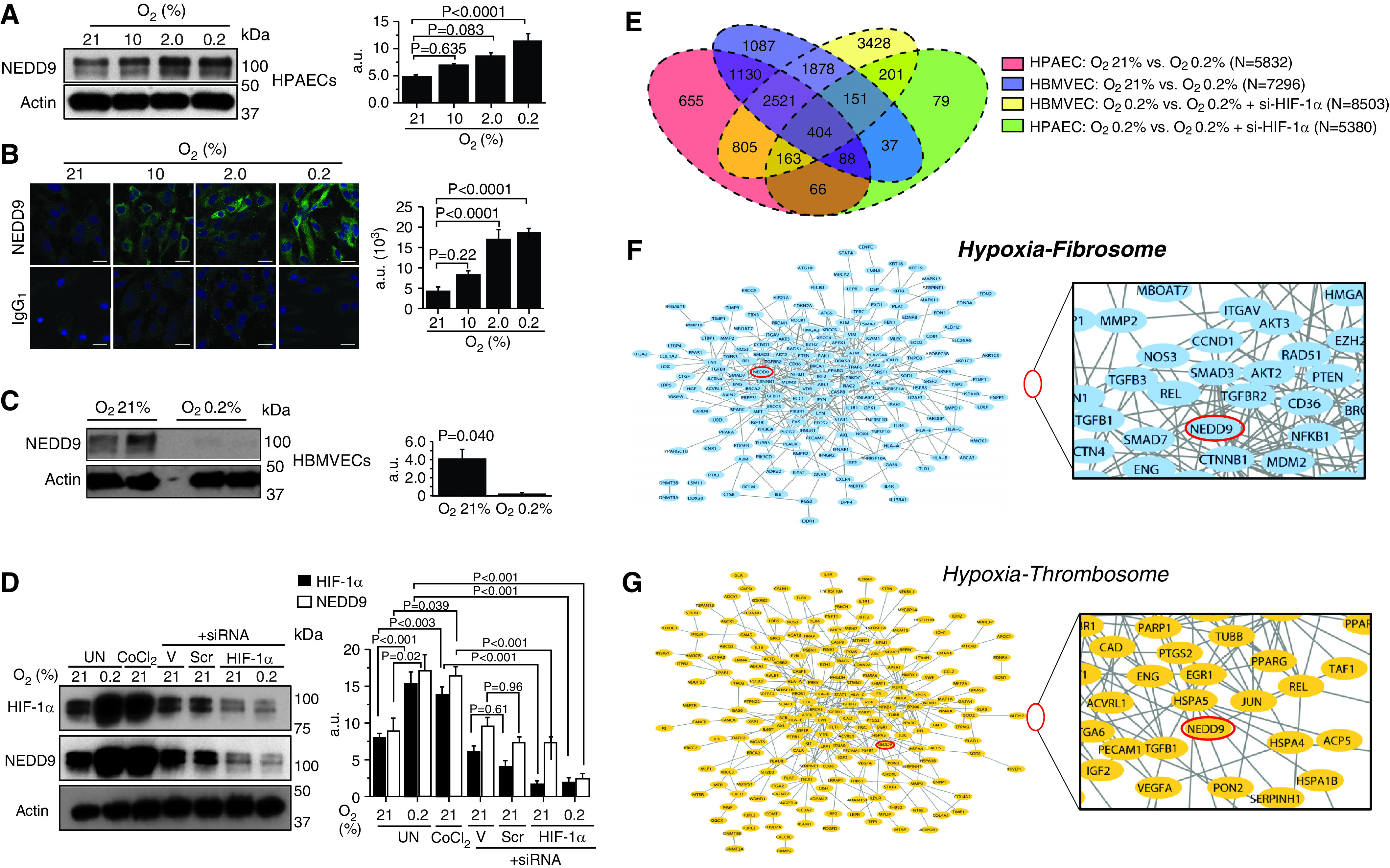
In human pulmonary artery endothelial cells (HPAECs), the protein NEDD9 is increased by hypoxia–HIF-1α (hypoxia-inducible factor-1α) signaling in vitro and is prothrombotic in silico. (A) Anti-NEDD9 immunoblotting and (B) immunofluorescence were performed on lysates from HPAECs treated with normoxia or hypoxia (10%, 2%, 0.2% O2) for 24 hours (n = 3). IgG1 is the negative control. (C) In contrast to our findings in HPAECs, hypoxia decreased NEDD9 in human brain microvascular endothelial cells (HBMVECs) (n = 3). (D) Inhibition of HIF-1α with siRNA decreases NEDD9 expression in HPAECs treated with hypoxia or cobalt chloride (CoCl2) (250 μmol/L) (n = 3). (E) Venn diagram illustrating the distribution of uniquely differentially expressed gene transcripts analyzed by RNA sequencing in HPAECs and HBMVECs treated with normoxia or hypoxia for 24 hours, and untransfected or transfected with si–HIF-1α (n = 3/condition). The genes related to fibrosis and thrombosis were collected from the curated literature. The gene products (proteins) of HPAEC transcripts that were differentially expressed between normoxia and hypoxia and that were associated with either fibrosis or thrombosis were mapped to the human protein–protein interactome (after Reference 10). NEDD9 (indicated by red oval) was identified in the (F) HPAEC hypoxia fibrosome and the (G) HPAEC hypoxia thrombosome. These findings suggest that NEDD9 is functionally relevant to endophenotypes involved in pulmonary thromboembolism. Scale bars, 20 μm. Data are presented as the mean ± SE. Representative immunoblots and micrographs are shown. a.u. = arbitrary units; Scr = scrambled siRNA (negative) control; si–HIF-1α = HIF-1α siRNA; UN = untreated; V = lipofectamine alone.
A prior gene probe analysis demonstrated previously that NEDD9 is increased 2.5-fold in HPAECs that constitutively express HIF 1-α (18), and hypoxia–NEDD9 signaling is regulated by HIF-1α in colorectal cancer cells (4). Thus, we next aimed to determine whether a similar mechanism could account for our findings involving NEDD9 in hypoxia-treated HPAECs. Compared with normoxia, cells treated with hypoxia (0.2% O2) or the hypoxia mimicker cobalt chloride (CoCl2) (19) (250 μmol/L) for 24 hours expressed increased HIF 1-α (4.9 ± 1.5 vs. 15.4 ± 1.5 vs. 13.9 ± 1.0 arbitrary units [AU]; n = 3–6/condition; P < 0.0001) and NEDD9 (9.0 ± 1.9 vs. 17.4 ± 2.2 vs. 16.5 ± 1.4 AU; n = 3–6/condition; P < 0.04). However, transfection with siRNA against HIF 1-α decreased NEDD9 significantly in hypoxia-treated cells (16.5 ± 1.4 vs. 2.4 ± 0.7 AU; n = 3/condition; P < 0.0001) and CoCl2-treated cells (16.5 ± 1.4 vs. 3.3 ± 0.7 AU; n = 3/condition; P < 0.0001) (Figure 1D). MicroRNA-145 is a HIF-1α target (20) linked to NEDD9 transcriptional regulation and pulmonary vascular disease (21). We observed that in HPAECs, microRNA-145 inhibition attenuated the effect of hypoxia on NEDD9 upregulation (Figure E1B).
The Hypoxia Transcriptomic Profiles Are Distinct between Endothelial Cell Types
Divergence in the effect of hypoxia on NEDD9 protein expression between HPAECs and human brain microvascular endothelial cells suggested endothelial cell type–specific differences in HIF-1α programming. We used RNA sequencing to identify n = 5,832 and n = 7,296 hypoxia-regulated genes in HPAECs and human brain microvascular endothelial cells, respectively, of which n = 721 and n = 2,965 were unique to each respective cell type. We also identified n = 66 and n = 1,878 hypoxia-regulated genes unique to HPAECs and the brain microvascular endothelial cells, respectively, that were differentially expressed in cells transfected with siRNA against HIF 1-α (Figure 1E and Data Files E2–E5 in the online supplement). Hypoxia increased NEDD9 mRNA transcription significantly in both HPAECs (+2.73-fold change vs. normoxia; P = 7.09 × 10−46; FDR = 7.00 × 10−44) and brain microvascular endothelial cells (+1.46-fold change vs. normoxia; P = 1.05 × 10−11; FDR = 1.53 × 10−10); however, the extent to which this effect was observed in the brain microvascular endothelial cells was significantly less compared with HPAECs (−1.62-fold change; P = 1.33 × 10−10; FDR = 1.06 × 10−9) (Figure E1C).
Thrombotic remodeling in the pulmonary vasculature hinges on increased collagen that stabilizes clotting, and increased NEDD9 is profibrotic in HPAECs (10). However, whether NEDD9 is functionally important in hypoxia–fibrosis or hypoxia–thrombosis signaling is not known. Therefore, we performed a network analysis to identify profibrotic and prothrombotic pathways using hypoxia-regulated transcripts from our RNA-sequencing experiments enriched for fibrosis or thrombosis genes. These data were used to generate the HPAEC hypoxia–fibrosis network and the HPAEC hypoxia–thrombosis network, which we termed as the HPAEC hypoxia fibrosome and HPAEC hypoxia thrombosome, respectively. We identified NEDD9 in the HPAEC hypoxia fibrosome (Figure 1F) and hypoxia thrombosome (Figure 1G), providing evidence in silico that pulmonary endothelial NEDD9 may be important in thrombotic remodeling under hypoxic conditions.
