

Abstract
BACKGROUND
Chimeric antigen receptor (CAR) T cells have emerged as an approach to treat malignant tumors. This strategy has also been proposed for the treatment of HIV-1 infection. We have developed a broadly neutralizing antibody–derived (bNAb-derived) CAR T cell therapy that can exert specific cytotoxic activity against HIV-1–infected cells.
METHODS
We conducted an open-label trial of the safety, side-effect profile, pharmacokinetic properties, and antiviral activity of bNAb-derived CAR T cell therapy in individuals infected with HIV-1 who were undergoing analytical interruption of antiretroviral therapy (ART).
RESULTS
A total of 14 participants completed only a single administration of bNAb-derived CAR T cells. CAR T cell therapy administration was safe and well tolerated. Six participants discontinued ART, and viremia rebound occurred in all of them, with a 5.3-week median time. Notably, the cell-associated viral RNA and intact proviruses decreased significantly after CAR T cell treatment. Analyses of HIV-1 variants before or after CAR T cell administration suggested that CAR T cells exerted pressure on rebound viruses, resulting in a selection of viruses with less diversity and mutations against CAR T cell–mediated cytotoxicity.
CONCLUSION
No safety concerns were identified with adoptive transfer of bNAb-derived CAR T cells. They reduced viral reservoir. All the rebounds were due to preexisting or emergence of viral escape mutations.
TRIAL REGISTRATION
ClinicalTrials.gov (NCT03240328).
FUNDING
Ministry of Science and Technology of China, National Natural Science Foundation of China, and Department of Science and Technology of Guangdong Province.
Keywords: AIDS/HIV, Clinical Trials
Keywords: Immunotherapy
Introduction
Antiretroviral therapy (ART) significantly suppresses HIV-1 to an undetectable level in the blood, improves immune function, delays progression of the disease, and decreases mortality in patients infected with HIV-1 (1). However, some HIV-1 replication-competent proviruses comprise a latent reservoir, which is quite stable, with a half-life of 44 months, requiring nearly 73.4 years for complete clearance (2, 3). In almost all individuals infected with HIV-1, plasma viral rebound predictably occurs within days after treatment interruption, resulting in the lifelong requirement for ART (4). To achieve durable suppression of viremia without daily therapy, various strategies have been proposed, including long-acting antiretroviral drugs (LA-ARVs), broadly neutralizing antibodies (bNAbs), and chimeric antigen receptor (CAR) T cells (5). In human clinical trials, viremic individuals who received bNAb therapies showed significant reductions in viremia (6–8). Moreover, individuals infected with HIV-1 who received multiple infusions of VRC01 or 3BNC117, 2 related bNAbs that target the CD4+ binding site on the HIV-1 envelope (Env) spike, showed significant viral suppression for 5.6 or 9.9 weeks, respectively, during analytical treatment interruption (ATI) of ART (9, 10). Furthermore, a combination therapy of 3BNC117 and 10-1074 maintained the suppression of virus rebound for a median of 21 weeks (11). These findings suggest that immunotherapy with CAR T cells, if HIV-1–specific and bNAb-derived, may also prevent virus rebound after ATI in individuals infected with HIV-1.
The CAR moiety is typically generated by coupling an antibody-derived, single-chain Fv domain to an intracellular T cell receptor zeta chain and costimulatory receptor-signaling domains. The clinical usage of CAR T cells resulted in complete remission in approximately 83% of patients with lymphocytic leukemia/lymphoma (12). Compared with CAR T cells targeting tumor-associated antigens, such as CD19+ and CD20+, which are also expressed in normal B lymphocytes, the HIV-1–specific CAR T cells target the HIV-1 Env protein, which is only expressed on the surface of virus-producing cells (13–15). Previously, a strategy that fuses the extracellular domain of CD4+ with the intracellular domain of the CD3ζ chain (CD4ζ-CAR) was shown to be safe and feasible in individuals infected with HIV-1. However, the antiviral efficacy was modest, and durable control of viral replication in clinical trials was not observed (16–20). In recent years, third and fourth generation intracellular CAR moieties have been developed (12). Moreover, a number of preclinical studies on bNAb-derived HIV-1–specific CAR T cells, in vitro and in animal models, have shown the suppression of viral replication or the reduction of virus-producing cells (21–26). In particular, we previously found that the VRC01-derived CAR T cells effectively reduced the reactivated viral reservoir isolated from individuals infected with HIV-1 who were receiving ART, and the engineered resistance to triple inhibitory receptors, including PD-1, Tim-3, and Lag-3, prevented CAR T cell exhaustion and improved their efficacy in vivo (22, 27).
Here, we report the results of a phase I clinical trial to investigate the effect of a single administration of bNAb-derived CAR T cells on virus rebound after the discontinuation of suppressive ART. We examined whether the adoptive transfer of bNAb-derived CAR T cells is safe and feasible, leads to long-term immune surveillance, and acts as a potential alternative to antiretroviral drugs to suppress viremia rebound after the discontinuation of ART.
Results
bNAb-derived CAR T cell treatment is safe and well tolerated.
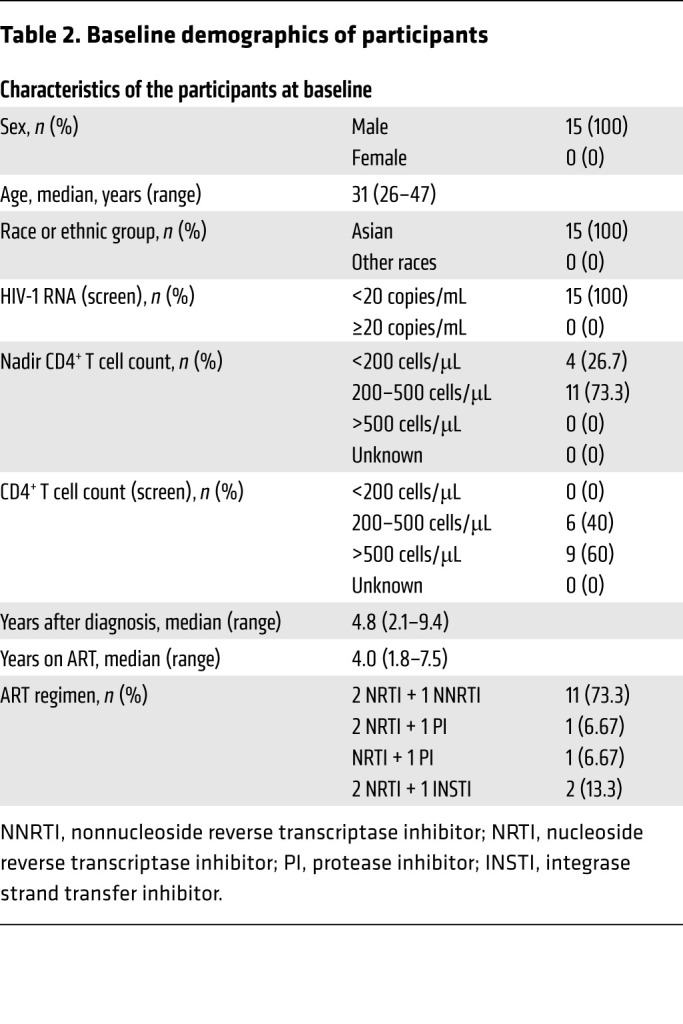
The clinical trial was divided into 4 parts: blood drawing and CAR T cell preparation, CAR T cell infusion, ATI, and ART reinitiation after viral rebound (Figure 1). The study enrolled 15 participants with chronic HIV-1 infection, all of whom were male, with a median CD4+ T cell count, at enrollment, of 597 cells/μL (range 380–842 cells/μL), and a median duration, from the initiation of ART to study entry, of 4 years (range 1.8–7.5 years) (Tables 1 and 2 and Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI150211DS1). All participants have not undergone any additional immunotherapeutic intervention besides ART and there were no conditioning regimens before CAR T cell administrations.
Figure 1. Schematic of the clinical study.

The clinical trial was divided into 4 parts: blood drawing and CAR T cell preparation, CAR T cell infusion, ATI, and ART reinitiation after viral rebound. The safety laboratory values and HIV-1 viral load were monitored at regular intervals throughout the study.
Table 1. Baseline clinical characteristics of enrolled participants.

Table 2. Baseline demographics of participants.
CAR T cells were successfully generated for 14 of the 15 enrolled patients by infecting the CD8+ T cells with a lentiviral vector expressing the VRC01-28BBz-shPTL CAR moiety (Figure 2A). The transduction efficiency ranged from 34.9% to 77.8%, and the doses of transferred CAR T cells ranged from 26.2 × 106 to 63.6 × 106 (Supplemental Table 2). patient 011 was ineligible to receive an infusion, because the cells failed to expand adequately (Supplemental Figure 1). Notably, CD8+ Fab+ CAR T cells had significantly less PD-1 expression compared with CD8+ Fab–cells, which suggested that the shRNA cluster can efficiently downregulate the expression of immune checkpoint (Supplemental Figures 2 and 3). The ex vivo–expanded CAR T cells exerted HIV-1 gp160-specific cytotoxicity in vitro, and some participant CAR T cells, such as those of patients 001, 005, 012, and 015, showed more effective lysis of HIV-1NL4-3 gp160-expressing cells than others at baseline (Supplemental Figure 4).
