

Abstract
Purpose of review:
In this review, we will describe how the combined ability of platelets and neutrophils to interact with each other drives ischemic stroke brain injury.
Recent findings:
Neutrophils are one of the first cells to respond during ischemic stroke. While animals stroke models have indicated targeting neutrophils improves outcomes, clinical trials have failed to yield successful strategies. Platelets play a critical role in recruiting neutrophils to sites of injury by acting as a bridge to the injured endothelium. After initial platelet adhesion, neutrophils can rapidly bind platelets through P-selectin and glycoprotein Ibα. In addition, recent data implicated platelet phosphatidylserine as a novel key regulator of platelet-neutrophil interactions in the setting of ischemic stroke. Inhibition of procoagulant platelets decreases circulating platelet-neutrophil aggregates and thereby reduces infarct size. Platelet binding alters neutrophil function, which contributes to the injury associated with ischemic stroke. This includes inducing the release of neutrophil extracellular traps (NETs), which are neurotoxic and pro-thrombotic, leading to impaired stroke outcomes.
Summary:
Platelet-neutrophil interactions significantly contribute to the pathophysiology of ischemic stroke brain injury. Better understanding the mechanisms behind their formation and the downstream consequences of their interactions will lead to improved therapies for stroke patients.
Keywords: Platelets, neutrophils, stroke, procoagulant platelets, neutrophil extracellular traps
Introduction
Annually, 800,000 Americans suffer from stroke and for 1 in 6 stroke patients this will be fatal. Globally, 15,000,000 individuals have a stroke each year with 5,000,000 stroke related deaths occuring1. The vast majority of strokes are ischemic and these are typically caused by a blood clot obstructing cerebral blood flow. Current acute ischemic stroke therapy focusses on the removal of the culprit thrombus via either pharmacological thrombolysis or mechanical thrombectomy. Due to a limited time window and other contra-indications, these therapies are only available for a small group of stroke patients2. Moreover, even when thrombolytic therapy or thrombectomy is initiated, this is often not effective. Indeed, successful thrombolysis occurs only in approximately 30% of treated patients3 and thrombectomy does not guarantee improved stroke outcomes, even when the thrombus is successfully removed4.
Once cerebral blood flow stops and ischemia occurs, a dynamic process takes place in the brain initiating a detrimental cascade of inflammation, microvascular thrombosis and neurotoxicity. While successful restoration of cerebral blood flow is critical to improve stroke outcomes, preclinical evidence suggests this might not be sufficient to maximally salvage brain tissue. In an effort to improve stroke outcomes, several studies have investigated neuroprotection via either anti-inflammatory5 or anti-thrombotic strategies6. However, none of these approaches have successfully translated to the clinic7. Limitations associated with anti-inflammatory strategies include post-stroke immunosuppression and an increased risk for lethal infections in stroke patients. Additionally, the human brain is very sensitive to bleeding associated with classic anti-thrombotic drugs. Importantly, recent evidence suggests in particular the interaction between inflammation and thrombosis is a key contributor to ischemic stroke brain injury8. Interestingly, conventional anti-inflammatory and anti-thrombotic drugs don’t interfere with these interactions. This highlights the need for new management strategies to target thrombo-inflammation in ischemic stroke.
Platelet-neutrophil interactions are at the interface between inflammation and thrombosis. In several thrombo-inflammatory disorders platelet-neutrophil aggregates (PNAs) have emerged as markers for disease severity and potential therapeutic targets9. Similarly, in ischemic stroke PNAs are increased acutely10–13, irrespective of use of anti-platelet drugs14,15. However, circulating PNAs don’t correlate with stroke severity11,14 or stroke etiology.12,13 In this review, we will discuss recent findings on novel ways platelets and neutrophils interact during ischemic stroke and how these interactions exacerbate ischemic stroke brain injury.
Neutrophil Activation and Recruitment in Ischemic Stroke
Neutrophils are among the first cells in the blood to respond after ischemic stroke, with the number of circulating neutrophils rising within the first few hours of stroke onset16. The increase in neutrophils contributes to disruption of the blood brain barrier (BBB), cerebral edema, and brain injury. Importantly, the increase in circulating neutrophils is associated with stroke severity, infarct volume, and worse functional outcomes17–19. Furthermore, the level of neutrophil accumulation in regions of brain ischemia correlates with stroke severity and worse stroke outcome in humans and mice20,21. While these studies indicate neutrophils are critical regulators of ischemic stroke injury, modulators of their activation and recruitment to the injured stroke brain remain unclear.
Many studies have focused on the role of the endothelium in regulating neutrophil recruitment and transmigration. Neutrophils express numerous endothelial adhesion receptors important in promoting neutrophil tethering and rolling on endothelial cells including P-selectin glycoprotein ligand-1 (PSGL-1), lymphocyte function associated antigen 1 (LFA-1) and macrophage-1 antigen (MAC-1)22,23. Many of these receptors increase shortly after ischemia thus promoting neutrophil adhesion and infiltration24–26. While many of these targets have been successful inhibited in animal models of ischemic stroke27,28, the translation to human studies has been less fruitful. Clinical studies targeting intracellular adhesion molecule 1 (ICAM-1), a key endothelial cell receptor regulating neutrophil binding, MAC-1, and LFA-1 have all failed in various clinical trials with results ranging from no benefit compared to placebo controls to increased rates of infection29–31. Given the importance of neutrophils in promoting neuroinflammation, resulting in worse outcomes in ischemic stroke injury, alternative mediators regulating neutrophil recruitment to the brain are important to consider.
