

Abstract
Glycosaminoglycans (GAGs) such as heparan sulfate and chondroitin sulfate decorate all mammalian cell surfaces. These mucopolysaccharides act as coreceptors for extracellular ligands, regulating cell signaling, growth, proliferation, and adhesion. In glioblastoma, the most common type of primary malignant brain tumor, dysregulated GAG biosynthesis results in altered chain length, sulfation patterns, and the ratio of contributing monosaccharides. These events contribute to the loss of normal cellular function, initiating and sustaining malignant growth. Disruption of the aberrant cell surface GAGs with small molecule inhibitors of GAG biosynthetic enzymes is a potential therapeutic approach to blocking the rogue signaling and proliferation in glioma, including glioblastoma. Previously, 4-azido-xylose-α-UDP sugar inhibited both xylosyltransferase (XYLT-1) and β−1,4-galactosyltransferase-7 (β-GALT-7)—the first and second enzymes of GAG biosynthesis—when microinjected into a cell. In another study, 4-deoxy-4-fluoro-β-xylosides inhibited β-GALT-7 at 1 mM concentration in vitro. In this work, we seek to solve the enduring problem of drug delivery to human glioma cells at low concentrations. We developed a library of hydrophobic, presumed prodrugs 4-deoxy-4-fluoro-2,3-dibenzoyl-(α- or β-) xylosides and their corresponding hydrophilic inhibitors of XYLT-1 and β-GALT-7 enzymes. The prodrugs were designed to be activatable by carboxylesterase enzymes overexpressed in glioblastoma. Using a colorimetric MTT assay in human glioblastoma cell lines, we identified a prodrug–drug pair (4-nitrophenyl-α-xylosides) as lead drug candidates. The candidates arrest U251 cell growth at an IC50 = 380 nM (prodrug), 122 μM (drug), and U87 cells at IC50 = 10.57 μM (prodrug). Molecular docking studies were consistent with preferred binding of the α- versus β-nitro xyloside conformer to XYLT-1 and β-GALT-7 enzymes.
Keywords: glycosaminoglycan biosynthesis inhibitors, α-xylosides, β-xylosides, glioblastoma, prodrugs, heparan sulfate, chondroitin sulfate, U251, U87
INTRODUCTION
All mammalian cell surfaces express membrane bound and secreted heparan sulfate (HS) and/or chondroitin sulfate (CS) proteoglycans (PGs), a class of protein–glycan complexes.1 HS and CS are glycosaminoglycans (GAGs) with repeating disaccharides composed of uronic acid (glucoronic acid or iduronic acid) and amino sugars (N-acetyl-glucosamine in HS and N-acetyl-galactosamine in CS).2 Biosynthesis of HS and CS begins in the endoplasmic reticulum (ER) of the cell with covalent attachment of the xylose sugar to the serine amino acid of a core protein.3 The xylosyltransferase-1 enzyme (XYLT-1) catalyzes the first step. β−1,4-Galactosyltransferase-7 (β-GALT-7) then transfers a galactose unit to the xylose sugar (Figure 1). Subsequent addition of another galactose and glucuronic acid residues result in the conserved tetrasaccharide structure of HS and CS.4 After this point, GAG biosynthesis diverges: addition of N-acetyl glucosamine results in HS, while addition of N-acetyl galactosamine produces CS (Figure 1). Numerous biosynthetic enzymes in the Golgi complex further modify HS and CS, resulting in heterogeneous, negatively charged HS and CS structures with distinct chain lengths, sulfation patterns, and glucuronic/iduronic acid ratios.5 These structural differences of the cell surface glycans impart divergent physiological functions related to cell adhesion, growth, and proliferation. The HS or CS act as coreceptors, mediating interactions between the core protein and extracellular ligands to induce cellular signaling.6,7
Figure 1.

Heparan sulfate and chondroitin sulfate biosynthesis.
Alterations in cell surface HS and CS are believed to contribute to cancer progression, proliferation, invasion, angiogenesis, and metastasis.8 Studies have shown that these alterations are related to changes in the expression and/or function of GAG biosynthetic enzyme actions, which trigger errors in the normal signaling cascades.9–14 Due to the role of GAGs in tumorigenesis, the inhibition of GAG biosynthesis may be a therapeutically useful strategy. Pan et al. showed increased expression of chondroitin 4-sulfate (CS4) and chondroitin 6-sulfate (CS6) in human glioma tissues compared to normal brain tissue. Inhibition of CS4 and CS6 expression through siRNAs reduced glioma malignancy.15 We hypothesize that aberrant cell surface HS and CS of glioma can be reduced by the chemical inhibition of target biosynthetic enzymes. The work presented in this manuscript focuses on the development of xyloside-derived small molecule inhibitors targeting the first two biosynthetic enzymes: XYLT-1 and β-GALT-7.
In developing these inhibitors, we were guided by two hypotheses: (a) benzoyl ester derivatives of inhibitors may show enhanced effect via a prodrug mechanism, whereby intracellular carboxylesterases release the active, hydrophilic drugs, and (b) α- or β-stereochemistry of xyloses is likely to show differential inhibition of XYLT-1/ β-GALT-7 with a corresponding decrease in glioma cell proliferation (Figure 2). Human brain expresses carboxylesterase enzyme (EC 3.1.1.1) that hydrolyzes a wide variety of esters.16 For example, in developing a peroxide-sensitive fluorescent dye, Chiu et al. showed liberation of a diacetate ester probe via carboxylesterases in U87 glioma cells.17 In another report, elevated carboxylesterases in glioma (U87MG, T87G) induced self-assembly of the hydrolyzed product to selectively kill cancer cells.18
Figure 2.

Proposed mechanism of prodrug action. Before drug treatment, the GAGs–protein complex, collectively known as proteoglycan (PG), decorates all cell surfaces and the extracellular matrix. The GAG coreceptor can extend to neighboring membrane proteins and presents various ligands such as growth factors, cytokines, and matrix proteins. When lipophilic prodrug is added to the cell, it undergoes passive diffusion through the membrane, hydrolysis by carboxylesterase (EC 3.1.1.1) enzymes, and inhibition of the GAG biosynthetic pathway. The drug thereby engenders truncation of the GAG coreceptor and disruption of cell signaling.
