

Abstract
Cigar tobacco leaves (CTLs) contain abundant bacteria and fungi that are vital to leaf quality during fermentation. In this study, artificial fermentation was used for the fermentation of CTLs since it was more controllable and efficient than natural aging. The bacterial and fungal community structure and composition in unfermented and fermented CTLs were determined to understand the effects of microbes on the characteristics of CTLs during artificial fermentation. The relationship between the chemical contents and alterations in the microbial composition was evaluated, and the functions of bacteria and fungi in fermented CTLs were predicted to determine the possible metabolic pathways. After artificial fermentation, the bacterial and fungal community structure significantly changed in CTLs. The total nitrate and nicotine contents were most readily affected by the bacterial and fungal communities, respectively. FAPROTAX software predictions of the bacterial community revealed increases in functions related to compound transformation after fermentation. FUNGuild predictions of the fungal community revealed an increase in the content of saprotrophic fungi after fermentation. These data provide information regarding the artificial fermentation mechanism of CTLs and will inform safety and quality improvements.
Keywords: artificial fermentation, chemical compositions, cigar tobacco leaves, function prediction, microbial community
Tobacco‐associated bacteria and fungi affect the aroma and quality of cigars. The microbial community structure significantly changed in cigar tobacco leaves after artificial fermentation. The total nitrate and nicotine contents were altered by the bacterial and fungal communities, respectively.

1. INTRODUCTION
Cigar tobacco has a long history of cultivation (Bauk, 2016). Unlike tobacco leaves for cigarettes, which are flue‐cured, tobacco leaves for cigars are air‐cured, and this difference is the main reason for the unique flavor and aroma characteristic of cigars (Burns, 1998; Sun et al., 2019). In general, after air‐curing, cigar tobacco leaves (CTLs) must additionally be fermented, to retard pungent and irritating odors, before being used for cigar products (Frankenburg, 1950; Huang et al., 2010). Tobacco leaves are fermented by natural aging or artificial fermentation (Lawrence & Bahnson, 1914). The quality of naturally aged tobacco is better than that of artificially fermented tobacco, but natural aging takes a long time (typically, 1–2 years) (Keller, 1929; Liu et al., 2016). The temperature and humidity of the aging room are easily influenced by external weather conditions, which may induce tobacco mildew and insect damage (Li et al., 2007). Compared with natural aging, artificial fermentation can shorten the fermentation cycle to 4–8 weeks, by controlling moisture and temperature (Keller, 1929). Thus, artificial fermentation can be beneficial in reducing storage costs compared with natural aging.
Tobacco aging is complex and involves the enzymatic action of microorganisms and chemical interactions in tobacco leaves (Zhao et al., 2007). The microbial community and its activity are important in improving tobacco quality. An integrated study of microbial community changes in CTLs may suggest the mechanism responsible for the sensory quality of CTLs during the fermentation process. Tobacco leaves contain many microorganisms (Zhou et al., 2020). Bacillus, Pseudomonas, Enterobacter, Sphingomonas, Pantoea, and Methylobacterium are the dominant genera in tobacco leaves (Huang et al., 2010; Zhou et al., 2020). These bacteria degrade macromolecular organic matter in tobacco leaves during fermentation. For example, Pseudomonas spp. can effectively degrade nicotine (Zhong et al., 2010) and Bacillus spp. can produce small fragrant substances by decomposing macromolecular substances, such as carotene (Maldonado‐Robledo et al., 2003). The fungal species identified in tobacco leaves are less diverse and less numerous than the bacterial species. Aspergillus, Phoma, Alternaria, Monographella, and Cladosporium are the major fungal genera in tobacco (Liu et al., 2020; Shanyi et al., 2018). Much of the recent literature concerning microorganisms associated with tobacco leaves has focused on the natural aging of flue‐cured tobacco leaves (Huang et al., 2010; Maldonado‐Robledo et al., 2003; Zhao et al., 2007; Zhou et al., 2020); CTLs have not been as extensively studied. Data from systematic research on the microbial community in CTLs during artificial fermentation would be valuable in cigar manufacturing.
In this study, the microbial community diversity of CTLs planted in Hainan, China, was investigated, and the major chemical contents of unfermented CTLs (UFD‐CTLs) and fermented CTLs (FD‐CTLs) were identified using the MiSeq platform (Illumina, San Diego, CA, USA) and continuous flow analyses, respectively. The functions of CTLs were assessed using the Functional Annotation of Prokaryotic Taxa (FAPROTAX) software. The data may inform quality improvements to CTL fermentation.
2. MATERIALS AND METHODS
2.1. Tobacco growth, curing conditions, and sample collection
Cigar tobacco (H382) was planted at Fan‐yang, Hainan, China, at the end of December 2018 and harvested in March. The cultivated area was 6.8 hm2. The freshly harvested CTLs were placed in a barn for air‐curing. After 10 weeks, 15 kg of CTLs of the same grade was randomly selected from the barn and sent to the laboratory immediately. Based on previous studies (Du et al., 2016; Yu et al., 2008), 5 kg samples of CTLs were evenly stacked and placed in separate cabinets (HWS‐P400C) at 40°C and 70% humidity. The experiments were repeated three times for three cabinets. Before artificial fermentation, CTLs were first wetted with sterile water within 2 days, until the moisture content was 35%–40% (Tian et al., 2018). The CTLs were sampled after 0 days (UFD‐CTLs) and 1 month (FD‐CTLs) of artificial fermentation (Yu et al., 2008). Each sample contained three independent biological replicates from the three cabinets, and for each biological replicate, 600 g CTLs were taken from the cabinet using a five‐point sampling method (Zhou et al., 2020). The samples were immediately stored at ˗80°C until further analysis.
2.2. Determination of chemical contents of CTLs
The chemical contents of CTLs were analyzed using a continuous flow analytical system (Lu et al., 2020). The contents of total nitrate (TN) and protein were determined according to the Tobacco Industry Standard YC/T161‐2002. The contents of water‐soluble sugar (WSS), nicotine, and starch were determined according to Tobacco Industry Standards YC/T159‐2002, YC/T468‐2013, and YC/T216‐2013, respectively. For each sample, three independent biological replicates from three cabinets were collected separately for chemical analysis, and 500 g of CTLs was used for each biological replicate (Furci et al., 2019). All the tests were conducted three times.
