

Abstract
In mammalian cells, changes in signaling networks and expressed proteins ensure the adequate detection and management of damaged macromolecules. Here, we review an emergent pathway of maintenance of homeostasis following genotoxic stress. The RNA-binding protein HuR associates with sirtuin (SIRT)1 mRNA and maintains constitutively elevated levels of SIRT1 protein, a deacetylase that elicits a prosurvival function. SIRT1 was recently shown to deacetylate the Nijmegen breakage syndrome (NBS1) protein, thereby rendering it phosphorylatable by ataxia telangiectasia mutated protein (ATM). A component of the MRN (MRE11–RAD50–NBS1) nuclease complex, NBS1 is crucial for sensing DNA damage and mounting a genotoxic response. This article covers the regulatory pathway of HuR→SIRT1→NBS1, through which post-transcriptional and post-translational effectors contribute to the maintenance of genomic integrity.
Introduction: HuR elicits an anti-apoptotic gene expression program
In response to stress-causing and metabolic signals, cells maintain homeostasis through the action of signaling pathways that implement specific gene expression programs. In addition to transcriptional processes, the collective of expressed proteins is strongly influenced by post-transcriptional events that control mRNA splicing, transport, stability and translation. The principal post-transcriptional regulators of gene expression are non-coding RNAs (including microRNAs) and RNA-binding proteins (RBPs) [1]. RBPs include distinct proteins that associate with specific mRNA sequences [typically at 5′- or 3′–untranslated regions (UTRs)] and regulate distinct subsets of mRNAs [2]. These proteins have been termed TTR–RBPs, given their function as turnover and translation regulatory proteins [3].
During the past decade, the TTR–RBP HuR has emerged as a crucial regulator of a group of mRNAs with which it associates through its three RNA-recognition motifs (RRMs) [4,5]. For many HuR target transcripts, including the sirtuin (SIRT)1 mRNA, association with HuR extends transcript half-life and consequently increases protein levels [4,6], whereas for other HuR target mRNAs, HuR instead influences the translation rate, either enhancing it or repressing it [7] (Box 1).
Box 1. Multiple post-transcriptional functions of HuR.
Mammalian HuR (also named HuA) is a ubiquitous member of the Hu family of RBPs originally identified as elav (embryonic lethal, abnormal vision) in D. melanogaster. Hu/elav proteins also include the primarily neuronal HuB (Hel-N1), HuC and HuD. The most extensively studied member of this family, HuR, is predominantly nuclear, but its influence on the expression of target mRNAs is linked to its localization in the cytoplasm. A variety of stimuli, including oxidants, growth factors, immune activators, genotoxins and the absence of specific nutrients, are inducers of HuR translocation to the cytoplasm, where it stabilizes or modulates the translation of target mRNAs.
Target mRNA stabilization
HuR was shown to increase the basal and/or stimulus-triggered half-lives of many target mRNAs, including those that encode c-Fos, c-Myc, p21, cyclins A2, B1 and D1, inducible nitric oxide synthase (iNOS), granulocyte macrophage-colony-stimulating factor (GM-CSF), vascular endothelial growth factor (VEGF), SIRT1, tumor necrosis factor (TNF)-α, cyclooxygenase (COX)-2, Bcl-2, Mcl-1, β-catenin, γ-glutamylcysteine synthetase heavy subunit (γ-GCSh), urokinase plasminogen activator (uPA) and its receptor (uPAR), myogenin, myoD and IL-3 [4,5,10,76–85]. On a molecular level, the stabilizing influence of HuR is not fully understood, but HuR probably exerts this effect by displacing decay-promoting RBPs and thereby lengthening the cytoplasmic presence of the target mRNAs.
Target mRNA translation
HuR was found to promote the translation of several target mRNAs, and to increase their association with actively translating polysomes. Target mRNAs whose translation is increased by HuR include those that encode prothymosin (ProTα), p53, hypoxia-inducible (HIF)1, cytochrome c, and the cationic amino acid transporter 1 (CAT-1) [8,9,68,86,87]. HuR was also shown to suppress the translation of p27, the type-I insulin-like growth factor receptor (IGF-IR), Wnt5a and several immune regulators [88–91]. The molecular mechanisms by which HuR modulates the translation of these target transcripts have not been elucidated in detail. Although they are probably also because of the displacement or recruitment of translation-modulatory proteins (such as other TTR-RBPs and translation factors), at least in one report (CAT-1 mRNA) HuR promoted the translation of a target mRNA by displacing an mRNA-associated microRNA [87].
Through its influence on target mRNAs, HuR functions as a key regulator of gene expression in cells responding to immune, stress-causing, metabolic, proliferative and differentiation stimuli. In addition, HuR is emerging as a potent enhancer of cell survival, an effect that is associated with its positive influence on several anti-apoptotic proteins. HuR was shown to bind to the mRNA encoding the apoptosome inhibitor prothymosin α (ProTα) and enhanced its translation and abundance [8]. Similarly, HuR promoted the translation of a target mRNA encoding the hypoxia-inducible factor (HIF-1α) [9], another antiapoptotic protein. HuR also increased the levels of mRNAs encoding Bcl-2 and Mcl-1, two major anti-apoptotic proteins [10]. Together with the finding that HuR elevated the stability and expression of a target mRNA encoding the pro-survival deacetylase SIRT1 [6], HuR seems to have a major role in coordinating a cellular pro-survival program [10]. In this article, we review recent evidence that suggests the HuR target SIRT1 could further contribute to the promotion of cell survival through its positive influence on Nijmegen breakage syndrome protein (NBS1) function, which in turn helps to recruit ATM to the site of damaged DNA, and thus initiates the DNA repair response. Accordingly, the HuR→ SIRT1→NBS1 regulatory pathway underscores the importance of post-transcriptional (mRNA stabilization) and post-translational (deacetylation, phosphorylation) processes towards ensuring the timely and accurate management of DNA damage at a time when transcriptional events are compromised.
