

Abstract
Neurodegenerative diseases encompass a large group of conditions that are clinically and pathologically diverse yet are linked by a shared pathology of misfolded proteins. The accumulation of insoluble aggregates is accompanied by a progressive loss of vulnerable neurons. For some patients, the symptoms are motor focused (ataxias), while others experience cognitive and psychiatric symptoms (dementias). Among the shared symptoms of neurodegenerative diseases is a disruption of the sleep/wake cycle that occurs early in the trajectory of the disease and may be a risk factor for disease development. In many cases, the disruption in the timing of sleep and other rhythmic physiological markers immediately raises the possibility of neurodegeneration-driven disruption of the circadian timing system. The aim of this Review is to summarize the evidence supporting the hypothesis that circadian disruption is a core symptom within neurodegenerative diseases, including Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease, and to discuss the latest progress in this field. The Review discusses evidence that neurodegenerative processes may disrupt the structure and function of the circadian system and describes circadian-based interventions as well as timed drug treatments that may improve a wide range of symptoms associated with neurodegenerative disorders. It also identifies key gaps in our knowledge.
Introduction
The term “neurodegenerative disease” (NDD) encompasses a large group of conditions that are clinically and pathologically diverse. Still, there is a shared biology, as these diseases are characterized by the accumulation of misfolded proteins into insoluble aggregates accompanied by a progressive loss of neurons. These protein aggregates are formed by mutant huntingtin in Huntington’s disease (HD); amyloid-β and tau in Alzheimer’s disease (AD); and misfolded α-synuclein in Parkinson’s disease (PD) and related disorders (1). These diseases share common elements due to the toxic nature of protein aggregates. There is also evidence that misfolded proteins can be transmitted from cell to cell with aggregates serving as a chemical template for their formation in recipient cells (2). The combination of selective vulnerability and spread may explain how NDDs influence different networks of cells.
Among the shared symptoms of NDD is a disruption of the sleep/wake cycle, which occurs early in the progression of the disease. In many cases, the timing of sleep and rhythmic physiological markers is disrupted, which raises the possibility of NDD-driven disruption of the circadian timing system. The circadian clock is a temporal program that generates a 24-hour structure on biological processes from gene expression to behavior. Circadian clocks are autonomous, producing rhythms even in the absence of daily environmental signals, and are widely distributed throughout the brain and body. The mammalian circadian timing system includes a central clock in the suprachiasmatic nucleus (SCN), which receives light information through a specialized pathway, as well as information about other relevant environmental cues, like the timing of food. The SCN communicates with the rest of the brain through several pathways, including arousal centers, the autonomic nervous system, and the hypothalamic-pituitary-adrenal (HPA) axis. Given this potent mix of output pathways, the central clock has the ability to regulate rhythms throughout the body, and most tissues have cell-autonomous molecular clocks. The result is a broad temporal structure that influences all biological systems. One of the central tenets of the circadian field is that this temporal structure is essential for our health (3, 4), and its disruption has costs felt throughout our bodies.
At the cellular and molecular level, the molecular clockwork drives the rhythmic transcription of a number of genes (4). This circadian clockwork drives the temporal regulation of key biological processes involved in protein processing that could impact the formation of aggregates (5) as well as the transmission of misfolded proteins (6). Importantly, the clearance of misfolded proteins is driven by sleep through the regulation of glymphatic flow (7). For these reasons, many researchers (8, 9) favor a bidirectional model in which the ongoing disease pathology can impact circadian rhythms, and the disrupted circadian rhythms may accelerate the pathology (Figure 1).
Figure 1. Illustration of the tight reciprocal relationships between circadian rhythms, sleep, and neurodegenerative disease.

Circadian disruption and sleep loss are likely to contribute to symptoms associated with NDD in humans. Work in animal models as well as in human subjects suggests that alterations in amplitude, regularity, and coherence of measured rhythms are common. Many researchers in this area favor a bidirectional model in which the ongoing disease pathology can impact circadian rhythms, and the disrupted circadian rhythms may accelerate the disease pathology. If this hypothesis is correct, future studies should be able to demonstrate that circadian-based interventions can improve a wide range of symptoms associated with NDDs.
The term “circadian disruption” is broadly used in the context of NDD but is not well defined by researchers (10). While there are a number of key rhythmic parameters driven by the circadian system (e.g., amplitude, phase, period), geneticists focus primarily on endogenous cycle length, and we have a detailed understanding of the underlying molecular mechanisms. Measured in constant conditions, the cycle length generated by the circadian timing system is precise and quantifiable, i.e., an excellent output to explore by mutational and reverse genetic strategies (11). By the narrowest definition, we could limit our discussion of circadian disruption to alterations in period due to defects in the core molecular mechanism, i.e., the network of transcriptional-translational feedback loops, including the core loop (BMAL1/CLOCK/NPAS2 and PERs/CRYs) and the stabilization loop (REV-ERBs and RORs). In experimental models like Drosophila and mice, circadian period is easily measured with automated equipment. Making these measurements in patients, however, is another matter altogether. A disease-driven change in circadian period can only be assessed by placing a patient in constant conditions, such as the constant routine protocol established by Czeisler and colleagues (12), and comparing with age-matched controls. In such a protocol, it is possible to evaluate older individuals and measure circadian period and amplitude (13). Still, it is extremely difficult to maintain people under these conditions (14), and, given the well-recognized psychiatric challenges experienced by patients with NDD, these measurements are unlikely. Some possible proxies can be used; e.g., recent studies have revealed that the characteristics of human skin fibroblast clocks, including period, may reflect the circadian phenotype of the person (15). The fibroblasts can be transfected with bioluminescent reporters that allow the measurement of period in vitro. In addition, studies have made use of postmortem tissue to infer disruption of circadian gene expression rhythms in patients with NDD (16), as well as in typically aging brains (17). The ideal here would be the development of assays in which measurements taken at a single time point from a patient’s blood could provide information about the full daily rhythm. Although we are still in the early days, there have been some real successes in this arena (18–20) that offer great promise for the future. Still, at this point, it is extremely challenging to measure NDD-driven changes in circadian period in patient populations.
Thus, focusing solely on circadian period is not likely a good strategy in considering NDDs. Other key rhythmic parameters, including amplitude and phase, are also driven by the circadian system and should be utilized. The most common deficits observed in patients and animal models include reduced amplitude, fragmentation (the loss of the normally consolidated sleep/wake states), and an increase in the cycle-to-cycle variability of daily rhythms. Unfortunately, these symptoms have a number of possible alternative explanations. For example, a reduction in rhythm amplitude may be caused by defects in many processes downstream of the circadian timing system. This is of particular concern in the case of HD and PD, in which the key symptom is a disruption in motor output through basal ganglia circuits. While we must be careful in our interpretations, disruptions in the temporal patterning of behavior or physiological rhythms raise, at a minimum, the possibility of deficits in the circadian timing system.
NDD-driven deficits in the circadian timing system
Classically, the circadian timing system consists of a light input pathway; a central clock located in the SCN; and multiple output pathways (Figure 2). Molecular circadian clocks are widely distributed and control the temporal pattern of transcription locally. It may be useful to consider possible NDD-driven dysfunction in each of these components.
Figure 2. Schematic representation of the pathways that may be compromised in the circadian system of patients with NDD.
