

Abstract
Rationale: Elexacaftor/tezacaftor/ivacaftor (ELX/TEZ/IVA) was shown to be efficacious and safe in patients ≥12 years of age with cystic fibrosis and at least one F508del-CFTR (cystic fibrosis transmembrane conductance regulator) allele, but it has not been evaluated in children <12 years of age.
Objectives: To assess the safety, pharmacokinetics, and efficacy of ELX/TEZ/IVA in children 6 through 11 years of age with F508del–minimal function or F508del-F508del genotypes.
Methods: In this 24-week open-label phase 3 study, children (N = 66) weighing <30 kg received 50% of the ELX/TEZ/IVA adult daily dose (ELX 100 mg once daily, TEZ 50 mg once daily, and IVA 75 mg every 12 h) whereas children weighing ⩾30 kg received the full adult daily dose (ELX 200 mg once daily, TEZ 100 mg once daily, and IVA 150 mg every 12 h).
Measurements and Main Results: The primary endpoint was safety and tolerability. The safety and pharmacokinetic profiles of ELX/TEZ/IVA were generally consistent with those observed in older patients. The most commonly reported adverse events included cough, headache, and pyrexia; in most of the children who had adverse events, these were mild or moderate in severity. Through Week 24, ELX/TEZ/IVA treatment improved the percentage of predicted FEV1 (10.2 percentage points; 95% confidence interval [CI], 7.9 to 12.6), Cystic Fibrosis Questionnaire–Revised respiratory domain score (7.0 points; 95% CI, 4.7 to 9.2), lung clearance index2.5 (−1.71 units; 95% CI, −2.11 to −1.30), and sweat chloride (−60.9 mmol/L; 95% CI, −63.7 to −58.2); body mass index-for-age z-score increased over the 24-week treatment period when compared with the pretreatment baseline.
Conclusions: Our results show ELX/TEZ/IVA is safe and efficacious in children 6 through 11 years of age with at least one F508del-CFTR allele, supporting its use in this patient population.
Clinical trial registered with www.clinicaltrials.gov (NCT03691779).
Keywords: cystic fibrosis, elexacaftor, tezacaftor, ivacaftor, child
At a Glance Commentary
Scientific Knowledge on the Subject
Previous studies showed that the CFTR (cystic fibrosis transmembrane conductance regulator) modulator elexacaftor/tezacaftor/ivacaftor (ELX/TEZ/IVA) is safe and efficacious in patients ⩾12 years of age with cystic fibrosis and at least one F508del-CFTR allele. Clinical benefits with ELX/TEZ/IVA exceeded those seen with previous CFTR modulators indicated for this patient population. As the clinical consequences of cystic fibrosis often begin to manifest in early childhood, it is crucial to initiate treatment as early as possible.
What This Study Adds to the Field
To evaluate the safety and efficacy of ELX/TEZ/IVA treatment in younger patients, we conducted a 24-week phase 3 open-label study in children 6 through 11 years of age with cystic fibrosis and at least one F508del-CFTR allele. Our results demonstrate that the safety and efficacy of ELX/TEZ/IVA in these children are consistent with those reported in adults and adolescents with cystic fibrosis, supporting use of ELX/TEZ/IVA in this younger patient population.
Cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in the CFTR (CF transmembrane conductance regulator) gene (1). In some regions of the world, nearly 90% of patients with CF have at least one F508del-CFTR mutation, resulting in decreased quantity and function of the CFTR protein at epithelial surfaces, including in the respiratory tract, pancreas, gastrointestinal system, and sweat glands (2, 3). The clinical consequences of impaired CFTR function generally appear in the first year of life, with pancreatic insufficiency, impaired growth, and progressive lung disease appearing as early childhood manifestations of CF (4–7). Prompt diagnosis in combination with effective treatment starting early in life improves clinical outcomes and life expectancy in patients with this multisystem disease (8, 9).
CFTR modulators are small-molecule therapeutics that treat the underlying cause of CF; correctors improve CFTR protein processing and trafficking, while potentiators enhance channel gating (10, 11). Recently, the efficacy and safety of a triple combination regimen composed of the CFTR correctors elexacaftor and tezacaftor and the CFTR potentiator ivacaftor (ELX/TEZ/IVA) was established in patients with CF ≥12 years of age with at least one F508del-CFTR allele (12, 13). In pivotal studies, treatment with ELX/TEZ/IVA led to robust improvements in lung function, respiratory symptoms, and CFTR function in patients with F508del-minimal function (F/MF) genotypes (13). As minimal function mutations result in either a complete absence of CFTR protein or a protein that cannot be modulated by TEZ and IVA, the responsiveness to ELX/TEZ/IVA in these patients is attributable to the single F508del allele (13). ELX/TEZ/IVA treatment was also found to be highly effective and provided substantially greater efficacy compared with the dual combination of TEZ/IVA in patients homozygous for F508del-CFTR (F/F) (12). These results demonstrate the ability of ELX/TEZ/IVA to address the disease-causing defect present in a large majority of patients with CF ⩾ 12 years of age.
The safety and efficacy of CFTR modulators have also been studied in children with CF. In children <12 years of age, the safety of CFTR modulators is consistent with the safety in patients ≥12 years of age (14–16). IVA treatment improved percentage of predicted FEV1 (ppFEV1) and decreased sweat chloride concentrations in children 6 through 11 years of age with CFTR gating mutations (14). In children 6 through 11 years of age homozygous for F508del, treatment with either TEZ/IVA or the dual combination modulator lumacaftor/IVA improved sweat chloride concentration and Cystic Fibrosis Questionnaire–Revised (CFQ-R) respiratory domain score (15, 16). Although these studies collectively indicate that early intervention with CFTR modulators in children <12 years of age can provide substantial clinical benefit, a critical need remains for more effective CFTR modulation in children <12 years of age who have at least one F508del allele.
Given the robust clinical improvements observed with ELX/TEZ/IVA treatment in patients ⩾12 years of age, as well as the unmet need in younger patients with at least one F508del allele, we evaluated the safety, pharmacokinetics (PK), and efficacy of ELX/TEZ/IVA in children 6 through 11 years of age with F/MF or F/F genotypes. As children in this age range who have the F/F genotype can be treated with approved dual-combination CFTR modulator regimens such as lumacaftor/IVA or TEZ/IVA, we used an open-label study design that prioritized safety data collection and within-group efficacy analysis rather than a blinded, randomized controlled trial design.
Methods
Patients, Trial Design, and Oversight
This phase 3, two-part, open-label, multicenter trial of ELX/TEZ/IVA enrolled children 6 through 11 years of age with CF and either F/MF or F/F genotypes. The CFTR genotype, part of the basis for the diagnosis of CF, was confirmed at screening. Qualifying minimal function mutations (see Table E1 in the online supplement) and other eligibility criteria are provided in the online supplement. All patients stopped or remained off CFTR modulators for ⩾28 days before the Day 1 visit.