Figure 2. Plasma viremia after discontinuation of ART in patients infected with HIV-1.

(A) Schematic representation of the lentiviral vectors carrying a gp120-specific CAR moiety containing CD28 and 4-1BB (CD137) costimulatory domains, followed by a herpes simplex virus-1 thymidine kinase (TK) and a truncated CD19 gene as the suicide genes. A combination of shRNAs, including sh-PD-1, sh-Lag-3, and sh-Tim-3, for preventing exhaustion and increasing the in vivo persistence of CAR T cells, was inserted into the vector. (B) Plasma viremia of participants in the study after the ATI of ART (n = 6). The limit of detection of HIV-1 RNA level in this assay was 20 copies/mL. (C) Kaplan-Meier curve of plasma viral suppression (< 200 copies/mL) after ATI in the trial participants.
The treatment regimen was generally well tolerated in all patients, and no serious adverse events occurred. Complete data on adverse events are provided (Supplemental Table 3). Discontinuation of ART is favorable to evaluate the antivirus effect of the CAR T cell therapy. Considering the potential risks for ATI, the ATI was performed when the several criteria were achieved and at least 4 weeks after CAR T cell administration (see Methods). Six participants (patients 002, 003, 004, 005, 006, and 015) met the requirements of ATI and consented to temporary suspension of ART. Notably, they showed significant reductions of cell-associated viral RNA and relatively high levels of in vivo CAR T cell persistence (Supplemental Figure 5 and Supplemental Table 4). Patients 001, 007, 010, and 013 also had significant reduction of cell-associated viral RNA and met the other requirements such as CAR T cell persistence and CD4+ T cell count; however, they dissented to interrupt ART (Supplemental Figure 5 and Supplemental Table 5). The cell-associated viral RNA in patients 008, 009, 012, and 014 had not shown significant reduction in the majority of testing points, therefore, the investigators decided not to proceed with the ATI for these participants, and they all agreed to maintain ART regimes (Supplemental Figure 5 and Supplemental Table 5). In 6 participants chosen for ATI, ART was reinitiated on confirmation of viral rebound (the plasma viral load exceeding 200 copies per milliliter), and their plasma viremia was suppressed again. No acute immune response was observed after the infusion of CAR T cells. The CD4+ T cell counts and the ratio of CD4+ to CD8+ T cells were maintained in the normal range at month 6 after adoptive transfer, while HIV-1 infection was under control with ART (Supplemental Figure 6). During the ATI, the peripheral CD4+ T cell counts decreased in patients 003 and 004, and the same trend was observed in the ratio of CD4+ cells to CD8+ T cells in patient 004 (Supplemental Figure 6).
CAR T cell treatment delayed the viral rebound after ATI.
In this clinical trial, 6 participants met the requirements of ATI and consented to temporary suspension of ART. Notably, they showed significant cell-associated viral RNA reduction and relatively high levels of in vivo CAR T cell persistence (Supplemental Table 4). After ATI, the inhibition of HIV-1 rebound in plasma was sustained as long as 10 weeks (patient 002). However, the therapy did not lead to long-term suppression of viremia. Viral rebound to more than 200 copies per milliliter occurred after ATI in all 6 participants, with a median time to rebound of 5.3 weeks (range 3–10 weeks). The longest time of viral suppression was in patient 002 (10 weeks), while the shortest was in patient 006 (3 weeks) (Figure 2, B and C). To determine whether the participants who received an infusion of CAR T cells had a delay in viral rebound after ATI, we introduced a historical control from ATI studies of the AIDS Clinical Trial Group (ACTG) (Supplemental Table 6). This historical control included 155 chronically infected participants with HIV-1 whose viral rebound data were captured in 4 ATI studies without additional immunologic interventions (ACTG 371, A5024, A5068, and A5197) (28–32). The control was selected on the basis of similar inclusion criteria to our study. As compared with 2.3 weeks to plasma viral rebound (HIV RNA level ≥200 copies per milliliter) in historical controls from the noninterventional ATI studies (28), the CAR T cell administration led to a longer time to rebound (≥200 copies per milliliter), and 67% of the participants versus 32% of the controls had viral suppression at week 4 (P < 0.0001 by a 2-sided χ2 test), and 17% versus 6%, respectively, had viral suppression at week 8 (P = 0.015 by a 2-sided χ2 test) (28). Meanwhile, as the ART was still used after the CAR T cell infusions, the plasma HIV-1 levels in the non-ATI group were lower than the detection limit during the observation period.
To further analyze other relevant factors impacting viral rebound, we stratified the ART regimens at screening, 4 of the 6 ATI participants (002, 004, 005, and 015) received NNRTI-containing regimens (2 NRTI + 1 NNRTI) (Table 1). Their percentages of virologic suppression (plasma viral load <200 copies/mL) after ATI were 100% and 25% at week 4 and week 8, respectively. Correspondingly, the percentages of virologic suppression in historical control (n = 99) receiving NNRTI-containing regimens were 44% and 9% at week 4 and week 8, respectively, at the same viral load threshold (28). When we directly compared the proportions of virologic suppression in our study with historical control receiving NNRTI-containing regime by χ2 test and Fisher’s exact test, the results still showed a significantly higher proportion of virologic suppression after CAR T administration in the NNRTI-receiving group than in historical control at week 4 (P < 0.0001) and week 8 (P < 0.0001) (Supplemental Figure 7). In participants 003 and 006, who were receiving non-NNRTI regimens at screening, the plasma viral rebound occurred at week 4 or week 3, respectively. Those receiving CAR T cell infusions on NNRTI background seemed to have a longer timing of virologic suppression. The reason might be associated to the prolonged half-life of NNRTIs. As the ATI cases are limited, we cannot conclude significant differences in durations of viral rebound among different ART regimes. When we compared the proportions of virologic suppression in our study (67% at week 4) to historical control by treatment during acute (28% at week 4, n = 32) or early infection (29% at week 4, n = 48), the results showed a significantly higher proportion of virologic suppression than in either the acute (P < 0.0001) or early (P < 0.0001) treatment group (Supplemental Figure 8).
Additionally, protective HLA-I alleles were detected in 6 participants including 001 (B*58:01; B*27), 003 (B*52:01), 005 (B*58:01; B*27), 010 (B*13:02), 013 (B*51), and 014 (B*51) (Supplemental Table 7) (33). The durations of viral suppression in participants 003 and 005 harboring protective HLA-B alleles were 4 and 3 weeks, respectively. They did not show longer durations of viral rebound.
Long-term in vivo persistence of CAR T cell after adoptive transfer.
In order to assess the duration of in vivo CAR T cell persistence, quantitative real-time PCR, using primers specific for the VRC01-28BBz-shPTL CAR transgene, was performed on genomic DNA from various time points after CAR T cell infusions. As previously described, we also detected the in vivo persistence of CAR T cells at an earlier time in some patients (Figure 3A and Supplemental Figure 9) (34, 35). The peak level of modified CAR T cells detected was 5.7% to 0.5% among CD8+ T cells for the 14 patients immediately after the infusions, and subsequently dropped to less than 0.2% (Figure 3A and Supplemental Figure 9). Nevertheless, the modified CAR T cells were persistently detectable for more than 44 weeks in all patients (Figure 3A).
Figure 3. Cell-associated viral RNA and in vivo CAR T cell persistence before and after adoptive transfer.

(A) Measured CAR+ cell concentrations (per million CD8+ T cells) for the 14 enrolled patients are shown in blue (log10 scale on left), and CA-RNA levels after adoptive transfer (per million CD4+ T cells) are shown in red (log10 scale shown on right). ATI period is shown by shades of gray. (B) HIV-1 RNA levels in plasma (copies/mL) are shown in black (log10 scale shown on right) and CA-RNA levels after ATI (per million CD4+ T cells) are shown in red (log10 scale shown on left). ATI periods are shown by shades of gray.
To further assess the persistence of HIV-1 Env-specific CAR T cells after adoptive transfer, interferon gamma (IFN-γ) ELISpot assays were performed by incubating the purified CD8+ T cells from participants infected with HIV-1 with the HIV-1NL4-3 Env-expressing cell line at a 1:1 ratio without any additional antigenic peptide addition. As shown in Supplemental Figure 10, the numbers of IFN-γ–secreting T cells from all 14 participants after CAR T cell administrations were much higher than those from pre-CAR T cell treatment, suggesting that they could develop HIV-1 Env-specific but MHC-I–independent T cell responses after adoptive transfers. With the extension of the observation period to more than 30 weeks after CAR T cell administrations, although the numbers of IFN-γ–secreting T cells were decreased compared with those at week 3, the HIV-1 Env-specific but MHC-I–independent T cell responses were still higher than that of pre-CAR T cell treatment (Supplemental Figure 10). Considering there were neither antigen presenting cells (APCs) nor any antigenic peptide in the coculture system, the MHC-I–independent secretion of IFN-γ was not ascribed to CTL response but the HIV-1 Env-specific CAR T cell response. Collectively, the PCR-based CAR assays and function-based analyses both showed that the long-term in vivo survival of anti–HIV-1 CAR T cells.
CAR T cell treatment effectively decreases the viral reservoir.