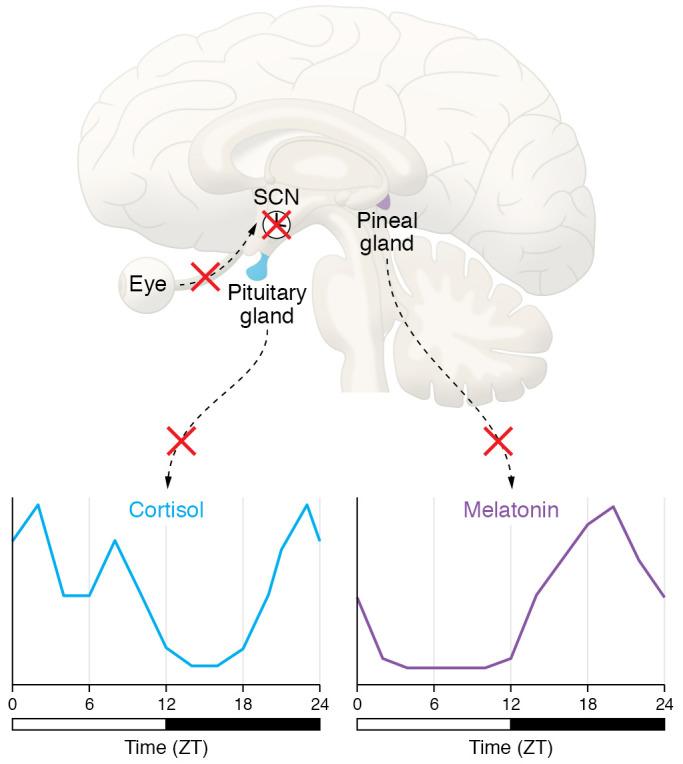
The light/dark (LD) cycle is the main external synchronizer of the central circadian pacemaker (via melanopsin and visual photoreceptors) and a direct retinohypothalamic tract. There is evidence for an NDD-driven loss of melanopsin-expressing intrinsically photoreceptive retinal ganglion cells (ipRGCs). The reduction in the ability of light to reset the circadian clock could provide an explanation for the reduction in rhythm amplitude under LD conditions, as well as the increase in cycle-to-cycle variability. The master clock in the SCN serves to synchronize central and peripheral oscillators to optimize the function of the organism relative to the 24-hour periodicities in the environment. There are a number of cell populations within the SCN as defined by gene expression and anatomical analysis. Some of these cell populations are vulnerable to AD-driven degeneration, and the data are at least consistent with the possibility that damage to the SCN itself underlies circadian disruption in NDD, at least in later stages of disease. SCN circuits send projections throughout the CNS and endocrine system, providing multiple pathways by which the SCN can convey temporal information to the brain and body. There is evidence that NDD can alter the amplitude and regularity of SCN-driven outputs such as rhythms in melatonin and cortisol. The weakening of the rhythms in these three key outputs would be expected to reduce the synchrony of molecular clocks found throughout the body, leading to a state of internal desynchronization. ZT, zeitgeber time.
NDD-driven deficits in circadian light detection
The daily light/dark (LD) cycle is a powerful regulator of behavior through both the direct effect of light and synchronization of the endogenous circadian clock (21). Light is detected by intrinsically photoreceptive retinal ganglion cells (ipRGCs) that make use of melanopsin as a photopigment (22, 23). The ipRGCs can also respond to rod- and cone-driven signals (24). The ipRGCs integrate this light information (25) and send a direct projection to the central circadian clock, where this signal synchronizes circadian oscillations. The ipRGCs also underlie, in part, the direct impact of light on mood and cognition (26, 27).
There is evidence that PD and AD patients have fewer ipRGCs with reduced dendritic complexity (28, 29). The contribution of rods, cones, and ipRGCs to the pupillary light reflex can be estimated by pupillometry. In particular, the post-illumination pupil response (PIPR) is a reliable metric of ipRGC function and is compromised in PD and AD patients (30–32). Patients at risk for AD have significant variability in the PIPR relative to healthy controls, suggesting that this light-evoked reflex could be predictive (32, 33). While more work is needed, this relatively simple assay may become an important clinical measure for disruption in the light input pathway to the circadian system.
In HD mouse models, the degeneration of ipRGCs occurs early in disease progression (34, 35). There are several ways to test how light impacts the circadian system. Perhaps the most definitive measure is the magnitude of light-evoked phase shifts when mice are held in constant darkness. Importantly, HD models show significant reductions in magnitude of light-induced phase delays, as well as longer times for re-entrainment to a shift in the LD cycle (35–37). Collectively, there are compelling preclinical data suggesting that the light input to the circadian system is compromised in HD. The situation in PD and AD is less clear. Behavioral data are mixed, with some groups finding no impact on the light input pathway in well-established mouse models for PD (38) or AD (39). On the other hand, the apolipoprotein E4–knockout (APOE4-KO) line, an AD model, showed a significant reduction in the magnitude of light-induced phase shifts (40), as well as increased activity during the light phase of the LD cycle (39, 41). While other mechanisms are possible, a reduction in the ability of light to reset the circadian clock could provide an explanation for the reduction in rhythm amplitude under LD conditions, as well as the increase in cycle-to-cycle variability.
NDD-driven deficits in the SCN
The SCN of the hypothalamus contains the so-called master oscillatory network necessary for coordinating circadian rhythms throughout the body. A number of studies have looked at postmortem structural changes in the SCN in NDD (42–47). These studies show a reduction in cell number and size, as well as a reduction in vasoactive intestinal peptide–expressing (VIP-expressing) and arginine vasopressin–expressing (AVP-expressing) cells in the SCN. A couple of particularly impactful studies managed to measure daily rhythms of subjects with AD and then examine the patients’ SCNs postmortem. The first study found that the specific loss of SCN neurotensin neurons was associated with loss of activity and temperature amplitude in AD patients (48). Work from the Saper laboratory (49) found that circadian rhythm amplitude in subjects with AD was correlated with the number of VIP-expressing SCN neurons. AD was additionally associated with delayed circadian phase in comparison with cognitively healthy subjects. Together these data are consistent with the possibility that damage to the SCN itself underlies circadian disruption in NDD, at least in later stages of disease.
There is some evidence of anatomical changes to the SCN in mouse models of NDD. For example, there have been reports of a reduction in VIP expression in the SCN in HD models (50, 51). In addition, there is a reduction in the Nissl-defined size of the male, but not female, SCN in an HD model (51). Perhaps the clearest case for SCN pathology comes from work in the APOE4-KO model (40). The authors found evidence for amyloidosis and tau deposition, as well as mitochondrial and synaptic deterioration, in the mutant SCN. Tauopathy was also observed in the SCN of aged Tg4510 mice (a model for AD) (52), highlighting the importance of age as a variable. These deficits in mouse models are all mostly observed in young and middle-aged animals (12 months or less), while the human data are all postmortem. It may well be that the deficits in mice are more pronounced toward the end of their lifespan.
A characteristic property of SCN neurons is that they generate a circadian rhythm of neural activity with higher spontaneous activity during the day than during the night. This neural activity rhythm is critical for the synchrony of cells within the SCN circuit, as well as the ability of this nucleus to drive outputs throughout the body. These rhythms in SCN electrical activity are impacted by age and already exhibit a reduction in amplitude and increase in variability by middle age (53). SCN neural activity rhythms are disrupted early in NDD and are accompanied by loss of normal daily variation in resting membrane potential in SCN neurons (36, 38, 51, 54). Thus, there is evidence for pathophysiology at the level of the SCN in mouse models of NDD.
NDD-driven alteration in SCN-driven outputs
As measured by actigraphy, many patients with NDD will exhibit increased fragmentation of their sleep/wake cycle as measured by increased arousals at night and increased sleep in the day (8, 9, 55, 56). The circuit through which the SCN controls locomotor activity is not well understood but certainly runs through the dopaminergic regulation of basal ganglion circuitry. Cell populations in this region are vulnerable to pathology driven by PD and HD. Understanding how NDDs impact the circadian regulation of the motor control regions is an important area for future research.
Melatonin.
One of the best-understood circadian circuits is the temporal control of melatonin secretion. The circuits involved in the nightly rise in melatonin secretion and autonomic function are relatively well defined (57). Melatonin receptors are expressed throughout the body (58) and have a broad impact on biological systems. The SCN itself expresses high levels of melatonin receptors, and the interaction between the SCN and the pineal gland forms a neuroendocrine feedback loop (59). From a clinical perspective, dim-light melatonin onset (DLMO) can be used as a laboratory measure to determine the phase of the circadian cycle (60). Melatonin levels are commonly measured in plasma or saliva, as this allows for frequent sampling for more precise determination of phase.
HD patients have been reported to have a delayed phase of melatonin secretion, which becomes more delayed with disease progression (61). Another study documented reduced melatonin levels in premanifest HD patients (62), with a further decline in melatonin levels with progression to manifest HD. In both groups, the intersubject DLMO was more variable in comparison with controls. However, another study of HD allele carriers did not find any change in DLMO, even among individuals with increased nightly awakenings (63). Among PD patients, there is evidence for a reduction in amplitude of melatonin secretion but mixed support for a difference in phase (64–66). A complicating factor is that dopaminergic drugs used to treat PD also impact melatonin secretion (67). Early work in AD patients found evidence for decreased melatonin levels (65–70). More recent work found that DLMO was delayed with reduced amplitude in patients with a mild cognitive impairment (71). Given that measurements can be made from saliva samples, the relative lack of data on the timing of melatonin secretion in patients with NDD is surprising. For the patients with NDD, it would be interesting to look at intrasubject variability in DLMO, as the data from activity suggest that the day-to-day variability is likely to be higher than in controls. Our lack of knowledge about the impact of NDD on melatonin rhythmicity extends to mouse models, as most are on the C57 background and thus do not produce rhythmic melatonin.
Core body temperature.