Part A of the study evaluated PK, safety, and tolerability of ELX/TEZ/IVA over a 2-week treatment period. Part B evaluated safety, tolerability, efficacy, and PK over a 24-week treatment period (Figure E1). Additional trial design details, including dosing and outcome measures for part A and eligibility criteria for both parts of the study, are provided in the online supplement.
Dosing for part B was based on weight at baseline. Children weighing <30 kg received 50% of the adult daily dose (ELX 100 mg once daily, TEZ 50 mg once daily, and IVA 75 mg every 12 h), whereas children weighing ⩾30 kg received the full adult daily dose (ELX 200 mg once daily, TEZ 100 mg once daily, and IVA 150 mg every 12 h).
The trial was designed by Vertex Pharmaceuticals in collaboration with the authors. For each enrolled child, informed consent was provided by a parent or legal guardian; assent was obtained from patients in accordance with local requirements. Safety was monitored by an independent data safety monitoring committee. Data collection and analysis were performed by Vertex Pharmaceuticals in collaboration with the authors and the VX18-445-106 Study Group. All authors had full access to the trial data after final database lock, critically reviewed the manuscript, and approved the manuscript for submission. The investigators vouch for the accuracy and completeness of the data generated at their respective sites, and the investigators and Vertex Pharmaceuticals vouch for the fidelity of the trial to the protocol. Confidentiality agreements were in place between the sponsor and each investigative site during the trial.
As part B of the study overlapped with the early months of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic, a global protocol addendum that enabled in-home assessments was implemented. Patient access to study drug therapy and collection of safety data were prioritized. Implemented measures were enabled based on country and local regulations, as well as site-level considerations, and included, as applicable, remote consent, shipment of the study drug to the patient’s home, virtual study visits conducted by site personnel via teleconference, home nursing visits for blood draws, in-home assessments, and remote monitoring. Additional details can be found in the online supplement.
The clinical trial protocol, SARS-CoV-2–related protocol addendum, and informed consent forms were approved by independent ethics committees for each region or site, as required by local regulations.
Outcome Measures
The primary endpoint for part B was safety and tolerability. Secondary endpoints included absolute change from baseline through Week 24 in ppFEV1, sweat chloride concentration, and CFQ-R respiratory domain score; absolute change from baseline at Week 24 in body mass index (BMI), weight, height, and corresponding for-age z-scores; number of pulmonary exacerbations and CF-related hospitalizations through Week 24; PK parameters of ELX, TEZ, and IVA; and absolute change from baseline through Week 24 in lung clearance index2.5 (LCI2.5). The proportion of patients achieving sweat chloride concentrations <60 mmol/L and <30 mmol/L was assessed as a post hoc analysis. Additional details are provided in the online supplement.
Statistical Analysis
Safety, efficacy, and PK analyses included children who received ⩾1 dose of ELX/TEZ/IVA. The main analyses were based on data collected up to Week 24; additional analyses were conducted to account for data collected at unscheduled visits beyond Week 24 as detailed in the online supplement. The main analyses for safety were based on all data collected up to Week 24, including both in-clinic and at-home assessments. The main analyses for all efficacy endpoints included only those data that had been collected in clinic through the Week 24 visit.
Absolute change from baseline in ppFEV1, sweat chloride concentration, CFQ-R respiratory domain score, LCI2.5, and nutritional parameters were analyzed using a mixed-effects model for repeated measures. Post hoc analyses of genotype subgroups were performed in a similar manner to the main efficacy analysis. Details of the analysis, including management of missing data resulting from the SARS-CoV-2 pandemic, are provided in the online supplement.
Results
Population
Part A enrollment details and baseline characteristics are provided in Table E2; there were 16 participants and all completed the 2-week treatment period. Part B of the trial was conducted at 21 sites in five countries from August 5, 2019, to August 7, 2020. Overall, 66 children were enrolled and received ⩾1 dose of ELX/TEZ/IVA; 64 children (97.0%) completed the treatment period (Figure 1). Two children discontinued treatment (one adverse event [AE] of erythematous rash and one withdrawal of consent). Baseline characteristics were similar between the F/MF (n = 37, 56.1%) and F/F genotype groups (n = 29, 43.9%) (Tables 1 and E3).
Figure 1.

Patient disposition diagram for part B. *Patient discontinued treatment because of erythematous rash.
Table 1.
Demographics and Clinical Characteristics of the Patients at Baseline in Part B
| ELX/TEZ/IVA |
|||
|---|---|---|---|
| All Patients (N = 66)* | F/MF (N = 37) | F/F (N = 29) | |
| Sex, F, n (%) | 39 (59.1) | 22 (59.5) | 17 (58.6) |
| Age at baseline, mean (SD), yr | 9.3 (1.9) | 9.7 (1.8) | 8.8 (1.9) |
| Race, n (%)† | |||
| White | 58 (87.9) | 33 (89.2) | 25 (86.2) |
| Black or African American | 0 | 0 | 0 |
| Asian | 1 (1.5) | 1 (2.7) | 0 |
| American Indian or Alaska Native | 0 | 0 | 0 |
| Native Hawaiian or other Pacific Islander | 0 | 0 | 0 |
| Other | 0 | 0 | 0 |
| Not collected per local regulations | 8 (12.1) | 4 (10.8) | 4 (13.8) |
| Geographic region, n (%) | |||
| North America | 47 (71.2) | 27 (73.0) | 20 (69.0) |
| Europe and Australia | 19 (28.8) | 10 (27.0) | 9 (31.0) |
| Weight, mean (SD), kg | 30.0 (7.7) | 31.4 (7.9) | 28.2 (7.3) |
| Weight distribution, n (%) | |||
| <30 kg | 36 (54.5) | 20 (54.1) | 16 (55.2) |
| ⩾30 kg | 30 (45.5) | 17 (45.9) | 13 (44.8) |
| Weight-for-age z-score, mean (SD) | −0.22 (0.76) | −0.20 (0.87) | −0.23 (0.59) |
| Height, mean (SD), cm | 134.1 (12.3) | 136.9 (12.0) | 130.4 (11.9) |
| Height-for-age z-score, mean (SD) | −0.11 (0.98) | −0.01 (1.03) | −0.23 (0.91) |
| BMI, mean (SD), kg/m2 | 16.39 (1.69) | 16.50 (1.77) | 16.26 (1.61) |
| BMI-for-age z-score, mean (SD) | −0.16 (0.74) | −0.21 (0.84) | −0.10 (0.61) |
| ppFEV1, mean (SD)‡ | 88.8 (17.7) | 89.8 (17.5) | 87.3 (18.3) |
| ppFEV1 category, n (%) | |||
| <70 | 10 (15.2) | 5 (13.5) | 5 (17.2) |
| ⩾70–⩽90 | 22 (33.3) | 15 (40.5) | 7 (24.1) |
| >90 | 30 (45.5) | 17 (45.9) | 13 (44.8) |
| Missing data | 4 (6.1) | 0 | 4 (13.8) |
| Sweat chloride concentration, mean (SD), mmol/L§ | 102.2 (9.1) | 104.4 (7.2) | 99.3 (10.8) |
| CFQ-R respiratory domain score, mean (SD) points‖ | 80.3 (15.2) | 79.1 (17.3) | 81.8 (12.0) |
| LCI2.5, mean (SD), units¶ | 9.77 (2.68) | 9.34 (1.82) | 10.26 (3.36) |
Definition of abbreviations: BMI = body mass index; CFQ-R = Cystic Fibrosis Questionnaire–Revised; CFTR = cystic fibrosis transmembrane conductance regulator; ELX/TEZ/IVA = elexacaftor/tezacaftor/ivacaftor; F/F = homozygous for the F508del-CFTR mutation; F/MF = heterozygous for the F508del-CFTR mutation and a minimal function CFTR mutation; LCI2.5 = lung clearance index2.5; ppFEV1 = percentage of predicted FEV1.
