

Abstract
Background
Animal disease models represent the cornerstone in basic cardiac arrest (CA) research. However, current experimental models of CA and resuscitation in mice are limited. In this study, we aimed to develop a mouse model of asphyxial CA followed by cardiopulmonary resuscitation (CPR), and to characterize the immune response after asphyxial CA/CPR.
Methods and Results
CA was induced in mice by switching from an O2/N2 mixture to 100% N2 gas for mechanical ventilation under anesthesia. Real‐time measurements of blood pressure, brain tissue oxygen, cerebral blood flow, and ECG confirmed asphyxia and ensuing CA. After a defined CA period, mice were resuscitated with intravenous epinephrine administration and chest compression. We subjected young adult and aged mice to this model, and found that after CA/CPR, mice from both groups exhibited significant neurologic deficits compared with sham mice. Analysis of post‐CA brain confirmed neuroinflammation. Detailed characterization of the post‐CA immune response in the peripheral organs of both young adult and aged mice revealed that at the subacute phase following asphyxial CA/CPR, the immune system was markedly suppressed as manifested by drastic atrophy of the spleen and thymus, and profound lymphopenia. Finally, our data showed that post‐CA systemic lymphopenia was accompanied with impaired T and B lymphopoiesis in the thymus and bone marrow, respectively.
Conclusions
In this study, we established a novel validated asphyxial CA model in mice. Using this new model, we further demonstrated that asphyxial CA/CPR markedly affects both the nervous and immune systems, and notably impairs lymphopoiesis of T and B cells.
Keywords: asphyxia, cardiac arrest, immunology, immunosuppression, lymphocyte, lymphopenia
Subject Categories: Basic Science Research, Ischemia, Animal Models of Human Disease, Inflammation
Nonstandard Abbreviations and Acronyms
- CA
cardiac arrest
- CBF
cerebral blood flow
- GFAP
glial fibrillary acidic protein
- Iba1
ionized calcium‐binding adaptor molecule 1
- LDF
laser Doppler flowmetry
- MV
mechanical ventilation
- pCO2
partial pressure of CO2
- pO2
partial pressure of O2
- ptO2
tissue oxygen tension
- ROSC
return of spontaneous circulation
Clinical Perspective
What Is New?
A clinically relevant mouse model of asphyxial cardiac arrest (CA) and cardiopulmonary resuscitation (CPR) has been successfully established and validated in both young and aged mice.
After asphyxial CA/CPR, a subacute immunosuppression phenotype, including severe lymphopenia, is evident in both young and aged mice.
After asphyxial CA/CPR, lymphopoiesis of T and B cells is markedly impaired, which likely accounts for the post‐CA lymphopenia.
What Are the Clinical Implications?
Our new CA/CPR model can serve as a valuable mouse tool to investigate the pathophysiology specific to asphyxial CA, a major clinical CA scenario.
Our findings of the immune defects after CA/CPR may be directly relevant to an increased risk for infection and poor prognosis observed in patients after CA.
Cardiac arrest (CA) is a life‐or‐death emergency in which the heart ceases to beat and blood circulation abruptly stops.1 Other than timely resuscitation and postresuscitation therapeutic hypothermia treatment, there is no specific intervention to ameliorate post‐CA mortality and morbidity. This dismal situation attests to the necessity for more research in CA.
Animal disease models are the cornerstone of CA research because they provide the means to a better understanding of CA pathophysiology and to exploring new treatment strategies.2 CA is associated with various causes, resulting in different disease progression processes. Understandably, no single animal model of CA can adequately recapitulate all aspects of CA in patients, and not all causes can be precisely modeled to induce CA in animals. In current experimental CA research, the most widely used animal types are pig and rat.2 Two common causes of CA in humans that have been successfully modeled in these animals are ventricular fibrillation and asphyxia.2 Capitalizing on these models, animal research has significantly contributed to our improved understanding of CA pathophysiology and potential treatments. For example, such studies have shed considerable light on how therapeutic hypothermia affects various aspects of the organ response to CA, including the heart, brain, and immune system.3, 4, 5, 6, 7 However, pig and rat models have critical limitations, which include difficulties in genetically modulating specific genes or pathways to perform detailed mechanistic studies. Consequently, mouse models have been increasingly used in experimental CA research.8, 9
Almost all current mouse models of CA and resuscitation rely on potassium chloride (KCl)‐induced asystolic arrest followed by cardiopulmonary resuscitation (CPR) that involves epinephrine dosing, mechanical ventilation, and chest compression.2 Surprisingly, asphyxia, a major noncardiac cause of clinical CA, has not yet been closely modeled in the mouse. In contrast to the immediate asystole induced by KCl, the progression of asphyxia‐induced CA causes a gradual increase in arterial CO2 and a decrease in oxygen delivery to all organs, leading to hypercapnia and tissue hypoxia in a prearrest period. This key difference in disease progression between asphyxial arrest and sudden CA results in distinct brain injury patterns, neurologic outcome, and dysfunction in various organs, even from equivalent arrest durations.10 Hence, to be specific to asphyxial CA, investigation must be conducted using a relevant animal model.10, 11, 12
In this study, we developed and characterized a novel validated murine asphyxial CA model. Because the immune response is a major component of the post‐CA syndrome,13, 14, 15, 16 we further performed a detailed analysis of the immune response using this new model. Importantly, the analysis revealed severely impaired T and B lymphopoiesis after asphyxial CA and resuscitation.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals
All experimental procedures were approved by the Duke University Animal Care and Use Committee. Young adult (2–4 months old) male C57Bl/6 mice were purchased from the Jackson Laboratory, and aged (21–22 months old) male C57Bl/6 mice were obtained from the National Institute of Ageing (Bethesda, MD). All animals had free access to food and water in a room maintained on a 14:10‐hour light/dark cycle. The online tool Quickcalcs was used to randomize animals for group assignments.
Surgical Procedures
The overall procedure sequence is shown in Figure 1A. First, after anesthesia induction with 5% isoflurane, mice were endotracheally intubated with a 20‐gauge intravenous catheter (noncuffed) and mechanically ventilated with a mixture of 1.5% isoflurane in 30% oxygen balanced with nitrogen. We used volume/pressure–adjusted controlled ventilation (RoVent Jr. rodent ventilator; Kent Scientific, Torrington, CT), automatically adjusted to the actual body weight of the animal. Usual respiratory rates were ≈145 breaths per minute, with peak pressures between 9 and 11 cm H2O, tidal volumes of 0.15 to 0.2 mL (≈6.6 mL/kg body weight), and minute volumes of 22 to 28 mL/min, achieving minimally invasive, pressure and volume feedback, weight‐adjusted ventilation. Mice were placed in the supine position. A 1‐cm skin incision was made in the right groin to expose the femoral artery and vein, which were cannulated with PE‐10 tubing for arterial blood pressure and blood gas measurements, and drug administration. Heparin (1:10, 50 μL, 5 U) was given through the arterial line. Mean arterial blood pressure (MAP) was measured through a digital blood pressure transducer connected to a Bridge Amp (ADInstruments, Dunedin, New Zealand). Of note, for experiments with behavioral tests, a right jugular vein was cannulated with PE‐10 tubing for drug administration, and blood pressure was not measured to preserve the femoral vessels. ECG needle probes were placed in both forearms and the left hind limb. The ECG was recorded via Animal Bio Amp (ADInstruments). Rectal temperature (monitored with a mouse rectal temperature probe) was maintained at 37.0±0.2°C with a heat lamp and a heating pad before CA. Furthermore, a needle probe was inserted under the right temporal muscle to monitor brain temperature. Of note, we have consistently observed that using this setup, brain temperature is 0.5 to 1°C higher than rectal temperature in healthy animals. Thus, we decided to actively maintain brain temperature at 38.0±0.2°C during the CA period using a homemade tubing coil with circulating warm water. The circular coil was made with 8 to 9 circles of the tubing (the internal diameter: 2 cm; shown in Figure 1B). Throughout the entire procedure, ECG, blood pressure, and temperature were continuously monitored, and values were recorded into a computer using PowerLab 8/35 (ADInstruments).
