

Abstract
Background
In sepsis, circulating cytokines and lipopolysaccharide elicit mitochondrial dysfunction and cardiomyopathy, a major cause of morbidity and mortality with this condition. Emerging research places the PHB1 (lipid raft protein prohibitin‐1) at the nexus of inflammation, metabolism, and oxidative stress. PHB1 has also been reported in circulation, though its function in this compartment is completely unknown.
Methods and Results
Using a wide‐ranging approach across multiple in vitro and in vivo models, we interrogated the functional role of intracellular and circulating PHB1 in the heart during sepsis, and elucidated some of the mechanisms involved. Upon endotoxin challenge or sepsis induction in rodent models, PHB1 translocates from mitochondria to nucleus in cardiomyocytes and is secreted into the circulation from the liver in a manner dependent on nuclear factor (erythroid‐derived 2)‐like 2, a key transcriptional regulator of the antioxidant response. Overexpression or treatment with recombinant human PHB1 enhances the antioxidant/anti‐inflammatory response and protects HL‐1 cardiomyocytes from mitochondrial dysfunction and toxicity from cytokine stress. Importantly, administration of recombinant human PHB1 blunted inflammation and restored cardiac contractility and ATP production in mice following lipopolysaccharide challenge. This cardioprotective, anti‐inflammatory effect of recombinant human PHB1 was determined to be independent of nuclear factor (erythroid‐derived 2)‐like 2, but partially dependent on PI3K/AKT signaling in the heart.
Conclusions
These findings reveal a previously unknown cardioprotective effect of PHB1 during sepsis, and illustrate a pro‐survival, protective role for PHB1 in the circulation. Exploitation of circulating PHB1 as a biomarker and/or therapeutic could have widespread benefit in the clinical management of sepsis and other severe inflammatory disorders.
Keywords: cardiomyopathy, circulating factor, inflammation, mitochondria, prohibitins, redox, shock
Subject Categories: Cardiopulmonary Resuscitation and Emergency Cardiac Care, Cardiomyopathy, Oxidant Stress, Pathophysiology, Inflammation
Nonstandard Abbreviations and Acronyms
- Nrf2
nuclear factor (erythroid‐derived 2)‐like 2
- PHB1/2
prohibitin 1/2
- ROS
reactive oxygen species
Clinical Perspective
What Is New?
We have discovered that the PHB‐1 (lipid raft scaffold protein prohibitin 1) is abundant in blood and behaves like an acute phase reactant protein during sepsis.
Hepatocytes are a major source of circulating PHB1 and its secretion, but not expression, is regulated by transcription factor nuclear factor erythroid 2‐related factor 2.
PHB1 overexpression or treatment with recombinant human PHB‐1 preserves mitochondrial oxidative phosphorylation and upregulates antioxidant capacity in cardiomyocytes undergoing inflammatory stress; dosing mice with recombinant human PHB‐1 blunts inflammation and induces pro‐survival signaling in heart, in part via PI3KAKT signaling.
What Are the Clinical Implications?
Circulating levels of PHB1 may be useful as a biomarker of cardiac and/or organ dysfunction in patients with sepsis.
Exploiting the pro‐survival, cardioprotective effect of PHB1 provides a blueprint for development of novel therapies to treat inflammatory disorders.
Overwhelming inflammatory stress imposed by circulating cytokines during the acute phase response in sepsis often precipitates rapid collapse of organ function, particularly within the cardiovascular system, where myocardial depression is associated with an ≈3‐fold increased risk of mortality.1, 2, 3 Efforts to identify the signals that govern the duration and magnitude of inflammatory stress during sepsis have uncovered novel pathways involving lipid mediators (eg, resolvins, protectins), small peptides, oligonucleotides, neurohumoral factors, and others.4, 5 Previous studies have also shown that pro‐inflammatory cytokines lead to myocardial depression in sepsis, at least in part, via their deleterious impact on mitochondrial energetics.6, 7, 8, 9, 10, 11 Unfortunately, although they have provided mechanistic insight, these studies have not translated into new therapies to combat organ dysfunction in sepsis, and mortality for patients in septic shock remains as high today (≈30%) as it was several decades ago.12, 13
PHB1 and PHB2 (prohibitins) are proteins that assemble in hetero‐oligomeric complexes within the mitochondrial inner membrane and in plasma membrane lipid rafts where they play critical roles in mitochondrial function and metabolism,14, 15, 16, 17, 18 cellular proliferation and tumorigenesis,19, 20, 21 inflammation/oxidative stress,22 and apoptosis.23, 24, 25 Little information exists regarding the function of PHBs in the heart, although overexpression of PHB1 was shown to protect against oxidative insults in cardiomyocytes.26, 27 PHB1 has antioxidant and protective effects in colonic epithelium,28 effects that are accompanied by sustained activation of the nuclear factor erythroid 2‐related factor 2 (Nrf2), a key transcriptional regulator of the antioxidant response system.29 There is evidence that PHBs are in the circulation, with 1 report showing that PHB1 is capable of binding to and activating complement,30 while others have documented the presence of PHB1/2 in serum of patients with cancer.31, 32, 33 Despite these reports, the function of PHBs in the circulation is completely unknown.
Since PHB1 is known to have pleiotropic protective effects, and because prior reports have outlined a role for this protein in regulating inflammation, we sought to determine the extent to which PHB1 regulates inflammation and metabolism in the heart during sepsis, and the mechanisms by which it may be acting. Our findings uncover a previously unrecognized role for PHB1 as a dynamically regulated circulatory protein that has pro‐survival, cardioprotective effects when administered as a recombinant biologic therapy in sepsis.
Methods
A detailed and expanded description of all methods and materials used in this study, including Tables of primers for quantitative real‐time polymerase chain reaction and antibodies used for protein detection and immunocytochemistry, is included in Data S1. All data related to the findings of this study are available from the corresponding author upon request.
Experimental Animals
All experiments in rodent models were conducted with approval from the Institutional Animal Care and Use Committee at East Carolina University and the University of Iowa, in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male Sprague‐Dawley rats (Charles River Laboratory, Wilmington, MA) weighing between 275 and 300 g (14–16 weeks old), and C57/Bl6J wild‐type (WT) mice (The Jackson Laboratory, Sacramento, CA) weighing between 20–25 grams (8–10 weeks old) were used. The Nrf2‐/‐ mice on a C57/Bl6J background were a kind gift from Thomas Kensler. Animals were housed in temperature‐ and light‐controlled conditions with free access to food and water. Unless otherwise specified, animals were deeply anesthetized with pharmaceutical grade ketamine (80–100 mg/kg) and xylazine (10–12.5 mg/kg), before euthanasia via exsanguination and cardiac dissection.
Primary Hepatocyte Isolation
Primary hepatocytes were isolated from WT and Nrf2‐/‐ mice according to procedures outlined previously34, 35 and described in detail in Data S1.
Induction of Endotoxemia and Dosing of Recombinant Human PHB1
Male rats (N=6 per group) and mice (N=8 per group) were given an intraperitoneal injection of lipopolysaccharide purified from Escherichia coli (Sigma‐Aldrich, St. Louis, MO) mixed in 5% dextrose‐H2O at various concentrations [rats: 0.5 and 7.5 mg/kg and mice: 12 mg/kg]. The control group received an injection of 5% dextrose‐H2O (Vehicle). Following the lipopolysaccharide injection, all animals were monitored every 4 hours until euthanasia. Approximately 25% of the animals in the lipopolysaccharide groups succumbed to shock and/or were euthanized for humane reasons before completion of the experiments. Purified recombinant human PHB1 (Origene, Rockville, MD) was diluted in saline immediately before injection. Mice received an intraperitoneal injection of recombinant PHB1 (rPHB1) (300 ng, corresponding to 9–10 μg/kg body weight) to achieve a final circulating concentration of 200 ng/mL at +2, +8, and +14 hours following lipopolysaccharide challenge. This concentration of rPHB1 was estimated by assuming a mouse blood volume/mass ratio of 58.5 mL/kg.36 For studies with the phosphoinositide‐3 kinase (PI3K) inhibitor buparlisib, mice were injected with buparlisib (30 mg/kg body weight) dissolved in 100 µL of the vehicle (0.1 M sulfobutylether‐β‐cyclodextrin, SBE‐β‐CD, MedChemExpress, in ddH2O) 1 hour before lipopolysaccharide challenge. Vehicle‐treated mice were treated with the SBE‐β‐CD alone before lipopolysaccharide. Dosing of rPHB1 was then administered in these mice as described above.