2.3. DNA extraction and sequencing
Genomic DNA was extracted based on previous studies (Su et al., 2011; Ye et al., 2017; Zhang, Acuña, et al., 2019). For each biological replicate, 60 g of CTLs was ground with liquid nitrogen and then divided into three equal parts, namely the technical replicates; then, the DNA extracted from the three technical replicates of each sample was pooled into one DNA sample to minimize any potential DNA extraction bias. The DNA extraction steps were performed following the manufacturer's instructions for the E. Z. N. A.® Soil DNA Kit (Omega Bio‐tek, Norcross, GA, USA). The integrity of the extracted DNA was checked on 1% agarose gel, and DNA concentration and purity were measured using a NanoDrop 2000 UV‐vis spectrophotometer (Thermo Scientific, Waltham, MA, USA).
The V5–V7 regions of the bacterial 16S rRNA gene were amplified using the universal primers 799F (5ʹ‐AACMGGATTAGATACCCKG‐3ʹ) and 1193R (5ʹ‐ACGTCATCCCCACCTTCC‐3ʹ) (Wang et al., 2018). The fungal internal transcribed spacer gene was amplified using the universal primers ITS1F (5ʹ‐CTTGGTCATTTAGAGGAAGTAA‐3ʹ) and ITS2R (5ʹ‐GCTGCGTTCTTCATCGATGC‐3ʹ). The PCR system and program were chosen based on a previous study (Ye et al., 2017). The PCR products were extracted and purified using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) following the manufacturer's instructions. Finally, the purified amplicons were sequenced (2 × 300) using the Illumina MiSeq platform following the standard protocols of Majorbio Bio‐Pharm Technology Co., Ltd. (Shanghai, China).
2.4. Processing of sequencing data
All raw gene sequencing reads were checked and merged using Trimmomatic and FLASH software, respectively (Bolger et al., 2014; Magoč & Salzberg, 2011). The sequences were identified as the same operational taxonomic units (OTUs) with 97% similarity, and the chimeric sequences were eliminated using UPARSE (version 7.1, http://drive5.com/uparse/) (Edgar, 2013). Based on the SILVA database, each sequence was classified and noted using the RDP Classifier (http://rdp.cme.msu.edu/) with a confidence threshold of 70% (Maidak et al., 2000; Pruesse et al., 2007).
2.5. Statistical analyses
All experimental results were expressed as mean values ±standard errors of the means (SEM). Data with a p‐value < 0.05 were considered statistically significant. Statistical calculations and analyses were performed using GraphPad Prism 6.0 software (GraphPad, La Jolla, CA, USA) and SPSS 19 software for Windows (IBM Corporation, Armonk, NY, USA). The bacterial functions of CTLs were predicted using FAPROTAX (http://www.zoology.ubc.ca/louca/FAPROTAX/lib/php/index.php?section=Instructions) (Liang et al., 2019). The trophic mode of the fungi was annotated using FUNGuild software (http://www.stbates.org/guilds/app.php) (Nguyen et al., 2016). Multivariate analysis was conducted, using CANOCO software (version 4.52), via detrended correspondence analysis (DCA) and redundancy analysis (RDA). DCA revealed first axis lengths <3.5 (1.9104 for bacterial analysis and 1.0841 for fungal analysis). Consequently, RDA was the appropriate linear model for analysis.
3. RESULTS
3.1. Major chemical component changes in CTLs after fermentation
The chemical contents, including those of TN, protein, nicotine, starch, and WSS, were analyzed before and after fermentation. The chemical composition of CTLs during fermentation changed under the action of microorganisms (Figure 1). All chemical contents decreased at the end of the CTL fermentation process. Significant decreases in the TN content occurred in FD‐CTLs, unlike in UFD‐CTLs (p < 0.01). The contents of protein, nicotine, starch, and WSS also significantly decreased after fermentation, compared with those in UFD‐CTLs (p < 0.05).
Figure 1.
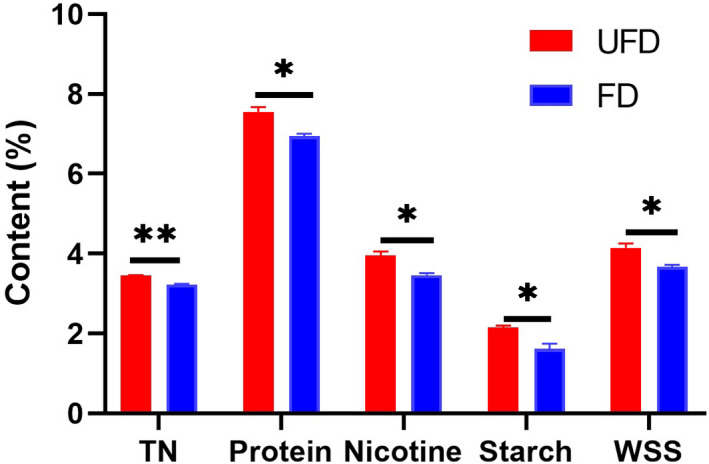
Chemical components in UFD‐CTLs and FD‐CTLs. Data are expressed as means ±SEM (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 versus UFD‐CTLs
3.2. Effects of fermentation on structural changes in CTL microbiota
A total of 273,699 bacterial sequencing reads and 430,876 fungal sequencing reads were obtained from all CTLs using Illumina sequence analysis. To account for potential biases between samples with uneven sequencing depth, all read counts from the remaining samples were rarefied to the read depth of the sample with the lowest read number. Specifically, bacterial and fungal communities in each sample were rarefied to 25,762 (the minimum number of bacterial sequences) and 68,080 sequences (the minimum number of fungal sequences), respectively (Glowska et al., 2020; Huang et al., 2020; Schloss & Handelsman, 2006); 154,572 valid bacterial sequences and 408,480 valid fungal sequences remained for subsequent analysis (Table A1 in Appendix 2). Illumina sequence analysis indicated 1,041 bacterial OTUs and 270 fungal OTUs with 97% similarity. The rarefaction curves (Figure A1 in Appendix 1) of all groups tended to be flat with an increase in the number of OTUs, and the coverage of all samples was more than 99% for the bacterial and fungal sequences (Table A1 in Appendix 2). The collective data revealed that the detection rate of the microbial community in CTLs was close to saturation and that most of the microorganisms in the CTLs were covered by the sequencing data. Community richness (ACE and Chao estimators) and diversity (Simpson and Shannon indices) were determined to reflect the alpha diversity of CTL microbiota. The decrease in ACE and Chao indices of bacteria and fungi indicated a decrease in the microbial richness of CTLs after fermentation (Figure 2). Additionally, the diversity of the microbial community in CTLs showed a downward trend, but this was not significant after fermentation.
Figure 2.