A few inhibitors of XYLT-1 or β-GALT-7 enzyme are reported. In previous work, a 4-azido-xylose-α-UDP sugar—an inhibitor of XYLT-1 and GALT-7 enzymes—microinjected in zebra fish resulted in HS and CS abrogation.19 X-ray crystallography suggested that the XYLT-1 enzyme catalyzed a nucleophilic substitution reaction (SN2-like mechanism) between the serine residue (nucleophile) of an acceptor peptide and xylose-α-UDP sugar (substrate with leaving group).20,21 However, substituting the 4-hydroxyl group of the α-substrate with an azide group prevented the β-GALT-7 enzyme from adding a galactose residue to it.
4-Deoxy-4-fluoro-β-xylosides inhibited GAG biosynthesis in Chinese Hamster Ovarian (CHO-K1) cells at 1 mM and bovine lung microvasculature endothelial cells (BLMVEC) at 300 μM.22,23 The α- and β-isomers of xylosides studied were limited only to aromatic, bulky substituents in the triazole ring. The separation of these two isomers required reverse phase high-performance liquid chromatography (HPLC) in the final step. These sugar derivatives contained two groups: (a) a 4-fluoro-xylose scaffold targeting the enzyme active site and (b) a hydrophobic aglycon group at the reducing end to enhance cell membrane permeation and binding affinity with the enzyme. The X-ray crystal structure of β-GALT-7 and the bound β-xyloside elucidated the presumed mechanism of inhibition.24,25
The reported inhibitors required either a high concentration (300 μM to 1 mM) or microinjection for GAG inhibition to affect the tumor phenotype. In this work, we devised a synthetic plan to access a library of α- or β-xylosides (prodrug and drug) for both aliphatic and aromatic aglycon groups, screened their therapeutic efficacy in glioblastoma cells through an in vitro MTT colorimetric assay, and discovered an α-prodrug–drug pair of lead candidates. Finally, a molecular modeling study with XYLT-1 and β-GALT-7 enzymes supported our observed success of α-xyloside.
EXPERIMENTAL SECTION
Materials and Reagents.
All reactions were done under dry and nitrogen atmosphere unless mentioned otherwise. Anhydrous chemicals and solvents were purchased from commercial suppliers. Column chromatography was done with manual column chromatography using silica gel 60 (36–71 μm, Alfa Aesar) as solid phase. Some purification was done with SilicaFlash R60 gel (20–45 μm, SiliCycle Inc.) as mentioned in the Table S1. Thin layer chromatography (TLC) was conducted on precoated polyester sheets (40 × 80 mm) from Machery-Nagel (POLYGRAM SIL G/UV254) with 0.2 mm silica gel 60 with fluorescent indicator. For visualization, a UV light source (254 and 366 nm) was used. The U251MG human cell line was purchased from Sigma (catalog 09063001). U87 MG cells (ATCC HTB-14) were purchased from the ATCC.
Instrumentation.
Nuclear magnetic resonance spectroscopy (NMR) was measured on a Bruker Avance III HD 400 equipped with a (1H/13C/19F) probe head. Chemical shifts (δ) are given in ppm; coupling constants J are in Hertz (Hz), and the multiplicities of the signals are designated as follows: s = singlet, bs = broad singlet, d = doublet, t = triplet, and m = multiplet. Reference for 1H, 13C is tetramethylsilane (TMS). The solvent signals of CDCl3 and CD3OD were calibrated on 7.26 and 3.34 ppm for 1H NMR and 77.0 and 49.15 ppm for 13C NMR spectra. Notre Dame Mass Spectrometry & Proteomics Facility recorded high-resolution mass spectra (HRMS). MTT assay was carried out using CellTiter 96 Non-Radioactive Cell Proliferation Assay (Promega, #G4100). A 96-well plate reader BIOTEK/SUNERGY 2 was used.
Synthesis of 1-Azido-2,3,4-tribenzoyl-l-arabinose (Scheme 1, Compounds 3A, 3B).
Scheme 1.

Synthesis of α- or β-Isomers of 4-Deoxy-4-fluoro-2,3-dibenzoyl-xylose Azide (5A, 5B)
To a solution of 2 (Scheme 1) (10.3 g, 17.65 mmol) in dry DCM (200 mL) was added azido-trimethylsilane (3.5 mL, 26.47 mmol) dropwise, followed by tin(IV) chloride (1.03 mL, 8.82 mmol) addition. The reaction was stirred at room temperature for 16 h. TLC analysis showed two products of Rf values 0.49 (α-isomer) and 0.31 (β-isomer) in 7:3 hexane:ethyl acetate (H:E) thin layer chromatography (TLC) solvent. DCM was evaporated off, and crude reaction was dissolved in ethyl acetate (250 mL). The organic layer was washed with saturated NaHCO3 (5 × 50 mL) and brine (2 × 50 mL), dried over anhydrous Na2SO4, and evaporated off through a Rotavap. Finally, the α- and β-isomers were separated through silica gel column chromatography in H:E (9:1). This yielded 3.67 g of 3A (42.6% overall yield) and 3.44 g of 3B (40% overall yield).
Compound 3A.
1HNMR (CDCl3, 400 MHz): δ 8.13–8.06 (m, 2H), 8.04–7.97 (m, 2H), 7.89–7.82 (m, 2H), 7.60 (d, J = 7.4 Hz, 1H), 7.57–7.37 (m, 6H), 7.33–7.24 (m, 2H), 5.88 (d, J = 3.8 Hz, 1H), 5.84 (dd, J = 10.2, 3.3 Hz, 1H), 5.82–5.72 (m, 2H), 4.36 (dd, J = 13.4, 1.6 Hz, 1H), 4.11 (dd, J = 13.3, 2.2 Hz, 1H). 13CNMR (CDCl3, 400 MHz): δ 165.82, 165.67, 165.46, 133.72, 133.55, 133.36, 129.96, 129.89, 129.74, 129.40, 129.02, 128.69, 128.62, 128.58, 128.38, 87.64, 69.54, 68.54, 67.61, 62.61. HRMS (ESI): calcd for C26H21N3NaO7 [M + Na]+ 510.1272, found 510.1268
Compound 3B.
1HNMR (CDCl3, 400 MHz): δ 8.11–8.04 (m, 2H), 8.04–7.97 (m, 2H), 7.92–7.85 (m, 2H), 7.65–7.38 (m, 7H), 7.36–7.24 (m, 3H), 5.76–5.67 (m, 2H), 5.61 (dd, J = 9.3, 3.4 Hz, 1H), 4.95 (d, J = 7.6 Hz, 1H), 4.41 (dd, J = 13.1, 3.3 Hz, 1H), 4.02 (dd, J = 13.2, 1.8 Hz, 1H). 13CNMR (CDCl3, 400 MHz): 165.62, 165.51, 165.21, 133.63, 133.60, 133.49, 129.91, 129.84, 129.24, 128.84, 128.78, 128.63, 128.53, 128.45, 88.66, 70.71, 69.22, 68.40, 65.38. HRMS (ESI): Calcd for C26H21N3NaO7 [M + Na]+ 510.1272, found 510.1271.
