

Abstract
Vitamin B-6 in the form of pyridoxine (PN) is commonly used by the general population. The use of PN-containing supplements has gained lots of attention over the past years as they have been related to the development of peripheral neuropathy. In light of this, the number of reported cases of adverse health effects due to the use of vitamin B-6 have increased. Despite a long history of study, the pathogenic mechanisms associated with PN toxicity remain elusive. Therefore, the present review is focused on investigating the mechanistic link between PN supplementation and sensory peripheral neuropathy. Excessive PN intake induces neuropathy through the preferential injury of sensory neurons. Recent reports on hereditary neuropathy due to pyridoxal kinase (PDXK) mutations may provide some insight into the mechanism, as genetic deficiencies in PDXK lead to the development of axonal sensory neuropathy. High circulating concentrations of PN may lead to a similar condition via the inhibition of PDXK. The mechanism behind PDXK-induced neuropathy is unknown; however, there is reason to believe that it may be related to γ-aminobutyric acid (GABA) neurotransmission. Compounds that inhibit PDXK lead to convulsions and reductions in GABA biosynthesis. The absence of central nervous system-related symptoms in PDXK deficiency could be due to differences in the regulation of PDXK, where PDXK activity is preserved in the brain but not in peripheral tissues. As PN is relatively impermeable to the blood–brain barrier, PDXK inhibition would similarly be confined to the peripheries and, as a result, GABA signaling may be perturbed within peripheral tissues, such as sensory neurons. Perturbed GABA signaling within sensory neurons may lead to excitotoxicity, neurodegeneration, and ultimately, the development of peripheral neuropathy. For several reasons, we conclude that PDXK inhibition and consequently disrupted GABA neurotransmission is the most plausible mechanism of toxicity.
Keywords: vitamin B-6, pyridoxine, toxicity, pyridoxal kinase, GABA, neuropathy
Statement of Significance: The number of reported cases of sensory peripheral neuropathy due to the use of vitamin B-6 supplements is increasing. Despite a long history of study, the pathogenic mechanism(s) associated with the toxicity of pyridoxine remain elusive. In our review, we reveal the most plausible mechanistic link between pyridoxine supplementation and sensory peripheral neuropathy.
Introduction
In nonruminant animals, vitamin B-6 is an essential, water-soluble vitamin (1), which is found in a variety of food sources including meat, poultry, fish, legumes, nuts, cereals, vegetables, and bananas (2). Vitamin B-6 is a generic term that refers to the pyridine-based compounds pyridoxine (PN), pyridoxamine (PM), pyridoxal (PL), and their phosphorylated derivatives. In human adults, the recommended daily oral intake of vitamin B-6 falls between 1.5 and 1.8 mg (3). Due to its abundance in food sources, vitamin B-6 deficiency in humans is rare and typically the result of a genetic deficiency or pharmaceutical interference (4). Nevertheless, the consumption of dietary supplements containing vitamin B-6 is common among the general population. PN is the form of vitamin B-6 most frequently used in dietary supplements and/or fortified food and beverage products, such as energy drinks (5). Previously, high doses of PN have been used to treat a number of conditions including vitamin B-6-dependent epilepsy, carpal tunnel syndrome, premenstrual syndrome, mushroom poisoning, autism, hyperkinesis, and even Parkinson disease (6). Similarly, high-dose PN supplementation is frequently used by athletes, such as bodybuilders (5).
Starting in the 1980s, a number of case reports described symptoms of sensory neuropathy in people taking large doses of PN for prolonged periods of time. PN-induced neuropathy is characterized by symmetric, progressive impairments in touch, pin-prick, temperature, vibrational, and positional sense at the extremities (6, 7). PN toxicity leads to both small- and large-fiber neuropathies (8, 9). In humans, there is strong evidence that small-fiber dysfunction is the earliest and predominant clinical manifestation (10). While the incidence of PN-induced neuropathy in humans is likely low, reports by neuropathic centers indicate that it may be more widespread than previously thought, with a prevalence in 2 cohorts of neuropathy patients between 2.5% and 12.4% (8, 11). Additionally, PN-induced small-fiber neuropathy may be underdiagnosed. Small-fiber neuropathies are more challenging to diagnose as nerve-conduction studies are often normal in these cases. More sensitive diagnostic procedures such as quantitative sensory threshold (QST) testing or skin biopsy are often not used (12, 13).
In their assessment of the evidence, the European Food Safety Authority (EFSA) established a Tolerable Upper Intake Level (UL) of 25 mg/d, while the US-based Food and Nutrition Board recommends a UL of 100 mg/d (6). Alarmingly, a number of more recent case reports have described neuropathic symptoms in patients taking PN at doses near or even below the EFSA UL (14, 15). In light of these reports, the UL for PN may require revision. The absence of neuropathic symptoms at these doses under more controlled observational settings could be related to large interindividual differences in sensitivity to PN toxicity. Additionally, small-fiber abnormalities may not have been detected. Remarkably, single-dose tablets with high doses of PN (20–100 mg) are widely available in stores. Considering the accessibility of these PN supplements to the general population, its safety profile should be further investigated.
In order to better characterize the safety profile of PN supplementation, the mechanism of toxicity has to be clarified. Despite a long history of study, the pathogenic mechanisms associated with PN toxicity remain elusive and there is even a lack of consensus over which vitamin B-6 vitamer leads to neuropathy. Several competing theories have been proposed to explain its mechanism of action. These include aldehyde toxicity through elevated pyridoxal 5′-phosphate (PLP) concentrations, the formation of reactive quinone methide-type intermediates, the competitive inhibition of PLP-dependent enzymes, and the saturation of pyridoxal kinase (PDXK) (4, 16–19). Many aspects of the proposed hypotheses are unexplored, and even the basic assumptions they are grounded in can be contested, particularly concerning PN pharmacokinetics, toxicological testing, and vitamin B-6 metabolism. Therefore, the present review is focused on investigating the mechanistic link between PN supplementation and sensory peripheral neuropathy.
Current Status of Knowledge
Function of vitamin B-6
PLP, the bioactive form of vitamin B-6, is a remarkably versatile cofactor that participates widely in metabolic pathways involving amino acid, oxoacid, or amine substrates. In humans, at least 56 PLP-dependent enzymes have been described (20). These PLP-dependent reactions contribute, for instance, to glycogenolysis, gluconeogenesis, and the biosynthesis of hemoglobin, sphingolipids, neurotransmitters, and hormones (1).
Metabolism
In humans, the major forms of vitamin B-6 include PN, PM, PL, their phosphorylated derivatives, and the metabolic end product 4-pyridoxic acid (4-PA). All forms of vitamin B-6 are present in food sources and are converted into PLP by the “B-6 salvage pathway” shown in Figure 1. Under normal dietary circumstances, the majority of absorbed PN and PM are converted into PLP and released as PL in the small intestine. With normal vitamin B-6 intake, there are negligible amounts of PN and PM in the portal vein and/or circulation (21–23). With higher doses, the liver is the primary site of PN metabolism (24, 25). In the blood, vitamin B-6 is present primarily as PLP and to a lesser extent as PL and 4-PA (26). Vitamin B-6 metabolism in humans has been discussed in greater detail in reviews by Parra et al. (27), Coburn (26) and EFSA (3). Recently, PL/PLP reductase activity was identified in humans. PLP-treated patients had increased PN concentrations in plasma and cerebrospinal fluid. The reduction of PL to PN has been observed in a large variety of mammalian cell types. This PL reductase activity is likely more important under conditions of high PL and/or unbound PLP concentrations (28).
FIGURE 1.
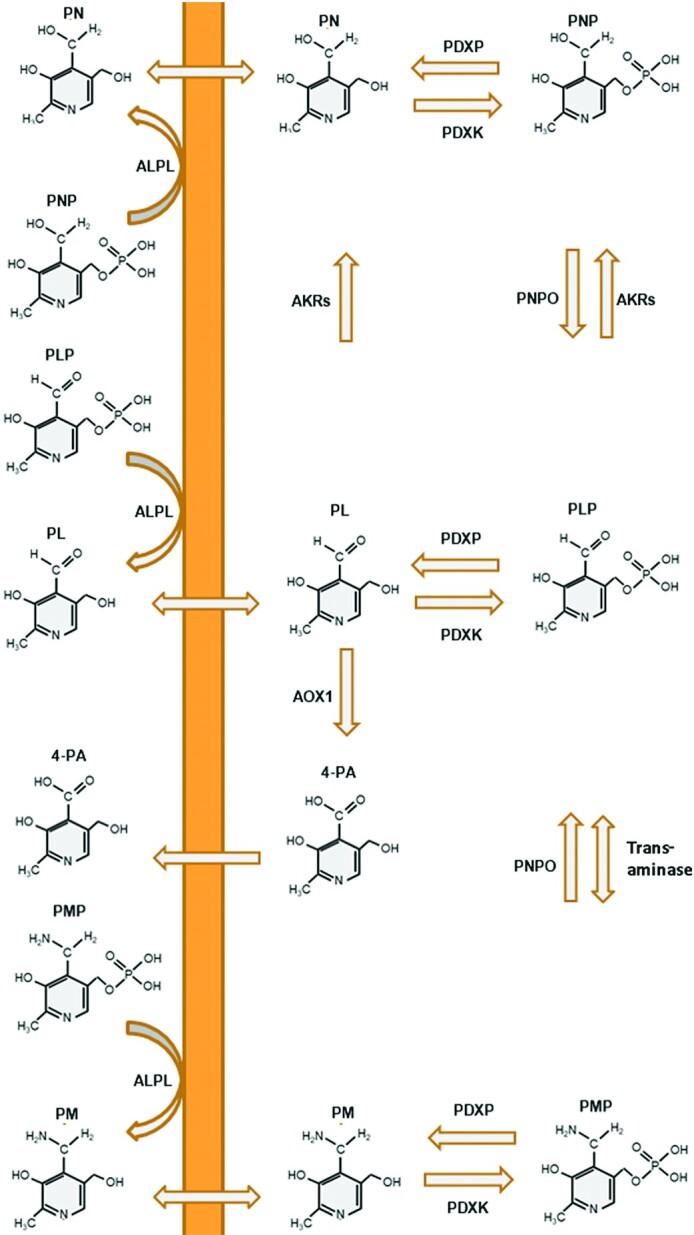
The vitamin B-6 salvage pathway. The different vitamers are converted into PLP through the “B-6 salvage pathway,” involving the enzymes PDXK, PNPO, and PDXP. PLP enters the circulation bound to lysine residues of proteins, mostly albumin. In order to enter the cell, the phosphorylated vitamers are hydrolyzed by ALPL. Once inside the cell, PDXK phosphorylates the different vitamers, yielding PNP, PMP, and PLP. PMP and PNP are converted into PLP by PNPO. Transaminase reactions may also interconvert PMP and PLP as a side reaction. PDXP, along with other phosphatases, hydrolyze phosphorylated vitamin B-6 vitamers, enabling them to exit the cell. PL is eliminated from the body following its conversion to 4-PA, catalyzed by AOX1. AKRs can convert PL and PLP into PN and PNP, respectively; however these activities likely play only a minor role in vitamin B-6 metabolism. AKR, aldo-keto reductase; ALPL, tissue-nonspecific alkaline phosphatase; AOX1, aldehyde oxidase; PDXK, pyridoxal kinase; PDXP, pyridoxal phosphatase; PL, pyridoxal; PLP, pyridoxal 5′-phosphate; PM, pyridoxamine; PMP, pyridoxamine 5'-phosphate; PN, pyridoxine; PNP, pyridoxine-5'-phosphate; PNPO, pyridoxine 5′-phosphate oxidase; 4-PA, 4-pyridoxic acid.
Intracellular trafficking
PLP is a highly reactive aldehyde and can form harmful, unproductive bonds with amine and thiol groups (4). As a result, its intracellular concentration and trafficking is under tight regulation. This ensures adequate supply to apo-enzymes while preventing harmful unproductive reactions. PLP availability in the cell is determined by the supply of PLP precursors, biosynthesis by PDXK and pyridoxine-5'-phosphate oxidase (PNPO), PLP scavenging by proteins and small molecules, and PLP hydrolysis by PDXP (4, 29). The expression of both PNPO and PDXK is regulated at the transcriptional level and likely depends on the activity of PLP-dependent enzymes and vitamin B-6 availability (30, 31). Intracellular PLP trafficking is facilitated by proteins such as PLP-binding protein (PLPBP), PDXK, and PNPO (32). Additionally, both PDXK and PNPO were shown to directly transfer PLP to vitamin B-6-dependent apo-enzymes and forgo releasing their product into the solvent.
The activation of vitamin B-6-dependent apo-enzymes was more efficient using PLP synthesized from PNPO and PDXK compared with free PLP (33, 34). In addition, exogenous PLP was found to form unproductive aldimines with cellular components (34). Interestingly, PNPO and PDXK have significantly different binding affinities and PLP transfer efficiencies with different vitamin B-6-dependent apo-enzymes (35, 36). These differences may be indicative of process-specific functions for PNPO and PDXK. Certain PLP-dependent processes may be more reliant on 1 enzyme system over the other. Additionally, the expression of PNPO and PDXK varies considerably between different tissues (37, 38). Hence, vitamin B-6 homeostasis is complex, and highly regulated.
Vitamin B-6 in disease
As vitamin B-6 plays an important role in many different processes, lacking sufficient amounts of the vitamin B-6 vitamers may lead to adverse health conditions. Vitamin B-6 deficiency in humans is associated with anemia, dermatitis, glossitis, electroencephalographic abnormalities, depression and confusion, and weakened immune function. In infants, a vitamin B-6 deficiency may lead to irritability, abnormally acute hearing, and convulsive seizures (39). Moreover, inborn errors in vitamin B-6 metabolism such as PNPO, PLPBP, alkaline phosphatase, tissue-nonspecific isozyme (ALPL), and antiquitin (ALDH7A1) are linked to neonatal epileptic seizures (40–45). Interestingly, genetic deficiencies in PDXK are associated with peripheral neuropathy (46, 47). Given the importance of PLP within neuronal tissue, disruptions in vitamin B-6 homeostasis likely have neurological consequences, such as with PN toxicity.
