

Abstract
Background
Migration of vascular smooth muscle cells (VSMCs) is the main contributor to neointimal formation. The Arp2/3 (actin‐related proteins 2 and 3) complex activates actin polymerization and is involved in lamellipodia formation during VSMC migration. Milk fat globule‐epidermal growth factor 8 (MFG‐E8) is a glycoprotein expressed in VSMCs. We hypothesized that MFG‐E8 regulates VSMC migration through modulation of Arp2/3‐mediated actin polymerization.
Methods and Results
To determine whether MFG‐E8 is essential for VSMC migration, a model of neointimal hyperplasia was induced in the common carotid artery of wild‐type and MFG‐E8 knockout mice, and the extent of neointimal formation was evaluated. Genetic deletion of MFG‐E8 in mice attenuated injury‐induced neointimal hyperplasia. Cultured VSMCs deficient in MFG‐E8 exhibited decreased cell migration. Immunofluorescence and immunoblotting revealed decreased Arp2 but not Arp3 expression in the common carotid arteries and VSMCs deficient in MFG‐E8. Exogenous administration of recombinant MFG‐E8 biphasically and dose‐dependently regulated the cultured VSMCs. At a low concentration, MFG‐E8 upregulated Arp2 expression. By contrast, MFG‐E8 at a high concentration reduced the Arp2 level and significantly attenuated actin assembly. Arp2 upregulation mediated by low‐dose MFG‐E8 was abolished by treating cultured VSMCs with β1 integrin function‐blocking antibody and Rac1 inhibitors. Moreover, treatment of the artery with a high dose of recombinant MFG‐E8 diminished injury‐induced neointimal hyperplasia and reduced VSMC migration.
Conclusions
MFG‐E8 plays a critical role in VSMC migration through dose‐dependent regulation of Arp2‐mediated actin polymerization. These findings suggest that high doses of MFG‐E8 may have therapeutic potential for treating vascular occlusive diseases.
Keywords: Arp2/3 complex, MFG‐E8, migration, neointimal formation, vascular smooth muscle cells
Subject Categories: Vascular Disease
Nonstandard Abbreviations and Acronyms
- Arp
actin‐related protein
- CCA
common carotid artery
- Del‐1
developmental endothelial locus‐1
- EGF
epidermal growth factor
- ERK
extracellular signal‐regulated kinas
- F‐actin
filamentous actin
- FAK
focal adhesion kinase
- G‐actin
globular actin
- MFG‐E8
milk fat globule‐epidermal growth factor 8
- rMFG‐E8
recombinant milk fat globule‐epidermal growth factor 8
- VSMC
vascular smooth muscle cell
- WT
wild‐type
Clinical Perspective
What Is New?
Milk fat globule‐epidermal growth factor 8 (MFG‐E8) plays a role in neointimal hyperplasia in response to vascular injury by promoting the proliferation and migration of vascular smooth muscle cells.
The effect of MFG‐E8 regulation on vascular smooth muscle cell migration is defined for the first time through dose‐dependent regulation of actin‐related protein 2–mediated actin polymerization.
Periadventitial delivery of high‐dose recombinant MFG‐E8 into mouse vessels attenuated ligation‐induced neointimal hyperplasia by limiting vascular smooth muscle cell migration.
What Are the Clinical Implications?
High‐dose MFG‐E8 may be a therapeutic strategy for the treatment of vascular occlusive diseases.
Vascular smooth muscle cells (VSMCs) are the major component of arterial walls and play crucial physiological and pathophysiological roles in blood vessels. Differentiated VSMCs proliferate slowly and exhibit a unique ability to contract.1 Unlike other fully differentiated skeletal or cardiac muscle cells, VSMCs retain their plasticity after differentiation and switch their phenotype from contractile to synthetic. In addition, they exhibit decreased contractility and increased proliferation and migration in response to vascular injury.2 The migration of VSMCs from the media to intima is a critical event that leads to the development of neointimal hyperplasia.3 The formation of lamellipodia at the leading edge of the migrating cells is driven by actin polymerization.4 The actin‐related proteins 2 and 3 (Arp2/3) complex, an actin nucleator, is composed of 2 actin‐related proteins, Arp2 and Arp3, and 5 other subunits, which are respectively named actin‐related protein complex 1 to 5.5, 6, 7 The Arp2/3 complex binds to the sides of the existing actin mother filaments and catalyzes filamentous actin (F‐actin) assembly from globular actin (G‐actin) in a branched manner.5, 6, 7, 8 Studies have revealed that the Arp2/3 complex translocates to the leading edge of motile VSMCs and facilitates actin assembly in protruding lamellipodia.9, 10, 11, 12 Milk fat globule‐epidermal growth factor 8 (MFG‐E8) is a secreted, integrin‐binding glycoprotein with multifunctional domains.13, 14 In healthy and diseased vessels, MFG‐E8 is expressed by endothelial cells, VSMCs, and macrophages.15, 16, 17 The ability of MFG‐E8 to facilitate the clearance of apoptotic cells in the vascular system has been well characterized.16, 18 Recently, its pathogenic role in VSMCs has also been revealed.15, 19, 20, 21 The level of MFG‐E8 is elevated in the serum and arteries of elderly people22 and has been identified as a biomarker for arterial aging.23 Several lines of evidence have demonstrated that MFG‐E8 contributes to intima–media thickening of the arterial wall by promoting VSMC proliferation and migration in aged arteries.17, 24 Our previous study further proved that MFG‐E8 upregulation in vessels promotes arterial aging by facilitating the proinflammatory phenotypic shift of aged VSMCs, thus rendering the VSMCs prone to proliferate and migrate.25 However, whether MFG‐E8 regulates actin dynamics in VSMC migration has not been explored. Evidence has shown that MFG‐E8 treatment triggers the reorganization of F‐actin in migrating intestinal epithelial cells,26 suggesting the involvement of Arp2/3‐mediated actin dynamics in MFG‐E8–regulated VSMC migration.
Herein, we report that MFG‐E8 is an essential mediator of VSMC migration and modulates VSMC migration by regulating Arp2‐mediated actin polymerization in a biphasic, dose‐dependent manner. We demonstrated that at low concentrations, MFG‐E8 upregulates Arp2 in VSMCs; by contrast, at high concentrations, MFG‐E8 downregulates Arp2 and functionally reduces the degree of actin polymerization and VSMC migration. Moreover, we revealed that β1 integrin and Rac1, the small GTPase, are involved in MFG‐E8–mediated Arp2 expression.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Complete Common Carotid Ligation
In the experiment, 6‐ to 8‐week‐old male and female wild‐type (WT) and MFG‐E8 knockout mice (Riken BioResource Center, Japan)27 backcrossed to the FVB/NJ background (Figure S1) were subjected to complete common carotid ligation on the left side. The animals were anesthetized through inhalation of 2% isoflurane throughout the procedure. The left common carotid artery (CCA) was exposed, separated from the surrounding connective tissue, and completely ligated using a 6‐0 silk suture proximal to the carotid bifurcation.28, 29, 30 In some WT mice, pluronic gel with recombinant MFG‐E8 (rMFG‐E8, R&D) was administered periadventitially to the carotid artery immediately after complete carotid artery ligation.25, 28, 29 All animal procedures were approved by the Institutional Animal Care and Use Committee of Chang Gung University (approval number: CGU12‐131) and were conducted in accordance with the Institutional Guidelines for Animal Research.
Polymerase Chain Reaction Genotyping
The mice were properly anesthetized with isoflurane (2%) before tail‐snipping for genomic DNA isolation. Briefly, sequences of polymerase chain reaction primers and a polymerase chain reaction protocol for genotyping MFG‐E8 knockout mice were provided by Riken BioResource Center. A sense primer (5′‐GTGAACCTTCTGCGGAAGAT‐3′) and an antisense primer (5′‐GGGCATAAACTCCAGCTCAC‐3′) were used for the WT MFG‐E8 allele. For the knockout allele, a phosphoglycerate kinase primer (5′‐CGTGGGATCATTGTTTTTCT‐3′) and an antisense primer (5′‐GGGCATAAACTCCAGCTCAC‐3′) were employed.
Tissue Collection and Morphometric Analysis
The mice were euthanized 10 and 21 days after surgery by overdose through inhalation of isoflurane followed by transcardial perfusion with 10% formalin. Formalin‐fixed CCAs harvested from the mice were embedded in paraffin and cut into 5‐μm–thick sections. Sections located at 200‐μm intervals within the first millimeter proximal to the ligature were selected for Verhoeff–van Gieson elastic staining. Then, the intimal area, medial area, and area within the external elastic lamina were analyzed with Image‐Pro Plus (Media Cybernetics) as described in the literature.28, 31
Immunohistochemistry and Quantitative Analysis
For each mouse, 3 sections located at 200‐μm intervals within the first millimeter proximal to the ligature were selected for immunohistochemistry analysis. Sections were incubated with the antibodies anti–MFG‐E8 (R&D systems), anti–Ki‐67 (Abcam), anti‐activated β1 integrin (1:1000, Merck Millipore), and anti–Rac1‐GTP antibody32 (1:1000, New East Biosciences). Subsequently, the sections were incubated with appropriate biotinylated secondary antibodies, and followed by analysis using an avidin–biotin immunoperoxidase system (Vector Laboratories). A liquid diaminobenzidine substrate chromogen system (Dako) was used for detection. To evaluate proliferating cells, digital images of the paraffin sections were captured, and the ratio of Ki‐67(+) cells to total cells or the number of Ki‐67(+) cells per square micrometer was determined. To assess the MFG‐E8, activated β1 integrin, and activated Rac1‐GTP levels, the average optical density in the intima–media area in each section was measured using Image‐Pro Plus (Media Cybernetics).
Cell Culture and Treatment
Rat aortic smooth muscle cells were obtained from Cell Applications, and A10 VSMCs were acquired from Bioresource Collection and Research Center. Rat aortic smooth muscle and A10 cells were grown in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum. All cells were cultured under standard conditions (5% CO2 in air in a humidified environment at 37°C). For MFG‐E8 silencing, small interfering RNAs (siRNAs; GE Healthcare Dharmacon) were delivered to rat aortic smooth muscle and A10 cells. For primary culture of VSMCs, mouse VSMCs were enzymatically isolated from the aortas of WT mice as described in our previous study.25 For MFG‐E8 dose experiments, primary VSMCs and A10 cells were incubated with various doses of rMFG‐E8 for the indicated periods. For the integrin function‐blocking assay, the A10 cells were incubated with either 50 μg/mL β1 inhibitory antibody (Ha2/5; BD Pharmingen) or isotype control for 4 hours before treatment with various doses of MFG‐E8 for the indicated periods. For the Rac1 inhibition assay, NSC23766 (200 μM, Tocris Bioscience), a Rac1 inhibitor,33 was added to the serum‐free medium 16 hours before incubation with various doses of MFG‐E8 for the indicated periods.