Baseline was defined as the most recent nonmissing measurement before the first dose of study drug.
All patients in the full analysis set.
The race categories may sum to >100% because each patient was able to indicate more than one race.
n = 62 for all patients; n = 37 for F/MF; n = 25 for F/F.
n = 62 for all patients; n = 36 for F/MF; n = 26 for F/F.
n = 65 for all patients; n = 37 for F/MF; n = 28 for F/F. Scores on the CFQ-R range from 0 to 100, with higher scores indicating a higher patient-reported quality of life with regard to respiratory status.
n = 53 for all patients; n = 28 for F/MF; n = 25 for F/F.
Safety and Pharmacokinetics
Safety and tolerability were the primary endpoints for part B of the study. Sixty-five children (98.5%) had AEs, which for most were mild (54.5%) or moderate (42.4%) in severity and generally consistent with manifestations of CF or common childhood infections (Table 2). The most commonly reported AEs included cough, headache, and pyrexia. One child experienced serious AEs in the study; these were concurrent events of rhinovirus infection, metapneumovirus infection, and pneumonia (which was resolved with intravenous antibiotics).
Table 2.
Adverse Events in Part B
| ELX/TEZ/IVA (N = 66) | |
|---|---|
| Any AE | 65 (98.5) |
| AE by maximum relatedness* | |
| Not related | 16 (24.2) |
| Unlikely related | 16 (24.2) |
| Possibly related | 29 (43.9) |
| Related | 4 (6.1) |
| AE by maximum severity* | |
| Mild | 36 (54.5) |
| Moderate | 28 (42.4) |
| Severe | 1 (1.5) |
| Serious AE | 1 (1.5) |
| AE leading to death | 0 |
| AE leading to discontinuation | 1 (1.5) |
| AE leading to interruption | 1 (1.5)† |
| Most common AEs‡ | |
| Cough | 28 (42.4) |
| Headache | 16 (24.2) |
| Pyrexia | 14 (21.2) |
| Oropharyngeal pain | 12 (18.2) |
| Upper respiratory tract infection | 11 (16.7) |
| Nasal congestion | 10 (15.2) |
| Rash | 8 (12.1) |
| Abdominal pain | 8 (12.1) |
| Rhinorrhea | 8 (12.1) |
| Viral upper respiratory tract infection | 8 (12.1) |
| Alanine aminotransferase increased | 7 (10.6) |
| Diarrhea | 7 (10.6) |
| Influenza | 7 (10.6) |
| Vomiting | 7 (10.6) |
Definition of abbreviations: AE = adverse event; ELX/TEZ/IVA = elexacaftor/tezacaftor/ivacaftor.
Data are presented as n (%).
A patient with multiple events within a category was counted only once in that category.
Relatedness to the trial regimen and severity were determined by the investigator observing the event.
The one AE leading to study drug interruption was because of diarrhea, vomiting, and fever.
Only AEs that occurred in ⩾10% of the patients are listed; listing is according to the preferred term (Medical Dictionary for Regulatory Activities version 23.0).
On the basis of previous experience with ELX/TEZ/IVA, including the phase 3 trials (12, 13), data related to aminotransferases, rash events, creatine kinase, and blood pressure were reviewed. Elevated levels of alanine aminotransferase and/or aspartate aminotransferase that were greater than three times the upper limit of the age-specific normal range occurred in seven children (10.6%); among these, only one child (1.5%) had an elevation that was greater than five times the upper limit, and no child had an elevation that was greater than eight times the upper limit (Table E4). No children had an elevated aminotransferase level greater than three times the upper limit of the normal range concurrent with an elevated bilirubin level greater than two times the upper limit of the normal range. Seven children had AEs of transaminase elevations, all of which were mild or moderate in severity. There were no study drug interruptions or discontinuations due to elevated transaminases.
Sixteen children (24.2%) had rash events (Table E5). Rash events constitute a group AE term that includes the preferred term of rash (Table 2) and other rash-related preferred terms (e.g., rash erythematous, rash maculopapular, rash papular, skin exfoliation, and urticaria). All rash events were mild or moderate in severity. Among these 16 children, 1 child had an erythematous rash that developed after the first dose of ELX/TEZ/IVA and led to study drug discontinuation; the rash was moderate in severity and resolved 2 days after study drug discontinuation. All other rash events resolved without change in study drug dosing. For eight of the children, the rash events were assessed as unlikely to be related or not related to the study drug. Of the remaining children with rash events, four had associated symptoms suggestive of an alternative etiology (e.g., concurrent infection or heat rash).
No children had creatine kinase levels greater than five times the upper limit of the age-specific normal range (Table E6), and two children had AEs related to creatine kinase. The mean change from baseline for systolic blood pressure through 24 weeks of ELX/TEZ/IVA treatment ranged from −1.4 mm Hg to 0.4 mm Hg, and that for diastolic blood pressure ranged from −0.3 mm Hg to 1.0 mm Hg (Table E7); no AEs related to blood pressure were reported. There were no relevant safety findings in other clinical or laboratory assessments.
Preliminary analysis of PK data from part A (online supplement) confirmed the planned part B dosing scheme. In part B, definitive population PK analyses for 6- through 11-year-old children with CF revealed that the area under the concentration-versus-time curve (AUC) values for ELX, TEZ, and IVA were within the range of exposures previously seen in those ⩾12 years of age (Figure 2), as were the mean observed trough and peak concentrations. These PK simulations confirmed that 30 kg was the optimal weight threshold for the transition from 50% of the adult daily dose to the full adult daily dose of ELX/TEZ/IVA.
Figure 2.