Clinical presentation of PN-induced neuropathy
The clinical presentation for PN-induced neuropathy in humans is largely drawn from a series of case reports involving large doses, typically in excess of 2 g/d, taken over many months to years (6). The case reports found that excessive PN intake led to progressive sensory peripheral neuropathy. Neurological symptoms included paresthesia, hyperesthesia, bone pains, muscle weakness, numbness, fasciculations, and loss of tendon reflexes. Symptoms primarily followed a stocking-glove distribution, although there are several reports of facial numbness and mild autonomic dysfunction (7, 9, 48–51). The initial symptoms typically involved paresthesia in the toes, and gradually progressed into symptoms related to sensory ataxia including gait, loss of balance and difficulty handling small objects. At these stages, sense of touch, temperature, and pain were typically less affected than the other modalities (7, 48). Electrophysiological studies found absent distal sensory nerve conduction action potentials with mild to preserved motor involvement. Somatosensory evoked potential studies found a severe to absent impairment in proximal nerve conduction. Sural nerve biopsy showed widespread axonal degeneration of both large- and small-fiber nerves. Following withdrawal from PN, symptoms often progressed before gradually improving. Dramatic improvements were usually observed within ∼6 mo; however, distal sensory perception was often diminished. During recovery, loss of vibrational sense persisted longer than with the other modalities. In those who were followed up, patients were almost fully recovered within 2–3 y (6, 7, 48).
These clinical observations were confirmed by a controlled study that investigated the dose-response of 12–56.9ímg PN·kg−¹·d−¹ in 5 healthy men. Within the study, the onset of symptoms was associated with the dose administered. The volunteer, receiving 56.9 mg PN·kg−¹·d−¹, complained of symptoms within 1.5 mo, while the participant, receiving the lowest dose of 12 mg PN·kg−¹·d−¹, had symptoms within 7 mo. In all of the volunteers with complaints, the severity of the symptoms, particularly paresthesia, worsened for 2–3 wk after discontinuing PN. In the volunteers receiving 1 g/d, the predominant symptom involved reduced thermal sensory thresholds, particularly in the toes. In contrast, the volunteers receiving 3 g/d had concurrent and equal vibrational and thermal sensory threshold reductions. At all doses, QST abnormalities preceded changes in nerve-conduction studies. These findings strongly support a dose-dependent relation for both the onset and vulnerability of different caliber axons in PN toxicity. Additionally, it indicates that QST testing may be more sensitive in detecting early changes in PN-induced neuropathy (10).
Dose–response relation
The minimum dose and treatment duration necessary to elicit neuropathy has not been firmly established (6). While many isolated case reports describe neuropathy in patients taking PN at doses well below 500 mg/d, PN toxicity at these doses may not be fully supported by the controlled studies. Multiple controlled studies found that PN doses of 100–500 mg/d were well tolerated; however, the duration of treatment was likely too short to assess neurological effects (6). Additional studies found that PN doses of 200–500 mg/d taken for several years did not lead to neurological symptoms. Mitwalli et al. (52) found that PN doses of 250–500 mg/d for 8 mo to 6 y produced no neurological effects in 22 patients treated for hyperoxaluria (52). Another study found that PN doses of 200–500 mg/d administered from birth for 7–24 y were well tolerated in 17 patients treated for homocystinuria (53). However, these studies may not be applicable to the general population because of possible interactions with the underlying health conditions. Furthermore, these observational studies were not subject to double-blind, placebo-controlled evaluation, and were limited by insufficient monitoring of PN intake, small group size, and inadequate assessment of adverse effects (6).
A number of more recent case reports have described symptoms of neuropathy in people taking PN at doses near or even below the EFSA UL of 25 mg/d. In 2005, de Kruijk and Notermans (54) reported on 2 patients who developed symptoms of sensory neuropathy after taking PN doses of 24 and 40 mg/d, who recovered following discontinuation. Another case report, involving a patient who self-administered 30 mg PN/d, confirmed a case of sensory ataxic ganglionopathy using electrophysiological studies. However, in this case, high consumption of PN-containing energy drinks may have been a contributing factor (55). Additionally, from 1991 until July 2017, the Netherlands Pharmacovigilance Centre Lareb received 90 reports of vitamin B-6-related neuropathy with doses ranging from 1.4 to 100 mg/tablet (14, 15). The most common symptoms involved numbness and paresthesia, which is consistent with the early symptoms described in previous case reports. In 82 of the cases, there was an observed latency in response. In 30 cases, symptoms diminished following discontinuation; however, in 29 cases, symptoms did not improve. A major limitation of this case series was the lack of information on PN intake. Vitamin B-6 content in products and whether the patient took more than the advised dosage could not be verified. Plasma vitamin B-6 concentrations were known in only 36 cases and dosage was only known in 37 cases. Given the inherent limitations of isolated case reports, it is not possible to infer a causal relation at these low doses (15). These reports do, however, indicate the possibility of significant interindividual differences in sensitivity to PN toxicity and toxicity at doses <25 mg/d within vulnerable individuals.
Animal studies
PN-induced neuropathy has been observed in many animals, including dogs, rats, mice, cats, chickens, rabbits, and guinea pigs (56–60). The dose–response and associated histopathological changes have been closely monitored in rats and dogs. In rats, 3 intraperitoneal dosing regimens were generally used including 1200 mg·kg−¹·d−¹ for 1–15 d, 600 mg·kg−¹·d−¹ for 1–15 d, and 100–300 mg·kg−¹·d−¹ for up to 12 wk (6, 61). In rats receiving acutely toxic doses, the expression of sensory ataxia was dependent on the cumulative dose. Symptoms of ataxia and neuronal necrosis manifested when the total PN dose reached about 4 g/kg. At doses of 300 mg·kg−¹·d−¹ or less, symptoms of ataxia were not observed even when the cumulative dose exceeded 25 g/kg. At doses of 300 mg·kg−¹·d−¹ and over, the initial changes involved damage to the cell bodies of neurons, while lower doses were initially characterized by axonal atrophy. At all doses, the initial changes occurred at the dorsal root ganglia (DRG) neurons in the L4–5 spinal regions. This was followed by secondary damage to their central and peripheral processes within the spinal nerves and spinal cord while sparing the ventral and lateral regions. Additionally, the nerve fibers derived from the L4–5 spinal region were damaged, including the sciatic, tibial, sural, and plantar nerves. At doses <200 mg·kg−¹·d−¹ over 12 wk, damage was limited to DRG axons and the endings of long nerve fibers (61, 62).
In contrast to rats, both acute and subacute toxicity produced proprioceptive deficits in dogs. Dogs receiving oral doses of 100–300 mg·kg−¹·d−¹ developed proprioceptive abnormalities in the hindquarters within 9–100 d. Tissue examination found extensive neuronal cell body degeneration in the cervical, thoracic, and lumbar DRG regions. Additionally, there was damage present at sensory nerve fibers, the descending spinal tract of the trigeminal nerve, dorsal roots, and the dorsal horn and funiculus of the spinal cord. Adjacent regions of the spinal cord were unaffected (56, 61, 63). Dogs receiving 50 mg·kg−¹·d−¹ over 100 d did not develop any observable clinical sequelae; however, modest histopathological changes were detected at the dorsal funiculus (63). Dogs receiving PN have a similar anatomical distribution and histopathological response as rats. The difference in sensitivity to PN toxicity between rats and dogs may be related to excretion. Rats were observed to excrete PN primarily in its unchanged form while, in dogs, 4-PA was the predominant metabolite in urine, as with humans (63, 64).
Compared with humans, animals were less sensitive to PN toxicity. Additionally, many of the animal studies found that PN preferentially targets large-diameter nerve fibers. Large-sized DRG neurons were reportedly more affected than other neurons in both dogs and rats (50, 65–67). At lower doses in humans, small-fiber deficits are the earliest and predominant indicator. This could be related to the dose-dependent vulnerability of different caliber axons (10). Most of the animal studies did not use diagnostic methods related to small-fiber dysfunction such as QST or skin biopsy. Use of these methods may have resulted in earlier detection of PN-induced neuropathy at lower doses. Significant proprioceptive deficits were not present in humans receiving doses of 56.9 mg·kg−¹·d−¹ over 1.5 mo (10). However, thermal latency response was reportedly less sensitive than proprioceptive changes in rats receiving 800 mg·kg−¹·d−¹ (68). Presently, it is unclear if larger-diameter nerve fibers are more vulnerable in animals than in humans. At later progressions in humans, small-fiber modalities are similarly less affected than positional or vibrational sense (7, 48). Considering that low-dose chronic exposure in animals consistently led to axonal damage, it is unlikely that these changes would not lead to clinical manifestations. There are, however, notable differences for a variety of DRG pain-processing and/or drug targets between rodents and humans (69–72). These differences may be relevant to the toxicological activity of PN. Clarification of the toxicological target sites and confirmation of target homology between animal models and humans may be needed to assess the relevance of animal studies to humans.
Classification
While there may be significant differences with humans, histopathological analysis of animal models is likely relevant. PN-induced neuropathies in animals share a common anatomical distribution and histopathological response as humans. Peripheral neuropathies are often classified as either myelinopathy, axonopathy, or neuronopathy. This refers to the primary site of damage (73). There is an indication that low levels of exposure to PN led to axonopathy with retrograde degeneration of distal axons. On the other hand, higher doses led to neuronopathy with necrosis of DRG neuronal cell bodies and secondary damage to nerve fibers. Morphometric analysis found that PN lesions were characterized by an increase in the total number of myelinated axons and significant decreases in the mean axon area and nerve fiber densities (65). Both chronic and acute PN intoxication involved nuclear and cytoplasmic aberrations in DRG neurons. The intensity of neuronal cell body alterations was dose dependent. This supports the view that axonal degeneration is a secondary effect from sublethal injury to neuronal cell bodies (5, 74, 75). Ultra-structurally, axonal degeneration was accompanied with reductions in cytoskeleton proteins, chromatolysis, mitochondria and satellite cell proliferations, and enlarged astrocytic processes (76). This is characteristic of Wallerian-type degeneration (5), which is a programmed axon death pathway and a common reaction to a large variety of insults (77).
These neuropathological changes and anatomical distribution are consistent with the majority of peripheral neurotoxicants and likely a consequence of the physiology of the sensory ganglia. While the peripheral nervous system (PNS) generally has a tight blood–nerve barrier, the DRG is densely vasculated and has a comparatively high blood-perfusion rate (78). Blood vessels in the cell body-rich area of the DRG have large fenestrations and fewer tight junction proteins. The length of sensory nerve fibers is proportional to the capillary surface area of the DRG they are derived from. The largest DRGs at the S1, L4–5, and C7–8 levels support the longest nerve fibers in the body (72). As a consequence, neurotoxic compounds, such as PN, preferentially accumulate and injure long primary sensory neurons (79). This leads to a predictable stocking-glove anatomical distribution. The extended lengths of DRG neurons make them particularly reliant on axoplasmic transport to meet the physiological requirements of distal axons. Axoplasmic transport is an energy- and resource-intensive process. Sublethal exposure to a neurotoxicant disrupts essential cellular processes, thereby interfering with axoplasmic transport and leading to a “dying back” of axons (80).
Toxicological screening of vitamin B-6 vitamers
There is a lack of consensus over which vitamin B-6 vitamer(s) lead to the above-described processes resulting in neuropathy. PLP is highly reactive, and along with PN has been proposed as a possible causative agent in PN neurotoxicity. Several pyridine-based compounds are neurotoxic, and previously it was suggested that the neurotoxicity of these compounds may be structurally dependent (5). In 1976, Frater-Schroder et al. (81) tested the effects of PM, PL, PN, and 7 other synthetic vitamin B-6 analogs in rats. Only PN and PL elicited the characteristic histopathological changes. A more recent study found that PN exclusively led to the morphological changes associated with PN-induced neuropathy. Rats injected with high doses of PM, PL, and PLP did not show an effect on the nerve fibers examined (82).
However, the specificity of PN toxicity was not settled by the in vitro toxicological assays. Windebank (18) tested the effects of PN, PL, PM, PMP, and 4-PA on the neurite outgrowth of rat DRG cells. The study found that PN, PL, and PM all inhibited neurite outgrowth at similar concentrations, between 0.1 and 1 mM, and had no effect at lower concentrations. The other compounds, 4-PA and PMP, produced no ill effects even at concentrations as high as 10 mM (18).
In contrast, Vrolijk et al. (16) found that PN, already at concentrations as low as 200 nM, increased cell death of SH-SY5Y neuroblastoma cells by 17%. At concentrations of 400 nM PN, cell death increased to 30%. Subsequent increases of PN, up to 5 μM, did not induce further increases in the cell death rate. Remarkably, at concentrations of 5 μM the other vitamers had no effect on cell viability. In Caco-2 cells, various concentrations of PN had no effect on cell viability, suggesting that PN is specifically toxic to neurons (16). The lack of increased toxicity at higher concentrations of PN can be explained by the fact that SH-SY5Y cells are observed to have variable PNPO activity. PNPO activity status may have an impact on cell proliferation, where some PNPO-positive variants were observed to divide more slowly. Using a culture medium supplemented with PN could select against PNPO-negative cells (83). The study design by Vrolijk et al. (16) used the DMEM medium, which contains mostly PL, and would have failed to select against PNPO negative variants. Even with higher concentrations, PN failed to increase the cell death rate over 30%. This may be because PN supplementation induced cell death in PNPO-negative variants, and selected for PNPO-positive cells, which may be then resistant to higher concentrations of PN. Possibly, cell death rates would have been higher when only PNPO-negative cells would have been selected. In general, cell culture experiments may not adequately model the underlying conditions necessary for PN-induced neuropathy. Thus, the effects observed in rats in vivo are the more reliable indicators.
The neurotoxicity of PN at low-dose chronic exposure is difficult to reconcile with the pharmacokinetics of PN, as it is normally rapidly eliminated (24, 84, 85). This implies that either brief intervals of exposure to PN are toxic to sensory neurons over the long term or that PN can accumulate following daily supplementation. The first case is unlikely considering that peripheral neurons readily regenerate and have a high capacity for recovery from injury. Daily supplementation with PN over an extended period could potentially alter the half-life of PN and lead to its accumulation. If the accumulation of PN is necessary to elicit neuropathy, then PN-induced neuropathic patients would be expected to have elevated concentrations of PN. At least 1 study found an association between elevated plasma PN concentrations and symptoms of neuropathy. Plasma PN values ranged from 140 to 1050 nmol/L. The majority of patients reported daily vitamin use and, as a result, elevated PN plasma concentrations are not necessarily indicative of PN accumulation. Patients may have consumed PN supplements shortly before blood samples were taken (86). In general, the time course of plasma vitamin B-6 vitamers has not been thoroughly investigated in patients with PN-induced neuropathy. Therefore, it is useful to consider the effects of daily supplementation on vitamin B-6 vitamer concentrations.