The NEDD9 Substrate Domain Localizes to the Extracellular Plasma Membrane of HPAECs
Immunofluorescence demonstrated distinct subcellular expression patterns on the basis of different NEDD9 Ab targets (Figure 2A). Specifically, NEDD9 was detected at the cell perimeter using NEDD9 Ab 1 (Figure 2B), which targets the p55-kD NEDD9 cleavage product, and includes the NEDD9 protein substrate domain (amino acid [AA], 82–398). Immunoprecipitation–liquid chromatography–mass spectrometry performed on HPAEC lysates confirmed that NEDD9 Ab 1 bound peptides exclusively in the NEDD9 p55-kD fragment (Figure 2C), which was in contrast to NEDD9 Ab 2, serving as a negative control, which targeted an alternative segment of the NEDD9 protein (Figure 2D) (Figure E2) (10, 22). Compared with treatment with normoxia, treatment with hypoxia enhanced colocalization of the NEDD9 p55-kD fragment with the endothelial plasma-membrane protein CD31 (PECAM-1 [platelet–endothelial cell adhesion molecule 1]) in nonpermeabilized HPAECs (Figure 2E). In addition, we demonstrated plasma-membrane expression of NEDD9 using an elution buffer–based method to isolate the plasma-membrane fraction of HPAECs (Figure 2F).
Figure 2.
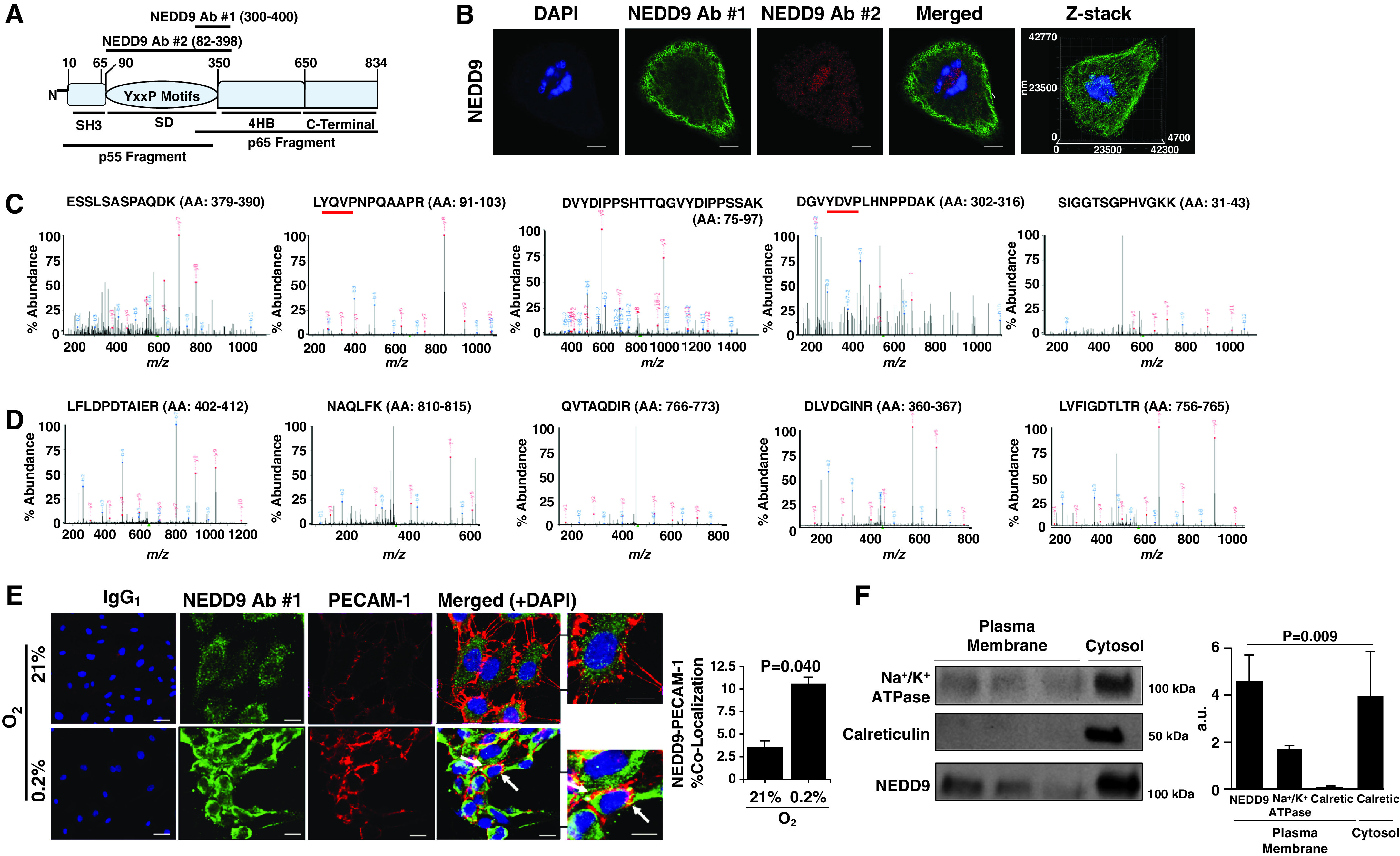
The NEDD9 substrate domain is expressed on the extracellular plasma membrane of human pulmonary endothelial cells (HPAECs). (A) NEDD9 is a scaffolding protein and in Homo sapiens is composed of 834 amino acids organized in four distinct domains: the SH3, substrate, 4HB, and C-terminal domains. Two NEDD9 cleavage peptide fragments (p55 kD and p65 kD) have been reported previously (22). To determine whether either cleavage product corresponded to differences in NEDD9 localization in HPAECs, anti-NEDD9 immunofluorescence was performed using NEDD9 antibody (Ab) 1 targeting the p55-kD fragment and NEDD9 Ab 2 targeting the p65-kD fragment. (B) Compared with NEDD9 Ab 2, NEDD9 expression detected using NEDD9 Ab 1 was localized predominantly to the perimeter of cells (n = 3). Scale bar = 10 μm. (C) The MS spectra from five abundant peptides detected in trypsin-digested HPAEC lysates immunoprecipitated using NEDD9 Ab 1 corresponded exclusively to the p55-kD fragment, whereas (D) NEDD9 Ab 2 identified NEDD9 peptides corresponding to the p65-kD fragment (n = 3). The red underlining denotes a tyrosine-x-x-proline (YxxP) sequence, where x is another amino acid. (E) Compared with normoxia, hypoxia (0.2% O2) for 24 hours increased colocalization of NEDD9 with the endothelial plasma-membrane protein PECAM-1 (platelet–endothelial cell adhesion molecule 1) analyzed using double immunofluorescence (n = 3) (colocalization indicated by white arrow). IgG1 represents the control. (F) Isolation of HPAEC plasma-membrane fractions was confirmed by Na+/K+ ATPase expression in the absence of (cytosolic) calreticulin, and NEDD9 was analyzed in the plasma-membrane fraction by immunoblotting (n = 5). Scale bar, 40 μm. Data are presented as the mean ± SE. Representative immunoblots and micrographs are presented. a.u. = arbitrary units; Calciretic = calreticulin; MS = mass spectrometry; m/z = mass-to-charge ratio.
P-Selectin Binds the NEDD9 Substrate Domain
Our data suggested that the NEDD9 p55-kD fragment, which includes the substrate domain, localizes to the HPAEC plasma membrane. The substrate domain is characterized by numerous tyrosine proline (YxxP) motifs. On the basis of older reports suggesting that tyrosine may underlie platelet P-selectin participation in platelet–endothelial interactions (23), we hypothesized that P-selectin targets the NEDD9 substrate domain in HPAECs. Plasma-membrane fractions incubated with recombinant P-selectin for 1 hour were immunoprecipitated with an anti–P-selectin Ab. Next, liquid chromatography–mass spectrometry performed on in-gel trypsin-digested lysates identified only two NEDD9 peptide sequences, both within the substrate domain: K.LYQVPNPQAAPR.D (AA, 91–102) (NEDD9 peptide sequence 1 [N9-P1]) and K.GPVFSVPVGEIKPQGVYDIPPTK.G (AA, 191–211) (NEDD9 peptide sequence [N9-P2]) (n = 2 replicates for n = 2 iterations) (Figure 3A). Plasma-membrane fractions from HPAECs were incubated with vehicle control or exogenous (recombinant) P-selectin for 1 hour, and NEDD9– P-selectin complex formation was assessed by coimmunoprecipitation. Compared with vehicle control, P-selectin (1.0 μg) increased NEDD9– P-selectin complex formation significantly by threefold (P = 0.030, n = 3) (Figure 3B). Microscale thermophoresis generated a dose titration curve indicative of a physical association between the receptor (NEDD9) and ligand (P-selectin) with an estimated Kd of 13.9 nM (n = 2) (Figures 3C–3E).
Figure 3.