Figure 1. Development of a novel murine asphyxial cardiac arrest (CA)/cardiopulmonary resuscitation (CPR) model.

A, The procedure sequence of the asphyxial CA/CPR model. B, Experimental setup diagram. A photo of our homemade tubing coil is shown. C, A representative trace of mean arterial pressure (MAP) before, during, and after CA. D, Representative traces of ECG at different stages of asphyxial CA/CPR. ABG indicates arterial blood gas; epi, epinephrine; FiO2, fraction of inspired oxygen; MAP, mean arterial pressure; MV, mechanical ventilation; and ROSC, return of spontaneous circulation.
Asphyxial CA was induced by abruptly changing the inspiratory gas mixture to 100% nitrogen. The mouse head was then placed into the tubing coil. At CA onset (ie, when the MAP decreased below 20 mm Hg), within ≈2.5 minutes, anesthesia (isoflurane), lung ventilation, and body heating lamp/heating pad were discontinued immediately. The mouse head remained in the tubing coil with brain temperature maintained at 38.0±0.2°C during CA, whereas the body temperature was allowed to spontaneously decrease. At 5 or 7 minutes following CA onset, the tubing coil was removed, and resuscitation was started by chest compression and epinephrine administration (Figure 1A). Chest compression was performed using a single finger at ≈300 strokes per minute. Intravenous epinephrine infusion was given with a bolus injection (100 μL of 40 μg/mL containing 5 U heparin) followed by 5 minutes of continuous infusion (40 μg/mL epinephrine) at the rate of 20 μL/min. The total dosage of epinephrine was ≈320 μg/kg for young adult mice (based on a body weight of 25 g) or 267 μg/kg for aged mice (based on a body weight of 30 g). Mechanical ventilation with 100% oxygen was started simultaneously with chest compressions (Figure 1A). When return of spontaneous circulation (ROSC) was achieved, defined as appearance of stable ECG sinus rhythm and MAP >60 mm Hg, chest compressions were stopped. If ROSC could not be achieved within 4 minutes after CPR onset, resuscitation was abandoned. After ROSC, mouse body temperature continued to be monitored and was maintained at 32.0±0.2°C using a heating pad and lamp. Of note, we also found that if we tried to actively reheat the animal back to 37°C during the recovery after ROSC, it required continuous heating and most mice died on day 0 or day 1 after CA. When mice became hemodynamically stable and recovered spontaneous ventilation, the catheters were removed. At this time point, mouse brain temperature was ≈33 to 34°C. Mice were then disconnected from the ventilator and transferred into a thermal incubator (32.0°C) to prevent hypothermia and help mice rewarm. When mice showed recovery of the righting reflex in the incubator, the trachea was extubated. During this incubation period, rectal temperature increased from 32°C to 37°C in ≈20 minutes and then slowly decreased again, and at the end of the incubation, the temperature was ≈35 to 36°C. After 2 hours in the incubator, mice were then returned to their home cages. Sham‐operated mice were subjected to the same surgical procedures until the point at which the femoral vessels or the jugular vein were exposed. For the postsurgical care, mice were given saline (0.5 mL) via subcutaneous injection daily to prevent dehydration, and soft food and extra bedding were placed in the cage.
Simultaneous Tissue Oxygen and Cerebral Blood Flow Measurements
Mice were placed in a stereotaxic frame, a midline incision over the skull was made, and scalp and periosteum were pulled aside. Burr holes were drilled under saline cooling over the left hemisphere at the following coordinates (millimeters from bregma and midline): (1) posterior 2, lateral 2 for monitoring tissue oxygen tension (ptO2) in the cortex with an oxygen electrode (Clarke‐style, 25‐μm tip, Picoammeter PA 2000; Unisense, Arhus, Denmark), and (2) posterior 5, lateral 2 for cerebral blood flow (CBF) via a laser Doppler flowmeter (VMS‐LDF1, 0.48‐mm tip diameter; Moor Instruments, Wilmington, DE). Oxygen measurements were made after standard calibration and stable baseline was achieved, and a second calibration was obtained at biological zero, after the animal had been euthanized. Because we were unable to perform CPR with this measurement setup, data were obtained only before and during CA.
Laser Speckle Flowmetry
Laser speckle imaging of the brain was performed using a full‐field laser perfusion imager RFLS III according to the manufacturer's instructions (RWD Life Science, Shenzhen, China).8 Mice were subjected to CA/CPR as described above. In addition, a midline scalp incision was made to expose the skull for laser speckle imaging by the imager positioned directly above the skull. The following imaging parameters were used: a display rate of 25 Hz, time constant of 1 second, camera exposure time of 10 milliseconds, camera frame rate of 37.59, laser intensity of 60 mA, and resolution of 2048×2048.
Functional Tests
Three days after CA/CPR, behavioral tests were performed by experimenters who were blinded to group assignments, whenever possible.16, 17
Open Field
Mice were gently placed into a square arena (50×50 cm; CleverSys, Reston, VA) and allowed to move freely. Spontaneous locomotor activity was recorded for 10 minutes, with a 10‐second delay. The distance traveled was calculated using Topscan software (CleverSys).
Rotarod Test
Mice were trained for 3 days before surgery. On the test day, mice were gently placed on the rod, and 15 seconds later, the machine (Med Associates, Fairfax, VT) was turned on to accelerate from 4 to 40 rpm over 300 seconds. The time at which the mouse fell off the rolling rod was recorded as the latency to fall. Three consecutive trials were conducted for each mouse, with an interval of 15 minutes between trials.
Neurologic Scoring
The performance of mice on the rope, bar, and screen was scored as 0 to 3, respectively, and the total neurologic score was computed (9 points=no deficits and 0 points=severe injury).
Immunofluorescent Histochemistry
Mice were transcardially perfused with 20 mL of cold saline followed by 20 mL of 4% paraformaldehyde. The brain was then removed and postfixed in the same fixative for 24 hours, followed by immersion in 30% sucrose at 4°C for 2 to 3 days. Coronal sections (30 μm) from the area located around 2.06 mm posterior to bregma were subsequently obtained using a cryostat. Sections were rinsed in PBS 3 times, and then blocked with 2% goat serum in PBS containing 0.2% Triton X‐100 for 2 hours at room temperature. Next, the sections were incubated overnight at 4°C with the primary antibodies: rabbit anti‐GFAP (glial fibrillary acidic protein) (1:300; Millipore Burlington, MA) and rabbit anti‐Iba1 (ionized calcium‐binding adaptor molecule 1) (1:300; Wako, Richmond, VA). After extensive washing, the sections were incubated with fluorescent secondary antibodies for 2 hours at room temperature. Images were captured on a Zeiss fluorescence microscope (Carl Zeiss, Oberkochen, Germany).
