

Abstract
Background
High‐fructose diet (HFr) induces hypertension and renal damage. However, it has been unknown whether the HFr‐induced hypertension and renal damage are exaggerated in subjects with salt sensitivity. We tested impacts of HFr in Dahl salt‐sensitive (DS) and salt‐resistant (DR) rats.
Methods and Results
Male DS and DR rats were fed control diet or HFr (60% fructose) with normal‐salt content. After 12 weeks, plasma and urinary parameters, renal histological characteristics, and renal expression of renin‐angiotensin system components were examined. Furthermore, effects of renin‐angiotensin system inhibitors were also examined in DS rats fed the HFr. HFr elevated blood pressure in DS rats but not in DR rats. HFr increased urinary albumin and liver type fatty acid binding protein excretions in both rats, but the excretions were exaggerated in DS rats. HFr increased plasma lipids and uric acid in both rats, whereas HFr increased creatinine clearance in DS rats but not DR rats. Although HFr decreased plasma renin activity in DS rats, HFr‐induced glomerular injury, afferent arteriolar thickening, and renal interstitial fibrosis were exaggerated in DS rats. HFr increased renal expression of angiotensinogen, renin, (pro)renin receptor, angiotensin‐converting enzyme, and angiotensin II type 1 receptor in DS rat, whereas HFr increased only angiotensin‐converting enzyme expression and decreased renin and angiotensin II type 1 receptor expressions in DR rats. Enalapril and candesartan attenuated the HFr‐induced hypertension, albuminuria, glomerular hyperfiltration, and renal damage in DS rats.
Conclusion
HFr‐induced hypertension and renal damage are exaggerated in DS rats via renal renin‐angiotensin system activation, which can be controlled by renin‐angiotensin system inhibitors.
Keywords: fructose, glomerular hyperfiltration, hypertension, renal damage, renin‐angiotensin system
Subject Categories: Hypertension
Nonstandard Abbreviations and Acronyms
- (P)RR
(pro)renin receptor
- AT1R
angiotensin II type 1 receptor
- AT2R
angiotensin II type 2 receptor
- Con
control diet
- Con‐DR
control diet–fed Dahl salt resistant
- Con‐DS
control diet–fed Dahl salt sensitive
- DR
Dahl salt resistant
- DS
Dahl salt sensitive
- HFr
high‐fructose diet
- HFr‐Can
high‐fructose diet and candesartan treatment
- HFr‐DR
high‐fructose diet–fed Dahl salt resistant
- HFr‐DS
high‐fructose diet–fed Dahl salt sensitive
- HFr‐Ena
high‐fructose diet and enalapril treatment
- HOMA‐IR
homeostasis model assessment for insulin resistance
- IGS
glomerulosclerosis index
- L‐FABP
liver type fatty acid binding protein
- PRA
plasma renin activity
- RAS
renin‐angiotensin system
- RIV
relative interstitial volume
- SBP
systolic blood pressure
Clinical Perspective
What Is New?
High‐fructose diet–induced blood pressure elevation, glomerular hyperfiltration, albuminuria, glomerular sclerosis, and renal interstitial fibrosis are exaggerated in Dahl salt‐sensitive rats compared with Dahl salt‐resistant rats.
High‐fructose diet activates renal renin‐angiotensin system despite suppression of circulatory renin‐angiotensin system in Dahl salt‐sensitive rats.
Renin‐angiotensin system inhibitors prevent the high‐fructose diet–induced hypertension, albuminuria, glomerular hyperfiltration, and renal damage in Dahl salt‐sensitive rats.
What Are the Clinical Implications?
Excessive fructose intake may be associated with a higher risk of developing hypertension and chronic kidney disease in salt‐sensitive people compared with salt‐resistant people.
In comparison to the low effectiveness in high‐salt–induced hypertension, renin‐angiotensin system inhibitors may be effective treatment against the high‐fructose diet–induced hypertension and renal damage regardless of salt sensitivity.
Metabolic syndrome, which includes obesity, insulin resistance, dyslipidemia, and hypertension, has become a major public health and clinical challenges worldwide. Fructose intake has increased dramatically since past decades, and consumption of fructose‐based sweetener contributes to the development of metabolic syndrome.1 Clinical and animal studies have reported that excessive fructose intake can induce metabolic syndrome. Excessive fructose intake resulted in insulin resistance, visceral obesity, and dyslipidemia in overweight or healthy adults.2, 3 High‐fructose diet (HFr) induced insulin resistance, hyperinsulinemia, hypertriglyceridemia, hyperuricemia, and elevated blood pressure in Sprague‐Dawley rats.4, 5, 6, 7 HFr caused renal hypertrophy, afferent arteriolar thickening, cortical vasoconstriction, and glomerular hypertension.5 In addition, HFr also developed a large amount of renal interstitial and perivascular collagen deposition and fibrosis.8
Fructose acutely stimulates Na+/H+ exchange activity in the proximal tubules and Na+‐K+‐Cl− cotransport in the thick ascending limbs.9, 10 Fructose also sensitizes the proximal tubules to angiotensin II.9 Therefore, the fructose‐stimulated sodium reabsorption is thought to cause salt‐sensitive hypertension. With regard to renin‐angiotensin system (RAS), HFr increased plasma renin activity (PRA), urinary angiotensin II levels,11 and renal expression of renin, angiotensin II, and angiotensin II type 1 receptor (AT1R)12 in Sprague‐Dawley rats. RAS blockade with an angiotensin‐converting enzyme (ACE) inhibitor or AT1R blocker attenuated the HFr‐induced blood pressure elevation13, 14, 15, 16 and prevented vascular dysfunction16 and left ventricular hypertrophy15 in Sprague‐Dawley rats. However, effects of RAS inhibitors on the HFr‐induced renal dysfunction and damage have not been reported to date.
Salt‐sensitive hypertension is more likely to cause multiple organ damage and to have a higher prevalence of cardiovascular and renal diseases among hypertensive subjects.17 Dahl salt‐sensitive (DS) rats have a genetic salt susceptibility with hypertriglyceridemia and insulin resistance,18, 19, 20 and high salt intake easily causes hypertension and multiple organ damage, compared with Dahl salt‐resistant (DR) rats.18, 21 Sharma et al reported that HFr induced hypertension and heart failure and increased mortality in DS rats.22 However, it is unclear whether the HFr‐induced hypertension and renal damage are exaggerated in DS rats compared with DR rats. Moreover, it is also unclear whether RAS inhibitors can ameliorate the HFr‐induced hypertension or renal damages in DS rats, because RAS inhibitors, even at a high dose, could not ameliorate hypertension or renal damage during a high‐salt diet in DS rats.23
We hypothesized that the HFr‐induced hypertension and renal damage may be exaggerated in DS rats compared with DR rats and that RAS may mediate the HFr‐induced hypertension and renal damage in DS rats. To test these hypotheses, the impacts of HFr on blood pressure, renal damages, and the RAS were examined in DS and DR rats under normal‐salt intake conditions. The effects of RAS inhibitors on the HFr‐induced hypertension and renal damage were also examined in DS rats.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Animals and Diets
All animal experiments were approved by the Tohoku University Committee for Animal Experiments and were performed in accordance with the Guidelines for Animal Experiments and Related Activities of Tohoku University, and the guiding principles of the Physiological Society of Japan and the US National Institutes of Health.
Five‐week‐old male Dahl‐Iwai S rats and Dahl‐Iwai R rats (Japan SLC Inc, Shizuoka, Japan) were housed in an animal care facility at Tohoku University Graduate School of Medicine at a controlled temperature, under a 12:12‐hour light‐dark cycle, and they were provided with standard rat chow and water. After 1 week of acclimation, their diet was changed to a control diet (Con) or a 60% HFr (TD.05075 and TD.89247, respectively; Envigo Teklad Diets, Madison, WI), which induced metabolic syndromes, as previously reported.4 The calorie (3.6 kcal/g) and the percentage of carbohydrate, protein, fat, and NaCl content (60.4%, 18.3%, 5.2%, and 1.25% by weight, respectively) are equivalent in Con and HFr. Two experiments were performed. The sample size calculations are provided by G Power 3.1.24 The effect size in this study was 0.8, considered to be large using Cohen (1988) criteria. With an α=0.05 and power=0.80, the projected total sample size needed with this effect size is approximately N=24 for this simplest between‐group comparison.