HuR regulates SIRT1 expression
HuR associated prominently with the 3′UTR of SIRT1 mRNA [6], leading to increases in SIRT1 mRNA half-life and steady-state SIRT1 mRNA and protein levels [6]. The resulting [HuR–SIRT1 mRNA] ribonucleoprotein (RNP) complex was readily identified in human cervical carcinoma HeLa cells and in early-passage, actively dividing WI-38 human diploid fibroblasts. SIRT1 mRNA stability and SIRT1 expression levels were enhanced following HuR overexpression and reduced when HuR expression was silenced [6]. In cells sustaining acute oxidative damage (following exposure to hydrogen peroxide), HuR and SIRT1 showed a protective influence that was mutually interdependent – HuR was fully protective only in the presence of SIRT1, whereas SIRT1 was only fully expressed and protective in the presence of elevated HuR [6].
Unexpectedly, however, the [HuR–SIRT1 mRNA] RNP complex dissociated following treatment with the potent oxidant [6]. The reduction in RNP levels was attributed to the phosphorylation of HuR by the checkpoint kinase Chk2, which is itself activated by phosphorylation in response to oxidative damage (Figure 1). Three Chk2 phosphorylation sites were identified on HuR, one within the HuR RRM1 (Ser-88), one within RRM2 (Ser-100), and one between RRM1 and RRM2 (Thr-118) [6,11]. In addition to HuR, Chk2 phosphorylates Cdc25A, Cdc25C, BRCA1 and p53 [12]. Although these proteins influence the cell division cycle, Chk2 does not seem to have a direct role in controlling cell cycle progression following genotoxic stress. Instead, studies using Chk2-deficient mice revealed that Chk2 was involved in p53-dependent apoptosis in response to ionizing radiation (IR) injury [13] because lymphocytes, thymocytes and neurons from Chk2−/−mice eserved following IR, whereas those derived from wild-type mice underwent p53-triggered apoptosis. In light of the pro-apoptotic function of Chk2, its ability to trigger the dissociation of HuR and SIRT1 mRNA is particularly significant, considering the dual roles of HuR and SIRT1 on cell survival (discussed in following section). In its capacity as an HuR kinase, we hypothesize that Chk2 could participate in the molecular decision to survive or perish following cellular damage. In normal growth conditions and in response to repairable damage to the cell, Chk2 activity is low, whereas [HuR–SIRT1 mRNA] RNP and SIRT1 protein levels are elevated, signaling survival. By contrast, lethal oxidative injury increases Chk2 function, which in turn dissociates [HuR–SIRT1 mRNA] RNPs and lowers SIRT1 expression [6], thus helping to trigger the death of cells that have sustained irreparable damage (Figure 2).
Figure 1.
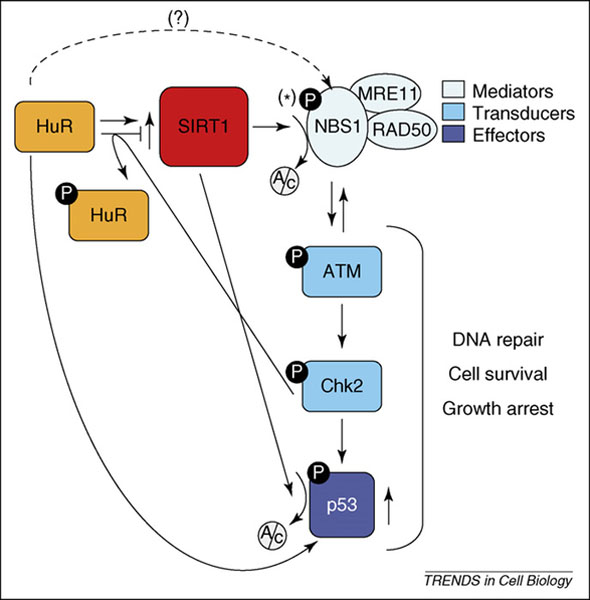
Schematic of the pathway HuR→SIRT1→NBS1. HuR upregulates SIRT1 expression levels post-transcriptionally by stabilizing the SIRT1 mRNA [6]. In turn, SIRT1 deacetylates (Ac) NBS1, rendering it a phosphorylation (P) substrate for ATM. Following its recruitment to the site of DNA damage, ATM is activated via mechanisms that are not fully understood, and phosphorylates Chk2, which can then phosphorylate HuR and thus inhibit its binding to SIRT1 mRNA, possibly creating a negative feedback regulatory loop. The Chk2 phosphorylation substrate, SIRT1 deacetylation target and HuR translational target p53 is also shown here because it impacts on the three major components of the genotoxic response: DNA repair, cell survival and growth arrest. The significance of NBS1 phosphorylation at Ser343 (*) awaits further study; the prediction that HuR associates with the NBS1 mRNA and influences its expression (broken line) needs to be tested experimentally.
Figure 2.