Effects of daily oral pyridoxine on plasma vitamin B-6 vitamer concentrations
Several pharmacokinetic studies that are representative of daily PN supplement use have been conducted and are summarized on Table 1. In general, the pharmacokinetic studies on daily PN supplementation reported consistent results. Edwards et al. (87) found that PLP concentrations did not increase with higher doses and this was corroborated by the other studies, where PLP concentrations ranged from 300 to 400 nmol/L. Similarly, 4-PA and PL concentrations were comparable to each other in all of the studies. Excluding the study by Speitling et al. (85), there was considerable interindividual variation for all of the vitamin B-6 vitamers measured. Significant outliers for 4-PA, PL, and PN were reported and PN concentrations were even found to be non–normally distributed.
TABLE 1.
Effects of daily oral pyridoxine on plasma vitamin B-6 vitamer concentrations in humans1
| Dose, mg/d | Participants, n | Supplementation Period, wk | Plasma concentration, nmol/L | |||||
|---|---|---|---|---|---|---|---|---|
| Study2 | PL | PLP | PN | PNP | 4-PA | |||
| Edwards et al. (87) | 10 | 6 | 1 | 473 ± 333 | 595 ± 287 | ND | ND | 295 ± 104 |
| Edwards et al. (87) | 25 | 9 | 1 | 231 ± 157 | 631 ± 158 | ND | ND | 371 ± 175 |
| Edwards et al. (87) | 100 | 9 | 1 | 1277 ± 678 | 518 ± 130 | 268 | 350 ± 60 | 1239 ± 464 |
| Edwards et al. (87) | 200 | 9 | 1 | 2441 ± 904 | 623 ± 138 | 124 | 269 ± 213 | 2200 ± 633 |
| Edwards et al. (87) | 400 | 8 | 1 | 4764 ± 1664 | 732 ± 202 | 389 | 120 | 3717 ± 922 |
| Edwards et al. (87) | 800 | 7 | 1 | 9484 ± 1616 | 644 ± 182 | 4664 | 245 | 7085 ± 1528 |
| Bor et al. (88) | 40 | 43 | 12 | 700 | 350 | 100 | NM | 1200 |
| Vrolijk et al. (14) | 50 | 6 | 1 | 1200 | 400 | 400 | NM | NM |
| Speitling et al. (85) | 300 | 10 | 2 | 140 | 330 | ND | NM | 190 |
ND, not detected; NM, not measured; PL, pyridoxal; PLP, pyridoxal 5′-phosphate; PN, pyridoxine; PNP, pyridoxine 5′-phosphate; 4-PA, 4-pyridoxic acid.
In the Edwards et al. (87) study, values are means ± SDs for all vitamin B-6 vitamers except for PN with all doses and for PNP with 400 and 800 mg/d, where medians are used. In the other studies, values are means for all vitamin B-6 vitamers.
The large interindividual variations in plasma vitamin B-6 vitamer concentrations were likely related to differences in the timing of blood samples, lack of control in the duration since the last dose, and differences in the rate at which PN is metabolized. Most studies did not wait longer than 4 h to draw blood samples or did not explicitly control for the time duration since the last dose. Speitling et al. (85) found that the rate at which PN was completely eliminated during a single dose varied between 6 and 13 h. Similarly, in the Vrolijk et al. (14) study, the maximum concentration for PN after a single dose ranged from 200 to 1687 nmol/L. At 4 h, PN concentrations could still be significant in individuals with slower PN metabolism, and the measured plasma vitamin B-6 vitamer concentrations would reflect a brief time point during rapid changes in the mass balances of PN metabolites. Small differences in the kinetic and/or expression parameters of all the enzymes involved in PN metabolism could lead to large variations in the metabolic flux of the vitamin B-6 vitamers.
Speitling et al. (85), the only study that used fasting blood samples, found that elevated steady-state concentrations were achieved for 4-PA, PLP, and PL and that PN did not accumulate. In comparison to the other studies, this study reported lower values for 4-PA and PL relative to the dose administered and found that PLP had the highest steady-state concentration. The values reported by Speitling et al. are likely more representative of exposure levels from daily PN supplementation because they are less dependent on the timing of blood samples. The collection of fasting samples would likely significantly lower interindividual variation.
The findings by Vrolijk et al. (14) do, however, suggest that, in some individuals, the clearance of PN and PL is delayed following daily PN administration. Three of the 6 individuals studied had increased PN concentrations at days 3 and 7. In 1 individual, the PN concentration reached 1023 nmol/L at day 7. This likely cannot be attributed to the lack of control in the timing of blood samples considering that, in the same volunteer, plasma concentrations for PN following a single dose fell to 211 nmol/L after 90 min. All of the volunteers with elevated PN concentrations at day 7 had concurrently high concentrations of PL. In 1 volunteer, days 3 and 7 PL concentrations exceeded the maximum concentration achieved during the single dose. Due to the lack of control in the time duration between dosing and sample collection, it is difficult to clearly evaluate these results. However, these findings indicate that, in some individuals, daily PN supplementation may extend the half-life of PN.
The delay in PL/PN elimination and increases in their peak concentration following daily PN administration was likely related to saturation of PDXK or PNPO. It is unclear if, in these individuals, the delay in PL/PN elimination would reach a new equilibrium or if the plasma time course of PL/PN would steadily increase. The study by Bor et al. (88) found that vitamin B-6 vitamer concentrations remained consistent over 84 d, which suggests that, under normal dosing conditions, the delay in PL/PN elimination is not significant. There is insufficient evidence to suggest that high PN plasma concentrations are maintained throughout the day following daily PN supplementation. However, over more extended time periods of administration and higher doses this may change.
The possibility remains that, under toxic dosing conditions, the steady-state concentration for the vitamin B-6 vitamers is altered. Through feedback inhibition and enzyme saturation, supraphysiological concentrations of PN and its metabolites could lead to gradual decreases in the activities of enzymes involved in PN metabolism. Both PDXK and PNPO were shown to be inhibited by their product PLP (89, 90). Gradual decreases in the activities of enzymes involved in PN metabolism could steadily delay the elimination of PN until elevated PN concentrations are retained up to the next dose. Additionally, enzymatic reduction in PL/PLP to PN/PNP by aldo-keto reductases (AKRs) may similarly lead to PN accumulation (28).
Mechanisms for toxicity
When considering mechanisms in toxic neuropathies, it is often difficult to identify a specific primary event or molecular target that can account for all of the observed neuropathological changes. Histopathological analysis may not be informative as many of the observed changes are likely secondary to an initial causal event. For most peripheral neurotoxicants, several plausible mechanisms exist that are not necessarily mutually exclusive (80). In the case of PN-induced neuropathy, observations in PN pharmacokinetics, vitamin B-6 vitamer toxicological testing, and tissue specificity of PN toxicity may be helpful in assessing the plausibility of the existing interpretations. Previously proposed mechanisms include the formation of reactive quinone methide, aldehyde toxicity through elevated PL/PLP concentrations, inhibition of PLP-dependent enzymes, and PDXK inhibition (4, 16–19).
Aldehyde toxicity by PLP
As a highly reactive electrophile, the 4′ aldehyde of PLP is attractive as a possible toxicological agent. The aldehyde of PL is less relevant as it is present almost entirely in its hemiacetal form at neutral pH (91). Many electrophilic compounds are neurotoxic. Electrophiles can form covalent adducts with macromolecules that impair the functional activity of enzymes, cytoskeleton proteins, and DNA, and induce cytotoxicity. In fact, the potential of an electrophile to form adducts with biomolecules is highly predictive of its neurotoxicity (92). However, there are several cellular mechanisms that prevent the buildup of PLP. Pharmacokinetic studies have shown that plasma PLP concentrations are independent of the PN dose and likely depend on protein-binding availability. The maximum plasma PLP concentrations are already achieved by relatively low daily PN doses, generally regarded as safe. Considering that PN toxicity is dose dependent, PN is the more likely candidate. Moreover, injections with high doses of PLP in rats failed to induce the morphological changes associated with PN-induced neuropathy (82).
Quinone methide-type intermediates
Due to their highly electrophilic reactivity and capacity to alkylate biomolecules, quinone methides could plausibly lead to the toxicological effects seen with PN toxicity. The formation of reactive quinone methide-type intermediates has been demonstrated following the irradiation of PN in aqueous solution at neutral pH (93). Previous studies have proposed that quinone methide-type intermediates may also form in vivo through interactions with an enzyme (94). However, this has not been thoroughly investigated. Moreover, quinone methide-type intermediates would be expected to have more broad toxicological activity and not specifically target neurons. The formation of quinone methide-type intermediates may explain PN phototoxicity and symptoms related to dermatitis and xeroderma pigmentosum. In fact, cells from PN-induced xeroderma pigmentosum patients showed similar survival curves as fibroblasts treated with irradiated PN (95). Unless there is a specific biological process within sensory neurons that generates quinone methide-type intermediates, it is unlikely that quinone methide-type molecules are responsible for PN neurotoxicity.
Inhibition of PLP-dependent enzymes
The clinical presentation of PN toxicity shares a few similarities with a vitamin B-6 deficiency. Both are associated with symptoms related to dermatitis and peripheral neuropathy. Multiple authors have proposed that elevated concentrations of PN could paradoxically lead to a vitamin B-6 deficiency by competitively inhibiting PLP (16, 17). Vrolijk et al. (16) found that PN inhibited the PLP-dependent enzymes tyrosine decarboxylase and alanine transaminase by 65% and 40%, respectively. Additionally, Speitling et al. (85) found that blood aspartate aminotransferase was most strongly inhibited at peak PN plasma concentrations. The mechanism by which PN inhibits PLP-dependent enzymes has not been clarified. PLP-dependent enzymes bind to PLP through the formation of a Schiff base with lysine residues of the protein and the aldehyde at the 4′ position of PLP. PN lacks an aldehyde, and as a result cannot directly occupy the PLP binding site. PN can, however, form coordination complexes with PL/PLP, amino acids, and metal ions. These coordination complexes could plausibly inhibit PLP-dependent enzymes (96). The study by Vrolijk et al. (16) did not determine the kinetic rate constants for PN inhibition, and it is unknown to what extent inhibition of PLP-dependent enzymes would occur at physiologically relevant concentrations.
However, there are several indications that vitamin B-6 function is retained following high doses of PN. The tryptophan load test, a sensitive index for vitamin B-6 status, found no differences between rats receiving PN doses of 1400 mg·kg−¹·d−¹ over 6 wk and controls (97). Moreover, a vitamin B-6 deficiency leads to several symptoms that are not observed in PN toxicity. The most common symptoms in humans include anemia, seborrheic dermatitis, glossitis, electroencephalographic abnormalities, depression and confusion, neutropenia, and weakened immune function. In experimental animal models, a vitamin B-6 deficiency induced similar changes, including a lowered growth rate, seborrheic dermatitis, acrodynia, convulsions, neurodegeneration in the brain, weakened immune function, anemia, atherosclerosis, weakness, lethargy, paralysis, alterations in tryptophan and alanine metabolism, and elevations in urinary creatinine and xanthurenic acid (39, 98). These effects have been observed in monkeys, rabbits, chickens, dogs, rats, and pigs (99–103). If the inhibition of PLP-dependent enzymes were responsible for toxicity, one would expect more systemic effects. Even in animal models receiving high doses, PN toxicity preferentially targets sensory neurons, and symptoms possibly related to a systemic PLP-dependent enzyme inhibition, such as anemia, are not observed. While symptoms of dermatitis have been reported with PN toxicity, there is evidence that this is related to a phototoxic reaction involving the formation of quinone methide-type intermediates (95, 104, 105), although the possibility of disrupted vitamin B-6 function cannot be dismissed.
In animal models of dietary vitamin B-6 deficiency, symptoms related to peripheral neuropathy are uncommon but have been observed in pigs and rats (106, 107). A vitamin B-6 deficiency in pigs led to the development of an ataxic gait and degeneration of sensory nerve fibers (106). Rats developed abnormal walking patterns and neuronal degeneration in the tibial nerve. Morphometric analysis characterized the lesion as a myelinopathy. There was a significant decrease in the mean myelin-axon ratio, while changes in the mean axon or nerve fiber diameter were not observed. This is distinct from the morphological changes associated with PN toxicity in rats, characterized as an axonopathy with significant increases in the mean myelin-axon ratio (65, 107).
In humans, there is insufficient evidence to support a link between a dietary vitamin B-6 deficiency and peripheral neuropathy. There are a few isolated case reports, but these were likely related to other underlying health conditions (107, 107). Previously, it was suggested that demyelinating peripheral neuropathy in elderly patients on chronic peritoneal dialysis may have been caused by a vitamin B-6 deficiency (108). However, peripheral neuropathy in patients on dialysis has also been attributed to uremic intoxication, hormonal imbalances, disruptions in ion concentration gradients, and/or other vitamin deficiencies (108–112). In rhesus monkeys, the animal model most relevant to humans, a vitamin B-6 deficiency did not induce neuropathy (99, 113, 114). Most of the evidence in favor of an association between vitamin B-6 deficiency and neuropathy in humans is related to pharmaceutical interventions (115). In a study involving the administration of the vitamin B-6 antagonist and PDXK inhibitor 4-deoxypyridoxine, 3 out of 50 people complained of sensory symptoms. The most common symptoms included nausea, listlessness, lethargy, dermatitis, cheilosis and conjunctivitis, glossitis, and anemia. Beyond patient complaints, evidence for peripheral neuropathy was not further investigated (116). There are no reports of neuropathy in animals exposed to 4-deoxypyridoxine and it primarily caused dermatitis, anemia, and seizures (117, 118). The antituberculosis drug isoniazid depletes vitamin B-6 and induces peripheral neuropathy at conventional doses and seizures from overdose in humans. Additional adverse reactions include hepatitis, dermatitis, fever, angiitis, anemia, autonomic neuropathy, and optic atrophy (119). Isoniazid disrupts vitamin B-6 activity by forming hydrazine complexes that inactivate PL/PLP. These complexes inhibit PDXK and lead to reductions in PLP formation (120). PN supplementation is effective in preventing and treating symptoms related to neuropathy, seizures, dermatitis, and anemia (121). Morphometric analysis determined that isoniazid-induced neuropathy in rats was characterized as an axonopathy and distinct from the myelinopathy associated with a vitamin B-6 deficiency (107, 122–124). While isoniazid-induced neuropathy is responsive to vitamin B-6 supplementation, it may not be directly caused by a vitamin B-6 deficiency and could be related to PDXK inhibition.