Immunofluorescence Analysis and Confocal Microscopy
To determine the expression of Arp2 and Arp3 in the mouse CCAs, the paraffinized sections within the first millimeter proximal to the ligature were deparaffinized, permeabilized with 0.1% Triton‐X 100 in PBS for 10 minutes, blocked with 5% normal goat serum in 0.1% Triton‐X 100 in PBS, and incubated with primary antibodies against Arp2 and Arp3 (1:200, Cell Signaling) overnight at 4°C; this was followed by an additional incubation with Alexa Fluor 594–conjugated secondary antibodies (1:100, Thermo Fisher Invitrogen) for 1 hour at room temperature. VSMCs derived from the aortas of WT mice were washed in warm PBS and fixed in 1.75% paraformaldehyde for 15 minutes after various treatments. After blocking, the cells were incubated with primary antibodies against Arp2 and Arp3, followed by incubation with the appropriate secondary antibodies. Cells were coincubated with Alexa Fluor 488–conjugated phalloidin (1:200, Thermo Fisher Invitrogen). To assess F‐actin assembly, A10 VSMCs were incubated for 1 hour at room temperature with Alexa Fluor 488–conjugated phalloidin (1:200, Thermo Fisher Invitrogen) or Alexa Fluor 594–conjugated deoxyribonuclease (DNase I, 1:100, Thermo Fisher Invitrogen). To evaluate the activation state of β1 integrin, A10 VSMCs were incubated with primary antibodies against activated β1 integrin (1:100, Merck Millipore) and focal adhesion kinase (FAK, 1:100, Cell Signaling) overnight at 4°C, followed by incubation with the appropriate secondary antibodies (1:100, Thermo Fisher Invitrogen). Confocal images were captured using a Zeiss LSM 780 confocal scanning microscope.
Quantification of Fluorescence Intensity
To quantitate the expression of Arp2 and Arp3 in CCAs, fluorescence images of CCAs derived from WT and MFG‐E8 knockout mice were captured with a 20× objective and transformed into grayscale images. The mean fluorescence intensities of Arp2 and Arp3 were analyzed using Image‐Pro Plus (Media Cybernetics). The VSMCs were subjected to quantitative analysis of fluorescence intensity for Arp2, Arp3, activated β1 integrin, phalloidin, and DNase I. Digital color images were captured using a 100× objective and transformed into grayscale images. Three independent experiments were performed. In a representative experiment, several fields of view of equal size per treatment were randomly selected, and the mean fluorescence intensities of Arp2, Arp3, activated β1 integrin, F‐actin, and G‐actin were analyzed using the Image‐Pro Plus.
Transwell Migration Assay
A10 cells were cultured in growth medium until 70% to 80% confluence was achieved. Then, the cells were treated overnight with various doses of rMFG‐E8, followed by serum starvation for 24 hours. The cells were subsequently trypsinized, and 3000 cells were seeded onto the upper chamber of transwells with a pore size of 8 μm in 24‐well plates (Corning). A total of 800 µL of PBS or platelet‐derived growth factor‐BB (10 ng/mL) was added into DMEM in the lower chamber to induce VSMC migration. After 6 hours of incubation, the VSMCs that migrated into the lower chambers were stained with calcein‐AM (Sigma‐Aldrich), and the fluorescence intensity of calcein‐AM was determined using a SpectraMax M5 microplate reader (Molecular Devices) set at 485/535 (excitation/emission).
Immunoblotting
The siRNA‐transfected and rMFG‐E8–treated cells were homogenized in 1× Laemmli sample buffer (Sigma‐Aldrich). Carotid arteries from MFG‐E8 knockout and WT mice were pooled and homogenized with cold lysis buffer (50 mM Tris‐HCl, pH 7.5, 150 mM NaCl, 1% NP‐40, and 5 mM ethylenediaminetetraacetic acid) supplemented with protease inhibitor cocktail (Sigma‐Aldrich). Cell and tissue lysates were electrophoresed on sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels under reducing conditions and then transferred onto nitrocellulose membranes. Primary antibodies of Arp2 and Arp3 (Cell Signaling), MFG‐E8 (R&D), and GAPDH or α‐tubulin (Sigma‐Aldrich) were employed for immunoblot analyses, as previously described.28, 34 Quantitative analysis was performed through densitometry by using ImageJ (National Institutes of Health).
Rac1 G‐LISA Assay
Rac1 activation in A10 VSMCs was determined using the G‐LISA Rac1‐activation assay (Cytoskeleton). A10 cells were starved and then treated with various doses of MFG‐E8 for 16 hours in the presence or absence of β1 integrin function‐blocking antibody. Subsequently, the cells were washed with ice‐cold PBS and extracted with lysis buffer. After collection of protein lysates, the activated GTP‐bound Rac1 was analyzed following standard procedures of the manufacturer. Bound active Rac1 was detected using a specific antibody, and the absorbance at 490 nm was quantified using a microplate reader (Molecular Device).
Statistical Analysis
After normality and equal variance tests, appropriate statistical analyses were performed, the results of which are indicated in the legend of each figure. For animal studies, data are presented as mean±SEM. For in vitro experiments, data are presented as mean±SD and are representative of at least 3 independent experiments. Data were analyzed using Student t test or 1‐way ANOVA and a post hoc test by using GraphPad Prism (GraphPad). For some experiments, the nonparametric Mann–Whitney U test was conducted when the normality test yielded negative results. Results with a P value of >.05 were considered nonsignificant.
Results
Genetic Deletion of MFG‐E8 Attenuates Neointimal Formation by Reducing Intimal and Medial Cell Growth and VSMC Migration
Studies have shown that MFG‐E8 is expressed by VSMCs and promotes VSMC proliferation and migration in aged aortas,17, 24 indicating that MFG‐E8 may participate in neointimal hyperplasia in response to vascular injury. To directly test the role of MFG‐E8 in a mouse model of neointimal formation, we evaluated the extent of intima–media thickening in MFG‐E8 knockout and WT mice 21 days after complete ligation of the CCA. The WT mice developed significant neointimal hyperplasia (Figure 1A). By contrast, in the MFG‐E8 knockout mice, the ligation‐induced intima–media thickening was considerably attenuated (Figure 1A). A morphometric analysis revealed significantly reduced intima–media area bound by the external elastic lamina (Figure S2) and intima/media ratio (Figure 1A) in knockout CCAs. To confirm that the decrease in neointimal hyperplasia in the MFG‐E8 knockout mice was not caused by any change in Del‐1 (developmental endothelial locus‐1), a protein homologous to MFG‐E8, we analyzed the immunohistochemistry intensity of Del‐1 in the MFG‐E8 knockout and WT mice 10 days after ligation (Figure S3). No difference was noted between the CCAs of the knockout and WT mice.
Figure 1. Milk fat globule‐epidermal growth factor (MFG‐E8) knockout mice exhibit reduced neointimal hyperplasia along with a decrease in vascular smooth muscle cell (VSMC) proliferation and migration.
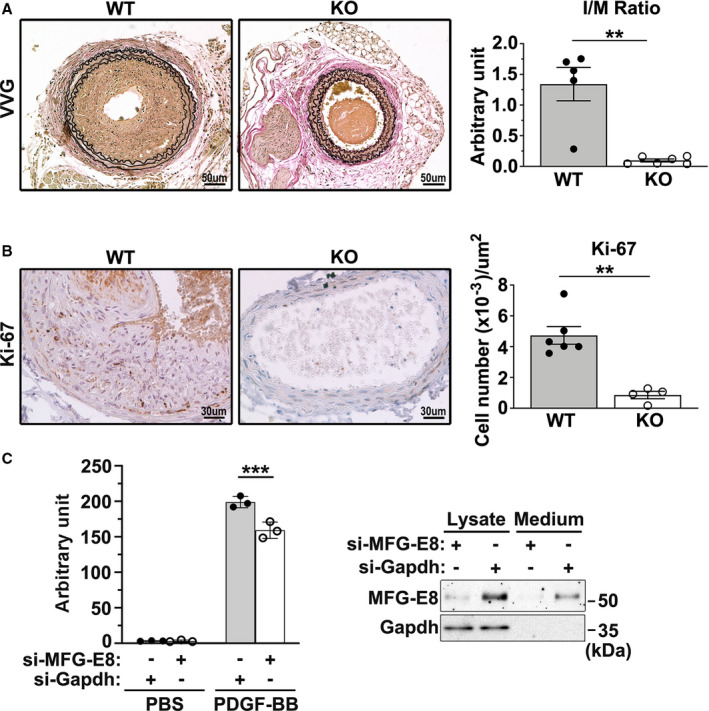
A, Representative images display the cross‐sectional areas of the ligated carotid artery of wild‐type (WT) and MFG‐E8 knockout (KO) mice 21 days after ligation. All sections were subjected to Verhoeff–van Gieson (VVG) staining. Bar, 50 μm. Morphometric analysis of the intima/media (I/M) ratio 21 days after ligation was conducted (WT: nmice=5, MFG‐E8 knockout: nmice=6). Results are presented as mean±SEM. Each point is derived from an assessment of 3 sections of an individual animal. **P<0.01, as obtained using t test. B, Photomicrographs depicting Ki‐67 immunostaining in the cross sections of the carotid arteries from WT and MFG‐E8 knockout mice 10 days after ligation. All sections were counterstained with hematoxylin. Bar, 30 μm. Quantitative immunohistochemistry analysis of Ki‐67(+) cells per square micrometer in the intima and media of the vessel (WT: nmice=6, knockout: nmice=4). Results are presented as mean±SEM. Each point is derived from an assessment of 3 sections of an individual animal. **P<0.01, as obtained using the nonparametric Mann–Whitney U test. C, Rat aortic smooth muscle (RASM) cells were transfected with small interfering RNA against MFG‐E8 (si‐MFG‐E8) or against GAPDH (si‐GAPDH) for 48 hours followed by 24‐hour serum starvation. On the following day, the cells were trypsinized and seeded on transwells with a pore size of 8 μm in 24‐well plates. A total of 800 µL of PBS or platelet‐derived growth factor‐BB (PDGF‐BB) (10 ng/mL) was added to Dulbecco's Modified Eagle Medium (DMEM) in the lower chamber to induce VSMC migration. RASM cells that migrated into the lower chambers were stained with calcein‐AM, and the fluorescence intensity was quantitated. Data represent the averages of triplicate samples of a representative experiment (n=3), and the error bars indicate the SD. ***P<0.001, as obtained using the t test. The efficiency of the MFG‐E8 knockdown in RASM cells was demonstrated using immunoblotting analysis.
We next performed immunohistochemistry analysis for Ki‐67, a cellular marker of proliferation, to confirm that the reduced neointimal hyperplasia was associated with changes in VSMC proliferation. Loss of MFG‐E8 significantly attenuated intimal and medial cell proliferation 10 days after ligation, as evidenced by the decreased number of Ki‐67‐positive cells (Figure 1B). To assess the involvement of MFG‐E8 in VSMC migration, VSMCs were treated with siRNA against MFG‐E8, and a transwell assay was performed. As shown in Figure 1C, siRNA treatment successfully attenuated the protein levels of MFG‐E8 in VSMCs, and the migration of VSMCs treated with siRNA against MFG‐E8 was significantly reduced compared with that of the control cells. Overall, our in vivo and in vitro findings revealed that MFG‐E8 participates in neointimal formation by promoting VSMC proliferation and migration.