Core body temperature (CBT) is another well-accepted marker of circadian rhythmicity that is high during the day and low during the night in humans. Circadian rhythms in CBT are independent of locomotor activity but dependent on an intact SCN (72–74). Projections travel from the SCN to the subparaventricular zone, and these neurons are necessary for driving circadian rhythms of CBT (75). New technologies allow CBT to be continuously measured through wireless capsules, and perhaps even wearable devices that should facilitate measurements in patient populations.
Presymptomatic HD patients were reported to have an elevated daytime CBT (76), but rhythms were not measured. In PD, the amplitude of the CBT rhythm was reduced in a subset of patients with rapid eye movement sleep behavior disorder (RBD) and dementia with Lewy bodies (77). The relationship between RBD and reduced amplitude of CBT rhythms has been seen in other work (78, 79). In AD, early studies found no difference in amplitude of the CBT rhythm, but phase was delayed in subjects with AD (80). A subset of patients with nocturnal agitation known as “sundowning behavior” actually showed a higher amplitude rhythm that was also phase-delayed (81). In one noteworthy study, CBT was measured in aged individuals, grouped into healthy aging or probable AD, and young subjects (82). The authors found a reduction in amplitude of circadian rhythms in CBT in both of the aged groups compared with the young. A phase delay was seen in the probable AD subjects, but not age-matched controls. Overall, the data suggest that some, but not all, patients with NDD exhibit either reductions in amplitude or a phase delay in these rhythms.
The hypothesis that CBT rhythms are essentially intact early in NDD disease progression and that those rhythms become blunted and phase-shifted in later stages could be easily tested in animal studies. For example, careful longitudinal studies of HD mouse models found intact CBT rhythms early in disease progression, and these rhythms exhibited reduced amplitude and increased cycle-to-cycle variability as the disease progressed (83–85). Another study used telemetry to measure CBT rhythms in young and middle-aged AD mice (3xTgAD) (86). The CBT rhythms were phase-advanced in young animals and, by middle age, actually showed an increase in amplitude. CBT as a measure of SCN-driven output is likely underutilized in mouse models of NDD.
Cortisol.
Release of cortisol from the adrenal cortex is under the regulation of the HPA axis, and multiple levels of this axis are under circadian control (59, 87). One of the most robust circadian rhythms is that of circulating cortisol that peaks just prior to waking. This hormone is as close as we have to a universal “wake” signal and may prepare the body for activity to come. Glucocorticoid and mineralocorticoid receptors have a widespread distribution. Interestingly, glucocorticoid receptors are not found within the SCN, suggesting that the daily increase in cortisol concentration could act as a unidirectional output signal from the SCN. Measurement of cortisol is routine, although mostly in serum, which makes frequent sampling difficult. Cortisol can also be measured in saliva, but the morning rise is thought to occur prior to waking, so the frequent sampling needed to capture the rhythm will disturb the sleep/wake cycle.
Despite these challenges, a number of studies have examined the impact of NDD on cortisol rhythms. An early study examined 24-hour cortisol secretion in early-stage, medication-free HD patients and found higher amplitude rhythms in the HD population (61). Another study found mild disturbance or no disturbance in morning cortisol secretion in HD carriers that preceded the onset of motor symptoms (63, 88, 89). Later states of HD have been reported to show a phase advance that was similar to that seen with aging (90), as well as elevated cortisol (91). In an important study, Hartmann and colleagues (92) directly compared PD and AD patients versus age-matched controls with 24-hour sampling every 15 minutes. Patients with AD and PD had significantly higher total plasma cortisol concentrations. The PD patients also exhibited a decreased amplitude in cortisol secretion rhythm, whereas the rhythm in AD patients appeared to be phase-advanced. A more recent study also found that PD patients had significantly elevated total plasma cortisol concentrations (93) with no change in the timing of cortisol onset or offset. However, a number of PD patients (41%) exhibited arrhythmic cortisol profiles. Other studies have found intact rhythms in cortisol secretion even in patients whose activity patterns are disrupted (94). Overall, many studies have found evidence for elevated cortisol in patients with NDD, but with rhythmic secretion generally intact.
Very little work has been done on possible disruption of the circadian rhythm in the HPA axis in NDD models. In one example, the R6/2 mouse model of HD exhibits elevated corticosterone levels, and, importantly, experimentally elevated corticosterone exacerbates HD symptomology, whereas reducing glucocorticoids improves symptoms (95). The question of whether the rhythm in glucocorticoid secretion is altered in NDD models remains largely unexplored.
NDD-driven deficits in the molecular clockwork
The rhythms in neural activity in the SCN are driven by cell-autonomous molecular feedback loops (96, 97). There is evidence that aging can impact clock gene expression in the human cortex. A detailed study from McClung’s laboratory used time-of-death information to examine the temporal pattern of gene expression from the human prefrontal cortex (17). The authors found that older people have disrupted PER1 expression patterns, whereas PER2 expression showed a phase advance. Broadly, the transcription of many genes exhibited phase shifts or loss of rhythmicity in the aged population. Interestingly, a subset of genes that were not rhythmic in tissue from younger adults became rhythmic in the older population. This type of complexity in gene expression is what we expect from patients with NDD. Another study examined clock gene expression in postmortem tissue (forebrain and pineal) and found evidence for alterations in the phase of clock gene rhythms in the brains of AD patients compared with controls (16). Similarly, the diurnal rhythmic pattern in the expression of the several clock genes was lost in the pineal gland in AD patients (98). Finally, the rhythmic methylation of BMAL1 is disrupted in AD, and this correlates with tau pathology, cognitive disturbance, and overnight wakefulness (99, 100). These results support the idea that rhythms in clock gene expression are disrupted in AD patients, but more work in this area is needed.
Clock gene expression can be measured from most cell populations, so it is possible to look at disease-driven changes in period and phase in peripheral tissues. This seems to be a very promising strategy, and one study examined clock gene expression in peripheral blood mononuclear cells taken from patients with early PD and discovered the loss of rhythms in BMAL1 and altered expression of both PER2 and REV-ERBα (93). To provide another example, rhythmic methylation of BMAL1 was altered in fibroblasts from AD patients (99). Measurement of rhythmic clock gene expression from peripheral tissues offers a number of advantages over postmortem tissue. It may have the limitation that at least in some tissues the phase will be highly influenced by the feed/fast cycle.
Clock gene expression in HD mouse models has been explored in some detail. In the more severely impacted R6/2 model, disruption of PER2 and BMAL1 rhythms was observed in the SCN and striatum in vivo (101). Interestingly, when clock gene expression was measured with PER2-driven bioluminescence, the main effects were on the phase of the rhythms, while the bioluminescence measured in the SCN appeared relatively intact (102, 103). This observation that the expression rhythms are worse in vivo than in vitro raises the possibility that the molecular clock in the SCN is relatively intact, but that extra-SCN signals are disrupting the rhythm in vivo. There has also been some related work with AD models. For example, in the APOE4 KO, the circadian pattern of core clock gene expression in the SCN was disrupted (40). There is also evidence for disrupted rhythms in clock gene expression outside of the SCN in AD models (52, 104, 105). Overall, the gene expression data from mouse models suggest that there is an internal desynchronization between the SCN and peripheral clocks in NDD.
Possible mechanisms underlying circadian disruption in NDDs
The molecular clockwork driving circadian oscillations is found throughout the body. Neurons and glial cells express daily oscillations that provide a temporal structure to transcription, cell biology, and network properties of all major CNSs that are impacted by NDD. Genome-wide analyses reveal global circadian control over processes involved in functionally important pathways, such as transcription, chromatin modification and remodeling, metabolism, inflammation, and cell signaling (Figure 3). Therefore, in trying to understand the mechanisms by which circadian disruption can accelerate NDD symptoms and pathology, there are a number of possible mediatory pathways (8, 9, 55, 106, 107), and three particularly promising intersections between circadian rhythms and NDD will be briefly considered here.
Figure 3. Circadian-regulated pathways essential to NDDs.