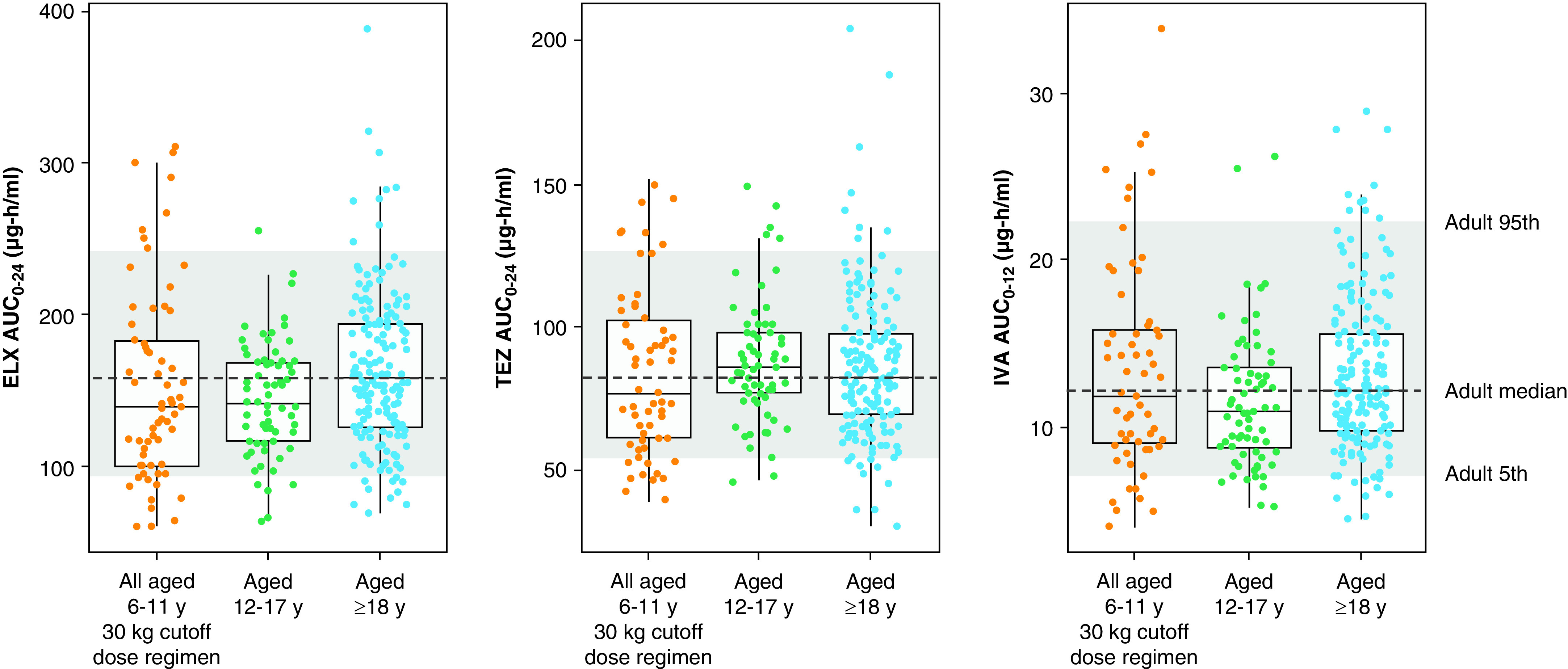
Pharmacokinetic exposure simulation. For each boxplot, the median is represented by a horizontal line, and the interquartile range is represented by the box. The whiskers mark the largest and smallest values within 1.5 × interquartile range. Dots represent individual empirical Bayes AUC estimate (EBE) values. In each panel, the dashed horizontal line represents the median of the ⩾18 years AUC values, and the gray shaded area indicates the 5th and 95th percentiles of the ⩾18 years AUC values. The EBE values for 6–11 years are from part B of this study. The EBE values for ⩾12 years are from studies 445-102 and 445-103 (12, 13). AUC = area under the concentration-versus-time curve; AUC0–12 = area under the concentration-versus-time curve (0–12 h after dose); AUC0–24 = area under the concentration-versus-time curve (0–24 h after dose); ELX = elexacaftor; IVA = ivacaftor; TEZ = tezacaftor.
Efficacy
Overall
Treatment with ELX/TEZ/IVA led to a mean absolute change from a baseline of 10.2 percentage points in ppFEV1 through Week 24 (95% confidence interval [CI], 7.9 to 12.6) (Table 3), with robust improvements seen as soon as 2 weeks after initiation (Figure 3A). Similarly, rapid and sustained improvements were seen in CFQ-R respiratory domain scores (mean absolute change from baseline of 7.0 points; 95% CI, 4.7 to 9.2) (Figure 3B and Table 3), LCI2.5 (mean absolute change from baseline of −1.71 units; 95% CI, −2.11 to −1.30) (Figure 3C and Table 3), and sweat chloride concentration (mean absolute change from baseline of −60.9 mmol/L through Week 24; 95% CI, −63.7 to −58.2) (Figure 3D and Table 3). BMI, BMI-for-age z-score, weight, weight-for-age z-score, and height increased over the 24-week treatment period without reaching a plateau, whereas height-for-age z-score was maintained (Figure 4 and Table E8).
Table 3.
Secondary Efficacy Endpoints from Part B
| ELX/TEZ/IVA |
|||
|---|---|---|---|
| All Patients (N = 66)* | F/MF (N = 37) | F/F (N = 29) | |
| ppFEV1, percentage points | |||
| Baseline, mean (SD)† | 88.8 (17.7) | 89.8 (17.5) | 87.3 (18.3) |
| Absolute change through Week 24, LS mean (95% CI) | 10.2 (7.9 to 12.6)‡ | 9.1 (6.3 to 11.9)‡ | 11.2 (7.2 to 15.2)‡ |
| CFQ-R respiratory domain score, points | |||
| Baseline, mean (SD)† | 80.3 (15.2) | 79.1 (17.3) | 81.8 (12.0) |
| Absolute change through Week 24, LS mean (95% CI) | 7.0 (4.7 to 9.2)‡ | 6.9 (3.2 to 10.6)‡ | 7.0 (3.9 to 10.1)‡ |
| LCI2.5, units | |||
| Baseline, mean (SD)† | 9.77 (2.68) | 9.34 (1.82) | 10.26 (3.36) |
| Absolute change through Week 24, LS mean (95% CI) | −1.71 (−2.11 to −1.30)‡ | −1.72 (−2.11 to −1.33)‡ | −1.64 (−2.34 to −0.94)‡ |
| Sweat chloride, mmol/L | |||
| Baseline, mean (SD)† | 102.2 (9.1) | 104.4 (7.2) | 99.3 (10.8) |
| Absolute change through Week 24, LS mean (95% CI) | −60.9 (−63.7 to −58.2)‡ | −55.1 (−59.0 to −51.2)‡ | −70.4 (−75.6 to −65.3)‡ |
Definition of abbreviations: CFQ-R = Cystic Fibrosis Questionnaire–Revised; CFTR = cystic fibrosis transmembrane conductance regulator; CI = confidence interval; ELX/TEZ/IVA = elexacaftor/tezacaftor/ivacaftor; F/F = homozygous for the F508del-CFTR mutation; F/MF = heterozygous for the F508del-CFTR mutation and a minimal function CFTR mutation; LCI2.5 = lung clearance index2.5; LS = least squares; ppFEV1 = percentage of predicted FEV1.