Alpha diversity index of microbial community in CTLs. Data are shown as means ±SEM (n = 3). * denotes significance level in bacterial community, *p < 0.05; # denotes significance level in fungal community, # p < 0.05, ## p < 0.01; & denotes significance level between the bacterial and fungal community, && p < 0.01, &&& p < 0.001
3.3. Differences in microbial communities before and after fermentation of CTLs
For bacterial diversity and succession, 1041 OTUs were classified into 21 phyla, 41 classes, 125 orders, 249 families, and 533 genera with 97% similarity. Proteobacteria and Actinobacteria were the predominant phyla in UFD‐CTL and FD‐CTL samples, with the latter accounting for a larger proportion before fermentation and the former accounting for a larger proportion after fermentation (Figure 3a). Before fermentation, the distribution of bacteria was relatively scattered at the order level. After fermentation, the distribution was dominated by the orders of Betaproteobacteriales and Corynebacteriales, accounting for 71.29% of the sequences, which increased by 41.66% compared with that of UFD‐CTLs (Figure 3c). For fungal diversity and succession (Figure 3b,d), 7 phyla, 19 classes, 45 orders, 88 families, and 139 genera were classified with 97% similarity. At the phylum level of fungal composition, Ascomycota was the dominant phylum throughout the fermentation period, whereas Capnodiales and Eurotiales were the most abundant orders in the fungal community of CTLs. Capnodiales showed a high relative abundance in UFD‐CTLs but were replaced by Eurotiales after fermentation.
Figure 3.

Bacterial (a) and fungal (b) compositions at the phylum level in CTLs. Bacterial (c) and fungal (d) compositions at the order level in CTLs
To assess the overall distribution of the microbial community at the genus level, the 50 most abundant genera of the bacterial and fungal communities were determined; they are depicted in Figures A2 and A3 in Appendix 1. These results further illustrated that fermentation changed the relative abundances of the microbial communities in CTLs. To better understand the effects of fermentation on the relative abundance at the genus level, a statistical comparison of the relative abundances of the predominant genera of UFD‐CTLs and FD‐CTLs (top 20) was conducted (Figure 4). The results revealed that the abundances of seven bacterial genera changed after fermentation. After fermentation, the relative abundances of Ralstonia and Pseudomonas increased, whereas those of Methylobacterium, Pseudonocardia, Aureimonas, Planococcus, and Nocardiopsis remarkably decreased. In the fungal community, the abundances of only four genera changed significantly. The relative abundance of Aspergillus significantly increased in FD‐CTLs by 40.48% compared with that in UFD‐CTLs. However, the relative abundances of Cladosporium, unclassified Basidiomycota, and Penicillium significantly decreased and that of Cladosporium decreased by 35.47% after fermentation. After the removal of OTUs with <0.01% relative abundance, the relative abundances of Ralstonia, Pseudomonas, Romboutsia, Chryseobacterium, and Comamonas in the bacterial community of CTLs significantly increased after fermentation (Table A2 in Appendix 2), whereas the relative abundances of Aspergillus and Phaeosphaeriaceae in the fungal community significantly increased and that of Cladosporium significantly decreased (Table A3 in Appendix 2).
Figure 4.

Mean proportions of 20 dominant genera in different groups of CTL samples: (a) bacteria and (b) fungi. Welch's t test was performed to assess the difference between two groups of CTL samples. *p < 0.05, **p < 0.01, ***p < 0.001
3.4. Relationships between microbial community and chemical components
RDA was performed to determine the relationship between the chemical components and the microbial communities (Figure 5). The chemical components explained most of the variations in the bacterial and fungal community structure in CTLs. The first ordination RDA axis that was strongly related to TN explained 67.23% of the variability in the bacterial community (Figure 5a). The first ordination RDA axis that was strongly related to nicotine explained 94.39% of the variability in the fungal community (Figure 5b). These findings indicated that a close relationship existed between the microbiome and chemical composition in the CTLs. TN and nicotine were the most probable factors to be influenced by microbial communities during the fermentation of CTLs.
Figure 5.

Ordination diagram of RDA for qualitative variables between microbial structure and major chemical compounds in CTLs for (a) bacteria and (b) fungi
3.5. Functional prediction of bacterial and fungal communities
A total of 52 functional assignments for 1041 OTUs were obtained using the FAPROTAX tool. Functional group chemoheterotrophy was most abundant in the bacterial community of CTLs, but its abundance decreased after fermentation (Figure 6a). In contrast, the abundant functions in unfermented samples, such as aromatic compound degradation, ligninolysis, hydrocarbon degradation, aliphatic non‐methane hydrocarbon degradation, and aromatic hydrocarbon degradation, which are related to compound transformation, increased in the fermentation process. Similarly, functions associated with the carbon cycle, such as phototrophy, photoheterotrophy, aerobic anoxygenic phototrophy, methylotrophy, and methanol oxidation, decreased after fermentation. Also, the number of plant pathogens increased during the fermentation process, and this might have negatively affected the quality of CTLs. However, animal parasites or symbionts, another functional group that may negatively affect CTLs, were maintained at a low level throughout the fermentation. At the end of fermentation, the functional groups related to chemoheterotrophy and carbon metabolism decreased, but those related to compound transformation and pathogens increased.
Figure 6.

Bacterial functional groups in CTLs using FAPROTAX. (a) Top 40 predicted gene families in UFD‐CTL and FD‐CTLs. (b) Relationship between functional communities and bacteria with significant changes in abundance after fermentation
Figure 6b shows the association between the functional groups and genera with significant changes in abundance after fermentation. Ralstonia and Pseudomonas were important for compound degradation, which accelerated the degradation of aromatics, hydrocarbons, aliphatic compounds, and lignin. Methylobacterium, Pseudonocardia, Aureimonas, Planococcus, and Nocardiopsis were important in the carbon and nitrogen cycles. Additionally, Ralstonia and Pseudomonas were closely associated with plant–pathogen function. Pseudomonas spp. were associated with pathogens in this study; previous studies have also demonstrated that in the process of plant growth, Pseudomonas could not only build a symbiotic relationship with plants as harmless bacteria but also infect them as pathogens (Hirano & Upper, 2000; Visnovsky et al., 2020). There are also many studies showing that Pseudomonas spp. are not mainly pathogens (Kim & Anderson, 2018; O'sullivan, & O'Gara, 1992). Further validation is needed to confirm whether Pseudomonas spp. are linked to pathogens or not.
FUNGuild was used to predict the trophic mode of the fungal community in CTLs. The fungal community was classified into four trophic mode groups, with saprophytic and pathotroph–saprotroph–symbiotroph being the dominant trophic modes in the fungal community during fermentation (Figure 7a). The abundance of saprophytic fungi increased from 37.06% to 74.52% after fermentation. The abundance of pathotroph–saprotroph–symbiotroph fungi was 61.35% and 23.44% in UFD‐CTLs and FD‐CTLs, respectively. Different guild modes in the fungal OTUs of CTL samples were classified (Figure 7b). As the most abundant primary guild mode, undefined saprotroph mode accounted for 37.10% and 74.54% abundance in UFD‐CTLs and FD‐CTLs, respectively. The second most abundant fungal guild of wood saprotroph accounted for 56.86% abundance of the fungal OTUs in UFD‐CTLs but decreased to 21.38% in FD‐CTLs.