At the beginning of the cycle, CLOCK and BMAL1 protein complexes bind DNA at specific promoter regions (E-box) to activate the transcription of a family of genes including the Period (Per1, Per2, and Per3) and Cryptochrome (Cry1 and Cry2) genes. The levels of the transcripts for Per and Cry reach their peak during mid- to late day, while the PER and CRY proteins peak in the early night. The PERs, CRYs, and other proteins form complexes that translocate back into the nucleus and turn off the transcriptional activity driven by CLOCK-BMAL1 with a delay (due to transcription, translation, dimerization, and nuclear entry). The proteins would be degraded by ubiquitination, allowing the cycle to begin again. Many cells contain this molecular feedback loop that regulates the rhythmic transcription of a number of genes. Other feedback loops within the cells serve to contribute to the precision and robustness of the core oscillation. Of particular importance, a rhythm in the transcription of BMAL1 is driven by a secondary feedback loop involving the activator retinoic acid receptor–related orphan receptor (ROR) and the repressor REV-ERBα/β. Mechanistically, this circadian clockwork drives a number of processes implicated in NDD. For example, the circadian clock regulates a number of pathways involved in proteostasis, including molecular chaperones as well as autophagy. In addition, many of the genes involved in control of excitability and secretion are rhythmically regulated by this molecular feedback loop. While the precise mechanisms involved in the transmission of misfolded proteins are not known, they are likely impacted by circadian disruption. Finally, it has long been appreciated that there is a close relationship between the circadian clock and the immune system, and disruptions of the circadian timing system drive neuroinflammation, mediated by glial cells. CCGs, circadian clock genes; RORE, ROR response element.
Proteostasis
Proteostasis pathways control the biogenesis, folding, trafficking, and degradation of proteins. These pathways are implicated in NDDs whose pathology is defined by the aggregation of hallmark proteins. A common hypothesis is that chronic expression of misfolded proteins early in life leads to accumulation of misfolded species and aggregates that overwhelm proteostasis (108). There is some evidence for circadian regulation of protein quality control systems, including molecular chaperones, the ubiquitin-proteasome system, and the autophagy-lysosomal degradation pathway in the brain. For example, gene ontology analysis of transcripts in the SCN found enrichment for genes involved in protein stabilization and folding that are rhythmically expressed across cell types (5). While relatively unexplored, proteostasis networks are likely to be important in understanding the links between circadian dysfunction and NDDs. In the liver, there is good evidence for circadian regulation of autophagy (109), and in fibroblasts, rhythms of proteasomal activity have been observed (110). In addition, a number of molecular chaperones, including heat shock proteins (Hsp70, Hsp90), are pivotal in protein folding and unfolding (111). The DNA binding of heat shock factor 1 (HSF1) is highly rhythmic (112) and drives the expression of heat shock proteins at the onset of activity. There is evidence that HSF1-dependent regulation of the heat shock response in vivo is impaired in HD mouse models (113). Using a Drosophila HD model, there is evidence for Hsp70/90 organizing protein in the regulation of mutant huntingtin (mHtt) aggregation and toxicity (114). Experiments to determine the role of the circadian system in regulating the proteostasis network in the CNS appear to be a particularly promising area for future work.
There has been growing evidence for sleep/wake regulation of the clearance of misfolded proteins in the brain glymphatic system, an astroglia-mediated interstitial fluid bulk flow (7). As described above, NDDs disrupt sleep, and it is logical to propose that a reduction in sleep drives a decline in clearance of brain waste. The activity of the glymphatic system is high during sleep and low during wakefulness (115). There are daily rhythms in both amyloid-β (Aβ) levels and extracellular levels of tau (116). The argument that these rhythms are sleep-driven comes from observations that anesthesia can acutely mimic the impact of sleep (117), whereas sleep deprivation increases Aβ plaque deposition, as well as tau pathology (118, 119). There is also evidence that disrupted sleep increases Aβ production (120). So present data suggest that sleep can regulate both generation and removal of Aβ.
While sleep may be a direct driver, there is also good reason to suspect the circadian system is involved as well. For example, deletion of Bmal1 causes severe circadian fragmentation, significantly blunts Aβ rhythms, and increases amyloid plaque deposition in a transgenic mouse model of AD (121). Also, there are data suggesting that glymphatic influx and clearance exhibit circadian rhythms that peak during the mid–rest phase of mice. Importantly, loss of the water channel aquaporin-4 (AQP4), which is localized to vascular end-feet of astrocytes, eliminates the day-night difference in both glymphatic influx and drainage to the lymph nodes (122). Finally, the circadian system regulates neural activity in many regions of the CNS, and these rhythms could impact Aβ plaque deposition.
Transmission of misfolded proteins
Many NDD-related pathological proteins undergo cell-to-cell transmission through mechanisms that are not clear. Injection and spread of synthetic Aβ fibrils or α-synuclein via anatomically defined circuits suggest that synaptic transmission of misfolded proteins occurs (2). The circadian system regulates neural activity, as well as synaptic transmission, so disruption of the circadian system could alter the spread of misfolded proteins through this mechanism. Recent work has highlighted the relationship between circadian rhythms, sleep, and synaptic transmission in the forebrain of mice (6, 123). In these studies, the authors examined the transcriptional, translational, and posttranslational regulation of molecules involved in synaptic transmission in forebrain synaptoneurosome preparations throughout the LD cycle. Most of the genes involved in synaptic signaling exhibited robust daily rhythms. These transcripts formed two sharp waves, with transcripts preceding dawn (sleep surge) related to metabolism and those anticipating dusk (wake surge) related to synaptic transmission. When mice were transiently sleep-deprived, the translation of proteins involved in synaptic signaling remained rhythmic, whereas genes associated with metabolism were suppressed. Sleep deprivation abolished almost all phosphorylation cycles in synaptoneurosomes. The authors concluded that the circadian clock directly regulates transcription of the network of genes involved in synaptic transmission and indirectly regulates translation through the behavioral state. Thus, the disruption of the sleep/wake cycle as seen in NDD would be expected to disrupt the temporal pattern of electrical activity and synaptic transmission seen in cortical circuits and alter the transmission of misfolded proteins.
Inflammation
The immune system is composed of a diverse network of cells, tissues, and organs (e.g., white blood cells, spleen, thymus, and bone marrow) that are linked together by function and synchronized in time. The temporal patterning of immune function is a general feature, with the circadian system driving a more activated immune system when the organism is awake and damping it down during times of sleep (124, 125). In peripheral organs, our understanding of the molecular interactions between the circadian clock and the immune system is fairly advanced. For example, the pulmonary inflammatory responses that have received so much attention in the time of COVID-19 are under circadian control. Recent work has implicated REV-ERBα as a critical regulator of fibrosis in mesenchymal cells (126). Other studies have found clear evidence that the molecular circadian clock drives the expression or repression of immune genes (127). In addition, the rhythmic acetylation or methylation of histones regulates transcription of inflammatory genes. There is even evidence that circadian clock proteins can engage in direct physical interactions with components of key inflammatory pathways, such as members of the NF-κB protein family. This intimate association between circadian rhythmicity and immune function likely serves to increase protection during times of day when we are out in the world, while limiting the cost of overstimulation during times of sleep.
Neuroinflammation, as measured by the chronic activation of astrocytes and microglia, is thought to be a major contributor to NDD (128–130). Like neurons, astrocytes exhibit robust circadian clock function (131). Brain-specific BMAL1 deletion weakened the blood-brain barrier, causing loss of pericytes, astrogliosis, microglia activation, and elevation of inflammatory gene expression (132–134). Similarly, microglia have functional circadian clocks, and the inflammatory response of microglia shows circadian variation (135, 136). One of the key discoveries in this area has been that brain-specific BMAL1 KO leads to dramatic microglia activation and synapse degeneration (137). Recent work implicates the circadian clock component REV-ERBα as a critical mediator of microglial activation and neuroinflammation. In mice, Rev-erbα deletion caused spontaneous microglial activation in the hippocampus and increased expression of proinflammatory transcripts, as well as secondary astrogliosis. Primary Rev-erbα–/– microglia exhibited proinflammatory phenotypes and increased basal NF-κB activation (138). Daily rhythms in microglial synaptic phagocytosis appear to be driven by Rev-erbα expression. Thus, the NDD-driven disruption in astrocytes and microglia provides a mechanism through which circadian disruption drives inflammation in the brain.
Future work
As described above, there is clear evidence that patients with NDD exhibit disruption in their sleep/wake cycle and that the loss of function in the circadian system is likely to contribute to these symptoms. Mouse and Drosophila models recapitulate the circadian dysfunction seen in patients and offer the opportunity for mechanistic studies. Circadian-based interventions are a critical test of the hypothesis that circadian disruption is an integral component of the disease (139–141). At a minimum, we would hope to be able to counter the decline in the sleep/wake cycle. Most optimistically, we could even delay the onset of symptoms by improving circadian regulation of proteostasis or reducing inflammation. Of course, it may be possible to improve the quality of life of patients with NDD without altering the core pathology of the disease. One special challenge in this population is the high degree of variability in the alterations of the circadian system. This work is in its early days (142), as we are still working out the mechanisms of interaction between circadian timing and NDD pathology. Still, the timed treatment of light, feeding, and exercise potentially offers low-cost ways to manage NDD. Given the heterogeneity in the symptoms experienced by the patients, the key to success likely lies in the stratification of subjects in clinical trials in order to focus on those most likely to benefit from a particular treatment.