Induction of Sepsis Via Cecal Ligation and Puncture
Male C57/Bl6J mice (N=8 per group, WT) were anesthetized under 2%–3% vaporized isoflurane in oxygen at a flow rate of 100 mL/min. The abdomen was shaved and prepped with Hibiclens followed by 70% isopropanol to sterilize the surgical field. Bupivacaine was applied topically to skin before incision. A surgical drape was made with Press & Seal Wrap and a fenestration cut to allow access to the incision site. A 5–8‐mm horizontal incision was made in the abdomen, and the cecum was identified and pulled out slightly from the cut muscle/skin. Ligation of the ileocecal junction was made with 4–0 silk suture. The cecum was punctured proximal to the ligation with a 27G needle and a small amount of cecal contents was allowed to spill out. Cecum was then returned to the abdomen and the incision was closed using silk suture and Vetbond. Sham animals underwent a similar procedure: laparotomy as described above with no cecal ligation or puncture. A bolus of 1 mL saline was then injected subcutaneously for fluid support, and animals were allowed to recover from surgery under a heat lamp and monitored for up to 48 hours, with cohorts being euthanized at time points indicated in Figure 1 under ketamine/xylazine as described above. Approximately 30% of the animals in the cecal ligation and puncture groups succumbed to shock and/or were euthanized for humane reasons before completion of the experiments.
Figure 1. PHB1 mobilizes within the heart and circulation during sepsis.
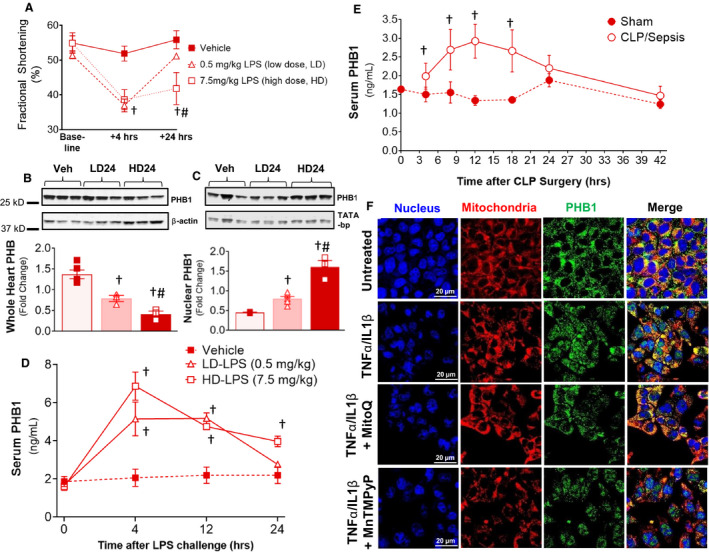
Endotoxemia was induced in Sprague‐Dawley rats with both low‐dose (0.5 mg/kg, LD) and high‐dose (HD) LPS (7.5 mg/kg, HD), and cardiac function parameters (fractional shortening) was assessed in the rats at +4 and +24 hours (A). Total levels of PHB1 protein were assessed in whole heart (B) and nuclear fraction of the heart (C), and in the serum (D) of the rats at time points indicated following LPS challenge. Serum PHB1 levels in mice at time points following CLP‐induced sepsis is indicated in (E). Localization of endogenous PHB1 in HL‐1 cardiomyocytes following a 4‐hour exposure to sepsis‐mimetic concentration of TNFα/IL1β, with or without the mitochondrial‐targeted antioxidant MitoQ, or the nontargeted free radical scavenger MnTMPyP, is shown in (F). N=4 rats/group for LPS experiments, N=7–10 mice per group for CLP. A 2‐tail Student t test was used to compare main effect of LPS or CLP vs Vehicle or Sham control, respectively, at each time point. † P<0.01 vs Vehicle or Sham, # P<0.001 vs low‐dose LPS for that respective time point/condition. LD4, low‐dose LPS at 4 hours postinjection, LD24, low‐dose LPS at 24 hours postinjection; HD24, high‐dose LPS at 24 hours postinjection. CLP indicates cecal ligation and puncture; .IL1β, interleukin 1β; LPS, lipopolysaccharide; PHB, prohibitin; TNFα, tumor necrosis factor α; and Veh, vehicle.
Echocardiography
Echocardiography in rats, WT, and Nrf2‐/‐ mice outlined in experiments shown in Figures 1, 3 and 4 were performed under controlled anesthesia (2%–3% vaporized isoflurane in oxygen at a flow rate of 100 mL/min) using a 30 MHz transducer (Vevo 2100, VisualSonics, Toronto, ON). Cardiac functional parameters were recorded at baseline, +4, and +24 hours following lipopolysaccharide in the rat model and at baseline, +4, +8, and +14 hours following lipopolysaccharide in the mouse model. Echocardiography in the mice used in buparlisib experiments was performed on conscious mice 16 hours following lipopolysaccharide challenge, by staff in the cardiovascular phenotyping core facility at University of Iowa.
Figure 3. Endotoxin‐induced secretion of PHB1 in liver is dependent on Nrf2.

Shown in (A) is a representative immunoblot of PHB1 in whole cell lysates, and in the conditioned media (B) obtained from primary murine hepatocytes exposed to LPS (2.5 ng/mL) for the time points indicated. Serum PHB1 from WT and Nrf2‐/‐ mice at +4 hours and +16 hours following a 12 mg/kg dose of LPS is shown in (C). Blots are representative of 2 independent experiments in hepatocytes. For mouse model serum PHB1, N=5. A 2‐tail Student t test was used to compare main effect of LPS vs vehicle control for each genotype at each time point. † P<0.05 vs control (vehicle‐treated) mice. LPS indicates lipopolysaccharide; Nrf2, nuclear factor (erythroid‐derived 2)‐like 2; rPHB1, recombinant human prohibitin 1; and WT, wild type.
Figure 4. Administration of rPHB1 restores cardiac systolic function and mitochondrial OxPHOS during endotoxemia independent of Nrf2.
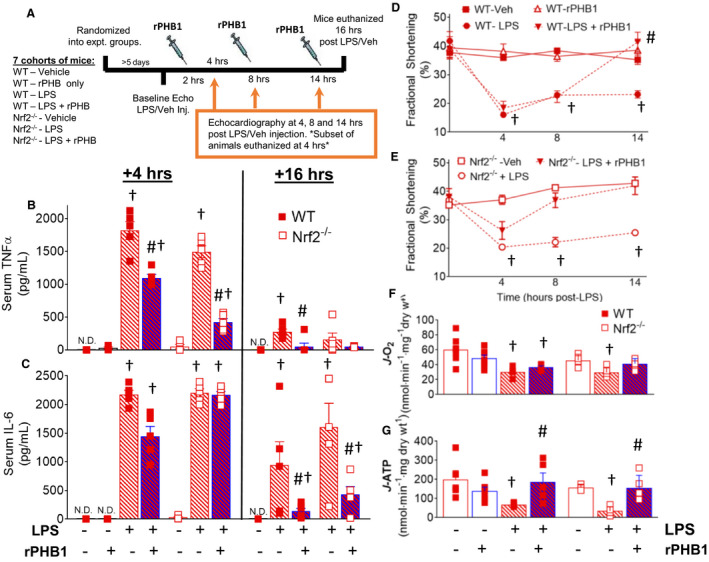
The broad experimental design and mouse genotypes/treatment groups is outlined in (A). Serum concentrations of TNFα (B) and IL‐6 (C) were measured in WT and Nrf2‐/‐ mice +4 hours and +16 hours after LPS challenge in mice administered either saline vehicle (red‐shaded bars), or rPHB1 (blue‐shaded bars) at +4, 10 and +16 hours after LPS challenge. Cardiac systolic function in WT (D) and Nrf2‐/‐ mice (E) were determined in response to LPS challenge and administration of vehicle or rPHB1 at time points indicated. Rates of pyruvate/malate (CI) ‐supported mitochondrial respiration (J‐O2) (F) and ATP production (J‐ATP) (G) in permeabilized ventricular myofibers prepared from the mice at +16 hours are also shown. N=4–6 mice in each group. A 1‐way ANOVA followed by Tukey’s post hoc multiple comparisons test between groups was used to test for differences in main effect of LPS and rPHB1 treatment within each genotype. † P<0.05 vs untreated mice (‐ LPS, ‐rPHB1), and # P<0.05 vs +LPS for that respective time point and genotype. LPS indicates lipopolysaccharide; N.D., not detectable; Nrf2, nuclear factor (erythroid‐derived 2)‐like 2; OxPHOS, oxidative phosphorylation; rPHB1, recombinant human prohibitin 1; TNFα, tumor necrosis factor α; and WT, wild type.
Mitochondrial Function in Permeabilized Left Ventricular Myofibers and HL‐1 Cardiomyocytes
Mitochondrial O2 consumption, ATP production, Ca2+ uptake, and H2O2 emission were measured in permeabilized left ventricular myofibers and HL‐1 cardiomyocytes according to previously published protocols from our group,37, 38, 39, 40, 41 and described in detail in Data S1.
Statistical Analysis
Statistical analysis was performed with PRISM (GraphPad, San Diego, CA). All data are expressed as means ± standard error of the mean (SEM) unless otherwise noted. For cell culture models, a 2‐way ANOVA (transfection × treatment) followed by Tukey’s post hoc multiple comparisons test was used to test for main effect of cytokine treatment between groups. For rodent models, student’s t test (two‐tail) or 1‐way ANOVA followed by Tukey’s test was used to test for differences in main effect of lipopolysaccharide and rPHB1 treatment between groups within each genotype, as described in each of the Figure legends. In all cases, P<0.05 indicated statistically significant differences between groups.