Platelet-Dependent Neutrophil Recruitment: A Missing Link
Platelets play a critical role in the formation of obstructive thrombi causing stroke with robust clinical data demonstrating acute reductions in recurrent ischemic stroke with the combination of antiplatelet agents aspirin and clopidogrel. Long-term benefits have also been demonstrated with aspirin, aspirin-dipyridamole, and clopidogrel32. Additionally, platelets exert pathological effects in the ischemic stroke brain that go beyond classical thrombus formation, which is the focus of this review. Similar to neutrophils, platelets are capable of interacting with the endothelium upon activation. Upon exposure of subendothelial matrix or activation of the endothelium platelets will quickly adhere. Importantly, the efficiency of platelet adhesion increases in the presence of high shear rates, which is common in cerebral arteries affected by stroke33,34. The principal regulator of platelet adhesion to injured or activated endothelium occurs through platelet glycoprotein Ibα binding to endothelial cell-bound von Willebrand factor (VWF), which is favored during high shear environments35. In contrast, direct neutrophil interactions with the endothelium are less favored during high shear environments36. Thus, the high shear environment of the cerebral arteries is primed for platelets to facilitate neutrophil adhesion to activated or injured endothelium. Once platelets are bound to the endothelium, platelets trap neutrophils using multiple receptors including platelet P-selectin binding to neutrophil PSGL-137 and platelet glycoprotein Ibα binding to neutrophil MAC-138,39 (Figure 1). Furthermore, neutrophil adhesion to activated platelets can proceed through flow-induced protrusions or FLIPRs that extend from adhered platelets and mediate neutrophil binding in in a P-selectin/PSGL-1-dependent manner40. In addition to P-selectin, other platelets receptors including αIIbβ341 and ICAM-242 are also critical in forming platelet-neutrophil aggregates by binding MAC-1 and LFA-1, respectively. Recently, a novel interaction between platelet αIIbβ3 and neutrophil SLC44A2 was identified43, which promotes venous thrombosis44,45. Interestingly, this interaction specifically occurs under flow and is VWF dependent, implying it could also play a role in ischemic stroke43,46. These findings highlight the need to further identify novel platelet-neutrophil binding partners that contribute to thrombosis.
Figure 1. Conventional and Novel Ligands Involved in Platelet-Neutrophil Aggregate Formation.

Platelets bind to neutrophils using classical receptors including αIIbβ3 and GPIbα which bind to MAC-1 as well as ICAM-2 which binds to LFA. Platelet alpha granule release results in P-selectin expression and the ability of the platelet to bind PSGL-1 on neutrophils. Besides these well-established pathways, the novel platelet ligand phosphatidylserine (PS) mediates platelet-neutrophil interactions through an unknown receptor to contribute to ischemic stroke outcomes. Neutrophil SLC44A2 has recently been shown to bind to platelet αIIbβ3 to mediate platelet-neutrophil aggregate formation.
Novel Regulators of Platelet-Neutrophil Aggregate Formation: Procoagulant Platelets
One particular subset of platelets that preferentially interacts with neutrophils are procoagulant platelets. Distinctive features of procoagulant platelets include high levels of externalized phosphatidylserine (PS), inactivation of integrin αIIbβ3 and their ability to generate microparticles47. These features result in a unique phenotype of platelets that can drive coagulation on their surface, but are less able to aggregate with other platelets. In recent years, an increasing body of work has described a predisposition of procoagulant platelets to interact with neutrophils and mediate inflammation and subsequent tissue injury21,40,48,49. Interestingly, ischemic stroke patients have increased levels of circulating procoagulant platelets50,51 and platelets from ischemic stroke patients have an increased ability to become procoagulant when tested in vitro52–57. Moreover, increased in vitro procoagulant platelet responses are associated with stroke severity and outcomes57, and have been shown to predict stroke recurrence53–56. Importantly, experimental stroke models have shown that targeting procoagulant platelets is protective21,58, implying a causal role between procoagulant platelet formation and ischemic stroke outcomes.
Procoagulant platelets are formed in response to a strong stimulus. In vitro this stimulus can be mimicked by dual agonist stimulation with thrombin and a GPVI agonist (i.e., convulxin or collagen related peptide). In stroke, the factors initiating procoagulant platelet formation are yet to be determined. Most likely, this is a combination of collagen exposed on damaged endothelium59 and locally increased levels of circulating thrombin60. Procoagulant platelet formation can then be potentiated by reactive oxygen species (ROS)61 released by ischemic brain tissue or by high shear62 occurring in the reperfused arteries, resulting in enhanced platelet PS exposure. Furthermore, we recently found that hyperglycemia, a common comorbidity for stroke patients, also potentiated procoagulant platelet formation and subsequent PNA formation in a mouse model of stroke63.
Importantly, a causal role for procoagulant platelets in the exacerbation of ischemic stroke outcomes was established in murine ischemic stroke models. Indeed, mice with platelets lacking the ability to become procoagulant (platelet specific cyclophilin D KO mice) were protected from ischemic stroke brain injury21. Similarly, when procoagulant platelets were targeted with AnnexinA158 or lactadherin64, ischemic stroke brain injury could be blocked. Mechanistically, these studies found that procoagulant platelets mediated the formation of detrimental platelet-neutrophil interactions in the reperfused ischemic brain. These interactions mediated trafficking of neutrophils to the brain, reduced local cerebral blood flow and thereby exacerbate stroke outcomes21,58. One clinical study recently underscored these observations. When procoagulant platelets were isolated from ischemic stroke patients and incubated with healthy neutrophils, these platelets induced neutrophil extracellular traps (NETs), which subsequently initiated coagulation and induced endothelial cell death in vitro51. Combined, these studies highlight the detrimental effects of the interaction between procoagulant platelets and neutrophils in the ischemic brain.
The mechanisms through which procoagulant platelets interact with neutrophils have yet to be elucidated; however, targeting either platelet P-selectin48 or PS on the platelet surface using recombinant AnnexinA148 was shown to abrogate interactions between procoagulant platelets and neutrophils.