For example, with our improved understanding of the role of melanopsin in the light input pathway, light interventions that enhance melanopsin-stimulation in the day while minimizing it at night are very promising. This type of dynamic lighting can improve sleep at night and reduce depression and agitation in patients with dementia living in controlled environments (143). This study focused on the subset of older patients who were experiencing sleep disturbances. In future work, PIPR could be used as a screen to exclude subjects with compromised circadian light detection. Similarly, the timed application of pharmacological agents that manipulate melatonin, histamine, orexin, or glucocorticoid signaling offers ways to get more use out of already established drugs. It is likely that dosing time can be optimized to maximize the therapeutic index (144–146). For example, nightly treatment with a drug that increased histamine levels improved several behavioral measures in an HD mouse model (147). Finally, there are new classes of pharmacological agents that directly act on the circadian timing system. For example, although much more work is needed, clock-enhancing small molecules like nobiletin, a citrus flavonoid, has been proposed as a treatment for NDD (148, 149). In addition, REV-ERB agonists have been reported to increase wakefulness and reduce sleep as well as anxiety-like behavior in mice (150, 151). To talk realistically about circadian medicine (4), we need to have a set of working tools that we can apply to treat the low-amplitude, fragmented, irregular rhythms that we see in so many patients with NDD.
Version 1. 10/01/2021
Electronic publication
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(19):e148288.https://doi.org/10.1172/JCI148288.
References
- 1.Soto C, Pritzkow S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat Neurosci. 2018;21(10):1332–1340. doi: 10.1038/s41593-018-0235-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peng C, et al. Protein transmission in neurodegenerative disease. Nat Rev Neurol. 2020;16(4):199–212. doi: 10.1038/s41582-020-0333-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roenneberg T, Merrow M. The circadian clock and human health. Curr Biol. 2016;26(10):R432–R443. doi: 10.1016/j.cub.2016.04.011. [DOI] [PubMed] [Google Scholar]
- 4.Cederroth CR, et al. Medicine in the fourth dimension. Cell Metab. 2019;30(2):238–250. doi: 10.1016/j.cmet.2019.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wen S, et al. Spatiotemporal single-cell analysis of gene expression in the mouse suprachiasmatic nucleus. Nat Neurosci. 2020;23(3):456–467. doi: 10.1038/s41593-020-0586-x. [DOI] [PubMed] [Google Scholar]
- 6.Noya SB, et al. The forebrain synaptic transcriptome is organized by clocks but its proteome is driven by sleep. Science. 2019;366(6462):eaav2642. doi: 10.1126/science.aav2642. [DOI] [PubMed] [Google Scholar]
- 7.Nedergaard M, Goldman SA. Glymphatic failure as a final common pathway to dementia. Science. 2020;370(6512):50–56. doi: 10.1126/science.abb8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Musiek ES, Holtzman DM. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science. 2016;354(6315):1004–1008. doi: 10.1126/science.aah4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fifel K, Videnovic A. Circadian alterations in patients with neurodegenerative diseases: neuropathological basis of underlying network mechanisms. Neurobiol Dis. 2020;144:105029. doi: 10.1016/j.nbd.2020.105029. [DOI] [PubMed] [Google Scholar]
- 10.Vetter C. Circadian disruption: what do we actually mean? Eur J Neurosci. 2020;51(1):531–550. doi: 10.1111/ejn.14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takahashi JS, et al. Searching for genes underlying behavior: lessons from circadian rhythms. Science. 2008;322(5903):909–912. doi: 10.1126/science.1158822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Czeisler CA, et al. Stability, precision, and near-24-hour period of the human circadian pacemaker. Science. 1999;284(5423):2177–2181. doi: 10.1126/science.284.5423.2177. [DOI] [PubMed] [Google Scholar]
- 13.Dijk DJ, et al. Contribution of circadian physiology and sleep homeostasis to age-related changes in human sleep. Chronobiol Int. 2000;17(3):285–311. doi: 10.1081/CBI-100101049. [DOI] [PubMed] [Google Scholar]
- 14.Amira SA, et al. Psychological screening for exceptional environments: laboratory circadian rhythm and sleep research. Clocks Sleep. 2020;2(2):13. doi: 10.3390/clockssleep2020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saini C, et al. Human peripheral clocks: applications for studying circadian phenotypes in physiology and pathophysiology. Front Neurol. 2015;6:95. doi: 10.3389/fneur.2015.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cermakian N, et al. Circadian clock gene expression in brain regions of Alzheimer’s disease patients and control subjects. J Biol Rhythms. 2011;26(2):160–170. doi: 10.1177/0748730410395732. [DOI] [PubMed] [Google Scholar]
- 17.Chen CY, et al. Effects of aging on circadian patterns of gene expression in the human prefrontal cortex. Proc Natl Acad Sci U S A. 2016;113(1):206–211. doi: 10.1073/pnas.1508249112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anafi RC. CYCLOPS reveals human transcriptional rhythms in health and disease. Proc Natl Acad Sci U S A. 2017;114(20):5312–5317. doi: 10.1073/pnas.1619320114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braun R, et al. Universal method for robust detection of circadian state from gene expression. Proc Natl Acad Sci U S A. 2018;115(39):E9247–E9256. doi: 10.1073/pnas.1800314115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wittenbrink N, et al. High-accuracy determination of internal circadian time from a single blood sample. J Clin Invest. 2018;128(9):3826–3839. doi: 10.1172/JCI120874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roenneberg T, et al. Light and the human circadian clock. Handb Exp Pharmacol. 2013;(217):311–331. doi: 10.1007/978-3-642-25950-0_13. [DOI] [PubMed] [Google Scholar]
- 22.Rollag MD, et al. Melanopsin, ganglion-cell photoreceptors, and mammalian photoentrainment. J Biol Rhythms. 2003;18(3):227–234. doi: 10.1177/0748730403018003005. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt TM, et al. Intrinsically photosensitive retinal ganglion cells: many subtypes, diverse functions. Trends Neurosci. 2011;34(11):572–580. doi: 10.1016/j.tins.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Diepen HC, et al. Distinct contribution of cone photoreceptor subtypes to the mammalian biological clock. Proc Natl Acad Sci U S A. 2021;118(22):e2024500118. doi: 10.1073/pnas.2024500118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lucas RJ. Mammalian inner retinal photoreception. Curr Biol. 2013;23(3):R125–R133. doi: 10.1016/j.cub.2012.12.029. [DOI] [PubMed] [Google Scholar]
- 26.LeGates TA, et al. Light as a central modulator of circadian rhythms, sleep and affect. Nat Rev Neurosci. 2014;15(7):443–454. doi: 10.1038/nrn3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.An K, et al. A circadian rhythm-gated subcortical pathway for nighttime-light-induced depressive-like behaviors in mice. Nat Neurosci. 2020;23(7):869–880. doi: 10.1038/s41593-020-0640-8. [DOI] [PubMed] [Google Scholar]
- 28.La Morgia C, et al. Retinal ganglion cells and circadian rhythms in Alzheimer’s disease, Parkinson’s disease, and beyond. Front Neurol. 2017;8:162. doi: 10.3389/fneur.2017.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ortuno-Lizaran I, et al. Degeneration of human photosensitive retinal ganglion cells may explain sleep and circadian rhythms disorders in Parkinson’s disease. Acta Neuropathol Commun. 2018;6(1):90. doi: 10.1186/s40478-018-0596-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joyce DS, et al. Melanopsin-mediated pupil function is impaired in Parkinson’s disease. Sci Rep. 2018;8(1):7796. doi: 10.1038/s41598-018-26078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chougule PS, et al. Light-induced pupillary responses in Alzheimer’s disease. Front Neurol. 2019;10:360. doi: 10.3389/fneur.2019.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oh AJ, et al. Pupillometry evaluation of melanopsin retinal ganglion cell function and sleep-wake activity in pre-symptomatic Alzheimer’s disease. PLoS One. 2019;14(12):e0226197. doi: 10.1371/journal.pone.0226197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Romagnoli M, et al. Chromatic pupillometry findings in Alzheimer’s disease. Front Neurosci. 