Cytokine Enzyme‐Linked Immunosorbent Assay
Blood samples were collected by cardiac puncture under anesthesia. For serum preparation, blood was allowed to clot for 30 minutes at room temperature followed by centrifugation (2000g) for 20 minutes at 4°C. Serum was collected and stored at −20°C. Quantification of interleukin‐1β in serum samples was performed using ELISA MAX Sets (BioLegend, San Diego, CA).
Cell Preparations and Flow Cytometry
Our standard protocols, as described previously,16 were followed. Single‐cell suspensions were obtained from the spleen, thymus, bone marrow, and brain. Spleens and thymi were triturated and filtered through a 70‐μm filter to obtain single‐cell suspensions. Red blood cells were lysed with 1×Red Blood Cell lysis buffer (BioLegend). Bone marrow from the femurs was flushed with FACS buffer (PBS+2% FBS) to collect single cells.
To collect immune cells from the brain, brain tissue without olfactory bulbs and cerebellum was harvested and minced into small pieces in ice‐cold DMEM medium supplemented with 2% FCS on ice. These small pieces were then incubated with collagenase D (1 mg/mL) and DNAse I (1 mg/mL) (Sigma‐Aldrich, St. Louis, MO) for 45 minutes at 37ºC, and the resultant homogenate was passed through a 70‐μm cell strainer. The filtered cells were then resuspended in a 70%/30% Percoll gradient (GE Healthcare Life Sciences, Marlborough, MA), followed by centrifugation at 500g. Immune cells at the interphase layer were collected. Total cell numbers were determined using a hemocytometer with trypan blue staining method for cell viability.
For flow cytometry, single cells were stained with the following antibodies (all from BioLegend) for 30 minutes at 4°C in 100‐μL FACS buffer. T, B, and natural killer cells were stained with the following fluorescently labeled antibodies: CD45‐FITC (103108), CD3‐APC (100312), CD4‐PE/cy7 (100422), CD8‐APC/cy7 (100714), CD‐NK1.1‐PE (108708), and CD19‐PE/cy5 (115510). Neutrophils and macrophages were stained with the following antibodies: CD45‐PE (103106), F4/80‐PE/cy5 (123112), Ly6G‐FITC (127606), and CD11b‐PE/cy7 (101216). For B‐cell progenitor staining, single‐cell suspensions were first stained with PE‐conjugated anti‐mouse antibody lineage cocktail including CD3 (100205), CD90.2 (140307), NK1.1 (108707), TER119 (116207), CD11b (101207), Ly6G (127607), CD115 (135505), and CD11c (117307). This was followed by a second staining with the antibodies: CD93‐FITC (136508) and B220‐BV711 (103222). Flow cytometry data were acquired on FACS Canto (BD Biosciences, San Jose, CA), and analyzed using FlowJo (Ashland, OR) software.
Statistical Analysis
Sample sizes were determined on the basis of our pilot studies. Because we compared between sham and CA groups, a relatively small sample size was required. Prism 8 software (GraphPad, La Jolla, CA) was used for data analysis. Data are presented as mean±SEM or median. The unpaired and paired Student t test or Mann‐Whitney U test was used to compare 2 groups. To compare >2 groups, 1‐way ANOVA with post hoc Holm‐Sidak correction for multiple comparisons was performed. The level of significance was set at P<0.05.
Results
Development and Evaluation of a Novel Murine Asphyxial CA Model
Our asphyxial CA mouse model and representative patterns of blood pressure, ECG, cerebral CBF, and ptO2 during the procedure are shown in Figures 1 and 2. Unlike rat asphyxial CA models,5 a muscle relaxant (eg, vecuronium) was not used in our model. Instead, asphyxia was induced by just switching from a 30% O2/70% N2 mixture to 100% N2 as the inspiratory gas for mechanical ventilation under anesthesia. We found that this was sufficient to induce progressive bradycardia with a sudden blood pressure reduction, pulseless electrical activity, and eventual asystole. Within ≈2.5 minutes, the MAP had decreased to <20 mm Hg, the threshold for defining CA onset in our model. Moreover, adapted from mouse KCl‐mediated CA models,16, 17, 18 a water‐circulating coil was used to maintain the brain temperature (38.0±0.2°C) during CA in this model, which helped to aggravate post‐CA brain injury and thus produce neurologic deficits that were readily measurable while allowing animal survival. Lastly, we calculated the dose of epinephrine based on a standard epinephrine dose used in humans. For adult patients with CA, the standard epinephrine dose is 1 mg every 3 to 5 minutes. Using an algometric scaling approach, we estimated that the mouse equivalent standard epinephrine dose is 4 μg (based on a body weight of 25 g).19
Figure 2. Cerebral blood flow and brain tissue oxygen in our asphyxial cardiac arrest (CA)/cardiopulmonary resuscitation model.

A, Representative images of laser speckle flowmetry depict cerebral blood flow at baseline and during CA (n=3). B, A representative screenshot depicting sequence of early events after start of 100% nitrogen (N2) ventilation (n=3). LDF indicates laser Doppler flowmetry; ptO2, tissue oxygen tension; and V, volt.
To initially characterize this model, CPR time, arterial blood gas, MAP, and ECG were monitored. It took an average of 2.24±0.61 minutes (mean±SD) of CPR to achieve ROSC. The physiologic parameters from arterial blood gas measurement (n=6 per group; Table) demonstrated that pH, partial pressure of CO2, and partial pressure of O2 were significantly changed at 20 minutes after ROSC. As expected, CA caused metabolic acidosis, as indicated by a decrease in pH and an increase in partial pressure of CO2. Because mice were ventilated with 100% O2 during resuscitation, the values of partial pressure of O2 were significantly higher than the baseline. Of note, arterial blood gas was not further measured beyond 20 minutes after ROSC. Figure 1C and 1D show typical patterns of arterial blood pressure monitoring values and ECG during the procedure. Furthermore, we monitored real‐time changes in blood flow in whole brain using laser speckle contrast imaging. As shown in Figure 2A, a complete cessation of cerebral blood flow in the brain was evident, indicating success of the circulatory arrest in this model. More importantly, we used another cohort of mice to further demonstrate that brain ptO2 decreased within 45 to 55 seconds after 100% nitrogen ventilation to ≈0 mm Hg, indicating primary asphyxia/hypoxia. Then it took ≈100 seconds for CBF to decrease to almost 0, depicting ischemia, which was accompanied with the MAP below 20 mm Hg and a complete stop of circulation (n=3; Figure 2B).
Table 1.
Arterial Blood Gas Parameters Before CA and After ROSC
| Before CA, Baseline | After ROSC, 20 min | P Value, n=6 | |
|---|---|---|---|
| pH | 7.34±0.02 | 6.71±0.05 | <0.0001 |
| pCO2, mm Hg | 36.90±2.34 | 54.35±6.11 | 0.018 |
| pO2, mm Hg | 143.67±7.78 | 231.83±34.37 | 0.045 |
| HCO3, mmol/L | 19.40±0.96 | 6.63±0.83 | <0.0001 |
| TCO2, mmol/L | 19.17±0.87 | 8.33±0.96 | 0.0003 |
| SO2, % | 99.17±0.17 | 97.50±0.85 | 0.080 |
CA indicates cardiac arrest; HCO3, bicarbonate; pCO2, partial pressure of CO2; pO2, partial pressure of O2; ROSC, return of spontaneous circulation; SO2, O2 saturation; and TCO2, total CO2.