Downstream Consequences of Platelet-Neutrophil Aggregate Formation
Platelet recruitment and activation of neutrophils alters the hemostatic balance in the brain to promote inflammation and create a procoagulant environment (Figure 2). Neutrophil activation results in the release granules containing cathepsin G, elastase and matrix metalloproteinases (MMP), all which disrupts the BBB65–67. Besides inducing alterations in the BBB, cathepsin G and elastase also are important regulators of coagulation. Both enzymes inactivate natural anticoagulants such as tissue factor pathway inhibitor, thrombomodulin, and antithrombin, thus promoting a procoagulant environment68–70. Cathepsin G also activates protease activated receptor-4 on endothelial cells71 and platelets72 to further amplify platelet activation and adhesion. In addition, platelet adhesion to neutrophils results in the generation and release of ROS from neutrophils73. ROS has cytotoxic effects on neural and endothelial cells, leading to increased BBB permeability74.
Figure 2. Platelet-Neutrophil Aggregate Formation Promotes Inflammation and a Procoagulant Environment After Ischemic Stroke.
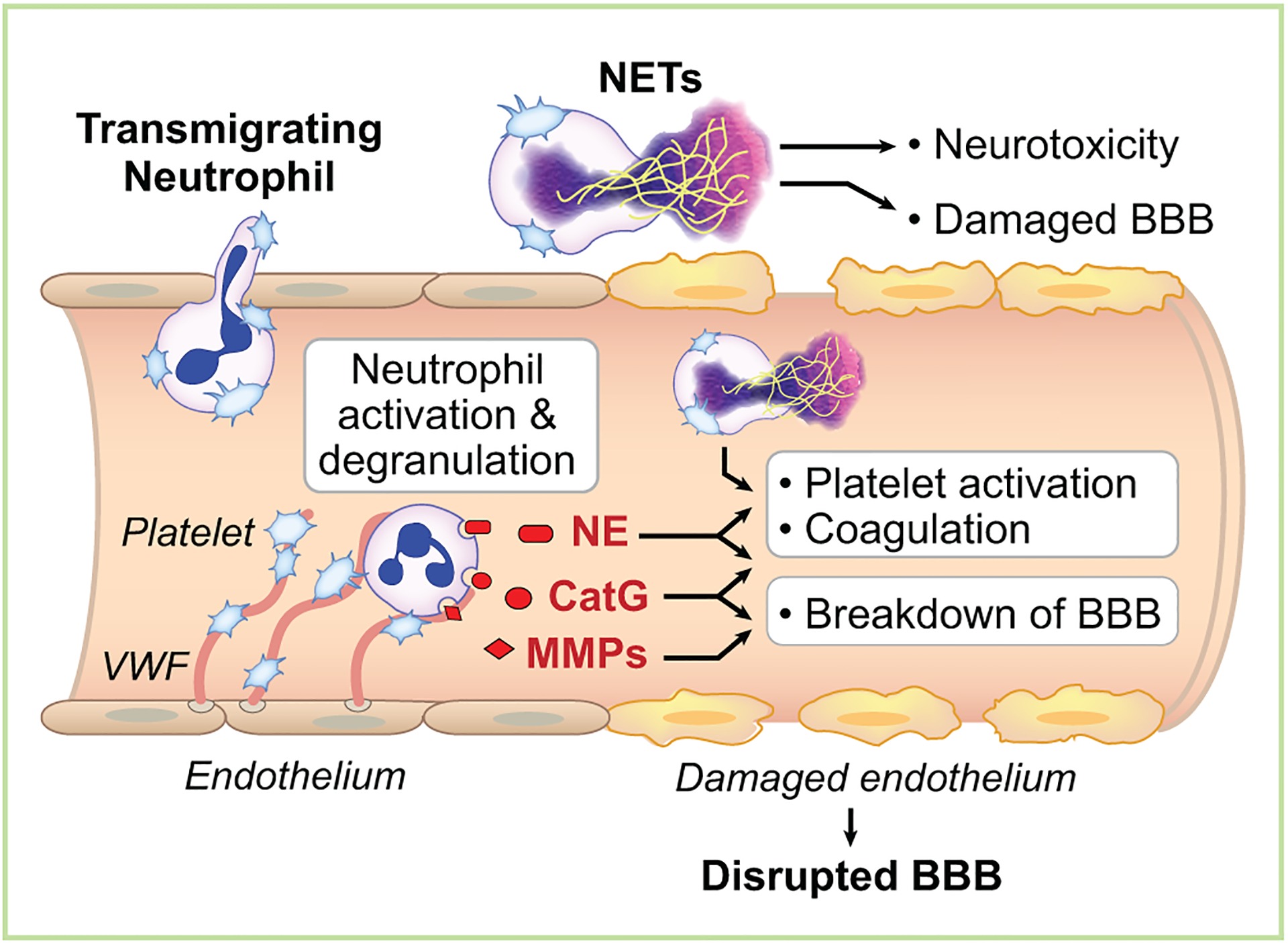
During ischemic stroke, endothelial von Willebrand factor (VWF) traps platelets under high shear stress resulting in release of platelet alpha granule content, including P-selectin. Furthermore, this environment promotes activation of platelet integrins and PS expression. The combination of platelet alpha granule release, integrin activation, and PS expression fosters the formation of platelet-neutrophil aggregates. Platelet binding activates neutrophils to release cathepsin G (CatG), neutrophil elastase (NE), and matrix metalloproteases (MMPs), which alter coagulation and propagate additional platelet activation and disruption of the blood-brain barrier (BBB). In addition, platelets can induce intravascular neutrophil extracellular traps (NETs) resulting in further recruitment of platelets and neutrophils to the ischemic brain. Platelet binding to neutrophils also induces transmigration of neutrophils into the ischemic brain tissue where extravascular neurotoxic NET formation can occur.