N indicates the total number of patients in each population; LS means were based on mixed-effects model for repeated measures; absolute changes through Week 24 were averages of the visits between Weeks 4 and 24 (inclusive).
All patients in the full analysis set.
Baseline was defined as the most recent nonmissing measurement before the first dose of study drug.
Nominal P value <0.001.
Figure 3.
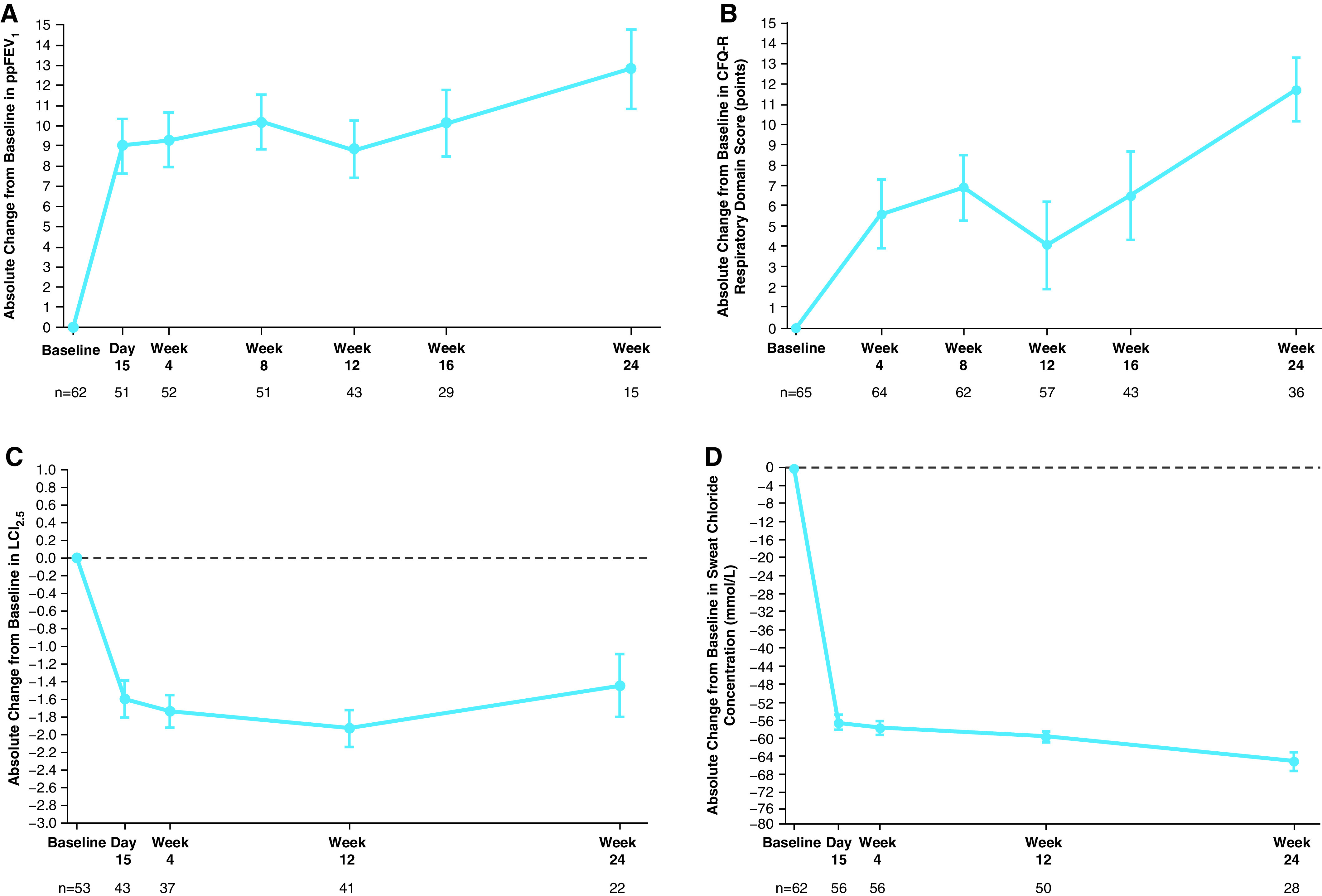
Efficacy results, by visit, in part B. (A) Absolute change from baseline at each visit in ppFEV1. (B) Absolute change from baseline at each visit in the respiratory domain score on the CFQ-R, child’s version; scores are normalized to a 100-point range, with higher scores indicating a higher patient-reported quality of life with regard to respiratory symptoms. (C) Absolute change from baseline at each visit in LCI2.5. (D) Absolute change from baseline at each visit in sweat chloride concentration; a reduction indicates improvement in CFTR (cystic fibrosis transmembrane conductance regulator) function. Data are least squares means based on a mixed-effects model for repeated measures, and error bars indicate SEMs; the dashed line indicates no change from baseline. Sample size shown under each x-axis is the number of patients at the time point with evaluable in-clinic data. CFQ-R = Cystic Fibrosis Questionnaire–Revised; LCI2.5 = lung clearance index2.5; ppFEV1 = percentage of predicted FEV1.
Figure 4.