2020;14:780. doi: 10.3389/fnins.2020.00780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ouk K, et al. Attenuated pupillary light responses and downregulation of opsin expression parallel decline in circadian disruption in two different mouse m-odels of Huntington’s disease. Hum Mol Genet. 2016;25(24):5418–5432. doi: 10.1093/hmg/ddw359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin MS, et al. Degeneration of ipRGCs in mouse models of Huntington’s disease disrupts non-image-forming behaviors before motor impairment. J Neurosci. 2019;39(8):1505–1524. doi: 10.1523/JNEUROSCI.0571-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kudo T, et al. Dysfunctions in circadian behavior and physiology in mouse models of Huntington’s disease. Exp Neurol. 2011;228(1):80–90. doi: 10.1016/j.expneurol.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loh DH, et al. The Q175 mouse model of Huntington’s disease shows gene dosage- and age-related decline in circadian rhythms of activity and sleep. PLoS One. 2013;8(7):e69993. doi: 10.1371/journal.pone.0069993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kudo T, et al. Circadian dysfunction in a mouse model of Parkinson’s disease. Exp Neurol. 2011;232(1):66–75. doi: 10.1016/j.expneurol.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 39.Sterniczuk R, et al. Characterization of the 3xTg-AD mouse model of Alzheimer’s disease: part 1. Circadian changes. Brain Res. 2010;1348:139–148. doi: 10.1016/j.brainres.2010.05.013. [DOI] [PubMed] [Google Scholar]
- 40.Zhou L, et al. Degeneration and energy shortage in the suprachiasmatic nucleus underlies the circadian rhythm disturbance in ApoE −/− mice: implications for Alzheimer’s disease. Sci Rep. 2016;6(1):36335. doi: 10.1038/srep36335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pugh PL, et al. Noncognitive behaviours in an APP/PS1 transgenic model of Alzheimer’s disease. Behav Brain Res. 2007;178(1):18–28. doi: 10.1016/j.bbr.2006.11.044. [DOI] [PubMed] [Google Scholar]
- 42.van Wamelen DJ, et al. Suprachiasmatic nucleus neuropeptide expression in patients with Huntington’s Disease. Sleep. 2013;36(1):117–125. doi: 10.5665/sleep.2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Pablo-Fernandez E, et al. A histologic study of the circadian system in Parkinson disease, multiple system atrophy, and progressive supranuclear palsy. JAMA Neurol. 2018;75(8):1008–1012. doi: 10.1001/jamaneurol.2018.0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swaab DF, et al. The suprachiasmatic nucleus of the human brain in relation to sex, age and senile dementia. Brain Res. 1985;342(1):37–44. doi: 10.1016/0006-8993(85)91350-2. [DOI] [PubMed] [Google Scholar]
- 45.Zhou JN, et al. VIP neurons in the human SCN in relation to sex, age, and Alzheimer’s disease. Neurobiol Aging. 1995;16(4):571–576. doi: 10.1016/0197-4580(95)00043-E. [DOI] [PubMed] [Google Scholar]
- 46.Stopa EG, et al. Pathologic evaluation of the human suprachiasmatic nucleus in severe dementia. J Neuropathol Exp Neurol. 1999;58(1):29–39. doi: 10.1097/00005072-199901000-00004. [DOI] [PubMed] [Google Scholar]
- 47.Baloyannis SJ, et al. The hypothalamus in Alzheimer’s disease: a Golgi and electron microscope study. Am J Alzheimers Dis Other Demen. 2015;30(5):478–487. doi: 10.1177/1533317514556876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harper DG, et al. Dorsomedial SCN neuronal subpopulations subserve different functions in human dementia. Brain. 2008;131(pt 6):1609–1617. doi: 10.1093/brain/awn049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang JL, et al. Suprachiasmatic neuron numbers and rest-activity circadian rhythms in older humans. Ann Neurol. 2015;78(2):317–322. doi: 10.1002/ana.24432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fahrenkrug J, et al. Decreased VIP and VPAC2 receptor expression in the biological clock of the R6/2 Huntington’s disease mouse. J Mol Neurosci. 2007;31(2):139–148. doi: 10.1385/jmn/31:02:139. [DOI] [PubMed] [Google Scholar]
- 51.Kuljis DA, et al. Sex differences in circadian dysfunction in the BACHD mouse model of Huntington’s disease. PLoS One. 2016;11(2):e0147583. doi: 10.1371/journal.pone.0147583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stevanovic K, et al. Disruption of normal circadian clock function in a mouse model of tauopathy. Exp Neurol. 2017;294:58–67. doi: 10.1016/j.expneurol.2017.04.015. [DOI] [PubMed] [Google Scholar]
- 53.Buijink MR, Michel SA. A multi-level assessment of the bidirectional relationship between aging and the circadian clock. J Neurochem. 2021;157(1):73–94. doi: 10.1111/jnc.15286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paul JR, et al. Behavioral and SCN neurophysiological disruption in the Tg-SwDI mouse model of Alzheimer’s disease. Neurobiol Dis. 2018;114:194–200. doi: 10.1016/j.nbd.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leng Y, et al. Association between circadian rhythms and neurodegenerative diseases. Lancet Neurol. 2019;18(3):307–318. doi: 10.1016/S1474-4422(18)30461-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duncan DJ. Interacting influences of aging and Alzheimer’s disease on circadian rhythms. Eur J Neurosci. 2020;51(1):310–325. doi: 10.1111/ejn.14358. [DOI] [PubMed] [Google Scholar]
- 57.Buijs RM, et al. The circadian system and the balance of the autonomic nervous system. Handb Clin Neurol. 2013;117:173–191. doi: 10.1016/B978-0-444-53491-0.00015-8. [DOI] [PubMed] [Google Scholar]
- 58.Liu C, et al. Molecular dissection of two distinct actions of melatonin on the suprachiasmatic circadian clock. Neuron. 1997;19(1):91–102. doi: 10.1016/S0896-6273(00)80350-5. [DOI] [PubMed] [Google Scholar]
- 59.Gamble KL, et al. Circadian clock control of endocrine factors. Nat Rev Endocrinol. 2014;10(8):466–475. doi: 10.1038/nrendo.2014.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reid K. Assessment of circadian rhythms. Neurol Clin. 2019;37(3):505–526. doi: 10.1016/j.ncl.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aziz NA, et al. Increased hypothalamic-pituitary-adrenal axis activity in Huntington’s disease. J Clin Endocrinol Metab. 2009;94(4):1223–1228. doi: 10.1210/jc.2008-2543. [DOI] [PubMed] [Google Scholar]
- 62.Kalliolia E, et al. Plasma melatonin is reduced in Huntington’s disease. Mov Disord. 2014;29(12):1511–1515. doi: 10.1002/mds.26003. [DOI] [PubMed] [Google Scholar]
- 63.Bartlett DM, et al. Investigating the relationships between hypothalamic volume and measures of circadian rhythm and habitual sleep in premanifest Huntington’s disease. Neurobiol Sleep Circadian Rhythms. 2018;6:1–8. doi: 10.1016/j.nbscr.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bordet R, et al. Study of circadian melatonin secretion pattern at different stages of Parkinson’s disease. Clin Neuropharmacol. 2003;26(2):65–72. doi: 10.1097/00002826-200303000-00005. [DOI] [PubMed] [Google Scholar]
- 65.Videnovic A, et al. Circadian melatonin rhythm and excessive daytime sleepiness in Parkinson disease. JAMA Neurol. 2014;71(4):463–469. doi: 10.1001/jamaneurol.2013.6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bolitho SJ, et al. Disturbances in melatonin secretion and circadian sleep-wake regulation in Parkinson disease. Sleep Med. 2014;15(3):342–347. doi: 10.1016/j.sleep.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 67.Kataoka H, et al. Melatonin secretion in patients with Parkinson’s disease receiving different-dose levodopa therapy. Sleep Med. 2020;75:309–314. doi: 10.1016/j.sleep.2020.07.043. [DOI] [PubMed] [Google Scholar]
- 68.Liu RY, et al. Decreased melatonin levels in postmortem cerebrospinal fluid in relation to aging, Alzheimer’s disease, and apolipoprotein E-epsilon4/4 genotype. J Clin Endocrinol Metab. 1999;84(1):323–327. doi: 10.1210/jcem.84.1.5394. [DOI] [PubMed] [Google Scholar]
- 69.Mishima K, et al. Melatonin secretion rhythm disorders in patients with senile dementia of Alzheimer’s type with disturbed sleep-waking. Biol Psychiatry. 1999;45(4):417–421. doi: 10.1016/S0006-3223(97)00510-6. [DOI] [PubMed] [Google Scholar]
- 70.Skene DJ, Swaab DF. Melatonin rhythmicity: effect of age and Alzheimer’s disease. Exp Gerontol. 2003;38(1–2):199–206. doi: 10.1016/s0531-5565(02)00198-5. [DOI] [PubMed] [Google Scholar]
- 71.Manni R, et al. Evening melatonin timing secretion in real life conditions in patients with Alzheimer disease of mild to moderate severity. Sleep Med. 2019;63:122–126. doi: 10.1016/j.sleep.2019.04.018. [DOI] [PubMed] [Google Scholar]
- 72.Stephan FK, Nunez AA. Elimination of circadian rhythms in drinking, activity, sleep, and temperature by isolation of the suprachiasmatic nuclei. Behav Biol. 1977;20(1):1–61. doi: 10.1016/S0091-6773(77)90397-2. [DOI] [PubMed] [Google Scholar]