It has been shown that HIV-1 unspliced (US) RNA represents intracellular transcripts to estimate the viral reservoir size and is usually much more abundant than the multiply spliced RNA (36–39). Therefore, the cell-associated viral US RNA (CA-RNA) was evaluated, using qRT-PCR, in peripheral CD4+ T cells (Figure 3, A and B, and Supplemental Figure 5). We compared the levels of CA-RNA between 14 participants prior to the infusions in our study and HIV-positive volunteers who were receiving ART-. There was no significant difference in CA-RNA between the 2 groups (Supplemental Figure 11A). We found that the levels of CA-RNA in samples from 2 weeks before adoptive transfers were not significantly different from those from week 0. However, they were significantly reduced 3 to 4 weeks after CAR T cell administration (Figure 4A). In particular, the CA-RNA was reduced approximately 100-fold after 4 weeks of CAR T cell administration in patient 002 (Figure 4A). These results suggested that CAR T cells effectively reduced these HIV-1 reservoirs. As the CA-RNA increased and the measurable plasma viruses rebounded during the periods of ART discontinuation, the transferred CAR T cells expanded accordingly (Figure 3, A and B). Unfortunately, CAR T cells failed to control viral rebound after ATI. The rebound viruses were suppressed again after ART was reintroduced, and the CAR T cell levels also gradually decreased (Figure 3A). Moreover, the increase in CA-RNA preceded the measurable plasma viral rebound in the periods of ART discontinuation, suggesting that it could serve as a sensitive marker to predict the viremia rebound during HIV-1 treatment interruption (Figure 3B). With the extension of the observation period to more than 30 weeks after CAR T cell administration, the CA-RNA levels had wide variance among different participants. However, the CA-RNA levels at week 30+ were still significantly lower than those at pre–CAR T cell time points (P = 0.0135) (Figure 4A, left panel). In 6 participants from ATI group, the CA-RNA levels at week 30+ were higher than those at week 3 or week 4 (Figure 4A, right panel). In contrast, in the non-ATI group, except in participant 007, further reductions of CA-RNA at week 30+ were observed in other 7 non-ATI participants compared with those at week 3 (Supplemental Figure 12). We followed up with 3 ART-receiving patients without any immunologic interventions. Their CA-RNA levels remained fluctuating but we did not detect a significant reduction over observation periods (Supplemental Figure 13). Therefore, the observations of reductions on CA-RNA in our study were most likely ascribed to CAR T cell infusions.
Figure 4. CAR T cell treatment decreased the CA-RNA and intact HIV-1 proviruses.

(A) Panels show the comparison of CA-RNA before and after administration of CAR T cells at indicated time points. Each point represents the mean of triplicate values. CA-RNA: 14 participants infected with HIV-1. CA-RNA (ATI): 6 ATI participants. P values were calculated using the Wilcoxon matched pairs signed-rank test. (B) Representative IPDA results from patients 002 and 015. Boxed areas are expanded to show individual positive droplets. (C) IPDA results in CD4+ T cells from 14 participants infected with HIV-1 (Intact proviral DNA) and 6 ATI participants (Intact proviral DNA [ATI]), before and after CAR T cell administration. P values were calculated using the Wilcoxon matched pairs signed-rank test.
Through a newly developed intact proviral DNA assay (IPDA) based on droplet digital PCR, we quantitatively analyzed the intact proviruses and defective proviruses in viral reservoirs from the clinical samples before and after CAR T cell administration, and found that intact proviruses were also significantly reduced at week 3 after CAR T cell administration (Figure 4, B and C, Supplemental Figure 14) (40). The same phenomena were also observed in both the ATI group and the non-ATI group (Figure 4C and Supplemental Figure 15). Interestingly, with the extension of the observation period to more than 30 weeks after CAR T cell administration, intact proviruses were further decreased compared with those at week 3 in 14 participants (Figure 4C and Supplemental Figure 15). Moreover, there was no difference in the levels of intact proviral DNA between 14 participants prior to CAR T cell infusions in our study and ART-receiving HIV-infected volunteers (Supplemental Figure 11B). Given that CAR T cells can be persistently detectable in peripheral blood for as long as 44 weeks, both CA-RNA and intact proviral DNA levels can be observed as decreased even 6 months after CAR T cell infusions. These results suggest that the long-term persistence of CAR T cells has the potential to undermine the viral reservoir.
CAR T cell treatment genetically restricts the rebound viruses.
To characterize the rebounding viral populations, single-genome sequencing of HIV-1 env genes from CD4+ T cell samples was performed before CAR T cell administration and the first and second weeks after detectable viremia (available in 6 participants). Previous studies have suggested that without any additional intervention besides ART, viral rebound after ATI is consistently polyclonal because of the reactivation of multiple latent viruses (41–43). In contrast, the CD4 binding site-directed neutralizing antibodies exert ongoing selection pressure on the conserved epitope of HIV-1 Env and rebound viruses clustered into relatively low-diversity lineages (44–46). Genetic evidence of CAR T cell–mediated restriction of viral rebound was assessed by analyzing the clonality of rebound virus or by enumerating genetically distinct virus populations composed of rebound viruses. In patients 002, 004, 006, and 015 (patients with chronic infection), the cell-associated viruses from pre–CAR T cell treatment formed characteristic diverse phylogenetic trees. In contrast, rebound viruses in cells were significantly distinct from those of pre–CAR T cell treatment and clustered into relatively low-diversity lineages (Figure 5 and Supplemental Figure 16). Additionally, the genetic diversities of full-length Env sequences from the post–CAR T cell sample were also significantly lower than those from the pre–CAR T cell sample in a non-ATI participant (patient 007) (Supplemental Figure 17). These findings suggested that HIV-1–specific CAR T cells were able to exert pressure on rebound viruses and that they reduced the number of HIV-1 intact proviruses at different locations, resulting in the emergence of genome-distinct viral mutations. In contrast, the rebound viruses in patient 003 clustered into multiple genetically distinct lineages that aligned throughout the pre–CAR T cell treatment virus phylogeny, indicating possible preexisting resistance (Figure 5) (41, 42).
Figure 5. Genetic comparison of the circulating latent reservoir and rebound viruses.

Panels show maximum likelihood phylogenetic trees of single‑genome sequencing–derived Env sequences from cell samples before CAR T cell administration and cell/plasma samples from the first and second weeks of detectable viremia. Sequences from pre-CAR T cell treatment are shown in blue, and the sequences from week 1 or week 2 of rebound viremia are shown in red. Genetic distance scale bars are shown for each tree; the bootstrap consensus trees were constructed based on HIV-1 sequences obtained from the corresponding patients. Sequences from before CAR T cell treatment were not available in patient 005.
CAR T cell treatment leads to the generation of resistant viruses.
According to previous reports, the VRC01 antibody–binding footprint was represented by Env residues in loop D, CD4+ binding sites, β20/β21 regions, and the base of the V5 loop, which are known as VRC01 contact residues, and many VRC01 antibody–resistant strains were identified in chronically infected patients (46–53). We generated sequence alignment and modified longitudinal logo plots to reveal mutations in the predicted VRC01 antibody–binding regions between pre–CAR T cell treatment and rebound virus populations (54). We also found a number of mutations in the rebound viruses in or near the VRC01 antibody–binding epitopes, mainly in the inner domain, V2 loop, loop D, CD4-binding site, and the V5 loop, from 5 of the 6 participants (Figure 6A). In patient 002, a change was found at position 197, a potential N-linked glycosylation (PNLG) site in the V2 region, where serine (S) was replaced by asparagine (N) (47). In patient 004, mutations were found at position 186 on the V2 loop and positions 279 and 280 on loop D (46, 47, 53). In patient 006, glycine (G) was replaced by alanine (A) at position 281 on loop D, isoleucine (I) was replaced by N at the PNLG site at position 461, and the residues 463–466 were also changed in the V5 loop (50, 53). These outcomes suggested bNAb-derived CAR T cell–mediated selective pressure on rebound viruses (Figure 6A and Supplemental Figure 18). However, in patient 003, some signature substitutions, such as threonine 278, of resistant strains were found in the viruses from pre–CAR T cell treatment, also suggesting a possible preexisting resistance (46).
Figure 6. Rebound virus clonality and resistance to bNAb-derived CAR T cell–mediated cytotoxicity.

(A) Clonal Env mutations on inner domain, V2 loop, loop D, CD4-binding site, and V5 loop after viral rebound in patients 002 and 006. All sequences were compared with the consensus of the rebound viruses. The residue numbers are based on HIV-1 HXB2 sequence. The top line shows amino acid differences in the pre–CAR T cell sequences from the rebound consensus. (B) PBMCs were isolated from healthy donors and divided into 2 populations. The CD8+ T lymphocytes were used to generate CAR T cells while the autologous CD4+ T cells were infected with outgrown HIV-1 from pre–CAR T cell latent reservoir (LR) or rebound reservoir (RD) (1 ng/mL p24). Six days after HIV-1 infection, antiretroviral compounds (azidothymidine and lopinavir) were added to the CD4+ T cell culture to inhibit virus production. Then the anti–HIV-1 drugs were withdrawn and CAR or control CD8+ T cells were mixed at a 1:1 ratio. Every 2 days the cultures were tested for the presence of p24 in the supernatant, using ELISA. Gray shade represents the addition of antiviral drugs. (C) HIV-1 Env derived from pre–CAR T cell latent reservoir or rebound reservoir was ectopically expressed on the HEK293T cell line. These target cell lines were compared for changes in sensitivity to CAR T cell–mediated specific cytotoxicity. Env derived from HIV-1NL4-3 served as the positive control. Direct killing of target cell lines was tested after 24-hour coculture by detecting LDH release. A 2‑sided P value for the estimated difference in pre–CAR T cell and rebound resistance was calculated. Data represent the mean of triplicate values, and error bars represent SEM. P values were calculated using the 2-tailed unpaired Student’s t test with equal variances.