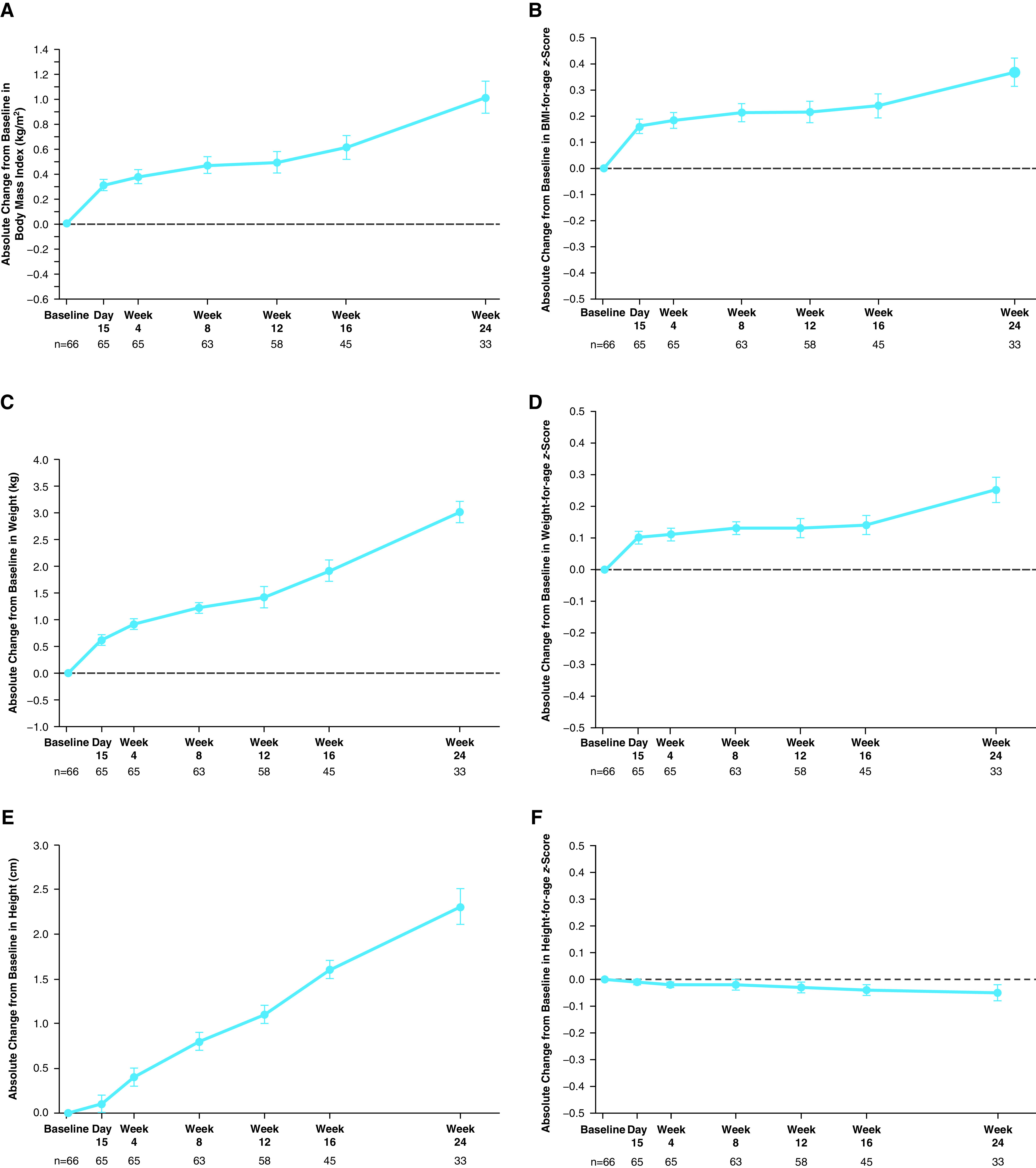
Absolute change from baseline at each visit in (A) BMI, (B) BMI-for-age z-score, (C) weight, (D) weight-for-age z-score, (E) height, and (F) height-for-age z-score in part B. Data are least squares means based on a mixed-effects model for repeated measures, and error bars indicate SEMs; the dashed line indicates no change from baseline. Sample size shown under each x-axis is the number of patients at the time point with evaluable in-clinic data. BMI = body mass index.
Subgroup analysis
The F/MF and F/F genotype groups showed similar improvements in ppFEV1, CFQ-R respiratory domain score, and LCI2.5 (Table 3). In contrast, the decrease in mean sweat chloride concentration was greater in the F/F genotype group (−70.4 mmol/L; 95% CI, −75.6 to −65.3) than in the F/MF genotype group (−55.1 mmol/L; 95% CI, −59.0 to −51.2) (Table 3). The distribution of individual changes from baseline in sweat chloride concentration showed clear separation by genotype, consistent with greater reductions among those with the F/F genotype (Figure E2). At baseline, no children had sweat chloride concentrations below the diagnostic threshold for CF of 60 mmol/L (Table E9) (5). For both the F/MF and F/F genotype groups, mean sweat chloride concentrations fell to below the diagnostic threshold of 60 mmol/L by Day 15, and for the F/F group, concentrations approached 30 mmol/L (Figure E3). Reductions in sweat chloride concentrations to <60 mmol/L and <30 mmol/L in response to ELX/TEZ/IVA treatment were assessed as a post hoc analysis and found to be more prevalent among children with the F/F genotype (100.0% and 42.9%, respectively) as compared with children with F/MF genotypes (80.0% and 5.7%, respectively) (Figure 5 and Table E9).
Figure 5.

Responder analysis for sweat chloride concentration by genotype group in part B. The percentage of children in each genotype group with sweat chloride concentrations <60 mmol/L or <30 mmol/L through Week 24 is shown. No children had sweat chloride concentrations <60 mmol/L at baseline. Percentages were calculated by dividing n (the number of patients with sweat chloride concentration below the indicated threshold) by N1, where N1 is the number of patients with evaluable data. Patients with missing data were considered missing at random and were not counted in the denominator. F/F = homozygous for the F508del-CFTR mutation; F/MF = heterozygous for the F508del-CFTR mutation and a minimal function CFTR mutation.
Discussion
We evaluated the safety and efficacy of ELX/TEZ/IVA in children 6 through 11 years of age with CF and at least one F508del-CFTR allele, the youngest patients treated with a three-drug CFTR modulator regimen to date. Treatment with ELX/TEZ/IVA was generally safe and well tolerated; the most common reported AEs were consistent with manifestations of CF or common childhood infections. The safety profile in children 6 through 11 years of age was consistent with the established profile in patients ⩾12 years of age with CF.
As ELX/TEZ/IVA is likely to be a long-term therapy in children with CF, understanding the incidence of laboratory and clinical events that may occur during drug exposure is important. Children in this study were found to have an incidence of aminotransferase elevation that was similar to the incidence previously observed in patients ⩾12 years of age treated with ELX/TEZ/IVA for 24 weeks (13) and in children aged 6 through 11 years treated with TEZ/IVA for 24 weeks (15). The rashes observed during this study were mild to moderate in severity, and the vast majority were transient and resolved with continued ELX/TEZ/IVA treatment.
Pharmacokinetic and clinical evaluation confirmed the appropriateness of a dose that is 50% of the adult daily dose of ELX/TEZ/IVA for children 6 through 11 years of age who are <30 kg and of the full adult dose for those ⩾30 kg. For these weight subgroups of children aged 6 through 11 years, exposures to ELX, TEZ, and IVA were within similar AUC ranges as those seen in patients ⩾12 years of age with CF.