Experiment 1
To investigate HFr, DS or DR rats were randomly divided into the following 4 groups: Con‐fed DR rat (Con‐DR) group, HFr‐fed DR rat (HFr‐DR) group, Con‐fed DS rat (Con‐DS) group, and HFr‐fed DS rat (HFr‐DS) group (n=8 in each group). The rats in all 4 groups were maintained on the Con or HFr for 12 weeks.
Experiment 2
To examine effects of RAS inhibitors in HFr‐fed DS rats, DS rats were randomly divided into the following 4 groups: Con group, HFr group, HFr and enalapril treatment (HFr‐Ena) group, and HFr and candesartan treatment (HFr‐Can) group (n=6 in each group). Enalapril (10 mg/kg per day; Sigma‐Aldrich, St. Louis, MO) or candesartan (1 mg/kg per day; Tokyo Chemical Industry, Tokyo, Japan) was given in the drinking water to the HFr‐Ena or HFr‐Can group, respectively, for 12 weeks.
To further examine effects of RAS inhibitors in Con‐fed DS rats, DS rats were randomly divided into the following 3 groups: Con group, Con and enalapril treatment group, and Con and candesartan treatment group (n=6 in each group). The rats in all 3 groups were maintained on Con for 12 weeks. Enalapril (10 mg/kg per day) or candesartan (1 mg/kg per day) was given in the drinking water to the Con and enalapril treatment and Con and candesartan treatment groups, respectively, for 12 weeks.
Systolic Blood Pressure Measurement and Plasma and Urinary Parameters
Systolic blood pressure (SBP) was measured every 2 weeks using the tail cuff method (MK‐2000A; Muromachi Kikai, Tokyo, Japan). On the last day of the experiments, urine samples were collected for 24 hours using metabolic cages, and urinary albumin and creatinine were measured (Oriental Yeast Co, Ltd, Nagahama, Japan). Urinary L‐FABP (liver type fatty acid binding protein), which is a biomarker of proximal tubular stress and tubulointerstitial disorder,25 was measured using a rat L‐FABP ELISA kit (CMIC, Tokyo, Japan).
On the 11th week of experiments, blood sample was collected from tail vein after rats were fasted for 8 hours. The homeostasis model assessment for insulin resistance (HOMA‐IR) was calculated with fasting plasma glucose and insulin.26
At the end of the experiments, all rats were anesthetized with sodium pentobarbital (50 mg/kg, IP) and euthanized by decapitation. Blood samples were collected into prechilled EDTA tubes. Collected blood samples were centrifuged at 2000g for 15 minutes, and the supernatant was collected and stored at −80°C. Plasma biochemical parameters were measured by standard auto‐analysis technique, and PRA was measured by double‐antibody radioimmunoassay (PRA [FR]; Fujirebio, Tokyo, Japan). Uric acid was measured using a kit (Wako c‐test; Fujifilm Wako Pure Chemical Co, Osaka, Japan).
Tissue Sample Preparation
After euthanasia, the kidneys were quickly dissected. The renal cortex was homogenized in a buffer containing 0.1 mol/L of KH2PO4, 0.4 mol/L of Na2HPO4, 10 mmol/L of EDTA, and 10 mmol/L of DL‐dithiothreitol. In addition, 0.1 mmol/L of phenylmethylsulfonyl fluoride was added before the homogenization. The homogenate was snap frozen in liquid nitrogen and stored at −80°C until further analysis. The sample protein concentration was measured using the Bradford method, with bovine‐globulin as a standard (number 500‐0005; Bio‐Rad Laboratories, Hercules, CA).
Histological Analysis
The kidney was quickly removed after decapitation and was fixed in 10% formalin for over 24 hours and then embedded in paraffin. Paraffin sections (3 µm thick) were cut, and stained with periodic acid–Schiff and Masson trichrome staining to determine the glomerulosclerosis index (IGS) and relative interstitial volume (RIV), respectively. The IGS and RIV were assessed in accordance with a previously described method.27 For histological analysis, 5 to 6 rats per group were prepared and 50 glomeruli in each rat were assessed for IGS. The percentages of interstitium of the renal cortex and outer medulla, except glomerulus, blood vessels, and tubules per unit area, were calculated; the values of RIV from 10 areas in each rat were measured, and the mean values were calculated. Quantifications were performed in a blinded manner.
Immunohistochemical Analysis
Immunohistochemical analyses of desmin and α‐smooth muscle actin were performed to determine the severity of podocyte injury and afferent arteriolar thickening, respectively, as described previously.28, 29 The deparaffinized tissues were rehydrated in graded ethanol, rinsed in PBS, then the sections were treated with 0.3% hydrogen peroxide in absolute ethanol for 5 minutes, and processed for immunostaining with an antibody targeting desmin (Abcam, Cambridge, UK) and α‐smooth muscle actin (Dako, Glostrup, Denmark). Thirty glomeruli in each rat were assessed for desmin immunostaining, and the ratio of the desmin‐expressing area/that of the whole glomerulus was determined using Image‐J (version 2.0.0; National Institutes of Health, Bethesda, MD).26 For each arteriole, the outline of the vessel and its internal lumen (excluding the endothelium) were traced using computer analysis to calculate the total medial area (outline‐inline), in 5 to 10 arterioles per slice. The media/lumen ratio was calculated using the outline/inline relationship.29 Quantifications were performed in a blinded manner.
Immunoblot Analysis and ACE Activity Measurement
Protein expression was examined using immunoblot analysis, which was described previously.26 Renal cortical homogenates (30 μg) were loaded onto 7.5% or 10% SDS‐polyacrylamide gels and separated by electrophoresis. Then, the proteins were transferred electrophoretically to nitrocellulose membranes at 100 V in cold, 20% methanol‐containing transfer buffer for 2 hours. After blocking in 5% bovine serum albumin, the membranes were rinsed in Tris‐buffered saline–Tween‐20 buffer several times, incubated with primary antibodies raised against angiotensinogen, renin, ACE, AT1R, angiotensin II type 2 receptor (AT2R) (Santa Cruz Biotechnology, Dallas, TX), (pro)renin receptor ([P]RR) (donation from Professor Totsune), and β‐actin (Sigma‐Aldrich). Membranes were rinsed with Tris‐buffered saline–Tween‐20 and incubated at room temperature for 1 hour with a horseradish peroxidase–conjugated anti‐goat, anti‐rabbit, or anti‐mouse IgG secondary antibody (Santa Cruz Biotechnology). After washing in Tris‐buffered saline–Tween‐20, immunoblots were developed using an enhanced chemiluminescent substrate (Super Signal; Thermo Fisher Scientific, Waltham, MA). The relative intensity of band was quantified using ImageJ. The intensity of band was normalized against β‐actin, which was used as an internal standard, and was assigned a value of 1 in the Con‐DR group or Con group.
ACE activity was measured using a fluorometric assay kit (K227‐100; Biovision, Milpitas, CA). Renal cortical homogenates (2 μg protein) were mixed with a synthetic peptide substrate to release a fluorophore by active ACE, and fluorometric assays (excitation, 330 nm; emission, 430 nm) were performed after 2 hours incubation at 37℃ using a microplate reader.
Statistical Analysis
The data are presented as mean±SEM. The data of were analyzed using 2‐way repeated‐measures ANOVA or a 1‐way ANOVA, followed by the Tukey test for multiple comparisons among the groups. P<0.05 was considered to indicate statistical significance.
Results
Experiment 1: Effects of HFr in DR and DS Rats
Blood Pressure and Parameters in Urine and Plasma
Figure 1A shows time course of SBP in the Con‐fed or HFr‐fed DR and DS rats. At the start of the experiment, the SBP was not different among the groups. After 6 weeks, the SBP was significantly higher in the Con‐DS group compared with the Con‐DR group (P<0.05). After 4 weeks, HFr significantly increased the SBP in DS rats (P<0.01), but it did not affect the SBP in DR rats (Figure 1A).
Figure 1. Effects of high‐fructose diet (HFr) on blood pressure and urinary parameters in Dahl salt‐resistant (DR) and Dahl salt‐sensitive (DS) rats.