- 73.Ruby NF, et al. The suprachiasmatic nucleus is essential for circadian body temperature rhythms in hibernating ground squirrels. J Neurosci. 2002;22(1):357–364. doi: 10.1523/JNEUROSCI.22-01-00357.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scheer FA, et al. Environmental light and suprachiasmatic nucleus interact in the regulation of body temperature. Neuroscience. 2005;132(2):465–477. doi: 10.1016/j.neuroscience.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 75.Lu J, et al. Contrasting effects of ibotenate lesions of the paraventricular nucleus and subparaventricular zone on sleep-wake cycle and temperature regulation. J Neurosci. 2001;21(13):4864–4874. doi: 10.1523/JNEUROSCI.21-13-04864.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schultz JL, et al. Autonomic dysregulation as an early pathologic feature of Huntington disease. Auton Neurosci. 2021;231:102775. doi: 10.1016/j.autneu.2021.102775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Raupach AK, et al. Assessing the role of nocturnal core body temperature dysregulation as a biomarker of neurodegeneration. J Sleep Res. 2020;29(5):e12939. doi: 10.1111/jsr.12939. [DOI] [PubMed] [Google Scholar]
- 78.Zhong G, et al. The relationship between thermoregulation and REM sleep behaviour disorder in Parkinson’s disease. PLoS One. 2013;8(8):e72661. doi: 10.1371/journal.pone.0072661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pierangeli G, et al. Nocturnal body core temperature falls in Parkinson’s disease but not in multiple-system atrophy. Mov Disord. 2001;16(2):226–232. doi: 10.1002/mds.1039. [DOI] [PubMed] [Google Scholar]
- 80.Satlin A, et al. Circadian locomotor activity and core-body temperature rhythms in Alzheimer’s disease. Neurobiol Aging. 1995;16(5):765–771. doi: 10.1016/0197-4580(95)00059-N. [DOI] [PubMed] [Google Scholar]
- 81.Volicer L, et al. Sundowning and circadian rhythms in Alzheimer’s disease. Am J Psychiatry. 2001;158(5):704–711. doi: 10.1176/appi.ajp.158.5.704. [DOI] [PubMed] [Google Scholar]
- 82.Harper DG, et al. Disturbance of endogenous circadian rhythm in aging and Alzheimer disease. Am J Geriatr Psychiatry. 2005;13(5):359–368. doi: 10.1097/00019442-200505000-00004. [DOI] [PubMed] [Google Scholar]
- 83.Fisher SP, et al. Longitudinal analysis of the electroencephalogram and sleep phenotype in the R6/2 mouse model of Huntington’s disease. Brain. 2013;136(Pt 7):2159–2172. doi: 10.1093/brain/awt132. [DOI] [PubMed] [Google Scholar]
- 84.Fisher SP, et al. Quantitative electroencephalographic analysis provides an early-stage indicator of disease onset and progression in the zQ175 knock-in mouse model of Huntington’s disease. Sleep. 2016;39(2):379–391. doi: 10.5665/sleep.5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cutler TS, et al. Neurocardiovascular deficits in the Q175 mouse model of Huntington’s disease. Physiol Rep. 2017;5(11):e13289. doi: 10.14814/phy2.13289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Knight EM, et al. Age-related changes in core body temperature and activity in triple-transgenic Alzheimer’s disease (3xTgAD) mice. Dis Model Mech. 2013;6(1):160–170. doi: 10.1242/dmm.010173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Oster H, et al. The functional and clinical significance of the 24-hour rhythm of circulating glucocorticoids. Endocr Rev. 2017;38(1):3–45. doi: 10.1210/er.2015-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van Duijn E, et al. Hypothalamic-pituitary-adrenal axis functioning in Huntington’s disease mutation carriers compared with mutation-negative first-degree controls. Brain Res Bull. 2010;83(5):232–237. doi: 10.1016/j.brainresbull.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 89.Adamczak-Ratajczak A, et al. Circadian rhythms of melatonin and cortisol in manifest Huntington’s disease and in acute cortical ischemic stroke. J Physiol Pharmacol. 2017;68(4):539–546. [PubMed] [Google Scholar]
- 90.Heuser IJ, et al. The limbic-hypothalamic-pituitary-adrenal axis in Huntington’s disease. Biol Psychiatry. 1991;30(9):943–952. doi: 10.1016/0006-3223(91)90007-9. [DOI] [PubMed] [Google Scholar]
- 91.Saleh N, et al. Neuroendocrine disturbances in Huntington’s disease. PLoS One. 2009;4(3):e4962. doi: 10.1371/journal.pone.0004962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hartmann A, et al. Twenty-four hour cortisol release profiles in patients with Alzheimer’s and Parkinson’s disease compared to normal controls: ultradian secretory pulsatility and diurnal variation. Neurobiol Aging. 1997;18(3):285–289. doi: 10.1016/S0197-4580(97)80309-0. [DOI] [PubMed] [Google Scholar]
- 93.Breen DP, et al. Sleep and circadian rhythm regulation in early Parkinson disease. JAMA Neurol. 2014;71(5):589–595. doi: 10.1001/jamaneurol.2014.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hatfield CF, et al. Disrupted daily activity/rest cycles in relation to daily cortisol rhythms of home-dwelling patients with early Alzheimer’s dementia. Brain. 2004;127(Pt 5):1061–1074. doi: 10.1093/brain/awh129. [DOI] [PubMed] [Google Scholar]
- 95.Dufour BD, McBride JL. Normalizing glucocorticoid levels attenuates metabolic and neuropathological symptoms in the R6/2 mouse model of huntington’s disease. Neurobiol Dis. 2019;121:214–229. doi: 10.1016/j.nbd.2018.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hogenesch JB, Ueda HR. Understanding systems-level properties: timely stories from the study of clocks. Nat Rev Genet. 2011;12(6):407–416. doi: 10.1038/nrg2972. [DOI] [PubMed] [Google Scholar]
- 97.Cox KH, Takahashi JS. Circadian clock genes and the transcriptional architecture of the clock mechanism. J Mol Endocrinol. 2019;63(4):R93–R102. doi: 10.1530/JME-19-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wu YH, et al. Pineal clock gene oscillation is disturbed in Alzheimer’s disease, due to functional disconnection from the “master clock”. FASEB J. 2006;20(11):1874–1876. doi: 10.1096/fj.05-4446fje. [DOI] [PubMed] [Google Scholar]
- 99.Cronin P, et al. Circadian alterations during early stages of Alzheimer’s disease are associated with aberrant cycles of DNA methylation in BMAL1. Alzheimers Dement. 2017;13(6):689–700. doi: 10.1016/j.jalz.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hulme B, et al. Epigenetic regulation of BMAL1 with sleep disturbances and Alzheimer’s disease. J Alzheimers Dis. 2020;77(4):1783–1792. doi: 10.3233/JAD-200634. [DOI] [PubMed] [Google Scholar]
- 101.Morton AJ, et al. Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J Neurosci. 2005;25(1):157–163. doi: 10.1523/JNEUROSCI.3842-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Maywood ES, et al. Disruption of peripheral circadian timekeeping in a mouse model of Huntington’s disease and its restoration by temporally scheduled feeding. J Neurosci. 2010;30(30):10199–10204. doi: 10.1523/JNEUROSCI.1694-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Whittaker DS, et al. Circadian-based treatment strategy effective in the BACHD mouse model of Huntington’s disease. J Biol Rhythms. 2018;33(5):535–554. doi: 10.1177/0748730418790401. [DOI] [PubMed] [Google Scholar]
- 104.Song H, et al. Aβ-induced degradation of BMAL1 and CBP leads to circadian rhythm disruption in Alzheimer’s disease. Mol Neurodegener. 2015;10:13. doi: 10.1186/s13024-015-0007-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Oyegbami O, et al. Abnormal clock gene expression and locomotor activity rhythms in two month-old female APPSwe/PS1dE9 mice. Curr Alzheimer Res. 2017;14(8):850–860. doi: 10.2174/1567205014666170317113159. [DOI] [PubMed] [Google Scholar]
- 106.Morton AJ. Circadian and sleep disorder in Huntington’s disease. Exp Neurol. 2013;243:34–44. doi: 10.1016/j.expneurol.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 107.Videnovic A, et al. The clocks that time us-circadian rhythms in neurodegenerative disorders. Nat Rev Neurol. 2014;10(12):683–693. doi: 10.1038/nrneurol.2014.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brehme M, et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014;9(3):1135–1150. doi: 10.1016/j.celrep.2014.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ma D, et al. Temporal orchestration of circadian autophagy rhythm by C/EBPβ. EMBO J. 2011;30(22):4642–4651. doi: 10.1038/emboj.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Desvergne A, et al. Circadian modulation of proteasome activity and accumulation of oxidized protein in human embryonic kidney HEK 293 cells and primary dermal fibroblasts. Free Radic Biol Med. 2016;94:195–207. doi: 10.1016/j.freeradbiomed.2016.02.037. [DOI] [PubMed] [Google Scholar]