To further validate the HIV-1–specific CAR T cell–mediated selective pressure on rebounding viruses, we examined the outgrowth of replication-competent viruses from pre–CAR T cell latent reservoir and rebound reservoir. PBMCs were isolated from healthy donors and divided into 2 populations, the CD8+ T lymphocytes were used to generate bNAb-derived CAR T cells, while the CD4+ T lymphocytes were used as target cells for outgrown HIV-1 infection. Six days after HIV-1 infection, the antiretroviral compounds were added to the CD4+ T cell culture to inhibit virus production and prevent further infection events. After approximately 8 days, the virus production substantially decreased to the lower limit for p24 detection, and infected CD4+ T cells were close to quiescence (22). We then withdrew anti–HIV-1 drugs to mimic the in vivo viral rebound process, and added autologous bNAb-derived CAR T cells. As shown in the groups infected by viruses from pre–CAR T cell latent reservoirs of patients 002, 004, and 015, viral production was significantly suppressed following coculture with CAR T cells (Figure 6B, left panel). However, in the groups infected by viruses from rebound reservoirs of the same patients, the CAR T cells exhibited only a limited inhibitory effect on viral propagation (Figure 6B, right panel). This experiment further validated the capacity of bNAb-derived CAR T cell therapy to drive resistant viruses during ATI. Moreover, the CAR T cells failed to inhibit viral propagation even in the CD4+ T cells infected by viruses from the pre–CAR T cell latent reservoir of patient 003, suggesting a possible preexisting resistance (Figure 6B, left panel). Notably, regardless of the time to rebound, resistance to HIV-1–specific CAR T cells occurred in 2 of 3 participants, as determined with a specific cytotoxicity assay (Figure 6C). Collectively, the sequence- and function-based analyses showed that the CAR T cell–resistant viruses could be preexisting or CAR T cell treatment–driven during ATI.
Discussion
CAR T cell therapy was introduced to HIV-1 clinical care in 1994, but little, if any, antiviral efficacy and durable control of viral replication were observed (16–20). However, these earlier studies were performed using CD4+ or less potent antibody-derived Fv as the extracellular domain. Additionally, CARs from the first generation had no costimulatory domains. Here, we investigated whether the bNAb-based third generation CAR T cell therapy can reduce the HIV-1 reservoir and maintain viral suppression during ATI in individuals infected with HIV-1. We found that a single adoptive transfer of bNAb-derived CAR T cells generated long-term in vivo persistence for more than 44 weeks in all 14 patients, and no safety concerns were identified. In chronically infected individuals with HIV-1 who were undergoing ATI, CAR T cell therapy delayed plasma viral rebound as compared with historical controls.
The viral reservoir is dynamically changed under the pressure of antiretroviral drugs or immune systems. At the early time, the CAR T cells expand in vivo and could efficiently reduce viral reservoir, consequently leading to a substantial decrease of CA-RNA. In the ATI group, as the replication-competent provirus could complete a life cycle after ART discontinuation, the viral escape mutations have the opportunity to quickly develop. When the plasma viremia rebounded due to the emergence of CAR T cell–resistant variants during the periods of ATI, the CA-RNA also significantly increased accordingly. Even several months after the plasma viral load was lower than the detection limit again after ART reinitiation, the levels of CA-RNA were still higher than the pre-ATI time points. However, within the non-ATI group, the development of resistant variants is quite difficult because the replication-competent provirus is unable to complete its replication. The surviving CAR T cells could keep cleaning the target cells, which occasionally express gp120 on the surface of latently infected cells, which could lead to the eventual decrease of CA-RNA and intact proviruses.
Although quantitative and qualitative viral outgrowth assays (Q2VOAs) have been frequently used to analyze the replication-competent latent viral reservoir in individuals infected with HIV-1 (55), these Q2VOA underestimate the reservoir size, because one round of activation does not induce all proviruses (56). Additionally, it is inadequate for long-term follow-up to perform the Q2VOA, which requires a large number of CD4+ T cells from patients. In order to measure the effect of CAR T cell therapy on the viral reservoir, we chose CA-RNA and IPDA as the biomarkers to measure HIV-1 reservoir size (39, 40). The CA-RNA has received much attention in recent years as a surrogate measure of the efficiency of HIV cure–related clinical trials and HIV-1 latency reversion (39). We notice that the levels of CA-RNA were relatively high and fluctuating in some of the participants, the wide variance of CA-RNA may be related to several aspects, such as infection time before diagnosis, treatment time, immunologic characteristics, and different ART regimes. The intermittent fluctuation and slight increase in viral RNA level may not lead to plasma viral rebound in participants without ATI, because a part of the transcripts is defective and production of infectious virions is regulated by multiple posttranscriptional levels (57). IPDA has recently been described as a more accurate method of measuring the HIV-1 reservoir that separately quantifies intact and defective proviruses, and there are strong correlations between IPDA and Q2VOA measurements in the same infected individuals (40). According to a previous study, the half-life of the intact provirus is 7.1 years under ART (58). In our clinical study, intact proviruses were significantly reduced at week 3 and further decreased at week 30+ after CAR T cell administration. These findings suggested that CAR T cell therapies could accelerate HIV-1 reservoir depletion based on ART regimes. Since the DNA samples of the ART-receiving HIV-infected volunteers from long-term follow-up visits were not available, we were unable to perform parallel comparison between participants receiving CAR T cells and a control cohort at multiple time points, which is a limitation of our current study. More quantitative IPDA analysis at multiple time points regarding the decay of viral reservoir under the pressure of cellular therapies merits further investigation.
Although we did not check the binding affinity of every single envelope protein from rebound viruses to VRC01-scFv, we found that at least several strains among the rebound viruses turned to resistance against the CD8+ CAR T cell–mediated cytotoxicity (Figure 6, B and C). As for whether the binding to escaped envelope may still occur or what level of binding affinity will be sufficient to trigger killing by the CAR T cells, further investigation and related research are required and will contribute to CAR design and cure-directed therapeutic strategies.
In our clinical study, participant 002 with the highest CD4+ T cell count had the longest time to viral rebound and participant 006 with minimal nadir CD4+ T cell count had the earliest viral rebound. The results suggest that the higher CD4+ T cell counts might lead to a longer time to viral rebound due to a kind of collaboration between CD4+ T cells and CD8+ CAR T cells (59–61). However, as the ATI cases are limited, we cannot conclude significant correlations in the study. A previous ATI study showed that viral rebound generally occurs quickly after treatment interruption and confirmed the rarity of posttreatment controllers, and there were no significant differences in screening CD4+ T cell count by timing of viral rebound (28). Nevertheless, the CD4+ T cell counts and the ratio of CD4+ to CD8+ T cells represent important indicators of the immune reconstitution. Increased CD4+ T cell counts during ART treatment were positively correlated with CD8+ T cell counts/function and HIV-1 DNA reduction (59). Thus, the correlations between the CD4+ T cell counts and the time of viral rebound during CAR T cell therapy need further exploration.
Both sequence-based and -specific cytotoxicity analyses seem to indicate that the rebound viruses after adoptive transfer are CAR T cell–resistant, suggesting that CAR T cells could effectively function by restricting viral replication, and force them to generate escape mutations. Alternatively, as mutations create HIV-1 resistance to CAR T cell therapy, future clinical trials may consider the administration of a combination of CAR T cells recognizing multiple distinct regions on the HIV-1 Env to potentiate their long-term surveillance on the viral reservoir. Additionally, the efficacy of CAR T cell therapy in chronically infected patients with HIV-1 will be dependent in part on whether the patients have resistant strains to that extracellular scFv of CAR moiety in persistent viral reservoirs. A preclinical screening to select appropriate patients may be necessary. Moreover, a combination with other testing methods is needed for an accurate measurement of viral reservoir size during HIV cure–related clinical trials, such as Q2VOA, HIV total, and integrated DNA and induced p24 SIMOA. (62).
Several clinical studies reported that infusion of CCR5 gene-edited hematopoietic stem and progenitor cells (HSPCs) or CD4+ T cells in individuals infected with HIV-1 (63–65). These studies showed that the replacement of part of the immune system through genetic engineering to produce CD4+ T cells resistant to HIV-1 infection was a feasible strategy. In contrast to replacement of the immune system, we used a different strategy to rebuild immune surveillance in patients infected with HIV-1 through adoptive transfer of ex vivo–expanded HIV-1–specific CD8+ CAR T cells, which can directly target virus-producing cells. Of note, in a nonhuman primate model of ART-suppressed HIV-1 infection, the investigators optimized CAR T cell production to maintain central memory subsets and further boosted robust in vivo expansion through supplemental infusion of HIV-1 Env or immune checkpoint blockade (66). The modifications significantly delayed viral rebound compared with controls and would provide guidance for future CAR T cell clinical studies aimed at HIV functional cure. Although so far only a modest delay in the time to viral rebound relative to historical controls was achieved by either therapy, the results provided certain directions for further investigation of therapeutic interventions. Given that the CCR5 gene-edited CD4+ T cell infusion could augment preexisting HIV-specific immune responses, the collaboration of CCR5 gene-edited CD4+ T and HIV-1–specific CD8+ CAR T cells might be developed as a therapeutic modality for eradicating viral reservoir in a clinical setting (65, 67).