MFG‐E8 Deficiency Reduces Arp2 Expression in Carotid Arteries and VSMCs
The Arp2/3 complex–mediated actin assembly is essential for cell migration. Our data revealed that deletion of MFG‐E8 impairs VSMC migration, suggesting that MFG‐E8 may mediate VSMC migration through regulation of the expression or activity of the Arp2/3 complex. Therefore, we evaluated the expression of Arp2 and Arp3, the actin‐related proteins of the complex, in the CCAs of WT and MFG‐E8 knockout mice through immunofluorescence and immunoblotting analyses. As depicted in Figure 2, the genetic deletion of MFG‐E8 significantly reduced the fluorescence intensity of Arp2 in the CCAs of knockout mice compared with the arteries of WT mice (Figure 2A). Immunoblotting of Arp2 revealed a consistent reduction in the protein level in the pooled CCAs of knockout mice relative to the CCAs of WT mice (Figure 2C). By contrast, no obvious reduction in Arp3 was noticed in the absence of MFG‐E8 in carotid arteries, as revealed by the immunofluorescence (Figure 2B) and immunoblotting (Figure 2C) analyses. Next, we investigated the role of MFG‐E8 in mediating the expression of Arp2 specifically in VSMCs, which are the cells responsible for neointimal formation in the arterial wall. Endogenous MFG‐E8 in A10 VSMCs was knocked down with siRNA, and the expression of Arp2 and Arp3 was evaluated through immunoblotting (Figure 2D). Consistent with our in vivo results, the deletion of MFG‐E8 significantly reduced the Arp2 protein level in VSMCs treated with siRNA against MFG‐E8 versus control VSMCs. By contrast, no obvious reduction in Arp3 was observed in the absence of MFG‐E8 (Figure 2D). Taken together, these results indicate that MFG‐E8 deficiency impairs the integrity of the Arp2/3 complex by reducing Arp2 expression, thereby leading to compromised migration of VSMCs.
Figure 2. Milk fat globule‐epidermal growth factor (MFG‐E8) deficiency reduces actin‐related protein 2 (Arp2) expression in carotid arteries and vascular smooth muscle cells (VSMCs).
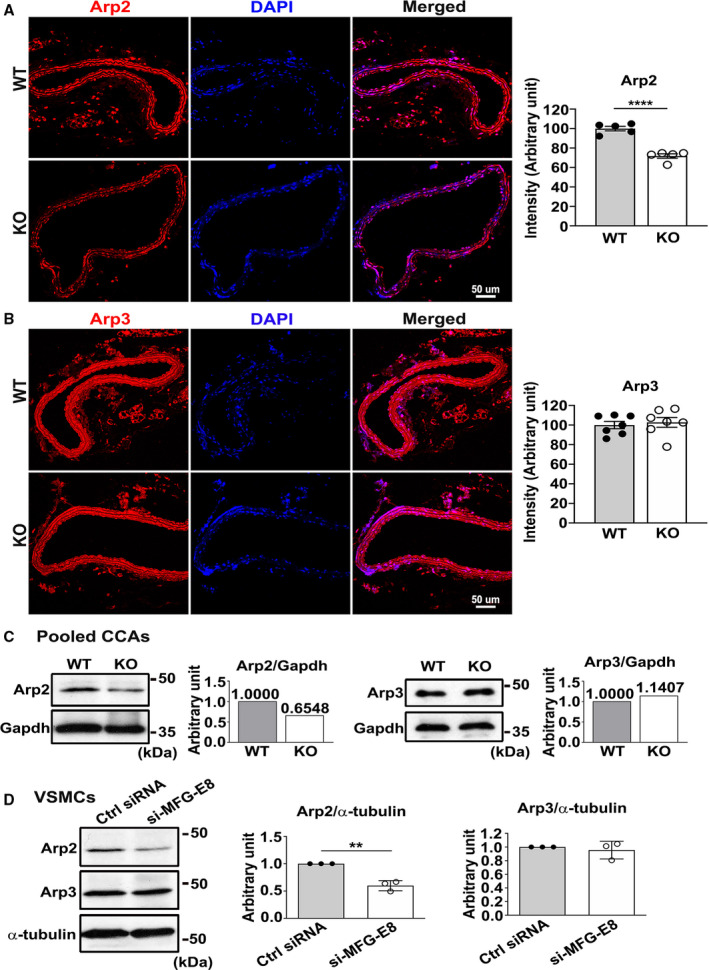
A and B, Representative immunofluorescence photographs of Arp2 (A) and actin‐related protein 3 (Arp3) (B) in the carotid arteries of wild‐type (WT) and MFG‐E8 knockout (KO) mice. Bar, 50 μm. Quantification of fluorescence intensities of Arp2 (WT: nmice=5, MFG‐E8 knockout: nmice=5) and Arp3 (WT: nmice=7, MFG‐E8 knockout: nmice=7) is shown in (A) and (B), respectively. Results are presented as mean±SEM. Each point is derived from an assessment of 3 sections of an individual animal. ****P<0.0001, as obtained using the t test. C, Proteins were extracted from 10 pooled common carotid arteries of WT and knockout mice for Western blotting and examined with antibodies against Arp2 and Arp3. In the quantitative analysis, the Arp2 and Arp3 levels were normalized to that of GAPDH. D, A10 VSMCs underwent 48‐hour transfection with small interfering RNA (siRNA) against MFG‐E8 (si‐MFG‐E8) or control siRNA (Ctrl siRNA). The expression of Arp2 and Arp3 was evaluated through Western blotting. The Arp2 (n=3) and Arp3 (n=3) levels were normalized to those of α‐tubulin. Three repeated experiments were performed. Data are presented as mean±SD. Each point is derived from each of the 3 repeated experiments. **P<0.01, as obtained using the t test. CCA indicates common carotid artery; and DAPI, 4',6‐diamidino‐2‐phenylindole.
MFG‐E8 Mediates the Protein Expression of Arp2 in a Biphasic, Dose‐Dependent Manner in VSMCs
Our previous study unexpectedly demonstrated that exogenous delivery of a large dose of rMFG‐E8 into the injured vessels of aged mice attenuated VSMC migration,25 indicating that MFG‐E8 regulates VSMC migration in a dose‐sensitive manner. Therefore, we next assessed whether the mediation of MFG‐E8 on Arp2 expression is dose dependent. We first assessed the protein expression of Arp2/3 in A10 VSMCs through immunoblotting after the addition of rMFG‐E8 for 16 hours. A low dose of MFG‐E8 (10 ng/mL) significantly upregulated Arp2 levels in the VSMCs (Figure 3A and 3B). By contrast, a high dose of MFG‐E8 (1000 ng/mL) elicited an obvious downregulation of Arp2 compared with the vehicle‐treated A10 cells. No changes in Arp3 protein expression were observed in the A10 cells treated with various doses of MFG‐E8 (Figure 3A and 3C). Next, we used early‐passage primary VSMCs derived from the aortas of WT mice to confirm the consistency of the biphasic regulation of Arp2 expression by MFG‐E8. As depicted in Figure 3D, treatment of VSMCs with a low dose of MFG‐E8 induced significantly higher Arp2 fluorescence intensity than treatment with PBS; cells treated with a high dose of MFG‐E8 exhibited obviously decreased Arp2 fluorescence intensity. Concurrently, we performed phalloidin staining to label assembled F‐actin in primary VSMCs. Notably, colocalization of Arp2 with F‐actin was more prominent in VSMCs incubated with low‐dose MFG‐E8 than in vehicle‐ and high‐dose MFG‐E8–treated cells. By contrast, no significant difference in Arp3 fluorescence intensities was noted among the PBS‐, low‐dose‐, and high‐dose–treated VSMCs (Figure 3E).
Figure 3. Milk fat globule‐epidermal growth factor (MFG‐E8) mediates actin‐related protein 2 (Arp2) expression in a biphasic, dose‐dependent manner in vascular smooth muscle cells (VSMCs).
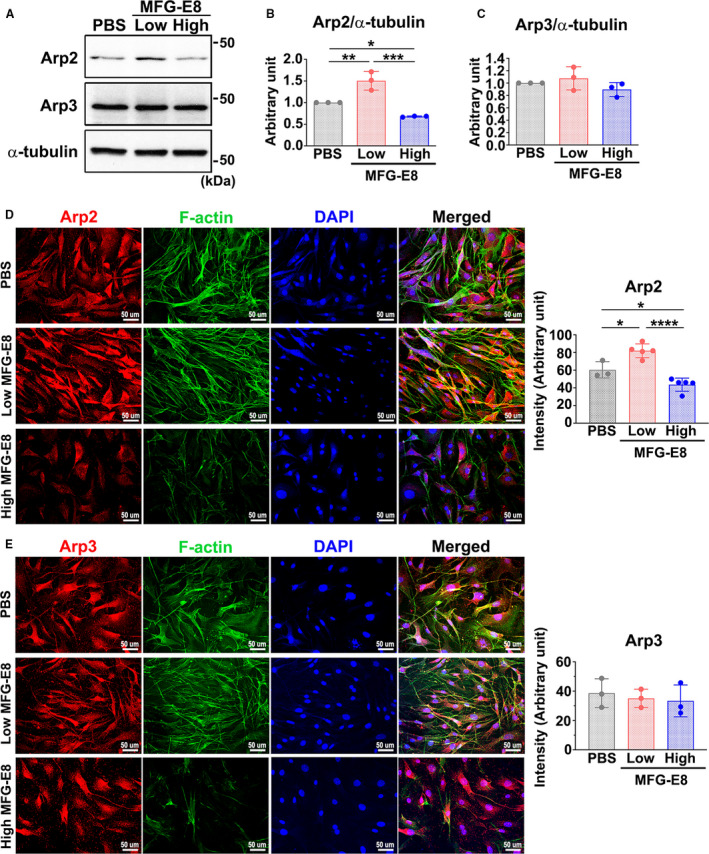
A through C, A10 VSMCs were treated with either a low (10 ng/mL) or high (1000 ng/mL) concentration of recombinant MFG‐E8 for 16 hours. A, Western blot analysis of the protein expression of Arp2 and actin‐related protein (Arp3) in A10 cells treated with low and high doses of MFG‐E8 for 16 hours. Quantitative analyses of Arp2 (B) and Arp3 (C) levels normalized to those of α‐tubulin were conducted (n=3). Data are presented as mean±SD. Three independent experiments were performed. Each point is derived from each of the 3 repeated experiments. *P<0.05, **P<0.01, ***P<0.001, as obtained using 1‐way ANOVA followed by Tukey multiple comparisons test. D through E, VSMCs isolated from aortas of wild‐type mice were immunostained with antibodies against Arp2 (D) and Arp3 (E) and costained with phalloidin to label filamentous actin (F‐actin). Bar, 50 μm. Three independent experiments were performed and each experiment was repeated with similar results. In a representative experiment, the fluorescence intensities of Arp2 (n=3–5) (D) and Arp3 (n=3) (E) were quantitated. Data are presented as mean±SD. Each point is derived from an assessment of each picture field. *P<0.05 and ****P<0.0001, as obtained using 1‐way ANOVA followed by Tukey multiple comparisons test. DAPI indicates 4',6‐diamidino‐2‐phenylindole.