PDXK inhibition
While one would expect more systemic effects, a PDXK mutation leads to the preferential injury of sensory neurons with similar symptoms as with PN-induced neuropathy. A PDXK mutation causes progressive, length-dependent, sensorimotor, predominantly axonal neuropathy with late-onset optic atrophy. Kinetic studies of the recombinant mutant enzyme found significant changes in the maximal rate (Vmax) and the Michaelis-Menten constant (Km) (Wild type: Km = 53.4 μmol/L, Vmax = 16.8 pmol/h; mutant: Km = 174.4 μmol/L, Vmax = 6.3 pmol/h). Plasma from affected patients showed a PLP deficiency with values ranging between 7.8 and 10.8 nmol/L (reference value: >30 nmol/L) (3). Clinical symptoms, such as gait or neuropathic pain, improved upon supplementation with PLP; however, electrophysiological studies found no progression even after 18 mo (47).
Several inherited genetic disorders related to vitamin B-6 metabolism are known to cause neurological symptoms. Inborn errors in PNPO, ALPL, PLPBP, and ALDH7A1 are linked to early-onset vitamin B-6-dependent epilepsy, which is thought to be related to disruptions in γ-aminobutyric acid (GABA) transmission (40–45). The mechanism linking PDXK deficiency to neuropathy is, however, unknown (47). Compounds that inhibit PDXK lead to convulsions and were shown to inhibit GABA biosynthesis and GABAergic neurotransmission (125, 126). Thus, there is reason to believe that PDXK-induced neuropathy may also be linked to GABA metabolism. The absence of central nervous system (CNS)-related symptoms in PDXK deficiency could be due to tissue-specific differences in the regulation of PDXK gene expression and activity levels. Interestingly, a vitamin B-6 deficiency in rats led to significant reductions in PDXK activity within the peripheries, while in the brain decreases were not as marked (127). Hence, reductions in PDXK activity, when confined to the peripheries, may lead to sensory neuropathy.
The neurological effects of PDXK inhibitors appear to be related to CNS bioavailability. At physiologically relevant concentrations, the 3 most potent PDXK inhibitors are ginkgotoxin, isoniazid, and theophylline (120, 128). Both ginkgotoxin and theophylline have a relatively high blood–brain barrier permeability and are primarily associated with CNS disturbances (129, 130). In contrast, isoniazid is associated with peripheral neuropathy at therapeutic doses and CNS disturbances at high doses. Log-P values determined for isoniazid (−0.64), ginkgotoxin (−0.229), and theophylline (−0.02) indicate that isoniazid is less lipophilic and likely has lower CNS permeability (128, 131, 132).
PN has a higher affinity toward PDXK compared with its other substrates and may act as a competitive inhibitor of the enzyme's capacity to convert PL to PLP (128). The degree of PDXK inhibition depends on the ratio of PL to PN (Figure 2) (133). Log-P values determined for PN (−1.10) indicate that PN is relatively impermeable across the blood–brain barrier (134). In rats fed high doses of PN, concentrations in the brain were indeed not increased significantly (97). In dogs fed 200 mg PN·kg−¹·d−¹, PN concentrations were slightly elevated in some, but not all, brain samples (63). Thus, PDXK inhibition by PN would likely be confined to the peripheries as with a PDXK deficiency.
FIGURE 2.
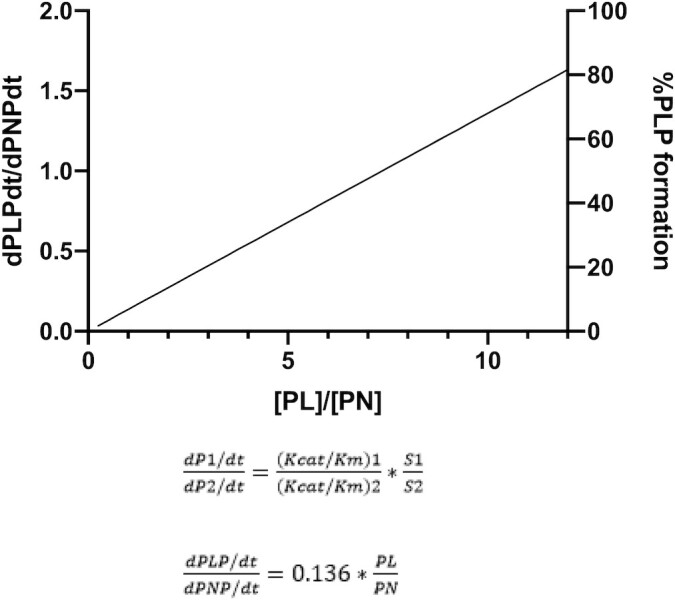
The ratio of reaction rates for alternative substrates when, in a common reaction, volume is proportional to the ratio of the substrate concentrations and their kcat/Km values. The ratio of kcat/Km of PL ( ) to kcat/Km of PN (
) to kcat/Km of PN ( ) predicts that PNP formation will be favored over PLP formation at PL-to-PN ratios <7.5. For a similar level of inhibition as the PDXK deficiency (37.5% capacity of WT Vmax), a PL:PN of 5.5 is necessary (47, 128, 133). dPLPdt/dPNPdt, the time derivative of PLP concentration over the time derivative of PNP concentration; kcat, catalytic constant; Km, Michaelis Menten constant; PDXK, pyridoxal kinase; PL, pyridoxal; PLP, pyridoxal 5′-phosphate; PN, pyridoxine; PNP, pyridoxine-5'-phosphate; Vmax, maximal rate; WT, wild type.
) predicts that PNP formation will be favored over PLP formation at PL-to-PN ratios <7.5. For a similar level of inhibition as the PDXK deficiency (37.5% capacity of WT Vmax), a PL:PN of 5.5 is necessary (47, 128, 133). dPLPdt/dPNPdt, the time derivative of PLP concentration over the time derivative of PNP concentration; kcat, catalytic constant; Km, Michaelis Menten constant; PDXK, pyridoxal kinase; PL, pyridoxal; PLP, pyridoxal 5′-phosphate; PN, pyridoxine; PNP, pyridoxine-5'-phosphate; Vmax, maximal rate; WT, wild type.
However, there is an important difference between PN toxicity and conditions of PDXK deficiency or inhibition. Both PDXK deficiency and isoniazid-induced neuropathy were responsive to vitamin B-6 treatment. Similarly, convulsions from ginkgotoxin coincided with an acute decrease in PLP concentrations (135, 136). This suggests that the neurotoxic effect of reduced PDXK activity is contingent on PLP depletion. High doses of PN are associated with significant elevations in plasma PLP concentrations. Moreover, PN toxicity is not associated with a systemic vitamin B-6 deficiency. Thus, PLP is likely bioavailable in most tissues.
There is evidence for large variation in the relative concentrations of vitamin B-6 vitamers across different tissues following high doses of PN. The distributions of vitamin B-6 vitamers in rats receiving 1400 mg·kg−¹·d−¹ and 7 mg·kg−¹·d−¹ over 6 wk are shown in Table 2. Generally, high doses of PN did not have an impact on PLP concentrations. As an exception, PLP concentrations were significantly elevated in RBCs. Interestingly, PLP concentrations decreased in the gastrocnemius. At all doses, PL concentrations were significantly higher than PN in most of the tissues examined, except for the kidney with high doses of PN and the gastrocnemius at all doses. The observed decrease in PLP within the gastrocnemius may have been related to both low PNPO activity and low PL-to-PN ratios. At low PL-to-PN ratios, PLP formation by PDXK is likely inhibited. Additionally, rats were observed to have low PNPO activity in skeletal muscle. In contrast, PNPO activity was significantly higher in the kidney and, as a result, PLP concentrations may have been less impacted by PDXK inhibition (37). Thus, high concentrations of PN may lead to a depletion in PLP via PDXK inhibition in tissues with low PNPO activity. As a result, tissues with low PNPO concentrations and enzyme activity favorable to a low PL:PN would be expected to be more susceptible to PN toxicity.
TABLE 2.
Tissue vitamin B-6 vitamer concentrations in rats fed 7 mg·kg−¹·d−¹ and 1400 mg·kg−¹·d−¹ for 6 wk1
| PN treatment, mg·kg−¹·d−¹ | Tissue concentration, nmol/g | PL-to-PN concentration ratio2 | ||||||
|---|---|---|---|---|---|---|---|---|
| Tissue | PLP | PMP | PL | PN | PM | 4-PA | ||
| Plasma3 | 7 | 588 (41) | 6.5 (1.3) | 685 (29) | 30.1 (4.4) | 16.5 (1.7) | 16.4 (6.1) | 22.76 |
| 1400 | 634 (52) | 9.1 (0.7)* | 4593 (269)*** | 1375.5 (308.33)*** | 38.2 (2.5)*** | 734.6 (92.8)*** | 3.34 | |
| RBCs3 | 7 | 203 (10) | 104.1 (3.3) | 939 (76) | 2.1 (2.1) | 3.9 (2) | ND | 447.14 |
| 1400 | 947 (43)*** | 100.6 (9.1) | 21,502 (1186)*** | 47.1 (47.1) | 7 (5.2) | 13.8 (8.7) | 456.52 | |
| Gastrocnemius4 | 7 | 14.9 (1.4) | 8.1 (0.5) | 0.12 (0.05) | 0.27 (0.07) | 0.49 (0.16) | ND | 0.44 |
| 1400 | 9.7 (0.8)*** | 6.9 (1.1)Ɨ | 2.47 (0.37)*** | 2.23 (0.77)ǂ | 0.8 (0.32) | ND | 1.11 | |
| Brain | 7 | 7.57 (0.4) | 11.3 (0.5) | 1.06 (0.33) | 0.15 (0.05) | 0.11 (0.04) | 0.25 (0.1) | 7.07 |
| 1400 | 7.32 (1.29) | 10.2 (0.7) | 3.81 (0.39)*** | 0.2 (0.07) | 0.14 (0.02) | 0.38 (0.16) | 19.05 | |
| Liver | 7 | 24.5 (1.9) | 25.1 (1.1) | 1 (0.3) | 0.31 (0.09) | 0.3 (0.17) | 0.92 (0.52) | 3.23 |
| 1400 | 24.4 (1.7) | 23.5 (0.8) | 2.6 (0.5)** | 0.38 (0.08) | 0.29 (0.14) | 0.71 (0.37) | 6.84 | |
| Kidney | 7 | 9.3 (1.3) | 28.4 (0.8) | 3.7 (0.4) | 0.27 (0.12) | 13.9 (0.6) | ND | 13.7 |
| 1400 | 10 (1.0) | 28.1 (1.9) | 13.8 (1.9)*** | 7.66 (2.34)** | 14.9 (1.9) | ND | 1.8 | |
Data were obtained from Schaeffer et al. (97). All values are means (SEM), n = 5–6, except for plasma and RBCs (SD), for which n = 11–12. *,**,***Different between diet groups within a tissue: *P < 0.11, **0.01 < P < 0.05, ***P < 0.01. ND, not detected; PL, pyridoxal; PLP, pyridoxal 5′-phosphate; PM, pyridoxamine; PMP, pyridoxamine 5′-phosphate; PN, pyridoxine; PNP, pyridoxine 5′-phosphate; 4-PA, 4-pyridoxic acid.
The concentration ratios of PL to PN were calculated using mean values.
Concentration values in plasma and RBCs are given in nmol/L.
Tissue concentrations are based on analysis of the right gastrocnemius only: ƗP = 0.1, ǂP = 0.06.
Factors influencing tissue-specific differences in PL:PN
The relative activities of PNPO and PDXK likely play a determining role in the ratio of PL to PN within specific tissues. Additionally, differences in the uptake of PN may contribute to this. There is evidence that PN uptake is mediated by both passive and facilitated diffusion. PN uptake is a saturable process that may be regulated at the transcriptional level and may vary between cell types (137). Currently, the uptake of PN in cells is not well understood. Tissues with higher activity in enzymes that oxidize PL, such as aldehyde oxidase (AOX1), or that reduce PL, such as AKRs, may similarly have a low PL:PN. A specific AKR with PL reductase activity has not been identified in humans, but application of the AKR family 1 member B1 (AKR1B1) and AKR family 1 member B10 (AKR1B10) inhibitor zopolrestat prevented the conversion of PL to PN in mammalian cells. However, genetic deletion of AKR1B1/AKR1B10 did not have an effect on PL reductase activity. Zopolrestat was also shown to inhibit AKR family 1 member A1 (AKR1A1) (138). Thus, AKR1A1 may participate in PL reductase activity, although many AKRs have low substrate selectivity, and other AKRs are likely involved (28). There is evidence for substantial variability in PL reduction activities between cell types. Upon supplementation with PL, the highest secretion of PN was observed in human hepatocellular carcinoma (HepG2) cells, followed by Caco2, mouse neuroblastoma (Neuro-2a), and human embryonic kidney (HEK293) cells. PN secretion rates in HepG2 cells were notably higher than the other cell types (35). Presently, both fluxomic data on PN metabolism and data on the relative activities of PNPO, PDXK, AOX1, and AKRs in different tissues are lacking. As a result, it is difficult to assess the contribution of these activities to the ratio of PL to PN within different tissues.
There are, however, indications that the testes may have enzyme activity favorable to a low PL:PN since the PDXK RNA expression is highest in the testes, while PNPO expression is comparatively low (38). Additionally, AOX1 protein expression is significantly elevated (139). Moreover, the testes likely have high levels of AKR activity, which could contribute to the low PL:PN. The AKR1C family enzymes (AKR1C1–AKR1C4) and AKR family 1 member D1 (AKR1D1) contribute to steroid metabolism and are elevated in endocrine organs (140). The AKR1C family enzymes have a low substrate selectivity and participate in the reduction of a wide variety of pharmaceutical compounds (141). Additionally, AKR1A1 RNA and protein expression was high in Sertoli cells and elongated spermatids (142). AKR1A1 has been shown to possess a broad substrate activity toward aldehydes (141). Additionally, the testes were shown to have a high level of methylglyoxal reduction capacity, an indicator of nonspecific AKR activity (142). Interestingly, PN was found to induce testicular damage in rats at similar doses to those that exhibit neurotoxicity. Intraperitoneal injections of PN in rats led to a delay in spermiation, degeneration of elongated spermatids, and Sertoli cell alterations. Additionally, germ cells were deformed, with multinucleate germ cells mingling with anisocytotic germ cells (143). Similar changes were observed in rats treated with isoniazid. Isoniazid led to testicular damage with germ cell malformation, including the formation of large multinucleate germ cells (144).