Children in this study had substantially higher mean baseline lung function (ppFEV1, 88.8 percentage points) and CFQ-R respiratory domain scores (80.3 points) than seen in the phase 3 pivotal studies in adults and adolescents (ppFEV1, ∼62 percentage points; CFQ-R respiratory domain scores, ∼70 points) (12, 13). Despite these higher baselines, ELX/TEZ/IVA treatment of these children for 24 weeks led to a 10.2–percentage point improvement in mean ppFEV1 and a 7.0-point improvement in mean CFQ-R respiratory domain score. Rapid improvements in ppFEV1 were seen within 2 weeks of ELX/TEZ/IVA initiation, consistent with results from other CFTR modulator studies (12, 13, 17–19). The improvements in ppFEV1 were corroborated by rapid and sustained improvements in LCI2.5, a measure of ventilation inhomogeneity that may be more sensitive than spirometry in detecting lung function changes during childhood. LCI2.5 has also previously been shown to improve with CFTR modulator treatment in children homozygous for the F508del allele (20). ELX/TEZ/IVA treatment over 24 weeks led to an LCI2.5 improvement of −1.71 units. In contrast, a longitudinal natural history study of children with CF not being treated with CFTR modulators showed an increase in LCI2.5 of 0.21 units per year (higher values of LCI2.5 reflect worse lung function) in children 6 through 11 years of age (21). Registry data have shown that long-term use of IVA slows lung function decline and lowers the risk of death (22, 23). Thus, early initiation of ELX/TEZ/IVA in children with CF is likely to improve lung function and minimize worsening of ppFEV1 and LCI2.5 associated with disease progression (7).
Maintaining or improving nutritional status is associated with better lung function and increased survival rates in patients with CF (24, 25). During this 24-week study, increases in both BMI-for-age and weight-for-age z-scores were seen, whereas height-for-age z-score was maintained. The improvements in BMI and weight, as well as their corresponding for-age z-scores, were greater than those observed in a previous 24-week trial of a dual-combination modulator regimen in children who were 6 through 11 years old and homozygous for F508del-CFTR (16), indicating ELX/TEZ/IVA may confer nutritional benefits greater than those of CFTR modulators that are currently approved for use in children.
Beyond respiratory and nutritional outcomes, changes in sweat chloride concentration provide a direct indicator of systemic CFTR function (3). In this study, ELX/TEZ/IVA treatment of children with CF led to greater improvements in sweat chloride than those previously seen in adults and adolescents (12, 13). Children homozygous for F508del-CFTR had a greater decrease in sweat chloride than those with a single F508del-CFTR allele, consistent with genotype-related differences seen in adolescents and adults (12, 13) and likely attributable to increased abundance of the F508del-CFTR protein that can be targeted for CFTR modulation in homozygotes. Reductions of sweat chloride concentrations to <60 mmol/L (the threshold for definitive diagnosis of CF [5]) and to <30 mmol/L (matching levels generally seen in the population of asymptomatic carriers with a single mutant CFTR allele [26], and a level below which a diagnosis of CF is unlikely [27]) were more prevalent in children with the F/F genotype than in those with F/MF genotypes. In a natural history study, sweat chloride concentrations <60 mmol/L were associated with improved survival in patients with CF (28), suggesting that substantial improvements in sweat chloride in response to CFTR modulator therapy may presage improvements in long-term clinical outcomes.
Because this study did not include a placebo group, the incidence of safety events attributable to the underlying disease process was not ascertained. Furthermore, the small sample size of this study limited the ability to detect uncommon and rare AEs. However, placebo groups in trials with similar populations provide perspective on safety and tolerability (20, 29), and the safety of ELX/TEZ/IVA is well established in children and adolescents ≥12 years of age (12, 13). In addition, the ongoing 96-week open-label extension study (NCT04183790) will assess the long-term safety and durability of efficacy of ELX/TEZ/IVA in children who began treatment at 6 through 11 years of age. Another potential limitation of this and other CF clinical studies is that patients from minority groups are less likely to be eligible because the F508del-CFTR mutation that was the focus of this study is less common in these populations (30).
As this study overlapped with the early months of the SARS-CoV-2 pandemic, a global protocol addendum was implemented that enabled in-home assessments. The safety results reported here were based on both in-clinic and in-home safety assessments. However, only a small number of in-home efficacy assessments were performed, and the efficacy results presented here were based solely on in-clinic assessments. Additional sensitivity analyses that pooled in-clinic and in-home efficacy data gave results consistent with the main efficacy analyses.
The efficacy and safety of ELX/TEZ/IVA in children are consistent with those reported in the controlled phase 3 pivotal studies in adults and adolescents with CF (12, 13) and further confirm the ability of ELX/TEZ/IVA to modulate a single F508del-CFTR allele in patients with CF. Taken together, these results strongly support the use of ELX/TEZ/IVA in children 6 through 11 years of age with at least one F508del-CFTR allele to provide effective treatment of CF at an early stage of disease when serious long-term complications may be averted.
Acknowledgments
Acknowledgment
The authors thank the patients and their families for participating in this trial; all site trial investigators and coordinators; the Australian, North American, and European Lung Clearance Index Central Over-Reading Centres teams for their contributions to the trial; the Cystic Fibrosis Foundation Therapeutics Development Network and the European Cystic Fibrosis Society Clinical Trials Network for their support of the trial sites; Emily Poulin, Ph.D., an employee of Vertex Pharmaceuticals, who may own stock or stock options in the company, for providing editorial coordination and support; Fanuel Hagos, Ph.D., an employee of Vertex Pharmaceuticals, who may own stock or stock options in the company, for providing support with the clinical pharmacological analysis; and Nathan Blow, Ph.D., of Vertex Pharmaceuticals, who may own stock or stock options in the company, and JoAnna Anderson, Ph.D., CMPP, of ArticulateScience LLC for providing editorial assistance under the guidance of the authors and with support from Vertex Pharmaceuticals.
Footnotes
Supported by Vertex Pharmaceuticals.