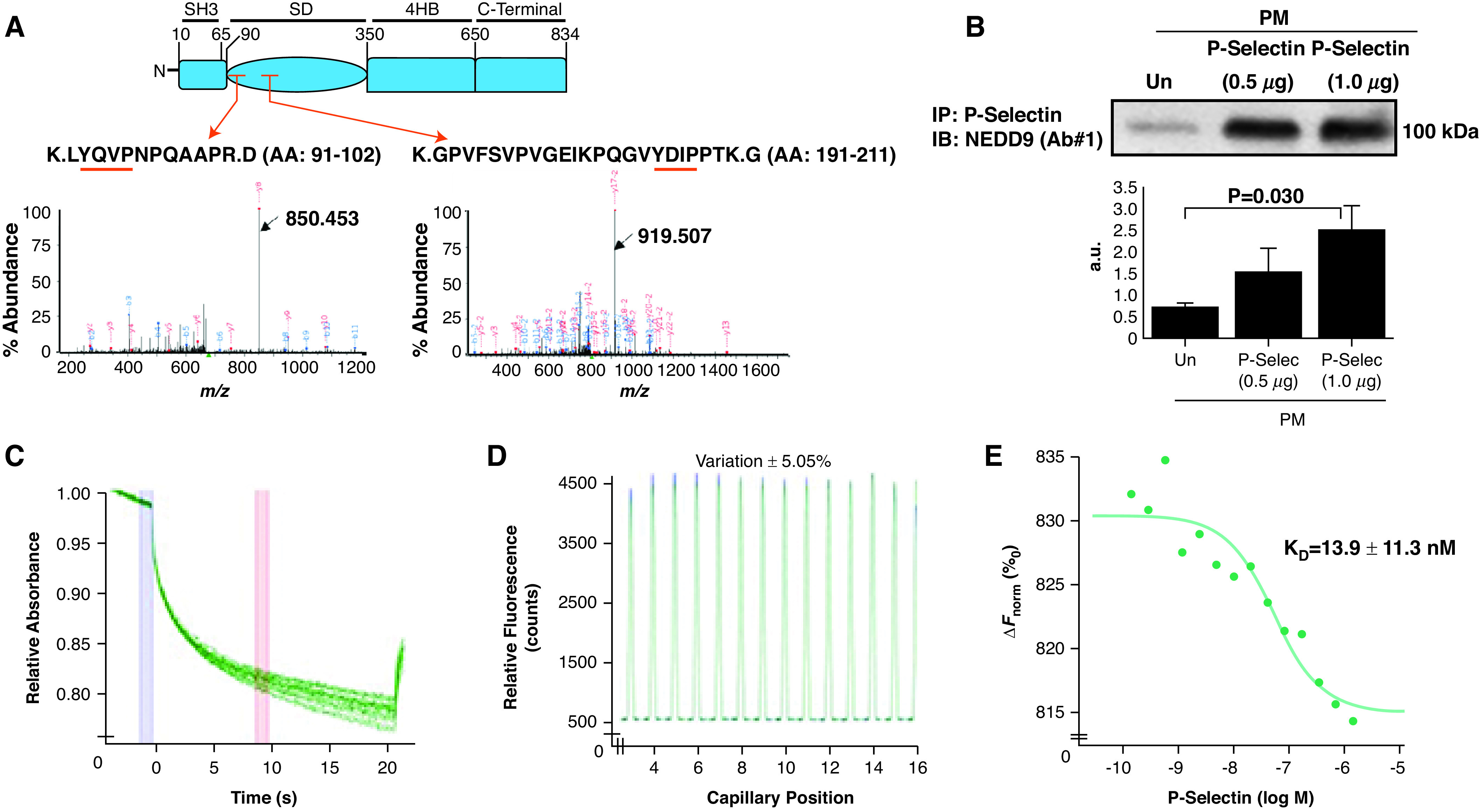
P-Selec (P-selectin) binds the NEDD9 substrate domain, and NEDD9 modulates platelet–endothelial adhesion. (A) Plasma-membrane fractions incubated with recombinant P-Selec for 1 hour were immunoprecipitated with an anti–P-Selec antibody (Ab). The fragmented ion MS spectra for each of the two detected NEDD9 peptide sequences, both within the substrate domain, are shown (black arrows): K.LYQVPNPQAAPR.D (amino acid [AA], 91–102) (m/z 677.36735 at retention time of 28.1 s) (NEDD9 peptide sequence 1) and K.GPVFSVPVGEIKPQGVYDIPPTK.G (AA, 191–211) (m/z 808.77731 at retention time 35.5 s) (NEDD9 peptide sequence 2) (n = 2 replicates for n = 2 iterations). The red arrows indicate the peptide location within the NEDD9 substrate domain (SD). The red underlining indicates a tyrosine-x-x-proline (YxxP) sequence, where x is another amino acid. (B) Human pulmonary-endothelial-cell plasma-membrane fractions were incubated with exogenous P-Selec, and coimmunoprecipitation was performed using an anti–P-Selec Ab followed by anti–NEDD9 immunoblot NEDD9. Varying concentrations of P-Selec (ligand) (2 μM–0.5 nM) were coincubated with fluorescently labeled NEDD9 (receptor) (20 nM), and microscale thermophoresis was performed to assess macromolecular interactions between these proteins. (C) Raw fluorescence tracings, (D) a capillary scan, and (E) a dose titration curve show a definitive protein–protein interaction between the receptor and ligand (Kd = 13.9 ± 11.3 nM) (n = 2). Data are presented as the mean ± SE. Representative immunoblot and titration curve are shown. a.u. = arbitrary units; ΔFnorm = change in normalized fluorescence; IB = immunoblot; IP = immunoprecipitation; M = molar; MS = mass spectrometry; m/z = mass-to-charge ratio; PM = plasma membrane; Un = untreated.
NEDD9 Is a Modifiable Target to Inhibit Platelet–Endothelial Adhesion In Vitro
Two model peptides corresponding to the N9-P1 and N9-P2 sequences were synthesized and used to develop an antihuman, monospecific polyclonal Ab (msAb) against each peptide (msAb–N9-P1 and msAb–N9-P2) (Figures E3A–E3D). Recombinant NEDD9 and P-selectin were incubated for 30 minutes in a cell-free system supplemented with either msAb–N9-P1 (10–20 μg/ml) or msAb–N9-P2 (10–20 μg/ml), and differences in NEDD9–P-selectin complex formation were analyzed by immunoprecipitation–immunoblot. We observed maximal inhibition of NEDD9–P-selectin complex formation by msAb–N9-P2 (20 μM) (Figure 4A).
Figure 4.
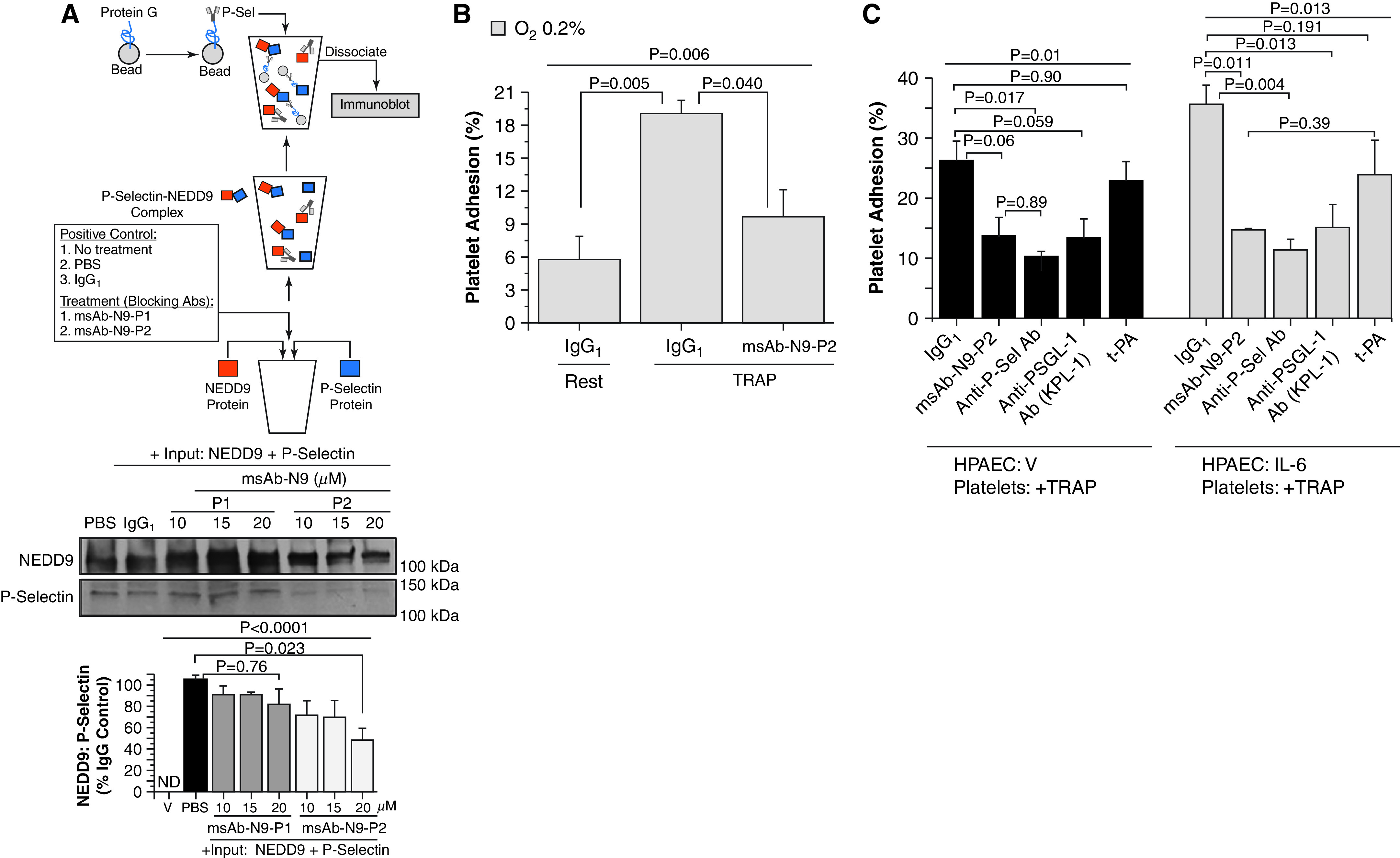
Inhibition of NEDD9 with monospecific polyclonal Ab (msAb)–NEDD9 peptide sequence 2 (N9-P2) decreases NEDD9–P-Sel (P-selectin) complex formation. Anti-NEDD9 antibodies raised against the NEDD9 target of P-Sel were developed in rabbits (msAb–NEDD9 peptide sequence 1 [N9-P1] and msAb–N9-P2). (A) The inhibitory effect of msAb–N9-P1 and msAb–N-P2 on NEDD9–P-Sel complex formation in vitro was analyzed (n = 3). V, a mixture of msAb–N9-P1 and msAb–N9-P2, was incubated without beads as an additional negative control (immunoblot data not shown). (B) In human pulmonary artery endothelial cells (HPAECs) treated with hypoxia (0.2%) for 24 hours, msAb–N9-P2 inhibited TRAP (thrombin receptor agonist peptide) (25 μM)–stimulated platelet–endothelial adhesion significantly (n = 3). IgG1 was the negative control. (C) Under normoxic conditions, HPAECs were treated with IL-6 (25 ng/ml) for 24 hours, and the effect of msAb–N9-P2 (20 μg/ml) on platelet–endothelial adhesion was compared with treatment with anti–P-Sel Ab (anti–P-Sel) (10 μg/ml), PSGL-1 (anti–P-Sel glycoprotein ligand-1 Ab from a KPL-1 clone) (15 μg/ml), and t-PA (tissue plasminogen activator) (15 ng/ml) (n = 3). Similar experiments were performed in untreated HPAECs after stimulation of platelets with TRAP. Data are presented as mean ± SEM. Representative immunoblots are shown. Ab = antibody; ND = not detected; PBS = phosphate-buffered saline; V = vehicle control.