The establishment of potent HIV-1–specific immune surveillance is a feasible approach for long-term suppression of the reactivated latent viral reservoir without continuation of ART. Although bNAb treatment, especially the combination of 2 bNAbs targeting different sites on gp120, has achieved durable control over the viral reservoir, it cannot persistently remain in vivo. In contrast, single administration of CAR T cells can potentially maintain long-term survival in vivo (68, 69). It has been reported that the half-life of CD4ζ CAR T cells in vivo could be as long as 10 years (19). Although we have no direct evidence yet for the role played by triple knockdown of PD-1, Tim-3, and Lag-3 with shRNA in CAR T cells, our data also suggested a long-term in vivo survival of anti–HIV-1 CAR T cells. In such situations, CAR T cells could function as a “living drug”. It is anticipated that superior scFv-based CAR T cells will be generated by improving the CAR moiety design, either by selecting specific scFvs targeting various sites of viral Env, or enhancing their capability of self-renewal, exhaustion prevention, and tissue distribution. Moreover, good manufacturing practice, manufacturing protocol compliancy, clinical safety, and efficacy require further optimization (70). We expect that the cellular therapies, in combination with application of bNAbs or LA-ARVs, would eradicate the persistent HIV-1–producing cells, by enhancing immune surveillance and maintaining a long-term viral suppression without the continuation of ART.
Methods
Study oversight.
The study was designed to mainly assess the safety and feasibility of the adoptive transfer of gp120-specific bNAb-derived CAR T cells into patients positive for HIV-1. This clinical trial recruited participants who had chronic HIV-1 infection with fully suppressed plasma viremia while receiving ART (clinically stable on ART regimen for at least 12 months with undetectable HIV-1 RNA level; screening CD4+ T cell count ≥ 350 cells/μL within 14 days prior to study entry). Detailed inclusion and exclusion criteria are listed in the clinical protocol in the Supplemental Material. Study participants were not prescreened for sensitivity of the infected cells to CAR T cell cytotoxicity and did not undergo any additional immunotherapeutic interventions besides ART.
Genetic modification and CAR T cell preparation.
Autologous PBMCs were collected from 50–60 mL peripheral blood of patients infected with HIV-1 and separated on the basis of CD8+ expression using the CliniMACS system (Miltenyi Biotec) following the manufacturer’s instructions. The sorted population was more than 95% CD8+ T cells. CD8+ T cell products were stimulated with paramagnetic antibodies (anti-CD3 and anti-CD28) and 130 IU IL-2 (SL Pharma Labs), then the cells were transduced with the VRC01-28BBz-shPTL transgene to express a gp120-specific bNAb-derived CAR containing CD28 and 4-1BB (CD137) costimulatory domains (22), followed by a herpes simplex virus-1 thymidine kinase (TK) and a truncated CD19 gene as the suicide genes (71). A combination of sh-PD-1, sh-Lag-3, and sh-Tim-3 was also inserted into the vector (27). Twelve hours after infection, the CAR-transduced CD8+ T cells were washed by saline and resuspended in Immuno-Cult-XF T cell Expansion medium (STEMCELL Technologies) with 130 IU IL-2 (SL Pharm) and the concentration of cells was maintained at 106 cells/mL. Every 2 days, the volume of the culture was adjusted according to the cell concentration (106 cells/mL) and supplemented with IL-2. The CAR autologous T cells were then expanded in flasks until the targeted cell dose was reached. The CAR T cells were infused after 2 to 3 weeks of expansion ex vivo, and the target cell dose range was at least 5 × 107 CD3+ CD8+ cells. Release criteria for the expanded CD8+ T cells were as follows: sterility, negative fungal and mycoplasma testing, negative Gram stain, endotoxin less than 5 EU/mL, more than 90% viability by Trypan Blue exclusion, CD3+ CD8+ T cells ≥ 95%, transduction efficiency (CAR+ CD8+) ≥ 30%, and gp120-specific cytotoxicity of more than 40% lysis at 100:1 E/T ratio. The doses of total transferred cells were between 5.00 × 107 to 1.00 × 108.
Treatment procedures, study objectives, and study outcomes.
Fifteen participants were enrolled in this study and 14 of them received single administration of bNAb-derived CAR T cells (5.00 × 107 to 1.00 × 108). There were no conditioning regimens before CAR T cell administrations. After the CAR T cell transfer, 6 participants discontinued ART and were followed at weekly intervals until they had a confirmed CD4+ T cell count of less than 350 cells/μL or viral rebound, which was defined as an HIV-1 RNA level of 200 copies or more per milliliter. On confirmation of viral rebound or a decrease in CD4+ T cells, participants reinitiated ART and were followed at weekly intervals until the HIV-1 viral load was less than 20 copies/μL.
The primary objective of the study was to assess the safety and side-effect profile of a single dose of bNAb-derived CAR T cells administered to individuals with viremia suppressed to below detectable levels. The secondary objective was to evaluate the pharmacokinetic characteristics of CAR T cell products. Key exploratory objectives were the size of the HIV-1 reservoir after CAR T cell administration and the genetic and phenotypic characterization of the rebound viruses. Post hoc analyses of the sequence diversity at the time of viremia rebound and the cytotoxic capacity of CAR T cells against autologous HIV-1 before and after adoptive transfer were performed.
Antiretroviral therapy interruption.
With informed consent from the patients, analytical interruptions of ART were performed to investigate the antivirus effect of bNAb-derived CAR T cell therapy. The ART interruption was performed when the following criteria were achieved: written informed consent was provided by the patient; no adverse events at the time of interruption; CD4+ T cell counts were maintained in a normal range (> 400 cells/μL peripheral blood); plasma viral load was under the detectable level (< 20 copies per milliliter); CA-RNA was reduced at least 50%; in vivo CAR T cells were persistently detectable (> 100 copies per million CD8+ T cells). To prevent the risk of efavirenz monotherapy and the emergence of resistant strains after stopping ART, the individuals whose regimens contained efavirenz stopped taking it 1 week before discontinuing the other agents. The individuals with integrase inhibitor–based or protease inhibitor–based regimes discontinued 3 ART agents simultaneously. When all the ART agents were discontinued, the time of plasma viral suppression was measured.
To protect the patient from adverse events induced by virus rebound, antivirus treatment would be reinitiated when any of the following criteria was met: plasma viral load exceeded 200 copies per milliliter; CD4+ T cells decreased under 350 cells/μL; any AIDS-related opportunistic infections were observed; occurrence of severe CAR T cell–related adverse events; request for reinitiation of ART by the patient.
Quantitative PCR to detect CAR T cells.
Patient PBMCs collected at baseline and at serial time points after CAR T cell infusions were collected and separated by Ficoll centrifugation and then cryopreserved. Batched cells were thawed and primary human CD8+ T cells were obtained from PBMCs by positive magnetic selection through BD IMag Human CD8+ T Lymphocyte Enrichment Set-DM (BD Biosciences). The genomic DNA was harvested using an AllPure Total DNA/RNA Micro Kit (Magen). The CAR transgene was detected by performing quantitative PCR as previously described, using either a primer amplifying the fragment spanning the junction of the CD3ζ domain and adjacent Flag domain (forward primer: 5′-GCCTTTACCAGGGTCTCA-3′, reverse primer: 5′-ACTTATCGTCGTCATCCTTG-3′), or a primer amplifying the fragment of VRC01 scFv (forward primer: 5′-ATTTTTTGGCCAGGGGACC-3′, reverse primer: 5′-AGGATTCTCCTCGACGTCACC-3′) (22). Quantitative real-time PCR was performed in triplicate using SYBR Premix ExTaq II Kit (Takara), in a C1000 Touch Thermal Cycler (BIO-RAD CFX96 Real-Time System) (22). Copy numbers per microgram of genomic DNA, generated from a standard curve of 10-fold serial dilutions of purified plasmid, were used to calculate the percentage of CAR+ cells among CD8+ T cells, assuming 1 copy per cell.
Quantitative real-time RT–PCR analysis.
Primary human CD4+ T cells were obtained from PBMCs by negative magnetic selection through BD IMag Human CD4+ T Lymphocyte Enrichment Set-DM (BD Biosciences). Total RNA was isolated with Trizol reagent (Life Technologies) and then subjected to cDNA synthesis using the PrimeScript RT reagent kit (Takara). All primers were annealed at 37°C and RT was processed at 42°C. Quantitative PCR was performed with SYBR Premix ExTaq II Kit (Takara) following the manufacturer’s instructions. The expressions of HIV-1 US RNAs were determined by real-time qRT-PCR with the primer pair SK38 (5′-ATAATCCACCTATCCCAGTAGGAGAAA-3′) and SK39 (5′-TTTGGTCCTTGTCTTATGTCCAGAATGC-3′). An in vitro–synthesized HIV-1 RNA, after quantification, was used as the external control for measuring CA-RNA (72). Quantification was normalized to the housekeeping gene GAPDH or β-actin.