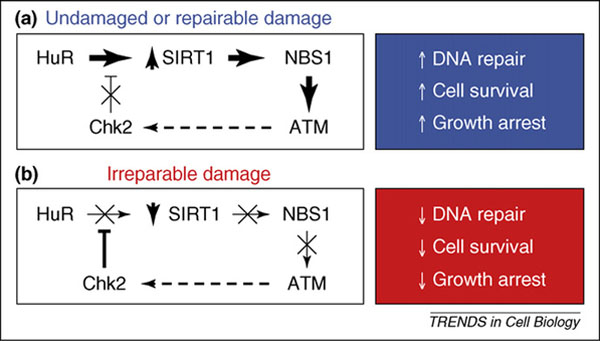
Dual model of SIRT1 action during the genotoxic response. (a) Under conditions of low-level or no cellular damage, HuR increases SIRT1 levels, which in turn deacetylates NBS1; ATM is recruited by the active NBS1-containing complex (MRN) to the site of the damaged DNA. The ensuing genotoxic response program includes the repair of DNA, inhibition of growth and avoidance of cell death. In this scenario the eventual activation of Chk2 by ATM could serve as a negative feedback loop (broken arrow), ensuring that the arrest and repair machineries do not remain in place indefinitely. (b) Under conditions of extreme genotoxic damage, HuR is phosphorylated, releasing the SIRT1 mRNA and thereby reducing SIRT1 expression levels. In turn, NBS1 remains acetylated, cannot be phosphorylated and is unable to activate ATM. The phenotypic consequences of this impaired response are a lack of DNA repair, an inability to activate control checkpoints and a reduction of cell survival. The converse negative feedback loop (broken arrow) is depicted here, as Chk2 eventually becomes inactive in the absence of ATM function.
In addition to the post-transcriptional processes, SIRT1 levels are regulated through transcriptional mechanisms. Transcription of the SIRT1 gene is inhibited by p53 in unstimulated cells, although the association of FoxO3a (forkhead-box transcription factor) with p53 relieved this suppression [14]. The tumor suppressor HIC1 associates with the SIRT1 protein and forms a transcriptional repressor complex that directly binds the SIRT1 promoter [15]. Overall, SIRT1 levels are controlled through a complex set of transcriptional and post-transcriptional processes, as well as by negative feedback autoregulation and regulation by p53 and FoxO3a, the very targets of SIRT1 deacetylase function, as discussed in the following section.
SIRT1 influences the cellular response to metabolic and damaging stress
SIRT1 belongs to the sirtuin protein family of class-III histone deacetylases (HDACs). In yeast, worms, flies and mammals, sirtuin expression and function are regulated by damaging agents and nutritional stress [16–19]. In turn, sirtuins exert a protective influence by modulating various aspects of cell physiology. Originally described in yeast as silent information regulator 2 protein (Sir2p), mammalian cells express seven sirtuin homologs (SIRT1–SIRT7), which catalyze two types of reactions: protein deacetylation (SIRT1, SIRT2, SIRT3 and SIRT5) and protein ADP-ribosylation (SIRT4, SIRT6) [20,21]. Given the requirement for NAD+ in both reactions, sirtuins connect the energy availability of the cell (NAD+) with its response to metabolic and environmental stresses [22–25].
The ubiquitous mammalian SIRT1 deacetylates many proteins other than histones. These substrates include transcription factors involved in regulating stress responses, energy metabolism and endocrine signaling, such as the peroxisome proliferator-activated receptor γ (PPARγ), PPARγ-coactivator 1α (PGC1α), nuclear factor-κB (NF-κB), FoxO proteins, liver X receptor (LXR) and p53 [26–33]. The activities of many of these transcription factors are determined by their acetylation status [34–40]. Because of its influence on transcriptional regulators and other deacetylation substrates, SIRT1 has been proposed to modulate apoptosis and cell proliferation in response to damage-causing agents and metabolic imbalances [19,40–44] (Box 2).
Box 2. SIRT1: integrating metabolic and stress responses.
A direct role for SIRT1 in mammalian longevity has not been demonstrated, but SIRT1 modulates stress resistance and adaptation to metabolic imbalances, two processes that are impaired with aging. Accordingly, SIRT1 is involved in responding to molecular damage (such as that elicited by oxidants and genotoxins), and to metabolic imbalances (such as those triggered by fasting, exercise, nutritional deficiencies and caloric restriction) (Figure I). In turn, SIRT1 deacetylates and modulates the activity of proteins implicated in transcription, cell division, energy metabolism and apoptosis. Through its action on these effectors, SIRT1 mediates responses that promote cell survival, enhance the repair of damaged DNA and reduce cell division. The net effect of these cellular functions is a protective role for SIRT1 against cancer, neurodegeneration, diabetes and other age-related processes, as reviewed recently [17,20,21].
Figure I.
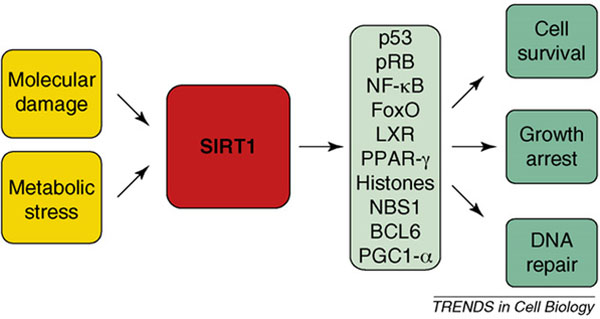
Convergence of stress signals on SIRT1, an upstream regulator of multiple effectors of the stress response.