Compared with normoxia, hypoxia for 24 hours increased HPAEC expression of the N9-P2 peptide (Figure E3E), and msAb–N9-P2 inhibited TRAP-stimulated platelet–endothelial adhesion in hypoxia-treated cells (Figure 4B). On the basis of these collective results, we focused on the effect of msAb–N9-P2 in further in vitro experiments. We also expanded our conditions to include HPAEC treatment with IL-6, which is an inflammatory cytokine that is increased in CTEPH (24). We observed that IL-6 increased NEDD9 expression and the adhesion of activated platelets to HPAECs (Figure E4). Compared with IgG1 control, the inhibitory effect of msAb–N9-P2 on the adhesion of activated platelets to HPAECs treated with IL-6 was equivalent to the effect of inhibiting P-selectin or P-selectin glycoprotein ligand-1, which is the P-selectin counter receptor (Figure 4C).
NEDD9 Does Not Regulate Platelet Aggregation
Using platelets isolated from healthy volunteers, we detected NEDD9 outside of platelet α-granules by anti-NEDD9 immunofluorescence and along the outer perimeter of platelets by electron microscopy (both experiments used NEDD9 Ab 3) (Figure 5A). We next isolated platelets from wild-type (WT) and NEDD9−/− mice and found no difference in global platelet aggregation between these two conditions in response to collagen or other potent murine platelet agonists (Figure 5B). We also observed no significant difference in whole-blood impedance aggregometry, plasma tissue plasminogen activator concentration, or plasminogen activator inhibitor-1 concentration between WT and NEDD9−/− mice, nor did we observe differences in impedance aggregometry observed between human blood samples incubated with or without msAb–N9-P2 (Figures 5C–5E). Furthermore, TRAP-stimulated platelet–endothelial adhesion was decreased significantly in HPAECs transfected with NEDD9 siRNA (Figure E5). These collective findings are in support of our hypothesis that platelet–pulmonary endothelial interactions are modulated by N9-P2 expressed in HPAECs, rather than NEDD9 expressed in platelets.
Figure 5.
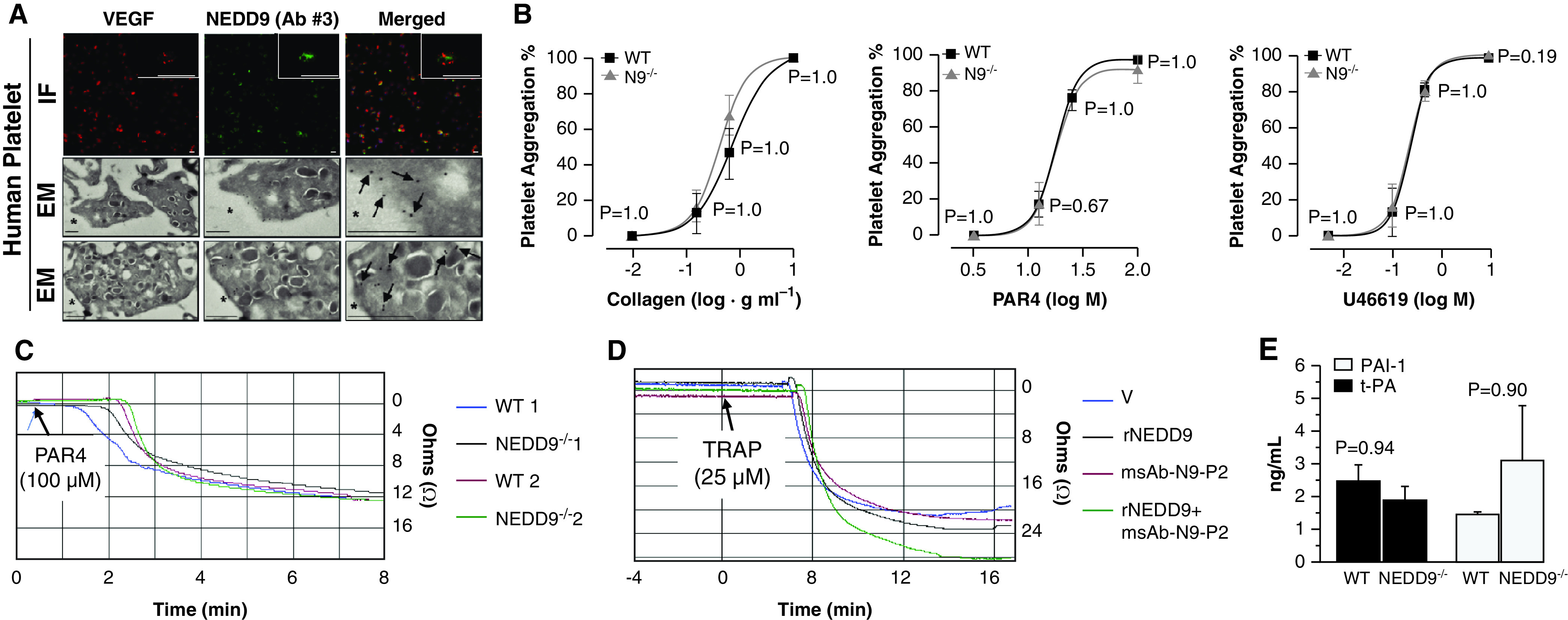
N9 (NEDD9) affects platelet–human pulmonary artery endothelial cell adhesion without affecting platelet aggregation. (A) Anti-N9 immunofluorescence (IF) and electron microscopy (EM) immunocytochemistry using N9 antibody (Ab) 3 were performed on platelets isolated from healthy human control donors. For IF, the scale bar was set at 1.5 μm, and the scale bar for the magnified inset was set at 5 μm. For EM, the black arrows identify N9 stain positivity (n = 3); scale bars, 500 nm. (B) No significant difference between wild-type (WT) and N9−/− mice was observed for global platelet aggregation in response to collagen, PAR4 (protease activator receptor) or PAR9, 11-dideoxy-9α, or 11α-methanoepoxy prostaglandin F2α (U46619) (N = 3 mice/condition). (C) Whole-blood impedance aggregometry was performed in samples acquired from WT and transgenic N9−/− mice stimulated with PAR4 (100 μM) (n = 4). (D) Compared with V control, biologically important differences in impedance aggregometry were not observed in human control blood incubated with rN9 (recombinant N9) (10 ng/ml) or our custom anti-N9 Ab (msAb–N9-P2) (0.08 μg) (n = 3). (E) Plasma expression of t-PA (tissue plasminogen activator) and PAI-1 (plasminogen activator inhibitor-1) in WT or N9−/− mice (n = 4 mice/condition). Data are presented as the mean ± SE. Representative micrographs are shown. M = molar; msAb = monospecific polyclonal Ab; P2 = peptide sequence 2; TRAP = thrombin receptor agonist peptide; V = vehicle; VEGF = vascular endothelial growth factor.