Figure 7.

Relative abundance of trophic modes (a) and guild modes (b) assigned by FUNGuild for fungal communities
4. DISCUSSION
Cigar tobacco is air‐cured; however, cured but unfermented tobacco is not suitable for cigar products because of its sharp and pungent flavor (Zhao et al., 2007). Artificial fermentation is a more controllable and efficient way of fermenting CTLs compared with natural aging. The microbial quality and functional profile of CTLs during artificial fermentation remain unclear. The data from this study enable a comprehensive understanding of the microbial diversity of UFD‐CTLs and FD‐CTLs with respect to the bacterial and fungal communities present. Also, the chemical components, bacterial functional groups, and fungal functional classifications in UFD‐CTLs and FD‐CTLs were analyzed.
The contents of chemical compounds change during fermentation. Primary chemical compounds decrease in content after fermentation. The present findings concerning the chemical components of CTLs were consistent with those of previous studies. After 24 months of natural aging, the content of WSS decreased by 2.63%, whereas the contents of TN, nicotine, starch, and protein decreased slightly (Zhou et al., 2020). Conversely, the contents of chemical components decreased significantly after short‐term artificial fermentation (Qiu et al., 2003), consistent with the findings of this study. We observed that the contents of TN, protein, nicotine, starch, and total sugar in CTLs significantly decreased after 30 days of artificial fermentation. These findings indicate that the chemical contents decrease faster in CTLs during artificial fermentation than during aging. This may be associated with the different activities of microorganisms during different types of fermentation; further research is needed to confirm this viewpoint. The transformation of chemical compounds is closely related to the microbial community in tobacco leaves during the fermentation process (Huang et al., 2010; Liu et al., 2016). The decrease in the contents of chemical components is associated with enzyme activity and microbial metabolism in tobacco leaves. The enzyme activity, such as amylase activity, is high during fermentation (Yamaguchi et al., 2013); this may be the reason for the decrease in starch content in tobacco leaves. The hydrolysis of starch produces WSSs, but the total sugar content does not increase, possibly because microorganisms use these sugars as nutrients to maintain their metabolism. Bacillus spp. degrade proteins in tobacco (Chen et al., 2015; Feng et al., 2012). A decrease in protein content can reduce the bitterness and astringent flavor after smoking (Ding et al., 2014; Pu et al., 2013). Nicotine in tobacco is degraded to niacin, methanesulfonic acid, oxidized nicotine, nicotinamide, n‐methylnicotinamide, and adamantane by Pseudomonas spp., Aspergillus fumigatus, and Rhodococcus spp. (Mu et al., 2020).
The richness and alpha diversity of the microbial community decreased after artificial fermentation in this study. This may be because some microorganisms are not suitable for artificial fermentation conditions and their growth is inhibited (Huang et al., 2010). Actinobacteria and Proteobacteria were the most abundant phyla in the bacterial communities of CTLs, suggesting that the bacteria were stable at the phylum level. Moreover, the dominant phyla were different from those in flue‐cured tobacco (Su et al., 2011). The abundance of Actinobacteria is less than that of other phyla in flue‐cured tobacco (Su et al., 2011). However, a large number of Actinobacteria were found in CTLs from Mexico (Zhang et al., 2018). The varieties, fertilization methods, growth environments, and curing methods of flue‐cured tobacco and cigar tobacco are different; these factors may account for the differences in the bacterial communities in their leaves (Yan et al., 2018). The relative abundances of Rhodococcus, Ralstonia, Bacillus, and Pseudomonas increased after fermentation, with those of Ralstonia and Pseudomonas increasing significantly. These findings indicate that these strains can adapt to fermentation and the complex chemical environment in tobacco leaves. Rhodococcus spp. are gram‐positive bacteria that readily degrade various organic matter. Some species of Rhodococcus and many species of Bacillus and Pseudomonas can degrade nicotine effectively (Liu et al., 2015; Mu et al., 2020; Ruan et al., 2005; Zhong et al., 2010). Bacillus spp. are widely distributed in nature and are commonly present in tobacco; they are important in tobacco fermentation (Carter, 1990; Huang et al., 2010). Bacillus spp. increase the formation of the desired aroma and enhance the quality of tobacco (English et al., 1967) and can degrade starch (Pen et al., 1992), cellulose (Li et al., 2006), and other compounds, making tobacco smoke less irritating. Bacillus thuringiensis can also control pests in stored products (Kaelin et al., 1994). Most species of Ralstonia are related to plant pathogens. For instance, Ralstonia solanacearum can cause bacterial wilt disease (Balamurugan et al., 2020). An increase in the abundance of Ralstonia may have been the reason for the increase in the abundance of plant–pathogen functional groups (Figure 6b). We also observed that the relative abundances of Methylobacterium, Pseudonocardia, Aureimonas, Planococcus, and Nocardiopsis notably decreased at the end of fermentation. These findings indicated that these bacteria could not survive well in the fermentation environment.
Understanding the functional group response to fermentation is essential for the accurate prediction of the potential effects of the fermentation process. Therefore, the functional capabilities of the bacterial communities in CTLs were assessed using FAPROTAX. The results indicated an extensive functional profile of the bacterial community in CTLs. The most abundant functional group was chemoheterotrophs. Chemoheterotrophic microorganisms mainly rely on energy and carbon sources in organic matter to maintain their growth (Glazer & Nikaido, 2007). Most bacteria depend on organic matter as their carbon and energy source in CTLs. Furthermore, many functional groups related to compound degradation participate in the fermentation of CTLs, and the abundance of these functional groups increased after fermentation; this result suggests a close connection between the chemical components and microbial community in CTLs during artificial fermentation, which is in agreement with the results of RDA. The changes in the chemical contents in CTLs can influence not only the quality of CTLs but also the safety (Wang & Ma, 2013) (TsoT, 1972). For instance, in addition to nicotine, a recognized harmful substance (Liu et al., 2015), the protein in tobacco could produce the smell of charred feather and harmful materials, such as quinoline, cyanic acid, and benzopyrene, during smoking when heated (Chen et al., 2015). The activities of microorganisms decreased the nicotine and protein contents in this study, and this was conducive to improving the safety of CTLs.