Intact proviral DNA assay.
The procedures for IPDA described previously were followed with minor modifications (40, 73). In general, the IPDA is performed on DNA from 2 × 106 CD4+ T cells. Genomic DNA is extracted using the AllPure Total DNA/RNA Micro Kit (Magen) with precautions to avoid excess DNA fragmentation. Quantification of intact, 5′ deleted, and 3′ deleted and/or hypermutated proviruses was carried out using primer/probe combinations optimized for subtype B HIV-1. The primer/probe mix consists of oligonucleotides for 2 independent hydrolysis probe reactions that interrogate conserved regions of the HIV-1 genome to discriminate intact from defective proviruses (Supplemental Table 8). HIV-1 reaction A targets the packaging signal (ψ) that is a frequent site of small deletions and is included in many large deletions in the proviral genome. The ψ amplicon is positioned at HXB2 coordinates 692–797. This reaction uses forward and reverse primers, as well as a 5′ 6-FAM-labeled hydrolysis probe. Successful amplification of HIV-1 reaction A produced FAM fluorescence in droplets containing ψ, detectable in channel 1 of the droplet reader. HIV-1 reaction B targets the rev-responsive element (RRE) of the proviral genome, with the amplicon positioned at HXB2 coordinates 7736–7851. This reaction used forward and reverse primers, as well as 2 hydrolysis probes: a 5′ VIC-labeled probe specific for WT proviral sequences, and a 5′ unlabeled probe specific for APOBEC3G/H hypermutated proviral sequences (Supplemental Material). Successful amplification of HIV-1 reaction B produced a VIC fluorescence in droplets containing a WT form of RRE, detectable in channel 2 of the droplet reader, whereas droplets containing a hypermutated form of RRE were not fluorescent.
Droplets containing HIV-1 proviruses were scored as follows. Droplets positive for FAM fluorescence only, which arises from amplification, were scored as containing 30 defective proviruses, with the defect attributable to either APOBEC3G-mediated hypermutation or 3′ deletion. Droplets positive for VIC fluorescence only, which arise from WT RRE amplification, were scored as containing 5′ defective proviruses, with the defect attributable to 5′ deletion. Droplets positive for both FAM and VIC fluorescence were scored as containing intact proviruses. Double-negative droplets contained no proviruses or rare proviruses with defects affecting both amplicons.
Viral outgrowth.
Recovery and amplification of replication-competent viruses were previously described (55, 74). Briefly, 1 × 106 CD4+ T cells from individuals infected with HIV-1 were stimulated by 1 × 107 irradiated allogeneic PBMCs from uninfected donors and 1 μg/mL PHA-M at day 1. Typically, 3 additions of 5 × 106 activated CD4+ lymphoblasts from uninfected donors as target cells were added for HIV-1 outgrowth at day 2, day 7, and day 14, respectively. The cells were cultured in RPMI-1640 media with IL-2 (10 ng/mL, recombinant human; R&D Systems) all the time. After 14 days of coculture, the recovered viruses were harvested and tested for HIV-1 p24 protein.
In vitro HIV-1 infection and drug withdrawal model.
In vitro HIV-1 infection model was previously described with minor modifications (22). Briefly, the PBMCs from healthy donors were stimulated by adding 1 mg/mL PHA and 10 ng/mL IL-2 to the conditioned RPMI 1640 media with 10% heat-inactivated fetal bovine serum and antibiotics for 2 days before isolation of CD4+ T cells. CD4+ T cells were infected with an outgrown HIV-1 from patients (p24 titer of 1 ng/mL). Three hours after infection, the culture media was changed by centrifugation. The infected CD4+ T cells were cultured in basal media with IL-2 (10 ng/mL, recombinant human; R&D Systems) and further incubated at 37°C in a humidified incubator with 5% CO2. Six days after HIV-1 infection, azidothymidine (Zidovudine, Sigma-Aldrich) and lopinavir (Sigma-Aldrich) were added into the CD4+ T cell culture (both at 50 μM) to inhibit virus production and prevent further infection events. The cells were then cultured in the presence of a low concentration of IL-2 (1 ng/mL). Anti–HIV-1 drugs were withdrawn when the viral production was substantially decreased to the marginal level for p24 detection (about 6–8 days after adding the drugs). Then, 0.5 × 106 CD4+ T cells were mixed with autologous CAR T cells or control CD8+ T cells at 1:1 ratio in the conditioned media plus IL-2 (10 ng/mL) at 1 mL in a 24-well plate. Every 2 days the cultures were tested for HIV-1 p24 antigen with the HIV-1 p24 Antigen Assay kit following the manufacturer’s instructions (KEY-BIO Biotech).
Genetic diversity analysis of activated HIV-1 viruses.
HIV-1 RNA extraction and single-genome amplification were performed as previously described (11, 75, 76). In brief, HIV-1 RNA was extracted from cell and plasma samples followed by first-strand cDNA synthesis using a HiScript II 1st Strand cDNA Synthesis KIT (Vazyme). cDNA synthesis for cell/plasma-derived HIV-1 RNA was performed using the antisense primer envB3out 5′-TTGCTACTTGTGATTGCTCCATGT-3′. The gp160 was amplified using envB5out 5′-TAGAGCCCTGGAAGCATCCAGGAAG-3′ and envB3out 5′-TTGCTACTTGTGATTGCTCCATGT-3′ in the first round, and envB5in 5′-CACCTTAGGCATCTCCTAT GGCAGGAAGAAG-3′ and envB3in 5′-GTCTCGAGATACTGCTCCCACCC-3′ in the second round with nested primers. PCRs were performed using Phanta Max Super-Fidelity DNA Polymerase (Vazyme) and run at 94°C for 2 minutes; 35 cycles of 94°C for 15 seconds, 55°C for 30 seconds, and 68°C for 4 minutes; and 68°C for 15 minutes. Second-round PCR was performed with 1 μL of the PCR product from the first round as template and Phanta Max Super-Fidelity DNA Polymerase (Vazyme) at 94°C for 2 minutes; 45 cycles of 94°C for 15 seconds, 55°C for 30 seconds, and 68°C for 4 minutes; and 68°C for 15 minutes. Amplicons were run on precast 1% agarose gels (BIOWESTE) and the PCR products proceeded to deoxyadenosine (A), tailing at the 3′ end, using Ex Taq DNA polymerase (Takara) without thermal cycling as follows: 95°C for 5 minutes; 72°C for 30 minutes. The A-tailed PCR products were TA-ligated into pMD-18 T vector (Takara).
Sequence and phylogenetic analysis.
Nucleotide alignments of intact env sequences were translation-aligned using MEGA 7 (https://megasoftware.net/). Sequences with premature stop codons and frameshift mutations that fell in the gp120 surface glycoprotein region were excluded from all analyses. The sequences from each group were aligned using MUSCLE. We computed the genetic distance of every single clone in the population based on the average of the relevant entire population by MEGA7. The phylogenetic bootstrap trees were generated for each sample using maximum likelihood method with 1000 bootstrap replications implemented in MEGA 7 to depict the global landscape of HIV-1 diversity. Logograms were generated using the Weblogo 3.0 tool. To analyze changes between latent reservoir and rebound viruses, Env sequences were aligned at the amino acid level to a HXB2 reference using BioEdit. Nucleotide sequences have been submitted to GenBank.
Cytotoxicity determination.
The specific killing activity of prestimulated CD8+ T cells towards Jurkat or HEK293T cells expressing HIV-1 Env glycoprotein at indicated ratios was measured after coculture for 8 hours by lactate dehydrogenase assay using the CytoTox 96 nonradioactive cytotoxicity kit (G1781, Promega), as described previously (22, 27). The manufacturer’s instructions were followed. Absorbance values of wells containing effector cells alone and target cells alone were combined and subtracted as background from the values of the cocultures. Wells containing target cells alone were mixed with a lysis reagent for 30 minutes at 37°C and the resulting luminescence was set as 100% lysis. Cytotoxicity was calculated by using the following formula: % Cytotoxicity = (Experimental — Effector spontaneous — Target spontaneous) / (Target maximum — Target spontaneous) × 100.
Cell lines.
HEK293T and Jurkat cell lines were obtained from ATCC. Jurkat-gp160NL4-3 cells were established by the infection of Jurkat cells with recombinant lentiviruses carrying HIV-1NL4-3 Env-IRES-GFP moiety, followed by sorting GFPhi cells.
Statistics.
We used the Pearson’s χ2 test or the Fisher’s exact test (2-sided) as appropriate to analyze differences in proportions of virologic suppression between the CAR T cell infusion group and historical controls. P less than 0.05 was considered significant. First, CA-RNA or IPDA values were assessed for whether they conformed to normal distribution by Shapiro-Wilk test. Since not all of the variables conformed to normal distribution, we used the multiple Wilcoxon matched pairs signed rank test to compare the data. Two-way ANOVAs were performed with Bonferroni’s correction for multiple comparisons. We have adjusted P values for multiple comparisons using Bonferroni’s corrections. Other P values of statistical analyses such as CAR T cell–mediated cytotoxicity were calculated using the 2-tailed unpaired Student’s t test with equal variances. We generated graphics with GraphPad Prism 5.0 software.