To further confirm that MFG‐E8 regulated Arp2 expression in a biphasic manner, A10 cells were treated with various doses of rMFG‐E8 from 10 ng/mL to 1000 ng/mL, and the levels of Arp2 were evaluated through immunoblotting. As shown in Figure S4A, 10‐ng/mL MFG‐E8 significantly induced Arp2 expression in A10 cells, but the trend of Arp2 induction by MFG‐E8 was reversed when the MFG‐E8 concentration was increased to >100 ng/mL. Immunofluorescence of Arp2 in primary VSMCs treated with various doses of MFG‐E8 demonstrated the same biphasic regulation of Arp2 expression by MFG‐E8 (Figure S4C). By contrast, results from immunoblotting revealed no obvious differences in Arp3 levels when the VSMCs were treated with various doses of MFG‐E8 (Figure S4B). Collectively, these data reveal that MFG‐E8 biphasically and dose‐dependently regulates the protein expression of Arp2 in VSMCs. Therefore, we continued to use 10 ng/mL and 1000 ng/mL of MFG‐E8 to represent low and high doses of treatment, respectively, in subsequent experiments.
High‐Dose MFG‐E8 Attenuates Actin Polymerization in VSMCs
We demonstrated that MFG‐E8 regulates Arp2 protein expression in a biphasic, dose‐dependent manner, and more colocalization of Arp2 and F‐actin was observed after treatment of the VSMCs with low‐dose MFG‐E8. We then determined whether MFG‐E8 modulation in actin assembly was also dose dependent. We treated A10 cells with low (10 ng/mL) and high (1000 ng/mL) doses of MFG‐E8 and then performed phalloidin and DNase I staining to label F‐actin and G‐actin, respectively. Quantitative image analysis revealed that the fluorescence intensities of F‐actin and G‐actin were not significantly altered in cells treated with low‐dose MFG‐E8, indicating that low‐dose MFG‐E8 did not significantly enhance F‐actin assembly from G‐actin (Figure 4B, 4E, 4G, and 4H). By contrast, high‐dose MFG‐E8 treatment substantially inhibited the polymerization of F‐actin and increased the G‐actin levels (Figure 4C, 4F, 4G, and 4H). From the findings taken together, we infer that treatment of VSMCs with low‐dose MFG‐E8, which only elevates Arp2 expression, is insufficient to drive actin polymerization. By contrast, treating VSMCs with high‐dose MFG‐E8 may impede VSMC migration through the attenuation of Arp2‐mediated actin polymerization.
Figure 4. Milk fat globule‐epidermal growth factor (MFG‐E8) modulates actin polymerization in vascular smooth muscle cells (VSMCs) in a dose‐dependent manner.
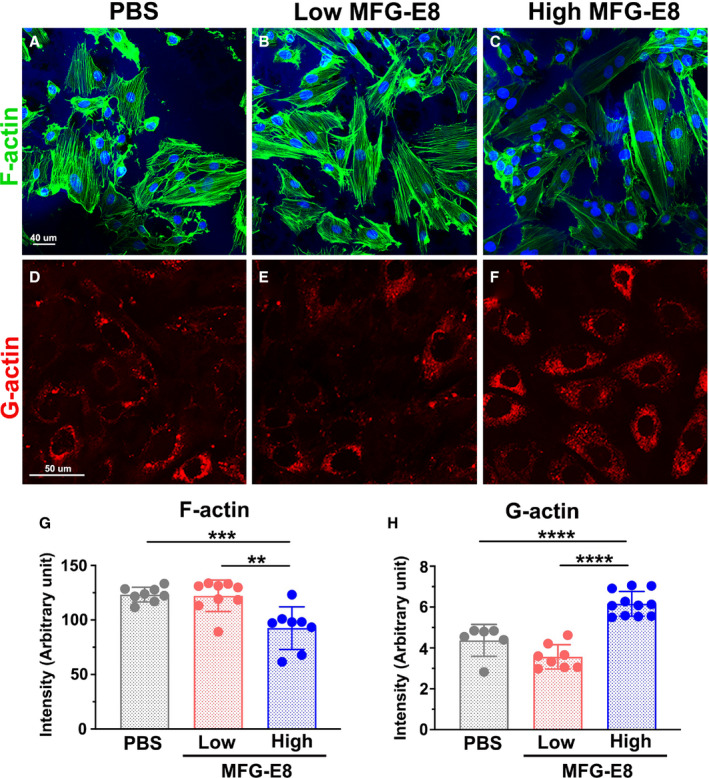
Representative confocal fluorescent images of phalloidin (filamentous actin [F‐actin], green) A through C, and DNase I (globular actin [G‐actin], red) (D) through (F) staining of A10 VSMCs incubated with low (10 ng/mL) and high (1000 ng/mL) doses of MFG‐E8 for 16 hours. Bar, 50 μm. Three independent experiments were performed, and each experiment was repeated with similar results. In a representative experiment, corresponding quantification of the mean fluorescence intensity of phalloidin for F‐actin (n=8–9) (G) and that of DNase I for G‐actin (n=6–11) H, was performed as described in the Methods section. Data are presented as mean±SD. Each point is derived from an assessment of each picture field. **P<0.01, ***P<0.001, ****P<0.0001, as obtained using 1‐way ANOVA followed by Tukey multiple comparisons test.
β1 Integrin–Rac1 Signaling Axis Is Involved in MFG‐E8–Mediated Arp2 Expression
β1 integrin is known to provide the critical link to the actin cytoskeleton from signals outside the cells during cell migration.35 β1 integrin can functionally pair with 12 different α chains.36 Recently, α8β1 integrin was proven to be a novel receptor for MFG‐E8 in smooth muscle cells.37 To determine whether MFG‐E8–mediated Arp2 expression is attributable to β1 integrin engagement, A10 cells were incubated with various doses of rMFG‐E8, and the activated state of β1 integrin was evaluated using immunofluorescence after the addition of MFG‐E8 for 16 hours; cell–extracellular matrix adhesions were labeled by costaining the cells with the anti‐FAK antibody. Upon stimulation with low‐dose rMFG‐E8, the fluorescence intensity of activated β1 integrin in the A10 cells was substantially increased compared with that in PBS‐ and high‐dose‐MFG‐E8–stimulated cells (Figure 5A and 5B). Enhanced FAK staining accompanied by increased colocalization of activated β1 integrin and FAK was noted after treatment with low‐dose MFG‐E8, indicating the clustering of β1 integrin in focal adhesions. By contrast, high‐dose MFG‐E8 significantly diminished the activation of β1 integrin (Figure 5A and 5B). Next, we performed an antibody‐blocking assay to confirm the involvement of β1 integrin in MFG‐E8–mediated Arp2 expression. Pretreatment of A10 cells with the neutralizing anti–β1 integrin antibody substantially restored the Arp2 protein expression induced by low‐dose MFG‐E8 to the basal level (Figure 6A and 6B); however, no further downregulation of Arp2 was observed after the addition of the β1 integrin–blocking antibody to cells treated with high‐dose MFG‐E8 (Figure 6A and 6B). No significant changes in Arp3 expression were observed in the A10 cells treated with the β1 integrin inhibitory antibody (Figure 6A and 6C). Additionally, the anti–β1 integrin antibody significantly inhibited F‐actin assembly from G‐actin (Figure 6D), which might be attributable to the further downregulation of Arp2. These data suggest that β1 integrin plays a major role in the enhanced protein expression of Arp2 mediated by low‐dose MFG‐E8. By contrast, high‐dose MFG‐E8 exhibited no engagement with β1 integrin.
Figure 5. Milk fat globule‐epidermal growth factor (MFG‐E8) regulates β1 integrin activation in a dose‐dependent manner.
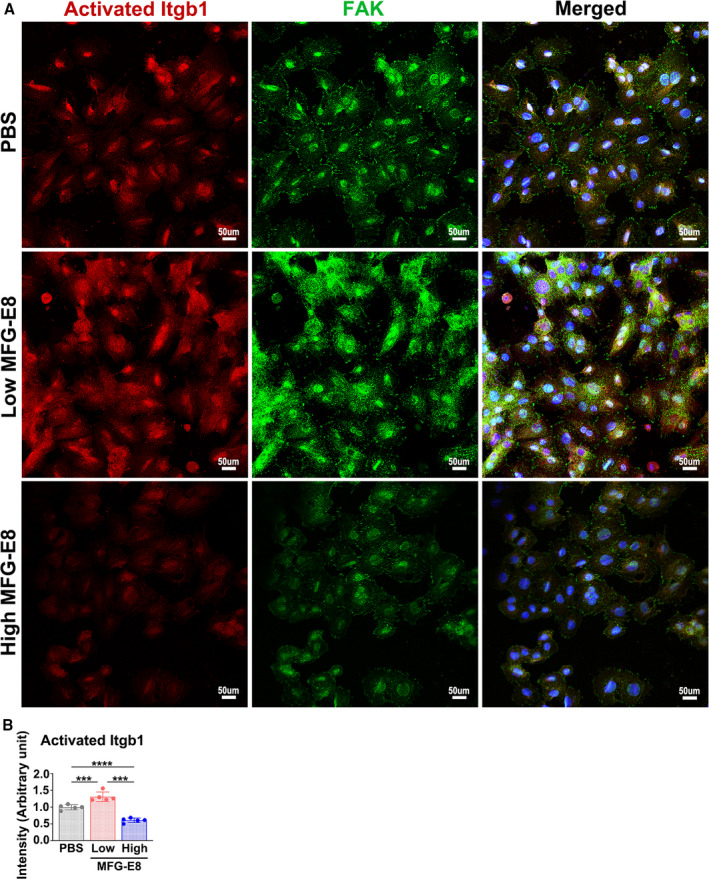
A10 vascular smooth muscle cells were stimulated with various doses of MFG‐E8 for 16 hours, followed by dual immunofluorescence of the activated state of β1 integrin (Itgb1) and focal adhesion kinase (FAK). Three independent experiments were performed, and each experiment was repeated with similar results. A, Representative immunofluorescence images showing activated β1 integrin and FAK in A10 cells treated with a low (10 ng/mL) or high (1000 ng/mL) concentration of recombinant MFG‐E8. The merged images present colocalization (yellow) of activated β1 integrin (red) and FAK (green). Bar, 50 μm. B, Fluorescence intensity of activated β1 integrin in A10 cells was quantitated using a representative experiment (n=5) as described in the Methods section. Data are presented as mean±SD. Each point is derived from an assessment of each picture field.***P<0.001 and ****P<0.0001, as obtained using 1‐way ANOVA followed by Tukey multiple comparisons test.
Figure 6. β1 integrin is involved in milk fat globule‐epidermal growth factor (MFG‐E8)–mediated actin‐related protein 2 (Arp2) expression and actin polymerization.