Although CTLs are mainly fermented by bacteria, fungi also play an important role. At the phylum level, Ascomycota were the most abundant fungi throughout the fermentation. Although Capnodiales and Eurotiales were the predominant orders, Aspergillus and Cladosporium were the predominant genera throughout the fermentation process. The relative abundances of these genera changed. Capnodiales and Cladosporium were the most abundant order and genus before fermentation, respectively. However, Eurotiales and Aspergillus became the dominant order and genus, respectively, after 30 days of fermentation. The changes in their relative abundances indicated that fermentation changed the fungal trophic mode; this was also reflected in the results of FUNGuild analysis. Previous studies have indicated that saprophytic fungi can promote the decomposition of organic matter (Chen et al., 2020; Zhang et al., 2019). Therefore, an increase in the content of saprophytic fungi would aid the degradation of the chemical components of tobacco leaves. RDA also revealed a strong link between the fungal community and the chemical components.
5. CONCLUSION
Chemical composition analysis showed that most of the metabolite levels in CTLs decreased after artificial fermentation. Fermentation changed the bacterial and fungal community structure. RDA revealed that the TN content was most readily affected by the bacterial community, whereas the nicotine content was most susceptible to the effects of the fungal community. FAPROTAX and FUNGuild analyses were used to predict the function of the microbial communities in CTLs. The dominant bacteria in cigars have important application prospects in the degradation of aromatic compounds, hydrocarbons, aliphatic compounds, and lignin. Fungi mainly have a saprotrophic function after fermentation. These results can be used as a basis to develop new strategies to improve the quality of CTLs through artificial fermentation.
CONFLICTS OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
Fang Liu: Conceptualization (equal); Data curation (lead); Formal analysis (lead); Investigation (lead); Visualization (equal); Writing‐original draft (lead); Writing‐review & editing (equal). Zhiyong Wu: Formal analysis (supporting); Writing‐review & editing (equal). Xiaoping Zhang: Software (supporting); Writing‐review & editing (supporting). Gaolei Xi: Writing‐review & editing (supporting). Zhe zhao: Resources (supporting). Miao Lai: Resources (supporting). Mingqin Zhao: Conceptualization (equal); Funding acquisition (lead); Project administration (lead); Supervision (lead).
ETHICS STATEMENT
None required.
ACKNOWLEDGMENTS
This work was supported by the State Tobacco Monopoly Administration, China National Tobacco Company [110200401014].
APPENDIX 1.
Figure A1.

Rarefaction curve of bacterial (a) and fungal (b) OTUs derived from CTLs.
Figure A2.

Community heat maps of CTLs at the genus level of the bacterial community (top 50).
Figure A3.

Community heat maps of CTLs at the genus level of the fungal community (top 50).
Table A1.
Numbers of valid sequences and coverage of 16S rRNA and 18S rRNA gene libraries at 97% similarity from the sequencing analysis
| Sample name | Bacteria | Fungi | ||
|---|---|---|---|---|
| Sequences | Coverage | Sequences | Coverage | |
| UFD‐CTLs−1 | 37735 | 0.999231 | 69640 | 0.999713 |
| UFD‐CTLs−2 | 57134 | 0.999387 | 73062 | 0.999822 |
| UFD‐CTLs−3 | 25762 | 0.998370 | 70339 | 0.999701 |
| FD‐CTLs−1 | 49260 | 0.999695 | 68080 | 0.999971 |
| FD‐CTLs−2 | 28435 | 0.999648 | 69618 | 0.999971 |
| FD‐CTLs−3 | 28516 | 0.999790 | 71840 | 0.999916 |
Table A2.
Mean proportions of the bacteria genera after removing OTUs with <0.01% in different groups of CTL samples
| Name | UFD‐mean | UFD‐sd | FD‐mean | FD‐sd | p‐value |
|---|---|---|---|---|---|
| Rhodococcus | 17.34 | 8.012 | 30.22 | 8.29 | 0.1838 |
| Ralstonia | 1.215 | 0.2086 | 24.22 | 4.008 | 0.004559** |
| Burkholderia–Caballeronia–Paraburkholderia | 6.488 | 3.11 | 14.95 | 3.005 | 0.05541 |
| Bacillus | 3.171 | 3.546 | 5.411 | 2.586 | 0.4654 |
| Sphingomonas | 7.554 | 8.141 | 0.1294 | 0.1171 | 0.1821 |
| Pseudomonas | 0.6618 | 0.3486 | 5.062 | 1.713 | 0.04211* |
| Brachybacterium | 4.186 | 4.559 | 0.02847 | 0.0493 | 0.182 |
| Romboutsia | 0.06405 | 0.008234 | 1.143 | 0.3311 | 0.02217* |
| Corynebacterium_1 | 1.114 | 1.016 | 0.5305 | 0.1728 | 0.3669 |
| Trichococcus | 0.1009 | 0.0549 | 0.9057 | 0.6644 | 0.2032 |
| Staphylococcus | 1.073 | 1.128 | 0.2018 | 0.0998 | 0.2418 |
| Aquamicrobium | 1.166 | 1.452 | 0.1359 | 0.1298 | 0.2738 |
| unclassified_f__Enterobacteriaceae | 0.9103 | 1.079 | 0.2018 | 0.09421 | 0.3042 |
| Stenotrophomonas | 0.2057 | 0.2306 | 0.2834 | 0.1321 | 0.6538 |
| Chryseobacterium | 0.01747 | 0.008234 | 0.4153 | 0.1614 | 0.04556* |
| norank_f__norank_o__SJA−28 | 0.1048 | 0.1098 | 0.2704 | 0.1793 | 0.338 |
| Comamonas | 0.07375 | 0.001261 | 0.3131 | 0.05439 | 0.0097** |
| Enterococcus | 0.04852 | 0.04666 | 0.2627 | 0.186 | 0.2257 |
| Leucobacter | 0.5124 | 0.5929 | 0.1617 | 0.08964 | 0.3527 |
| norank_f__Bacillaceae | 0.06987 | 0.06038 | 0.1708 | 0.1688 | 0.4935 |
| Dyella | 0.1863 | 0.1372 | 0.06728 | 0.03522 | 0.2196 |
| Bradyrhizobium | 0.13 | 0.04666 | 0.09445 | 0.06614 | 0.5644 |
| Caulobacter | 0.06211 | 0.04392 | 0.1255 | 0.05051 | 0.2469 |
| Luteimonas | 0.2523 | 0.247 | 0.185 | 0.02642 | 0.6446 |
| Micrococcus | 0.1378 | 0.06313 | 0.05176 | 0.02913 | 0.1189 |
| Cutibacterium | 0.1087 | 0.1043 | 0.06211 | 0.03703 | 0.5039 |
| Sulfuritalea | 0.03494 | 0.00549 | 0.1061 | 0.03607 | 0.07819 |
| Clostridium_sensu_stricto_1 | 0.06599 | 0.02196 | 0.4542 | 0.2912 | 0.1721 |
p < 0.05,
p < 0.01.