NEDD9 Inhibition Prevents the Formation of Platelet–Pulmonary Endothelial Aggregates and Pulmonary Hypertension In Vivo
We next turned to the established murine model of ADP-induced platelet activation (Figures E6 and E7) (25). Compared with WT mice, NEDD9−/− mice were resistant to the formation of ADP-induced platelet–pulmonary endothelial aggregates (Figures 6A and E8A) and had a significantly blunted increase in right ventricular systolic pressure (Figures 6B and E8B). To determine whether NEDD9 antagonism affects platelet–endothelial adhesion in vivo, WT mice were pretreated with msAb–N9-P2 or msAb–N9-P1 (as a negative control) for 10 minutes before ADP infusion. Compared with IgG1 control, treatment with msAb–N9-P2 decreased ADP-induced vascular occlusion and pulmonary hypertension significantly (Figures 6C and 6D).
Figure 6.
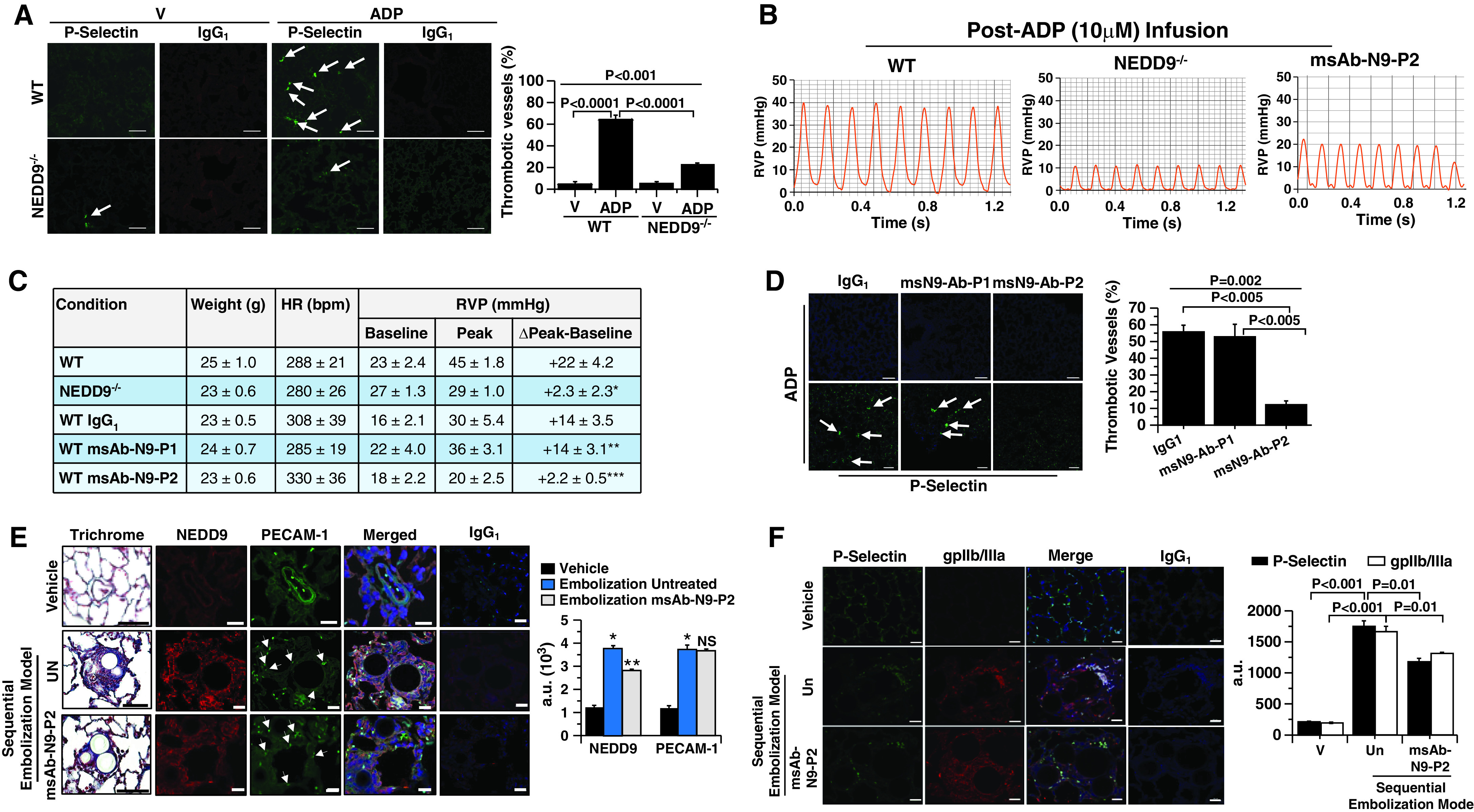
The effect of N9 (NEDD9) inhibition with msAb–N9-P2 on platelet–pulmonary endothelial aggregate formation, pulmonary hypertension, and chronic thromboembolic pulmonary arterial remodeling in vivo. (A–D) Compared with wild-type (WT) mice, N9−/− mice were resistant to ADP-induced pulmonary arteriolar thrombotic occlusion (arrows) and increased right-ventricular systolic pressure analyzed by anti–P-selectin immunofluorescence and cardiac catheterization, respectively. IgG1 was used as a negative control. Similar effects were observed in WT mice treated with msAb–N9-P2 compared with IgG1 administered 10 minutes before ADP infusion. n = 3–6 mice/condition. For C, *P = 0.005 versus WT, **P = 0.99 versus WT IgG1, and ***P = 0.037 versus WT IgG1; scale bars, 5 μm. Hemodynamic tracings are after administration of maximal ADP dose (10 μM), and full-time course data are presented in Figure E8B. (E) Male and female Wistar rats were randomized to treatment with V (saline) control (n = 3), sequential prothrombotic bead embolization (n = 5), or sequential embolization plus treatment with msAb–N9-P2 (20 μg/ml) (n = 5) (an illustration of the protocol is provided in Figure E6). Paraffin-embedded lung sections were isolated, affected vessels were analyzed for collagen quantity by using Masson trichrome staining, and endothelial N9 and PECAM-1 (platelet–endothelial cell adhesion molecule 1) expression was quantified by using immunofluorescence. Arrows indicate endothelial cells. Scale bars, 20 μm. *P < 0.001, embolization untreated versus vehicle; **P < 0.001 embolization msAb-N9-P2 versus vehicle. (F) Thrombotic remodeling was assessed further by anti–P-selectin and anti-gpIIb/IIIa immunofluorescence. P < 0.001 by ANOVA for P-selectin and P < 0.001 by ANOVA for gpIIb/IIIa. Representative micrographs and hemodynamic tracings are shown. Data are presented as the mean ± SE. Ab = antibody; a.u. = arbitrary units; bpm = beats per minute; gp = glycoprotein; HR = heart rate; ms = monospecific polycolonal; NS = not significant; P1 = peptide sequence 1; P2 = peptide sequence 2; RVP = right ventricular pressure; Un = untreated; V = vehicle.
Figure 7.