Study approval.
This clinical trial was approved by the IRB of Guangzhou Eighth People’s Hospital and Sun Yat-sen University (protocol 201803040002). Written informed consent was obtained from all patients prior to their enrollment in the clinical trial. This clinical trial is registered at ClinicalTrials.gov (NCT03240328). The study was conducted in accordance with legal and regulatory requirements, as well as the general principles set forth in the International Ethical Guidelines for Biomedical Research Involving Human Patients, Guidelines for Good Clinical Practice, and the Declaration of Helsinki. In addition, the study was conducted in accordance with the protocol and applicable local regulatory requirements and laws.
Data and materials availability.
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplemental Materials.
Author contributions
BL, L Li, and HZ designed the experiments, performed most of the experiments, analyzed the data, and wrote the manuscript. WZ, BX, SJ, YD, FZ, RL, L Lu, SC, YL, QH, YL, YZ, ZH, and XZ made substantial contributions to the acquisition of data and data analyses. XC, XT, T Peng, and WC provided scientific expertise and supervised the analysis of clinical data. T Pan, L Li, and HZ had full access to all data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. All authors reviewed and approved the final version of the report.
Supplementary Material
Acknowledgments
This work was supported by the National Special Research Program of China for Important Infectious Diseases (2017ZX10202102 and 2018ZX10302103 to HZ, L Li, and T Pan); the Important Key Program of the Natural Science Foundation of China (81730060 to HZ); the Natural Science Foundation of China (82072265 to L Li and 81971918 to T Pan); the Joint-Innovation Program in Healthcare for Special Scientific Research Projects of Guangzhou (201803040002 to HZ, L Li, and WC); the Natural Science Foundation of Guangdong Province (2020A1515011108 to BL); and the Shenzhen Science and Technology Program (JSGG20200225150431472 and JCYJ20200109142601702 to T Pan).
Version 1. 08/10/2021
In-Press Preview
Version 2. 10/01/2021
Electronic publication
Footnotes
Conflict of interest: Aspects of the CAR design are the subject of patent “VC-CAR molecule and use thereof in removing HIV-1 infected cells” (US 10,633,432 B2), on which BL, FZ, and HZ are listed as inventors.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(19):e150211.https://doi.org/10.1172/JCI150211.
See the related Commentary at HIV-specific CAR T cells return to the clinic.
Contributor Information
Bingfeng Liu, Email: liubf5@mail.sysu.edu.cn.
Wanying Zhang, Email: 273793055@qq.com.
Baijin Xia, Email: 2415708935@qq.com.
Shuliang Jing, Email: 1969182136@qq.com.
Yingying Du, Email: 275063508@qq.com.
Fan Zou, Email: zoufan76@163.com.
Rong Li, Email: 610420477@qq.com.
Lijuan Lu, Email: lulj2010@126.com.
Shaozhen Chen, Email: 569914699@qq.com.
Yonghong Li, Email: 512967304@qq.com.
Qifei Hu, Email: huqifei@outlook.com.
Yingtong Lin, Email: 313140408@qq.com.
Yiwen Zhang, Email: zhangyw57@mail.sysu.edu.cn.
Zhangping He, Email: qianchengjing@163.com.
Xu Zhang, Email: zhang18xun@163.com.
Xiejie Chen, Email: gz8hcxj@126.com.
Tao Peng, Email: pengtao@gdscvc.com.
Xiaoping Tang, Email: xtang@21cn.com.
Weiping Cai, Email: gz8hcwp@126.com.
Ting Pan, Email: pant8@mail.sysu.edu.cn.
Linghua Li, Email: llheliza@126.com.
Hui Zhang, Email: zhangh92@mail.sysu.edu.cn.
References
- 1.Sengupta S, Siliciano RF. Targeting the latent reservoir for HIV-1. Immunity. 2018;48(5):872–895. doi: 10.1016/j.immuni.2018.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siliciano JD, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med. 2003;9(6):727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 3.Buzon MJ, et al. HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat Med. 2014;20(2):139–142. doi: 10.1038/nm.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chun TW, et al. Re-emergence of HIV after stopping therapy. Nature. 1999;401(6756):874–875. doi: 10.1038/44755. [DOI] [PubMed] [Google Scholar]
- 5.Liu B, et al. Development of CAR-T cells for long-term eradication and surveillance of HIV-1 reservoir. Curr Opin Virol. 2019;38:21–30. doi: 10.1016/j.coviro.2019.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Lynch RM, et al. Virologic effects of broadly neutralizing antibody VRC01 administration during chronic HIV-1 infection. Sci Transl Med. 2015;7(319):319ra206. doi: 10.1126/scitranslmed.aad5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caskey M, et al. Antibody 10-1074 suppresses viremia in HIV-1-infected individuals. Nat Med. 2017;23(2):185–191. doi: 10.1038/nm.4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caskey M, et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature. 2015;522(7557):487–491. doi: 10.1038/nature14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bar KJ, et al. Effect of HIV antibody VRC01 on viral rebound after treatment interruption. N Engl J Med. 2016;375(21):2037–2050. doi: 10.1056/NEJMoa1608243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheid JF, et al. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature. 2016;535(7613):556–560. doi: 10.1038/nature18929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mendoza P, et al. Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature. 2018;561(7724):479–484. doi: 10.1038/s41586-018-0531-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.June CH, et al. CAR T cell immunotherapy for human cancer. 2018;359(6382):1361–1365. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 13.Roberts MR, et al. Targeting of human immunodeficiency virus-infected cells by CD8+ T lymphocytes armed with universal T-cell receptors. Blood. 1994;84(9):2878–2889. [PubMed] [Google Scholar]
- 14.Tran AC, et al. Chimeric zeta-receptors direct human natural killer (NK) effector function to permit killing of NK-resistant tumor cells and HIV-infected T lymphocytes. J Immunol. 1995;155(2):1000–1009. [PubMed] [Google Scholar]
- 15.Masiero S, et al. T-cell engineering by a chimeric T-cell receptor with antibody-type specificity for the HIV-1 gp120. Gene Ther. 2005;12(4):299–310. doi: 10.1038/sj.gt.3302413. [DOI] [PubMed] [Google Scholar]
- 16.Mitsuyasu RT, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood. 2000;96(3):785–793. [PubMed] [Google Scholar]
- 17.Deeks SG, et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol Ther. 2002;5(6):788–797. doi: 10.1006/mthe.2002.0611. [DOI] [PubMed] [Google Scholar]
- 18.Walker RE, et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood. 2000;96(2):467–474. [PubMed] [Google Scholar]
- 19.Scholler J, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4(132):132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colovos C, et al. Safety and stability of retrovirally transduced chimeric antigen receptor T cells. Immunotherapy. 2012;4(9):899–902. doi: 10.2217/imt.12.91. [DOI] [PubMed] [Google Scholar]
- 21.Liu L, et al. Novel CD4-based bispecific chimeric antigen receptor designed for enhanced anti-HIV potency and absence of HIV entry receptor activity. J Virol. 2015;89(13):6685–6694. doi: 10.1128/JVI.00474-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu B, et al. Chimeric antigen receptor T cells guided by the single-chain Fv of a broadly neutralizing antibody specifically and effectively eradicate virus reactivated from latency in CD4+ T lymphocytes isolated from HIV-1-infected individuals receiving suppressive combined antiretroviral therapy. J Virol. 2016;90(21):9712–9724. doi: 10.1128/JVI.00852-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ali A, et al. HIV-1-specific chimeric antigen receptors based on broadly neutralizing antibodies. J Virol. 2016;90(15):6999–7006. doi: 10.1128/JVI.00805-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hale M, et al. Engineering HIV-resistant, anti-HIV chimeric antigen receptor T cells. Mol Ther. 2017;25(3):570–579. doi: 10.1016/j.ymthe.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghanem MH, et al. Bispecific chimeric antigen receptors targeting the CD4 binding site and high-mannose Glycans of gp120 optimized for anti-human immunodeficiency virus potency and breadth with minimal immunogenicity. Cytotherapy. 2018;20(3):407–419. doi: 10.1016/j.jcyt.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Anthony-Gonda K, et al. Multispecific anti-HIV duoCAR-T cells display broad in vitro antiviral activity and potent in vivo elimination of HIV-infected cells in a humanized mouse model. Sci Transl Med. 2019;11(504):eaav5685. doi: 10.1126/scitranslmed.aav5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou F, et al. Engineered triple inhibitory receptor resistance improves anti-tumor CAR-T cell performance via CD56. Nat Commun. 2019;10(1):4109. doi: 10.1038/s41467-019-11893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li JZ, et al. The size of the expressed HIV reservoir predicts timing of viral rebound after treatment interruption. AIDS. 2016;30(3):343–353. doi: 10.1097/QAD.0000000000000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kilby JM, et al. A randomized, partially blinded phase 2 trial of antiretroviral therapy, HIV-specific immunizations, and interleukin-2 cycles to promote efficient control of viral replication (ACTG A5024) J Infect Dis. 2006;194(12):1672–1676. doi: 10.1086/509508. [DOI] [PubMed] [Google Scholar]
- 30.Jacobson JM, et al. Evidence that intermittent structured treatment interruption, but not immunization with ALVAC-HIV vCP1452, promotes host control of HIV replication: the results of AIDS Clinical Trials Group 5068. J Infect Dis. 2006;194(5):623–632. doi: 10.1086/506364. [DOI] [PubMed] [Google Scholar]
- 31.Skiest DJ, et al. Interruption of antiretroviral treatment in HIV-infected patients with preserved immune function is associated with a low rate of clinical progression: a prospective study by AIDS Clinical Trials Group 5170. J Infect Dis. 2007;195(10):1426–1436. doi: 10.1086/512681. [DOI] [PubMed] [Google Scholar]