The relative activities of PDXK, PNPO, AKRs, and AOX1 have not been investigated in peripheral nerves. Interestingly, there is evidence for elevated activity of AKRs. The distribution of diabetes complications is thought to be related to elevated AKR1A1/AKR1B1 expression in the peripheral nerves, ocular lenses, retinas, and kidney (145). In peripheral nerves, AKR1A1 is predominantly localized in Schwann cells (146). One purpose of AKRs is to deactivate toxic carbonyl compounds (147, 148). Considering that neurons are particularly sensitive to aldehydes and DRG neurons have an incompetent blood–nerve barrier, it is plausible that sensory neurons have a high level of AKR activity. Similar arguments are applicable to PL oxidase activity. While information on the expression or activity of PNPO is limited, there are indications that PNPO activity may be low within sensory neurons. PNPO is developmentally regulated within neurons. In rats, PNPO activity was significantly lower in fetal brain tissue than found in adult brain tissue (83). Neural stem cells with retentive multipotency were shown to be preserved in the adult PNS (149). Moreover, Schwann cells have remarkable plasticity and the ability to differentiate into progenitor-like states (150). The differentiation status of these cell types and the general regenerative capacity of the PNS suggest that PNPO activity may be low within sensory neurons.
PN toxicity to sensory neurons: disruption of GABA signaling?
Reduced PLP formation by PDXK in sensory neurons demonstrably leads to axonal degeneration. The reason behind this is unclear; however, there is reason to believe that it may be related to GABA neurotransmission. Compounds that inhibit PDXK lead to reductions in GABA biosynthesis and seizures. Moreover, genetic disruptions in vitamin B-6 metabolic enzymes consistently have an impact on GABA activity. Mutations in PNPO, ALPL, PLPBP, and ALDH7A1 are associated with convulsions and decreases in GABA biosynthesis. Mice deficient in PDXP have elevated GABA concentrations in the brain and mild anxiety-like behavior (29). Additionally, PN toxicity was enhanced in zebrafish larvae exposed to the epileptogenic compounds ginkgotoxin, caffeine, and pentylenetetrazol. No effect was observed when ginkgotoxin and PN were added to culture medium at the same time. Interestingly, adding PN to the medium pretreated with ginkgotoxin caused cardiac arrest and necrotic death in a time- and dose-dependent manner. All larvae in the ginkgotoxin control group survived, while 20% died in the PN control group. Similar effects were observed using PN with caffeine and pentylenetetrazol. PLP alleviated the convulsions associated with ginkgotoxin treatments. Adding PLP and PN simultaneously prevented PN-induced larvae death (151). Thus, it is plausible that PN toxicity may be related to disruptions in GABA activity via PDXK inhibition.
GABA actions are mediated by the ionotropic receptors GABAA/GABAC and the metabotropic receptors GABAB. GABA concentrations are determined by both GABA production and catabolism involving the PLP-dependent enzymes glutamate decarboxylase (GAD) and GABA transaminase, respectively. There are 2 isoforms of GAD expressed by different genes. The constitutively active form GAD67 provides the steady basal supply of GABA in the cells, while GAD65 is primarily activated during GABA neurotransmission. GAD65 is membrane bound and transiently binds to PLP following its activation by phosphorylation (152). Interestingly, PDXK has been shown to directly interact with apo-GAD and likely facilitates the transfer of PLP to the enzyme (33). GAD67 is more saturated with PLP. Mice deficient in GAD67 show a lethal development with cleft palate and respiratory defects. In contrast, mice deficient in GAD65 appear normal at birth but are susceptible to seizures (153). As GAD65 exists primarily as the apo-enzyme, it may be more vulnerable to PDXK inhibition (154). GAD65 dysfunction is implicated in epilepsy, stiff person syndrome, and cerebellar ataxia (155, 156). There are reports of elevated GAD65 autoantibodies and peripheral neuropathy; however, a causal association cannot be made (157).
In the PNS, GABA-mediated presynaptic inhibition of afferent inputs plays a crucial role in signal processing and modulating sensory neurotransmission (Figure 3). DRG neurons have fully functioning local GABAergic signaling, including transporters, receptors, and metabolic enzymes (158). Additionally, glial cells, including satellite cells and Schwann cells, likely serve as a source of GABA for DRG neurons (159–161). Because of differences in chloride-homeostasis, GABAA receptor activation has a depolarizing effect on DRG neurons. This primary afferent depolarization inhibits the generation of action potentials and leads to presynaptic inhibition. In DRG neurons, GABA signaling may be nonsynaptic and localized in the soma and T-junction. Action potentials generated peripherally enter the soma, thereby activating GAD65 through phosphorylation and, as a result, triggering extracellular GABA release. GABAA receptor activation via paracrine or autocrine signaling of the same or neighboring nerve fibers depolarizes the T-junction. Depolarization lowers the action potential safety factor, which results in action potential failure at the T-junction. GABA-mediated presynaptic inhibition has been observed in subpopulations of small-, medium-, and large-fiber DRG neurons (158).
FIGURE 3.

(A) Normally, action potentials generated peripherally enter the soma of DRG neurons, and thereby activate GAD65 through phosphorylation. As GAD65 can only bind to PLP following its activation, it depends on rapid PLP biosynthesis. PDXK has been shown to directly interact with apo-GAD and it may facilitate the transfer of PLP to GAD. Following its release, GABA causes paracrine or autocrine inhibition of the same or neighboring nerve fibers through increased action potential filtering at T-junctions. GABA is also synthesized and released by glial cells, such as Schwann and satellite cells. (B) When the ratio of PL: PN is low, PLP formation by PDXK is inhibited and PLP binding to apo-GAD65 is reduced. As a result, GAD65 is unable to synthesize GABA in sufficient quantities following its activation by the incoming action potential. This disrupts the action potential filtering capacity of DRG neurons and leads to aberrant changes in the membrane potential at T-junctions/DRG soma. Excessive depolarization may induce calcium release and lead to excitotoxicity. DRG, dorsal root ganglia; GABA, γ-aminobutyric acid; GABAA, γ-aminobutyric acid A receptor; GAD, glutamate decarboxylase; GAT, GABA transporter; PDXK, pyridoxal kinase; PL, pyridoxal; PLP, pyridoxal 5′-phosphate; PN, pyridoxine.
The inhibition of GABAergic-mediated presynaptic inhibition leads to the increased excitability of DRG neurons (158). Genetic disruptions in GABAA receptors in primary nociceptors led to a hyperalgesic phenotype within mice (162). The possibility that local GABA inhibition and/or DRG excitability may cause peripheral neuropathy has not been investigated. It is generally known that overactivation of excitatory neurotransmitters leads to neuronal degeneration through excitotoxicity. Excitotoxicity has been shown to induce axonopathy with a Wallerian-type degeneration pattern in the optic nerve and spinal motor neurons (163, 164). Most experimental studies on excitotoxicity involve excessive glutamate receptor activation. Inhibitory neurotransmission contributes to the regulation of neuronal excitability and its inhibition may similarly lead to excitotoxicity. Chronic infusion with bicuculline, a GABAA receptor antagonist, in the spinal cord of rats led to gait alterations and significant motor neuron degeneration (165). Thus, it is plausible that interference with GABA neurotransmission in DRG neurons would lead to a similar effect. Figure 2 provides a graphical description of the proposed mechanism linking PDXK inhibition by PN to GABA inhibition and excitotoxicity. There is evidence that the primary changes associated with PN toxicity originate at or near the T-junction. Early PN lesions at these sites were characterized by a swollen membranous appearance (166). Similar changes were observed in retinal ganglion cells exposed to N-methyl-D-aspartate (NMDA) (164, 167). Considering that action potentials are generated at the T-junction, it is plausible that this could be related to excitotoxicity or neuronal misfiring. Excitotoxicity may, however, not exclusively involve GABA neurotransmission as PLP-dependent enzymes are involved in the biosynthesis of most neurotransmitters.
Impairments in GABA neurotransmission play a pivotal role in the development of neuropathic pain (162, 168). Following peripheral nerve injury, reductions in GABA activity have been observed in the spinal cord, dorsal horn, and DRG neurons (169–171). Because changes in GABAergic neurotransmission are associated with neuropathy, it is difficult to interpret findings relevant to the role of GABA in PN toxicity. A study found that the administration of chicory extract, an upregulator of GABAA receptors, ameliorated the symptoms of PN-induced neuropathy in rats. Improvements were observed in the hot plate test, rotarod, foot fault, and histological analysis. Conversely, pretreatment with picrotoxin, a GABAA receptor antagonist, exacerbated the severity of symptoms and inhibited the protective effects of chicory extract (172). These effects are not unique to PN-induced neuropathy. Similar findings were reported with surgically induced neuropathy (170, 171, 173).
PDXK inhibition may have deleterious effects on a large number of processes, including energy metabolism, redox balance, and/or hormone activities. As PLP-dependent enzymes have a wide range of activity, metabolite analysis may be needed for future investigations. Compared with other PLP-dependent enzymes, GAD65 may be particularly sensitive to PDXK inhibition and decreases in PLP biosynthesis as it exists primarily as the apo-enzyme and binds to PLP during neurotransmission. Using co-expression network analysis, Chelban et al. (47) found that PDXK in peripheral nerves was associated with oxidation-reduction processes and suggested that PDXK-induced neuropathy may be related to mitochondrial dysfunction. Xiong et al. (174) identified a co-expression relation between PDXK and growth-associated protein 43 (GAP43). GAP43 is an axonal membrane protein that contributes to neuronal regeneration, axon pathfinding, growth cone formation, and synapse formation (175–178). PDXK may cooperate with GAP43 and its inhibition may disrupt these activities. However, gene co-expression networks typically do not provide information relevant to causality (179). Thus, given the available evidence, PN toxicity is most plausibly related to reductions GABA activity via GAD65 inhibition in sensory neurons.
Conclusions
In summary, despite the fact that the number of case reports describing neuropathic symptoms in patients taking PN is increasing, the mechanisms behind PN toxicity are not yet known. Previously proposed mechanisms include aldehyde toxicity by PLP, the formation of quinone methide-type intermediates, competitive inhibition of PLP-dependent enzymes, and PDXK inhibition. For several reasons, we conclude that PDXK inhibition and consequently disrupted GABA neurotransmission is the more plausible hypothesis.
Genetic deficiencies in PDXK lead to the development of axonal sensory neuropathy. The neurotoxic effect of reduced PDXK activity is contingent on PLP depletion. PN has a higher affinity toward PDXK compared with its other substrates and likely acts as a competitive inhibitor of the enzyme. The degree of PDXK inhibition therefore depends on the ratio of PL to PN. Generally, PL concentrations are much higher than PN in most tissues. Tissues with comparatively low PNPO activity and high PL reductase and oxidase activity are more likely to have a low PL:PN. In these tissues, PLP biosynthesis is likely reduced as with a PDXK deficiency. There are, for instance, indications that the testes have low PNPO activity and elevated PL reductase/oxidase activity. The testes are therefore susceptible to PN toxicity. Future investigations are needed to examine the relation between PL:PN and PN toxicity in testicular tissue. There is a paucity of information related to relative gene and protein expression in sensory neurons; however, there is evidence for high AKR activity.
Presently, there is insufficient information to infer a mechanistic link between PDXK inhibition and toxicity. PDXK inhibition likely has a number of pleiotropic effects. Mutations in vitamin B-6 metabolic enzymes consistently have an impact on GABA activity. Additionally, compounds that inhibit PDXK lead to reductions in GABA biosynthesis. Therefore, PDXK-induced neuropathy is most likely related to GABA activity. While no direct evidence exists in DRG neurons, GABA inhibition may lead to excitotoxicity and neurodegeneration. More investigations are needed to test the effects of PN, PDXK, and GABA inhibition on sensory neurons.
ACKNOWLEDGEMENTS
The authors’ responsibilities were as follows—all authors: performed the study selection and read and approved the final manuscript.
Notes
The authors reported no funding received for this work.
Author disclosures: The authors report no conflicts of interest.
Abbreviations used: AKR, aldo-keto reductase; AKR1A1, aldo-keto reductase family 1 member A1; AKR1B1, aldo-keto reductase family 1 member B1; AKR1B10, aldo-keto reductase family 1 member B10; AKR1C1–AKR1C4, aldo-keto reductase 1C family enzymes; ALDH7A1, antiquitin; ALPL, alkaline phosphatase, tissue-nonspecific isozyme; AOX1, aldehyde oxidase; CNS, central nervous system; DRG, dorsal root ganglia; EFSA, European Food Safety Authority; GABA, γ-aminobutyric acid; GAD, glutamate decarboxylase; GAP43, growth-associated protein 43; Km, Michaelis-Menten constant; PDXK, pyridoxal kinase; PDXP, pyridoxal 5′-phosphate phosphatase; PL, pyridoxal; PLP, pyridoxal 5′-phosphate; PLPBP, pyridoxal 5'-phosphate–binding protein; PM, pyridoxamine; PMP, pyridoxamine 5'-phosphate; PN, pyridoxine; PNP, pyridoxine-5'-phosphate; PNPO, pyridoxine-5'-phosphate oxidase; PNS, peripheral nervous system; QST, quantitative sensory threshold; UL, Tolerable Upper Intake Level; Vmax, maximal rate; 4-PA, 4-pyridoxic acid.
Contributor Information
Felix Hadtstein, University College Venlo, Campus Venlo, Maastricht University, Maastricht, The Netherlands.
Misha Vrolijk, University College Venlo, Campus Venlo, Maastricht University, Maastricht, The Netherlands.