Results
Dynamic Changes in PHB1 Levels in the Heart and Blood During Sepsis
Cardiac electromechanical dysfunction is a well‐described complication of severe sepsis, including both systolic and diastolic dysfunction.1, 2 A classic experimental model of severe sepsis involves injection of lipopolysaccharide at sufficiently high dose to elicit myocardial depression and the “cytokine storm.” We first examined cardiac PHB1 in the context of severe sepsis using lipopolysaccharide at 2 doses—a low, sublethal (0.5 mg/kg, LD) and high, lethal dose (7.5 mg/kg, HD), in Sprague‐Dawley rats. As expected, both doses of lipopolysaccharide caused a sharp decrease in systolic function within 4 hours, which was sustained in the HD group at 24 hours following lipopolysaccharide injection (Figure 1A). Derangements in mitochondrial function were also present at both doses, with decreased respiration and elevated reactive oxygen species (ROS) production (Figure S1). Cardiac PHB1 in whole tissue lysates decreased in a dose‐dependent manner with lipopolysaccharide (Figures 1B), but the remaining PHB1 accumulated in the nucleus (Figures 1C). Interestingly, circulating PHB1 levels were highly dynamic in the acute phase following lipopolysaccharide challenge at both doses, showing peak elevations in serum at 4 to 12 hours post‐lipopolysaccharide (Figure 1D). To determine whether this elevation in serum PHB1 was exclusive to lipopolysaccharide challenge and/or the rat model, we used cecal ligation‐puncture to induce severe sepsis in WT mice and observed a time‐dependent rise and fall of serum PHB1 that followed a very similar time course to the endotoxemia model in the rats (Figure 1E).
In order to more closely delineate the factors regulating the mobilizability of PHB1 in the heart during sepsis, we developed an in vitro model of acute inflammatory stress using a sepsis‐mimetic cocktail of the pro‐inflammatory cytokines tumor necrosis factor/interleukin‐1β (TNFα/IL‐1β) in HL‐1 cardiomyocytes (Figure S2). Just as was observed in the rat heart following lipopolysaccharide challenge, a similar mitochondria‐to‐nucleus translocation of PHB1 occurred in HL‐1 cardiomyocytes in response to a 4‐hour treatment with TNFα/IL‐1β. This nuclear translocation of PHB1 was significantly blunted by the mitochondrial lipid peroxide‐scavenger Mito‐quinone, although a cell‐permeable superoxide‐radical scavenger (MnTMPyP) had no effect (Figure 1F) and was much less effective at blunting the nuclear translocation.
PHB1 Protects HL‐1 Cardiomyocytes From Inflammatory Stress Via Preservation of Mitochondrial Function and Upregulation of Antioxidant Capacity
Previous studies have reported that PHB1 protects cells against various stressors, including oxidative stress and inflammation.22, 28, 42 To examine whether PHB1 is acting in similar manner during acute inflammation, HL‐1 cardiomyocytes were transfected with plasmid encoding PHB1 with a GFP (green fluorescent protein) tag under control of CMV promoter (overexpression of PHB1), or GFP vector alone (Vec). As expected, localization of PHB1‐GFP in normal, unstressed HL‐1 cardiomyocytes was primarily mitochondrial (Figure 2A). Overexpression of PHB1‐GFP in these cells did not alter mitochondrial function, but did attenuate the deleterious effects of TNFα/IL‐1β on mitochondrial respiration, ATP production, ROS production, and Ca2+‐sensitization of the permeability transition pore (Figure 2B through 2F), supporting a role for PHB1 in preserving mitochondrial function and integrity under inflammatory stress.
Figure 2. PHB1 preserves mitochondrial energetics and viability during inflammatory and oxidative stress in HL‐1 cardiomyocytes.

Cells were transiently transfected with plasmid DNA containing PHB1‐GFP for 24 hours, then labeled with MitoTracker Red to confirm mitochondrial localization (A). The mitochondrial‐protective effect of overexpressing PHB1 (oPHB1) in HL‐1 cardiomyocytes exposed to TNFα/IL1β (B) is illustrated with respiratory flux (J‐O2) supported by pyruvate/malate (complex I substrate, CI), or pyr/mal + succinate (CI and CII substrates), ATP production (JATP) with pyruvate/malate (Pyr/Mal) (C), Ca2+ sensitivity of permeability transition pore (D), and H2O2 production/emission (E). Representative images of mitochondrial ROS staining with MitoSox in HL‐1 cardiomyocytes exposed to a sepsis‐mimetic concentration of TNFα/IL1β in GFP‐vector control, oPHB1 and with recombinant human PHB1 (rPHB1) is shown in (F) and quantified in (G). Cytotoxicity was assessed by LDH release in HL1 cardiomyocytes following exposure to TNFα/IL1β (H) and H2O2 (I). For oPHB1 experiments, N=3–4 with technical replicates for each condition. A 2‐way ANOVA (transfection × treatment) followed by Tukey’s post hoc multiple comparisons test was used to compare main effect of cytokine treatment on end points indicated. *P<0.0001 vs untreated, GFP‐vector Control. For gene expression and cytotoxicity analysis, N=4 per group with technical replicates for each condition. *P<0.0001 vs untreated Control, † P<0.0001 vs TNFα/IL1β, or H2O2‐treated groups. AU indicates arbitrary units ; GFP, green fluorescent protein; IL1β, interleukin 1 β; LDH, lactate dehydrogenase; P/M, Pyruvate/Malate; ROS, reactive oxygen species; and TNFα, tumor necrosis factor α.
Overexpression of PHB1 was shown in a prior report to protect cardiomyocytes against oxidative stress in prior studies, although the mechanisms of protection were not clearly determined.26, 27 Interestingly, recombinant human PHB1 (rPHB1) protected pancreatic cells against oxidative stress induced by ethanol,22 suggesting that PHB1 also acts via exogenous mechanisms. To gain mechanistic insight into these protective effects of PHB1, HL‐1 cardiomyocytes were transfected with a lentiviral‐based reporter system containing tandem repeats of the antioxidant response element promoter controlling expression of GFP. Cells treated with rPHB1 exhibited robust activation of antioxidant response element promoter, even in the presence of TNFα/IL‐1β (Figure S3A and S3B). Moreover, both overexpression of PHB1 and rPHB1‐treated cells had increased nuclear localization of the antioxidant response element transcriptional activator Nrf2 (not shown) and upregulation of antioxidant/phase II detoxifying genes (Figure S3C). In parallel with this upregulation of antioxidant enzymes, overexpression of PHB1 and rPHB1 abrogated inflammatory gene expression in HL‐1 cardiomyocytes following stimulation with TNFα/IL‐1β (Figure S4). These antioxidant/anti‐inflammatory effects of PHB1 were indeed cytoprotective, as overexpression of PHB1 and rPHB1 mitigated mitochondrial ROS (Figure 2F and 2G), and cytotoxicity induced by overnight exposure to TNFα/IL‐1β (Figure 2H) and H2O2 (Figure 2I).
Endotoxin‐Induced Secretion of PHB1 Is Mediated by Nrf2 in Hepatocytes
A principal source of acute phase proteins during sepsis is the liver.43, 44 Additionally, Nrf2 is known to have a key role in regulating the innate immune response to sepsis, and Nrf2‐/‐ mice are more susceptible to sepsis‐induced injury and mortality as compared with WT mice.45 Given that rPHB1 has a potent cytoprotective effect, we sought to determine whether hepatocytes are a potential source of blood‐borne PHB1 and whether expression and/or secretion of PHB1 is dependent on Nrf2. Both intracellular and extracellular (in conditioned media) PHB1 increased dramatically in primary hepatocytes from WT mice in time‐dependent manner following exposure to low, sublethal dose (2.5 ng/mL) of lipopolysaccharide (Figure 3A and 3B). Surprisingly, in Nrf2‐/‐ hepatocytes, upregulation of PHB1 expression occurred to similar or slightly greater extent than in WT, but secretion of PHB1 was almost completely abrogated. Moreover, this effect was reproducible in mice following lipopolysaccharide challenge, whereby serum PHB1 levels increased considerably at 4 hours following lipopolysaccharide in WT mice, but not in Nrf2‐/‐ mice (Figure 3C), although a very modest increase above baseline was observed in the Nrf2‐/‐ mice 16 hours after lipopolysaccharide.
Intraperitoneal Dosing of rPHB1 Suppresses Inflammation and Preserves Mitochondrial Energetics and Cardiac Function During Endotoxemia Independent of Nrf2
Given the anti‐inflammatory and cytoprotective effects of rPHB1 seen in the cardiomyocytes, we next examined whether these protective effects of rPHB1 also occur in vivo during endotoxemia, and whether these effects are dependent on Nrf2. Both WT and Nrf2‐/‐ mice were given an intraperitoneal injection of a high dose of lipopolysaccharide (12 mg/kg b.w.), then 3 doses of rPHB1 (300 ng, corresponding to 9–10 μg/kg b.w.) were administered to the mice via intraperitoneal injection at +2, +8, and +14 hours following lipopolysaccharide. Echocardiography was performed at 3 time points following lipopolysaccharide, immediately after the 3 intraperitoneal doses of rPHB1 each animal received, and a subset of mice were euthanized 2 hours after the first rPHB1 injection for analysis (Figure 4A).