- 32.Schooley RT, et al. AIDS Clinical Trials Group 5197: a placebo-controlled trial of immunization of HIV-1-infected persons with a replication-deficient adenovirus type 5 vaccine expressing the HIV-1 core protein. J Infect Dis. 2010;202(5):705–716. doi: 10.1086/655468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goulder PJ, Walker BD. HIV and HLA class I: an evolving relationship. Immunity. 2012;37(3):426–440. doi: 10.1016/j.immuni.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Till BG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119(17):3940–3950. doi: 10.1182/blood-2011-10-387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fraietta JA, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558(7709):307–312. doi: 10.1038/s41586-018-0178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fischer M, et al. Residual cell-associated unspliced HIV-1 RNA in peripheral blood of patients on potent antiretroviral therapy represents intracellular transcripts. Antivir Ther. 2002;7(2):91–103. [PubMed] [Google Scholar]
- 37.Fischer M, et al. Attenuated and nonproductive viral transcription in the lymphatic tissue of HIV-1-infected patients receiving potent antiretroviral therapy. J Infect Dis. 2004;189(2):273–285. doi: 10.1086/380797. [DOI] [PubMed] [Google Scholar]
- 38.Pasternak AO, et al. Cellular levels of HIV unspliced RNA from patients on combination antiretroviral therapy with undetectable plasma viremia predict the therapy outcome. PLoS One. 2009;4(12):e8490. doi: 10.1371/journal.pone.0008490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pasternak AO, Berkhout B. What do we measure when we measure cell-associated HIV RNA. Retrovirology. 2018;15(1):13. doi: 10.1186/s12977-018-0397-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bruner KM, et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature. 2019;566(7742):120–125. doi: 10.1038/s41586-019-0898-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rothenberger MK, et al. Large number of rebounding/founder HIV variants emerge from multifocal infection in lymphatic tissues after treatment interruption. Proc Natl Acad Sci U S A. 2015;112(10):E1126–E1134. doi: 10.1073/pnas.1414926112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kearney MF, et al. Origin of rebound plasma HIV includes cells with identical proviruses that are transcriptionally active before stopping of antiretroviral therapy. J Virol. 2016;90(3):1369–1376. doi: 10.1128/JVI.02139-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bednar MM, et al. Diversity and tropism of HIV-1 rebound virus populations in plasma level after treatment discontinuation. J Infect Dis. 2016;214(3):403–407. doi: 10.1093/infdis/jiw172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou T, et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. 2010;329(5993):811–817. doi: 10.1126/science.1192819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu X, et al. Selection pressure on HIV-1 envelope by broadly neutralizing antibodies to the conserved CD4-binding site. J Virol. 2012;86(10):5844–5856. doi: 10.1128/JVI.07139-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lynch RM, et al. HIV-1 fitness cost associated with escape from the VRC01 class of CD4 binding site neutralizing antibodies. J Virol. 2015;89(8):4201–4213. doi: 10.1128/JVI.03608-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Utachee P, et al. Two N-linked glycosylation sites in the V2 and C2 regions of human immunodeficiency virus type 1 CRF01_AE envelope glycoprotein gp120 regulate viral neutralization susceptibility to the human monoclonal antibody specific for the CD4 binding domain. J Virol. 2010;84(9):4311–4320. doi: 10.1128/JVI.02619-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shang H, et al. Genetic and neutralization sensitivity of diverse HIV-1 env clones from chronically infected patients in China. J Biol Chem. 2011;286(16):14531–14541. doi: 10.1074/jbc.M111.224527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo D, et al. A single residue within the V5 region of HIV-1 envelope facilitates viral escape from the broadly neutralizing monoclonal antibody VRC01. J Biol Chem. 2012;287(51):43170–43179. doi: 10.1074/jbc.M112.399402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liao HX, et al. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature. 2013;496(7446):469–476. doi: 10.1038/nature12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou T, et al. Quantification of the impact of the HIV-1-glycan shield on antibody elicitation. Cell Rep. 2017;19(4):719–732. doi: 10.1016/j.celrep.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parks KR, et al. Overcoming steric restrictions of VRC01 HIV-1 neutralizing antibodies through immunization. Cell Rep. 2019;29(10):3060–3072. doi: 10.1016/j.celrep.2019.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou P, et al. Broadly resistant HIV-1 against CD4-binding site neutralizing antibodies. PLoS Pathog. 2019;15(6):e1007819. doi: 10.1371/journal.ppat.1007819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hraber P, et al. Longitudinal antigenic sequences and sites from intra-host evolution (LASSIE) identifies immune-selected HIV variants. Viruses. 2015;7(10):5443–5475. doi: 10.3390/v7102881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Laird GM, et al. Rapid quantification of the latent reservoir for HIV-1 using a viral outgrowth assay. PLoS Pathog. 2013;9(5):e1003398. doi: 10.1371/journal.ppat.1003398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ho YC, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155(3):540–551. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chun TW, et al. Gene expression and viral prodution in latently infected, resting CD4+ T cells in viremic versus aviremic HIV-infected individuals. Proc Natl Acad Sci U S A. 2003;100(4):1908–1913. doi: 10.1073/pnas.0437640100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gandhi RT, et al. Selective decay of intact HIV-1 proviral DNA on antiretroviral therapy. J Infect Dis. 2021;223(2):225–233. doi: 10.1093/infdis/jiaa532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang LX, et al. HIV reservoir decay and CD4 recovery associated with high CD8 counts in immune restored patients on long-term ART. Front Immunol. 2020;11:1541. doi: 10.3389/fimmu.2020.01541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bedoui S, et al. CD4(+) T-cell help amplifies innate signals for primary CD8(+) T-cell immunity. Immunol Rev. 2016;272(1):52–64. doi: 10.1111/imr.12426. [DOI] [PubMed] [Google Scholar]
- 61.Zander R, et al. CD4+ T cell help is required for the formation of a cytolytic CD8+ T cell subset that protects against chronic infection and cancer. Immunity. 2019;51(6):1028–1042. doi: 10.1016/j.immuni.2019.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abdel-Mohsen M, et al. Recommendations for measuring HIV reservoir size in cure-directed clinical trials. Nat Med. 2020;26(9):1339–1350. doi: 10.1038/s41591-020-1022-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu L, et al. CRISPR-edited stem cells in a patient with HIV and acute lymphocytic leukemia. N Engl J Med. 2019;381(13):1240–1247. doi: 10.1056/NEJMoa1817426. [DOI] [PubMed] [Google Scholar]
- 64.Tebas P, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370(10):901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tebas P, et al. CCR5-edited CD4+ T cells augment HIV-specific immunity to enable post-rebound control of HIV replication. J Clin Invest. 2021;131(7):144486. doi: 10.1172/JCI144486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rust BJ, et al. Robust expansion of HIV CAR T cells following antigen boosting in ART-suppressed nonhuman primates. Blood. 2020;136(15):1722–1734. doi: 10.1182/blood.2020006372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu C, et al. HIV-1 functional cure: will the dream come true? BMC Med. 2015;13:284. doi: 10.1186/s12916-015-0517-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muul LM, et al. Persistence and expression of the adenosine deaminase gene for 12 years and immune reaction to gene transfer components: long-term results of the first clinical gene therapy trial. Blood. 2003;101(7):2563–2569. doi: 10.1182/blood-2002-09-2800. [DOI] [PubMed] [Google Scholar]
- 69.Heslop HE, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115(5):925–935. doi: 10.1182/blood-2009-08-239186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang X, Riviere I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics. 2016;3:16015. doi: 10.1038/mto.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bonini C, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276(5319):1719–1724. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 72.Zhang Y, et al. A novel HIV-1-encoded microRNA enhances its viral replication by targeting the TATA box region. Retrovirology. 2014;11:23. doi: 10.1186/1742-4690-11-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pan T, et al. USP49 potently stabilizes APOBEC3G protein by removing ubiquitin and inhibits HIV-1 replication. Elife. 2019;8:e48318. doi: 10.7554/eLife.48318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pentikainen PJ, et al. Comparative pharmacokinetics of lovastatin, simvastatin and pravastatin in humans. J Clin Pharmacol. 1992;32(2):136–140. doi: 10.1002/j.1552-4604.1992.tb03818.x. [DOI] [PubMed] [Google Scholar]
- 75.Salazar-Gonzalez JF, et al. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J Virol. 2008;82(8):3952–3970. doi: 10.1128/JVI.02660-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jordan MR, et al. Comparison of standard PCR/cloning to single genome sequencing for analysis of HIV-1 populations. J Virol Methods. 2010;168(1–2):114–120. doi: 10.1016/j.jviromet.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.