Synthesis of Prodrug (1-(2,3-Di-O-benzoyl-4-deoxy-4-fluoro-d-xylopyranosyl)-4-(4-nitrophenyl)-1,2,3-triazole) (Scheme 2, Compound 6A-NO2).
Scheme 2.

Synthesis of Copper(I) Mediated 6A-NO2 Prodrug and 6A′-NO2 Drug
Compound 5A (50 mg, 0.13 mmol) and an alkyne (0.194 mmol) (Scheme 2) were charged into a 50 mL RB flask equipped with a stir bar. A cocktail of acetone/water/THF (1:1:1, 3 mL) was added and stirred vigorously. Freshly prepared 1 M sodium ascorbate (52 μL, 0.052 mmol) was added to the above reaction. After 5 min, a freshly prepared aqueous solution of 1 M copper sulfate (26 μL, 0.026 mmol) was added. The reaction mixture was stirred at room temperature for three days depending on monitoring via TLC (see Supporting Table S1). The organic compound was extracted with ethyl acetate (3 × 5 mL), washed with brine (2 × 5 mL), dried over anhydrous Na2SO4, and evaporated in a Rotavap. The crude reaction was purified through column chromatography in silica gel (20–45 μm) hexane:ethyl acetate 4:1 (250 mL) to 7:3 (500 mL). Percentage yield: 25%. 1HNMR (CDCl3, 400 MHz): δ 8.25–8.17 (m, 2H), 8.14–8.06 (m, 3H), 8.00–7.93 (m, 2H), 7.85–7.78 (m, 2H), 7.69–7.46 (m, 4H), 7.51–7.38 (m, 2H), 6.55 (d, J = 3.1 Hz, 1H), 6.12 (dt, J = 9.5, 5.0 Hz, 1H), 5.65 (dd, J = 5.2, 3.1 Hz, 1H), 4.87 (dtd, J = 45.7, 4.6, 2.8 Hz, 1H), 4.46 (tdd, J = 11.7, 4.4, 1.1 Hz, 1H), 4.35 (dd, J = 13.2, 2.9 Hz, 1H), 4.28 (dd, J = 13.2, 2.9 Hz, 1H). 13CNMR (CDCl3, 400 MHz): δ 164.78, 164.35, 147.48, 145.58, 136.21, 134.09, 134.05, 129.98, 129.94, 128.77, 128.45, 128.24, 126.22, 124.27, 121.36, 84.93, 83.53, 83.09, 68.26, 68.24, 67.92, 67.65, 66.06, 65.82. 19FNMR (CDCl3, 400 MHz): δ −194.64. HRMS (ESI): Calcd for C27H22FN4O7 [M + H]+ 533.1467, found 533.1468.
Synthesis of Drug (1-(4-Deoxy-4-fluoro-β-d-xylopyranosyl)-4-(4-nitrophenyl)-1,2,3-triazole) (Scheme 2, Compound 6A′-NO2).
Compound 6A-NO2 (33 mg, 0.062 mmol) was treated with sodium methoxide (0.156 mmol) in methanol (5 mL) in a glass vial for 4 h at room temperature. The pH of the reaction was brought to neutral by adding Amberlite IR-120 hydrogen form and immediately filtered through glass wool. Methanol was evaporated through a Rotavap, and the crude reaction was purified in silica gel (20–45 μm) column chromatography using hexane:ethyl acetate 1:1 solvent system. Percentage yield: 95%. 1HNMR (CD3OD, 400 MHz): δ 8.74 (s, 1H), 8.41–8.25 (m, 2H), 8.21–8.05 (m, 2H), 6.23 (d, J = 2.9 Hz, 1H), 4.72–4.44 (m, 1H), 4.36 (dt, J = 9.8, 4.9 Hz, 1H), 4.29–4.12 (m, 2H), 3.99 (dd, J = 5.2, 3.0 Hz, 1H). 13CNMR (CD3OD, 400 MHz): δ 147.40, 144.74, 136.76, 132.85, 129.14, 128.15, 127.69, 125.97, 123.91, 123.19, 88.78, 85.41, 72.06, 69.69, 69.00, 68.78, 65.71, 65.47. 19FNMR (CD3OD, 400 MHz): δ −194.64. HRMS (ESI): Calcd for C13H14FN4O5 [M + H]+ 325.0943, found 325.0943
MTT Assay.
U251 (or U87) cells were plated in a 96-well plate (2000 cells/well/100 μL) and incubated overnight. The next day, media was aspirated and fresh media containing the desired compounds (prodrug or drug stock solutions in DMSO) were added. The cells were incubated for days 1, 2, 3, or 4 and MTT proliferation assay was performed as recommended by the manufacturer (Promega, #G4100).
The data: x-axis = concentration, y-axis = corrected absorbance (absorbance at 570 nm – background at 650 nm) are plotted in Prism software (version 8.4.3).
RESULTS AND DISCUSSION
Synthesis of 4-Deoxy-4-fluoro-xylose-derived Inhibitors.
During GAG biosynthesis, the replacement of the carbon-4 hydroxyl group of xyloside with other groups prevents catalytic transfer via β-GALT-7 enzyme.24,26 4-Deoxy-4-fluoro-xylose sugar linked to 4-methylumbelliferone (IC50 = 0.06 mM, Ki = 0.03 mM) inhibits human recombinant β-GALT-7 at a significantly lower concentration than its 4-deoxy analogue (IC50 = 1.28 mM, Ki = 0.53 mM).24 Such a dramatic difference in activity stems from the equatorial 4-fluoro atom’s ability to bind the enzyme tightly through its oxygen-like electronegativity and van der Waals radius. Siegbahn et al. synthesized a library of carbon-4 modified xylosides and studied their inhibitory potential. 4-Deoxy-4-fluoro xyloside was one of the top inhibitors.26 These studies motivated us to consider 4-fluoro-triazole-xylosides as inhibitors of XYLT-1 and β-GALT-7 enzymes.
In considering xyloside-derived inhibitors of XYLT-1 and β-GALT-7, we elaborated a series of fluorine-modified molecules, suggested by both previous structural data and literature precedent for biochemically inert 19F drugs (Scheme 1).