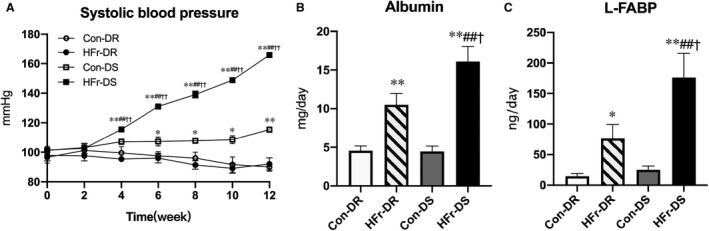
DR and DS rats were maintained on a control diet (Con) or HFr for 12 weeks. A, The time course of systolic blood pressure among the Con‐fed DR (Con‐DR) rat (n=8), HFr‐fed DR (HFr‐DR) rat (n=8), Con‐fed DS (Con‐DS) rat (n=8), and HFr‐fed DS (HFr‐DS) rat (n=8) groups. Urinary albumin excretion (B) and urinary L‐FABP (liver type fatty acid binding protein) excretion (C) were compared among the Con‐DR (n=8), HFr‐DR (n=8), Con‐DS (n=7), and HFr‐DS (n=8) groups. Data are presented as mean±SEM. *P<0.05, **P<0.01 compared with the Con‐DR group; ## P<0.01 compared with the Con‐DS group; † P<0.05, †† P<0.01 compared with the HFr‐DR group.
Urinary excretion of albumin or L‐FABP was not different between the Con‐DR and Con‐DS groups (Figure 1B and 1C). HFr significantly increased the urinary excretion of albumin and L‐FABP in both DR and DS rats (HFr‐DR, P<0.01 and P<0.01; HFr‐DS, P<0.01 and P<0.01, respectively), and the urinary albumin and L‐FABP excretions were significantly higher in the HFr‐DS group compared with the HFr‐DR group (P<0.05 and P<0.05). Diet and strain showed a significant interaction and affected urinary albumin excretion (interaction: P<0.05) but not urinary L‐FABP excretion (Table S1).
Table 1 shows body weight and plasma parameters at the end of the experiment. The body weight is no different between the groups at the start of the experiment (Con‐DR, 188±6 g; HFr‐DR, 192±5 g; Con‐DS, 175±4 g; HFr‐DS, 178±5 g), but significantly lower in the Con‐DS group than the Con‐DR group at the end of the experiment, although food intake was not significantly different between the groups. HFr did not affect the body weight in DS and DR rats. Among the parameters, HOMA‐IR and triglyceride were higher in the Con‐DS group compared with the Con‐DR group, and creatinine clearance was lower in the Con‐DS group compared with the Con‐DR group. HFr significantly increased HOMA‐IR, triglyceride, total cholesterol, free fatty acid, glucose, and uric acid in DR and DS rats, and the HOMA‐IR, total cholesterol, free fatty acid, and glucose were significantly higher in the HFr‐DS group compared with the HFr‐DR group. HFr did not affect creatinine or creatinine clearance in DR rats, but it significantly decreased creatinine and increased creatinine clearance in DS rats. HFr significantly increased PRA in DR rats, but it significantly decreased PRA in DS rats. Among the parameters, diet and strain showed a significant interaction for total cholesterol, creatinine, creatinine clearance, and PRA (Table S1).
Table 1.
Impacts of High‐Fructose Intake on Body Weight and Plasma Parameters in DR Rats and DS Rats
| Variable | Con‐DR Rats (n=8) | HFr‐DR Rats (n=8) | Con‐DS Rats (n=8) | HFr‐DS Rats (n=8) | Diet | Strain | Interaction |
|---|---|---|---|---|---|---|---|
| Body weight, g | 475±9 | 468±8 | 402±6† | 397±6§ | NS | <0.05 | NS |
| HOMA‐IR | 0.30±0.02 | 0.43±0.03† | 0.55±0.03† | 0.68±0.04†,§,‖ | <0.05 | <0.05 | NS |
| Triglyceride, mg/dL | 178±13 | 775±32† | 272±27* | 831±45†,¶ | <0.05 | <0.05 | NS |
| Total cholesterol, mg/dL | 82±2 | 91±1† | 77±3 | 102±3†,§,¶ | <0.05 | NS | <0.05 |
| Free fatty acid, mEq/L | 399±37 | 581±51* | 259±32* | 377±39‡ | <0.05 | <0.05 | NS |
| Glucose, mg/dL | 161±2 | 179±3† | 172±8 | 201±8†,‡,‖ | <0.05 | <0.05 | NS |
| Creatinine, mg/dL | 0.35±0.01 | 0.34±0.01 | 0.34±0.02 | 0.24±0.02†,§,¶ | <0.05 | <0.05 | <0.05 |
| Uric acid, mg/dL | 1.66±0.03 | 2.02±0.04† | 1.64±0.04 | 2.01±0.07†,¶ | <0.05 | NS | NS |
| CCr, mL/min | 3.53±0.12 | 3.28±0.10 | 3.07±0.14* | 4.17±0.34‡,‖ | <0.05 | NS | <0.05 |
| Renin activity, ng/mL per h | 6.2±0.6 | 9.2±0.7* | 5.2±0.8 | 2.9±0.4†,§,‖ | NS | <0.05 | <0.05 |
Data are presented as mean±SEM. CCr indicates creatinine clearance; Con‐DR, control diet–fed DR; Con‐DS, control diet–fed DS; DR, Dahl salt resistant; DS, Dahl salt sensitive; HFr‐DR, high‐fructose–fed DR; HFr‐DS, high‐fructose–fed DS; HOMA‐IR, homeostasis model assessment for insulin resistance; and NS, no significance.
*P<0.05, †P<0.01 compared with the Con‐DR group.
‡P<0.05, §P<0.01 compared with the HFr‐DR group.
‖P<0.05, ¶P<0.01 compared with the Con‐DS group.
Renal Histological Characteristics
IGS, desmin immunostaining, and RIV were significantly greater in the Con‐DS group compared with the Con‐DR group (P<0.01, P<0.01, and P<0.01, respectively), but the media/lumen ratio was not different between these groups. HFr increased the IGS, desmin immunostaining, media/lumen ratio, and RIV in both DR and DS rats (HFr‐DR, P<0.01, P<0.01, P<0.01, and P<0.05; HFr‐DS, P<0.01, P<0.01, P<0.01, and P<0.01, respectively). The IGS, desmin immunostaining, media/lumen ratio, and RIV were significantly higher in the HFr‐DS group compared with the HFr‐DR group (P<0.05, P<0.01, P<0.01, and P<0.01, respectively) (Figure 2B through 2E). Diet and strain showed a significant interaction for desmin immunostaining, media/lumen ratio, and RIV (Table S1).
Figure 2. Effects of high‐fructose diet (HFr) on renal histological characteristics in Dahl salt‐resistant (DR) and Dahl salt‐sensitive (DS) rats.
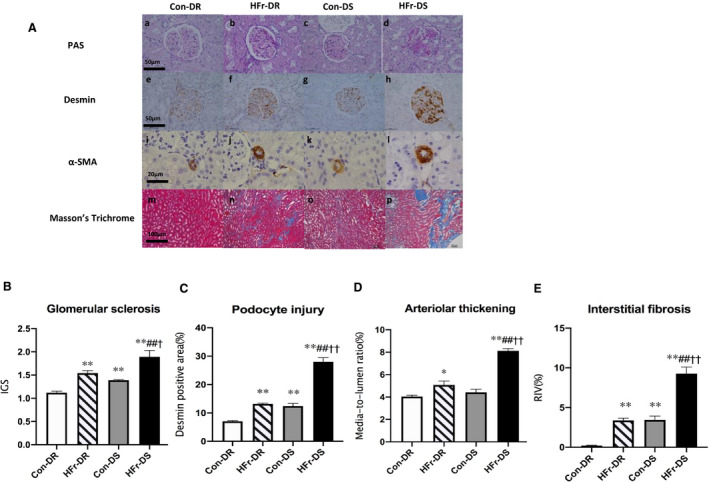
A, Representative images of the glomerulus and the renal cortex stained with periodic acid–Schiff (PAS) (top panel), against desmin (second panel), against α‐smooth muscle actin (α‐SMA) (third panel), and Masson trichrome (bottom panel) in the control diet (Con)–fed DR (Con‐DR) rat, HFr‐fed DR (HFr‐DR) rat, Con‐fed DS (Con‐DS) rat, and HFr‐fed DS (HFr‐DS) rat groups. Index of glomerular sclerosis (IGS) (B), glomerular desmin‐positive area (C), media/lumen ratio (D), and relative interstitial volume (RIV) (E) were compared among the Con‐DR (n=6), HFr‐DR (n=6), Con‐DS (n=6), and HFr‐DS (n=6) groups. Data are presented as mean±SEM. *P<0.05, **P<0.01 compared with the Con‐DR group; ## P<0.01 compared with the Con‐DS group; † P<0.05, †† P<0.01 compared with the HFr‐DR group.