Then, we used a cohort of young adult mice to evaluate functional deficits after CA/CPR. In this experiment, a total of 12 mice were subjected to CA/CPR. Two mice failed to achieve ROSC, and were excluded for analysis. The mortality rate was 20% (2/10) in the CA/CPR group during the 3‐day observation period. On day 3 after CA, the body weight loss in the CA group was 15.31% of baseline. Compared with sham‐operated mice (n=7), the surviving CA/CPR mice (n=8) showed significant functional deficits in all 3 behavioral tests (Figure 3). Specifically, CA/CPR mice exhibited significantly lower neurologic scores and impairment in rotarod performance and spontaneous locomotor activity (open field test).
Figure 3. Functional deficits after asphyxial cardiac arrest (CA)/cardiopulmonary resuscitation (CPR) in young adult mice.
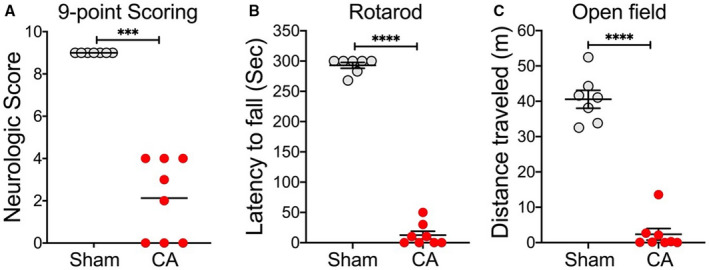
Aged mice were subjected to 7 minutes CA or sham surgery. Functional outcome assessments on day 3 after CA/CPR included neurologic scores (A), rotarod test (B), and open field test (C). Data are presented as median or mean±SEM (n=7–8/group). ***P<0.001. ****P<0.0001.
Application of the Asphyxial CA/CPR Model in Aged Mice
To test whether this new model is applicable in aged animals as well, we initially performed 7 minutes of CA in aged mice. However, most of the mice failed to achieve ROSC or died shortly after resuscitation. Thus, we shortened the CA duration to 5 minutes. With this CA duration, the majority of the aged mice can be successfully resuscitated after CA. Thus, we subjected a cohort of 10 aged mice to this CA condition. Two mice failed to achieve ROSC and were excluded for analysis. The mortality rate was 37.5% (3 out of 8) over 3 days after CA, which is considered acceptable for aged animals. On day 3 after CA, the body weight loss in the CA group was 9.71% of baseline. We then evaluated functional deficits after this short CA duration in aged mice. After CA/CPR, aged mice exhibited significant impairment in all 3 behavioral tests compared with aged sham mice (n=5 per group; Figure 4), demonstrating the suitability of this model to study the effects of aging in asphyxial CA.
Figure 4. Functional deficits after asphyxial cardiac arrest (CA)/cardiopulmonary resuscitation (CPR) in aged mice.

Mice were subjected to 5 minutes CA or sham surgery. Functional outcome assessments on day 3 after CA/CPR included neurologic scores (A), rotarod test (B), and open field test (C). Data are presented as median or mean±SEM (n=5/group). **P<0.01. ***P<0.001. ****P<0.0001.
Immune Responses After Asphyxial CA/CPR
After having successfully established and validated our new murine asphyxial CA/CPR model, we set out to examine the post‐CA immune response after asphyxial CA, because the immune response after resuscitation is a key component of post‐CA syndrome, and it has not yet been systemically assessed in the context of asphyxial CA. We thus performed a relatively comprehensive characterization of the post‐CA immune response in young adult mice. Then, we further used aged mice to determine whether aging has any major effect on the key immune changes observed in young adult mice after CA/CPR.
We first analyzed neuroinflammation in the brain, because of its critical importance in CA outcome. Consistent with other CA/CPR models, astrocytes and microglia were markedly activated, as confirmed by increased immunostaining signals of GFAP and Iba1, respectively (Figure 5A through 5D). We also analyzed the infiltration of peripheral immune cells into the post‐CA brain. In agreement with our recent finding in a KCl‐mediated CA model,16 most of the infiltrating immune cells were monocytes and neutrophils (Figure 5E), and there was no evidence that the number of T or B cells was increased in the post‐CA brain on day 3 after asphyxial CA/CPR.
Figure 5. Immune response in the brain after asphyxial cardiac arrest (CA)/cardiopulmonary resuscitation (CPR).
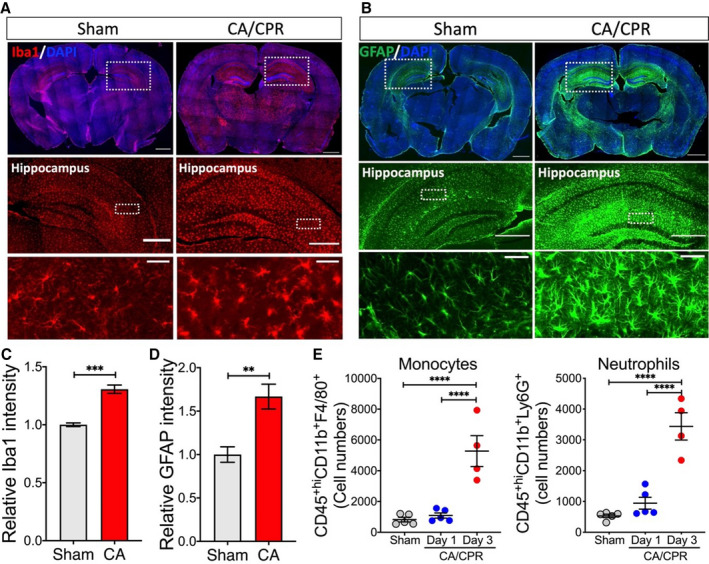
Young adult mice were subjected to 7 minutes CA or sham surgery. Immune response in the brain was evaluated on day 3 after CA/CPR. A and B, Representative images of immunofluorescence staining for Iba1 (ionized calcium‐binding adaptor molecule 1, microglial marker) (A) and GFAP (glial fibrillary acidic protein, astrocyte marker) (B), with enlarged images shown below. C and D, Relative immunofluorescence signal in the hippocampus regions. Iba1 (C) and GFAP (D) signals in the hippocampus from both hemispheres were measured (2 sham mice and 3 CA mice). E, Quantification of infiltrating monocytes/macrophages (CD45+hiCD11b+F4/80+) and neutrophils (CD45+hiCD11b+Ly6G+) in the post‐CA brain (n=4–5/group). Data are presented as mean±SEM. **P<0.01. ***P<0.001. ****P<0.0001. DAPI indicates 4′,6‐diamidino‐2‐phenylindole.