Platelets binding to neutrophils also promote the formation of NETs75,76. NETs are extracellular DNA lattices generated upon neutrophil activation77. While NETs primarily are thought to trap bacteria and pathogens to help fight infection, they are neurotoxic when they are formed in the brain parenchyma78,79. NETs appear to promote thrombotic complications in animal models as they trap other blood cells including platelets and neutrophils as well as serve as a scaffolding to support thrombin generation80. This combination leads to decreased blood flow and further exacerbates ischemia in the tissue. Mice deficient in PAD4, an enzyme critical for a histone modification required for chromatin decondensation, are unable to make NETs and are protected in a venous thrombosis model81. Dissolving NETs after their formation using DNase also reduces thrombosis, including during ischemic stroke82.
Platelet-neutrophil aggregates, themselves, also regulate blood flow and ischemia. Using intravital microscopy, Yuan and colleagues were able to demonstrate that rolling neutrophils extract large membrane fragments from procoagulant platelets in multiple organs after gut ischemia-reperfusion injury48. In their model, procoagulant platelets bridged neutrophils to form macroaggregates and subsequently plug vessels in distal organs. Plugging of the cerebral microvasculature is a common observation in both clinical and experimental stroke and contributes to the no-reflow phenomenon after reperfusion, but also exacerbates outcomes in permanent stroke models83–87. In line with this, a recent report observed platelets and neutrophils in high shear environments alter blood rheology to promote microvascular clotting, which could be critical in ischemic stroke injury88.
Targeting Platelet-Neutrophil Aggregates Therapeutically in Stroke
Platelet-neutrophil aggregates are critical in the pathophysiology of ischemic stroke; therefore, therapeutic targeting may provide clinical benefit to stroke patients. Previous studies have directed therapies against PSGL-1 and P-selectin in the setting of myocardial infarction (MI). However, these studies demonstrated little benefit towards MI outcomes89,90. Importantly, these therapies have not been examined in the setting of ischemic stroke, which has a distinctive pathophysiology compared to MI. Indeed, whereas targeting platelet αIIbβ3 was protective in MI91, this was ineffective and induced severe bleeding complications in stroke patients.92 While inhibition of platelet specific receptors that bind neutrophils has not been examined in clinical trials for stroke patients, several recent preclinical studies have suggested inhibition of procoagulant platelets plays a significant role in dampening ischemic stroke injury. In animal models deletion of cyclophilin D a key regulator of procoagulant platelet formation or administration of Annexin A1 in a cerebral ischemia-reperfusion model reduces platelet PS expression, platelet-neutrophil interactions and infarct size21,58. Furthermore, in stroke patients the administration of cyclosporine, which inhibits procoagulant platelet formation, improved outcomes in patients where reperfusion was achieved after cerebral ischemia93. Besides targeting therapies against neutrophil and platelets receptors, therapeutics directed against downstream consequences of platelets-neutrophil aggregates such as NET formation could be viable as well. NET formation is a known regulator of venous and arterial thrombosis, including ischemia/reperfusion injury and NETs have previously been demonstrated to be embedded in thrombi retrieved from ischemic stroke patients94–96. Targeting NETs could be achieved by directing agents against the DNA lattice generated when NETs are released through DNase administration82. Furthermore, inhibition of molecular regulators of NET formation, including PAD4, could block DNA release altogether81. Targeting procoagulant platelet formation or NET release during ischemic stroke would be more beneficial compared to targeting platelet receptors as these interventions have less impact on basic hemostatic platelet or normal neutrophil function, meaning there would be minimal risk for bleeding or infections.
Conclusion
The unsuccessful translation of anti-neutrophil strategies from bench to bedside stimulated stroke research to further elucidate the precise mechanisms by which neutrophils mediate ischemic stroke brain damage. In recent years, the focus shifted from neutrophil transmigration and neutrophil-endothelial cell interactions, to platelet-neutrophil interactions and the associated cerebral microvascular complications. Novel therapeutic interventions targeting procoagulant platelets or the formation of NETs hold promise as safe ischemic stroke therapies and warrant further investigation.
Key points:
Neutrophils are detrimental during acute ischemic stroke brain injury but critical to fight infections after stroke.
Platelets-neutrophil interactions are critical in mediating injurious neutrophil function in the brain after stroke.
Targeting novel mediators of platelet-neutrophil interactions such as SLC44A2 and phosphatidyl serine hold promise for stroke treatment.
Acknowledgements
The authors want to thank Diana Lim for her excellent figure preparation.
Financial support and sponsorship
This work was supported by the NIA and NINDS (K01AG059892 to R.A.C and Utah StrokeNET U24NS107228 to F.D.).