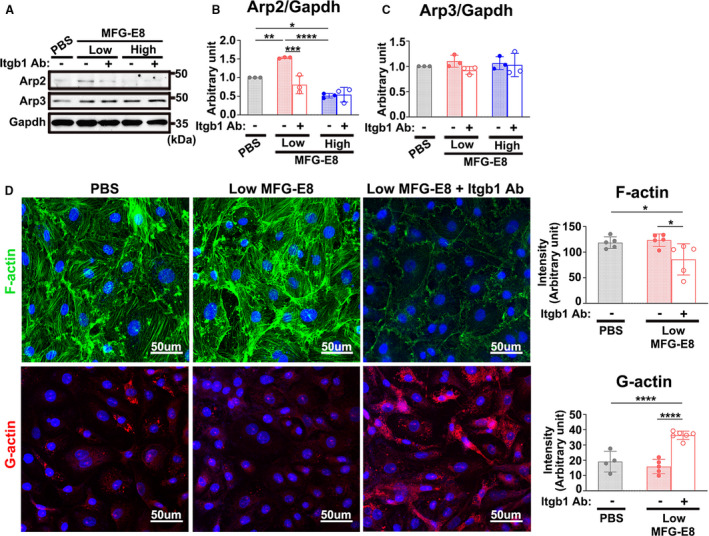
A through C, A10 vascular smooth muscle cells (VSMCs) incubated with either 50 μg/mL β1 inhibitory antibody (Itgb1 Ab) or isotype control 4 hours before treatment with either a low or high concentration of recombinant MFG‐E8 for 16 hours. A, Immunoblotting was performed to assess the protein expression of Arp2 and actin‐related protein 3 (Arp3) in A10 cells in a representative experiment. Quantitative analyses of Arp2 (B) and Arp3 (C) normalized to GAPDH were conducted (n=3). Data are presented as mean±SD. Three repeated experiments were performed. Each point is derived from each of the 3 repeated experiments. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, as obtained using 1‐way ANOVA followed by Tukey multiple comparisons test. D, Representative confocal fluorescent images of phalloidin for filamentous actin (F‐actin) (green) and DNase I for globular actin (G‐actin) (red) staining of A10 VSMCs stimulated with low (10 ng/mL) and high (1000 ng/mL) doses of MFG‐E8 for 16 hours. Bar, 50 μm. In a representative experiment, corresponding quantification of the mean fluorescence intensity of phalloidin for F‐actin (n=5) and that of DNase I for G‐actin (n=4–6) was performed. Data are presented as mean±SD. Each point is derived from an assessment of each picture field. **P<0.01 and ****P<0.0001, as obtained using 1‐way ANOVA followed by Tukey multiple comparisons test.
We next determined the downstream signaling molecule between β1 integrin and Arp2 after treatment with low‐dose MFG‐E8. Arp2/3 complex–mediated actin assembly is activated by the small Rho GTPase Rac1.10, 38 Several lines of evidence have indicated that Rac1 activation is promoted by β1 integrin–FAK signaling cascades,39 leading to the rearrangement of the actin cytoskeleton in VSMCs.40 Notably, a study indicated that Rac1 is required for Arp2 and Arp3 production.41 We therefore investigated whether Rac1 is the essential signaling component downstream to β1 integrin for MFG‐E8–mediated Arp2 expression. A10 VSMCs were treated with low and high doses of MFG‐E8 in the presence or absence of anti–β1 integrin antibody, and the Rac1 activity was evaluated using the G‐LISA assay. As shown in Figure 7A, low‐dose MFG‐E8 treatment significantly activated Rac1, whereas high‐dose MFG‐E8 treatment obviously attenuated it. Treatment of VSMCs with the β1 integrin–blocking antibody significantly attenuated Rac1 activity induced by low‐dose MFG‐E8 (Figure 7A), indicating that Rac1 is the downstream molecule to MFG‐E8–β1 integrin engagement. Additionally, no further downregulation of Rac1 activity was observed after the addition of the β1 integrin–blocking antibody to cells treated with high‐dose MFG‐E8. To further verify whether Rac1 is involved in MFG‐E8–mediated Arp2 expression, Rac1 was pharmacologically inhibited with an inhibitor, NSC23766,33 in A10 VSMCs 16 hours before treatment with various MFG‐E8 doses. Low‐dose MFG‐E8–induced Arp2 upregulation was significantly attenuated in A10 cells treated with NSC23766 (Figure 7B and 7C). A slight decrease in the Arp2 level was observed upon addition of NSC23766 to high‐dose MFG‐E8–treated A10 cells (Figure 7B and 7C). Taken together, these findings suggest that Rac1 is the downstream effector of MFG‐E8–induced β1 integrin activation and the β1 integrin–Rac1 signaling axis is involved in Arp2 upregulation induced by low‐dose MFG‐E8. By contrast, the inhibitory effect of high‐dose MFG‐E8 on Arp2 levels is associated with decreased β1 integrin engagement and Rac1 activation.
Figure 7. Rac1 is activated by milk fat globule‐epidermal growth factor (MFG‐E8)–β1 engagement and is involved in MFG‐E8–mediated actin‐related protein 2 (Arp2) expression.
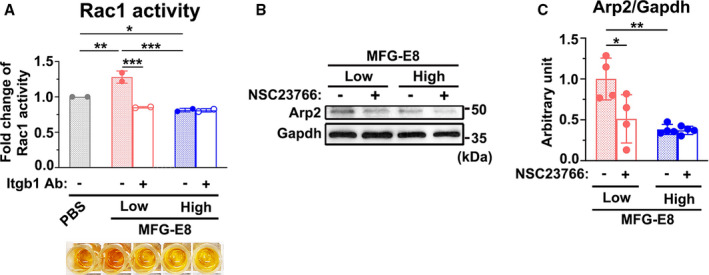
A, A10 vascular smooth muscle cells (VSMCs) incubated with either 50 μg/mL β1 inhibitory antibody (Itgb1 Ab) or isotype control 4 hours before treatment with either a low or high concentration of recombinant MFG‐E8 for 16 hours. Rac1 activity was measured using the G‐LISA (Cytoskeleton) assay. Data represent the averages of duplicate samples (n=2), and the error bars indicate the SD. *P<0.05, **P<0.01, ***P<0.001, as obtained using 1‐way ANOVA followed by Tukey multiple comparisons test. B and C, A10 cells were incubated with the Rac1 inhibitor NSC23766 (200 μM, Tocris) 16 hours before treatment with low and high doses of MFG‐E8 for 16 hours. B, Immunoblotting analysis of the Arp2 protein expression in A10 VSMCs was performed. C, Quantitative analysis of Arp2 normalized to GAPDH was performed (n=4). Data are presented as mean±SD. Four repeated experiments were performed. Each point is derived from each of the 4 repeated experiments. *P<0.05 and **P<0.01, as obtained using 1‐way ANOVA followed by Tukey multiple comparisons test.
High‐Dose MFG‐E8 Treatment Attenuates Ligation‐Induced Arp2 Expression and Alleviates Neointimal Hyperplasia in Injured Vessels by Limiting VSMC Migration
Injury‐induced VSMC migration and proliferation are the main contributors to neointimal formation.3 Our in vitro data revealed that high‐dose MFG‐E8 attenuated actin polymerization by reducing the Arp2 expression in VSMCs, suggesting that treatment of injured vessels with high‐dose MFG‐E8 might alleviate neointimal hyperplasia. To validate our inference, neointimal formation was induced in the CCAs of WT mice through vessel ligation, and high‐dose rMFG‐E8 (2 μg/mL) was periadventitially administered to the ligated arteries by using pluronic gel. We previously demonstrated that a recombinant peptide could be delivered periadventitially by using pluronic gel into the intima and media layers of a mouse carotid artery after surgery.25, 28, 31 Here, we found that the expression of MFG‐E8 was abundantly induced in the neointima and media of the CCAs 21 days after ligation (Figure S5). Ligation injury also significantly induced Arp2 expression in the intimal and medial VSMCs of CCAs compared with that in sham‐operated vessels, as evaluated through immunofluorescence (Figure 8A). Immunoblotting of Arp2 revealed a consistent upregulation in the protein level in the pooled CCAs of WT mice (Figure 8C). The application of high‐dose rMFG‐E8 to the ligated CCAs significantly attenuated ligation‐induced Arp2 expression in the arterial wall, as evidenced by immunofluorescence and immunoblotting results (Figure 8A and 8C). By contrast, immunofluorescence evaluation revealed no obvious alteration in Arp3 levels in the sham‐operated CCAs, ligated CCAs, and ligated CCAs treated with rMFG‐E8 (Figure 8B).
Figure 8. High‐dose milk fat globule‐epidermal growth factor (MFG‐E8) treatment attenuates ligation‐induced actin‐related protein 2 (Arp2) expression.
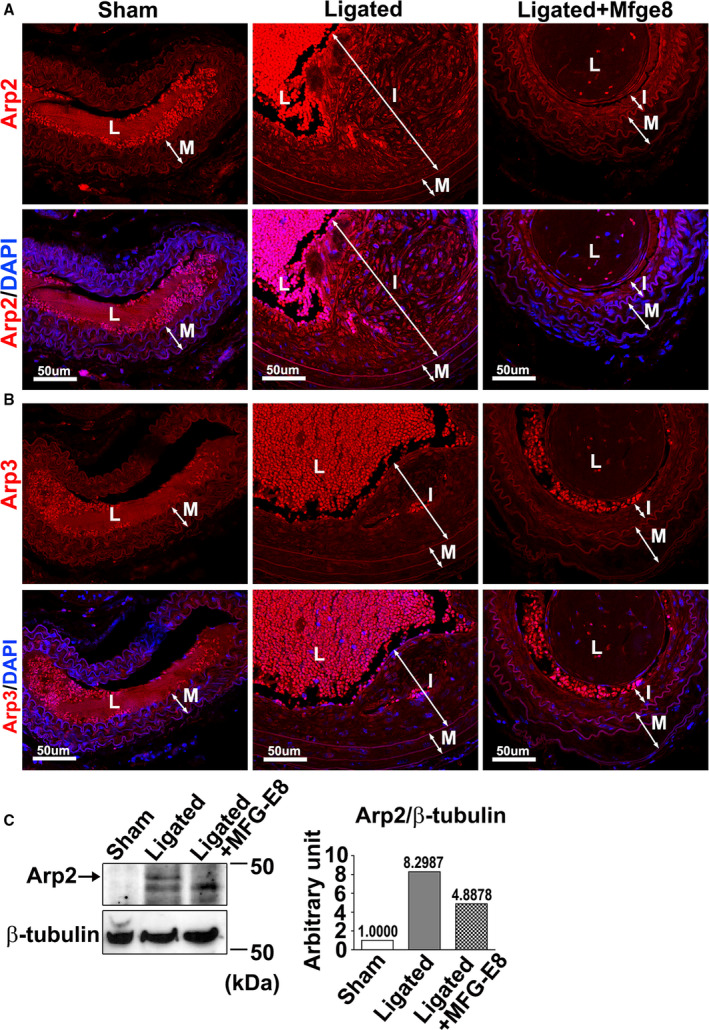
Common carotid arteries (CCAs) of wild‐type (WT) mice were ligated, and recombinant MFG‐E8 (2 μg/mL) was delivered using pluronic gel into the vessels. A and B, Representative immunofluorescence images display the expression of Arp2 (A) and Arp3 (B) in the sham‐operated CCAs, ligated CCAs, and ligated CCAs treated with high‐dose MFG‐E8 in WT mice 21 days after ligation. Lumen (L), neointima (I), and media (M) of the vessels are indicated. Bar, 50 μm. C, Protein levels of Arp2 were quantified through immunoblotting. Proteins were extracted from 10 pooled CCAs per group for Western blotting and examined using antibodies against Arp2. In the quantitative analysis, the Arp2 and actin‐related protein 3 (Arp3) levels were normalized to that of β‐tubulin. DAPI indicates 4',6‐diamidino‐2‐phenylindole.