Renal Expression of RAS Components
Compared with the Con‐DR group, ACE and AT2R expressions and ACE activity were significantly higher in the Con‐DS group (P<0.01, P<0.01, and P<0.05, respectively), and renin and AT1R expressions were significantly lower in the Con‐DS group (P<0.01 and P<0.01, respectively). Angiotensinogen or (P)RR expression was not different between the Con‐DR and Con‐DS groups (Figure 3A through 3G).
Figure 3. Effects of high‐fructose diet (HFr) on the expression of renin‐angiotensin system components in the renal cortex of Dahl salt‐resistant (DR) and Dahl salt‐sensitive (DS) rats.
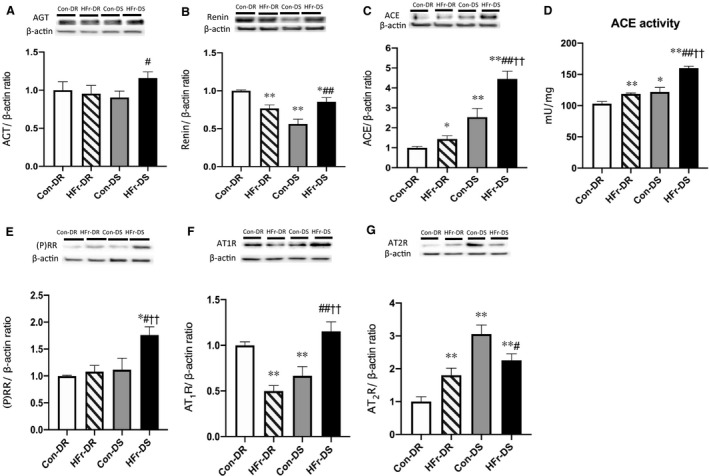
Angiotensinogen (AGT) (A), renin (B), angiotensin‐converting enzyme (ACE) (C), (pro)renin receptor ([P]RR) (E), angiotensin II type 1 receptor (AT1R) (F), angiotensin II type 2 receptor (AT2R) (G) protein expressions and ACE activity (D) were compared among the control diet (Con)–fed DR (Con‐DR) rat (n=7), HFr‐fed DR (HFr‐DR) rat (n=8), Con‐fed DS (Con‐DS) rat (n=7), and HFr‐fed DS (HFr‐DS) rat (n=8) groups. Top panels in A through C and E through G depict representative immunoblots from the different groups. The intensities of each specific protein band were normalized to that of β‐actin, and the mean intensities of the values for the Con‐DR group. Data are presented as mean±SEM. *P<0.05, **P<0.01 compared with the Con‐DR group; # P<0.05, ## P<0.01 compared with the Con‐DS group; †† P<0.01 compared with the HFr‐DR group.
In DR rats, HFr significantly decreased the renin and AT1R expressions (P<0.01 and P<0.01, respectively) (Figure 3B and 3F), increased the ACE and AT2R expressions and the ACE activity (P<0.05, P<0.01, and P<0.01, respectively) (Figure 3C, 3D, and 3G), and did not affect the angiotensinogen or (P)RR expression (Figure 3A and 3E). In DS rats, HFr significantly increased the angiotensinogen, renin, (P)RR, ACE, and AT1R expressions and the ACE activity (P<0.01, P<0.01, P<0.05, P<0.01, P<0.01, and P<0.01, respectively) (Figure 3A through 3F) and decreased the AT2R expression (P<0.05) (Figure 3G). Although HFr significantly increased the ACE expression and the ACE activity in both DR and DS rats, the ACE expression and activity were significantly higher in the HFr‐DS group compared with the HFr‐DR group (P<0.01 and P<0.01, respectively) (Figure 3C and 3D). Diet and strain showed significant interactions for the renin, AT1R, and AT2R expressions and the ACE activity, but there was no significant interaction for the angiotensinogen, (P)RR, or ACE expression (Table S1).
Experiment 2: Effects of RAS Inhibitors in HFr‐Fed DS Rats
Blood Pressure and Parameters in Urine and Plasma
Figure 4A and Figure S1A show the time courses of SBP in RAS inhibitor–treated DS rats fed HFr or Con. After 2 weeks, both enalapril and candesartan significantly decreased the SBP in HFr‐fed or Con‐fed DS rats. The RAS inhibitors completely blocked the SBP elevation in HFr‐fed DS rats (P<0.01) (Figure 4A), and the RAS inhibitors slightly but significantly decreased SBP in Con‐fed DS rats at the end of the experiment (P<0.01) (Figure S1A).
Figure 4. Effects of enalapril and candesartan on blood pressure and urinary parameters in high‐fructose diet (HFr)–fed Dahl salt‐sensitive (DS) rats.
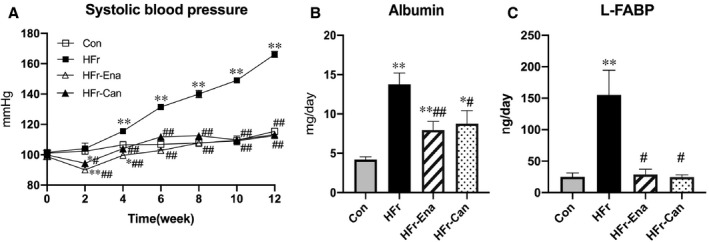
The HFr‐fed DS rats were treated with enalapril (HFr‐Ena; 10 mg/kg per day) or candesartan (HFr‐Can; 1 mg/kg per day) for 12 weeks. A, The time course of systolic blood pressure among the control diet (Con), HFr, HFr‐Ena, and HFr‐Can groups. Urinary albumin excretion (B) and urinary L‐FABP (liver type fatty acid binding protein) excretion (C) were compared among the Con (n=6), HFr (n=6), HFr‐Ena (n=6), and HFr‐Can (n=6) groups. Data are presented as mean±SEM. *P<0.05, **P<0.01 compared with the Con group; # P<0.05, ## P<0.01 compared with the HFr group.
At the end of the experiment, both enalapril and candesartan significantly decreased the HFr‐increased urinary albumin excretion (P<0.01 and P<0.05, respectively), but the levels were still higher compared with those in the Con group (Figure 4B). Enalapril but not candesartan significantly decreased the urinary albumin excretion in Con‐fed DS rats (P<0.01) (Figure S1B). Both enalapril and candesartan significantly decreased the HFr‐increased urinary L‐FABP excretion to the levels of the Con group (P<0.05 and P<0.05, respectively) (Figure 4C), but the RAS inhibitors did not affect urinary L‐FABP excretion in Con‐fed DS rats (Figure S1C).
Table 2 and Table S2 show plasma parameters at the end of the experiment. In HFr‐fed DS rats, both enalapril and candesartan significantly decreased the HFr‐increased triglyceride, but these levels were still higher compared with those in the Con group. Enalapril significantly decreased the HFr‐increased total cholesterol, and candesartan significantly decreased the HFr‐increased free fatty acid, but the levels were still higher than those in the Con group. Neither enalapril nor candesartan affected the HOMA‐IR or glucose levels. Both enalapril and candesartan significantly decreased creatinine clearance to the levels observed in the Con group. Candesartan but not enalapril significantly decreased uric acid. Both enalapril and candesartan significantly increased PRA. In Con‐fed DS rats, enalapril significantly decreased the total cholesterol, and both enalapril and candesartan significantly increased free fatty acid. Both enalapril and candesartan significantly increased PRA. The RAS inhibitor–increased PRA was greater in the Con‐fed DS rats compared with the HFr‐fed DS rats (Table 2 and Table S2).
Table 2.