Footnotes
Conflicts of interest
None
References
- 1.Virani SS, Alonso A, Aparicio HJ, et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation. 2021;143(8):e254–e743. [DOI] [PubMed] [Google Scholar]
- 2.Vanacker P, Lambrou D, Eskandari A, Mosimann PJ, Maghraoui A, Michel P. Eligibility and Predictors for Acute Revascularization Procedures in a Stroke Center. Stroke. 2016;47(7):1844–1849. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez RG, Furie KL, Goldmacher GV, et al. Good outcome rate of 35% in IV-tPA-treated patients with computed tomography angiography confirmed severe anterior circulation occlusive stroke. Stroke. 2013;44(11):3109–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Badhiwala JH, Nassiri F, Alhazzani W, et al. Endovascular Thrombectomy for Acute Ischemic Stroke: A Meta-analysis. JAMA. 2015;314(17):1832–1843. [DOI] [PubMed] [Google Scholar]
- 5.Levard D, Buendia I, Lanquetin A, Glavan M, Vivien D, Rubio M. Filling the gaps on stroke research: Focus on inflammation and immunity. Brain Behav Immun. 2021;91:649–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burkard P, Vogtle T, Nieswandt B. Platelets in Thrombo-Inflammation: Concepts, Mechanisms, and Therapeutic Strategies for Ischemic Stroke. Hamostaseologie. 2020;40(2):153–164. [DOI] [PubMed] [Google Scholar]
- 7.Dirnagl U, Endres M. Found in translation: preclinical stroke research predicts human pathophysiology, clinical phenotypes, and therapeutic outcomes. Stroke. 2014;45(5):1510–1518. [DOI] [PubMed] [Google Scholar]
- 8.Stoll G, Nieswandt B. Thrombo-inflammation in acute ischaemic stroke - implications for treatment. Nat Rev Neurol. 2019;15(8):473–481. [DOI] [PubMed] [Google Scholar]
- 9.Dib PRB, Quirino-Teixeira AC, Merij LB, et al. Innate immune receptors in platelets and platelet-leukocyte interactions. J Leukoc Biol. 2020;108(4):1157–1182. [DOI] [PubMed] [Google Scholar]
- 10.Marquardt L, Anders C, Buggle F, Palm F, Hellstern P, Grau AJ. Leukocyte-platelet aggregates in acute and subacute ischemic stroke. Cerebrovasc Dis. 2009;28(3):276–282. [DOI] [PubMed] [Google Scholar]
- 11.Htun P, Fateh-Moghadam S, Tomandl B, et al. Course of platelet activation and platelet-leukocyte interaction in cerebrovascular ischemia. Stroke. 2006;37(9):2283–2287. [DOI] [PubMed] [Google Scholar]
- 12.Zeller JA, Lenz A, Eschenfelder CC, Zunker P, Deuschl G. Platelet-leukocyte interaction and platelet activation in acute stroke with and without preceding infection. Arterioscler Thromb Vasc Biol. 2005;25(7):1519–1523. [DOI] [PubMed] [Google Scholar]
- 13.Tao L, Changfu W, Linyun L, Bing M, Xiaohui H. Correlations of platelet-leukocyte aggregates with P-selectin S290N and P-selectin glycoprotein ligand-1 M62I genetic polymorphisms in patients with acute ischemic stroke. J Neurol Sci. 2016;367:95–100. [DOI] [PubMed] [Google Scholar]
- 14.Schmalbach B, Stepanow O, Jochens A, Riedel C, Deuschl G, Kuhlenbaumer G. Determinants of platelet-leukocyte aggregation and platelet activation in stroke. Cerebrovasc Dis. 2015;39(3–4):176–180. [DOI] [PubMed] [Google Scholar]
- 15.McCabe DJ, Harrison P, Mackie IJ, et al. Increased platelet count and leucocyte-platelet complex formation in acute symptomatic compared with asymptomatic severe carotid stenosis. J Neurol Neurosurg Psychiatry. 2005;76(9):1249–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ross AM, Hurn P, Perrin N, Wood L, Carlini W, Potempa K. Evidence of the peripheral inflammatory response in patients with transient ischemic attack. J Stroke Cerebrovasc Dis. 2007;16(5):203–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim J, Song TJ, Park JH, et al. Different prognostic value of white blood cell subtypes in patients with acute cerebral infarction. Atherosclerosis. 2012;222(2):464–467. [DOI] [PubMed] [Google Scholar]
- 18.Buck BH, Liebeskind DS, Saver JL, et al. Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke. 2008;39(2):355–360. [DOI] [PubMed] [Google Scholar]
- 19.Kumar AD, Boehme AK, Siegler JE, Gillette M, Albright KC, Martin-Schild S. Leukocytosis in patients with neurologic deterioration after acute ischemic stroke is associated with poor outcomes. J Stroke Cerebrovasc Dis. 2013;22(7):e111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akopov SE, Simonian NA, Grigorian GS. Dynamics of polymorphonuclear leukocyte accumulation in acute cerebral infarction and their correlation with brain tissue damage. Stroke. 1996;27(10):1739–1743. [DOI] [PubMed] [Google Scholar]
- *21.Denorme F, Manne BK, Portier I, et al. Platelet necrosis mediates ischemic stroke outcome in mice. Blood. 2020;135(6):429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study was the first to demonstrate a detrimental role for procoagulant platelets in a murine model of stroke and highlights the safe therapeutic potential of targeting procoagulant platelets for stroke.