Dosing of rPHB1 following lipopolysaccharide injection triggered a rapid and systemic anti‐inflammatory effect. Serum TNFα levels were diminished in both WT and Nrf2‐/‐ mice by just a single dose of rPHB1 at 4 hours, and were completely back to normal after 3 doses of rPHB1 at 16 hours (Figure 4B). A similar effect of rPHB1 on circulating IL‐6 was observed, although a single dose of rPHB1 had no effect on circulating IL‐6 in the Nrf2‐/‐ mice (Figure 4C). Importantly, dosing of rPHB1 led to improvements in cardiac function. Every animal that received rPHB1 after lipopolysaccharide challenge had systolic function and other major hemodynamic parameters return to normal by 16 hours following lipopolysaccharide (Figure 4D and 4E and Table). This cardioprotective effect occurred to similar extent in both WT and Nrf2‐/‐ mice, and may even have been more rapid in the Nrf2‐/‐ mice than in WT, suggesting the cardioprotective effect of rPHB1 is independent of Nrf2.
Table 1.
Cardiac Function Parameters for WT and Nrf2‐/‐ Mice With Endotoxemia
| WT | Nrf2‐/‐ | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Veh | rPHB1 only | LPS only | rPHB1 +LPS | Veh | LPS only | rPHB1 +LPS | |||||||||||
| Age, wks | 11.60±0.5 | 12.25±1.0 | 11.60±0.5 | 12.33±0.8 | 9.50± 0.7 | 9.25±0.5 | 9.50±0.6 | ||||||||||
| Weight, g | 26.16±2.3 | 27.60±2.0 | 26.96±1.0 | 26.65±1.3 | 24.35±1.5 | 23.96±1.0 | 24.20±1.6 | ||||||||||
| 4 h | 8 h | 14 h | 4 h | 8 h | 14 h | 4 h | 8 h | 14 h | 4 h | 8 h | 14 h | 4 h | 8 h | 14 h | |||
| LVeSD, mm | 2.53±0.3 | 2.46±0.4 | 2.53±0.3 | 2.39±0.4 | 3.49±0.4* | 3.01±0.2* | 3.19±0.3* | 3.29±0.3* | 3.15±0.4* | 2.12±0.5 | 2.65±0.3 | 3.24±0.2* | 3.03±0.3* | 2.79±0.3 | 2.82±0.1 | 2.33±0.2 | 2.11±0.5 |
| LVeDD, mm | 3.99±0.3 | 3.95±0.4 | 3.97±0.3 | 3.86±0.3 | 4.16±0.4 | 3.90±0.2 | 4.14±0.3 | 4.03±0.2 | 4.05±0.3 | 3.57±0.4 | 4.34±0.3 | 4.07±0.2 | 3.88±0.3 | 3.75±0.3 | 3.84±0.3 | 3.69±0.2 | 3.77±0.3 |
| Heart rate, bpm | 463.98±42.6 | 531.14±24.1 | 526.04±28.9 | 505.99±35.1 | 552.34±40.8* | 459.69±22.4 | 412.35±52.3 | 565.26±36.0* | 497.13±57.9 | 482.52±46.4 | 435.33±40.1 | 527.55±16.1* | 365.11±33.8 | 370.89±44.3 | 523.14±17.0* | 433.76±46.7 | 431.49±31.3 |
| Stroke volume, μL | 46.82±7.3 | 46.38±6.8 | 45.64±6.2 | 44.10±5.3 | 26.00±4.3* | 30.78±4.5* | 35.45±4.9* | 27.37±7.6* | 32.27±6.3* | 38.33±6.6 | 59.17±7.0 | 30.78±3.4* | 29.06±3.3* | 30.10±6.7* | 33.68±11.5* | 38.87±5.9* | 45.56±5.6 |
A 1‐way ANOVA followed by Tukey’s post hoc multiple comparisons test between groups was used to test for differences in main effect of treatment between groups. N, 4–7 mice per group. bpm indicates beats per minute; LPS, lipopolysaccharide; LVeDD, left ventricular end diastolic diameter; LVeSD, left ventricular end systolic diameter; rPHB1, recombinant human prohibitin 1; Veh, vehicle; and WT, wild type.
P<0.05 vs untreated mice (Vehicle), for that respective time point and genotype.
We also assessed the effect of rPHB1 on mitochondrial respiration and ATP production during endotoxemia using permeabilized left ventricular myofibers prepared from WT and Nrf2‐/‐ mice after the third dose of rPHB1 (16 hours post‐lipopolysaccharide). Maximal ADP‐stimulated respiration in both WT and Nrf2‐/‐ mice was diminished by lipopolysaccharide and was unaffected by rPHB1 (Figure 4F and Figure S5). However, ATP production rates were significantly improved with rPHB1 in both WT and Nrf2‐/‐ mice (Figure 4G), suggesting that rPHB1 may have a specific effect on the mitochondrial phosphorylation system.
The anti‐inflammatory effect of rPHB1 was also observed in the myocardium of both WT and Nrf2‐/‐ mice, as nuclear activation of the pro‐inflammatory transcription factors nuclear factor κ light‐chain‐enhancer of activated B cells and phospho‐signal transducer and activator of transcription 3 (Tyr705), and pro‐inflammatory gene markers were blunted in the heart after just a single dose of rPHB1 (Figure 5). To ascertain whether the anti‐inflammatory effects of rPHB1 were limited to the heart, we measured expression of pro‐inflammatory cytokines in liver, kidney, and lungs in the WT mice after 3 successive doses of rPHB1. Treatment with rPHB1 suppressed lipopolysaccharide‐induced TNFα expression only in kidney tissue (Figure S6A), while rPHB1 completely blunted lipopolysaccharide‐induced IL‐6 expression in liver, kidney, and lung tissue (Figure S6B). Lipopolysaccharide‐induced IL‐1β expression was not affected by rPHB1 in any tissues (Figure S6C). We also assessed markers of tissue damage and necrosis in a subset of the mice. As expected, lipopolysaccharide challenge increased circulating lactate dehydrogenase (LDH) and NADH levels, but rPHB1 completely attenuated these markers (Figure S7).
Figure 5. rPHB1 attenuates cardiac inflammation during endotoxemia independent of Nrf2.

Representative immunoblots of p65 (NFkB) and phospho‐STAT3 from nuclear extracts prepared from WT and Nrf2‐/‐ mouse hearts dissected 4 hours after LPS challenge and 2 hours after 1 dose of rPHB1 (300 ng) are shown in (A), and quantified via densitometry using the nuclear marker TATA‐binding protein as loading control (B, C). Expression of pro‐inflammatory cytokines IL6, TNFα, IL1β and iNOS (D through G) were also measured in whole heart tissue from this same cohort of mice. N=4–6 mice in each group. A 1‐way ANOVA followed by Tukey’s post hoc multiple comparisons test between groups was used to test for differences in main effect of LPS and rPHB1 treatment within each genotype. † P<0.05 vs untreated mice (Baseline), for that respective time point and genotype; # P<0.05 vs LPS alone, for that respective time point and genotype. IL1β indicates interleukin β; IL6, interleukin 6; i NOS, inducible nitric oxide synthase; LPS, lipopolysaccharide; Nrf2, nuclear factor (erythroid‐derived 2)‐like 2; rPHB1, recombinant human prohibitin 1; TNFα, tumor necrosis factor α; and WT, wild type.
Mitochondria produce ROS continuously in cardiomyocytes, necessitating a robust antioxidant network to maintain redox balance. During sepsis, mitochondrial ROS production increases because of many factors, including disruptions in oxidative phosphorylation, mitochondrial fission/mitophagy, and loss of antioxidant capacity.9, 46, 47, 48 In the present study, rPHB1 dosing, on its own, significantly diminished cardiac mitochondrial ROS production in WT, but not Nrf2‐/‐ mice (Figure S8A). To determine the contribution of the mitochondrial redox enzyme thioredoxin reductase‐2, we used the thioredoxin reductase inhibitor auranofin, a gold thiolate salt that we and others have used to elucidate the role of thioredoxin reductase‐2 in the mitochondrial redox adaptations to exercise and cardiometabolic diseases.38, 49, 50 Auranofin significantly increased mitochondrial ROS in all experimental groups. Interestingly, the auranofin‐stimulated ROS was diminished with lipopolysaccharide challenge in both WT and Nrf2‐/‐ mice, and substantially more so in the Nrf2‐/‐ mice (Figure S8A). Conversely, rPHB1 dosing increased the auranofin‐stimulated effect. This corresponded with increased rPHB1‐induced expression of thioredoxin reductase‐2 enzyme in WT, but not Nrf2‐/‐ mice (Figure S8B). Treatment with rPHB1 also upregulated glutathione reductase enzyme in WT but not Nrf2‐/‐ mice (Figure S8C).