References
- 1.Mooney S, Leuendorf J-E, Hendrickson C, Hellmann H. Vitamin B6: a long known compound of surprising complexity. Molecules. 2009;14:329–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ueland PM, Ulvik A, Rios-Avila L, Midttun Ø, Gregory JF. Direct and functional biomarkers of vitamin B6 status. Annu Rev Nutr. 2015;35:33–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA). Dietary reference values for vitamin B6. EFSA J. 2016;14:e04485. [Google Scholar]
- 4.Salvo ML, di Salvo ML, Contestabile R, Safo MK. Vitamin B6 salvage enzymes: mechanism, structure and regulation. Biochimica et Biophysica Acta. 2011;1814(11):1597–608. [DOI] [PubMed] [Google Scholar]
- 5.Jortner BS. Mechanisms of toxic injury in the peripheral nervous system: neuropathologic considerations. Toxicol Pathol. 2000;28:54–69. [DOI] [PubMed] [Google Scholar]
- 6.EFSA Scientific Committee on Food; Scientific Panel on Dietetic Products, Nutrition and Allergies; Tolerable Upper Intake Levels for vitamins and minerals. EFSA J. 2006. [Google Scholar]
- 7.Schaumburg H, Kaplan J, Windebank A, Vick N, Rasmus S, Pleasure D, Brown MJ. Sensory neuropathy from pyridoxine abuse. A new megavitamin syndrome. N Engl J Med. 1983;309:445–8. [DOI] [PubMed] [Google Scholar]
- 8.Farhad K, Traub R, Ruzhansky KM, Brannagan TH. Causes of neuropathy in patients referred as “idiopathic neuropathy”. Muscle Nerve. 2016:856–61. [DOI] [PubMed] [Google Scholar]
- 9.Bacharach R, Lowden M, Ahmed A. Pyridoxine toxicity small fiber neuropathy with dysautonomia: a case report. J Clin Neuromuscul Dis. 2017;19:43–6. [DOI] [PubMed] [Google Scholar]
- 10.Berger AR, Schaumburg HH, Schroeder C, Apfel S, Reynolds R. Dose response, coasting, and differential fiber vulnerability in human toxic neuropathy: a prospective study of pyridoxine neurotoxicity. Neurology. 1992;42:1367–70. [DOI] [PubMed] [Google Scholar]
- 11.Latov N, Vo ML, Chin RL, Carey BT, Langsdorf JA, Feuer NT. Abnormal nutritional factors in patients evaluated at a neuropathy center. J Clin Neuromusc Dis. 2016;17:212–4. [DOI] [PubMed] [Google Scholar]
- 12.Haroutounian S, Todorovic MS, Leinders M, Campagnolo M, Gewandter JS, Dworkin RH, Freeman R. Diagnostic criteria for idiopathic small fiber neuropathy: a systematic review. Muscle Nerve. 2020;63(2):170–7. [DOI] [PubMed] [Google Scholar]
- 13.Sène D. Small fiber neuropathy: diagnosis, causes, and treatment. Joint Bone Spine. 2018;85:553–9. [DOI] [PubMed] [Google Scholar]
- 14.Vrolijk MF, Hageman GJ, van de Koppel S, van Hunsel F, Bast A. Inter-individual differences in pharmacokinetics of vitamin B6: a possible explanation of different sensitivity to its neuropathic effects. PharmaNutrition. 2020;12:100188. [Google Scholar]
- 15.van Hunsel F, van de Koppel S, van Puijenbroek E, Kant A. Vitamin B6 in health supplements and neuropathy: case series assessment of spontaneously reported cases. Drug Saf. 2018;41:859–69. [DOI] [PubMed] [Google Scholar]
- 16.Vrolijk MF, Opperhuizen A, Jansen E, Hageman GJ, Bast A, Haenen G. The vitamin B6 paradox: supplementation with high concentrations of pyridoxine leads to decreased vitamin B6 function. Toxicol In Vitro. 2017;44:206–12. [DOI] [PubMed] [Google Scholar]
- 17.Rudman D, Williams PJ. Megadose vitamins. N Engl J Med. 1983;309:488–90. [DOI] [PubMed] [Google Scholar]
- 18.Windebank AJ. Neurotoxicity of pyridoxine analogs is related to coenzyme structure. Neurochem Pathol. 1985;3:159–67. [DOI] [PubMed] [Google Scholar]
- 19.Krinke G, Schaumburg HH, Spencer PS, Suter J, Thomann P, Hess R. Pyridoxine megavitaminosis produces degeneration of peripheral sensory neurons (sensory neuronopathy) in the dog. Neurotoxicology. 1981;2:13–24. [PubMed] [Google Scholar]
- 20.Percudani R, Peracchi A. The B6 database: a tool for the description and classification of vitamin B6-dependent enzymatic activities and of the corresponding protein families. BMC Bioinformatics. 2009;10:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakurai T, Asakura T, Matsuda M. Transport and metabolism of pyridoxine in the intestine of the mouse. J Nutr Sci Vitaminol. 1988;34:179–87. [DOI] [PubMed] [Google Scholar]
- 22.Sakurai T, Asakura T, Matsuda M. Transport and metabolism of pyridoxine and pyridoxal in mice. J Nutr Sci Vitaminol. 1987;33:11–9. [DOI] [PubMed] [Google Scholar]
- 23.Albersen M, Bosma M, Nine VVA, de Ruiter BHB, Diekman EF, de Ruijter J, Visser WF, de Koning TJ, Verhoeven-Duif NM. The intestine plays a substantial role in human vitamin B6 metabolism: a Caco-2 cell model. PLoS One. 2013;8(1):e54113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zempleni J. Pharmacokinetics of vitamin B6 supplements in humans. J Am Coll Nutr. 1995;14:579–86. [DOI] [PubMed] [Google Scholar]
- 25.Merrill AH Jr, Henderson JM, Wang E, McDonald BW, Millikan WJ. Metabolism of vitamin B-6 by human liver. J Nutr. 1984;114:1664–74. [DOI] [PubMed] [Google Scholar]
- 26.Coburn SP. Vitamin B-6 metabolism and interactions with TNAP. Subcell Biochem. 2015;76:207–38. [DOI] [PubMed] [Google Scholar]
- 27.Parra M, Stahl S, Hellmann H. Vitamin B₆ and its role in cell metabolism and physiology. Cells. 2018;7:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramos RJ, Albersen M, Vringer E, Bosma M, Zwakenberg S, Zwartkruis F, Jans JJM, Verhoeven-Duif NM. Discovery of pyridoxal reductase activity as part of human vitamin B6 metabolism. Biochim Biophys Acta Gen Subj. 2019;1863:1088–97. [DOI] [PubMed] [Google Scholar]
- 29.Jeanclos E, Albersen M, Ramos RJJ, Raab A, Wilhelm C, Hommers L, Lesch K-P, Verhoeven-Duif NM, Gohla A. Improved cognition, mild anxiety-like behavior and decreased motor performance in pyridoxal phosphatase-deficient mice. Biochim Biophys Acta Mol Basis Dis. 2019;1865:193–205. [DOI] [PubMed] [Google Scholar]
- 30.Kwak S-E, Kim J-E, Kim D-W, Kwon O-S, Choi S-Y, Kang T-C. Pyridoxine 5′-phosphate oxidase, not pyridoxal kinase, involves in long-term potentiation induction in the rat dentate gyrus. Hippocampus. 2009;19(1):45–56. [DOI] [PubMed] [Google Scholar]
- 31.Huang S, Yao L, Zhang J, Huang L. Direct and indirect effects of RNA interference against pyridoxal kinase and pyridoxine 5′-phosphate oxidase genes in Bombyx mori. Gene. 2016;587(1):48–52. [DOI] [PubMed] [Google Scholar]
- 32.Whittaker JW. Intracellular trafficking of the pyridoxal cofactor: implications for health and metabolic disease. Arch Biochem Biophys. 2016;592:20–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheung P-Y, Fong C-C, Ng K-T, Lam W-C, Leung Y-C, Tsang C-W, Yang M, Wong M-S. Interaction between pyridoxal kinase and pyridoxal-5-phosphate-dependent enzymes. J Biochem (Tokyo). 2003;134:731–8. [DOI] [PubMed] [Google Scholar]
- 34.Yang ES, Schirch V. Tight binding of pyridoxal 5'-phosphate to recombinant Escherichia coli pyridoxine 5'-phosphate oxidase. Arch Biochem Biophys. 2000;377:109–14. [DOI] [PubMed] [Google Scholar]
- 35.Karve S. Role of pyridoxine 5’-phosphate oxidase in metabolism and transfer of pyridoxal 5'-phosphate. [Internet]. Virginia Commonwealth University; 2010; [cited 2021 Jan 18]. Available from: https://scholarscompass.vcu.edu/etd/2253/. [Google Scholar]
- 36.Desai J. Pyridoxal kinase: its role in vitamin B6 metabolism. [Internet]. Virginia Commonwealth University; 2010; [cited 2021 Jan 18]. Available from: https://scholarscompass.vcu.edu/etd/2254/?mode=full. [Google Scholar]
- 37.Fonda ML. Pyridoxamine (pyridoxine) phosphate oxidase activity in mammalian tissues. Comp Biochem Physiol B. 1988;90:731–7. [DOI] [PubMed] [Google Scholar]
- 38.Kang JH, Hong M-L, Kim DW, Park J, Kang T-C, Won MH, Baek N-I, Moon BJ, Choi SY, Kwon O-S. Genomic organization, tissue distribution and deletion mutation of human pyridoxine 5’-phosphate oxidase. Eur J Biochem. 2004;271:2452–61. [DOI] [PubMed] [Google Scholar]
- 39.Abosamak NER, Gupta V. Vitamin B6 (pyridoxine). StatPearls. Treasure Island (FL): StatPearls Publishing; 2020. [PubMed] [Google Scholar]
- 40.Waymire KG, Dennis Mahuren J, Michael Jaje J, Guilarte TR, Coburn SP, MacGregor GR. Mice lacking tissue non-specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat Genet. 1995;11(1):45–51. [DOI] [PubMed] [Google Scholar]
- 41.Darin N, Reid E, Prunetti L, Samuelsson L, Husain RA, Wilson M, El Yacoubi B, Footitt E, Chong WK, Wilson LCet al. Mutations in PROSC disrupt cellular pyridoxal phosphate homeostasis and cause vitamin-B6-dependent epilepsy. Am J Hum Genet. 2016;99(6):1325–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mills PB, Surtees RAH, Champion MP, Beesley CE, Dalton N, Scambler PJ, Heales SJR, Briddon A, Scheimberg I, Hoffmann GFet al. Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5′-phosphate oxidase. Hum Mol Genet. 2005;14(8):1077–86. [DOI] [PubMed] [Google Scholar]
- 43.Clayton PT. B6-responsive disorders: a model of vitamin dependency. J Inherit Metab Dis. 2006;29(2-3):317–26. [DOI] [PubMed] [Google Scholar]
- 44.Staufner C. Disorders of vitamin B6 metabolism. Nyhan WF, Hoffman GF, Al-Aqeel AI, Barshop BAeditors. In: Atlas of inherited metabolic diseases, Boca Raton, Fl: CRC Press; 2020. p. 763–70. [Google Scholar]
- 45.Stockler S, Plecko B, Gospe SM Jr, Coulter-Mackie M, Connolly M, van Karnebeek C, Mercimek-Mahmutoglu S, Hartmann H, Scharer G, Struijs Eet al. Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Mol Genet Metab. 2011;104:48–60. [DOI] [PubMed] [Google Scholar]
- 46.Keller N, Mendoza-Ferreira N, Maroofian R, Chelban V, Khalil Y, Mills PB, Boostani R, Torbati PN, Karimiani EG, Thiele Het al. Hereditary polyneuropathy with optic atrophy due to PDXK variant leading to impaired vitamin B6 metabolism. Neuromuscul Disord. 2020;30:583–9. [DOI] [PubMed] [Google Scholar]
- 47.Chelban V, Wilson MP, Warman Chardon J, Vandrovcova J, Zanetti MN, Zamba-Papanicolaou E, Efthymiou S, Pope S, Conte MR, Abis Get al. PDXK mutations cause polyneuropathy responsive to pyridoxal 5’-phosphate supplementation. Ann Neurol. 2019;86:225–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dalton K, Dalton MJ. Characteristics of pyridoxine overdose neuropathy syndrome. Acta Neurol Scand. 1987;76:8–11. [DOI] [PubMed] [Google Scholar]
- 49.Ohnishi A, Ishibashi H, Ohtani K, Matsunaga K, Yamamoto T. [Peripheral sensory neuropathy produced by a megadose of vitamin B6] J UOEH. 1985;7:201–5. [DOI] [PubMed] [Google Scholar]
- 50.Parry GJ, Bredesen DE. Sensory neuropathy with low-dose pyridoxine. Neurology. 1985;35:1466–8. [DOI] [PubMed] [Google Scholar]
- 51.Albin RL, Albers JW, Greenberg HS, Townsend JB, Lynn RB, Burke JM Jr, Alessi AG. Acute sensory neuropathy-neuronopathy from pyridoxine overdose. Neurology. 1987;37:1729–32. [DOI] [PubMed] [Google Scholar]
- 52.Mitwalli A, Blair G, Oreopoulos DG. Safety of intermediate doses of pyridoxine. Can Med Assoc J. 1984;131:14. [PMC free article] [PubMed] [Google Scholar]
- 53.Mpofu C, Alani SM, Whitehouse C, Fowler B, Wraith JE. No sensory neuropathy during pyridoxine treatment in homocystinuria. Arch Dis Child. 1991;66:1081–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Kruijk JR, Notermans NC. [Sensory disturbances caused by multivitamin preparations.]. Ned Tijdschr Geneeskd. 2005;149:2541–4. [PubMed] [Google Scholar]
- 55.Malek E, Doumiati H, Salameh JS. Pyridoxine-induced sensory ataxic ganglionopathy: a case report and literature review. Acta Neurol Belg. 2020;120:413–4. [DOI] [PubMed] [Google Scholar]
- 56.Hoover DM, Carlton WW, Henrikson CK. Ultrastructural lesions of pyridoxine toxicity in beagle dogs. Vet Pathol. 1981;18:769–77. [DOI] [PubMed] [Google Scholar]
- 57.Pearson KG, Misiaszek JE, Hulliger M. Chemical ablation of sensory afferents in the walking system of the cat abolishes the capacity for functional recovery after peripheral nerve lesions. Exp Brain Res. 2003;150:50–60. [DOI] [PubMed] [Google Scholar]
- 58.Stapley PJ, Ting LH, Hulliger M, Macpherson JM. Automatic postural responses are delayed by pyridoxine-induced somatosensory loss. J Neurosci. 2002;22:5803–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chiba S, Edamura M, Ito M, Kikuchi S, Matsumoto H. [Experimental pyridoxine neuropathy—an electrophysiological and histological study in rabbits]. Rinsho Shinkeigaku. 1985;25:939–43. [PubMed] [Google Scholar]
- 60.Sharp AA, Fedorovich Y. Teratogenic effects of pyridoxine on the spinal cord and dorsal root ganglia of embryonic chickens. Neuroscience. 2015;289:233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cohen M, Bendich A. Safety of pyridoxine–a review of human and animal studies. Toxicol Lett. 1986;34:129–39. [DOI] [PubMed] [Google Scholar]