Effects of Enalapril and Candesartan on Body Weight and Plasma Parameters in High‐Fructose–Fed DS Rats
| Variable | Con‐DS Rats (n=6) | HFr‐DS Rats (n=6) | HFr‐Ena DS Rats (n=6) | HFr‐Can DS Rats (n=6) |
|---|---|---|---|---|
| Body weight, g | 398±6 | 397±6 | 402±3 | 393±4 |
| HOMA‐IR | 0.56±0.04 | 0.70±0.04* | 0.66±0.05 | 0.62±0.04 |
| Triglyceride, mg/dL | 267±27 | 882±49† | 639±50†,§ | 618±30† |
| Total cholesterol, mg/dL | 76±1 | 105±2† | 95±3†,‡ | 98±4† |
| Free fatty acid, mEq/L | 218±16 | 386±37† | 303±23* | 284±18*,‡ |
| Glucose, mg/dL | 152±9 | 194±8† | 175±3* | 180±2† |
| Creatinine, mg/dL | 0.35±0.02 | 0.22±0.02† | 0.26±0.01† | 0.25±0.01† |
| Uric acid, mg/dL | 1.64±0.05 | 2.13±0.08† | 1.89±0.09* | 1.46±0.06§ |
| CCr, mL/min | 3.03±0.16 | 3.91±0.27* | 3.14±0.13‡ | 3.22±0.13‡ |
| Renin activity, ng/mL per h | 5.7±0.8 | 3.0±0.3† | 17.1±1.9†,§ | 18.0±0.6†,§ |
Data are presented as mean±SEM. CCr indicates creatinine clearance; Con‐DS, control diet–fed DS; DS, Dahl salt sensitive; HFr‐Can, high‐fructose diet and candesartan treatment; HFr‐DS, high‐fructose–fed DS; HFr‐Ena, high‐fructose diet and enalapril treatment; and HOMA‐IR, homeostasis model assessment for insulin resistance.
*P<0.05, †P<0.01 compared with the Con‐DS group.
‡P<0.05, §P<0.01 compared with the HFr‐DS group.
Renal Histological Characteristics
In HFr‐fed DS rats, both enalapril and candesartan significantly lowered the HFr‐increased IGS, desmin immunostaining, media/lumen ratio, and RIV (HFr‐Ena: P<0.05, P<0.01, P<0.05, and P<0.01; HFr‐Can: P<0.05, P<0.01, P<0.01, and P<0.01, respectively). RAS inhibitors significantly decreased the desmin immunostaining to the levels of the Con group. However, IGS and the media/lumen ratio were still higher in the HFr‐Ena and HFr‐Can groups compared with the Con group (Figure 5B through 5E).
Figure 5. Effects of enalapril and candesartan on renal histological characteristics in high‐fructose diet (HFr)–fed Dahl salt‐sensitive (DS) rats.
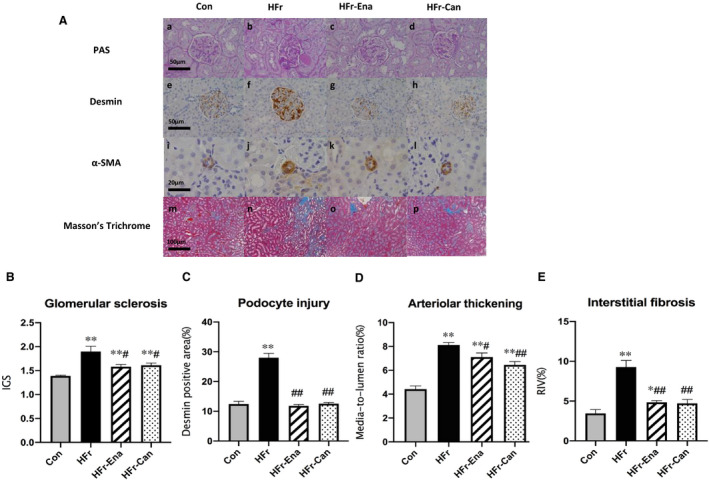
The HFr‐fed DS rats were treated with enalapril (HFr‐Ena; 10 mg/kg per day) or candesartan (HFr‐Can; 1 mg/kg per day) for 12 weeks. A, Representative images of the glomerulus and the renal cortex stained with periodic acid–Schiff (PAS) (top panel), against desmin (second panel), against α‐smooth muscle actin (α‐SMA) (third panel), and Masson trichrome (bottom panel) in the control diet (Con), HFr, HFr‐Ena, and HFr‐Can groups. Index of glomerular sclerosis (IGS) (B), glomerular desmin‐positive area (C), media/lumen ratio (D), and relative interstitial volume (RIV) (E) were compared among the Con (n=6), HFr (n=6), HFr‐Ena (n=6), and HFr‐Can (n=6) groups. Data are presented as mean±SEM. *P<0.05, **P<0.01 compared with the Con group; # P<0.05, ## P<0.01 compared with the HFr group.
Renal Expression of RAS Components
In HFr‐fed DS rats, both enalapril and candesartan significantly decreased the HFr‐increased renin, ACE, and AT1R expressions (HFr‐Ena: P<0.05, P<0.05, and P<0.01; HFr‐Can: P<0.05, P<0.05, and P<0.01, respectively) (Figure 6B, 6C, and 6F), and the levels were not different from those in the Con group. Enalapril and candesartan also significantly decreased the HFr‐increased ACE activity (HFr‐Ena, P<0.01; HFr‐Can, P<0.01), and the levels were lower than those in the Con group (Figure 6D). Neither enalapril nor candesartan affected the HFr‐increased angiotensinogen or (P)RR expression (Figure 6A and 6E). Both enalapril and candesartan further decreased the HFr‐decreased AT2R expression (HFr‐Ena, P<0.01; HFr‐Can, P<0.01) (Figure 6G). In Con‐fed DS rats, both enalapril and candesartan significantly increased the renin and (P)RR expressions (Con and enalapril treatment, P<0.05 and P<0.01; Con and candesartan treatment, P<0.05 and P<0.01, respectively) (Figure S2B and S2E), but decreased the ACE and AT2R expressions and the ACE activity (Con and enalapril treatment: P<0.05, P<0.01, and P<0.01; Con and candesartan treatment: P<0.05, P<0.01, and P<0.05, respectively) (Figure S2C, S2D, and S2G).
Figure 6. Effects of enalapril and candesartan on the expression of renin‐angiotensin system components in the renal cortex of high‐fructose diet (HFr)–fed Dahl salt‐sensitive (DS) rats.
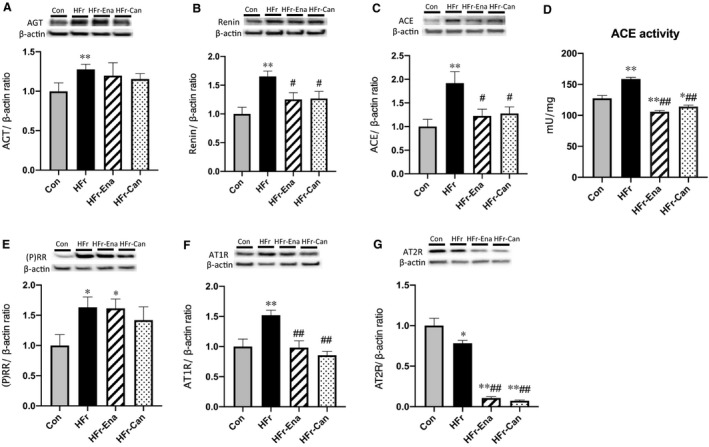
The HFr‐fed DS rats were treated with enalapril (HFr‐Ena; 10 mg/kg per day) or candesartan (HFr‐Can; 1 mg/kg per day) for 12 weeks. Angiotensinogen (AGT) (A), renin (B), angiotensin‐converting enzyme (ACE) (C), (pro)renin receptor ([P]RR) (E), angiotensin II type 1 receptor (AT1R) (F), angiotensin II type 2 receptor (AT2R) (G) protein expressions and ACE activity (D) were compared among the control diet (Con) (n=6), HFr (n=6), HFr‐Ena (n=6), and HFr‐Can (n=6) groups. Top panels in A through C and E through G depict representative immunoblots from the different groups. The intensities of each specific protein band were normalized to that of β‐actin, and the mean intensities of the values for the Con group. Data are presented as mean±SEM. *P<0.05, **P<0.01 compared with the Con group; # P<0.05, ## P<0.01 compared with the HFr group.
Discussion
The present study compared the impacts of HFr on blood pressure and renal damage between DS and DR rats and investigated the role of RAS in the HFr‐induced hypertension and renal damage in DS rats. HFr induced insulin resistance, dyslipidemia, and hyperuricemia and increased urinary albumin and L‐FABP excretion and renal damage in both DS and DR rats, but these changes were even greater in DS rats compared with DR rats. HFr induced hypertension and glomerular hyperfiltration in DS rats but not in DR rats. Although HFr increased PRA in DR rats, it decreased PRA in DS rats. HFr increased renal angiotensinogen, renin, (P)RR, ACE, and AT1R expression and ACE activity in DS rats, whereas it increased only ACE expression and activity in the DR rats. RAS inhibitors prevented the HFr‐induced hypertension, glomerular hyperfiltration, and renal damage in DS rats. This is the first report to show differences in the impacts of HFr between DS and DR rats and the effectiveness of RAS inhibitors against the HFr‐induced hypertension and renal damage in DS rats.