SIRT1 function increases in response to a broad variety of metabolic and damaging stresses. Among the metabolic inducers of SIRT1 are calorie restriction, fasting and insulin action [19,45–49]. In turn, SIRT1 activates gluconeogenesis and represses glycolysis in the liver via deacetylation of PGC1-α, induces the mobilization of fat from white adipose tissue by repressing PPARγ, and represses uncoupling protein 2 (UCP2) transcription in the pancreas, augmenting its sensitivity to glucose levels [27,32,48,49]. Among its responses to damage-causing stimuli, SIRT1 influences cell division and stress resistance by deacetylating FoxO, p53 and Ku70 and thereby reducing their ability to trigger apoptosis [26,29,30,35,42]. The SIRT1-mediated deacetylation of p53 attenuates stress-induced apoptosis by reducing the ability of p53 to transcriptionally induce the expression of the pro-apoptotic factor Bax [34,35]. Similarly, deacetylation of FoxO3a by SIRT1 blocked its apoptosis-promoting activity, which is elicited in part via the positive influence of FoxO3a on pro-apoptotic proteins Bim and p53 [26,29,50,51]. By deacetylating Ku70, SIRT1 further inhibited Bax pro-apoptotic function because it sequestered Bax in the cytoplasm, prevented its relocation to mitochondria and blocked its ability to initiate apoptosis [42]. The central role of SIRT1 in the integration of metabolic and damaging stresses is discussed in Box 2.
Despite the widespread protection engendered by SIRT1, no specific SIRT1 substrates had been linked to the maintenance of genomic integrity until the publication by Yuan et al.. During the DNA damage response, SIRT1 deacetylated p53 and thus suppressed its apoptotic effects. However, whether or not SIRT1 functions upstream of p53-mediated effects on the repair of damaged DNA remains unclear; according to a recent study, SIRT1 directly modulates the p53-elicited genotoxic response [15] but other reports indicate that deacetylation of p53 by SIRT1 does not alter p53 functions [52,53]. As discussed below, SIRT1 was recently shown to deacetylate NBS1, a protein implicated in the cellular response to DNA damage. These findings identify an alternative effector through which SIRT1 helps to preserve genomic integrity [54].
NBS1 is a novel deacetylation target of SIRT1
A recent report demonstrated that SIRT1 associates with NBS1, the regulatory subunit of the MRN (MRE11–RAD50–NBS1) nuclease complex, which participates in sensing DNA damage and in mounting the cellular response to DNA double-strand breaks (DSBs) [54]. This study also showed that the SIRT1-mediated deacetylation of NBS1 enabled the phosphorylation of NBS1 at Ser-343 by the kinase ATM. Named after its discovery in the human genetic disorder ataxia-telangiectasia (Box 3), ATM is activated by DNA damage and in turn phosphorylates many substrate proteins that affect growth arrest checkpoints, regulate the repair of DSBs and modulate apoptosis [55]. Phosphorylation of NBS1 at Ser-343 and Ser-278 was previously shown to be required for the activation of an intra-S phase checkpoint [56–59]. However, primary lymphocytes from mice expressing NBS1 with non-phosphorylatable Ser-278 and Ser-343 residues did not exhibit significant checkpoint defects and showed efficient phosphorylation of ATM substrates Chk2 and SMC1 (structural maintenance of chromosomes 1) in response to ionizing radiation (IR) [60]. Together with similar findings reported earlier [61], this evidence brings into question the requirement for NBS1 phosphorylation at Ser-278 and Ser-343 for activating an intra-S phase checkpoint in primary mouse lymphocytes. However, the possibilities remain that other NBS1 phosphorylation sites contribute to the intra-S phase checkpoint and that NBS1 has distinct phosphorylation requirements in different cell types. According to a model proposed recently, the DNA-damage response following DSBs begins with the detection and association of the MRN complex at the site of the DNA break; the ATM kinase is then recruited to the DSB site via multiple associations with the MRN complex; ATM is subsequently activated through mechanisms that are incompletely understood but that might be linked to changes in chromatin structure. Active ATM in turn phosphorylates other proteins to initiate the DNA repair process [62]. In this process, the N-terminal segment of NBS1 is important for the implementation of checkpoints and DNA repair and the C-terminal segment interacts with ATM and is implicated in apoptosis [62].
Box 3. ATM and NBS1.
Deficiency in ATM causes the human genetic disorder ataxia telangiectasia, associated with genomic instability, increased cancer susceptibility and neurodegeneration [92,93]. The precise mechanisms that activate ATM remain unclear but the MRN complex has a crucial role in recruiting ATM to the sites of DNA damage and in the activation of ATM [94]. Autophosphorylation at Ser-1981 was reported to be necessary to activate ATM [95] but further studies found this modification to be dispensable and suggested that autophosphorylation might be a consequence of rather than a cause for ATM activation [96]. Active ATM has been reported to phosphorylate several dozen proteins, although it probably has many more substrates [97]. As recently reviewed by Lavin and Kozlov [55], the ATM-regulated effectors of cell cycle checkpoints include phosphorylation targets Mdm2, Mdmx, p53 and Chk2 for progression through G1 and S; NBS1, ATF2 and SMC1 for progression through S phase; and BRCA1 and Chk1 for progression through G2 and M. ATM also promotes DNA repair through phosphorylation substrates that include DNA-PKcs and BRCA1, and influences apoptosis via targets such as p53, E2F1 and TRF1/Pin2.