We then assessed the activation of β1 integrin and Rac1 in the CCAs by immunostaining the vessels with the antibodies specific to activated β1 integrin and Rac1‐GTP, respectively. As shown in Figure 9, ligation injury increased the levels of activated β1 integrin (Figure 9A and 9B) and Rac1 (Figure 9C and 9D). Consistent with our in vitro results, high‐dose treatment with MFG‐E8 significantly reduced the levels of activated β1 integrin and Rac1 in the intima and media of the ligated vessels. We next determined whether high‐dose treatment with rMFG‐E8 attenuates neointimal hyperplasia as expected. The intima, but not the media, was obviously attenuated (Figure 9E and Figure S6), and the intima/media ratio (Figure 9E and 9F) was significantly decreased in the ligated CCAs treated with high‐dose rMFG‐E8 21 days after ligation compared with the untreated ligated arteries. To determine the mechanism through which high‐dose MFG‐E8 attenuates the intima area, we performed immunohistochemistry analysis for Ki‐67 to evaluate the proliferation of VSMCs, and negative control for the antibody in the vessel is shown in Figure S7. The number of Ki‐67(+) cells in the ligated WT CCAs treated with high‐dose MFG‐E8 was higher than that in the untreated WT vessels (Figure 9G and 9H). Notably, the Ki‐67(+) cells were crowded in the thinner intima of the MFG‐E8–treated CCAs (Figure 9G), suggesting that the migratory ability of these VSMCs was impeded. We then performed a transwell assay to determine whether high‐dose MFG‐E8 inhibited VSMC migration. The VSMC migration was significantly reduced in A10 cells treated with high‐dose MFG‐E8 (1000 ng/mL) in response to platelet‐derived growth factor‐BB compared with cells treated with a vehicle or low‐dose MFG‐E8 (10 ng/mL; Figure 9I). By contrast, low‐dose MFG‐E8 only slightly accelerated VSMC migration. Overall, these data suggest that high‐dose MFG‐E8 can attenuate ligation‐induced neointimal hyperplasia by restraining VSMC migration, which was associated with downregulated Arp2 expression and β1 integrin and Rac1 activation.
Figure 9. High‐dose milk fat globule‐epidermal growth factor (MFG‐E8) treatment attenuates ligation‐induced β1 integrin and Rac1 activation and alleviates neointimal hyperplasia after vascular injury by limiting vascular smooth muscle cell (VSMC) migration.
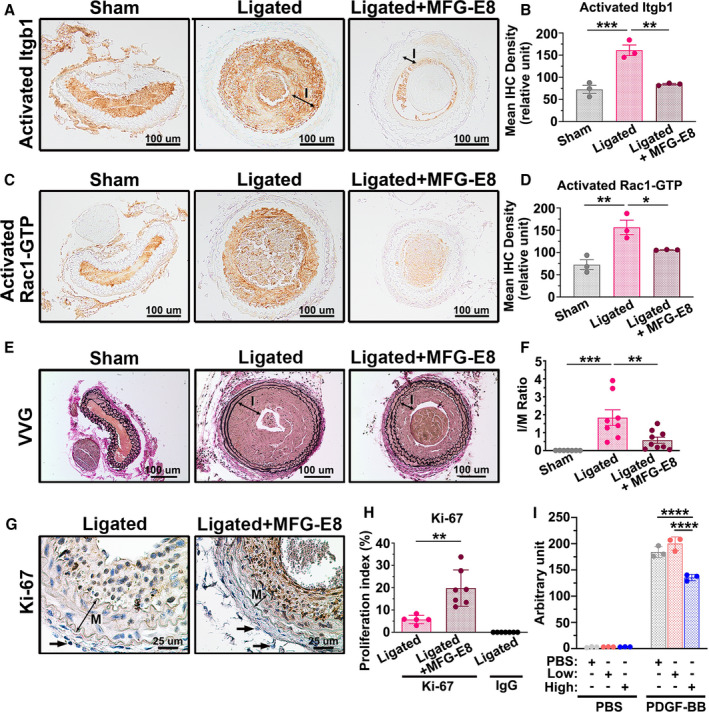
Recombinant MFG‐E8 (rMFG‐E8) (2 μg/mL) was delivered using pluronic gel into the carotid artery after complete ligation of the vessel. A, Representative immunohistochemistry images of activated β1 integrin (Itgb1) in sham‐operated common carotid arteries (CCAs), ligated CCAs, and ligated CCAs treated with high‐dose MFG‐E8 in wild‐type (WT) mice 21 days after ligation. Bar, 100 μm. B, Quantitative analysis of the immunostaining intensities of active β1 integrin in the intimal–medial area was performed (nmice=3 for each experimental group). Data are presented as mean±SEM. Each point is derived from an assessment of 3 sections of an individual animal. ** P<0.01 and ***P<0.001, as obtained using 1‐way ANOVA followed by Tukey multiple comparisons test. C, Representative immunohistochemistry photomicrographs of activated Rac1‐GTP in sham‐operated CCAs, ligated CCAs, and ligated CCAs treated with high‐dose MFG‐E8 in WT mice 21 days after ligation. Bar, 100 μm. D, Immunostaining intensity of the activated Rac1‐GTP in the intimal–medial area (nmice=3 for each experimental group) was quantified. Data are presented as mean±SEM. Each point is derived from an assessment of 3 sections of an individual animal. *P<0.05 and **P<0.01, as obtained using 1‐way ANOVA followed by Tukey multiple comparisons test. E, Representative images displaying the cross‐sectional areas of sham‐operated CCAs, ligated CCAs, and ligated CCAs treated with rMFG‐E8 in WT mice 21 days after ligation. Verhoeff–van Gieson (VVG) staining was performed on all sections. The intima (labeled I) in the ligated artery is indicated. Bar, 100 μm. F, Morphometric analysis of the intima/media (I/M) ratio was conducted 21 days after ligation (sham: nmice=7, ligated: nmice=8, ligated+MFG‐E8: nmice=9). Data are presented as mean±SEM. Each point is derived from an assessment of 3 sections of an individual animal. **P<0.01 and ***P<0.001, as obtained using 1‐way ANOVA followed by Tukey multiple comparisons test. G, Photomicrographs displaying immunostaining of Ki‐67 in cross sections of the ligated CCAs and ligated CCAs treated with rMFG‐E8 at 10 days after ligation. Negative control for the antibody in the intima–media of the vessel 10 days after ligation is shown in Figure S7. The media (labeled M) in the ligated artery is indicated. Bar, 25 μm. Arrows denote the fibroblasts in the adventitia. The similar sizes of the fibroblasts in ligated CCAs and MFG‐E8–treated CCAs affirm the same magnification in the 2 figures. H, Quantitative immunohistochemistry analysis of the proliferation index (Ki‐67[+] cells per total cell number) in the intima–media of the vessel 10 days after ligation (ligated: nmice=5, ligated+MFG‐E8: nmice=7, IgG: nmice=7). Data are presented as mean±SEM. Each point is derived from an assessment of 3 sections of an individual animal. **P<0.01, as obtained using the t test. I, A10 VSMCs were treated overnight with vehicle or various doses of rMFG‐E8. The cells were trypsinized and seeded on transwells with a pore size of 8 μm in 24‐well plates; thereafter, a total of 800 µL of PBS or platelet‐derived growth factor‐BB (PDGF‐BB) (10 ng/mL) was added to Dulbecco's Modified Eagle Medium (DMEM) in the lower chamber to induce VSMC migration. The A10 cells that migrated into the lower chambers were stained with calcein‐AM, and the fluorescence intensity was quantitated. Data represent the averages of triplicate samples of a representative experiment (n=3), and the error bars indicate the SD. ****P<0.0001, as obtained using 1‐way ANOVA followed by Tukey multiple comparisons test. IHC indicates immunohistochemistry; Itgb1, beta1 Integrin; and MFG‐E8, milk fat globule‐epidermal growth factor 8.
Discussion
In this study, we evaluated the role of MFG‐E8 as a mediator of actin polymerization involved in VSMC migration. MFG‐E8 knockout mice exhibited less intima–media thickening than did WT mice, indicating that MFG‐E8 is essential for VSMC proliferation and migration (Figure 1). We demonstrated that MFG‐E8 deficiency is accompanied by decreased Arp2 expression in carotid arteries and VSMCs (Figure 2). Notably, our data suggest that MFG‐E8 regulates Arp2 expression through biphasic, dose‐dependent modulation in VSMCs. Exogenous application of a low dose of rMFG‐E8 resulted in higher Arp2 expression in VSMCs (Figure 3); however, this dose was insufficient to enhance significant actin polymerization (Figure 4) and cell migration (Figure 9). By contrast, exogenous rMFG‐E8 application at a high concentration reduced Arp2 expression (Figure 3) and functionally attenuated actin assembly (Figure 4) and VSMC migration (Figure 9). Our in vivo experiments demonstrated that periadventitial delivery of high‐dose rMFG‐E8 into injured vessels ameliorated ligation‐induced neointimal hyperplasia (Figure 9), which further supports our in vitro finding that treatment with high‐dose MFG‐E8 impedes VSMC migration. Moreover, we further validated that β1 integrin engagement and downstream Rac1 activation were involved in Arp2 upregulation induced by low‐dose MFG‐E8 and that the inhibitory effects of high‐dose MFG‐E8 on Arp2 expression were associated with decreased β1 integrin engagement and Rac1 activation (Figures 5, 6, 7).
Together with 5 other subunits (actin‐related proteins 1 to 5), Arp2 and Arp3 form a 7‐subunit complex, the Arp2/3 complex,5, 6 which plays a critical role in the organization of the actin cytoskeleton involved in VSMC proliferation, migration, differentiation, and contraction.42, 43, 44, 45 A study demonstrated that MFG‐E8 induced intracellular movement of the Arp2/3 complex into the leading edge of lamellipodia, resulting in epithelial cell migration.26 However, whether MFG‐E8 modulates Arp2 and Arp3 expression has not been elucidated. In the present study, we revealed that high‐dose MFG‐E8 or knockout downregulated the expression of Arp2 but not that of Arp3 in VSMCs and carotid arteries (Figure 2). The lack of Arp2 compromises the integrity of the Arp2/3 complex, leading to the impairment of Arp2/3 complex–mediated actin assembly and VSMC migration. Notably, treatment with low‐dose MFG‐E8 only slightly enhanced actin polymerization (Figure 4) and VSMC migration (Figure 9), even when MFG‐E8 incubation increased Arp2 expression in A10 cells. We speculate that the excess production of Arp2 induced by exogenous MFG‐E8 did not result in the formation of additional Arp2/3 complexes because of the limited concentration of Arp3 and other proteins in VSMCs; therefore, more F‐actin and cell migration were not generated after MFG‐E8 treatment.