Next, we examined changes in the immune system after CA/CPR. As a marker of acute inflammatory response after CA, serum levels of interleukin‐1β were significantly increased on day 1 after asphyxial CA/CPR, consistent with previous findings.16 Interestingly, the levels of interleukin‐1β returned to baseline on day 3 after CA (Figure 6A), supporting a shift from an acute proinflammatory immune response to subacute immunosuppression, as proposed by our recent study.16 On day 3 after CA/CPR, we observed marked atrophy and cell loss in both the spleen and thymus, 2 key organs in maintaining the body's immune homeostasis (Figure 6B and 6C). Specifically, total cell numbers decreased from 43.16±4.49×106 to 11.68±2.70×106 and from 14.39±1.81×106 to 0.43±0.06×106 in the spleen and thymus, respectively. Similar changes were observed in aged mice (Figure 6D). Of note, as expected, the size of the thymus was much smaller in aged mice than in young adult mice. In general, these findings are consistent with what we discovered in our KCl‐mediated CA model,16 thus strongly indicating that immunosuppression is a common immunologic consequence after CA/CPR.
Figure 6. Lymphopenia after asphyxial cardiac arrest (CA)/cardiopulmonary resuscitation (CPR).
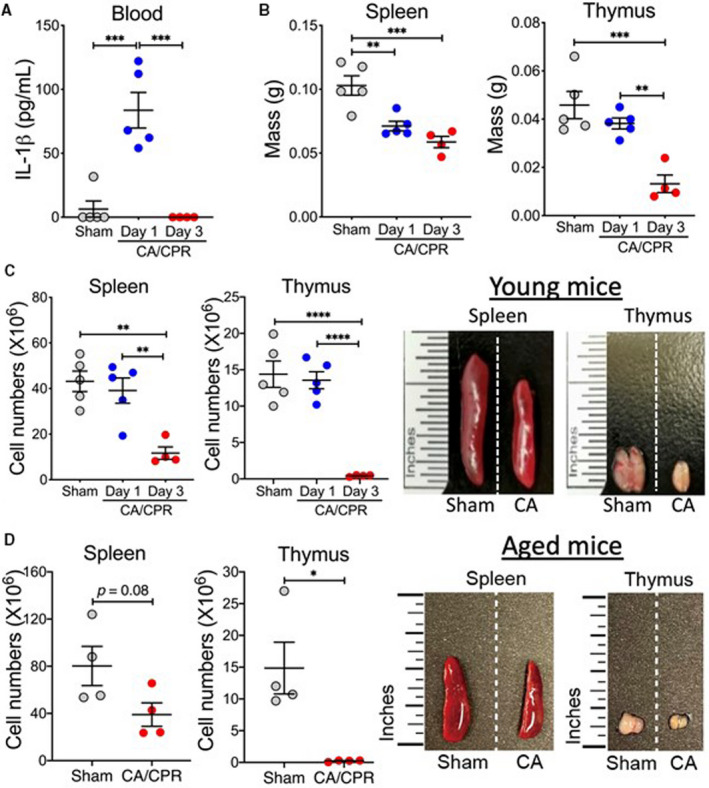
A through C, Young adult mice were subjected to 7 minutes CA or sham surgery. Analyses were performed on day 1 and day 3 after CA/CPR. A, Interleukin (IL)‐1β. Levels of the proinflammatory cytokine IL‐1β in the blood were measured with ELISA. B and C, Atrophy of the spleen and thymus after asphyxial CA/CPR. Time course of changes in tissue mass (B) and cell numbers (C) of the spleen and thymus after CA/CPR (n=4–5/group). Representative images of spleens and thymi on day 3 after CA/CPR are shown. D, Aged mice were subjected to 5 minutes CA or sham surgery (n=4/group). Cell numbers of the spleen and thymus were counted on day 3 after CA/CPR. Representative images of spleens and thymi on day 3 after CA/CPR are shown. Data are presented as mean±SEM. *P<0.05. **P<0.01. ***P<0.001. ****P<0.0001.
We then performed flow cytometric analysis of time‐course changes in the composition of major immune cell populations (T cells, B cells, monocytes, neutrophils, and natural killer cells) in the spleen after CA/CPR using the gating strategy depicted in Figure 7A. As shown in Figure 7B, no significant changes were observed on day 1 after CA/CPR compared with sham mice. However, on day 3 after CA/CPR, the percentage of B cells in the spleen decreased significantly from 58.35±2.82% to 34.00±4.78%, whereas the percentages of monocytes and T cells increased, with T cells increasing markedly from 22.84±2.71% to 52.83±5.84% (Figure 7B). However, it is important to note that considering a dramatic reduction in the total number of cells in the spleen on day 3 after CA/CPR (Figure 6C), the absolute numbers of all immune cell types under investigation were decreased. Using aged mice, we observed similar changes in immune cell populations in the spleen after CA/CPR (Figure 8).
Figure 7. Flow cytometric analysis of immune cell populations in the spleen after asphyxial cardiac arrest (CA)/cardiopulmonary resuscitation (CPR) in young adult mice.
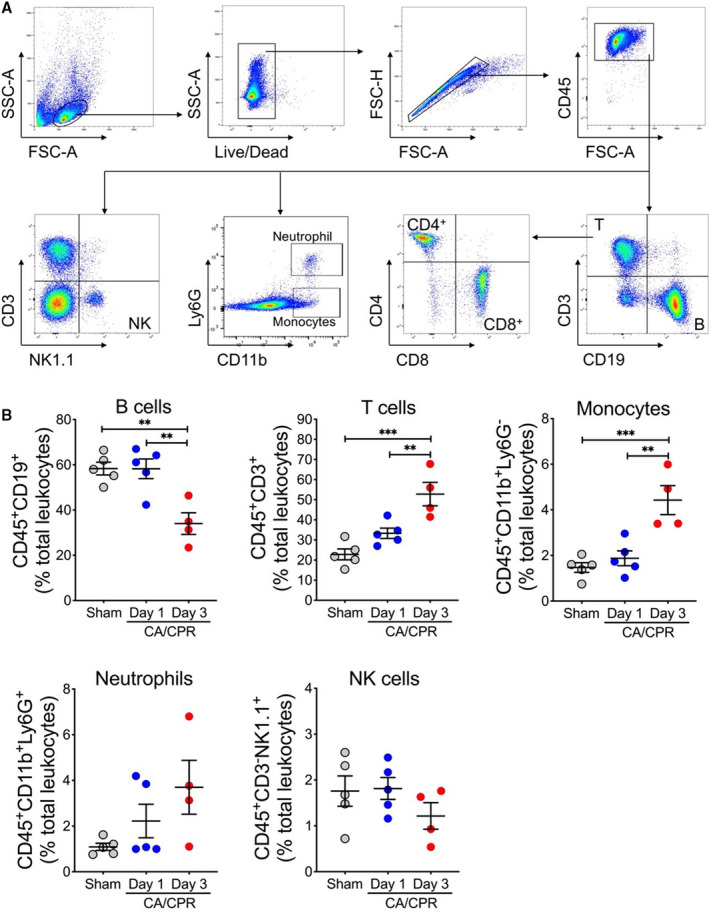
A, Gating strategy. B, Young adult mice (n=6–7/group) were subjected to 7 minutes CA or sham surgery. On day 1 and day 3 after CA, single‐cell suspensions were prepared from the spleen. The following leukocyte subpopulations are shown here: B cells (CD45+CD19+), T cells (CD45+CD3+), monocytes (CD45+CD11b+Ly6G−), neutrophils (CD45+CD11b+Ly6G+), and natural killer cells (NK; CD45+CD3−NK1.1+). Data are presented as mean±SEM. **P<0.01. ***P<0.001.