Table A3.
Mean proportions of the fungi genera after removing OTUs with <0.01% in different groups of CTL samples
| Name | UFD‐mean | UFD‐sd | FD‐mean | FD‐sd | p‐value |
|---|---|---|---|---|---|
| Aspergillus | 28.47 | 21.4 | 68.95 | 13.08 | 0.04902* |
| Cladosporium | 56.85 | 19.48 | 21.38 | 5.234 | 0.03816* |
| Wallemia | 5.753 | 9.144 | 4.88 | 7.625 | 0.9051 |
| unclassified_o__Tremellales | 2.525 | 1.985 | 0.966 | 0.7917 | 0.2751 |
| Hannaella | 0.6101 | 0.7858 | 0.3746 | 0.3374 | 0.6583 |
| Cutaneotrichosporon | 0.0803 | 0.05276 | 0.5352 | 0.3973 | 0.1208 |
| unclassified_f__Mycosphaerellaceae | 0.1689 | 0.1827 | 0.1552 | 0.2112 | 0.9363 |
| Sporobolomyces | 0.1214 | 0.1009 | 0.212 | 0.06445 | 0.2603 |
| Uwebraunia | 0.08911 | 0.0656 | 0.1772 | 0.2537 | 0.5914 |
| unclassified_f__Phaeosphaeriaceae | 0.04309 | 0.0221 | 0.09548 | 0.007344 | 0.01759* |
| unclassified_f__Nectriaceae | 0.06659 | 0.07476 | 0.04798 | 0.06152 | 0.756 |
p < 0.05.
Contributor Information
Xiaoping Zhang, Email: zhaomingqin@henau.edu.cn.
Mingqin Zhao, Email: zhaomingqin@henau.edu.cn.
DATA AVAILABILITY STATEMENT
All the Illumina 16S rRNA and ITS gene sequence data obtained in this study are available in the NCBI database under the BioProject accession number PRJNA644460: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA644460. All other data are provided in the results section and the appendix of this article.
REFERENCES
- Balamurugan, A., Muthamilan, M., Kamalakannan, A., Shanthi, A., & Arumugam, T. (2020). Characterization of Ralstonia solanacearum causing bacterial wilt disease of tomato in Coimbatore district of Tamil Nadu, India. International Journal of Current Microbiology and Applied Sciences, 9(2), 3010–3016. [Google Scholar]
- Bauk, M. (2016). The history of Tobacco. Filozofski fakultet. [Google Scholar]
- Bolger, A. M., Lohse, M., & Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns, D. M. (1998). Cigar smoking: Overview and current state of the science. Smoking and Tobacco Control Monograph, 9, 1–19. [Google Scholar]
- Carter, G. (1990). Bacillus. Diagnostic procedure in veterinary bacteriology and mycology (pp. 221–228). London: Academic Press. [Google Scholar]
- Chen, X., Wang, X., Zeng, X., Yang, Y., Ling, J., & Duan, Y. (2015). Screening of protein degrading bacterial strains and their effects on tobacco. Acta Agriculturae Jiangxi, 27(6), 90–91. 10.19386/j.cnki.jxnyxb.2015.06.022 [DOI] [Google Scholar]
- Chen, Y., Tian, W., Shao, Y., Li, Y.‐J., Lin, L.‐A., Zhang, Y.‐J., Han, H., & Chen, Z.‐J. (2020). Miscanthus cultivation shapes rhizosphere microbial community structure and function as assessed by Illumina MiSeq sequencing combined with PICRUSt and FUNGUIld analyses. Archives of Microbiology, 202(5), 1157–1171. 10.1007/s00203-020-01830-1 [DOI] [PubMed] [Google Scholar]
- Ding, R., Ma, Y., Zhou, H., Li, H., Tang, H., & Zhang, D. (2014). Research overview of protein in tobacco. Guangzhou Chemical Industry, 42(5), 5–8. [Google Scholar]
- Du, J., Zhang, X., Wu, G., Zhou, R., Cui, Y., & Shi, X. (2016). Studies on leaf surface microflora of cigar‐wrapper during artificial fermentation. Current Biotechnology, 6(3), 188–192. 10.3969/j.issn.2095-2341.2016.03.07 [DOI] [Google Scholar]
- Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10(10), 996–998. 10.1038/nmeth.2604 [DOI] [PubMed] [Google Scholar]
- English, C. F., Bell, E. J., & Berger, A. J. (1967). Isolation of thermophiles from broadleaf tobacco and effect of pure culture inoculation on cigar aroma and mildness. Applied and Environmental Microbiology, 15(1), 117–119. 10.1128/aem.15.1.117-119.1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Z., Chen, T., Hu, X., Guo, Z., & An, D. (2012). Isolation, screen and identification of Bacillus with high proteolytic activity from tobacco leaf surfaces. Acta Tabacaria Sinica, 01, 101–105.1004‐5708(2012)01‐0101‐05 [Google Scholar]
- Frankenburg, W. G. (1950). Chemical changes in the harvested tobacco leaf. Part II. Chemical and Enzymic Conversions during Fermentation and Aging. Advances in Enzymology, 10(351), 10. [DOI] [PubMed] [Google Scholar]
- Furci, L., Jain, R., Stassen, J., Berkowitz, O., Whelan, J., Roquis, D., Baillet, V., Colot, V., Johannes, F., & Ton, J. (2019). Identification and characterisation of hypomethylated DNA loci controlling quantitative resistance in Arabidopsis . Elife, 8, e40655. 10.7554/eLife.40655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazer, A. N., & Nikaido, H. (2007). Microbial biotechnology: Fundamentals of applied microbiology. :Cambridge University Press. [Google Scholar]
- Glowska, E., Filutowska, Z. K., Dabert, M., & Gerth, M. (2020). Microbial composition of enigmatic bird parasites: Wolbachia and Spiroplasma are the most important bacterial associates of quill mites (Acariformes: Syringophilidae). MicrobiologyOpen, 9(5), e964–e964. 10.1002/mbo3.964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano, S. S., & Upper, C. D. (2000). Bacteria in the leaf ecosystem with emphasis on Pseudomonas syringae—a pathogen, ice nucleus, and epiphyte. Microbiology and Molecular Biology Reviews, 64(3), 624–653. 10.1128/mmbr.64.3.624-653.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, J., Yang, J., Duan, Y., Gu, W., Gong, X., Zhe, W., Su, C., & Zhang, K.‐Q. (2010). Bacterial diversities on unaged and aging flue‐cured tobacco leaves estimated by 16S rRNA sequence analysis. Applied Microbiology and Biotechnology, 88(2), 553–562. 10.1007/s00253-010-2763-4 [DOI] [PubMed] [Google Scholar]