Rac1, a member of the Rho GTPase family, is required for lamellipodium‐driven cell migration.46, 47 Studies have demonstrated that Rac1 activation is biphasically dependent on the substratum concentration in the migrating fibroblasts and that reduced transient membrane protrusion with attenuated Rac1 activation is observed when cells migrate to a high concentration of substratum.48 In the present study, we revealed a biphasic relationship between Rac1 activation and MFG‐E8 concentration in VSMCs; the inhibitory effect of high‐dose MFG‐E8 on Rac1 activation may result in decreased VSMC migration attributable to Arp2 deficit (Figure 7A). Additionally, Arp2/3 complex–mediated actin polymerization in protruding lamellipodium is activated by the Rac1–WAVE complex signaling axis.46, 47 In addition to activation of the Arp2/3 complex, a recent study revealed that the knockout of small GTPase Rac1 with siRNA suppressed Arp2 and Arp3 expression in endothelial cells,41 indicating that Rac1 is required for Arp2 and Arp3 production. Coincidently, we proved that low‐dose MFG‐E8–induced Arp2 upregulation is mediated by Rac1 activation (Figure 7). By contrast, high‐dose MFG‐E8 attenuated Rac1 activation, leading to the downregulation of Arp2 and a subsequent decrease in VSMC migration. Our in vitro data revealed that β1 integrin was involved in the dose‐dependent mediation of Arp2 expression by MFG‐E8 (Figures 5 and 6). MFG‐E8 contains 2 N‐terminal epidermal growth factor (EGF)–like domains followed by 2 C‐terminal discoidin domains.13 The second EGF domain harbors an integrin‐binding RGD motif that binds to integrins.13 We infer that the engagement of low‐dose MFG‐E8 with β1 integrin can trigger a downstream effector, Rac1, to induce Arp2 expression. MFG‐E8 may form a dimer or oligomer through the antiparallel pairing of EGF domains.49 We postulate that exogenous high‐dose MFG‐E8 may form dimers or oligomers by itself or even sequester the endogenous MFG‐E8 that masks the RGD domain, preventing ligation to β1 integrin and subsequent Arp2 expression.
Actin remodeling plays a crucial role in regulating VSMC proliferation, migration, and contraction.43 Our present in vivo results demonstrate that the genetic deletion of MFG‐E8 impeded VSMC migration and proliferation (Figure 1). However, upon exogenous administration of high‐dose MFG‐E8 in the ligated carotid artery, only VSMC migration was inhibited, with VSMC proliferation unaffected. A study indicated that VSMCs treated with rMFG‐E8 exhibited dose‐dependent cell proliferation through αvβ5 integrin/extracellular signal‐regulated kinase 1/2 (ERK1/2) signaling,24 indicating that MFG‐E8 regulates VSMC proliferation through mechanisms other than actin reorganization. We postulate that although high‐dose MFG‐E8 impairs Arp2‐mediated actin polymerization, VSMC proliferation can be activated by other MFG‐E8–regulated signaling cascades such as the integrin‐mediated phosphorylation of ERK1/2. Our previous study demonstrated that exogenous application of MFG‐E8 (250 ng/mL) to aged VSMCs attenuated platelet‐derived growth factor‐BB–induced migration.25 Because high‐dose MFG‐E8 (1000 ng/mL) reduced VSMC migration in this study, we postulate that the migration of aged VSMCs is more sensitive to the concentration of MFG‐E8.
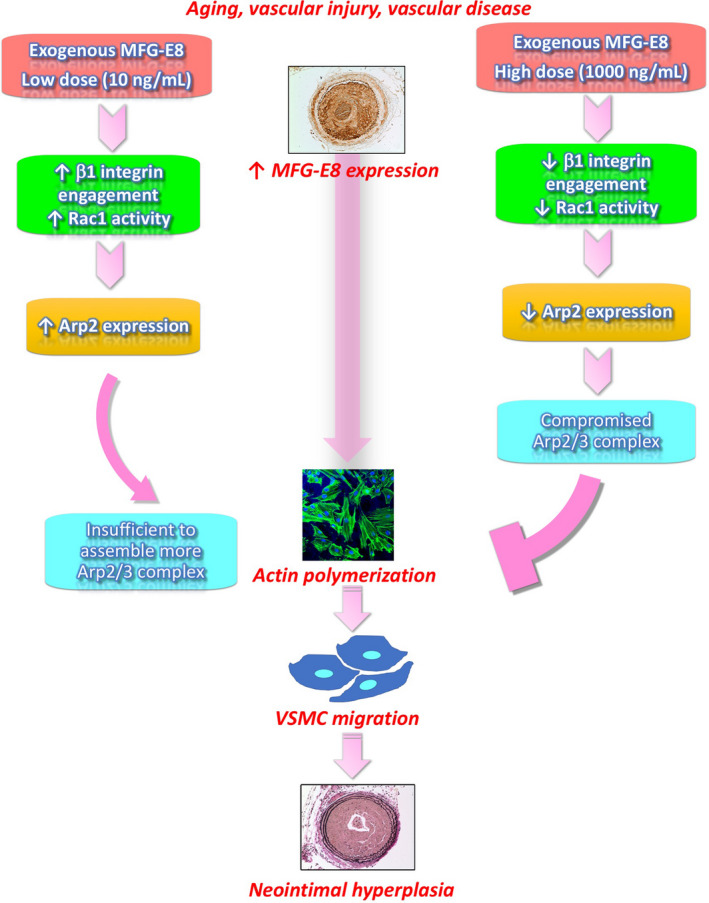
In summary, MFG‐E8 was identified to play a critical role in VSMC migration, and molecular links were established between MFG‐E8 and actin remodeling. We demonstrated the dose‐dependent effect of MFG‐E8 on the regulation of Arp2‐mediated assembly of actin cytoskeletons through β1 integrin–Rac1 signaling (Figure 10). We also revealed that the exogenous application of high‐dose MFG‐E8 to arteries attenuates injury‐induced neointimal hyperplasia. These findings suggest that high doses of MFG‐E8 have potential therapeutic application in treating vascular occlusive diseases.
Figure 10. Proposed model in which milk fat globule‐epidermal growth factor (MFG‐E8) regulates vascular smooth muscle cell (VSMC) migration and neointimal hyperplasia through mediation of actin‐related protein 2 (Arp2)–mediated actin assembly in a biphasic, dose‐dependent manner.
Sources of Funding
This work was supported by research grants from the Ministry of Science and Technology, Taiwan (108‐2320‐B‐182‐042 to T.‐H.L., 109‐2314‐B‐182A‐155 to P.‐H.C., and 102‐2320‐B‐182‐007‐MY3 and 105‐2320‐B‐182‐022 to H.‐Y.C.) and Chang Gung Memorial Hospital (CMRPD1J0291, CMRPD1J0292, and BMRPE27 to T.‐H.L. and CMRPD1F0261, CMRPD1I0101, CMRPD1I0102, CMRPD1I0103, and BMRPC00 to H.‐Y.C.).
Disclosures
None.
Supporting information
Figure S1–S7
Acknowledgments
We thank Dr Shigekazu Nagata (Osaka University) for providing MFG‐E8 knockout mice from Riken BioResource Center.
(J Am Heart Assoc. 2021;10:e06289. DOI: 10.1161/JAHA.121.020870.)
Supplementary Materials for this article are available at https://www.ahajournals.org/doi/suppl/.
For Sources of Funding and Disclosures, see page 18.
Contributor Information
Hou‐Yu Chiang, Email: tingheinlee@mail.cgu.edu.tw.
Pao‐Hsien Chu, Email: tingheinlee@mail.cgu.edu.tw.
Ting‐Hein Lee, Email: tingheinlee@mail.cgu.edu.tw.