Figure 8. Flow cytometric analysis of immune cell populations in the spleen after asphyxial cardiac arrest (CA)/cardiopulmonary resuscitation (CPR) in aged mice.

Aged mice (n=4/group) were subjected to 5 minutes CA or sham surgery. On day 3 after CA, single‐cell suspensions were prepared from the spleen. The following leukocyte subpopulations are shown here: B cells (CD45+CD19+), T cells (CD45+CD3+), monocytes (CD45+CD11b+Ly6G−), neutrophils (CD45+CD11b+Ly6G+), and natural killer cells (NK; CD45+CD3−NK1.1+). A, Representative flow cytometry plots. B, Changes in percentages of immune cell populations after CA/CPR. Data are presented as mean±SEM. *P<0.05.
Finally, the massive loss of lymphocytes (T and B cells) after asphyxial CA/CPR can be expected to undermine the capacity of both the innate and adaptive immune responses, which may account for the high incidence of infections in postresuscitation patients.20, 21, 22 In an initial attempt to dissect the mechanisms underlying these extremely low lymphocyte counts, we hypothesized that CA/CPR impairs lymphopoiesis in respective lymphoid organs. To test this hypothesis, we analyzed CA‐induced changes in the premature lymphocytes in the thymus (T cells) and bone marrow (B cells). In the thymus, CD4+CD8+ T cells (also known as double‐positive T cells) are the last stage of T development before undergoing positive and negative selections, and maturing into CD4+ helper T cells and CD8+ cytotoxic T cells.23 As shown in Figure 9A, double‐positive T cells were massively depleted in the thymus on day 3 after CA/CPR in young adult mice. B cells mature in the bone marrow, and early B‐cell progenitors can be identified as a lineage−B220+CD93+ population. We found a significant loss of bone marrow B‐cell progenitors after CA/CPR (Figure 9B). Similarly, in aged mice after CA/CPR, the percentages of double‐positive T cells in the thymus and early B‐cell progenitors in the bone marrow were significantly reduced (Figure 9C and 9D). Together, these data provide the first evidence suggesting that CA/CPR‐induced lymphopenia results, at least in part, from an impaired supply of mature T and B cells, as indicated by reduced numbers and frequencies of premature lymphocytes in lymphoid organs.
Figure 9. Detrimental effects of asphyxial cardiac arrest (CA)/cardiopulmonary resuscitation (CPR) on the development of lymphocytes in the thymus and bone marrow.

A and B, Young adult mice (n=4–6/group) were subjected to 7 minutes CA or sham surgery. On day 3 after CA, single‐cell suspensions were prepared from the thymus and bone marrow. A, Percentages of double‐positive T cells (CD4+CD8+) were dramatically reduced in the thymus on day 3 after CA. Shown are representative flow cytometry plots. B, Percentages of B progenitors (lineage−B220+CD93+) were markedly reduced in the bone marrow on day 3 after CA/CPR. Shown are representative flow cytometry plots. C and D, Aged mice (n=4/group) were subjected to 5 minutes CA or sham surgery. Analyses were performed on day 3 after CA. C, Reduced percentages of double‐positive T cells in the spleen of aged mice after CA/CPR. D, Markedly reduced percentages of B progenitors in the bone marrow on day 3 after CA/CPR. Data are presented as mean±SEM. *P<0.05. ***P<0.001. ****P<0.0001.
Discussion
Animal models of CA/CPR are essential to basic CA research. Notably, murine CA/CPR models have become increasingly used because the animals are relatively inexpensive, a wide array of behavioral tests have been well developed in mice, and numerous genetically modified mouse strains are available. In this study, we successfully established and validated an asphyxial CA/CPR mouse model that can also be used in aged mice. Moreover, using this novel model, we performed a systematic analysis of the immune response in young adult and aged mice after asphyxial CA/CPR, and importantly, discovered that CA/CPR markedly impaired lymphopoiesis of T and B cells, which likely accounts for the severe lymphopenia observed during the postresuscitation subacute phase.
Almost all published murine CA/CPR models use KCl to induce immediate circulatory arrest. The advantages of KCl models include that there is no special device required, and the ischemic duration is highly controlled. Our group previously developed a modified KCl‐induced CA model by reducing the KCl dosage, which shortens the duration of CPR necessary to achieve ROSC, and substantially improves survival rates while still producing detectable neurologic deficits.24 In addition to KCl models, few other murine CA/CPR models have been reported, but their utility in experimental CA research has not been well established.25, 26 In one such model, CA was induced by bolus injections of vecuronium (neuromuscular blockade) and esmolol (a β‐adrenergic receptor blocker) followed by cessation of mechanical ventilation. Of note, circulatory arrest occurred in only about 10 seconds after the esmolol injection, thus indicating a sudden CA model.25 Chen et al have attempted to develop a ventricular fibrillation–induced CA model in mice26; however, it appears to have been difficult to achieve a sustained ventricular fibrillation in their study, probably attributable to a high heart rate (>600 bpm) in the mice. Moreover, it is not clear whether their model is suitable for behavioral tests, because they only monitored the resuscitated mice for an additional 30 minutes after ROSC.26
As discussed above and to the best of our knowledge, all existing mouse CA models mimic sudden‐onset CA, which typically occurs in patients with a coronary artery disease, the most common cause of CA in the clinic. In the present study, we modeled asphyxia, a major noncardiac cause of clinical CA. Clinical causes of asphyxial CA include suffocation, drowning, and drug overdose. Although the ultimate event in all CA models, including our model here, is the cessation of functional heart contractility resulting in whole‐body ischemia, and asphyxial CA causes a pathophysiologic sequence that is different from CA models with immediate CA onset.10 Specifically, because of a progressive hypoxic condition preceding the complete cessation of heart contractions, a bradyasystolic circulatory arrest occurs within minutes following apnea onset, eventually leading to asystole. This asphyxial CA‐specific phenomenon was evident in our model (Figure 2B). As such, unlike KCl‐mediated CA models, the arrest duration cannot be strictly controlled, which may introduce large variability in CA outcome such as neurologic function. To minimize this variability, we measured ptO2 in the brain, CBF, and MAP in addition to ECG monitoring, and analyzed correlations between these parameters. Based on our analyses, we determined that the MAP <20 mm Hg is a reliable criterion for defining arrest onset, because at this point, CBF and ptO2 had decreased to 4.97±0.22% of the baseline and almost 0 mm Hg, respectively, and ECG rhythm depicted functional asystole. Of note, catheterization of the femoral artery to measure blood pressure, the method used in this study during model development, is invasive, and inevitably leads to a degree of paralysis of the hind leg, which impedes the subsequent behavioral tests. Therefore, for experiments that required behavioral tests after CA/CPR, we did not monitor MAP during surgery, but rather chose the time point of 2.5 minutes after start of ventilation with 100% N2 for CA onset, because our data indicated that the time lapse for MAP to decrease to <20 mm Hg in most mice was ≈2.5 minutes. Under this surgical condition, we have obtained consistent behavioral data using our model.