In agreement with the influence of the MRN complex on ATM function, lowering SIRT1 or otherwise interfering with the deacetylation of NBS1 led to a decrease in cell survival following IR-induced DNA damage [54]. NBS1 is not only a downstream effector of ATM, but it is also an upstream activator of ATM [63–65]. In an interesting convergence of pathways, NBS1 phosphorylation was found to be necessary for the activation of the intra-S phase checkpoint by promoting the phosphorylation of the HuR kinase Chk2 by ATM [66]. This regulatory step might establish a negative feedback loop to suppress SIRT1 expression because phosphorylation of HuR by Chk2 leads to the release of SIRT1 mRNA and consequently lowers SIRT1 mRNA stability and SIRT1 levels [6] (Figure 2). The possibility that SIRT1 also deacetylates and thereby regulates other components of the DNA damage detection and repair machineries remains to be studied.
DNA damage response by HuR→SIRT1→NBS1 pathway
The heterogeneous collection of HuR target mRNAs is in keeping with the broad spectrum of HuR effects in the cell (Box 1) [7,67]. Similarly, the diverse subset of proteins deacetylated by SIRT1 elicits the pleiotropic influences of SIRT1 on cellular processes (Box 2) [20,21]. In addition, HuR and SIRT1 have both been implicated in the cellular response to genotoxic stress, but the specific downstream mediators are only now beginning to emerge.
One of the common downstream mediators of HuR and SIRT1 action during the genotoxic response is p53. HuR increases the translation of p53 [68]; however, HuR does not trigger apoptosis, perhaps because it concomitantly elevates the levels of SIRT1, which in turn deacetylates p53 and, together with the effects of SIRT1 on Ku70 and FoxO activity, it protects against unscheduled p53-mediated apoptosis. Because it remains unclear whether or not SIRT1 modulates the cellular response of p53 to genotoxic injury [15,52,53], the identification of NBS1 as a SIRT1 deacetylation substrate provides an alternative downstream effector of SIRT1 action during the DNA damage response. Interestingly, HuR is also predicted to bind the NBS1 mRNA (NM_002485) because the NBS1 3′UTR contains eight occurences of a previously reported motif present in HuR target mRNAs [5]. The existence of a putative [HuR–NBS1 mRNA] complex remains to be assessed experimentally, but a positive influence of HuR on NBS1 expression levels raises the possibility that HuR elevate the expression of both components of this regulatory pathway, possibly leading to a further enhancement of MRN function.
In addition to the induction of SIRT1 by DNA damage leading to the activation of NBS1 and ATM [6,54,69], recent reports link other sirtuins to the genotoxic stress response. SIRT6 is a chromatin-associated sirtuin that was proposed to function in base excision repair and which therefore promotes constitutive resistance to DNA damage and suppresses genomic instability. Accordingly, SIRT6-deficient mice are viable but exhibit genomic abnormalities that resemble accelerated aging [70]. SIRT2 is also likely to participate in the DNA damage response. SIRT2 colocalizes with microtubules and deacetylates α-tubulin, thereby regulating cell mitosis and protecting genomic integrity [71–74]; moreover, stress treatment increases SIRT2 levels and function in deacetylating FoxO3a, consequently altering the apoptotic response [72]. In addition, injury to DNA triggers the translocation of SIRT3 to mitochondria, suggesting that this sirtuin might also participate in the genotoxic stress response [75]. Together, this evidence suggests that sirtuins influence the cellular response to genotoxic damage in a coordinated manner.
Regulation of cell survival through the HuR→SIRT1→NBS1 pathway
The cytoprotective influence of HuR has been observed under normal culture conditions, in cells sustaining moderate injury and shortly after exposure to acute damaging agents [10]. In the absence of genotoxic stress, HuR bound the SIRT1 mRNA and enhanced SIRT1 expression. Even though strong oxidative damage disrupted this interaction [6], the existence of [HuR–SIRT1 mRNA] RNP complexes following mild genotoxic damage has not been assessed experimentally. Without overt cellular damage or under conditions of repairable damage, the deacetylase function of SIRT1 has been shown to predominate over the function of acetylases sharing the same apoptosis-regulatory substrates (Ku70, FoxO, p53). In this scenario, HuR positively regulates SIRT1 expression, which in turn deacetylates NBS1 and enables it to activate ATM and the DNA damage repair pathway [54]. The ATM substrate Chk2 is then activated and phosphorylates HuR, thereby dissociating the [HuR–SIRT1 mRNA] complex and turning off the induction of SIRT1 in a negative feedback loop (Figure 2).
However, beyond the response of individual cells, the maintenance of homeostasis at the organism level requires that cells bearing irreparable genotoxic injury be eliminated to preserve tissue and organ function. In keeping with this notion, a dual mode of action is proposed whereby the prosurvival pathway HuR→SIRT1→NBS1 could become impaired in response to overwhelming, irreparable damage. After potent oxidative damage (e.g. high hydrogen peroxide doses), Chk2 phosphorylated HuR at RRMs, leading to the release of SIRT1 mRNA and the reduction of SIRT1 expression. In this scenario, the suppression of SIRT1 expression by dissociation from HuR is accompanied by a loss of cell viability [6] (Figure 2).