References
- 1.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. DOI: 10.1152/physrev.00041.2003 [DOI] [PubMed] [Google Scholar]
- 2.Rensen S, Doevendans P, van Eys G. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J. 2007;15:100–108. DOI: 10.1007/BF03085963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerthoffer WT. Mechanisms of vascular smooth muscle cell migration. Circ Res. 2007;100:607–621. DOI: 10.1161/01.RES.0000258492.96097.47 [DOI] [PubMed] [Google Scholar]
- 4.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. DOI: 10.1016/S0092-8674(03)00120-X [DOI] [PubMed] [Google Scholar]
- 5.Goley ED, Welch MD. The ARP2/3 complex: an actin nucleator comes of age. Nat Rev Mol Cell Biol. 2006;7:713–726. DOI: 10.1038/nrm2026 [DOI] [PubMed] [Google Scholar]
- 6.Rotty JD, Wu C, Bear JE. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat Rev Mol Cell Biol. 2013;14:7–12. DOI: 10.1038/nrm3492 [DOI] [PubMed] [Google Scholar]
- 7.Welch MD, DePace AH, Verma S, Iwamatsu A, Mitchison TJ. The human Arp2/3 complex is composed of evolutionarily conserved subunits and is localized to cellular regions of dynamic actin filament assembly. J Cell Biol. 1997;138:375–384. DOI: 10.1083/jcb.138.2.375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L, Zhang Y, Lei Y, Wei Z, Li Y, Wang Y, Bu Y, Zhang C. Proline‐rich 11 (PRR11) drives F‐actin assembly by recruiting the actin‐related protein 2/3 complex in human non‐small cell lung carcinoma. J Biol Chem. 2020;295:5335–5349. DOI: 10.1074/jbc.RA119.012260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lv P, Zhang F, Yin YJ, Wang YC, Gao M, Xie XL, Zhao LL, Dong LH, Lin YL, Shu YN, et al. SM22α inhibits lamellipodium formation and migration via Ras‐Arp2/3 signaling in synthetic VSMCs. Am J Physiol Cell Physiol. 2016;311:C758–C767. DOI: 10.1152/ajpcell.00033.2016 [DOI] [PubMed] [Google Scholar]
- 10.Afewerki T, Ahmed S, Warren D. Emerging regulators of vascular smooth muscle cell migration. J Muscle Res Cell Motil. 2019;40:185–196. DOI: 10.1007/s10974-019-09531-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suraneni P, Rubinstein B, Unruh JR, Durnin M, Hanein D, Li R. The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J Cell Biol. 2012;197:239–251. DOI: 10.1083/jcb.201112113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swaney KF, Li R. Function and regulation of the Arp2/3 complex during cell migration in diverse environments. Curr Opin Cell Biol. 2016;42:63–72. DOI: 10.1016/j.ceb.2016.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raymond A, Ensslin MA, Shur BD. SED1/MFG‐E8: a bi‐motif protein that orchestrates diverse cellular interactions. J Cell Biochem. 2009;106:957–966. DOI: 10.1002/jcb.22076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oshima K, Yasueda T, Nishio S, Matsuda T. MFG‐E8: origin, structure, expression, functions and regulation. In: Wang P, ed. MFG‐E8 and Inflammation. Netherlands: Springer; 2014:1–31. [Google Scholar]
- 15.Li BY, Li XL, Cai Q, Gao HQ, Cheng M, Zhang JH, Wang JF, Yu F, Zhou RH. Induction of lactadherin mediates the apoptosis of endothelial cells in response to advanced glycation end products and protective effects of grape seed procyanidin B2 and resveratrol. Apoptosis. 2011;16:732–745. DOI: 10.1007/s10495-011-0602-4 [DOI] [PubMed] [Google Scholar]
- 16.Ait‐Oufella H, Kinugawa K, Zoll J, Simon T, Boddaert J, Heeneman S, Blanc‐Brude O, Barateau V, Potteaux S, Merval R, et al. Lactadherin deficiency leads to apoptotic cell accumulation and accelerated atherosclerosis in mice. Circulation. 2007;115:2168–2177. DOI: 10.1161/CIRCULATIONAHA.106.662080 [DOI] [PubMed] [Google Scholar]
- 17.Fu Z, Wang M, Gucek M, Zhang J, Wu J, Jiang L, Monticone RE, Khazan B, Telljohann R, Mattison J, et al. Milk fat globule protein epidermal growth factor‐8. Circ Res. 2009;104:1337–1346. DOI: 10.1161/CIRCRESAHA.108.187088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brissette MJ, Lepage S, Lamonde AS, Sirois I, Groleau J, Laurin LP, Cailhier JF. MFG‐E8 released by apoptotic endothelial cells triggers anti‐inflammatory macrophage reprogramming. PLoS One. 2012;7:e36368. DOI: 10.1371/journal.pone.0036368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu F, Li BY, Li XL, Cai Q, Zhang Z, Cheng M, Yin M, Wang JF, Zhang JH, Lu WD, et al. Proteomic analysis of aorta and protective effects of grape seed procyanidin B2 in db/db mice reveal a critical role of milk fat globule epidermal growth factor‐8 in diabetic arterial damage. PLoS One. 2012;7:e52541. DOI: 10.1371/journal.pone.0052541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z, Li BY, Li XL, Cheng M, Yu F, Lu WD, Cai Q, Wang JF, Zhou RH, Gao HQ, et al. Proteomic analysis of kidney and protective effects of grape seed procyanidin B2 in db/db mice indicate MFG‐E8 as a key molecule in the development of diabetic nephropathy. Biochim Biophys Acta Mol Basis Dis. 2013;1832:805–816. DOI: 10.1016/j.bbadis.2013.02.022 [DOI] [PubMed] [Google Scholar]
- 21.Cheng M, Li BY, Li XL, Wang Q, Zhang JH, Jing XJ, Gao HQ. Correlation between serum lactadherin and pulse wave velocity and cardiovascular risk factors in elderly patients with type 2 diabetes mellitus. Diabetes Res Clin Pract. 2012;95:125–131. DOI: 10.1016/j.diabres.2011.09.030 [DOI] [PubMed] [Google Scholar]
- 22.Wang M, Wang HH, Lakatta EG. Milk fat globule epidermal growth factor VIII signaling in arterial wall remodeling. Curr Vasc Pharmacol. 2013;11:768–776. DOI: 10.2174/1570161111311050014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang M, Monticone RE, McGraw KR. Proinflammation, profibrosis, and arterial aging. Aging Med. 2020;3:159–168. DOI: 10.1002/agm2.12099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang M, Fu Z, Wu J, Zhang J, Jiang L, Khazan B, Telljohann R, Zhao M, Krug AW, Pikilidou M, et al. MFG‐E8 activates proliferation of vascular smooth muscle cells via integrin signaling. Aging Cell. 2012;11:500–508. DOI: 10.1111/j.1474-9726.2012.00813.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiang HY, Chu PH, Lee TH. MFG‐E8 mediates arterial aging by promoting the proinflammatory phenotype of vascular smooth muscle cells. J Biomed Sci. 2019;26:61. DOI: 10.1186/s12929-019-0559-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bu HF, Zuo XL, Wang X, Ensslin MA, Koti V, Hsueh W, Raymond AS, Shur BD, Tan XD. Milk fat globule–EGF factor 8/lactadherin plays a crucial role in maintenance and repair of murine intestinal epithelium. J Clin Invest. 2007;117:3673–3683. DOI: 10.1172/JCI31841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, Nagata S. Autoimmune disease and impaired uptake of apoptotic cells in MFG‐E8‐deficient mice. Science. 2004;304:1147–1150. DOI: 10.1126/science.1094359 [DOI] [PubMed] [Google Scholar]
- 28.Lee TH, Sottile J, Chiang HY. Collagen inhibitory peptide R1R2 mediates vascular remodeling by decreasing inflammation and smooth muscle cell activation. PLoS One. 2015;10:e0117356. DOI: 10.1371/journal.pone.0117356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Satoh K, Matoba T, Suzuki J, O'Dell MR, Nigro P, Cui Z, Mohan A, Pan S, Li L, Jin ZG, et al. Cyclophilin A mediates vascular remodeling by promoting inflammation and vascular smooth muscle cell proliferation. Circulation. 2008;117:3088–3098. DOI: 10.1161/CIRCULATIONAHA.107.756106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee TH, Chen J, Miano JM. Functional characterization of a putative serine carboxypeptidase in vascular smooth muscle cells. Circ Res. 2009;105:271–278. DOI: 10.1161/CIRCRESAHA.109.199869 [DOI] [PubMed] [Google Scholar]
- 31.Chiang HY, Korshunov VA, Serour A, Shi F, Sottile J. Fibronectin is an important regulator of flow‐induced vascular remodeling. Arterioscler Thromb Vasc Biol. 2009;29:1074–1079. DOI: 10.1161/ATVBAHA.108.181081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rao J, Zhou ZH, Yang J, Shi YU, Xu SL, Wang B, Ping YF, Chen LU, Cui YH, Zhang X, et al. Semaphorin‐3F suppresses the stemness of colorectal cancer cells by inactivating Rac1. Cancer Lett. 2015;358:76–84. DOI: 10.1016/j.canlet.2014.12.040 [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Kunit T, Ciotkowska A, Rutz B, Schreiber A, Strittmatter F, Waidelich R, Liu C, Stief CG, Gratzke C, et al. Inhibition of prostate smooth muscle contraction and prostate stromal cell growth by the inhibitors of Rac, NSC23766 and EHT1864. Br J Pharmacol. 2015;172:2905–2917. DOI: 10.1111/bph.13099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiang HY, Chu PH, Lee TH. R1R2 peptide ameliorates pulmonary fibrosis in mice through fibrocyte migration and differentiation. PLoS One. 2017;12:e0185811. DOI: 10.1371/journal.pone.0185811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wehrle‐Haller B. The role of integrins in cell migration. In: Danen E, ed. Integrins in Development. Georgetown, TX: Landes Bioscience; 2005:25–48. [Google Scholar]
- 36.Labus J, Wöltje K, Stolte KN, Häckel S, Kim KS, Hildmann A, Danker K. IL‐1β promotes transendothelial migration of PBMCs by upregulation of the FN/α5β1 signalling pathway in immortalised human brain microvascular endothelial cells. Exp Cell Res. 2018;373:99–111. DOI: 10.1016/j.yexcr.2018.10.002 [DOI] [PubMed] [Google Scholar]
- 37.Khalifeh‐Soltani A, Ha A, Podolsky MJ, McCarthy DA, McKleroy W, Azary S, Sakuma S, Tharp KM, Wu N, Yokosaki Y, et al. α8β1 integrin regulates nutrient absorption through an Mfge8‐PTEN dependent mechanism. eLife. 2016;5:e13063. DOI: 10.7554/eLife.13063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang Y, Yi X, Kang C, Wu C. Arp2/3‐branched actin maintains an active pool of GTP‐RhoA and controls RhoA abundance. Cells. 2019;8:1264. DOI: 10.3390/cells8101264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boehm M, Krause‐Gruszczynska M, Rohde M, Tegtmeyer N, Takahashi S, Oyarzabal O, Backert S. Major host factors involved in epithelial cell invasion of Campylobacter jejuni: role of fibronectin, integrin Beta1, FAK, Tiam‐1, and DOCK180 in activating Rho GTPase Rac1. Front Cell Infect Microbiol. 2011;10:17. DOI: 10.3389/fcimb.2011.00017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sundberg LJ, Galante LM, Bill HM, Mack CP, Taylor JM. An endogenous inhibitor of focal adhesion kinase blocks Rac1/JNK but not Ras/ERK‐dependent signaling in vascular smooth muscle cells. J Biol Chem. 2003;278:29783–29791. DOI: 10.1074/jbc.M303771200 [DOI] [PubMed] [Google Scholar]
- 41.Tang Y, Zhao J, Shen L, Jin Y, Zhang Z, Xu G, Huang X. ox‐LDL induces endothelial dysfunction by promoting Arp2/3 complex expression. Biochem Biophys Res Commun. 2016;475:182–188. DOI: 10.1016/j.bbrc.2016.05.068 [DOI] [PubMed] [Google Scholar]
- 42.Tang DD, Anfinogenova Y. Physiologic properties and regulation of the actin cytoskeleton in vascular smooth muscle. J Cardiovasc Pharmacol Ther. 2008;13:130–140. DOI: 10.1177/1074248407313737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang D. Critical role of actin‐associated proteins in smooth muscle contraction, cell proliferation, airway hyperresponsiveness and airway remodeling. Respir Res. 2015;16:134. DOI: 10.1186/s12931-015-0296-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen L, DeWispelaere A, Dastvan F, Osborne WRA, Blechner C, Windhorst S, Daum G. Smooth muscle‐alpha actin inhibits vascular smooth muscle cell proliferation and migration by inhibiting Rac1 activity. PLoS One. 2016;11:e0155726. DOI: 10.1371/journal.pone.0155726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Albinsson S, Nordström I, Hellstrand P. Stretch of the vascular wall induces smooth muscle differentiation by promoting actin polymerization. J Biol Chem. 2004;279:34849–34855. DOI: 10.1074/jbc.M403370200 [DOI] [PubMed] [Google Scholar]
- 46.Ridley AJ. Rho GTPase signalling in cell migration. Curr Opin Cell Biol. 2015;36:103–112. DOI: 10.1016/j.ceb.2015.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sit ST, Manser E. Rho GTPases and their role in organizing the actin cytoskeleton. J Cell Sci. 2011;124:679–683. DOI: 10.1242/jcs.064964 [DOI] [PubMed] [Google Scholar]
- 48.Cox EA, Sastry SK, Huttenlocher A. Integrin‐mediated adhesion regulates cell polarity and membrane protrusion through the Rho family of GTPases. Mol Biol Cell. 2001;12:265–277. DOI: 10.1091/mbc.12.2.265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ensslin MA, Shur BD. Identification of mouse sperm SED1, a bimotif EGF repeat and discoidin‐domain protein involved in sperm‐egg binding. Cell. 2003;114:405–417. DOI: 10.1016/S0092-8674(03)00643-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1–S7