Finally, we used our new model to examine the immune response of the brain and the immune organs to asphyxial CA/CPR. Consistent with previous reports that used other CA/CPR models,16, 25 we found strong activation of astrocytes and microglia, especially in the hippocampal region, and a considerable number of monocytes and neutrophils infiltrated from the blood into the brain after CA/CPR. Critically, a characteristic immunosuppression phenotype, as revealed in our recent report using a KCl‐mediated CA model, and which is corroborated by clinical immune manifestations,16, 27, 28, 29 is also evident in young adult and aged mice after asphyxial CA/CPR. We further discovered that in the thymus, the number of double‐positive T cells became largely depleted, and similarly in the bone marrow, the number of B progenitors was massively decreased. These findings may reveal a new primary mechanism that underpins post‐CA lymphopenia observed in both experimental and clinical CA studies.16, 27 Future research should be directed to clarify the underlying mechanisms responsible for these marked changes in lymphocyte progenitors and their impact on CA outcome.
Several limitations in the study were noted. First, in our model, because we need to keep animals under anesthesia before CA onset, we ventilated mice for a brief period of time (≈2.5 minutes) during the N2 delivery until MAP decreased below 20 mm Hg, which likely prevented CO2 accumulation to some extent and did not exactly reflect the clinical situation. However, the effect of this step on pathophysiology of asphyxial CA could be modest, because our blood gas data indicated a significant CO2 increase and low pH value after ROSC in our model. Second, we did not actively rewarm the animal after the naturally occurring temperature loss during CA in our model. Because patients do not remain at normothermic body temperature during CA and resuscitation, we allowed the temperature to decrease spontaneously during and after injury in our mouse model. However, because we needed to control the experimental condition for all experiments, we decided to prevent any decrease of body temperature below 32°C by active intervention. Also, after being disconnected from the ventilator, mice were kept in a warm incubator to prevent hypothermia and allow spontaneous body temperature recovery. Third, we understand that epinephrine dosing is a highly debatable issue, and there is no full consensus on an optimal dosage for experimental CA with rodents. We determined 4 μg of epinephrine as a standard dose for mice, and we used 2 doses in our model. Using this dosing regimen, we were able to reproducibly resuscitate both young adult and aged mice. Our dosage is comparable to those in published experimental CA studies.30, 31 However, future studies need to investigate the effects of different epinephrine dosing regimens on CA outcome, especially considering the increased concerns of potentially detrimental effects of the epinephrine use during resuscitation on neurologic recovery.32 Lastly, although we analyzed the post‐CA immune response in both young adult and aged mice, it must be noted that a short CA duration was applied to aged mice. Therefore, this critical difference should be considered when comparing the immune changes between young adult and aged mice. We observed that the extent of some responses, such as changes in the percentages of B and T cells in the spleen after CA/CPR, appeared to be less in aged mice versus young adult mice. It remained to be clarified whether these differences were the effects of CA duration or aging. Overall, our data demonstrated that post‐CA–induced changes in the immune system were similar between both young adult and aged mice. However, in this respect, more research is warranted.
In summary, using a novel murine asphyxial CA model established in this study, we demonstrated that asphyxial CA/CPR severely affects both the nervous and immune systems, and impairs lymphopoiesis of T and B lymphocytes. This model and the findings reported here could serve as a basis for future basic CA research that is aimed at deciphering the mechanisms involved in asphyxial CA‐induced neuro‐ and immunopathology, and to assess the efficacy of new therapeutics. It is noteworthy that one descriptive analysis study concluded that overall investment for CA research is remarkably low compared with research for other diseases such as diabetes mellitus, stroke, and ischemic heart disease, suggesting an urgent need for increasing resources for CA research.33 We echo that need.
Sources of Funding
This work was supported by funds from the Department of Anesthesiology (Duke University Medical Center), an American Heart Association grant (18CSA34080277), and a National Institutes of Health grant (NS117973).
Disclosures
None.
Acknowledgments
The authors thank P. Miao for her excellent technical support, and K. Gage for her excellent editorial contributions.
(J Am Heart Assoc. 2021;10:e019142. DOI: 10.1161/JAHA.120.019142.)
For Sources of Funding and Disclosures, see page 14.
Contributor Information
Huaxin Sheng, Email: huaxin.sheng@duke.edu.
Wei Yang, Email: wei.yang@duke.edu.
References
- 1.Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, et al. Heart disease and stroke statistics‐2020 update: a report from the american heart association. Circulation. 2020;141:e139–e596. DOI: 10.1161/CIR.0000000000000757. [DOI] [PubMed] [Google Scholar]
- 2.Vognsen M, Fabian‐Jessing BK, Secher N, Lofgren B, Dezfulian C, Andersen LW, Granfeldt A. Contemporary animal models of cardiac arrest: a systematic review. Resuscitation. 2017;113:115–123. DOI: 10.1016/j.resuscitation.2017.01.024. [DOI] [PubMed] [Google Scholar]
- 3.Piktel JS, Cheng A, McCauley M, Dale Z, Nassal M, Maleski D, Pawlowski G, Laurita KR, Wilson LD. Hypothermia modulates arrhythmia substrates during different phases of resuscitation from ischemic cardiac arrest. J Am Heart Assoc. 2017;6:e006472. DOI: 10.1161/JAHA.117.006472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suh GJ, Kwon WY, Kim KS, Lee HJ, Jeong KY, Jung YS, Lee JH. Prolonged therapeutic hypothermia is more effective in attenuating brain apoptosis in a swine cardiac arrest model. Crit Care Med. 2014;42:e132–e142. DOI: 10.1097/CCM.0b013e3182a668e4. [DOI] [PubMed] [Google Scholar]
- 5.Wang Q, Miao P, Modi HR, Garikapati S, Koehler RC, Thakor NV. Therapeutic hypothermia promotes cerebral blood flow recovery and brain homeostasis after resuscitation from cardiac arrest in a rat model. J Cereb Blood Flow Metab. 2019;39:1961–1973. DOI: 10.1177/0271678X18773702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gong P, Zhao S, Wang J, Yang Z, Qian J, Wu X, Cahoon J, Tang W. Mild hypothermia preserves cerebral cortex microcirculation after resuscitation in a rat model of cardiac arrest. Resuscitation. 2015;97:109–114. DOI: 10.1016/j.resuscitation.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 7.Callaway CW, Rittenberger JC, Logue ES, McMichael MJ. Hypothermia after cardiac arrest does not alter serum inflammatory markers. Crit Care Med. 2008;36:2607–2612. DOI: 10.1097/CCM.0b013e318184443b. [DOI] [PubMed] [Google Scholar]