Perspectives
Following genotoxic injury, cells respond rapidly through the post-translational modification (phosphorylation, acetylation, ubiquitination, etc.) of pre-existing proteins. These proteins are responsible for locating and assessing the extent of the damage, transducing appropriate signals and directing the cell’s repair, division and apoptotic machineries. The recent identification of SIRT1 as a deacetylase for NBS1 underscores the complex network of post-translational modifications elicited during the genotoxic response. Although the functional role of NBS1 deacetylation and Ser-343 phosphorylation awaits further study, the implication of SIRT1 in the DNA damage response is significant in several respects. First, it creates an important link between the genotoxic stress response and energy metabolism (Box 2). Second, it introduces NBS1 as an additional downstream effector of SIRT1 action, which together with numerous other SIRT1 substrates, contributes to the pleiotropic functions of SIRT1 during the cellular stress response. Third, it identifies a novel influence of SIRT1 on ATM activity and consequently on the DNA repair process, the cell division cycle and cell survival.
The post-transcriptional regulation of SIRT1 expression also ensures that functional SIRT1 protein can be synthesized from pre-existing mRNA without requiring de novo transcription. This level of regulation reduces the chance of SIRT1 expression being suppressed by the global reduction in transcription that typically follows genotoxic damage, and it prevents the propagation of possible mutations arising from the newly damaged DNA. However, several aspects of the HuR→SIRT1→NBS1 regulatory pathway await further study. For example, HuR was not reported to affect gene expression patterns following IR. In light of the signaling paradigm discussed here, further study of HuR phosphorylation and binding to target mRNAs following IR is warranted. It is also worth systematically examining the influence of HuR on SIRT1 expression. This analysis should cover lower doses of oxidative damage and also other genotoxic stress agents. The hypothesis that HuR has higher affinity for SIRT1 mRNA under conditions of low, repairable oxidative damage also deserves immediate testing. Similarly, whether severe DNA damage causes HuR to dissociate from other target mRNAs encoding genotoxic stress-response proteins remains to be formally studied. It will also be important to ascertain if such a bimodal mechanism of action characterizes HuR binding to other target mRNAs, with HuR–RNP associations increasing in cells that will ultimately survive and decreasing in cells that will ultimately succumb to the damage.
Ultimately, however, studying the phenotypic consequences of the HuR→SIRT1→NBS1 pathway awaits the development of suitable mammalian genetic models, particularly for the analysis of HuR. In such animal models, we anticipate finding that perturbations of this pathway will have a broad range of pathophysiologic consequences because multiple upstream pathways converge on HuR, SIRT1 and NBS1, and these proteins in turn impact on broad collections of substrate molecules. However, as these studies move forward, we also expect to gain increasing support for the notion that the regulatory axis HuR→SIRT1→NBS1 promotes the establishment and maintenance of cell, organ and organism homeostasis.
Acknowledgements
We thank A. Nussenzweig and D.M. Wilson 3rd for insight into this subject. M.G. and R.d.C. are supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health.
References
- 1.Moore MJ (2005) From birth to death: the complex lives of eukaryotic mRNAs. Science 309, 1514–1518 [DOI] [PubMed] [Google Scholar]
- 2.Keene JD (2007) RNA regulons: coordination of post-transcriptional events. Nat. Rev. Genet. 8, 533–543 [DOI] [PubMed] [Google Scholar]
- 3.Pullmann R Jr et al. (2007) Analysis of turnover and translation regulatory RNA-binding protein expression through binding to cognate mRNAs. Mol. Cell. Biol. 27, 6265–6278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brennan CM and Steitz JA (2001) HuR and mRNA stability. Cell. Mol. Life Sci. 58, 266–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.López de Silanes I et al. (2004) Identification of a target RNA motif for RNA-binding protein HuR. Proc. Natl. Acad. Sci. U. S. A. 101, 2987–2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abdelmohsen K et al. (2007) Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol. Cell 25, 543–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorospe M (2003) HuR in the mammalian genotoxic response: post-transcriptional multitasking. Cell Cycle 2, 412–414 [PubMed] [Google Scholar]
- 8.Lal A et al. (2005) Antiapoptotic function of RNA-binding protein HuR effected through prothymosin alpha. EMBO J. 24, 1852–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gálban S et al. (2008) RNA-binding proteins HuR and PTB promote the translation of hypoxia-inducible factor-1. Mol. Cell. Biol. 28, 93–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdelmohsen K et al. (2007) Posttranscriptional orchestration of an anti-apoptotic program by HuR. Cell Cycle 6, 1288–1292 [DOI] [PubMed] [Google Scholar]
- 11.Wilusz CJ and Wilusz J (2007) HuR-SIRT: the hairy world of posttranscriptional control. Mol. Cell 25, 485–487 [DOI] [PubMed] [Google Scholar]
- 12.Bartek J and Lukas J (2003) Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3, 421–429 [DOI] [PubMed] [Google Scholar]
- 13.Takai H et al. (2002) Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J. 21, 5195–5205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nemoto S et al. (2004) Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science 306, 2105–2108 [DOI] [PubMed] [Google Scholar]
- 15.Chen WY et al. (2005) Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell 123, 437–448 [DOI] [PubMed] [Google Scholar]
- 16.Rogina B and Helfand SL (2004) Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. U. S. A. 101, 15998–16003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang T et al. (2006) SIRT1 and endocrine signaling. Trends Endocrinol. Metab. 17, 186–191 [DOI] [PubMed] [Google Scholar]