Consistent with previous studies,18, 19, 20 insulin resistance and hypertriglyceridemia were observed in Con‐fed DS rats. Although HFr induced insulin resistance and dyslipidemia in both DS and DR rats, it induced hypertension and glomerular hyperfiltration in DS rats but not in DR rats. High‐salt–induced renal injury was associated with a marked elevation in glomerular pressure in DS rats, suggesting an impairment in long‐term autoregulation of renal blood flow and the glomerular filtration rate.30 Impaired glomerular filtration rate autoregulation in DS rats is thought to result from impaired myogenic and tubuloglomerular feedback responses in the afferent arterioles.31 The impaired myogenic and tubuloglomerular feedback responses may also contribute to the HFr‐induced glomerular hyperfiltration in DS rats. Future study is necessary to investigate impacts of HFr on autoregulation of glomerular filtration rate and tubuloglomerular feedback response in DS rats.
Xu et al reported that HFr increased PRA, renal (P)RR, and renin expression in parallel with increased urinary angiotensin II levels in Sprague‐Dawley rats,11 which suggests that there is an interaction between HFr and RAS. The present study assessed impacts of HFr on circulatory and renal RAS and revealed that HFr decreased PRA in DS rats, whereas it increased PRA in DR rats. In contrast to PRA, HFr increased renal angiotensinogen, renin, (P)RR, ACE, and AT1R expressions and ACE activity in DS rats, whereas HFr increased only the renal ACE expression and activity and decreased the renin and AT1R expressions in DR rats. DS rats have been reported to have lower PRA and renal renin activity compared with DR rats.32 HFr increased renal renin expression despite lowering PRA in DS rats, suggesting that the HFr‐induced renal damage might be attributable to activation of renal RAS. Because HFr also induced albuminuria, it is possible that glomerular‐filtrated angiotensinogen might affect the activation of renal RAS in HFr‐fed DS rats. To clarify the role of glomerular‐filtrated angiotensinogen, we further examined the urinary excretion of total angiotensinogen in DS rats. Although the urinary total angiotensinogen excretion was significantly increased by HFr (P<0.05), the HFr‐increased urinary total angiotensinogen excretion was not decreased by enalapril or candesartan (Con, 508±46 ng/d; HFr, 844±108 ng/d; HFr‐Ena, 737±53 ng/d; HFr‐Can, 855±79 ng/d). The changes in urinary total angiotensinogen excretion were same as the changes in renal angiotensinogen expression but not in urine albumin, suggesting that effects of HFr on renal RAS might be mainly caused by changes in inherent intrarenal RAS than glomerular filtration of plasma angiotensinogen. Previous studies indicated beneficial roles for the AT2R in controlling blood pressure and renal function and the development of interstitial fibrosis.33, 34 Although HFr decreased the renal AT2R expression, RAS inhibitors further reduced the HFr‐decreased AT2R expression in DS rats, suggesting that AT2R might not mediate the HFr‐induced hypertension or renal damage.
Consistent with the previous studies that reported that RAS inhibitors prevented HFr‐induced insulin resistance, dyslipidemia, and blood pressure elevation in Sprague‐Dawley and Wistar rats,13, 14, 15, 16, 35 both enalapril and candesartan prevented HFr‐induced hypertension, albuminuria, glomerular hyperfiltration, and renal damage in DS rats. Sugimoto et al23 reported that enalapril or candesartan did not attenuate the increase in blood pressure or change PRA during a high‐salt diet (8% NaCl) in DS rats and that the RAS inhibitors greatly reduced blood pressure and increased PRA after switching from the high‐salt diet to a low‐salt diet (0.3% NaCl). This suggests that the RAS was involved in the control of blood pressure in salt‐depleted DS rats. The present study shows that the RAS inhibitors greatly reduced blood pressure and increased PRA in the HFr‐fed DS rats as well as in the salt‐depleted DS rats.23 However, the RAS inhibitor–induced increase in PRA was lower in the HFr‐fed DS rats compared with the Con‐fed DS rats. Fructose is reported to stimulate sodium reabsorption in the proximal tubules and thick ascending limbs and sensitize the proximal tubules to angiotensin II.9, 10 Long‐term treatment with hydrochlorothiazide (2–5 mg/kg per day) for 12 weeks prevented the HFr‐induced hypertension, glomerular hyperfiltration, and podocyte injury, partially inhibited the HFr‐induced albuminuria, glomerular sclerosis, and arteriolar thickening, but did not affect the HFr‐induced interstitial fibrosis in DS rats (Figure S3 and Table S3). Thus, the HFr‐induced hypertension, glomerular hyperfiltration, and glomerular damages might be mediated by sodium reabsorption in addition to renal RAS activation in DS rats.
Because enalapril and candesartan decreased SBP in DS rats, it is possible that the antihypertensive effects of RAS inhibitors might contribute to the renal protective effects. Thus, an additional experiment was performed. Long‐term treatment with hydralazine (4 mg/kg per day) for 12 weeks decreased blood pressure to similar levels as the RAS inhibitors but did not significantly prevent the HFr‐induced albuminuria, glomerular hyperfiltration, and renal damage in the HFr‐fed rats (Figure S3 and Table S3). These results suggest that the renal protective effects of RAS inhibitors might be independent of their antihypertensive effects in DS rats.
Under the control diet, the plasma insulin concentration was 6.5 times higher in DS rats and DR rats (2.05±0.22 and 2.21±0.11 ng/mL, respectively) than in Sprague‐Dawley rats (0.33±0.05 ng/mL), which was reported in our previous report.36 HFr increased the plasma insulin concentration by 20% and 10% in DS rats and DR rats (2.72±0.13 and 2.26±0.13 ng/mL), respectively, whereas HFr increased the plasma insulin concentration by 7‐fold in Sprague‐Dawley rats (2.18±0.34 ng/mL).36 HFr also increased the SBP by 30 and 50 mm Hg in Sprague‐Dawley rats and DS rats, respectively, but did not change the SBP in DR rats. The 7‐fold increase in the plasma insulin concentration might cause the HFr pressor response in Sprague‐Dawley rats.36 In addition, it is also possible the 20% increase in the plasma insulin concentration might affect the pressor response of HFr in DS rats, because DS rats have an exaggerated chloride reabsorption in the Henle loop and the thick ascending limb,37, 38 where insulin stimulates chloride reabsorption,39, 40 and insulin administration increased blood pressure in DS rats.37, 38, 39, 41
HFr increased plasma triglyceride and free fatty acid in both DR and DS rats. It is possible that dyslipidemia and renal metabolism of fatty acid might affect the HFr‐induced renal damage in DS rats, because we previously reported that HFr increased triglyceride content in the kidney of Sprague‐Dawley rats.36 However, lipid‐lowering treatment with fibrate for 12 weeks did not ameliorate the HFr‐induced hypertension or albuminuria in DS rats despite normalizing plasma triglycerides and free fatty acid (data not shown).
Uric acid is a related product of fructose metabolism, and it plays an important role in hypertension and renal damage.4, 42 Previous studies indicate that uric acid increases blood pressure and is correlated with renal RAS activation in humans and rats.43, 44 However, plasma uric acid levels were not different between the HFr‐DR and HFr‐DS rats. In addition, candesartan but not enalapril significantly decreased plasma uric acid, although the antihypertensive and renal protective effects of both drugs were similar. We further examined the activity of xanthine oxidase, an enzyme that metabolizes hypoxanthine to xanthine and uric acid. HFr increased the xanthine oxidase activity in the kidney of both DR and DS rats, but this renal activity was lower in the HFr‐DS rats compared with the HFr‐DR rats (data not shown). These results suggest that uric acid and xanthine oxidase might have minimal effects on the HFr‐induced hypertension or renal damage in DS rats.
This study had several limitations. We used Dahl‐Iwai S rats, in which salt sensitivity is relatively weak compared with Dahl SS/Jr rats. Future study is necessary to examine whether the HFr‐induced hypertension and organ damage are further exaggerated in Dahl SS/Jr rats. We measured SBP using the tail‐cuff method, which is not the most accurate method. Blood pressure measurement via telemetry might be more accurate. Plasma and intrarenal angiotensin II levels were not examined. Although sodium reabsorption affects the HFr‐induced hypertension and renal damage,45 tubular sodium transport was not examined. In addition, the HFr‐induced renal dysfunction and damage were reported to be associated with oxidative stress, alterations in metabolism of nitric oxide system, and a proinflammatory condition in Sprague‐Dawley rats.8 Further studies are required to investigate other mechanisms of the HFr‐induced hypertension and renal damages.