- 62.Xu Y, Sladky JT, Brown MJ. Dose-dependent expression of neuronopathy after experimental pyridoxine intoxication. Neurology. 1989;39(8):1077–83. [DOI] [PubMed] [Google Scholar]
- 63.Phillips WE, Mills JH, Charbonneau SM, Tryphonas L, Hatina GV, Zawidzka Z, Bryce FR, Munro IC. Subacute toxicity of pyridoxine hydrochloride in the beagle dog. Toxicol Appl Pharmacol. 1978;44:323–33. [DOI] [PubMed] [Google Scholar]
- 64.Johannsson S, Tiselius HG. Metabolism of tritium- and 14C-labeled pyridoxine in the rat. Scand J Clin Lab Invest. 1973;32:9–14. [DOI] [PubMed] [Google Scholar]
- 65.Perry TA, Weerasuriya A, Mouton PR, Holloway HW, Greig NH. Pyridoxine-induced toxicity in rats: a stereological quantification of the sensory neuropathy. Exp Neurol. 2004;190:133–44. [DOI] [PubMed] [Google Scholar]
- 66.Helgren ME, Cliffer KD, Torrento K, Cavnor C, Curtis R, DiStefano PS, Wiegand SJ, Lindsay RM. Neurotrophin-3 administration attenuates deficits of pyridoxine-induced large-fiber sensory neuropathy. J Neurosci. 1997;17:372–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schaeppi U, Krinke G. Pyridoxine neuropathy: correlation of functional tests and neuropathology in beagle dogs treated with large doses of vitamin B6. Agents Actions. 1982;12:575–82. [DOI] [PubMed] [Google Scholar]
- 68.Arkaravichien T, Sattayasai N, Daduang S, Sattayasai J. Dose-dependent effects of glutamate in pyridoxine-induced neuropathy. Food Chem Toxicol. 2003;41:1375–80. [DOI] [PubMed] [Google Scholar]
- 69.Shiers S, Klein RM, Price TJ. Quantitative differences in neuronal subpopulations between mouse and human dorsal root ganglia demonstrated with RNAscope in situ hybridization. Pain. 2020;161:2410–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Han C, Estacion M, Huang J, Vasylyev D, Zhao P, Dib-Hajj SD, Waxman SG. Human Na(v)1.8: enhanced persistent and ramp currents contribute to distinct firing properties of human DRG neurons. J Neurophysiol. 2015;113:3172–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Castro J, Harrington AM, Garcia-Caraballo S, Maddern J, Grundy L, Zhang J, Page G, Miller PE, Craik DJ, Adams DJet al. α-Conotoxin Vc1.1 inhibits human dorsal root ganglion neuroexcitability and mouse colonic nociception via GABA receptors. Gut. 2017;66:1083–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haberberger RV, Barry C, Dominguez N, Matusica D. Human dorsal root ganglia. Front Cell Neurosci. 2019;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jortner BS. Common structural lesions of the peripheral nervous system. Toxicol Pathol. 2020;48:96–104. [DOI] [PubMed] [Google Scholar]
- 74.Krinke G, Naylor DC, Skorpil V. Pyridoxine megavitaminosis. J Neuropathol Exp Neurol. 1985;44(2):117–29. [PubMed] [Google Scholar]
- 75.Krinke GJ, Fitzgerald RE. The pattern of pyridoxine-induced lesion: difference between the high and the low toxic level. Toxicology. 1988;49(1):171–8. [DOI] [PubMed] [Google Scholar]
- 76.Montpetit VJ, Clapin DF, Tryphonas L, Dancea S. Alteration of neuronal cytoskeletal organization in dorsal root ganglia associated with pyridoxine neurotoxicity. Acta Neuropathol (Berl). 1988;76:71–81. [DOI] [PubMed] [Google Scholar]
- 77.Conforti L, Gilley J, Coleman MP. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci. 2014;15:394–409. [DOI] [PubMed] [Google Scholar]
- 78.Godel T, Pham M, Heiland S, Bendszus M, Bäumer P. Human dorsal-root-ganglion perfusion measured in-vivo by MRI. Neuroimage. 2016;141:81–7. [DOI] [PubMed] [Google Scholar]
- 79.Jimenez-Andrade JM, Herrera MB, Ghilardi JR, Vardanyan M, Melemedjian OK, Mantyh PW. Vascularization of the dorsal root ganglia and peripheral nerve of the mouse: implications for chemical-induced peripheral sensory neuropathies. Mol Pain. 2008;4:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Valentine WM. Toxic peripheral neuropathies: agents and mechanisms. Toxicol Pathol. 2020;48:152–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frater-Schroder M, Alder S, Zbinden G. Neurotoxic effects of pyridoxine and analogs in rats. [Internet]. Proc Eur Soc Toxicol. 1976. Available from: http://agris.fao.org/agris-search/search.do?recordID=US201303065826. [Google Scholar]
- 82.Levine S, Saltzman A. Pyridoxine (vitamin B6) neurotoxicity: enhancement by protein-deficient diet. J Appl Toxicol. 2004;24:497–500. [DOI] [PubMed] [Google Scholar]
- 83.Ngo EO, LePage GR, Thanassi JW, Meisler N, Nutter LM. Absence of pyridoxine-5’-phosphate oxidase (PNPO) activity in neoplastic cells: isolation, characterization, and expression of PNPO cDNA. Biochemistry. 1998;37:7741–8. [DOI] [PubMed] [Google Scholar]
- 84.Zempleni J, Kübler W. The utilization of intravenously infused pyridoxine in humans. Clin Chim Acta. 1994;229:27–36. [DOI] [PubMed] [Google Scholar]
- 85.Speitling A, Heseker H, Kübler W. Pharmacokinetic properties of the plasma B6 vitamers after single and chronic oral pyridoxine mega doses. Ann NY Acad Sci. 1990;585:557–9. [Google Scholar]
- 86.Scott K, Zeris S, Kothari MJ. Elevated B6 levels and peripheral neuropathies. Electromyogr Clin Neurophysiol. 2008;48:219–23. [PubMed] [Google Scholar]
- 87.Edwards P, Liu PK, Rose GA. Liquid chromatographic studies of vitamin B6 metabolism in man. Clin Chim Acta. 1990;190:67–80. [DOI] [PubMed] [Google Scholar]
- 88.Bor MV, Refsum H, Bisp MR, Bleie Ø, Schneede J, Nordrehaug JE, Ueland PM, Nygard OK, Nexø E. Plasma vitamin B6 vitamers before and after oral vitamin B6 treatment: a randomized placebo-controlled study. Clin Chem. 2003;49:155–61. [DOI] [PubMed] [Google Scholar]
- 89.Musayev FN, Di Salvo ML, Ko T-P, Schirch V, Safo MK. Structure and properties of recombinant human pyridoxine 5′-phosphate oxidase. Protein Sci. 2003;12(7):1455–63.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Salvo ML, di Salvo ML, Nogués I, Parroni A, Tramonti A, Milano T, Pascarella S, Contestabile R. On the mechanism of Escherichia coli pyridoxal kinase inhibition by pyridoxal and pyridoxal 5′-phosphate. Biochimica et Biophysica Acta Proteins Proteomics. 2015;1854(9):1160–6. [DOI] [PubMed] [Google Scholar]
- 91.Argoudelis CJ. In vitro reaction of L-tryptophan and vitamin B6: synthesis of the corresponding .beta.-tetrahydrocarbolines. J Agric Food Chem. 1994;42(11):2372–5. [Google Scholar]
- 92.LoPachin RM, Geohagen BC, Nordstroem LU. Mechanisms of soft and hard electrophile toxicities. Toxicology. 2019;418:62–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brousmiche DW, Wan P. Photogeneration of quinone methide-type intermediates from pyridoxine and derivatives. J Photochem Photobiol A. 2002;149(1-3):71–81. [Google Scholar]
- 94.Frater-Schröder M, Mahrer-Busato M. A model reaction demonstrating alkylating properties of pyridoxol, involving an o-quinone methide intermediate. Bioorg Chem. 1975;4(4):332–41. [Google Scholar]
- 95.Sato K, Taguchi H, Maeda T, Yoshikawa K. Pyridoxine toxicity to cultured fibroblasts caused by near-ultraviolet light. J Invest Dermatol. 1993;100:266–70. [DOI] [PubMed] [Google Scholar]
- 96.Casas JS, Couce MD, Sordo J. Coordination chemistry of vitamin B6 and derivatives: a structural overview. Coord Chem Rev. 2012;256(23-24):3036–62. [Google Scholar]
- 97.Schaeffer MC, Sampson DA, Skala JH, Gietzen DW, Grier RE. Evaluation of vitamin B-6 status and function of rats fed excess pyridoxine. J Nutr. 1989;119:1392–8. [DOI] [PubMed] [Google Scholar]
- 98.Spinneker A, Sola R, Lemmen V, Castillo MJ, Pietrzik K, González-Gross M. Vitamin B6 status, deficiency and its consequences–an overview. Nutr Hosp. 2007;22:7–24. [PubMed] [Google Scholar]
- 99.Rinehart JF, Greenberg LD. Vitamin B6 deficiency in the rhesus monkey. Am J Clin Nutr. 1956;4(4):318–28. [DOI] [PubMed] [Google Scholar]
- 100.Attar MW, Daghir NJ, Asmar J. Influence of vitamin B6 deficiency on certain serum components in mature female chickens. Poult Sci. 1967;46:838–43. [DOI] [PubMed] [Google Scholar]
- 101.Street HR, Cowgill GR, Zimmerman HM. Some observations of vitamin B6 deficiency in the dog. J Nutr. 1941;27(3):275–90. [Google Scholar]
- 102.Sumi Y, Miyakawa M, Kanzaki M, Kotake Y. Vitamin B-6 deficiency in germfree rats. J Nutr. 1977;107:1707–14. [DOI] [PubMed] [Google Scholar]
- 103.Hove EL, Herndon JF. Vitamin B6 deficiency in rabbits. J Nutr. 1957;61(1):127–36. [DOI] [PubMed] [Google Scholar]
- 104.Murata Y, Kumano K, Ueda T, Araki N, Nakamura T, Tani M. Photosensitive dermatitis caused by pyridoxine hydrochloride. J Am Acad Dermatol. 1998;39:314–7. [DOI] [PubMed] [Google Scholar]
- 105.Rezaković S, Mokos ZB, Paštar Z. Pyridoxine induced rosacea-like dermatitis. Acta Clin Croat. 2015;54:99–102. [PubMed] [Google Scholar]
- 106.Wintrobe MM, Mushatt C, Miller JL, Kolb LC, Stein HJ, Lisco H. The prevention of sensory neuron degeneration in the pig, with specific reference to the role of liver fractions. J Clin Invest. 1942;21(1):71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dellon AL, Dellon ES, Tassler PL, Ellefson RD, Hendrickson M. Experimental model of pyridoxine (B6) deficiency-induced neuropathy. Ann Plast Surg. 2001;47:153–60. [DOI] [PubMed] [Google Scholar]
- 108.Moriwaki K, Kanno Y, Nakamoto H, Okada H, Suzuki H. Vitamin B6 deficiency in elderly patients on chronic peritoneal dialysis. Adv Perit Dial. 2000;16:308–12. [PubMed] [Google Scholar]
- 109.Espinoza M, Aguilera A, Auxiliadora Bajo M, Codoceo R, Caravaca E, Cirugeda A, del Peso G, Hevia C, Selgas R. Tumor necrosis factor alpha as a uremic toxin: correlation with neuropathy, left ventricular hypertrophy, anemia, and hypertriglyceridemia in peritoneal dialysis patients. Adv Perit Dial. 1999;15:82–6. [PubMed] [Google Scholar]
- 110.Krishnan AV, Kiernan MC. Uremic neuropathy: clinical features and new pathophysiological insights. Muscle Nerve. 2007;35:273–90. [DOI] [PubMed] [Google Scholar]
- 111.Hamed SA. Neurologic conditions and disorders of uremic syndrome of chronic kidney disease: presentations, causes, and treatment strategies. Expert Rev Clin Pharmacol. 2019;12:61–90. [DOI] [PubMed] [Google Scholar]
- 112.Hung SC, Hung SH, Tarng DC, Yang WC, Chen TW, Huang TP. Thiamine deficiency and unexplained encephalopathy in hemodialysis and peritoneal dialysis patients. Am J Kidney Dis. 2001;38:941–7. [DOI] [PubMed] [Google Scholar]
- 113.Victor M, Adams RD. The neuropathology of experimental vitamin B6 deficiency in monkeys. Am J Clin Nutr. 1956;4:346–53. [DOI] [PubMed] [Google Scholar]
- 114.Greenberg LD, McIvor B, Moon HD. Experimental arteriosclerosis in pyridoxine-deficient rhesus monkeys. Am J Clin Nutr. 1958;6:635–7. [DOI] [PubMed] [Google Scholar]
- 115.Ghavanini AA, Kimpinski K. Revisiting the evidence for neuropathy caused by pyridoxine deficiency and excess. J Clin Neuromuscul Dis. 2014;16:25–31. [DOI] [PubMed] [Google Scholar]
- 116.Vilter RW, Mueller JF, Glazer HS, Jarrold T, Abraham J, Thompson C, Hawkins VR. The effect of vitamin B6 deficiency induced by desoxypyridoxine in human beings. J Lab Clin Med. 1953;42:335–57. [PubMed] [Google Scholar]
- 117.Meldrum BS, Horton RW. Convulsive effects of 4-deoxypyridoxine and of bicuculline in photosensitive baboons (Papio papio) and in rhesus monkeys (Macaca mulatta). Brain Res. 1971;35:419–36. [DOI] [PubMed] [Google Scholar]
- 118.Coburn SP, Mahuren JD, Schaltenbrand WE, Wostmann BS, Madsen D. Effects of vitamin B-6 deficiency and 4′-deoxypyridoxine on pyridoxal phosphate concentrations, pyridoxine kinase and other aspects of metabolism in the rat. J Nutr. 1981;111:391–8. [DOI] [PubMed] [Google Scholar]
- 119.Goldman AL, Braman SS. Isoniazid: a review with emphasis on adverse effects. Chest. 1972;62:71–7. [DOI] [PubMed] [Google Scholar]
- 120.Lainé-Cessac P, Cailleux A, Allain P. Mechanisms of the inhibition of human erythrocyte pyridoxal kinase by drugs. Biochem Pharmacol. 1997;54:863–70. [DOI] [PubMed] [Google Scholar]
- 121.Snider DE Jr. Pyridoxine supplementation during isoniazid therapy. Tubercle. 1980;61:191–6. [DOI] [PubMed] [Google Scholar]
- 122.Chua CL, Ohnishi A, Tateishi J, Kuroiwa Y. Morphometric evaluation of degenerative and regenerative changes in isoniazid-induced neuropathy. Acta Neuropathol (Berl). 1983;60:183–93. [DOI] [PubMed] [Google Scholar]
- 123.Beuche W, Friede RL. Remodelling of nerve structure in experimental isoniazid neuropathy in the rat. Brain. 1986;109(Pt 4):759–69. [DOI] [PubMed] [Google Scholar]