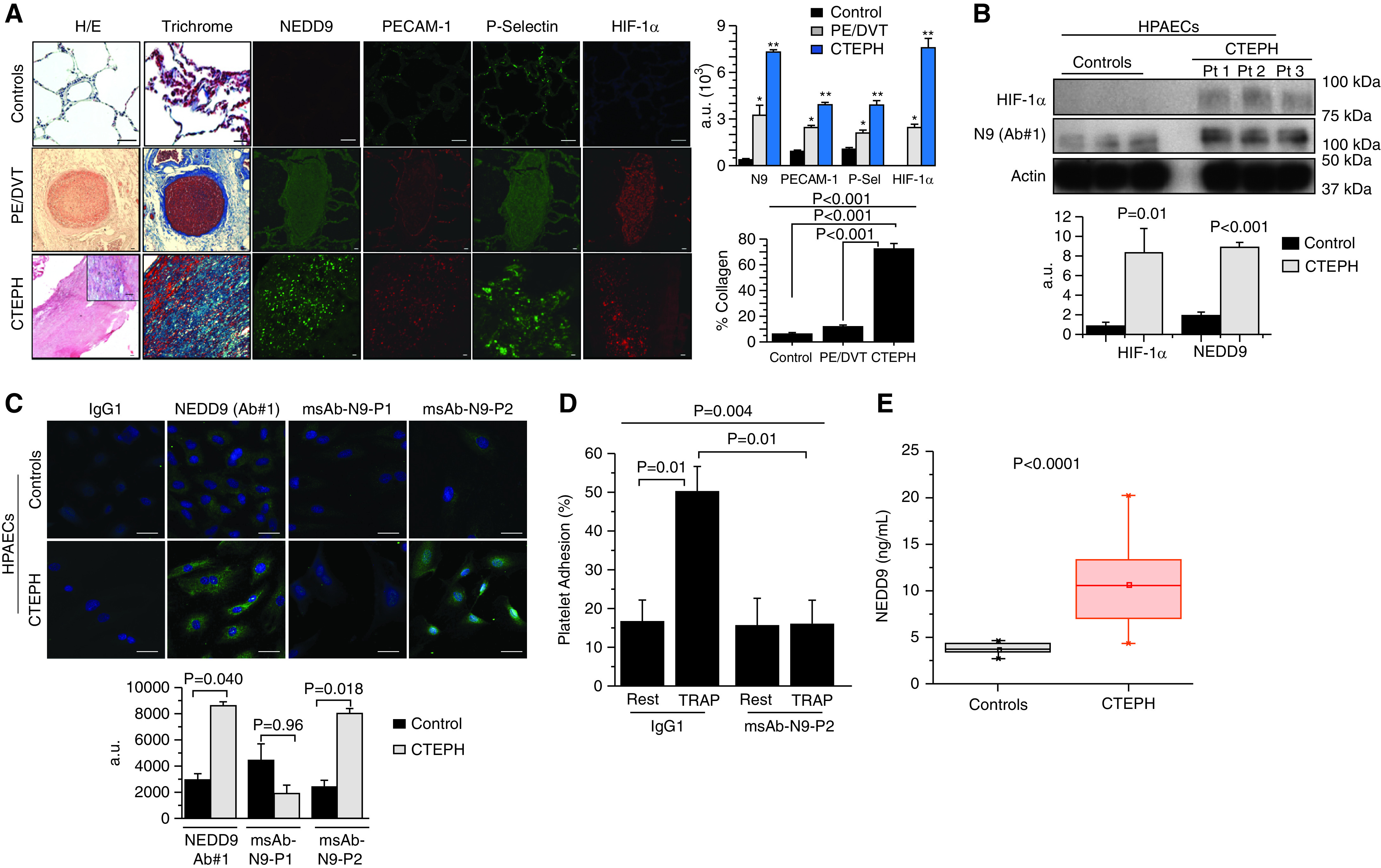
N9 (NEDD9) is increased in chronic thromboembolic pulmonary hypertension (CTEPH). (A) Compared with healthy control donor lung specimens, a step-wise increase was observed in acute pulmonary embolism/deep vein thrombosis (PE/DVT) specimens and CTEPH specimens for vascular fibrosis and HIF-1α (hypoxia-inducible factor-1α), N9, and P-Sel (P-selectin) expression. n = 3–6 samples per condition. Control scale bars, 20 μm; PE/DVT and CTEPH scale bars, 40 μm. *P < 0.01 versus internal control and **P < 0.01 versus PE/DVT. (B) Cultured control human pulmonary artery endothelial cells (HPAECs) and HPAECs from CTEPH specimens were analyzed using anti–HIF-1α and anti-N9 (Ab 1) immunoblotting (n = 6). (C) Anti-N9 immunofluorescence was performed on cultured CTEPH HPAECs using N9 Ab 1, msAb–N9-P1, as a negative control or msAb–N9-P2 (n = 3/condition). IgG1 was used as a negative control. (D) Platelet–endothelial adhesion was analyzed in CTEPH HPAECs and control HPAECs incubated with platelets from healthy donors under basal conditions and after stimulation with TRAP (thrombin receptor agonist peptide) (10 μM) in the presence or absence of msAb–N9-P2 (n = 4). (E) Plasma NEDD9 was increased significantly in patients with CTEPH (n = 27) compared with age-matched and sex-matched healthy controls (n = 7). Scale bars, 20 μm. Representative micrographs and immunoblots are shown. For A–D, data are presented as mean ± SE. For E, the means were represented by squares; the medians were represented by horizontal lines; the interquartile ranges were represented by the box distribution; and the maximum and minimum were represented by the y-axis lines. a.u. = arbitrary units; H/E = hematoxylin and eosin; msAb–N9-P1 = monospecific anti-N9 Ab against substrate domain P1; msAb–N9-P2 = monospecific anti-N9 Ab against substrate domain P2; P1 = peptide sequence 1; P2 = peptide sequence 2; Pt = patient.
NEDD9 Inhibition Prevents Chronic Thromboembolic Remodeling In Vivo
To determine whether msAb–N9-P2 was effective at inhibiting chronic thromboembolic pulmonary arterial remodeling in vivo, we used a lung-embolization disease model in which beads containing fibrinogen-coated and collagen-coated microspheres in thrombin solution were administered sequentially to rats (Figure E6). Compared with untreated rats, sequential bead embolization increased PECAM-1 expression (indicative of endothelial proliferation), vascular NEDD9 expression, and fibrotic and thrombotic pulmonary arterial remodeling without significantly affecting right-ventricular fibrosis quantity. Using a disease-prevention strategy, treatment with msAb–N9-P2 significantly inhibited the platelet-rich thrombi burden as well as vascular fibrosis in vivo (Figures 6E and E9).
Pulmonary endothelial apoptosis and sloughing are reported for monocrotaline-treated rats (26), which we studied next as a negative-control in vivo model on the basis of our findings in vitro indicating that pulmonary endothelial NEDD9 was required for modulating NEDD9-dependent platelet–HPAEC interactions. In disease prevention and reversal studies (Figure E6), we observed, respectively, that pulmonary arterial PECAM-1 was decreased significantly in monocrotaline-treated rats compared with normal control rats (60 ± 13 vs. 63 ± 17 vs. 437 ± 25 AU; n = 3; P < 0.0001). In line with this finding, msAb–N9-P2 had no significant effect on the platelet-rich thrombi burden, cardiopulmonary hemodynamics, or right-ventricular fibrosis in monocrotaline-treated rats (Figure E10).
NEDD9 Is Increased in Patients with CTEPH
Compared with healthy (rejected donor) control lungs, a graded increase in vascular fibrosis was observed for acute deep vein thrombosis and pulmonary embolism and CTEPH pulmonary endarterectomy specimens (Figure 7A). Immunofluorescence analyses showed a directionally similar pattern of expression across control donor lung, deep vein thrombosis or pulmonary embolism, and CTEPH samples for HIF-1α and NEDD9, as well as P-selectin and NEDD9 (Figure 7A). Analyzing the pulmonary embolism and deep vein thrombosis and CTEPH specimens collectively, NEDD9 correlated strongly with P-selectin (r = +0.86; P = 0.004) and HIF-1α (r = +0.89; P = 0.04) (Figure E11).
These data were consistent with our findings in HPAECs isolated from patients with CTEPH, which expressed increased HIF-1α and NEDD9 compared with control donor HPAECs (Figure 7B). Immunofluorescence analyses of HPAECs from patients with CTEPH also confirmed increased NEDD9 expression using msAb–N9-P2 (Figure 7C). Akin to older reports indicating that CTEPH is associated with a prothrombotic endothelium (27), platelet–endothelial adhesion was increased in HPAECs from patients with CTEPH compared with control donor HPAECs under basal conditions as well as after stimulation of healthy donor platelets with TRAP (Figure E12A). Despite enhanced thrombogenicity in HPAECs from patients with CTEPH, msAb–N9-P2 inhibited TRAP-stimulated platelet adhesion to these cells significantly (Figure 7D). In plasma from patients with CTEPH, increased platelet activation and NEDD9 concentration were observed compared with age-matched and sex-matched healthy control volunteers (Figures 7E and Figure E12B). An illustrative summary of these data is provided in Figure E13.
Discussion
These data demonstrate HIF-1α–dependent upregulation of NEDD9 expression on the extracellular surface of HPAECs. A functional NEDD9 peptide participates in a previously unrecognized protein–protein interaction with platelet P-selectin. The sequence of this NEDD9 peptide was used to develop a novel anti-NEDD9 Ab (msAb–N9-P2), and msAb–N9-P2 inhibited the adhesion of activated platelets to HPAECs under conditions of hypoxia or inflammation in vitro, prevented the acute formation of platelet–endothelial aggregates and pulmonary hypertension in vivo, and decreased chronic thromboembolic remodeling in rats in vivo. Expression of NEDD9 was increased in endarterectomy specimens from patients with CTEPH, and msAb–N9-P2 inhibited adhesion of activated platelets to pulmonary artery endothelial cells ex vivo from patients with CTEPH. Overall, our findings identify NEDD9 as a modifiable mediator of platelet–endothelial interactions, with relevance to pulmonary vascular diseases characterized by hypoxia, activated platelets, and thromboembolic remodeling.
These data expand NEDD9 binding targets to include P-selectin, which, to our knowledge, has not been shown previously. We focused on P-selectin because it is an established mediator of pulmonary arterial thrombosis (28, 29), supported further by our histomicrographs showing increased NEDD9 and P-selectin in endarterectomy specimens. The mechanisms implicated in P-selectin biofunctionality vary (30); however, in one report, P-selectin binding affinity was determined by a critical tyrosine residue in its counter receptor (23), and it is notable that the NEDD9 peptides targeted by P-selectin in this study included YxxP motifs. Microscale thermophoresis yielded a Kd for NEDD9–P-selectin binding akin to those of other clinically relevant platelet–endothelial protein–protein interactions, including glycoprotein IIb/IIIa–von Willebrand Factor (31) and P-selectin–P-selectin glycoprotein ligand-1 (32). Importantly, inhibition of P-selectin glycoprotein ligand-1 induces major pulmonary vascular injury (33), suggesting NEDD9 may be an attractive alternative therapeutic target to limit pathogenic platelet–pulmonary endothelial aggregates.
The network analyses provided key data in silico implicating NEDD9 involvement in hypoxia, fibrosis, and thrombosis, which are three endophenotypes underlying pulmonary vascular thrombotic remodeling (34). Indeed, a continuum in pulmonary vascular fibrosis, endothelial hypoxia–inducible factor-1α, clot extent, and NEDD9 expression was observed across negative control, pulmonary embolism, and CTEPH specimens. This parallels older reports showing that vascular remodeling correlates positively with persistent hypoxemia and HIF-1α cell positivity after endarterectomy (7, 35). In a second disease-relevant experimental system, msAb–N9-P2 inhibited platelet–endothelial adhesion induced by the proinflammatory CTEPH cytokine IL-6.