- 18.Wang Y and Tissenbaum HA (2006) Overlapping and distinct functions for a Caenorhabditis elegans SIR2 and DAF-16/FOXO. Mech. Ageing Dev. 127, 48–56 [DOI] [PubMed] [Google Scholar]
- 19.Cohen HY et al. (2004) Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305, 390–392 [DOI] [PubMed] [Google Scholar]
- 20.Michan S and Sinclair D (2007) Sirtuins in mammals: insights into their biological function. Biochem. J. 404, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haigis MC and Guarente LP (2006) Mammalian sirtuins–emerging roles in physiology, aging, and calorie restriction. Genes Dev. 20, 2913–2921 [DOI] [PubMed] [Google Scholar]
- 22.Imai S et al. (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 [DOI] [PubMed] [Google Scholar]
- 23.Landry J et al. (2000) The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. U. S. A. 97, 5807–5811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith JS et al. (2000) A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. U. S. A. 97, 6658–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grubisha O et al. (2006) Metabolite of SIR2 reaction modulates TRPM2 ion channel. J. Biol. Chem. 281, 14057–14065 [DOI] [PubMed] [Google Scholar]
- 26.Motta MC et al. (2004) Mammalian SIRT1 represses forkhead transcription factors. Cell 116, 551–563 [DOI] [PubMed] [Google Scholar]
- 27.Picard F et al. (2004) Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 429, 771–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giannakou ME and Partridge L (2004) The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol. 14, 408–412 [DOI] [PubMed] [Google Scholar]
- 29.Brunet A et al. (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015 [DOI] [PubMed] [Google Scholar]
- 30.Kobayashi Y et al. (2005) SIRT1 is critical regulator of FOXO-mediated transcription in response to oxidative stress. Int. J. Mol. Med. 16, 237–243 [PubMed] [Google Scholar]
- 31.Kitamura YI et al. (2005) FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2, 153–163 [DOI] [PubMed] [Google Scholar]
- 32.Rodgers JT et al. (2005) Nutrient control of glucose homeostasis through a complex of PGC and SIRT1. Nature 434, 113–118 [DOI] [PubMed] [Google Scholar]
- 33.Li X et al. (2007) SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol. Cell 28, 91–106 [DOI] [PubMed] [Google Scholar]
- 34.Vaziri H et al. (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159 [DOI] [PubMed] [Google Scholar]
- 35.Luo J et al. (2001) Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 107, 137–148 [DOI] [PubMed] [Google Scholar]
- 36.Cheng HL et al. (2003) Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1) deficient mice. Proc. Natl. Acad. Sci. U. S. A. 100, 10794–10799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Heide LP and Smidt MP (2005) Regulation of FoxO activity by CBP/p300-mediated acetylation. Trends Biochem. Sci. 30, 81–86 [DOI] [PubMed] [Google Scholar]
- 38.Vo N and Goodman RH (2001) CREB-binding protein and p300 in transcriptional regulation. J. Biol. Chem. 276, 13505–13508 [DOI] [PubMed] [Google Scholar]
- 39.Bouras T et al. (2005) SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J. Biol. Chem. 280, 10264–10276 [DOI] [PubMed] [Google Scholar]
- 40.Leibiger IB and Berggren PO (2006) Sirt1: a metabolic master switch that modulates lifespan. Nat. Med. 12, 34–36 [DOI] [PubMed] [Google Scholar]
- 41.Chua KF et al. (2005) Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell Metab. 2, 67–76 [DOI] [PubMed] [Google Scholar]
- 42.Cohen HY et al. (2004) Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol. Cell 13, 627–638 [DOI] [PubMed] [Google Scholar]
- 43.Langley E (2002) Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 21, 2383–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin SJ and Guarente L (2003) Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr. Opin. Cell Biol. 15, 241–246 [DOI] [PubMed] [Google Scholar]
- 45.Rodgers JT and Puigserver P (2007) Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc. Natl. Acad. Sci. U. S. A. 104, 12861–12866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun C et al. (2007) SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab. 6, 307–319 [DOI] [PubMed] [Google Scholar]
- 47.Zabolotny JM and Kim YB (2007) Silencing insulin resistance through SIRT1. Cell Metab. 6, 247–249 [DOI] [PubMed] [Google Scholar]
- 48.Bordone L et al. (2005) Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 4, e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moynihan KA et al. (2005) Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2, 105–117 [DOI] [PubMed] [Google Scholar]
- 50.Gilley J et al. (2003) FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J. Cell Biol. 162, 613–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.You H et al. (2006) FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J. Exp. Med. 203, 1657–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kamel C et al. (2006) SirT1 fails to affect p53-mediated biological functions. Aging Cell 5, 81–88 [DOI] [PubMed] [Google Scholar]
- 53.Solomon JM et al. (2006) Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell. Biol. 26, 28–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yuan Z et al. (2007) SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol. Cell 27, 149–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lavin MF and Kozlov S (2007) ATM activation and DNA damage response. Cell Cycle 6, 931–942 [DOI] [PubMed] [Google Scholar]
- 56.Gatei M et al. (2000) ATM dependent phosphorylation of nibrin in response to radiation exposure. Nat. Genet. 25, 115–119 [DOI] [PubMed] [Google Scholar]
- 57.Lim DS et al. (2000) ATM phosphorylates p95/nbs1 in an S phase checkpoint pathway. Nature 404, 613–617 [DOI] [PubMed] [Google Scholar]
- 58.Wu X et al. (2000) ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature 405, 477–482 [DOI] [PubMed] [Google Scholar]
- 59.Zhao S et al. (2000) Functional link between ataxia telangiectasia and Nijmegen breakage syndrome gene products. Nature 405, 473–477 [DOI] [PubMed] [Google Scholar]