- 22.Neelamegham S, Taylor AD, Shankaran H, Smith CW, Simon SI. Shear and time-dependent changes in Mac-1, LFA-1, and ICAM-3 binding regulate neutrophil homotypic adhesion. J Immunol. 2000;164(7):3798–3805. [DOI] [PubMed] [Google Scholar]
- 23.Ma YQ, Plow EF, Geng JG. P-selectin binding to P-selectin glycoprotein ligand-1 induces an intermediate state of alphaMbeta2 activation and acts cooperatively with extracellular stimuli to support maximal adhesion of human neutrophils. Blood. 2004;104(8):2549–2556. [DOI] [PubMed] [Google Scholar]
- 24.Kataoka H, Kim SW, Plesnila N. Leukocyte-endothelium interactions during permanent focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2004;24(6):668–676. [DOI] [PubMed] [Google Scholar]
- 25.Hallenbeck JM, Dutka AJ, Tanishima T, et al. Polymorphonuclear leukocyte accumulation in brain regions with low blood flow during the early postischemic period. Stroke. 1986;17(2):246–253. [DOI] [PubMed] [Google Scholar]
- 26.Watcharotayangul J, Mao L, Xu H, et al. Post-ischemic vascular adhesion protein-1 inhibition provides neuroprotection in a rat temporary middle cerebral artery occlusion model. J Neurochem. 2012;123Suppl 2:116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kitagawa K, Matsumoto M, Mabuchi T, et al. Deficiency of intercellular adhesion molecule 1 attenuates microcirculatory disturbance and infarction size in focal cerebral ischemia. J Cereb Blood Flow Metab. 1998;18(12):1336–1345. [DOI] [PubMed] [Google Scholar]
- 28.Soriano SG, Coxon A, Wang YF, et al. Mice deficient in Mac-1 (CD11b/CD18) are less susceptible to cerebral ischemia/reperfusion injury. Stroke. 1999;30(1):134–139. [DOI] [PubMed] [Google Scholar]
- 29.Krams M, Lees KR, Hacke W, et al. Acute Stroke Therapy by Inhibition of Neutrophils (ASTIN): an adaptive dose-response study of UK-279,276 in acute ischemic stroke. Stroke. 2003;34(11):2543–2548. [DOI] [PubMed] [Google Scholar]
- 30.Enlimomab Acute Stroke Trial I. Use of anti-ICAM-1 therapy in ischemic stroke: results of the Enlimomab Acute Stroke Trial. Neurology. 2001;57(8):1428–1434. [DOI] [PubMed] [Google Scholar]
- 31.Becker KJ. Anti-leukocyte antibodies: LeukArrest (Hu23F2G) and Enlimomab (R6.5) in acute stroke. Curr Med Res Opin. 2002;18 Suppl 2:s18–22. [DOI] [PubMed] [Google Scholar]
- 32.Hackam DG, Spence JD. Antiplatelet Therapy in Ischemic Stroke and Transient Ischemic Attack. Stroke. 2019;50(3):773–778. [DOI] [PubMed] [Google Scholar]
- 33.Remuzzi A, Languino LR, Costantini V, Guardabasso V, de Gaetano G, Dejana E. Platelet adhesion to subendothelium--effect of shear rate, hematocrit and platelet count on the dynamic equilibrium between platelets adhering to and detaching from the surface. Thromb Haemost. 1985;54(4):857–861. [PubMed] [Google Scholar]
- 34.Houdijk WP, de Groot PG, Nievelstein PF, Sakariassen KS, Sixma JJ. Subendothelial proteins and platelet adhesion. von Willebrand factor and fibronectin, not thrombospondin, are involved in platelet adhesion to extracellular matrix of human vascular endothelial cells. Arteriosclerosis. 1986;6(1):24–33. [DOI] [PubMed] [Google Scholar]
- 35.Qiu Y, Ciciliano J, Myers DR, Tran R, Lam WA. Platelets and physics: How platelets “feel” and respond to their mechanical microenvironment. Blood Rev. 2015;29(6):377–386. [DOI] [PubMed] [Google Scholar]
- 36.Kuijper PH, Gallardo Torres HI, van der Linden JA, et al. Platelet-dependent primary hemostasis promotes selectin- and integrin-mediated neutrophil adhesion to damaged endothelium under flow conditions. Blood. 1996;87(8):3271–3281. [PubMed] [Google Scholar]
- 37.Hamburger SA, McEver RP. GMP-140 mediates adhesion of stimulated platelets to neutrophils. Blood. 1990;75(3):550–554. [PubMed] [Google Scholar]
- 38.Simon DI, Chen Z, Xu H, et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med. 2000;192(2):193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Gao H, Shi C, et al. Leukocyte integrin Mac-1 regulates thrombosis via interaction with platelet GPIbalpha. Nat Commun. 2017;8:15559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tersteeg C, Heijnen HF, Eckly A, et al. FLow-induced PRotrusions (FLIPRs): a platelet-derived platform for the retrieval of microparticles by monocytes and neutrophils. Circ Res. 2014;114(5):780–791. [DOI] [PubMed] [Google Scholar]
- 41.Flick MJ, Du X, Witte DP, et al. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest. 2004;113(11):1596–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuijper PH, Gallardo Tores HI, Lammers JW, Sixma JJ, Koenderman L, Zwaginga JJ. Platelet associated fibrinogen and ICAM-2 induce firm adhesion of neutrophils under flow conditions. Thromb Haemost. 1998;80(3):443–448. [PubMed] [Google Scholar]
- **43.Constantinescu-Bercu A, Grassi L, Frontini M, Salles C II, Woollard K, Crawley JT. Activated alphaIIbbeta3 on platelets mediates flow-dependent NETosis via SLC44A2. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrated a novel interaction between platelets and neutrophils that can mediate NET formation under conditions of shear stress. These findings shed new light on how platelets mediate NET formation and offer insights into how NETs are formed in the blood stream during thrombosis.
- 44.Bennett JA, Mastrangelo MA, Ture SK, et al. The choline transporter Slc44a2 controls platelet activation and thrombosis by regulating mitochondrial function. Nat Commun. 2020;11(1):3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tilburg J, Coenen DM, Zirka G, et al. SLC44A2 deficient mice have a reduced response in stenosis but not in hypercoagulability driven venous thrombosis. J Thromb Haemost. 2020;18(7):1714–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zirka G, Robert P, Tilburg J, et al. Impaired adhesion of neutrophils expressing Slc44a2/HNA-3b to VWF protects against NETosis under venous shear rates. Blood. 2021;137(16):2256–2266. [DOI] [PubMed] [Google Scholar]
- 47.Agbani EO, Poole AW. Procoagulant platelets: generation, function, and therapeutic targeting in thrombosis. Blood. 2017;130(20):2171–2179. [DOI] [PubMed] [Google Scholar]
- 48.Yuan Y, Alwis I, Wu MCL, et al. Neutrophil macroaggregates promote widespread pulmonary thrombosis after gut ischemia. Sci Transl Med. 2017;9(409). [DOI] [PubMed] [Google Scholar]
- 49.Kulkarni S, Woollard KJ, Thomas S, Oxley D, Jackson SP. Conversion of platelets from a proaggregatory to a proinflammatory adhesive phenotype: role of PAF in spatially regulating neutrophil adhesion and spreading. Blood. 2007;110(6):1879–1886. [DOI] [PubMed] [Google Scholar]
- 50.Yao Z, Wang L, Wu X, et al. Enhanced Procoagulant Activity on Blood Cells after Acute Ischemic Stroke. Transl Stroke Res. 2017;8(1):83–91. [DOI] [PubMed] [Google Scholar]
- *51.Zhou P, Li T, Jin J, et al. Interactions between neutrophil extracellular traps and activated platelets enhance procoagulant activity in acute stroke patients with ICA occlusion. EBioMedicine. 2020;53:102671. [DOI] [PMC free article] [PubMed] [Google Scholar]; This was the first study to demonstrate the presence of procoagulant platelets and NETs in the cerebral circulation of ischemic stroke patients.