The deleterious role of nitric oxide synthase in septic cardiomyopathy has been well‐characterized.51, 52, 53 To determine whether the rPHB1‐mediated rescue of myocardial dysfunction in our model was associated with altered cardiac inducible nitric oxide synthase, we examined this enzyme in hearts of the mice used in this study. With rPHB1 dosing there was substantial downregulation of inducible nitric oxide synthase expression at both the mRNA and protein level in WT mice (Figure S9). No significant effect of rPHB1 dosing on inducible nitric oxide synthase was observed in Nrf2‐/‐ mice, although these mice had diminished expression of inducible nitric oxide synthase at baseline compared with WT.
Cardioprotective Effects of rPHB1 Are Mediated in Part by PI3K/AKT Pathway Activation
Previous reports have linked PHB1 with coordination of intracellular signaling at plasma membrane lipid rafts via recruitment and activation (ie, phosphorylation) of serine/threonine kinase AKT, the master regulator of cell survival and metabolism.20, 54 We next examined whether the cardioprotective effect of rPHB1 acts via similar mechanism and found that rPHB1 caused a significant increase in AKT phosphorylation in human AC16 cardiomyocytes after 1 hour (Figure 6A). Importantly, this rPHB1‐induced phosphorylation of AKT was not significantly affected by C‐Raf inhibitor AZ628, but was completely abrogated by the PI3K inhibitor buparlisib, suggesting that rPHB1 activates AKT via PI3K‐dependent mechanism (Figure 6A). To determine whether the protective effect of rPHB1 in the heart during endotoxemia was also dependent on PI3K/AKT pathway, WT mice were subjected to lipopolysaccharide challenge (12 mg/kg) and rPHB1 dosing in absence and presence of buparlisib. PI3K inhibition in this model blunted the cardioprotective effect afforded by rPHB1 (Figure 6B). Immunoblot analysis showed that rPHB1 dosing caused an ≈3‐fold upregulation of phospho‐AKT levels in the hearts of these mice, which was attenuated with buparlisib (Figure 6C and 6D), consistent with what was observed in the AC16 cardiomyocytes. Interestingly, despite the severe cardiac dysfunction caused by buparlisib when combined with lipopolysaccharide + rPHB1, expression of pro‐inflammatory cytokines TNFα and IL‐6, but not IL‐1β, were substantially lower with buparlisib (Figure 6E through 6G), suggesting that the cardioprotection afforded by rPHB1 is not exclusively because of its anti‐inflammatory effect.
Figure 6. Protective effect of rPHB1 during endotoxemia is mediated by PI3K/AKT pathway in heart.

Effect of 1‐hour treatment of rPHB1 (100 ng) on Akt phosphorylation in AC16 cardiomyocytes in absence and presence of the C‐Raf inhibitor, AZ628, or PI3K inhibitor, buparlisib, is shown in (A). The effect of 3 doses of rPHB1 (300 ng) on cardiac systolic function in WT mice measured 18 hours after LPS challenge (12 mg/kg) in absence and presence of buparlisib is shown in (B), with levels of Akt phosphorylation in hearts dissected from this same cohort of mice (C, D). Expression of pro‐inflammatory cytokines TNFα (E), IL1β (F) and IL‐6 (G) were also measured in whole heart tissue. Cell culture immunoblots shown are representative of 2 independent experiments. For mouse models, N=5–7 mice in each group. A 1‐way ANOVA followed by Tukey’s post hoc multiple comparisons test between groups was used to test for differences in main effect of treatment between groups. † P<0.05 vs Vehicle‐treated mice, # P<0.05 vs LPS + rPHB1. Bupar indicates buparlisib; IL1β, interleukin β; IL6, interleukin 6; LPS, lipopolysaccharide; rPHB1, recombinant human prohibitin 1; TNFα, tumor necrosis factor α; and WT, wild type.
Discussion
PHBs have pleiotropic roles in mammalian cells, and emerging evidence has implicated a role for these proteins in a wide variety of diseases including cancer, obesity/diabetes mellitus, and neurodegenerative and chronic inflammatory diseases.55 Our findings have revealed a role for PHB1 in mitigating systemic inflammation and cardiac dysfunction during sepsis. These findings are important and distinct because we demonstrate that (1) PHB1 is a highly dynamic protein that mobilizes within the cell and bloodstream during the acute phase response; (2) overexpression or administration of human rPHB1 preserves mitochondrial integrity and energetics in cardiomyocytes during inflammatory stress; (3) endogenous PHB1 behaves like an acute phase reactant in the bloodstream during endotoxemia and severe sepsis, and its secretion from hepatocytes is mediated by Nrf2; (4) dosing of human rPHB1 mitigates inflammation in vital organs during endotoxemia, and completely restores cardiac function, preserves cardiac mitochondrial energetics, and augments antioxidant capacity during endotoxemia independent of Nrf2; and (5) the cardioprotective effect of rPHB1 during endotoxemia is mediated, at least in part, by PI3K/AKT signaling. A schematic summarizing the overall findings of this study is shown in Figure 7. These findings are translational and potentially of clinical importance as they suggest that PHB1 and its downstream signaling effectors are not only provocative drug targets, but PHB1 itself may, in certain contexts, be therapeutic.
Figure 7. Model of cardioprotective, anti‐inflammatory effect of circulating PHB1 during sepsis.

A summary schematic of the overall findings uncovered in the present study are shown. Upon exposure to endotoxin, hepatocytes secrete pro‐inflammatory acute phase reactant cyto/chemokines such as TNFα and IL1β. Our findings suggest that in parallel with this canonical pro‐inflammatory response to endotoxin, Nrf2 mediates an anti‐inflammatory response in hepatocytes via secretion of PHB1, either directly or indirectly via transcriptional activation of downstream target genes. As a circulating paracrine factor, PHB1 has systemic anti‐inflammatory effects and acts directly on cardiomyocytes to upregulate pro‐survival signaling and preserve mitochondrial OxPHOS independently of Nrf2, mediated in part via PI3K/AKT activation. IL1β indicates interleukin β; Nrf2, nuclear factor (erythroid‐derived 2)‐like 2; OxPHOS, oxidative phosphorylation; PHB1, prohibitin 1; and TNFα, tumor necrosis factor α.
Acute phase reactants have been defined as proteins whose concentration in the blood change from baseline (up or down) by >25% during an inflammatory response.43, 56 While it may be premature to call PHB1 a bona fide acute phase reactant at this point, it is clear that PHB1 fits these criteria as shown in Figure 1. The principal source of acute phase reactants during sepsis is the liver, and, while we have not definitively shown that liver is the only source of circulating PHB1, it is clear from Figure 3 that hepatocytes dramatically ramp up PHB1 expression and secretion in response to lipopolysaccharide, and PHB1 secretion, but not expression, is regulated by Nrf2. Acute phase proteins have diverse actions in the host, ranging from coordination of both pro‐inflammatory and anti‐inflammatory responses, to recruitment of innate immune cells and regulation of vascular permeability and hemostasis. Some of these proteins possess both pro‐ and anti‐inflammatory effects, depending on dose and time. An example of this is CRP (C‐reactive protein), which is essential for coordinating clearance of foreign pathogens and activation of the complement system.57, 58 However, the overproduction of CRP in transgenic mice can also be anti‐inflammatory and protective in models of septic shock.59, 60 This is thought to be due in large part to the inhibiting effect of CRP and its degradation products on neutrophil activation and extravasation.61 Whether circulating PHB1 is having a similar effect on complement and neutrophils remains to be determined, since recent studies on PHBs in the innate immune system have focused solely on the intracellular signaling roles of these proteins, ignoring their role in the circulation.62, 63
A link between PHBs and protection from oxidative/inflammatory stress has been well‐documented in numerous cell types, including cardiomyocytes,22, 26, 27, 28, 29 although a clear mechanism for how PHBs are functionally involved in this antioxidant/anti‐inflammatory effect has not been fully elucidated. A detailed mechanism for how intracellular PHB1 inhibits TNFα‐induced nuclear translocation of nuclear factor κ light‐chain‐enhancer of activated B cells was outlined in colonic epithelial cells by Theiss and colleagues, who demonstrated that PHB1 downregulates expression of importin α3, a protein required for nuclear factor κ light‐chain‐enhancer of activated B cells nuclear import.42 Further work by this group outlined a role for PHB1 in protection against inflammatory bowel disease by upregulation of Nrf2 in colonic epithelium, corresponding to increased antioxidant gene expression.29 However, in subsequent work, Nrf2 was found to be nonessential for the protective effect of PHB1, as there was a similar protective effect of PHB1 overexpression on colitis in Nrf2‐/‐ mice as in WT.64 Our findings show that Nrf2 is essential for secretion of PHB1 from hepatocytes in endotoxemia (Figure 3), but Nrf2 is nonessential for the rescue of cardiac dysfunction and inflammation with human rPHB1 in endotoxemia (Figures 4 and 5). Much work remains to delineate the mechanisms by which PHB1 is regulated and secreted from cells during the acute phase, as it is clear that Nrf2 target gene(s) are likely involved.