In summary, HFr‐induced blood pressure elevation and renal damage were exaggerated in DS rats compared with DR rats. Although it suppressed circulatory RAS, HFr activated renal RAS in DS rats, and RAS inhibitors attenuated the HFr‐induced hypertension and renal damage in DS rats. These results suggest that the HFr‐induced hypertension and renal damage were mediated by renal RAS activation and that they could be controlled by RAS inhibitors. Excessive fructose intake may be associated with a higher risk of developing hypertension and renal damage in salt‐sensitive people compared with salt‐resistant people. In contrast to the low effectiveness of RAS inhibitors in high‐salt–induced hypertension, RAS inhibitors may be effective drugs against the HFr‐induced hypertension and renal damage regardless of salt sensitivity.
Sources of Funding
This work was supported by Grants‐in‐Aid for Scientific Research (Japan society for the promotion of science KAKENHI numbers 17H0211, 20H04054, and 20H04034).
Disclosures
None.
Supporting information
Tables S1–S3
Figures S1–S3
Acknowledgments
We would like to thank the Institute for Animal Experimentation of Tohoku University Graduate School of Medicine for animal care, and the Biomedical Research Unit of Tohoku University Hospital and the Biomedical Research Core of Tohoku University Graduate School of Medicine for technical assistance. We are also grateful to Professor Kazuhito Totsune for his generous donation of the (pro)renin receptor antibody. We would like to thank Edanz Group (www.edanzediting.com/ac) for editing a draft of this article.
(J Am Heart Assoc. 2021;10:e016543. DOI: 10.1161/JAHA.120.016543.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.016543
For Sources of Funding and Disclosures, see page 12.
References
- 1.Hannou SA, Haslam DE, McKeown NM, Herman MA. Fructose metabolism and metabolic disease. J Clin Invest. 2018;545–555. DOI: 10.1172/JCI96702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, Hatcher B, Cox CL, Dyachenko A, Zhang W, et al. Consuming fructose‐sweetened, not glucose‐sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. 2009;1322–1334. DOI: 10.1172/JCI37385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perez‐Pozo SE, Schold J, Nakagawa T, Sanchez‐Lozada LG, Johnson RJ, Lillo JL. Excessive fructose intake induces the features of metabolic syndrome in healthy adult men: role of uric acid in the hypertensive response. Int J Obes (Lond). 2010;454–461. DOI: 10.1038/ijo.2009.259. [DOI] [PubMed] [Google Scholar]
- 4.Nakagawa T, Hu H, Zharikov S, Tuttle KR, Short RA, Glushakova O, Ouyang X, Feig DI, Block ER, Herrera‐Acosta J, et al. A causal role for uric acid in fructose‐induced metabolic syndrome. Am J Physiol Renal Physiol. 2006;F625–F631. DOI: 10.1152/ajprenal.00140.2005. [DOI] [PubMed] [Google Scholar]
- 5.Sánchez‐Lozada LG, Tapia E, Jiménez A, Bautista P, Cristóbal M, Nepomuceno T, Soto V, Ávila‐Casado C, Nakagawa T, Johnson RJ, et al. Fructose‐induced metabolic syndrome is associated with glomerular hypertension and renal microvascular damage in rats. Am J Physiol Renal Physiol. 2007;F423–F429. DOI: 10.1152/ajprenal.00124.2006. [DOI] [PubMed] [Google Scholar]
- 6.Hwang IS, Ho H, Hoffman BB, Reaven GM. Fructose‐induced insulin resistance and hypertension in rats. Hypertension. 1987;512–516. DOI: 10.1161/01.HYP.10.5.512. [DOI] [PubMed] [Google Scholar]
- 7.Nakayama T, Kosugi T, Gersch M, Connor T, Sanchez‐Lozada LG, Lanaspa MA, Roncal C, Perez‐Pozo SE, Johnson RJ, Nakagawa T. Dietary fructose causes tubulointerstitial injury in the normal rat kidney. Am J Physiol Renal Physiol. 2010;F712–F720. DOI: 10.1152/ajprenal.00433.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prince PD, Lanzi CR, Toblli JE, Elesgaray R, Oteiza PI, Fraga CG, Galleano M. Dietary (‐)‐epicatechin mitigates oxidative stress, no metabolism alterations, and inflammation in renal cortex from fructose‐fed rats. Free Radic Biol Med. 2016;35–46. DOI: 10.1016/j.freeradbiomed.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 9.Cabral PD, Hong NJ, Hye Khan MA, Ortiz PA, Beierwaltes WH, Imig JD, Garvin JL. Fructose stimulates Na/H exchange activity and sensitizes the proximal tubule to angiotensin II. Hypertension. 2014;e68–e73. DOI: 10.1161/HYPERTENSIONAHA.113.02564. [DOI] [PubMed] [Google Scholar]
- 10.Ares GR, Kassem KM, Ortiz PA. Fructose acutely stimulates NKCC2 activity in rat thick ascending limbs by increasing surface NKCC2 expression. Am J Physiol Renal Physiol. 2019;F550–F557. DOI: 10.1152/ajprenal.00136.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu C, Lu A, Lu X, Zhang L, Fang H, Zhou L, Yang T. Activation of renal (pro)renin receptor contributes to high fructose‐induced salt sensitivity. Hypertension. 2017;339–348. DOI: 10.1161/HYPERTENSIONAHA.116.08240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dhar I, Dhar A, Wu L, Desai KM. Increased methylglyoxal formation with upregulation of renin angiotensin system in fructose fed Sprague Dawley rats. PLoS One. 2013;10:e74212. DOI: 10.1371/journal.pone.0074212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furuhashi M, Ura N, Takizawa H, Yoshida D, Moniwa N, Murakami H, Higashiura K, Shimamoto K. Blockade of the renin‐angiotensin system decreases adipocyte size with improvement in insulin sensitivity. J Hypertens. 2004;1977–1982. DOI: 10.1097/00004872-200410000-00021. [DOI] [PubMed] [Google Scholar]
- 14.Okada K, Hirano T, Ran J, Adachi M. Olmesartan medoxomil, an angiotensin II receptor blocker ameliorates insulin resistance and decreases triglyceride production in fructose‐fed rats. Hypertens Res. 2004;293–299. DOI: 10.1291/hypres.27.293. [DOI] [PubMed] [Google Scholar]
- 15.Kamide K, Rakugi H, Higaki J, Okamura A, Nagai M, Moriguchi K, Ohishi M, Satoh N, Tuck ML, Ogihara T. The renin‐angiotensin and adrenergic nervous system in cardiac hypertrophy in fructose‐fed rats. Am J Hypertens. 2002;66–71. DOI: 10.1016/S0895-7061(01)02232-4. [DOI] [PubMed] [Google Scholar]
- 16.Nyby MD, Abedi K, Smutko V, Eslami P, Tuck ML. Vascular angiotensin type 1 receptor expression is associated with vascular dysfunction, oxidative stress and inflammation in fructose‐fed rats. Hypertens Res. 2007;451–457. DOI: 10.1291/hypres.30.451. [DOI] [PubMed] [Google Scholar]
- 17.Morimoto A, Uzu T, Fujii T, Nishimura M, Kuroda S, Nakamura S, Inenaga T, Kimura G. Sodium sensitivity and cardiovascular events in patients with essential hypertension. Lancet. 1997;1734–1737. DOI: 10.1016/S0140-6736(97)05189-1. [DOI] [PubMed] [Google Scholar]
- 18.Rapp JP. Dahl salt‐susceptible and salt‐resistant rats: a review. Hypertension. 1982;753–763. [DOI] [PubMed] [Google Scholar]
- 19.Reaven GM, Twersky J, Chang H. Abnormalities of carbohydrate and lipid metabolism in Dahl rats. Hypertension. 1991;630–635. DOI: 10.1161/01.HYP.18.5.630. [DOI] [PubMed] [Google Scholar]