- 52.Prodan CI, Vincent AS, Dale GL. Coated-platelet levels are elevated in patients with transient ischemic attack. Transl Res. 2011;158(1):71–75. [DOI] [PubMed] [Google Scholar]
- 53.Kirkpatrick AC, Vincent AS, Dale GL, Prodan CI. Coated-platelets predict stroke at 30 days following TIA. Neurology. 2017;89(2):125–128. [DOI] [PubMed] [Google Scholar]
- 54.Prodan CI, Stoner JA, Cowan LD, Dale GL. Higher coated-platelet levels are associated with stroke recurrence following nonlacunar brain infarction. J Cereb Blood Flow Metab. 2013;33(2):287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kirkpatrick AC, Stoner JA, Dale GL, Prodan CI. Elevated coated-platelets in symptomatic large-artery stenosis patients are associated with early stroke recurrence. Platelets. 2014;25(2):93–96. [DOI] [PubMed] [Google Scholar]
- 56.Kirkpatrick AC, Vincent AS, Dale GL, Prodan CI. Increased platelet procoagulant potential predicts recurrent stroke and TIA after lacunar infarction. J Thromb Haemost. 2020;18(3):660–668. [DOI] [PubMed] [Google Scholar]
- 57.Kirkpatrick AC, Stoner JA, Dale GL, Rabadi M, Prodan CI. Higher Coated-Platelet Levels in Acute Stroke are Associated with Lower Cognitive Scores at Three Months Post Infarction. J Stroke Cerebrovasc Dis. 2019;28(9):2398–2406. [DOI] [PubMed] [Google Scholar]
- *58.Senchenkova EY, Ansari J, Becker F, et al. Novel Role for the AnxA1-Fpr2/ALX Signaling Axis as a Key Regulator of Platelet Function to Promote Resolution of Inflammation. Circulation. 2019;140(4):319–335. [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study, reducing levels of procoagulant platelets with recombinat AnnexinA1 reduced platelet-neutrophil interactions and improved ischemic stroke outcomes in mice. This study demonstrated the fact that procoagulant platelets can be targeted by therapeutic interventions to improve ischemic stroke outcomes.
- 59.Kleinschnitz C, Pozgajova M, Pham M, Bendszus M, Nieswandt B, Stoll G. Targeting platelets in acute experimental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation. 2007;115(17):2323–2330. [DOI] [PubMed] [Google Scholar]
- 60.Ye F, Garton HJL, Hua Y, Keep RF, Xi G. The Role of Thrombin in Brain Injury After Hemorrhagic and Ischemic Stroke. Transl Stroke Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choo HJ, Saafir TB, Mkumba L, Wagner MB, Jobe SM. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arterioscler Thromb Vasc Biol. 2012;32(12):2946–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pang A, Cui Y, Chen Y, et al. Shear-induced integrin signaling in platelet phosphatidylserine exposure, microvesicle release, and coagulation. Blood. 2018;132(5):533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Denorme F, Portier I, Kosaka Y, Campbell RA. Hyperglycemia exacerbates ischemic stroke outcome independent of platelet glucose uptake. J Thromb Haemost. 2021;19(2):536–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen Z, Chopp M, Zacharek A, et al. Brain-Derived Microparticles (BDMPs) Contribute to Neuroinflammation and Lactadherin Reduces BDMP Induced Neuroinflammation and Improves Outcome After Stroke. Front Immunol. 2019;10:2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Castellanos M, Sobrino T, Millan M, et al. Serum cellular fibronectin and matrix metalloproteinase-9 as screening biomarkers for the prediction of parenchymal hematoma after thrombolytic therapy in acute ischemic stroke: a multicenter confirmatory study. Stroke. 2007;38(6):1855–1859. [DOI] [PubMed] [Google Scholar]
- 66.Ikegame Y, Yamashita K, Hayashi S, Yoshimura S, Nakashima S, Iwama T. Neutrophil elastase inhibitor prevents ischemic brain damage via reduction of vasogenic edema. Hypertens Res. 2010;33(7):703–707. [DOI] [PubMed] [Google Scholar]
- 67.Armao D, Kornfeld M, Estrada EY, Grossetete M, Rosenberg GA. Neutral proteases and disruption of the blood-brain barrier in rat. Brain Res. 1997;767(2):259–264. [DOI] [PubMed] [Google Scholar]
- 68.von Bruhl ML, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(4):819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.MacGregor IR, Perrie AM, Donnelly SC, Haslett C. Modulation of human endothelial thrombomodulin by neutrophils and their release products. Am J Respir Crit Care Med. 1997;155(1):47–52. [DOI] [PubMed] [Google Scholar]
- 70.Jordan RE, Nelson RM, Kilpatrick J, Newgren JO, Esmon PC, Fournel MA. Inactivation of human antithrombin by neutrophil elastase. Kinetics of the heparin-dependent reaction. J Biol Chem. 1989;264(18):10493–10500. [PubMed] [Google Scholar]
- 71.Kataoka H, Hamilton JR, McKemy DD, et al. Protease-activated receptors 1 and 4 mediate thrombin signaling in endothelial cells. Blood. 2003;102(9):3224–3231. [DOI] [PubMed] [Google Scholar]
- 72.Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, Coughlin SR. Cathepsin G activates protease-activated receptor-4 in human platelets. J Biol Chem. 2000;275(10):6819–6823. [DOI] [PubMed] [Google Scholar]
- 73.Lindsberg PJ, Siren AL, Feuerstein GZ, Hallenbeck JM. Antagonism of neutrophil adherence in the deteriorating stroke model in rabbits. J Neurosurg. 1995;82(2):269–277. [DOI] [PubMed] [Google Scholar]