- 8.Li R, Shen Y, Li X, Lu L, Wang Z, Sheng H, Hoffmann U, Yang W. Activation of the XBP1s/O‐GlcNAcylation pathway improves functional outcome after cardiac arrest and resuscitation in young and aged mice. Shock. 2021. Jan 21 [epub ahead of print]. DOI: 10.1097/SHK.0000000000001732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayashida K, Bagchi A, Miyazaki Y, Hirai S, Seth D, Silverman MG, Rezoagli E, Marutani E, Mori N, Magliocca A, et al. Improvement in outcomes after cardiac arrest and resuscitation by inhibition of s‐nitrosoglutathione reductase. Circulation. 2019;139:815–827. DOI: 10.1161/CIRCULATIONAHA.117.032488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Varvarousis D, Varvarousi G, Iacovidou N, D'Aloja E, Gulati A, Xanthos T. The pathophysiologies of asphyxial vs dysrhythmic cardiac arrest: implications for resuscitation and post‐event management. Am J Emerg Med. 2015;33:1297–1304. DOI: 10.1016/j.ajem.2015.06.066. [DOI] [PubMed] [Google Scholar]
- 11.Drabek T, Foley LM, Janata A, Stezoski J, Hitchens TK, Manole MD, Kochanek PM. Global and regional differences in cerebral blood flow after asphyxial versus ventricular fibrillation cardiac arrest in rats using ASL‐MRI. Resuscitation. 2014;85:964–971. DOI: 10.1016/j.resuscitation.2014.03.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vaagenes P, Safar P, Moossy J, Rao G, Diven W, Ravi C, Arfors K. Asphyxiation versus ventricular fibrillation cardiac arrest in dogs. Differences in cerebral resuscitation effects—a preliminary study. Resuscitation. 1997;35:41–52. DOI: 10.1016/S0300-9572(97)01108-8. [DOI] [PubMed] [Google Scholar]
- 13.Adrie C, Adib‐Conquy M, Laurent I, Monchi M, Vinsonneau C, Fitting C, Fraisse F, Dinh‐Xuan AT, Carli P, Spaulding C, et al. Successful cardiopulmonary resuscitation after cardiac arrest as a "sepsis‐like" syndrome. Circulation. 2002;106:562–568. DOI: 10.1161/01.CIR.0000023891.80661.AD. [DOI] [PubMed] [Google Scholar]
- 14.Hassager C, Nagao K, Hildick‐Smith D. Out‐of‐hospital cardiac arrest: in‐hospital intervention strategies. Lancet. 2018;391:989–998. DOI: 10.1016/S0140-6736(18)30315-5. [DOI] [PubMed] [Google Scholar]
- 15.Mai N, Miller‐Rhodes K, Knowlden S, Halterman MW. The post‐cardiac arrest syndrome: a case for lung‐brain coupling and opportunities for neuroprotection. J Cereb Blood Flow Metab. 2019;39:939–958. DOI: 10.1177/0271678X19835552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Q, Shen Y, Li R, Wu J, Lyu J, Jiang M, Lu L, Zhu M, Wang W, Wang Z, et al. Cardiac arrest and resuscitation activates the hypothalamic‐pituitary‐adrenal axis and results in severe immunosuppression. J Cereb Blood Flow Metab. 2021;41:1091–1102. DOI: 10.1177/0271678X20948612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen Y, Yan B, Zhao Q, Wang Z, Wu J, Ren J, Wang W, Yu S, Sheng H, Crowley SD, et al. Aging is associated with impaired activation of protein homeostasis‐related pathways after cardiac arrest in mice. J Am Heart Assoc. 2018;7:e009634. DOI: 10.1161/JAHA.118.009634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allen D, Nakayama S, Kuroiwa M, Nakano T, Palmateer J, Kosaka Y, Ballesteros C, Watanabe M, Bond CT, Lujan R, et al. SK2 channels are neuroprotective for ischemia‐induced neuronal cell death. J Cereb Blood Flow Metab. 2011;31:2302–2312. DOI: 10.1038/jcbfm.2011.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27–31. DOI: 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perbet S, Mongardon N, Dumas F, Bruel C, Lemiale V, Mourvillier B, Carli P, Varenne O, Mira JP, Wolff M, et al. Early‐onset pneumonia after cardiac arrest: characteristics, risk factors and influence on prognosis. Am J Respir Crit Care Med. 2011;184:1048–1054. DOI: 10.1164/rccm.201102-0331OC. [DOI] [PubMed] [Google Scholar]
- 21.Dankiewicz J, Nielsen N, Linder A, Kuiper M, Wise MP, Cronberg T, Erlinge D, Gasche Y, Harmon MB, Hassager C, et al. Infectious complications after out‐of‐hospital cardiac arrest‐a comparison between two target temperatures. Resuscitation. 2017;113:70–76. DOI: 10.1016/j.resuscitation.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 22.Gajic O, Festic E, Afessa B. Infectious complications in survivors of cardiac arrest admitted to the medical intensive care unit. Resuscitation. 2004;60:65–69. DOI: 10.1016/j.resuscitation.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 23.Shah DK, Zuniga‐Pflucker JC. An overview of the intrathymic intricacies of T cell development. J Immunol. 2014;192:4017–4023. DOI: 10.4049/jimmunol.1302259. [DOI] [PubMed] [Google Scholar]
- 24.Liu H, Yu Z, Li Y, Xu B, Yan B, Paschen W, Warner DS, Yang W, Sheng H. Novel modification of potassium chloride induced cardiac arrest model for aged mice. Aging Dis. 2018;9:31–39. DOI: 10.14336/AD.2017.0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang C, Brandon NR, Koper K, Tang P, Xu Y, Dou H. Invasion of peripheral immune cells into brain parenchyma after cardiac arrest and resuscitation. Aging Dis. 2018;9:412–425. DOI: 10.14336/AD.2017.0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen MH, Liu TW, Xie L, Song FQ, He T, Mo SR, Zeng ZY. A simpler cardiac arrest model in the mouse. Resuscitation. 2007;75:372–379. DOI: 10.1016/j.resuscitation.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 27.Villois P, Grimaldi D, Spadaro S, Shinotsuka CR, Fontana V, Scolletta S, Franchi F, Vincent JL, Creteur J, Taccone FS. Lymphopaenia in cardiac arrest patients. Ann Intensive Care. 2017;7:85. DOI: 10.1186/s13613-017-0308-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beurskens CJ, Horn J, de Boer AM, Schultz MJ, van Leeuwen EM, Vroom MB, Juffermans NP. Cardiac arrest patients have an impaired immune response, which is not influenced by induced hypothermia. Crit Care. 2014;18:R162. DOI: 10.1186/cc14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang M, Li R, Lyu J, Li X, Wang W, Wang Z, Sheng H, Zhang W, Karhausen J, Yang W. MCC950, a selective NLPR3 inflammasome inhibitor, improves neurologic function and survival after cardiac arrest and resuscitation. J Neuroinflammation. 2020;17:256. DOI: 10.1186/s12974-020-01933-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng GY, Orfila JE, Dietz RM, Moreno‐Garcia M, Rodgers KM, Coultrap SJ, Quillinan N, Traystman RJ, Bayer KU, Herson PS. Autonomous CaMKII activity as a drug target for histological and functional neuroprotection after resuscitation from cardiac arrest. Cell Rep. 2017;18:1109–1117. DOI: 10.1016/j.celrep.2017.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rutledge CA, Chiba T, Redding K, Dezfulian C, Sims‐Lucas S, Kaufman BA. A novel ultrasound‐guided mouse model of sudden cardiac arrest. PLoS One. 2020;15:e0237292. DOI: 10.1371/journal.pone.0237292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bornstein K, Long B, Porta AD, Weinberg G. After a century, epinephrine's role in cardiac arrest resuscitation remains controversial. Am J Emerg Med. 2021;39:168–172. DOI: 10.1016/j.ajem.2020.08.103. [DOI] [PubMed] [Google Scholar]
- 33.Coute RA, Panchal AR, Mader TJ, Neumar RW. National institutes of health‐funded cardiac arrest research: a 10‐year trend analysis. J Am Heart Assoc. 2017;6:e005239. DOI: 10.1161/JAHA.116.005239. [DOI] [PMC free article] [PubMed] [Google Scholar]