We also adopted prodrug approach to address concerns about inhibitor delivery at the optimum concentration required for biological effect. We designed a synthetic route to distinct α- or β-isomers of 4-deoxy-4-fluoro-2,3-dibenzoyl xylose azides (Scheme 1, compounds 5A and 5B). We reasoned that benzoyl esters of the prodrug enhance their cellular uptake, retention, and target carboxylesterase enzymes simultaneously.27 We used the azido group at the reducing end to initiate copper(I) aided azide–alkyne click chemistry to build α- or β-isomers of 4-deoxy-4-fluoro-2,3-dibenzoyl xylose azide (prodrug) and their corresponding hydrophilic, debenzoylated 4-deoxy-4-fluoro-xyloside (drug) library (see Supporting Information for experimental details). Copper(I) catalyzed azide–alkyne click chemistry afforded 1,4-disubstituted triazole rings on the reducing end of the sugar with a hydrophobic aglycon group on carbon-4. Cyclopentyl, phenyl, and 4-nitrophenyl groups served as aglycon groups in our study.
The chemical synthesis began with the protection of the hydroxyl groups (−OH) of l-(+)-arabinose as benzoyl ester supplied by benzoyl chloride; for a full description of syntheses, please see the Supporting Information. The crude product (2) of this reaction was reacted with azido-trimethylsilane in the presence of tin(IV) chloride catalyst to afford azidation at the anomeric carbon. Surprisingly, we observed two products on the thin layer chromatography (TLC) plate of Rf values 0.66 (α-isomer) and 0.54 (β-isomer) in 7:3 hexane:ethyl acetate in hexane:ethyl acetate solvent mixture at 7:3 ratio (see Supporting Table S2). The polarity difference was enough to purify the products through silica gel (0.036–0.071 mm, 215–400 mesh) column chromatography.
1H and 13C NMR and HPLC confirmed pure α- (Scheme 1, compound 3A) and β-azides (Scheme 1, compound 3B). The α-isomer was distinguished from the β-partner based on the differences in the chemical shift and coupling constant of the anomeric protons: δ 5.88 ppm, 3JH-1,H-2 = 3.8 Hz equatorial-axial coupling for compound 3A and δ 4.98 ppm, 3JH-1,H-2 = 7.5 Hz, axial–axial coupling for compound 3B (Figure 3A, B).23,28 The dihedral angle θ between H-1 and H-2 dictates the proton–proton coupling constant 3J in compounds 3A and 3B. The anomeric effect in the α-isomer (compound 3A) shifts the anomeric proton downfield. This stems from hyperconjugation between the nonbonding sp3 orbital of oxygen (highest occupied molecular orbital) and empty antibonding σ* orbital of C1–N (lowest unoccupied molecular orbital). This phenomenon reduces the electron density at the anomeric proton H-1 resulting downfield shift in the NMR spectrum (resonance structure III, Supporting Figure S1A).29,30 Karplus equation predicts coupling constant of 4 Hz for compound 3A (the dihedral angle of 60°) and 13 Hz in compound 3B (the dihedral angle of 180°) (Supporting Figure S1B).31–33
Figure 3.
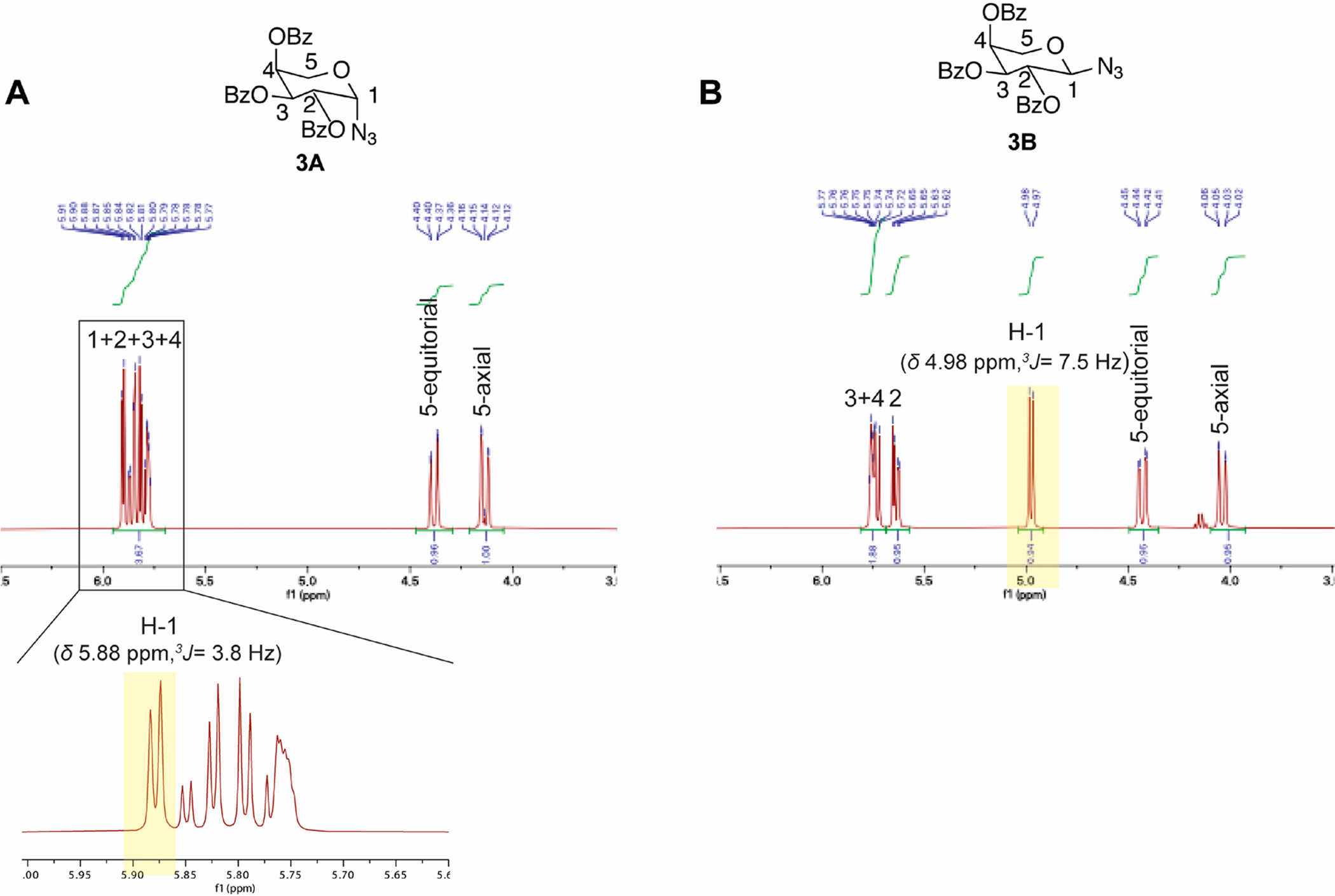
1HNMR of the xylose residue in compounds 3A and 3B. Significant difference in the chemical shifts of (A) anomeric proton H-1 δ 5.84 in compound 3A and (B) δ 4.98 compound 3B indicates the synthesis of α- and β-azides.