These observations suggest that chronic overexpression of pulmonary endothelial NEDD9 due to hypoxic and/or inflammatory vascular injury after pulmonary embolism may offer novel mechanistic insight underpinning the phenotype transition to CTEPH (Figure E13). However, the pathogenesis of CTEPH is complex and undoubtedly extends beyond the effects of hypoxic and/or inflammatory signaling on NEDD9 (36, 37). For example, platelet access to an intact endothelium appeared necessary for preventing NEDD9-mediated pulmonary arterial thrombi in vivo. Topological characteristics of thrombus that affect its clearance and variability in collagen-dependent clot stabilization are also likely to influence NEDD9-dependent and NEDD9-independent mechanistic drivers of the CTEPH pathophenotype.
There were 1,812 more hypoxia-regulated, HIF-1α–dependent genes unique to human brain microvascular endothelial cells compared with HPAECs. Cell type–specific hypoxia-response patterns may, thus, be useful for understanding mechanisms that differentially affect platelet–endothelial adhesion in the pulmonary versus cerebral circulations. We also observed that hypoxia increased NEDD9 protein expression in saphenous vein endothelial cells and HPAECs but not in brain microvascular endothelial cells, implying that NEDD9-modulating therapies may correspond to different off-target side-effect profiles across vascular beds. These findings present an opportunity to explore endothelial dysfunction in prothrombotic clinical phenotypes characterized by hypoxia, such as acute respiratory distress syndrome and ischemic stroke, which are central to critical care medicine (38).
P-selectin–independent mechanisms involving NEDD9 that could affect pulmonary vascular thrombosis were not studied here, and future research should focus on established thrombotic treatment targets such as integrins or glycoprotein IIb/IIIa (39). Similarly, the effect of SMAD3 (mothers against decapentaplegic homolog 3) on hemostasis was not a focus of this study, although SMAD3 was connected to NEDD9 in the hypoxia fibrosome and SMAD3 upregulates plasminogen activator inhibitor-1 (10, 40). Our findings affirm older reports suggesting microRNA-145 is a HIF-1α target that regulates NEDD9 transcription (21); however, alternative molecular mechanisms that regulate NEDD9 transcription may exist. For example, SOX13 and HDAC1 were HIF-1α–dependent genes, transcription factors predicted to target NEDD9, and established already in the pathobiology of pulmonary vascular disease (41). Uncoupling between NEDD9 mRNA and protein concentration may occur because of post-transcriptional events affecting NEDD9 degradation, as shown previously (10, 42). Exploring the molecular mechanisms that accounted for this phenomenon in hypoxia-treated brain microvascular endothelial cells is needed and may identify additional hypoxia-sensitive vasoactive signaling pathways that are cell-type specific.
The inclusion of patients with CTEPH biospecimens in network analyses is likely to offer particularly valuable insight on disease-specific signaling pathways, and understanding NEDD9 biofunctionality in platelet–endothelial adhesion could be enhanced by using systems that incorporate dynamic (flow) conditions. Dedicated analysis of platelet interactions with neutrophils, which express NEDD9 (43) and are known to modulate pulmonary microthrombi formation (particularly sickle cell disease) (44) and platelet sequestration, is warranted. Data showing that msAb–N9-P2 reverses thrombotic pathophenotypes are needed before characterizing the full clinical translational potential of our findings.
In conclusion, this study identifies NEDD9 as a previously unrecognized mediator of platelet–endothelial adhesion and expands the range of protein–protein interactions involved in the pathobiology of cardiopulmonary diseases. We also show that NEDD9-mediated platelet–endothelial interactions are modifiable pharmacologically, which was accomplished through the development of a novel anti-NEDD9 Ab targeting the extracellular NEDD9 peptide that binds P-selectin. Overall, these data illustrate a novel prothrombotic pathogenetic molecular mechanism that is relevant to diseases characterized by hypoxia, activated platelets, and endothelial dysfunction, including CTEPH.
Acknowledgments
Acknowledgment
The authors thank Ross Tomaino, Ph.D., at Harvard Medical School (HMS) for assistance with liquid chromatography–mass spectrometry; the Biopolymers Facility Next-Generation Sequencing Core Facility at HMS for their expertise and instrument availability that supported RNA-sequencing analyses; Kelly Arnett, Ph.D., at HMS for assistance with the microscale thermophoresis experiments; Maria Ericsson, Ph.D., at HMS for assistance with the scanning electron microscopy experiment; and Nour Nazo at Duke University Medical Center for technical assistance in CTEPH endothelial cell acquisition.
Footnotes
This work was conducted with the support of a KL2 award (5KL2TR002542-02) from Harvard Catalyst | The Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences, NIH Award KL2 TR002542). The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic healthcare centers, or the NIH. This work was also supported by NIH grants U54HL119145 (G.A.A.); 5T32HL007633-32 (B.M.W.); R56HL131787, R01HL153502, 1R01HL139613-01, R21HL145420, and U54HL119145 (B.A.M.); and HL061795, HL119145, HG007690, and GM107618; and by American Heart Association grants D700382 (J.L.) and 15GRNT25080016 (B.A.M.); the National Scleroderma Foundation (B.A.M.); the Cardiovascular Medical Research and Education Foundation (B.A.M.); and the McKenzie Family Charitable Trust (B.A.M.).
Author Contributions: G.A.A., E.M.B., and B.A.M.: study conception/design; acquisition, analysis, and interpretation of the data; and drafting and revising the manuscript. A.O.S., R.-S.W., Y.-Y.Z., B.M.W., and E.A.: acquisition and analysis of data and revising the data. J.L.: interpretation of the data and revising the manuscript. E.A.G., M.H.L.S., J.R.C., K.J.H., F.P.B., Y.-R.Y., J.C.H., G.E., R.N.M., A.S., S.O.V., S.S., P.J.L., and S.R.: acquisition and analysis of data and revising the manuscript. R.N.C.: study conception, acquisition of data, and revising the manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202003-0719OC on February 1, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Kroll MH, Afshar-Kharghan V. Platelets in pulmonary vascular physiology and pathology. Pulm Circ. 2012;2:291–308. doi: 10.4103/2045-8932.101398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rhodes CJ, Im H, Cao A, Hennigs JK, Wang L, Sa S, et al. RNA sequencing analysis detection of a novel pathway of endothelial dysfunction in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192:356–366. doi: 10.1164/rccm.201408-1528OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maron BA, Oldham WM, Chan SY, Vargas SO, Arons E, Zhang YY, et al. Upregulation of steroidogenic acute regulatory protein by hypoxia stimulates aldosterone synthesis in pulmonary artery endothelial cells to promote pulmonary vascular fibrosis. Circulation. 2014;130:168–179. doi: 10.1161/CIRCULATIONAHA.113.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim S-H, Xia D, Kim S-W, Holla V, Menter DG, Dubois RN. Human enhancer of filamentation 1 Is a mediator of hypoxia-inducible factor-1alpha-mediated migration in colorectal carcinoma cells. Cancer Res. 2010;70:4054–4063. doi: 10.1158/0008-5472.CAN-09-2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. 2011;11:123–134. doi: 10.1038/nrc3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moore FE, Osmundson EC, Koblinski J, Pugacheva E, Golemis EA, Ray D, et al. The WW-HECT protein Smurf2 interacts with the docking protein NEDD9/HEF1 for Aurora A activation. Cell Div. 2010;5:22. doi: 10.1186/1747-1028-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu W, Zhang Y, Lu L, Wang L, Chen M, Hu T. Expression and correlation of hypoxia-inducible factor-1α (HIF-1α) with pulmonary artery remodeling and right ventricular hypertrophy in experimental pulmonary embolism. Med Sci Monit. 2017;23:2083–2088. doi: 10.12659/MSM.900354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Robbins IM, Pugh ME, Hemnes AR. Update on chronic thromboembolic pulmonary hypertension. Trends Cardiovasc Med. 2017;27:29–37. doi: 10.1016/j.tcm.2016.05.010. [DOI] [PubMed] [Google Scholar]
- 9. Bochenek ML, Rosinus NS, Lankeit M, Hobohm L, Bremmer F, Schütz E, et al. From thrombosis to fibrosis in chronic thromboembolic pulmonary hypertension. Thromb Haemost. 2017;117:769–783. doi: 10.1160/TH16-10-0790. [DOI] [PubMed] [Google Scholar]
- 10. Samokhin AO, Stephens T, Wertheim BM, Wang R-S, Vargas SO, Yung L-M, et al. NEDD9 targets COL3A1 to promote endothelial fibrosis and pulmonary arterial hypertension. Sci Transl Med. 2018;10:445. doi: 10.1126/scitranslmed.aap7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lo EH, Hara H, Rogowska J, Trocha M, Pierce AR, Huang PL, et al. Temporal correlation mapping analysis of the hemodynamic penumbra in mutant mice deficient in endothelial nitric oxide synthase gene expression. Stroke. 1996;27:1381–1385. doi: 10.1161/01.str.27.8.1381. [DOI] [PubMed] [Google Scholar]