A number of elegant studies have illustrated that PHBs are critical for optimal mitochondrial cristae organization and morphology, in part via regulation of cardiolipin and membrane fission‐fusion. The latter process has been reported to be regulated by a partnership between PHBs and the dynamin‐like GTPase, OPA1.24, 65, 66, 67 Following exposure to TNFα/IL‐1β, mitochondrial PHB1 depletion (and nuclear accumulation) occurred simultaneously with mitochondrial fission, loss of ATP, production and decreased respiration in cardiomyocytes (Figures 1 and 2). One of our more surprising findings is that rPHB1 improved cardiac ATP production in endotoxemia (Figure 4G) while having no effect at all on improving the decrease in ADP‐stimulated respiration following lipopolysaccharide challenge (Figure 4F and Figure S5). It is possible that rPHB1 is mitigating an inhibitory effect of inflammatory cyto/chemokines on enzymes in the phosphorylation system (eg, adenine nucleotide translocase, H+‐ATPase), rather than components of the electron transport system or TCA cycle. Given the effect of rPHB1 alone on cardiac mitochondrial ROS, and that rPHB1 increases thioredoxin reductase‐2 expression and auranofin‐inhibitable ROS in the heart (Figure S8), it is plausible that some of the cardioprotective effects of rPHB1 are coming from enhanced mitochondrial antioxidant capacity. In any case, much more work is needed to identify the mechanisms by which cytokine‐induced mitochondrial PHB1 depletion leads to impaired ATP production, and in turn, how recombinant PHB1 restores it.
Our preliminary findings implicate a role for PI3K/AKT pathway in mediating the paracrine‐like protective effect of rPHB1 in the heart (Figure 6), though we cannot exclude the possibility that some uptake of rPHB1 into the cardiomyocytes is mediating this effect. In a pancreatic cell line, human rPHB1 treatment protected the cells from ethanol toxicity, and the authors provided evidence that cellular uptake of rPHB1 was responsible for the effect.22 PHBs are on plasma membrane as well as mitochondrial membranes, and molecules from both bacterial68 and synthetic origin69 are capable of targeting cell surface PHB to facilitate cellular internalization. Though we were not able to comprehensively define the mechanisms by which rPHB1 protects the heart during endotoxemia, the fact that pro‐inflammatory cytokine expression was actually blunted by buparlisib (Figure 6E through 6G) suggests that the protective effects of rPHB1 in heart are mediated by additional mechanisms unrelated to its anti‐inflammatory effects.
Collectively, our findings suggest that PHB1 has an important role in the acute phase response to sepsis. Furthermore, we provide evidence that circulating PHB1 is anti‐inflammatory and cardioprotective in a model of endotoxemia and severe sepsis, but only when administered at doses corresponding to 10‐fold increase over physiological levels. This suggests that circulating PHB1 is only one part of a complex cascade of acute phase reactants that have inflammation‐resolving effects. The protective effects of rPHB1 are complex and may be highly dependent on the tissue, underlying metabolic factors, and the time during progression of sepsis at which the effect is observed. Studies directed at establishing more detailed insight into how intracellular PHB1 mobilization and trafficking occurs, in addition to identifying interactors of PHB1 in the blood, are beyond the scope of the present study but will be necessary for therapeutic exploitation of these pathways to treat inflammatory disorders. The use of circulating PHB1 as a biomarker of organ dysfunction in patients with sepsis may also be of clinical significance, and these studies are ongoing.
Sources of Funding
Sources of funding for this project came from Department of Defense grant W81XWH‐19‐1‐0037 (E.J.A.), National Institutes of Health grants R01HL122863 and R21AG057006 (E.J.A), R01ES028829 (K.M.G.), T32HL007121 (K.W.) American Heart Association Strategically‐Focused Research Network grant 20SFRN35200003 (E.J.A.), American Association of Immunologists Careers in Immunology Fellowship (C.E.P.), and from East Carolina University and the University of Iowa‐College of Pharmacy.
Disclosures
None.
Supporting information
Acknowledgments
The authors would like to thank Kathy Zimmerman and other staff within the Cardiovascular Phenotyping core facility at University of Iowa, Carver College of Medicine, for their assistance with echocardiography.
(J Am Heart Assoc. 2021;10:e019877. DOI: 10.1161/JAHA.120.019877.)
This manuscript was sent to Hossein Ardehali, MD, PhD, Guest Editor, for review by expert referees, editorial decision, and final disposition.
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.019877
For Sources of Funding and Disclosures, see page 14.
References
- 1.Antonucci E, Fiaccadori E, Donadello K, Taccone FS, Franchi F, Scolletta S. Myocardial depression in sepsis: from pathogenesis to clinical manifestations and treatment. J Crit Care. 2014;29:500–511. DOI: 10.1016/j.jcrc.2014.03.028. [DOI] [PubMed] [Google Scholar]
- 2.Sanfilippo F, Corredor C, Fletcher N, Landesberg G, Benedetto U, Foex P, Cecconi M. Diastolic dysfunction and mortality in septic patients: a systematic review and meta‐analysis. Intensive Care Med. 2015;41:1004–1013. DOI: 10.1007/s00134-015-3748-7. [DOI] [PubMed] [Google Scholar]
- 3.Palmieri V, Innocenti F, Guzzo A, Guerrini E, Vignaroli D, Pini R. Left ventricular systolic longitudinal function as predictor of outcome in patients with sepsis. Circ Cardiovasc Imaging. 2015;8:e003865. DOI: 10.1161/CIRCIMAGING.115.003865. [DOI] [PubMed] [Google Scholar]
- 4.Prucha M, Bellingan G, Zazula R. Sepsis biomarkers. Clin Chim Acta. 2015;440:97–103. DOI: 10.1016/j.cca.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 5.van Engelen TSR, Wiersinga WJ, Scicluna BP, van der Poll T. Biomarkers in sepsis. Crit Care Clin. 2018;34:139–152. DOI: 10.1016/j.ccc.2017.08.010. [DOI] [PubMed] [Google Scholar]
- 6.Vincent JL, Bakker J, Marecaux G, Schandene L, Kahn RJ, Dupont E. Administration of anti‐TNF antibody improves left ventricular function in septic shock patients. Results of a pilot study. Chest. 1992;101:810–815. DOI: 10.1378/chest.101.3.810. [DOI] [PubMed] [Google Scholar]
- 7.Bujak M, Frangogiannis NG. The role of IL‐1 in the pathogenesis of heart disease. Arch Immunol Ther Exp (Warsz). 2009;57:165–176. DOI: 10.1007/s00005-009-0024-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pathan N, Franklin JL, Eleftherohorinou H, Wright VJ, Hemingway CA, Waddell SJ, Griffiths M, Dennis JL, Relman DA, Harding SE, et al. Myocardial depressant effects of interleukin 6 in meningococcal sepsis are regulated by p38 mitogen‐activated protein kinase. Crit Care Med. 2011;39:1692–1711. DOI: 10.1097/CCM.0b013e3182186d27. [DOI] [PubMed] [Google Scholar]
- 9.Cimolai MC, Alvarez S, Bode C, Bugger H. Mitochondrial mechanisms in septic cardiomyopathy. Int J Mol Sci. 2015;16:17763–17778. DOI: 10.3390/ijms160817763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arulkumaran N, Deutschman CS, Pinsky MR, Zuckerbraun B, Schumacker PT, Gomez H, Gomez A, Murray P, Kellum JA, Workgroup AX. Mitochondrial function in sepsis. Shock. 2016;45:271–281. DOI: 10.1097/SHK.0000000000000463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alvarez S, Vico T, Vanasco V. Cardiac dysfunction, mitochondrial architecture, energy production, and inflammatory pathways: interrelated aspects in endotoxemia and sepsis. Int J Biochem Cell Biol. 2016;81:307–314. DOI: 10.1016/j.biocel.2016.07.032. [DOI] [PubMed] [Google Scholar]
- 12.Kumar G, Kumar N, Taneja A, Kaleekal T, Tarima S, McGinley E, Jimenez E, Mohan A, Khan RA, Whittle J, et al. Nationwide trends of severe sepsis in the 21st century (2000–2007). Chest. 2011;140:1223–1231. DOI: 10.1378/chest.11-0352. [DOI] [PubMed] [Google Scholar]
- 13.Lagu T, Rothberg MB, Shieh MS, Pekow PS, Steingrub JS, Lindenauer PK. Hospitalizations, costs, and outcomes of severe sepsis in the United States 2003 to 2007. Crit Care Med. 2012;40:754–761. DOI: 10.1097/CCM.0b013e318232db65. [DOI] [PubMed] [Google Scholar]
- 14.Ande SR, Mishra S. Prohibitin interacts with phosphatidylinositol 3,4,5‐triphosphate (PIP3) and modulates insulin signaling. Biochem Biophys Res Commun. 2009;390:1023–1028. DOI: 10.1016/j.bbrc.2009.10.101. [DOI] [PubMed] [Google Scholar]