- 60.Difilippantonio S et al. (2007) Distinct domains in Nbs1 regulate irradiation-induced checkpoints and apoptosis. J. Exp. Med. 204, 1003–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Horejsi Z et al. (2004) Distinct functional domains of Nbs1 modulate the timing and magnitude of ATM activation after low doses of ionizing radiation. Oncogene 23, 3122–3127 [DOI] [PubMed] [Google Scholar]
- 62.Difilippantonio S and Nussenzweig A (2007) The NBS1-ATM connection revisited. Cell Cycle 6, 2366–2370 [DOI] [PubMed] [Google Scholar]
- 63.Lee JH and Paull TT (2005) ATM activation by DNA double-strand breaks through the Mre11Rad50-Nbs1 complex. Science 308, 551–554 [DOI] [PubMed] [Google Scholar]
- 64.You Z et al. (2005) ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell. Biol. 25, 5363–5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cerosaletti K et al. (2006) Active role for nibrin in the kinetics of atm activation. Mol. Cell. Biol. 26, 1691–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee JH and Paull TT (2004) Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304, 93–96 [DOI] [PubMed] [Google Scholar]
- 67.López de Silanes I et al. (2005) HuR: post-transcriptional paths to malignancy. RNA Biol. 2, 11–13 [DOI] [PubMed] [Google Scholar]
- 68.Mazan-Mamczarz K et al. (2003) RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc. Natl. Acad. Sci. U. S. A. 100, 8354–8359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang C et al. (2006) Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat. Cell Biol. 8, 1025–1031 [DOI] [PubMed] [Google Scholar]
- 70.Mostoslavsky R et al. (2006) Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329 [DOI] [PubMed] [Google Scholar]
- 71.Dryden SC et al. (2003) Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol. Cell. Biol. 23, 3173–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang F et al. (2007) SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6, 505–514 [DOI] [PubMed] [Google Scholar]
- 73.North BJ and Verdin E (2007) Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS ONE 2, e784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.North BJ and Verdin E (2007) Mitotic regulation of SIRT2 by cyclin-dependent kinase 1-dependent phosphorylation. J. Biol. Chem. 282, 19546–19555 [DOI] [PubMed] [Google Scholar]
- 75.Scher MB (2007) SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 21, 920–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lal A et al. (2004) Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 23, 3092–3102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fan XC and Steitz JA (1998) Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE containing mRNAs. EMBO J. 17, 3448–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Peng SS et al. (1998) RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J. 17, 3461–3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen CY et al. (2002) Highly selective actions of HuR in antagonizing AU-rich element-mediated mRNA destabilization. Mol. Cell. Biol. 22, 7268–7278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang W et al. (2000) HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J. 19, 2340–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sengupta S et al. (2003) The RNA-binding protein HuR regulates the expression of cyclooxygenase-2. J. Biol. Chem. 278, 25227–25233 [DOI] [PubMed] [Google Scholar]
- 82.Ming XF et al. (2001) Parallel and independent regulation of interleukin-3 mRNA turnover by phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase. Mol. Cell. Biol. 21, 5778–5789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Levy NS et al. (1998) Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA binding protein HuR. J. Biol. Chem. 273, 6417–6423 [DOI] [PubMed] [Google Scholar]
- 84.Tran H et al. (2003) Stabilization of urokinase and urokinase receptor mRNAs by HuR is linked to its cytoplasmic accumulation induced by activated mitogen-activated protein kinase-activated protein kinase. Mol. Cell. Biol. 23, 7177–7188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Song IS et al. (2005) Delayed mechanism for induction of gamma-glutamylcysteine synthetase heavy subunit mRNA stability by oxidative stress involving p38 mitogen-activated protein kinase signaling. J. Biol. Chem. 280, 28230–28240 [DOI] [PubMed] [Google Scholar]
- 86.Kawai T et al. (2006) Translational control of cytochrome c by RNA-binding proteins TIA-1 and HuR. Mol. Cell. Biol. 26, 3295–3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bhattacharyya SN et al. (2006) Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 125, 1111–1124 [DOI] [PubMed] [Google Scholar]
- 88.Kullmann M et al. (2002) ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5′UTR. Genes Dev. 16, 3087–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meng Z et al. (2005) The ELAV RNA-stability factor HuR binds the 5′-untranslated region of the human IGF-IR transcript and differentially represses cap-dependent and IRES-mediated translation. Nucleic Acids Res. 33, 2962–2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Leandersson K et al. (2006) Wnt-5a mRNA translation is suppressed by the Elav-like protein HuR in human breast epithelial cells. Nucleic Acids Res. 34, 3988–3999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Katsanou V et al. (2005) HuR as a negative posttranscriptional modulator in inflammation. Mol. Cell 19, 777–789 [DOI] [PubMed] [Google Scholar]
- 92.Lavin MF and Shiloh Y (1997) The genetic defect in ataxia-telangiectasia. Annu. Rev. Immunol. 15, 177–202 [DOI] [PubMed] [Google Scholar]
- 93.Shiloh Y and Kastan MB (2001) ATM: genome stability, neuronal development, and cancer cross paths. Adv. Cancer Res. 83, 209–254 [DOI] [PubMed] [Google Scholar]
- 94.Zhou J et al. (2006) The role of NBS1 in the modulation of PIKK family proteins ATM and ATR in the cellular response to DNA damage. Cancer Lett. 243, 9–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bakkenist CJ and Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506 [DOI] [PubMed] [Google Scholar]
- 96.Pellegrini M et al. (2006) Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature 443, 222–225 [DOI] [PubMed] [Google Scholar]
- 97.Matsuoka S et al. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 [DOI] [PubMed] [Google Scholar]