- 20.Mondon CE, Plato PA, Dall'Aglio E, Sztalryd C, Reaven G. Mechanism of hypertriglyceridemia in Dahl rats. Hypertension. 1993;373–379. DOI: 10.1161/01.HYP.21.3.373. [DOI] [PubMed] [Google Scholar]
- 21.Nagase M, Shibata S, Yoshida S, Nagase T, Gotoda T, Fujita T. Podocyte injury underlies the glomerulopathy of Dahl salt‐hypertensive rats and is reversed by aldosterone blocker. Hypertension. 2006;1084–1093. DOI: 10.1161/01.HYP.0000222003.28517.99. [DOI] [PubMed] [Google Scholar]
- 22.Sharma N, Okere IC, Duda MK, Johnson J, Yuan CL, Chandler MP, Ernsberger P, Hoit BD, Stanley WC. High fructose diet increases mortality in hypertensive rats compared to a complex carbohydrate or high fat diet. Am J Hypertens. 2007;403–409. DOI: 10.1016/j.amjhyper.2006.09.022. [DOI] [PubMed] [Google Scholar]
- 23.Sugimoto K, Fujimura A, Takasaki I, Tokita Y, Iwamoto T, Takizawa T, Gotoh E, Shionoiri H, Ishii M. Effects of renin‐angiotensin system blockade and dietary salt intake on left ventricular hypertrophy in Dahl salt‐sensitive rats. Hypertens Res. 1998;163–168. DOI: 10.1291/hypres.21.163. [DOI] [PubMed] [Google Scholar]
- 24.Faul F, Erdfelder E, Lang AG, Buchner A. G*power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;175–191. DOI: 10.3758/BF03193146. [DOI] [PubMed] [Google Scholar]
- 25.Kamijo‐Ikemori A, Sugaya T, Kimura K. Urinary fatty acid binding protein in renal disease. Clin Chim Acta. 2006;1–7. DOI: 10.1016/j.cca.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 26.Ito D, Cao P, Kakihana T, Sato E, Suda C, Muroya Y, Ogawa Y, Hu G, Ishii T, Ito O, et al. Chronic running exercise alleviates early progression of nephropathy with upregulation of nitric oxide synthases and suppression of glycation in Zucker diabetic rats. PLoS One. 2015;10:e0138037. DOI: 10.1371/journal.pone.0138037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanazawa M, Kawamura T, Li L, Sasaki Y, Matsumoto K, Kataoka H, Ito O, Minami N, Sato T, Ootaka T, et al. Combination of exercise and enalapril enhances renoprotective and peripheral effects in rats with renal ablation. Am J Hypertens. 2006;80–86. DOI: 10.1016/j.amjhyper.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 28.Zou J, Yaoita E, Watanabe Y, Yoshida Y, Nameta M, Li H, Qu Z, Yamamoto T. Upregulation of nestin, vimentin, and desmin in rat podocytes in response to injury. Virchows Arch. 2006;485–492. DOI: 10.1007/s00428-005-0134-9. [DOI] [PubMed] [Google Scholar]
- 29.Sanchez‐Lozada LG, Tapia E, Santamaria J, Avila‐Casado C, Soto V, Nepomuceno T, Rodriguez‐Iturbe B, Johnson RJ, Herrera‐Acosta J. Mild hyperuricemia induces vasoconstriction and maintains glomerular hypertension in normal and remnant kidney rats. Kidney Int. 2005;237–247. DOI: 10.1111/j.1523-1755.2005.00074.x. [DOI] [PubMed] [Google Scholar]
- 30.Williams JM, Sarkis A, Hoagland KM, Fredrich K, Ryan RP, Moreno C, Lopez B, Lazar J, Fenoy FJ, Sharma M, et al. Transfer of the CYP4A region of chromosome 5 from Lewis to Dahl S rats attenuates renal injury. Am J Physiol Renal Physiol. 2008;F1764–F1777. DOI: 10.1152/ajprenal.90525.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ge Y, Murphy SR, Fan F, Williams JM, Falck JR, Liu R, Roman RJ. Role of 20‐HETE in the impaired myogenic and TGF responses of the Af‐Art of Dahl salt‐sensitive rats. Am J Physiol Renal Physiol. 2014;F509–F515. DOI: 10.1152/ajprenal.00273.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwai J, Dahl LK, Knudsen KD. Genetic influence on the renin‐angiotensin system: low renin activities in hypertension‐prone rats. Circ Res. 1973;678–684. DOI: 10.1161/01.RES.32.6.678. [DOI] [PubMed] [Google Scholar]
- 33.Kaschina E, Unger T. Angiotensin AT1/AT2 receptors: regulation, signalling and function. Blood Press. 2003;70–88. DOI: 10.1080/08037050310001057. [DOI] [PubMed] [Google Scholar]
- 34.Kaschina E, Namsolleck P, Unger T. AT2 receptors in cardiovascular and renal diseases. Pharmacol Res. 2017;39–47. DOI: 10.1016/j.phrs.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 35.Rabie EM, Heeba GH, Abouzied MM, Khalifa MM. Comparative effects of aliskiren and telmisartan in high fructose diet‐induced metabolic syndrome in rats. Eur J Pharmacol. 2015;145–153. DOI: 10.1016/j.ejphar.2015.04.019. [DOI] [PubMed] [Google Scholar]
- 36.Hu G, Xu L, Ma Y, Kohzuki M, Ito O. Chronic exercise provides renal‐protective effects with upregulation of fatty acid oxidation in the kidney of high fructose‐fed rats. Am J Physiol Renal Physiol. 2020;F826–F834. DOI: 10.1152/ajprenal.00444.2019. [DOI] [PubMed] [Google Scholar]
- 37.Roman RJ, Kaldunski ML. Enhanced chloride reabsorption in the loop of Henle in Dahl salt‐sensitive rats. Hypertension. 1991;1018–1024. DOI: 10.1161/01.HYP.17.6.1018. [DOI] [PubMed] [Google Scholar]
- 38.Ito O, Roman RJ. Role of 20‐HETE in elevating chloride transport in the thick ascending limb of Dahl SS/Jr rats. Hypertension. 1999;419–423. DOI: 10.1161/01.HYP.33.1.419. [DOI] [PubMed] [Google Scholar]
- 39.Kirchner KA. Insulin increases loop segment chloride reabsorption in the euglycemic rat. Am J Physiol. 1988;F1206–F1213. DOI: 10.1152/ajprenal.1988.255.6.F1206. [DOI] [PubMed] [Google Scholar]
- 40.Ito O, Kondo Y, Takahashi N, Kudo K, Igarashi Y, Omata K, Imai Y, Abe K. Insulin stimulates NaCl transport in isolated perfused MTAL of Henle's loop of rabbit kidney. Am J Physiol. 1994;F265–F270. DOI: 10.1152/ajprenal.1994.267.2.F265. [DOI] [PubMed] [Google Scholar]
- 41.Tomiyama H, Kushiro T, Abeta H, Kurumatani H, Taguchi H, Kuga N, Saito F, Kobayashi F, Otsuka Y, Kanmatsuse K, et al. Blood pressure response to hyperinsulinemia in salt‐sensitive and salt‐resistant rats. Hypertension. 1992;596–600. DOI: 10.1161/01.HYP.20.5.596. [DOI] [PubMed] [Google Scholar]
- 42.Sanchez‐Lozada LG, Tapia E, Bautista‐Garcia P, Soto V, Avila‐Casado C, Vega‐Campos IP, Nakagawa T, Zhao L, Franco M, Johnson RJ. Effects of febuxostat on metabolic and renal alterations in rats with fructose‐induced metabolic syndrome. Am J Physiol Renal Physiol. 2008;F710–F718. DOI: 10.1152/ajprenal.00454.2007. [DOI] [PubMed] [Google Scholar]
- 43.Mazzali M, Kanellis J, Han L, Feng L, Xia Y‐Y, Chen Q, Kang D‐H, Gordon KL, Watanabe S, Nakagawa T, et al. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure‐independent mechanism. Am J Physiol Renal Physiol. 2002;F991–F997. DOI: 10.1152/ajprenal.00283.2001. [DOI] [PubMed] [Google Scholar]
- 44.Perlstein TS, Gumieniak O, Hopkins PN, Murphey LJ, Brown NJ, Williams GH, Hollenberg NK, Fisher ND. Uric acid and the state of the intrarenal renin‐angiotensin system in humans. Kidney Int. 2004;1465–1470. DOI: 10.1111/j.1523-1755.2004.00909.x. [DOI] [PubMed] [Google Scholar]
- 45.Oudot C, Lajoix AD, Jover B, Rugale C. Dietary sodium restriction prevents kidney damage in high fructose‐fed rats. Kidney Int. 2013;674–683. DOI: 10.1038/ki.2012.478. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S3
Figures S1–S3