- Huang, K., Jiang, Q., Liu, L., Zhang, S., Liu, C., Chen, H., Ding, W., & Zhang, Y. (2020). Exploring the key microbial changes in the rhizosphere that affect the occurrence of tobacco root‐knot nematodes. Amb Express, 10(1), 11. 10.1186/s13568-020-01006-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin, P., Morel, P., & Gadani, F. (1994). Isolation of Bacillus thuringiensis from stored Tobacco and Lasioderma serricorne (F.). Applied and Environmental Microbiology, 60(1), 19–25. 10.1128/aem.60.1.19-25.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, T. H. (1929). Process of artificially aging tobacco. In: Google Patents. [Google Scholar]
- Kim, Y. C., & Anderson, A. J. (2018). Rhizosphere pseudomonads as probiotics improving plant health. Molecular Plant Pathology, 19(10), 2349–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, L. W., & Bahnson, F. F. (1914). Process of treating tobacco. In: Google Patents. [Google Scholar]
- Li, X., Huang, S., Huang, C., Xu, M., Zhou, X., & Zhong, W. (2007). Main existing problems and countermeasures on management techniques for leaf tobacco storage. Guangxi Agriculture Science, 38(1), 84–87. [Google Scholar]
- Li, Y., Ding, M., Wang, J., Xu, G., & Zhao, F. (2006). A novel thermoacidophilic endoglucanase, Ba‐EGA, from a new cellulose‐degrading bacterium, Bacillus sp.AC‐1. Applied Microbiology and Biotechnology, 70(4), 430–436. 10.1007/s00253-005-0075-x. [DOI] [PubMed] [Google Scholar]
- Liang, S., Deng, J., Jiang, Y., Wu, S., Zhou, Y., & Zhu, W.‐X. (2019). Functional distribution of bacterial community under different land use patterns based on FaProTax function prediction. Polish Journal of Environmental Studies, 29(2), 1245–1261. 10.15244/pjoes/108510 [DOI] [Google Scholar]
- Liu, F., Zhang, X., Wang, M., Guo, L., Yang, Y., & Zhao, M. (2020). Biosorption of sterols from tobacco waste extract using living and dead of newly isolated fungus Aspergillus fumigatus strain LSD‐1. Bioscience, Biotechnology, and Biochemistry, 84(7), 1521–1528. 10.1080/09168451.2020.1742089 [DOI] [PubMed] [Google Scholar]
- Liu, F., Zhao, Z., & Zhao, M. (2016). Detection and quantitative analysis of dominant bacteria on aging flue‐cured tobacco leaves. Agricultural Science & Technology, 17(11), 2611–2614. 10.16175/j.cnki.1009-4229.2016.11.041 [DOI] [Google Scholar]
- Liu, J., Ma, G., Chen, T., Hou, Y., Yang, S., Zhang, K. Q., & Yang, J. (2015). Nicotine‐degrading microorganisms and their potential applications. Applied Microbiology and Biotechnology, 99(9), 3775–3785. 10.1007/s00253-015-6525-1 [DOI] [PubMed] [Google Scholar]
- Lu, S., Zhang, J.‐W., Zhao, Z., Zhong, Q., & Zhao, M.‐Q. (2020). Effects of air‐curing humidity on key enzyme activities related to carbon and nitrogen metabolism in and quality of cigar tobacco leaves. Acta Tabacaria Sinica, 26(04), 26–34. 10.16472/j.chinatobacco.2019.369 [DOI] [Google Scholar]
- Magoč, T., & Salzberg, S. L. (2011). FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics, 27(21), 2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maidak, B. L., Cole, J. R., Lilburn, T. G., Parker, C. T.Jr, Saxman, P. R., Stredwick, J. M., & Tiedje, J. M. (2000). The RDP (Ribosomal Database Project) continues. Nucleic Acids Research, 28(1), 173–174. 10.1093/nar/28.1.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado‐Robledo, G., Rodriguez‐Bustamante, E., Sanchez‐Contreras, A., Rodriguez‐Sanoja, R., & Sanchez, S. (2003). Production of tobacco aroma from lutein. Specific role of the microorganisms involved in the process. Applied Microbiology and Biotechnology, 62(5–6), 484–488. 10.1007/s00253-003-1315-6 [DOI] [PubMed] [Google Scholar]
- Mu, Y., Chen, Q., Parales, R. E., Lu, Z., Hong, Q., He, J., Qiu, J., & Jiang, J. (2020). Bacterial catabolism of nicotine: Catabolic strains, pathways and modules. Environmental Research, 183, 13. 10.1016/j.envres.2020.109258 [DOI] [PubMed] [Google Scholar]
- Nguyen, N. H., Song, Z., Bates, S. T., Branco, S., Tedersoo, L., Menke, J., Schilling, J. S., & Kennedy, P. G. (2016). FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecology, 20, 241–248. [Google Scholar]
- O'sullivan, D. J., & O'gara, F. (1992). Traits of fluorescent Pseudomonas spp. involved in suppression of plant root pathogens. Microbiology and Molecular Biology Reviews, 56(4), 662–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pen, J., Molendijk, L., Quax, W. J., Sijmons, P. C., van Ooyen, A. J. J., van den Elzen, P. J. M., Rietveld, K., & Hoekema, A. (1992). Production of active Bacillus licheniformis alpha‐amylase in tobacco and its application in starch liquefaction. Nature Biotechnology, 10(3), 292–296. 10.1038/nbt0392-292 [DOI] [PubMed] [Google Scholar]
- Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., & Glockner, F. O. (2007). SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Research, 35(21), 7188–7196. 10.1093/nar/gkm864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu, Y., Bao, X., Wang, S., & Li, X. (2013). Study on reducing protein content in tobacco leaves by flavorzyme. Food Industry 8, 146–149. [Google Scholar]
- Qiu, L., Zhao, M., Li, F., Qi, W., Zhang, W., Yue, X., & Cui, J. (2003). Changes in biological activity during artificial fermentation of flue‐cured tobacco. Tobacco Science, 46, 24–27. 10.3381/0082-4623-46.1.24 [DOI] [Google Scholar]
- Ruan, A., Min, H., Peng, X., & Huang, Z. (2005). Isolation and characterization of Pseudomonas sp. strain HF‐1, capable of degrading nicotine. Research in Microbiology, 156(5), 700–706. 10.1016/j.resmic.2005.02.010 [DOI] [PubMed] [Google Scholar]