Author Contributions: The study sponsor (Vertex Pharmaceuticals Incorporated) designed the protocol in collaboration with the academic authors. Site investigators collected the data, which were analyzed by the sponsor. All authors had full access to the study data. E.T.Z., J.L.T.-C., C.E.W., and S.A.M. developed the initial draft of the manuscript, with writing assistance from the sponsor. All authors participated in subsequent revisions. All authors approved the final version submitted for publication.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202102-0509OC on March 18, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
Contributor Information
Collaborators: for the VX18-445-106 Study Group
References
- 1. Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 2.Cystic Fibrosis Foundation. Bethesda, MD: Cystic Fibrosis Foundation; 2020. https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2019-Patient-Registry-Annual-Data-Report.pdf [Google Scholar]
- 3. Elborn JS. Cystic fibrosis. Lancet. 2016;388:2519–2531. doi: 10.1016/S0140-6736(16)00576-6. [DOI] [PubMed] [Google Scholar]
- 4. Bell SC, Mall MA, Gutierrez H, Macek M, Madge S, Davies JC, et al. The future of cystic fibrosis care: a global perspective. Lancet Respir Med. 2020;8:65–124. doi: 10.1016/S2213-2600(19)30337-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ratjen F, Bell SC, Rowe SM, Goss CH, Quittner AL, Bush A. Cystic fibrosis. Nat Rev Dis Primers. 2015;1:15010. doi: 10.1038/nrdp.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schlüter DK, Griffiths R, Adam A, Akbari A, Heaven ML, Paranjothy S, et al. Impact of cystic fibrosis on birthweight: a population based study of children in Denmark and Wales. Thorax. 2019;74:447–454. doi: 10.1136/thoraxjnl-2018-211706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. VanDevanter DR, Kahle JS, O’Sullivan AK, Sikirica S, Hodgkins PS. Cystic fibrosis in young children: a review of disease manifestation, progression, and response to early treatment. J Cyst Fibros. 2016;15:147–157. doi: 10.1016/j.jcf.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 8. Farrell PM, Kosorok MR, Rock MJ, Laxova A, Zeng L, Lai HC, et al. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Pediatrics. 2001;107:1–13. doi: 10.1542/peds.107.1.1. [DOI] [PubMed] [Google Scholar]
- 9. Lopes-Pacheco M. CFTR modulators: the changing face of cystic fibrosis in the era of precision medicine. Front Pharmacol. 2020;10:1662. doi: 10.3389/fphar.2019.01662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boyle MP, De Boeck K. A new era in the treatment of cystic fibrosis: correction of the underlying CFTR defect. Lancet Respir Med. 2013;1:158–163. doi: 10.1016/S2213-2600(12)70057-7. [DOI] [PubMed] [Google Scholar]
- 11. Mall MA, Mayer-Hamblett N, Rowe SM. Cystic fibrosis: emergence of highly effective targeted therapeutics and potential clinical implications. Am J Respir Crit Care Med. 2020;201:1193–1208. doi: 10.1164/rccm.201910-1943SO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, et al. VX17-445-103 Trial Group. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet. 2019;394:1940–1948. doi: 10.1016/S0140-6736(19)32597-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, et al. VX17-445-102 Study Group. Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. 2019;381:1809–1819. doi: 10.1056/NEJMoa1908639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, et al. VX08-770-103 (ENVISION) Study Group. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187:1219–1225. doi: 10.1164/rccm.201301-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Walker S, Flume P, McNamara J, Solomon M, Chilvers M, Chmiel J, et al. VX15-661-113 Investigator Group. A phase 3 study of tezacaftor in combination with ivacaftor in children aged 6 through 11 years with cystic fibrosis. J Cyst Fibros. 2019;18:708–713. doi: 10.1016/j.jcf.2019.06.009. [DOI] [PubMed] [Google Scholar]
- 16. Milla CE, Ratjen F, Marigowda G, Liu F, Waltz D, Rosenfeld M. VX13-809-011 Part B Investigator Group. Lumacaftor/ivacaftor in patients aged 6–11 years with cystic fibrosis and homozygous for F508del-CFTR. Am J Respir Crit Care Med. 2017;195:912–920. doi: 10.1164/rccm.201608-1754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al. VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taylor-Cousar JL, Munck A, McKone EF, van der Ent CK, Moeller A, Simard C, et al. Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med. 2017;377:2013–2023. doi: 10.1056/NEJMoa1709846. [DOI] [PubMed] [Google Scholar]
- 19. Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. TRAFFIC Study Group; TRANSPORT Study Group. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373:220–231. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ratjen F, Hug C, Marigowda G, Tian S, Huang X, Stanojevic S, et al. VX14-809-109 investigator group. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir Med. 2017;5:557–567. doi: 10.1016/S2213-2600(17)30215-1. [DOI] [PubMed] [Google Scholar]
- 21. Frauchiger BS, Binggeli S, Yammine S, Spycher B, Krüger L, Ramsey KA, et al. Longitudinal course of clinical lung clearance index in children with cystic fibrosis. Eur Respir J. doi: 10.1183/13993003.02686-2020. [DOI] [PubMed] [Google Scholar]
- 22. Bessonova L, Volkova N, Higgins M, Bengtsson L, Tian S, Simard C, et al. Data from the US and UK cystic fibrosis registries support disease modification by CFTR modulation with ivacaftor. Thorax. 2018;73:731–740. doi: 10.1136/thoraxjnl-2017-210394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sawicki GS, McKone EF, Pasta DJ, Millar SJ, Wagener JS, Johnson CA, et al. Sustained Benefit from ivacaftor demonstrated by combining clinical trial and cystic fibrosis patient registry data. Am J Respir Crit Care Med. 2015;192:836–842. doi: 10.1164/rccm.201503-0578OC. [DOI] [PubMed] [Google Scholar]
- 24. Konstan MW, Butler SM, Wohl ME, Stoddard M, Matousek R, Wagener JS, et al. Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Growth and nutritional indexes in early life predict pulmonary function in cystic fibrosis. J Pediatr. 2003;142:624–630. doi: 10.1067/mpd.2003.152. [DOI] [PubMed] [Google Scholar]
- 25. Stallings VA, Stark LJ, Robinson KA, Feranchak AP, Quinton H. Clinical Practice Guidelines on Growth and Nutrition Subcommittee; Ad Hoc Working Group. Evidence-based practice recommendations for nutrition-related management of children and adults with cystic fibrosis and pancreatic insufficiency: results of a systematic review. J Am Diet Assoc. 2008;108:832–839. doi: 10.1016/j.jada.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 26. Wilschanski M, Dupuis A, Ellis L, Jarvi K, Zielenski J, Tullis E, et al. Mutations in the cystic fibrosis transmembrane regulator gene and in vivo transepithelial potentials. Am J Respir Crit Care Med. 2006;174:787–794. doi: 10.1164/rccm.200509-1377OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farrell PM, White TB, Ren CL, Hempstead SE, Accurso F, Derichs N, et al. Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation J Pediatr 2017181SS4–S15, e1.. [Published erratum appears in J Pediatr 184:243.] [DOI] [PubMed] [Google Scholar]
- 28. McKone EF, Velentgas P, Swenson AJ, Goss CH. Association of sweat chloride concentration at time of diagnosis and CFTR genotype with mortality and cystic fibrosis phenotype. J Cyst Fibros. 2015;14:580–586. doi: 10.1016/j.jcf.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 29. Davies JC, Sermet-Gaudelus I, Naehrlich L, Harris RS, Campbell D, Ahluwalia N, et al. VX16-661-115 Investigator Group. A phase 3, double-blind, parallel-group study to evaluate the efficacy and safety of tezacaftor in combination with ivacaftor in participants 6 through 11 years of age with cystic fibrosis homozygous for F508del or heterozygous for the F508del-CFTR mutation and a residual function mutation. J Cyst Fibros. 2021;20:68–77. doi: 10.1016/j.jcf.2020.07.023. [DOI] [PubMed] [Google Scholar]
- 30. McGarry ME, McColley SA. Cystic fibrosis patients of minority race and ethnicity less likely eligible for CFTR modulators based on CFTR genotype. Pediatr Pulmonol. 2021;56:1496–1503. doi: 10.1002/ppul.25285. [DOI] [PMC free article] [PubMed] [Google Scholar]