The syntheses split into α- and β-routes after the azidation reaction. Sodium methoxide treatment of the azido compounds 3A and 3B removed three benzoyl ester groups, resulting in trihydroxylated α- and β-azides and methyl benzoate side products, which were removed via repeated washing with hexane. Benzoyl chloride was again used to protect the 2,3-dihydroxyl groups of these semipure azides as benzoyl esters (Scheme 1, compounds 4A and 4B), leaving 4-hydroxy group intact for the final fluorination. The side products of this step, including tri- and undesired di- and mono-benzoyl esters were first recycled back to the trihydroxylated α- and β-azides (with sodium methoxide treatment) followed by selective 2,3-dibenzoylation (Scheme 1, compounds 4A and 4B). Diethylaminosulfur trifluoride (DAST) furnished the fluoride nucleophile (F−) to replace the −OH group on the carbon-4 of compounds 4A and 4B via the SN2 pathway, resulting 4-deoxy-4-fluoro-2,3-dibenzoyl-xylose-(α- or β-) azides (Scheme 1, compounds 5A and 5B) with an inversion of configuration at carbon-4. Finally, copper(I) catalyzes 1,3-dipolar cycloaddition reactions between azides and alkynes to generate a library of click-fluoro-xyloside prodrugs (section 3 of Supporting Information, compounds 6A and 6B).34 Treatment with sodium methoxide revealed the hydrophilic inhibitors of XYLT-1 and β-GALT-7 enzymes (Scheme 2, compounds 6A′ and 6B′).
An MTT Colorimetric Assay in U251 and U87 Glioma Cells Identified a Potential Prodrug/Drug Inhibitor Pair.
To screen this library of xylose-derived inhibitors targeting GAG, the inhibitors and their corresponding benzoyl adducts were applied to U251 cells, and MTT cell viability assays were performed (Supporting Figure S2). This study identified compound 6A-NO2, an α-prodrug with nanomolar IC50 value (380 nM). Its corresponding hydrophilic drug 6A′-NO2 showed effectiveness at high micromolar concentration (IC50 = 122 μM) with the observed difference between the compounds potentially related to cell permeability (Figure 4A–D and Supporting Figure S3). The hydrophobicity of the xyloside-derived prodrugs—imparted through benzoyl esters—likely plays a crucial role in cell penetration through the phospholipid bilayer. In the absence of any xylose specific ion channel, we hypothesized that the hydrophobic effect of the prodrug determines the rate of cellular entry. The accelerated cytotoxicity of the prodrug (6A-NO2) supports this hypothesis.
Figure 4.

MTT assay in glioblastoma cell lines. U251 cells (A–D). (A) compound 6A-NO2, a prodrug, shows concentration and time-dependent cell toxicity. The 2 μM prodrug showed 1.41-fold reduction in the absorbance when compared with the DMSO control (P = 0.0157, unpaired t test for day 2) and (B) hydrophilic drug 6A′-NO2 showed concentration and time (day) dependent cell toxicity. The 200 μM drug showed 2.27-fold reduction in the absorbance when compared with the DMSO control (P < 0.0001, unpaired t test for day 4). Corresponding IC50 of (C) compound 6A-NO2 and (D) compound 6A′-NO2 pooled from panels A and B, respectively. MTT assay in U87 cells (E, F). (E) Compound 6A-NO2 shows correlation between concentration (or time (days)) and cytotoxicity with 20 μM prodrug, producing a 1.37-fold reduction in the absorbance when compared with the DMSO control (P = 0.0010, unpaired t test for day 2). Corresponding IC50 plot (F) compound 6A-NO2 pooled from panel E. The plotting of concentration and normalized absorbance furnished the IC50 values of the compounds. The concentrations were expressed in logarithm scale, normalized, and plotted in Prism software (version 8.4.3). The data were fitted with nonlinear regression (curve fit) followed by log (inhibitor) v response-variable slope (four parameters).
We also used our lead inhibitors (6A-NO2 and 6A′-NO2) to perform MTT assay on U87 cells. The prodrug (6A-NO2) produced 50% cell death at 10.57 μM (Figure 4E, F). On the other hand, the drug (6A′-NO2) produced a more modest effect with 7% cell death at 200 μM (Supporting Figure S4). We attribute this effect to heterogeneity of cancer cells, their uptake potency, and expression of XYLT-1 and β-GALT-7 enzymes.35–37
Compound 6A-Ph (prodrug) and its hydrophilic partner 6A′-Ph (drug) also inhibited U251 cell viability, reinforcing the potential potency of the α-xylosides (Supporting Figure S2 E, F). To our knowledge, this is the first report of a xyloside-derived, GAG-targeted inhibitor inhibiting glioma cell viability. The β-partners of these molecules were ineffective (Supporting Figure S2), as were the other compounds screened. Most compounds in the library had no effect on the viability of U251 cells. Overall, MTT assay results provide a starting point for further mechanistic study and validation of this class of xyloside-derived inhibitors.
Molecular Modeling Provides Insight into Lead Compound Binding.
We used computational docking to understand the mode of binding of 6A′-NO2 to XYLT-1 and β-GALT-7. For XYLT-1, we used a recent crystal structure (PDB 6EJ7)21 containing an acceptor peptide with residue S215 mutated to A215. The predicted binding mode of 6A′-NO2 (Figure 5A) shows a hydrogen-bonding interaction between the hydroxyl groups of 6A′-NO2 and acidic residues (D494 and E529) of XYLT-1 as well as pi-stacking interaction between the triazole and nitrophenyl groups of 6A′-NO2 and W392 and W495 side chains of XYLT-1. The fluorine atom of 6A′-NO2 is only 3.5 Å from the methyl group of A215, the site of catalytic transport. Occupation of the F atom in this space likely inhibits the SN2-like nucleophilic transfer of xyloside (Figure 5A, Supporting Figure S5). In the model of 6A′-NO2 binding to the structurally unrelated β-GALT-7 enzyme (PDB 4IRQ), the binding is again predicted to be stabilized by pi-stacking involving the nitrophenyl and triazole rings, in this case with tyrosine residues (Y194, Y196, Y199) in the active site (Figure 5B).24
Figure 5.