- 15.Ande SR, Nguyen KH, Padilla‐Meier GP, Wahida W, Nyomba BL, Mishra S. Prohibitin overexpression in adipocytes induces mitochondrial biogenesis, leads to obesity development, and affects glucose homeostasis in a sex‐specific manner. Diabetes. 2014;63:3734–3741. DOI: 10.2337/db13-1807. [DOI] [PubMed] [Google Scholar]
- 16.Osman C, Haag M, Potting C, Rodenfels J, Dip PV, Wieland FT, Brugger B, Westermann B, Langer T. The genetic interactome of prohibitins: coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J Cell Biol. 2009;184:583–596. DOI: 10.1083/jcb.200810189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Supale S, Thorel F, Merkwirth C, Gjinovci A, Herrera PL, Scorrano L, Meda P, Langer T, Maechler P. Loss of prohibitin induces mitochondrial damages altering beta‐cell function and survival and is responsible for gradual diabetes development. Diabetes. 2013;62:3488–3499. DOI: 10.2337/db13-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei Y, Chiang WC, Sumpter R Jr, Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017;168:224–238 e10. DOI: 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rajalingam K, Wunder C, Brinkmann V, Churin Y, Hekman M, Sievers C, Rapp UR, Rudel T. Prohibitin is required for Ras‐induced Raf‐MEK‐ERK activation and epithelial cell migration. Nat Cell Biol. 2005;7:837–843. DOI: 10.1038/ncb1283. [DOI] [PubMed] [Google Scholar]
- 20.Chiu CF, Ho MY, Peng JM, Hung SW, Lee WH, Liang CM, Liang SM. Raf activation by Ras and promotion of cellular metastasis require phosphorylation of prohibitin in the raft domain of the plasma membrane. Oncogene. 2013;32:777–787. DOI: 10.1038/onc.2012.86. [DOI] [PubMed] [Google Scholar]
- 21.Martin B, Sanz R, Aragues R, Oliva B, Sierra A. Functional clustering of metastasis proteins describes plastic adaptation resources of breast‐cancer cells to new microenvironments. J Proteome Res. 2008;7:3242–3253. DOI: 10.1021/pr800137w. [DOI] [PubMed] [Google Scholar]
- 22.Lee JH, Nguyen KH, Mishra S, Nyomba BL. Prohibitin is expressed in pancreatic beta‐cells and protects against oxidative and proapoptotic effects of ethanol. FEBS J. 2010;277:488–500. DOI: 10.1111/j.1742-4658.2009.07505.x. [DOI] [PubMed] [Google Scholar]
- 23.Zhu B, Zhai J, Zhu H, Kyprianou N. Prohibitin regulates TGF‐beta induced apoptosis as a downstream effector of Smad‐dependent and ‐independent signaling. Prostate. 2010;70:17–26. DOI: 10.1002/pros.21033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Lower B, Wunderlich FT, von Kleist‐Retzow JC, Waisman A, Westermann B, Langer T. Prohibitins control cell proliferation and apoptosis by regulating OPA1‐dependent cristae morphogenesis in mitochondria. Genes Dev. 2008;22:476–488. DOI: 10.1101/gad.460708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joshi B, Ko D, Ordonez‐Ercan D, Chellappan SP. A putative coiled‐coil domain of prohibitin is sufficient to repress E2F1‐mediated transcription and induce apoptosis. Biochem Biophys Res Commun. 2003;312:459–466. DOI: 10.1016/j.bbrc.2003.10.148. [DOI] [PubMed] [Google Scholar]
- 26.Muraguchi T, Kawawa A, Kubota S. Prohibitin protects against hypoxia‐induced H9c2 cardiomyocyte cell death. Biomed Res. 2010;31:113–122. DOI: 10.2220/biomedres.31.113. [DOI] [PubMed] [Google Scholar]
- 27.Liu X, Ren Z, Zhan R, Wang X, Wang X, Zhang Z, Leng X, Yang Z, Qian L. Prohibitin protects against oxidative stress‐induced cell injury in cultured neonatal cardiomyocyte. Cell Stress Chaperones. 2009;14:311–319. DOI: 10.1007/s12192-008-0086-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Theiss AL, Idell RD, Srinivasan S, Klapproth JM, Jones DP, Merlin D, Sitaraman SV. Prohibitin protects against oxidative stress in intestinal epithelial cells. FASEB J. 2007;21:197–206. DOI: 10.1096/fj.06-6801com. [DOI] [PubMed] [Google Scholar]
- 29.Theiss AL, Vijay‐Kumar M, Obertone TS, Jones DP, Hansen JM, Gewirtz AT, Merlin D, Sitaraman SV. Prohibitin is a novel regulator of antioxidant response that attenuates colonic inflammation in mice. Gastroenterology. 2009;137:199–208, 208 e1–6. DOI: 10.1053/j.gastro.2009.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mishra S, Moulik S, Murphy LJ. Prohibitin binds to C3 and enhances complement activation. Mol Immunol. 2007;44:1897–1902. DOI: 10.1016/j.molimm.2006.09.025. [DOI] [PubMed] [Google Scholar]
- 31.Mengwasser J, Piau A, Schlag P, Sleeman JP. Differential immunization identifies PHB1/PHB2 as blood‐borne tumor antigens. Oncogene. 2004;23:7430–7435. DOI: 10.1038/sj.onc.1207987. [DOI] [PubMed] [Google Scholar]
- 32.Mojtahedi Z, Safaei A, Yousefi Z, Ghaderi A. Immunoproteomics of HER2‐positive and HER2‐negative breast cancer patients with positive lymph nodes. OMICS. 2011;15:409–418. DOI: 10.1089/omi.2010.0131. [DOI] [PubMed] [Google Scholar]
- 33.Ren H, Du N, Liu G, Hu HT, Tian W, Deng ZP, Shi JS. Analysis of variabilities of serum proteomic spectra in patients with gastric cancer before and after operation. World J Gastroenterol. 2006;12:2789–2792. DOI: 10.3748/wjg.v12.i17.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tompkins SC, Sheldon RD, Rauckhorst AJ, Noterman MF, Solst SR, Buchanan JL, Mapuskar KA, Pewa AD, Gray LR, Oonthonpan L, et al. Disrupting mitochondrial pyruvate uptake directs glutamine into the TCA cycle away from glutathione synthesis and impairs hepatocellular tumorigenesis. Cell Rep. 2019;28:2608–2619 e6. DOI: 10.1016/j.celrep.2019.07.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gray L, Sultana M, Rauckhorst A, Oonthonpan L, Tompkins S, Sharma A, Fu X, Miao R, Pewa A, Brown K, et al. Hepatic mitochondrial pyruvate carrier 1 is required for efficient regulation of gluconeogenesis and whole‐body glucose homeostasis. Cell Metab. 2015;22:669–681. DOI: 10.1016/j.cmet.2015.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prescott MJ, Lidster K. Improving quality of science through better animal welfare: the NC3Rs strategy. Lab Anim (NY). 2017;46:152–156. DOI: 10.1038/laban.1217. [DOI] [PubMed] [Google Scholar]
- 37.Anderson EJ, Thayne K, Harris M, Carraway K, Shaikh SR. Aldehyde stress and up‐regulation of Nrf2‐mediated antioxidant systems accompany functional adaptations in cardiac mitochondria from mice fed n‐3 polyunsaturated fatty acids. Biochem J. 2012;441:359–366. DOI: 10.1042/BJ20110626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fisher‐Wellman KH, Mattox TA, Thayne K, Katunga LA, La Favor JD, Neufer PD, Hickner RC, Wingard CJ, Anderson EJ. Novel role for thioredoxin reductase‐2 in mitochondrial redox adaptations to obesogenic diet and exercise in heart and skeletal muscle. J Physiol. 2013;591:3471–3486. DOI: 10.1113/jphysiol.2013.254193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lark DS, Torres MJ, Lin CT, Ryan TE, Anderson EJ, Neufer PD. Direct real‐time quantification of mitochondrial oxidative phosphorylation efficiency in permeabilized skeletal muscle myofibers. Am J Physiol Cell Physiol. 2016;311:C239–C245. DOI: 10.1152/ajpcell.00124.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson EJ, Rodriguez E, Anderson CA, Thayne K, Chitwood WR, Kypson AP. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial‐dependent pathways. Am J Physiol Heart Circ Physiol. 2011;300:H118–H124. DOI: 10.1152/ajpheart.00932.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boyle KE, Zheng D, Anderson EJ, Neufer PD, Houmard JA. Mitochondrial lipid oxidation is impaired in cultured myotubes from obese humans. Int J Obes (Lond). 2012;36:1025–1031. DOI: 10.1038/ijo.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Theiss AL, Jenkins AK, Okoro NI, Klapproth JM, Merlin D, Sitaraman SV. Prohibitin inhibits tumor necrosis factor alpha‐induced nuclear factor‐kappa B nuclear translocation via the novel mechanism of decreasing importin alpha3 expression. Mol Biol Cell. 2009;20:4412–4423. DOI: 10.1091/mbc.e09-05-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gabay C, Kushner I. Acute‐phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–454. DOI: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 44.Vary TC, Kimball SR. Regulation of hepatic protein synthesis in chronic inflammation and sepsis. Am J Physiol. 1992;262:C445–C452. DOI: 10.1152/ajpcell.1992.262.2.C445. [DOI] [PubMed] [Google Scholar]