- Schloss, P. D., & Handelsman, J. (2006). Introducing SONS, a tool for operational taxonomic unit‐based comparisons of microbial community memberships and structures. Applied and Environmental Microbiology, 72(10), 6773–6779. 10.1128/aem.00474-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanyi, C., Jingjing, L., Jian, L., Kexiang, B., Jianqiang, F., Ruiqiang, Z., & Wei, H. (2018). High‐throughput sequencing fungal community structures in aging tobacco strips from different growing areas and stalk positions. Tobacco Science and Technology, 51, 12–19. 10.16135/j.issn1002-0861.2017.0270 [DOI] [Google Scholar]
- Su, C., Gu, W., Zhe, W., Zhang, K., Duan, Y., & Yang, J. (2011). Diversity and phylogeny of bacteria on Zimbabwe tobacco leaves estimated by 16S rRNA sequence analysis. Applied Microbiology and Biotechnology, 92(5), 1033–1044. [DOI] [PubMed] [Google Scholar]
- Sun, Y., Liu, H., Gao, H., Lin, B., Xiang, X., Lu, H., & Ma, X. (2019). Effects of transplanting period on the growth, development, yield and quality of Hainan cigar wrapper tobacco. Chinese Tobacco Science, 40(3), 91. 10.13496/j.issn.1007-5119.2019.03.013 [DOI] [Google Scholar]
- Tian, Y., Joe, B. M., Liu, X., Wu, F., Tan, S., Liu, X., & Chen, C. (2018). Preliminary study on wetting method of cigar tobacco before fermentation. Science & Technology Information, 16(9), 3. 10.16661/j.cnki.1672-3791.2018.09.252 [DOI] [Google Scholar]
- TsoT (1972). Physiology and biochemistry of tobacco plants. Physiology & Biochemistry of Tobacco Plants. [Google Scholar]
- Visnovsky, S. B., Panda, P., Everett, K. R., Lu, A., Butler, R. C., Taylor, R. K., & Pitman, A. R. (2020). A PCR diagnostic assay for rapid detection of plant pathogenic pseudomonads. Plant Pathology, 69(7), 1311–1330. 10.1111/ppa.13204 [DOI] [Google Scholar]
- Wang, F., Zhao, H., Xiang, H., Wu, L., Men, X., Qi, C., Chen, G., Zhang, H., Wang, Y. I., & Xian, M. O. (2018). Species diversity and functional prediction of surface bacterial communities on aging flue‐cured tobaccos. Current Microbiology, 75(10), 1306–1315. 10.1007/s00284-018-1525-x [DOI] [PubMed] [Google Scholar]
- Wang, L., & Ma, R. (2013). Major chemical components and quality and style and their relationships in full flavor tobacco growing regions. Chinese Tobacco Science, 34(5), 3. 10.3969/j.issn.1007-5119.2013.05.006 [DOI] [Google Scholar]
- Yamaguchi, N., Suzuki, S., & Makino, A. (2013). Starch degradation by alpha‐amylase in tobacco leaves during the curing process. Soil Science and Plant Nutrition, 59(6), 904–911. 10.1080/00380768.2013.842884 [DOI] [Google Scholar]
- Yan, X., Wang, Z., Mei, Y. U., Wang, L., Wang, X. U., Xu, Q., Peng, S. U., Zhou, Y. U., & Wei, C. (2018). Isolation, diversity, and growth‐promoting activities of endophytic bacteria from tea cultivars of Zijuan and Yunkang‐10. Frontiers in Microbiology, 9, 1848. 10.3389/fmicb.2018.01848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, J., Yan, J. I., Zhang, Z., Yang, Z., Liu, X., Zhou, H., Wang, G., Hao, H., Ma, K. E., Ma, Y., Mao, D., & Yang, X. (2017). The effects of threshing and redrying on bacterial communities that inhabit the surface of tobacco leaves. Applied Microbiology and Biotechnology, 101(10), 4279–4287. 10.1007/s00253-017-8143-6 [DOI] [PubMed] [Google Scholar]
- Yu, J., Yang, Y., Li, L., Wang, B., Liu, X., Pang, T., & Guo, W. (2008). Effects of different temperatures and moistures on neutral aroma components of sun‐cured yellow tobacco during fermentation. Transactions of the CSAE, 24(12), 4. [Google Scholar]
- Zhang, G., Liang, K., Zhang, H., Xin, Y., & Liu, H. (2018). Diversity and succession of bacteria during the fermentation of a cigar wrapper using high throughput sequencing technology and traditional isolation. Chinese Journal of Applied & Environmental Biology, 24(4), 6. 10.19675/j.cnki.1006-687x.2017.11014. [DOI] [Google Scholar]
- Zhang, Q., Acuña, J. J., Inostroza, N. G., Mora, M. L., Radic, S., Sadowsky, M. J., & Jorquera, M. A. (2019). Endophytic bacterial communities associated with roots and leaves of plants growing in Chilean extreme environments. Scientific Reports, 9(1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W.‐H., Sun, R.‐B., Xu, L., Liang, J.‐N., & Zhou, J. (2019). Assessment of bacterial communities in Cu‐contaminated soil immobilized by a one‐time application of micro‐/nano‐hydroxyapatite and phytoremediation for 3 years. Chemosphere, 223, 240–249. 10.1016/j.chemosphere.2019.02.049 [DOI] [PubMed] [Google Scholar]
- Zhao, M., Wang, B., Li, F., Qiu, L., Li, F., Wang, S., & Cui, J. (2007). Analysis of bacterial communities on aging flue‐cured tobacco leaves by 16S rDNA PCR‐DGGE technology. Applied Microbiology and Biotechnology, 73(6), 1435–1440. 10.1007/s00253-006-0625-x [DOI] [PubMed] [Google Scholar]
- Zhong, W., Zhu, C., Shu, M., Sun, K., Zhao, L., Wang, C., Ye, Z., & Chen, J. (2010). Degradation of nicotine in tobacco waste extract by newly isolated Pseudomonas sp. ZUTSKD. Bioresource Technology, 101(18), 6935–6941. 10.1016/j.biortech.2010.03.142 [DOI] [PubMed] [Google Scholar]
- Zhou, J., Yu, L., Zhang, J., Zhang, X., Xue, Y., Liu, J., & Zou, X. (2020). Characterization of the core microbiome in tobacco leaves during aging. MicrobiologyOpen, 9(3), e984. 10.1002/mbo3.984 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the Illumina 16S rRNA and ITS gene sequence data obtained in this study are available in the NCBI database under the BioProject accession number PRJNA644460: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA644460. All other data are provided in the results section and the appendix of this article.