- 12. Gómez-Outes A, Terleira-Fernández AI, Lecumberri R, Suárez-Gea ML, Vargas-Castrillón E. Direct oral anticoagulants in the treatment of acute venous thromboembolism: a systematic review and meta-analysis. Thromb Res. 2014;134:774–782. doi: 10.1016/j.thromres.2014.06.020. [DOI] [PubMed] [Google Scholar]
- 13. Italiano JE, Jr, Richardson JL, Patel-Hett S, Battinelli E, Zaslavsky A, Short S, et al. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood. 2008;111:1227–1233. doi: 10.1182/blood-2007-09-113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevens JM.Platelet adhesion assays performed under static conditionsGibbins JM, Mahaut-Smith MP.Platelets and megakaryocytes, vol. 1: functional assays. Methods in molecular biology 272Totowa, NJ: Humana Press Inc; 2004. [DOI] [PubMed] [Google Scholar]
- 15. Korporaal SJA, Molenaar TJM, Lutters BCH, Meurs I, Drost-Verhoef S, Kuiper J, et al. Peptide antagonists for P-selectin discriminate between sulfatide-dependent platelet aggregation and PSGL-1-mediated cell adhesion. J Clin Med. 2019;8:E1266. doi: 10.3390/jcm8081266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schleimer RP, Rutledge BK. Cultured human vascular endothelial cells acquire adhesiveness for neutrophils after stimulation with interleukin 1, endotoxin, and tumor-promoting phorbol diesters. J Immunol. 1986;136:649–654. [PubMed] [Google Scholar]
- 17. Comhair SA, Xu W, Mavrakis L, Aldred MA, Asosingh K, Erzurum SC. Human primary lung endothelial cells in culture. Am J Respir Cell Mol Biol. 2012;46:723–730. doi: 10.1165/rcmb.2011-0416TE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 19. Boucherat O, Peterlini T, Bourgeois A, Nadeau V, Breuils-Bonnet S, Boilet-Molez S, et al. Mitochondrial HSP90 accumulation promotes vascular remodeling in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2018;198:90–103. doi: 10.1164/rccm.201708-1751OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Blick C, Ramachandran A, McCormick R, Wigfield S, Cranston D, Catto J, et al. Identification of a hypoxia-regulated miRNA signature in bladder cancer and a role for miR-145 in hypoxia-dependent apoptosis. Br J Cancer. 2015;113:634–644. doi: 10.1038/bjc.2015.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Han T, Yi X-P, Liu B, Ke M-J, Li Y-X. MicroRNA-145 suppresses cell proliferation, invasion and migration in pancreatic cancer cells by targeting NEDD9. Mol Med Rep. 2015;11:4115–4120. doi: 10.3892/mmr.2015.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Law SF, Estojak J, Wang B, Mysliwiec T, Kruh G, Golemis EA. Human enhancer of filamentation 1, a novel p130Cas-like docking protein, associates with focal adhesion kinase and induces pseudohyphal growth in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:3227–3237. doi: 10.1128/mcb.16.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sako D, Comess KM, Barone KM, Camphausen RT, Cumming DA, Shaw GD. A sulfated peptide segment at the amino terminus of PSGL-1 is critical for P-selectin binding. Cell. 1995;83:323–331. doi: 10.1016/0092-8674(95)90173-6. [DOI] [PubMed] [Google Scholar]
- 24. Zabini D, Heinemann A, Foris V, Nagaraj C, Nierlich P, Bálint Z, et al. Comprehensive analysis of inflammatory markers in chronic thromboembolic pulmonary hypertension patients. Eur Respir J. 2014;44:951–962. doi: 10.1183/09031936.00145013. [DOI] [PubMed] [Google Scholar]
- 25. Jin RC, Mahoney CE, Coleman Anderson L, Ottaviano F, Croce K, Leopold JA, et al. Glutathione peroxidase-3 deficiency promotes platelet-dependent thrombosis in vivo. Circulation. 2011;123:1963–1973. doi: 10.1161/CIRCULATIONAHA.110.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thomas HC, Lamé MW, Dunston SK, Segall HJ, Wilson DW. Monocrotaline pyrrole induces apoptosis in pulmonary artery endothelial cells. Toxicol Appl Pharmacol. 1998;151:236–244. doi: 10.1006/taap.1998.8458. [DOI] [PubMed] [Google Scholar]
- 27. Mercier O, Arthur Ataam J, Langer NB, Dorfmüller P, Lamrani L, Lecerf F, et al. Abnormal pulmonary endothelial cells may underlie the enigmatic pathogenesis of chronic thromboembolic pulmonary hypertension. J Heart Lung Transplant. 2017;36:305–314. doi: 10.1016/j.healun.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 28. Chung T, Connor D, Joseph J, Emmett L, Mansberg R, Peters M, et al. Platelet activation in acute pulmonary embolism. J Thromb Haemost. 2007;5:918–924. doi: 10.1111/j.1538-7836.2007.02461.x. [DOI] [PubMed] [Google Scholar]
- 29. Novoyatleva T, Kojonazarov B, Owczarek A, Veeroju S, Rai N, Henneke I, et al. Evidence for the fucoidan/P-selectin axis as a therapeutic target in hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med. 2019;199:1407–1420. doi: 10.1164/rccm.201806-1170OC. [DOI] [PubMed] [Google Scholar]
- 30. Johansson MW, Han S-T, Gunderson KA, Busse WW, Jarjour NN, Mosher DF. Platelet activation, P-selectin, and eosinophil β1-integrin activation in asthma. Am J Respir Crit Care Med. 2012;185:498–507. doi: 10.1164/rccm.201109-1712OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Federici AB, Bader R, Pagani S, Colibretti ML, De Marco L, Mannucci PM. Binding of von Willebrand factor to glycoproteins Ib and IIb/IIIa complex: affinity is related to multimeric size. Br J Haematol. 1989;73:93–99. doi: 10.1111/j.1365-2141.1989.tb00226.x. [DOI] [PubMed] [Google Scholar]
- 32. Mehta P, Cummings RD, McEver RP. Affinity and kinetic analysis of P-selectin binding to P-selectin glycoprotein ligand-1. J Biol Chem. 1998;273:32506–32513. doi: 10.1074/jbc.273.49.32506. [DOI] [PubMed] [Google Scholar]
- 33. González-Tajuelo R, de la Fuente-Fernández M, Morales-Cano D, Muñoz-Callejas A, González-Sánchez E, et al. Spontaneous pulmonary hypertension associated with systemic sclerosis in P-selectin glycoprotein ligand 1-deficient mice. Arthritis Rheumatol. 2020;72:477–487. doi: 10.1002/art.41100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kellermair J, Redwan B, Alias S, Jabkowski J, Panzenboeck A, Kellermair L, et al. Platelet endothelial cell adhesion molecule 1 deficiency misguides venous thrombus resolution. Blood. 2013;122:3376–3384. doi: 10.1182/blood-2013-04-499558. [DOI] [PubMed] [Google Scholar]
- 35. Jujo T, Tanabe N, Sakao S, Ishibashi-Ueda H, Ishida K, Naito A, et al. Severe pulmonary arteriopathy is associated with persistent hypoxemia after pulmonary endarterectomy in chronic thromboembolic pulmonary hypertension. PLoS One. 2016;11:e0161827. doi: 10.1371/journal.pone.0161827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tapson VF, Platt DM, Xia F, Teal SA, de la Orden M, Divers CH, et al. Monitoring for pulmonary hypertension following pulmonary embolism: the INFORM Study. Am J Med. 2016;129:978–985.e2. doi: 10.1016/j.amjmed.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 37. Lang IM, Pesavento R, Bonderman D, Yuan JX-J. Risk factors and basic mechanisms of chronic thromboembolic pulmonary hypertension: a current understanding. Eur Respir J. 2013;41:462–468. doi: 10.1183/09031936.00049312. [DOI] [PubMed] [Google Scholar]
- 38. Brummel NE, Hughes CG, Thompson JL, Jackson JC, Pandharipande P, McNeil JB, et al. Inflammation and coagulation during critical illness and long-term cognitive impairment and disability. Am J Respir Crit Care Med. doi: 10.1164/rccm.201912-2449OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shah PB, Lilly CM. Interventional therapy for coronary artery disease. Am J Respir Crit Care Med. 2002;166:791–796. doi: 10.1164/rccm.200203-220UP. [DOI] [PubMed] [Google Scholar]
- 40. Zhang Y, Handley D, Kaplan T, Yu H, Bais AS, Richards T, et al. High throughput determination of TGFβ1/SMAD3 targets in A549 lung epithelial cells. PLoS One. 2011;6:e20319. doi: 10.1371/journal.pone.0020319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao L, Chen CN, Hajji N, Oliver E, Cotroneo E, Wharton J, et al. Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation. 2012;126:455–467. doi: 10.1161/CIRCULATIONAHA.112.103176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O’Neill GM, Golemis EA. Proteolysis of the docking protein HEF1 and implications for focal adhesion dynamics. Mol Cell Biol. 2001;21:5094–5108. doi: 10.1128/MCB.21.15.5094-5108.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nakamoto T, Seo S, Sakai R, Kato T, Kutsuna H, Kurokawa M, et al. Expression and tyrosine phosphorylation of Crk-associated substrate lymphocyte type (Cas-L) protein in human neutrophils. J Cell Biochem. 2008;105:121–128. doi: 10.1002/jcb.21799. [DOI] [PubMed] [Google Scholar]
- 44. Bennewitz MF, Jimenez MA, Vats R, Tutuncuoglu E, Jonassaint J, Kato GJ, et al. Lung vaso-occlusion in sickle cell disease mediated by arteriolar neutrophil-platelet microemboli. JCI Insight. 2017;2:e89761. doi: 10.1172/jci.insight.89761. [DOI] [PMC free article] [PubMed] [Google Scholar]