- 45.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. DOI: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suliman HB, Welty‐Wolf KE, Carraway M, Tatro L, Piantadosi CA. Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc Res. 2004;64:279–288. DOI: 10.1016/j.cardiores.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 47.Turner A, Tsamitros M, Bellomo R. Myocardial cell injury in septic shock. Crit Care Med. 1999;27:1775–1780. DOI: 10.1097/00003246-199909000-00012. [DOI] [PubMed] [Google Scholar]
- 48.Gonzalez AS, Elguero ME, Finocchietto P, Holod S, Romorini L, Miriuka SG, Peralta JG, Poderoso JJ, Carreras MC. Abnormal mitochondrial fusion‐fission balance contributes to the progression of experimental sepsis. Free Radic Res. 2014;48:769–783. DOI: 10.3109/10715762.2014.906592. [DOI] [PubMed] [Google Scholar]
- 49.Aon MA, Stanley BA, Sivakumaran V, Kembro JM, O'Rourke B, Paolocci N, Cortassa S. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: an experimental‐computational study. J Gen Physiol. 2012;139:479–491. DOI: 10.1085/jgp.201210772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stanley BA, Sivakumaran V, Shi S, McDonald I, Lloyd D, Watson WH, Aon MA, Paolocci N. Thioredoxin reductase‐2 is essential for keeping low levels of H(2)O(2) emission from isolated heart mitochondria. J Biol Chem. 2011;286:33669–33677. DOI: 10.1074/jbc.M111.284612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barth E, Radermacher P, Thiemermann C, Weber S, Georgieff M, Albuszies G. Role of inducible nitric oxide synthase in the reduced responsiveness of the myocardium to catecholamines in a hyperdynamic, murine model of septic shock. Crit Care Med. 2006;34:307–313. DOI: 10.1097/01.CCM.0000199070.46812.21. [DOI] [PubMed] [Google Scholar]
- 52.Boyle WA III, Parvathaneni LS, Bourlier V, Sauter C, Laubach VE, Cobb JP. iNOS gene expression modulates microvascular responsiveness in endotoxin‐challenged mice. Circ Res. 2000;87:E18–E24. DOI: 10.1161/01.RES.87.7.e18. [DOI] [PubMed] [Google Scholar]
- 53.Xu C, Yi C, Wang H, Bruce IC, Xia Q. Mitochondrial nitric oxide synthase participates in septic shock myocardial depression by nitric oxide overproduction and mitochondrial permeability transition pore opening. Shock. 2012;37:110–115. DOI: 10.1097/SHK.0b013e3182391831. [DOI] [PubMed] [Google Scholar]
- 54.Wu Q, Wu S. The role of lipid raft translocation of prohibitin in regulation of Akt and Raf‐protected apoptosis of HaCaT cells upon ultraviolet B irradiation. Mol Carcinog. 2017;56:1789–1797. DOI: 10.1002/mc.22636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thuaud F, Ribeiro N, Nebigil CG, Desaubry L. Prohibitin ligands in cell death and survival: mode of action and therapeutic potential. Chem Biol. 2013;20:316–331. DOI: 10.1016/j.chembiol.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morley JJ, Kushner I. Serum C‐reactive protein levels in disease. Ann N Y Acad Sci. 1982;389:406–418. DOI: 10.1111/j.1749-6632.1982.tb22153.x. [DOI] [PubMed] [Google Scholar]
- 57.Ballou SP, Lozanski G. Induction of inflammatory cytokine release from cultured human monocytes by C‐reactive protein. Cytokine. 1992;4:361–368. DOI: 10.1016/1043-4666(92)90079-7. [DOI] [PubMed] [Google Scholar]
- 58.Cermak J, Key NS, Bach RR, Balla J, Jacob HS, Vercellotti GM. C‐reactive protein induces human peripheral blood monocytes to synthesize tissue factor. Blood. 1993;82:513–520. DOI: 10.1182/blood.V82.2.513.513. [DOI] [PubMed] [Google Scholar]
- 59.Xia D, Samols D. Transgenic mice expressing rabbit C‐reactive protein are resistant to endotoxemia. Proc Natl Acad Sci USA. 1997;94:2575–2580. DOI: 10.1073/pnas.94.6.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ahmed N, Thorley R, Xia D, Samols D, Webster RO. Transgenic mice expressing rabbit C‐reactive protein exhibit diminished chemotactic factor‐induced alveolitis. Am J Respir Crit Care Med. 1996;153:1141–1147. DOI: 10.1164/ajrccm.153.3.8630558. [DOI] [PubMed] [Google Scholar]
- 61.Heuertz RM, Ahmed N, Webster RO. Peptides derived from C‐reactive protein inhibit neutrophil alveolitis. J Immunol. 1996;156:3412–3417. [PubMed] [Google Scholar]
- 62.Kim DK, Kim HS, Kim AR, Jang GH, Kim HW, Park YH, Kim B, Park YM, Beaven MA, Kim YM, et al. The scaffold protein prohibitin is required for antigen‐stimulated signaling in mast cells. Sci Signal. 2013;6:ra80. DOI: 10.1126/scisignal.2004098. [DOI] [PubMed] [Google Scholar]
- 63.Lucas CR, Cordero‐Nieves HM, Erbe RS, McAlees JW, Bhatia S, Hodes RJ, Campbell KS, Sanders VM. Prohibitins and the cytoplasmic domain of CD86 cooperate to mediate CD86 signaling in B lymphocytes. J Immunol. 2013;190:723–736. DOI: 10.4049/jimmunol.1201646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kathiria AS, Butcher MA, Hansen JM, Theiss AL. Nrf2 is not required for epithelial prohibitin‐dependent attenuation of experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2013;304:G885–G896. DOI: 10.1152/ajpgi.00327.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Merkwirth C, Martinelli P, Korwitz A, Morbin M, Bronneke HS, Jordan SD, Rugarli EI, Langer T. Loss of prohibitin membrane scaffolds impairs mitochondrial architecture and leads to tau hyperphosphorylation and neurodegeneration. PLoS Genet. 2012;8:e1003021. DOI: 10.1371/journal.pgen.1003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tatsuta T, Model K, Langer T. Formation of membrane‐bound ring complexes by prohibitins in mitochondria. Mol Biol Cell. 2005;16:248–259. DOI: 10.1091/mbc.e04-09-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Richter‐Dennerlein R, Korwitz A, Haag M, Tatsuta T, Dargazanli S, Baker M, Decker T, Lamkemeyer T, Rugarli EI, Langer T. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab. 2014;20:158–171. DOI: 10.1016/j.cmet.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 68.Sharma A, Qadri A. Vi polysaccharide of Salmonella typhi targets the prohibitin family of molecules in intestinal epithelial cells and suppresses early inflammatory responses. Proc Natl Acad Sci USA. 2004;101:17492–17497. DOI: 10.1073/pnas.0407536101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim DH, Woods SC, Seeley RJ. Peptide designed to elicit apoptosis in adipose tissue endothelium reduces food intake and body weight. Diabetes. 2010;59:907–915. DOI: 10.2337/db09-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saks VA, Veksler VI, Kuznetsov AV, Kay L, Sikk P, Tiivel T, Tranqui L, Olivares J, Winkler K, Wiedemann F, et al. Permeabilized cell and skinned fiber techniques in studies of mitochondrial function in vivo. Mol Cell Biochem. 1998;184:81–100. [PubMed] [Google Scholar]
- 71.Anderson EJ, Neufer PD. Type II skeletal myofibers possess unique properties that potentiate mitochondrial H(2)O(2) generation. Am J Physiol Cell Physiol. 2006;290:C844–C851. [DOI] [PubMed] [Google Scholar]
- 72.Perry CG, Kane DA, Lin CT, Kozy R, Cathey BL, Lark DS, Kane CL, Brophy PM, Gavin TP, Anderson EJ, et al. Inhibiting myosin‐ATPase reveals a dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem J. 2011;437:215–222. DOI: 10.1042/BJ20110366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Claycomb WC, Lanson NA Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ Jr. HL‐1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA. 1998;95:2979–2984. DOI: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.White SM, Claycomb WC. Cardiac cell transplantation: protocols and applications. Methods Mol Biol. 2003;219:83–95. DOI: 10.1385/1-59259-350-x:83. [DOI] [PubMed] [Google Scholar]
- 75.White SM, Constantin PE, Claycomb WC. Cardiac physiology at the cellular level: use of cultured HL‐1 cardiomyocytes for studies of cardiac muscle cell structure and function. Am J Physiol Heart Circ Physiol. 2004;286:H823–H829. DOI: 10.1152/ajpheart.00986.2003. [DOI] [PubMed] [Google Scholar]
- 76.Deryckere F, Gannon F. A one‐hour minipreparation technique for extraction of DNA‐binding proteins from animal tissues. Biotechniques. 1994;16:405. [PubMed] [Google Scholar]
- 77.Tsien RY. Fluorescence measurement and photochemical manipulation of cytosolic free calcium. Trends Neurosci. 1988;11:419–424. DOI: 10.1016/0166-2236(88)90192-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.