- 124.Sea CP, Peterson RG. Ultrastructure and biochemistry of myelin after isoniazid-induced nerve degeneration in rats. Exp Neurol. 1975;48:252–60. [DOI] [PubMed] [Google Scholar]
- 125.Kasaragod VB, Pacios-Michelena A, Schaefer N, Zheng F, Bader N, Alzheimer C, Villmann C, Schindelin H. Pyridoxal kinase inhibition by artemisinins downregulates inhibitory neurotransmission. Proc Natl Acad Sci USA. 2020;117:33235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jang H-S, Roh SY, Jeong EH, Kim B-S, Sunwoo MK. Ginkgotoxin induced seizure caused by vitamin B6 deficiency. J Epilepsy Res. 2015;5:104–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Meisler NT, Thanassi JW. Pyridoxine kinase, pyridoxine phosphate phosphatase and pyridoxine phosphate oxidase activities in control and B-6-deficient rat liver and brain. J Nutr. 1980;110(10):1965–75. [DOI] [PubMed] [Google Scholar]
- 128.Kästner U, Hallmen C, Wiese M, Leistner E, Drewke C. The human pyridoxal kinase, a plausible target for ginkgotoxin from Ginkgo biloba. FEBS J. 2007;274:1036–45. [DOI] [PubMed] [Google Scholar]
- 129.Könczöl Á, Rendes K, Dékány M, Müller J, Riethmüller E, Balogh GT. Blood-brain barrier specific permeability assay reveals N-methylated tyramine derivatives in standardised leaf extracts and herbal products of Ginkgo biloba. J Pharm Biomed Anal. 2016;131:167–74. [DOI] [PubMed] [Google Scholar]
- 130.Habgood MD, Knott GW, Dziegielewska KM, Saunders NR. Permeability of the developing and mature blood-brain barriers to theophylline in rats. Clin Exp Pharmacol Physiol. 1998;25:361–8. [DOI] [PubMed] [Google Scholar]
- 131.Fernandes G, Salgado HRN, Santos JLD. Isoniazid: a review of characteristics, properties and analytical methods. Crit Rev Anal Chem. 2017;47:298–308. [DOI] [PubMed] [Google Scholar]
- 132.Hansch C, Leo A; Heller, SR; Exploring QSAR: Volume 1: fundamentals and applications in chemistry and biology, American Chemical Society, Washington DC; 1995. [Google Scholar]
- 133.Anderson VE. Multiple alternative substrate kinetics. Biochim Biophys Acta. 2015;1854:1729–36. [DOI] [PubMed] [Google Scholar]
- 134.Golovenko NY, Larionov VB, Karpova OV. Physical-chemical properties and the reactivity of pyridoxine and pyrrolidone carboxylate and their protolytic forms. Ukr Biochem J. 2016;88:73–81. [DOI] [PubMed] [Google Scholar]
- 135.Kosaki Y, Naito H, Nojima T, Nakao A. Epileptic seizure from ginkgo nut intoxication in an adult. Case Rep Emerg Med. 2020;2020:5072954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sado T, Nakata S, Tsuno T, Sato M, Misawa Y, Yamauchi S, Inaba Y, Kobayashi D, Wada K. Concentrations of various forms of vitamin B6 in ginkgo seed poisoning. Brain Dev. 2019;41(3):292–5. [DOI] [PubMed] [Google Scholar]
- 137.Said HM, Ortiz A, Ma TY. A carrier-mediated mechanism for pyridoxine uptake by human intestinal epithelial Caco-2 cells: regulation by a PKA-mediated pathway. Am J Physiol Cell Physiol. 2003;285:C1219–25. [DOI] [PubMed] [Google Scholar]
- 138.Zhang L, Zhang H, Zhao Y, Li Z, Chen S, Zhai J, Chen Y, Xie W, Wang Z, Li Qet al. Inhibitor selectivity between aldo-keto reductase superfamily members AKR1B10 and AKR1B1: role of Trp112 (Trp111). FEBS Lett. 2013;587:3681–6. [DOI] [PubMed] [Google Scholar]
- 139.Thul PJ, Lindskog C. The human protein atlas: a spatial map of the human proteome. Protein Sci. 2018;27(1):233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rižner TL, Penning TM. Role of aldo-keto reductase family 1 (AKR1) enzymes in human steroid metabolism. Steroids. 2014;79:49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Barski OA, Tipparaju SM, Bhatnagar A. The Aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab Rev. 2008;40(4):553–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kobayashi T, Kaneko T, Iuchi Y, Matsuki S, Takahashi M, Sasagawa I, Nakada T, Fujii J. Localization and physiological implication of aldose reductase and sorbitol dehydrogenase in reproductive tracts and spermatozoa of male rats. J Androl. 2002;23:674–83. [PubMed] [Google Scholar]
- 143.Kaido M, Mori K, Ide Y, Inoue N, Koide O. Testicular damage by high doses of vitamin B6 (pyridoxine) in rats: a light and electron microscopical study. Exp Mol Pathol. 1991;55:63–82. [DOI] [PubMed] [Google Scholar]
- 144.Shayakhmetova GM, Bondarenko LB, Voronina AK, Anisimova SI, Matvienko AV, Kovalenko VM. Induction of CYP2E1 in testes of isoniazid-treated rats as possible cause of testicular disorders. Toxicol Lett. 2015;234:59–66. [DOI] [PubMed] [Google Scholar]
- 145.Tang WH, Martin KA, Hwa J. Aldose reductase, oxidative stress, and diabetic mellitus. Front Pharmacol. 2012;3:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kern TS, Engerman RL. Immunohistochemical distribution of aldose reductase. Histochem J. 1982;14:507–15. [DOI] [PubMed] [Google Scholar]
- 147.Laskar AA, Younus H. Aldehyde toxicity and metabolism: the role of aldehyde dehydrogenases in detoxification, drug resistance and carcinogenesis. Drug Metab Rev. 2019;51(1):42–64.. [DOI] [PubMed] [Google Scholar]
- 148.Chen W-D, Zhang Y. Regulation of aldo-keto reductases in human diseases. Front Pharmacol. 2012;3:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Singh RP, Cheng Y-H, Nelson P, Zhou FC. Retentive multipotency of adult dorsal root ganglia stem cells. Cell Transplant. 2009;18:55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Stierli S, Imperatore V, Lloyd AC. Schwann cell plasticity—roles in tissue homeostasis, regeneration, and disease. Glia. 2019;67:2203–15. [DOI] [PubMed] [Google Scholar]
- 151.Chen P-Y, Tsai Y-W, Chang AY, Chang H-H, Hsiao Y-H, Huang C-W, Sung P-S, Chen B-H, Fu T-F. Increased leptin-b expression and metalloprotease expression contributed to the pyridoxine-associated toxicity in zebrafish larvae displaying seizure-like behavior. Biochem Pharmacol. 2020;182:114294. [DOI] [PubMed] [Google Scholar]
- 152.Tillakaratne NJ, Medina-Kauwe L, Gibson KM. gamma-Aminobutyric acid (GABA) metabolism in mammalian neural and nonneural tissues. Comp Biochem Physiol A Physiol. 1995;112:247–63. [DOI] [PubMed] [Google Scholar]
- 153.Kash SF, Johnson RS, Tecott LH, Noebels JL, Mayfield RD, Hanahan D, Baekkeskov S. Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci. 1997;94:14060–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hsu C-C, Davis KM, Jin H, Foos T, Floor E, Chen W, Tyburski JB, Yang C-Y, Schloss JV, Wu J-Y. Association of l-glutamic acid decarboxylase to the 70-kDa heat shock protein as a potential anchoring mechanism to synaptic vesicles. J Biol Chem. 2000;275(27):20822–8. [DOI] [PubMed] [Google Scholar]
- 155.Honnorat J, Saiz A, Giometto B, Vincent A, Brieva L, de Andres C, Maestre J, Fabien N, Vighetto A, Casamitjana Ret al. Cerebellar ataxia with anti-glutamic acid decarboxylase antibodies: study of 14 patients. Arch Neurol. 2001;58:225–30. [DOI] [PubMed] [Google Scholar]
- 156.Saiz A, Blanco Y, Sabater L, González F, Bataller L, Casamitjana R, Ramió-Torrentà L, Graus F. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain. 2008;131:2553–63. [DOI] [PubMed] [Google Scholar]
- 157.Termsarasab P, Thammongkolchai T, Katirji B. Anti-glutamic acid decarboxylase 65 (GAD65)-associated syndromes. Stiff-Person Syndrome Rel Disord. 2020:55–71. [Google Scholar]
- 158.Du X, Hao H, Yang Y, Huang S, Wang C, Gigout S, Ramli R, Li X, Jaworska E, Edwards Iet al. Local GABAergic signaling within sensory ganglia controls peripheral nociceptive transmission. J Clin Invest. 2017;127:1741–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Vargas-Parada A, Loeza-Alcocer E, González-Ramírez R, Rodríguez-Sánchez M, Raya-Tafolla G, Florán B, Felix R, Delgado-Lezama R. γ-Aminobutyric acid (GABA) from satellite glial cells tonically depresses the excitability of primary afferent fibers. Neurosci Res. 2020. [DOI] [PubMed] [Google Scholar]
- 160.Magnaghi V, Parducz A, Frasca A, Ballabio M, Procacci P, Racagni G, Bonanno G, Fumagalli F. GABA synthesis in Schwann cells is induced by the neuroactive steroid allopregnanolone. J Neurochem. 2010;112:980–90. [DOI] [PubMed] [Google Scholar]
- 161.Serrano-Regal MP, Bayón-Cordero L, Ordaz RP, Garay E, Limon A, Arellano RO, Matute C, Sánchez-Gómez MV. Expression and function of GABA receptors in myelinating cells. Front Cell Neurosci. 2020;14:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chen JT-C, Guo D, Campanelli D, Frattini F, Mayer F, Zhou L, Kuner R, Heppenstall PA, Knipper M, Hu J. Presynaptic GABAergic inhibition regulated by BDNF contributes to neuropathic pain induction. Nat Commun. 2014;5:5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.King AE, Dickson TC, Blizzard CA, Foster SS, Chung RS, West AK, Chuah MI, Vickers JC. Excitotoxicity mediated by non-NMDA receptors causes distal axonopathy in long-term cultured spinal motor neurons. Eur J Neurosci. 2007;26:2151–9. [DOI] [PubMed] [Google Scholar]
- 164.Saggu SK, Chotaliya HP, Blumbergs PC, Casson RJ. Wallerian-like axonal degeneration in the optic nerve after excitotoxic retinal insult: an ultrastructural study. BMC Neurosci. 2010;11:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ramírez-Jarquín UN, Tapia R. Chronic GABAergic blockade in the spinal cord in vivo induces motor alterations and neurodegeneration. Neuropharmacology. 2017;117:85–92.. [DOI] [PubMed] [Google Scholar]
- 166.Krinke G, Naylor DC, Skorpil V. Pyridoxine megavitaminosis: an analysis of the early changes induced with massive doses of vitamin B6 in rat primary sensory neurons. J Neuropathol Exp Neurol. 1985;44:117–29. [PubMed] [Google Scholar]
- 167.Saggu SK, Chotaliya HP, Cai Z, Blumbergs P, Casson RJ. The spatiotemporal pattern of somal and axonal pathology after perikaryal excitotoxic injury to retinal ganglion cells: a histological and morphometric study. Exp Neurol. 2008;211:52–8. [DOI] [PubMed] [Google Scholar]
- 168.Janssen SP, Truin M, Van Kleef M, Joosten EA. Differential GABAergic disinhibition during the development of painful peripheral neuropathy. Neuroscience. 2011;184:183–94. [DOI] [PubMed] [Google Scholar]
- 169.Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci. 2002;22:6724–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Naik AK, Pathirathna S, Jevtovic-Todorovic V. GABAA receptor modulation in dorsal root ganglia in vivo affects chronic pain after nerve injury. Neuroscience. 2008;154:1539–53. [DOI] [PubMed] [Google Scholar]
- 171.Lever I, Cunningham J, Grist J, Yip PK, Malcangio M. Release of BDNF and GABA in the dorsal horn of neuropathic rats. Eur J Neurosci. 2003;18:1169–74. [DOI] [PubMed] [Google Scholar]
- 172.Hasannejad F, Ansar MM, Rostampour M, Mahdavi Fikijivar E, Khakpour Taleghani B. Improvement of pyridoxine-induced peripheral neuropathy by Cichorium intybus hydroalcoholic extract through GABAergic system. J Physiol Sci. 2019;69:465–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hwang JH, Yaksh TL. The effect of spinal GABA receptor agonists on tactile allodynia in a surgically-induced neuropathic pain model in the rat. Pain. 1997;70(1):15–22. [DOI] [PubMed] [Google Scholar]
- 174.Xiong L-L, Qin Y-X, Xiao Q-X, Jin Y, Al-Hawwas M, Ma Z, Wang Y-C, Belegu V, Zhou X-F, Xue L-Let al. MicroRNA339 targeting PDXK improves motor dysfunction and promotes neurite growth in the remote cortex subjected to spinal cord transection. Front Cell Dev Biol. 2020;8:577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Shen Y, Meiri K. GAP-43 dependency defines distinct effects of netrin-1 on cortical and spinal neurite outgrowth and directional guidance. Int J Dev Neurosci. 2013;31:11–20. [DOI] [PubMed] [Google Scholar]
- 176.Denny JB. Molecular mechanisms, biological actions, and neuropharmacology of the growth-associated protein GAP-43. Curr Neuropharmacol. 2006;4:293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Strittmatter SM, Fankhauser C, Huang PL, Mashimo H, Fishman MC. Neuronal pathfinding is abnormal in mice lacking the neuronal growth cone protein GAP-43. Cell. 1995;80:445–52. [DOI] [PubMed] [Google Scholar]
- 178.Dent EW, Meiri KF. Distribution of phosphorylated GAP-43 (neuromodulin) in growth cones directly reflects growth cone behavior. J Neurobiol. 1998;35:287–99. [DOI] [PubMed] [Google Scholar]
- 179.van Dam S, Võsa U, van der Graaf A, Franke L, de Magalhães JP. Gene co-expression analysis for functional classification and gene-disease predictions. Brief Bioinform. 2018;19:575–92. [DOI] [PMC free article] [PubMed] [Google Scholar]