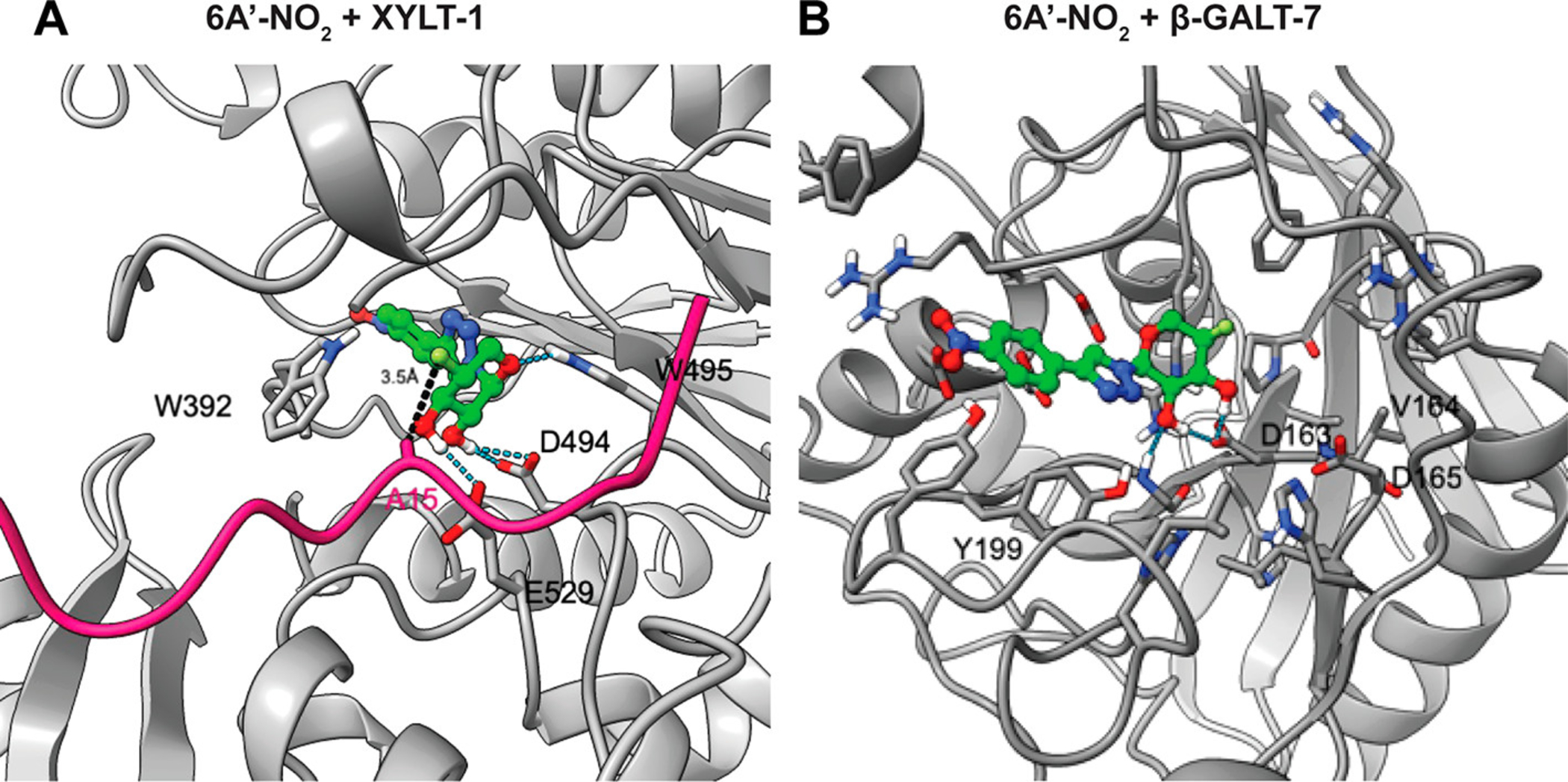
Docking model of 6A′-NO2 bound to (A) XYLT-1 (PDB 6EJ7) with the acceptor peptide from the crystal structure (with S215 mutated to A215) shown in pink color and (B) β-GALT-7 enzyme (PDB 4IRQ). Protein is shown in gray, and 6A′-F-NO2 carbon, nitrogen, oxygen, hydrogen, and fluorine atoms are shown in green, blue, red, white, and lime colors. Distance between β-carbon atom of A215 and the inhibitor F atom is also shown. Protein structures of XYLT-1 (PDB 6EJ7) and β-GALT-7 (PDB 4IRQ) enzymes were subjected to a protein preparation step using the Protein Preparation Wizard (Schrodinger Inc.). The coordinates of the cocrystallized sugar-donor substrate were used to define the center of the docking grid in both cases. Structures of our presumed inhibitors were prepared using Edit/Built panel of Maestro software (Schrodinger Inc.), energy minimized using LigPrep software (v.5.40749, Schrodinger Inc.), and subsequently docked to the active sites of XYLT-1 and β-GALT-7 using Glide docking software (v8.7, Schrodinger Inc.).
CONCLUSIONS
In this proof-of-concept study, we designed a synthetic pathway that resulted in a library of prodrugs and drugs consisting of α- and β-isomers of 4-deoxy-4-fluoro-xylosides and identified nanomolar to micromolar inhibitors of U251 and U87 glioma cell proliferation. For the 6A′-NO2 xylosides, the benzolyl ester adduct demonstrated enhanced performance relative to its hydrophilic adduct, suggesting improved cell membrane permeability. The compounds 6A-NO2 and 6A′-NO2 inhibited the growth of cells, likely mediated via HS and CS, consistent with numerous published studies. This effect likely occurs via disruption of the tumor microenvironment by altering the interactions between ligands (growth factors, cytokines, chemokines) or exosomes and GAG. A computational docking study supports binding of the α-xyloside in the active sites of XYLT-1 and β-GALT-7 enzymes. Future work will focus on mechanistic validation of this class of inhibitors and the development of more potent analogues with lower IC50 values for in vivo targeting of brain tumors. Successful separation of α- and β-azides at early stage of synthesis also gives us a handle for functional group transformation of azide to amines, amide (for amino acid coupling), and C–H insertion by nitrenes beyond click chemistry.38,39
Supplementary Material
Funding
Funding was received from the following sources: NIH R01 NS081117 (J.J.P.), U01CA168878 (J.J.P.), R01EB025985, R01EB024014, and DOD A132172.