- 74.Gasche Y, Copin JC, Sugawara T, Fujimura M, Chan PH. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2001;21(12):1393–1400. [DOI] [PubMed] [Google Scholar]
- 75.Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463–469. [DOI] [PubMed] [Google Scholar]
- 76.Caudrillier A, Kessenbrock K, Gilliss BM, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012;122(7):2661–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. [DOI] [PubMed] [Google Scholar]
- 78.Allen C, Thornton P, Denes A, et al. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J Immunol. 2012;189(1):381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Euler M, Hoffmann MH. The double-edged role of neutrophil extracellular traps in inflammation. Biochem Soc Trans. 2019;47(6):1921–1930. [DOI] [PubMed] [Google Scholar]
- 80.Gould TJ, Vu TT, Swystun LL, et al. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler Thromb Vasc Biol. 2014;34(9):1977–1984. [DOI] [PubMed] [Google Scholar]
- 81.Martinod K, Demers M, Fuchs TA, et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A. 2013;110(21):8674–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.De Meyer SF, Suidan GL, Fuchs TA, Monestier M, Wagner DD. Extracellular chromatin is an important mediator of ischemic stroke in mice. Arterioscler Thromb Vasc Biol. 2012;32(8):1884–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.del Zoppo GJ, Schmid-Schonbein GW, Mori E, Copeland BR, Chang CM. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke. 1991;22(10):1276–1283. [DOI] [PubMed] [Google Scholar]
- 84.Perez-de-Puig I, Miro-Mur F, Ferrer-Ferrer M, et al. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol. 2015;129(2):239–257. [DOI] [PubMed] [Google Scholar]
- *85.El Amki M, Gluck C, Binder N, et al. Neutrophils Obstructing Brain Capillaries Are a Major Cause of No-Reflow in Ischemic Stroke. Cell Rep. 2020;33(2):108260. [DOI] [PubMed] [Google Scholar]; In a thrombotic mouse model, the authors of this study demonstrated a significant contribution of neutrophils to cappilary plugging after t-PA induced thrombolysis. Depletion of neutrophils improved stroke outcomes when combined with t-PA.
- 86.Erdener SE, Tang J, Kilic K, et al. Dynamic capillary stalls in reperfused ischemic penumbra contribute to injury: A hyperacute role for neutrophils in persistent traffic jams. J Cereb Blood Flow Metab. 2021;41(2):236–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rolfes L, Riek-Burchardt M, Pawlitzki M, et al. Neutrophil granulocytes promote flow stagnation due to dynamic capillary stalls following experimental stroke. Brain Behav Immun. 2021;93:322–330. [DOI] [PubMed] [Google Scholar]
- 88.Puhr-Westerheide D, Schink SJ, Fabritius M, et al. Neutrophils promote venular thrombosis by shaping the rheological environment for platelet aggregation. Sci Rep. 2019;9(1):15932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mertens P, Maes A, Nuyts J, et al. Recombinant P-selectin glycoprotein ligand-immunoglobulin, a P-selectin antagonist, as an adjunct to thrombolysis in acute myocardial infarction. The P-Selectin Antagonist Limiting Myonecrosis (PSALM) trial. Am Heart J. 2006;152(1):125 e121–128. [DOI] [PubMed] [Google Scholar]
- 90.Stahli BE, Gebhard C, Duchatelle V, et al. Effects of the P-Selectin Antagonist Inclacumab on Myocardial Damage After Percutaneous Coronary Intervention According to Timing of Infusion: Insights From the SELECT-ACS Trial. J Am Heart Assoc. 2016;5(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Investigators E Use of a monoclonal antibody directed against the platelet glycoprotein IIb/IIIa receptor in high-risk coronary angioplasty. N Engl J Med. 1994;330(14):956–961. [DOI] [PubMed] [Google Scholar]
- 92.Ciccone A, Motto C, Abraha I, Cozzolino F, Santilli I. Glycoprotein IIb-IIIa inhibitors for acute ischaemic stroke. Cochrane Database Syst Rev. 2014(3):CD005208. [DOI] [PubMed] [Google Scholar]
- 93.Nighoghossian N, Berthezene Y, Mechtouff L, et al. Cyclosporine in acute ischemic stroke. Neurology. 2015;84(22):2216–2223. [DOI] [PubMed] [Google Scholar]
- 94.Laridan E, Denorme F, Desender L, et al. Neutrophil extracellular traps in ischemic stroke thrombi. Ann Neurol. 2017;82(2):223–232. [DOI] [PubMed] [Google Scholar]
- **95.Novotny J, Oberdieck P, Titova A, et al. Thrombus NET content is associated with clinical outcome in stroke and myocardial infarction. Neurology. 2020;94(22):e2346–e2360. [DOI] [PubMed] [Google Scholar]; This was the first study to compared ischemic stroke thrombi with clots obtained from patients with an acute myocaridal infarction. NETs were abundantly present in all ischemic stroke clots, while only 20% of acute myocaridal infarction thrombi contained NETs. Interestingly, the amount of NETs in the thrombi associated with long term stroke outcomes.
- 96.Ducroux C, Di Meglio L, Loyau S, et al. Thrombus Neutrophil Extracellular Traps Content Impair tPA-Induced Thrombolysis in Acute Ischemic Stroke. Stroke. 2018;49(3):754–757. [DOI] [PubMed] [Google Scholar]