- 111.Saibil HR. The PDB and protein homeostasis: from chaperones to degradation and disaggregase machines. J Biol Chem. 2021;296:100744. doi: 10.1016/j.jbc.2021.100744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Reinke H, et al. Differential display of DNA-binding proteins reveals heat-shock factor 1 as a circadian transcription factor. Genes Dev. 2008;22(3):331–345. doi: 10.1101/gad.453808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Neueder A, et al. HSF1-dependent and -independent regulation of the mammalian in vivo heat shock response and its impairment in Huntington’s disease mouse models. Sci Rep. 2017;7(1):12556. doi: 10.1038/s41598-017-12897-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xu F, et al. Circadian clocks function in concert with heat shock organizing protein to modulate mutant Huntingtin aggregation and toxicity. Cell Rep. 2019;27(1):59–70. doi: 10.1016/j.celrep.2019.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xie L, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373–377. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Holth JK, et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science. 2019;363(6429):880–884. doi: 10.1126/science.aav2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hablitz LM, et al. Increased glymphatic influx is correlated with high EEG delta power and low heart rate in mice under anesthesia. Sci Adv. 2019;5(2):eaav5447. doi: 10.1126/sciadv.aav5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kang JE, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326(5955):1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Di Meco A, et al. Sleep deprivation impairs memory, tau metabolism, and synaptic integrity of a mouse model of Alzheimer’s disease with plaques and tangles. Neurobio Aging. 2014;35(8):1813–1820. doi: 10.1016/j.neurobiolaging.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 120.Lucey BP, et al. Effect of sleep on overnight cerebrospinal fluid amyloid β kinetics. Ann Neurol. 2018;83(1):197–204. doi: 10.1002/ana.25117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kress GJ, et al. Regulation of amyloid-β dynamics and pathology by the circadian clock. J Exp Med. 2018;215(4):1059–1068. doi: 10.1084/jem.20172347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hablitz LM, et al. Circadian control of brain glymphatic and lymphatic fluid flow. Nat Commun. 2020;11(1):4411. doi: 10.1038/s41467-020-18115-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brüning F, et al. Sleep-wake cycles drive daily dynamics of synaptic phosphorylation. Science. 2019;11;366(6462):eaav3617. doi: 10.1126/science.aav3617. [DOI] [PubMed] [Google Scholar]
- 124.Man K, et al. Immunity around the clock. Science. 2016;354(6315):999–1003. doi: 10.1126/science.aah4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Baxter M, Ray DW. Circadian rhythms in innate immunity and stress responses. Immunology. 2020;161(4):261–267. doi: 10.1111/imm.13166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cunningham PS, et al. The circadian clock protein REVERBα inhibits pulmonary fibrosis development. Proc Natl Acad Sci U S A. 2020;117(2):1139–1147. doi: 10.1073/pnas.1912109117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hergenhan S, et al. Molecular interactions between components of the circadian clock and the immune system. J Mol Biol. 2020;432(12):3700–3713. doi: 10.1016/j.jmb.2019.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Irwin MR, Vitiello MV. Implications of sleep disturbance and inflammation for Alzheimer’s disease dementia. Lancet Neurol. 2019;18(3):296–306. doi: 10.1016/S1474-4422(18)30450-2. [DOI] [PubMed] [Google Scholar]
- 129.Akiyama H, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21(3):383–421. doi: 10.1016/S0197-4580(00)00124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lananna BV, Musiek ES. The wrinkling of time: aging, inflammation, oxidative stress, and the circadian clock in neurodegeneration. Neurobiol Dis. 2020;139:104832. doi: 10.1016/j.nbd.2020.104832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Prolo LM, et al. Circadian rhythm generation and entrainment in astrocytes. J Neurosci. 2005;25(2):404–408. doi: 10.1523/JNEUROSCI.4133-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Musiek ES, et al. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J Clin Invest. 2013;123(12):5389–5400. doi: 10.1172/JCI70317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nakazato R, et al. Disruption of Bmal1 impairs blood-brain barrier integrity via pericyte dysfunction. J Neurosci. 2017;37(42):10052–10062. doi: 10.1523/JNEUROSCI.3639-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lananna BV, et al. Cell-autonomous regulation of astrocyte activation by the circadian clock protein BMAL1. Cell Rep. 2018;25(1):1–9. doi: 10.1016/j.celrep.2018.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hayashi Y, et al. The intrinsic microglial molecular clock controls synaptic strength via the circadian expression of cathepsin S. Sci Rep. 2013;3:2744. doi: 10.1038/srep02744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fonken LK, et al. Microglia inflammatory responses are controlled by an intrinsic circadian clock. Brain Behav Immun. 2015;45:171–179. doi: 10.1016/j.bbi.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Griffin P, et al. Circadian clock protein Rev-erbα regulates neuroinflammation. Proc Natl Acad Sci U S A. 2019;116(11):5102–5107. doi: 10.1073/pnas.1812405116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Griffin P, et al. REV-ERBα mediates complement expression and diurnal regulation of microglial synaptic phagocytosis. Elife. 2020;9:e58765. doi: 10.7554/eLife.58765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schroeder AM, Colwell CS. How to fix a broken clock. Trends Pharmacol Sci. 2013;34(11):605–619. doi: 10.1016/j.tips.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fifel K, Videnovic A. Chronotherapies for Parkinson’s disease. Prog Neurobiol. 2019;174:16–27. doi: 10.1016/j.pneurobio.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Steele TA, et al. Circadian rhythm sleep-wake disorders: a contemporary review of neurobiology, treatment, and dysregulation in neurodegenerative disease. Neurotherapeutics. 2021;18(1):53–74. doi: 10.1007/s13311-021-01031-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Duffy JF, et al. Workshop report. Circadian rhythm sleep-wake disorders: gaps and opportunities. Sleep. 2021;44(5):zsaa281. doi: 10.1093/sleep/zsaa281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Figueiro MG, et al. Effects of a tailored lighting intervention on sleep quality, rest-activity, mood, and behavior in older adults with Alzheimer disease and related dementias: a randomized clinical trial. J Clin Sleep Med. 2019;15(12):1757–1767. doi: 10.5664/jcsm.8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Allada R, Bass J. Circadian mechanisms in medicine. N Engl J Med. 2021;384(6):550–561. doi: 10.1056/NEJMra1802337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cardinali DP, et al. Chronotherapy. Handb Clin Neurol. 2021;179:357–370. doi: 10.1016/B978-0-12-819975-6.00023-6. [DOI] [PubMed] [Google Scholar]
- 146.Ruan W, et al. Circadian rhythm as a therapeutic target. Nat Rev Drug Discov. 2021;20(4):287–307. doi: 10.1038/s41573-020-00109-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Whittaker DS, et al. Possible use of a H3R antagonist for the management of nonmotor symptoms in the Q175 mouse model of Huntington’s disease. Pharmacol Res Perspect. 2017;5(5):e00344. doi: 10.1002/prp2.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gloston GF, et al. Clock-enhancing small molecules and potential applications in chronic diseases and aging. Front Neurol. 2017;8:100. doi: 10.3389/fneur.2017.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nakajima A, Ohizumi Y. Potential benefits of nobiletin, a citrus flavonoid, against Alzheimer’s disease and Parkinson’s disease. Int J Mol Sci. 2019;20(14):3380. doi: 10.3390/ijms20143380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Banerjee S, et al. Pharmacological targeting of the mammalian clock regulates sleep architecture and emotional behaviour. Nat Commun. 2014;5:5759. doi: 10.1038/ncomms6759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chung S, et al. Impact of circadian nuclear receptor REV-ERBα on midbrain dopamine production and mood regulation. Cell. 2014;157(4):858–868. doi: 10.1016/j.cell.2014.03.039. [DOI] [PubMed] [Google Scholar]