ABBREVIATIONS
- GAG
glycosaminoglycan
- HS
heparan sulfate
- CS
chondroitin sulfate
- NMR
nuclear magnetic resonance
- TLC
thin layer chromatography
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.0c00839.
Detailed experimental procedures, including chemical syntheses, MTT assays, IC50 plots, and molecular modeling descriptions (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.molpharmaceut.0c00839
The authors declare the following competing financial interest(s): M.P.J. is a consultant to and shareholder of Schrodinger LLC, which licenses the software used in this work.
Contributor Information
Mausam Kalita, Department of Radiology and Biomedical Imaging, University of California, San Francisco, San Francisco, California 94158, United States; Department of Neurological Surgery, Brain Tumor Center University of California, San Francisco, San Francisco, California 94158, United States;.
Javier Villanueva-Meyer, Department of Radiology and Biomedical Imaging, University of California, San Francisco, San Francisco, California 94158, United States.
Yuki Ohkawa, Department of Neurological Surgery, Brain Tumor Center University of California, San Francisco, San Francisco, California 94158, United States.
Chakrapani Kalyanaraman, Department of Pharmaceutical Chemistry, University of California, San Francisco, San Francisco, California 94158, United States.
Katharine Chen, Department of Neurological Surgery, Brain Tumor Center University of California, San Francisco, San Francisco, California 94158, United States.
Esraa Mohamed, Department of Neurological Surgery, Brain Tumor Center University of California, San Francisco, San Francisco, California 94158, United States.
Matthew F. L. Parker, Department of Radiology and Biomedical Imaging, University of California, San Francisco, San Francisco, California 94158, United States
Matthew P. Jacobson, Department of Pharmaceutical Chemistry, University of California, San Francisco, San Francisco, California 94158, United States;.
Joanna J. Phillips, Department of Neurological Surgery, Brain Tumor Center University of California, San Francisco, San Francisco, California 94158, United States; Helen Diller Family Comprehensive Cancer Center, University of California, San Francisco, San Francisco, California 94158, United States; Department of Pathology, Division of Neuropathology University of California, San Francisco, San Francisco, California 94143, United States
Michael J. Evans, Department of Radiology and Biomedical Imaging, University of California, San Francisco, San Francisco, California 94158, United States;.
David M. Wilson, Department of Radiology and Biomedical Imaging, University of California, San Francisco, San Francisco, California 94158, United States;.
REFERENCES
- (1).Bishop JR; Schuksz M; Esko JD Heparan Sulphate Proteoglycans Fine-Tune Mammalian Physiology. Nature 2007, 446, 1030–1037. [DOI] [PubMed] [Google Scholar]
- (2).Kjellén L; Lindahl U Proteoglycans: Structures and Interactions. Annu. Rev. Biochem 1991, 60, 443–475. [DOI] [PubMed] [Google Scholar]
- (3).Prydz K; Dalen KT Synthesis and Sorting of Proteoglycans. J. Cell Sci 2000, 113 (2), 193–205. [DOI] [PubMed] [Google Scholar]
- (4).Kreuger J; Kjellén L Heparan Sulfate Biosynthesis: Regulation and Variability. J. Histochem. Cytochem 2012, 60, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sheng J; Liu R; Xu Y; Liu J The Dominating Role of N-Deacetylase/N-Sulfotransferase 1 in Forming Domain Structures in Heparan Sulfate. J. Biol. Chem 2011, 286, 19768–19776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Esko JD; Lindahl U Molecular Diversity of Heparan Sulfate. J. Clin. Invest 2001, 108, 169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sarrazin S; Lamanna WC; Esko JD Heparan Sulfate Proteoglycans. Cold Spring Harbor Perspect. Biol 2011, 3, a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Fuster MM; Esko JD The Sweet and Sour of Cancer: Glycans as Novel Therapeutic Targets. Nat. Rev. Cancer 2005, 5, 526–542. [DOI] [PubMed] [Google Scholar]
- (9).Su G; Meyer K; Nandini CD; Qiao D; Salamat S; Friedl A Glypican-1 Is Frequently Overexpressed in Human Gliomas and Enhances FGF-2 Signaling in Glioma Cells. Am. J. Pathol 2006, 168, 2014–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Qiao D; Meyer K; Mundhenke C; Drew SA; Friedl A Heparan Sulfate Proteoglycans as Regulators of Fibroblast Growth Factor-2 Signaling in Brain Endothelial Cells. Specific Role for Glypican-1 in Glioma Angiogenesis. J. Biol. Chem 2003, 278, 16045–16053. [DOI] [PubMed] [Google Scholar]
- (11).Phillips JJ; Huillard E; Robinson AE; Ward A; Lum DH; Polley M-Y; Rosen SD; Rowitch DH; Werb Z Heparan Sulfate Sulfatase SULF2 Regulates PDGFRα Signaling and Growth in Human and Mouse Malignant Glioma. J. Clin. Invest 2012, 122, 911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Christianson HC; Svensson KJ; van Kuppevelt TH; Li J-P; Belting M Cancer Cell Exosomes Depend on Cell-Surface Heparan Sulfate Proteoglycans for Their Internalization and Functional Activity. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 17380–17385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hu F; Dzaye OD; Hahn A; Yu Y; Scavetta RJ; Dittmar G; Kaczmarek AK; Dunning KR; Ricciardelli C; Rinnenthal JL; Heppner FL; Lehnardt S; Synowitz M; Wolf SA; Kettenmann H Glioma-Derived Versican Promotes Tumor Expansion via Glioma-Associated Microglial/Macrophages Toll-like Receptor 2 Signaling. Neuro. Oncol 2015, 17, 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hu B; Kong LL; Matthews RT; Viapiano MS The Proteoglycan Brevican Binds to Fibronectin after Proteolytic Cleavage and Promotes Glioma Cell Motility. J. Biol. Chem 2008, 283, 24848–24859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Pan H; Xue W; Zhao W; Schachner M Expression and Function of Chondroitin 4-Sulfate and Chondroitin 6-Sulfate in Human Glioma. FASEB J. 2020, 34, 2853–2868. [DOI] [PubMed] [Google Scholar]
- (16).Hojring N; Svensmark O Carboxylesterases with Defferent Substrate Specificity in Human Brain Extracts. J. Neurochem 1976, 27, 523–528. [DOI] [PubMed] [Google Scholar]
- (17).Chiu W-T; Shen S-C; Chow J-M; Lin C-W; Shia L-T; Chen Y-C Contribution of Reactive Oxygen Species to Migration/Invasion of Human Glioblastoma Cells U87 via ERK-Dependent COX-2/PGE(2) Activation. Neurobiol. Dis 2010, 37, 118–129. [DOI] [PubMed] [Google Scholar]
- (18).Li J; Shi J; Medina JE; Zhou J; Du X; Wang H; Yang C; Liu J; Yang Z; Dinulescu DM; Xu B Selectively Inducing Cancer Cell Death by Intracellular Enzyme-Instructed Self-Assembly (EISA) of Dipeptide Derivatives. Adv. Healthc Mater 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Beahm BJ; Dehnert KW; Derr NL; Kuhn J; Eberhart JK; Spillmann D; Amacher SL; Bertozzi CR A Visualizable Chain-Terminating Inhibitor of Glycosaminoglycan Biosynthesis in Developing Zebrafish. Angew. Chem., Int. Ed 2014, 53, 3347–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kuhn J; Kleesiek K; Götting C Determination of B4-Galactosyltransferase-7 Activity Using High-Performance Liquid Chromatography–electrospray Ionization Tandem Mass Spectrometry. Clin. Biochem 2009, 42, 521–527. [DOI] [PubMed] [Google Scholar]
- (21).Briggs DC; Hohenester E Structural Basis for the Initiation of Glycosaminoglycan Biosynthesis by Human Xylosyltransferase 1. Structure 2018, 26, 801–809. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Garud DR; Tran VM; Victor XV; Koketsu M; Kuberan B Inhibition of Heparan Sulfate and Chondroitin Sulfate Proteoglycan Biosynthesis. J. Biol. Chem 2008, 283, 28881–28887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tsuzuki Y; Nguyen TKN; Garud DR; Kuberan B; Koketsu M 4-Deoxy-4-Fluoro-Xyloside Derivatives as Inhibitors of Glycosaminoglycan Biosynthesis. Bioorg. Med. Chem. Lett 2010, 20, 7269–7273. [DOI] [PubMed] [Google Scholar]
- (24).Saliba M; Ramalanjaona N; Gulberti S; Bertin-Jung I; Thomas A; Dahbi S; Lopin-Bon C; Jacquinet J-C; Breton C; Ouzzine M; Fournel-Gigleux S Probing the Acceptor Active Site Organization of the Human Recombinant B1,4-Galactosyltransferase 7 and Design of Xyloside-Based Inhibitors. J. Biol. Chem 2015, 290, 7658–7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tsutsui Y; Ramakrishnan B; Qasba PK Crystal Structures of β−1,4-Galactosyltransferase 7 Enzyme Reveal Conformational Changes and Substrate Binding. J. Biol. Chem 2013, 288, 31963–31970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Siegbahn A; Manner S; Persson A; Tykesson E; Holmqvist K; Ochocinska A; Rönnols J; Sundin A; Mani K; Westergren-Thorsson G; Widmalm G; Ellervik U Rules for Priming and Inhibition of Glycosaminoglycan Biosynthesis; Probing the B4GalT7 Active Site. Chem. Sci 2014, 5, 3501. [Google Scholar]
- (27).Sampathkumar S-G; Jones MB; Meledeo MA; Campbell CT; Choi SS; Hida K; Gomutputra P; Sheh A; Gilmartin T; Head SR; Yarema KJ Targeting Glycosylation Pathways and the Cell Cycle: Sugar-Dependent Activity of Butyrate-Carbohydrate Cancer Prodrugs. Chem. Biol 2006, 13, 1265–1275. [DOI] [PubMed] [Google Scholar]
- (28).Gaitonde V; Sucheck SJ Synthesis of β-Glycosyl Amides from N-Glycosyl Dinitrobenzenesulfonamides. J. Carbohydr. Chem 2012, 31, 353–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Greenway KT; Bischoff AG; Pinto BM Probing Hyperconjugation Experimentally with the Conformational Deuterium Isotope Effect. J. Org. Chem 2012, 77, 9221–9226. [DOI] [PubMed] [Google Scholar]
- (30).Alabugin IV Stereoelectronic Interactions in Cyclohexane, 1,3-Dioxane, 1,3-Oxathiane, and 1,3-Dithiane: W-Effect, σC-X ↔ Σ*C-H Interactions, Anomeric EffectWhat Is Really Important? J. Org. Chem 2000, 65, 3910–3919. [DOI] [PubMed] [Google Scholar]
- (31).Abronina PI; Kachala VV; Kononov LO A Novel Synthesis of Beta-D-Mannopyranosyl Azide by Phase Transfer Catalysis. Carbohydr. Res 2009, 344, 240–244. [DOI] [PubMed] [Google Scholar]
- (32).Bubb WA NMR Spectroscopy in the Study of Carbohydrates: Characterizing the Structural Complexity. Concepts Magn. Reson 2003, 19A, 1–19. [Google Scholar]
- (33).Karplus M Vicinal Proton Coupling in Nuclear Magnetic Resonance. J. Am. Chem. Soc 1963, 85, 2870–2871. [Google Scholar]
- (34).Rostovtsev VV; Green LG; Fokin VV; Sharpless KB A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem., Int. Ed 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- (35).Skaga E; Kulesskiy E; Fayzullin A; Sandberg CJ; Potdar S; Kyttälä A; Langmoen IA; Laakso A; Gaál-Paavola E; Perola M; Wennerberg K; Vik-Mo EO Intertumoral Heterogeneity in Patient-Specific Drug Sensitivities in Treatment-Naïve Glioblastoma. BMC Cancer 2019, 19, 628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Tan SK; Jermakowicz A; Mookhtiar AK; Nemeroff CB; Schürer SC; Ayad NG Drug Repositioning in Glioblastoma: A Pathway Perspective. Front. Pharmacol 2018, 9, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Motaln H; Koren A; Gruden K; Ramšak Ž; Schichor C; Lah TT Heterogeneous Glioblastoma Cell Cross-Talk Promotes Phenotype Alterations and Enhanced Drug Resistance. Oncotarget 2015, 6, 40998–41017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Molander GA; Rönn M Total Synthesis of (−)-Cylindricine C. J. Org. Chem 1999, 64, 5183–5187. [DOI] [PubMed] [Google Scholar]
- (39).Okano K; Fujiwara H; Noji T; Fukuyama T; Tokuyama H Total Synthesis of Dictyodendrin A and B. Angew. Chem., Int. Ed 2010, 49, 5925–5929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.