

Abstract
The recent outbreak of COVID-19, caused by the novel pathogen SARS-coronavirus 2 (SARS-CoV-2) is a severe health emergency. In this pandemic, drug repurposing seems to be the most promising alternative to identify effective therapeutic agents for immediate treatment of infected patients. The present study aimed to evaluate all the drugs present in drug bank as potential novel SARS-CoV-2 inhibitors, using computational drug repurposing studies. Docking-based virtual screening and binding energy prediction were performed, followed by Absorption Distribution Metabolism Excretion calculation. Hydroxychloroquine and Nelfinavir have been identified as the best potential inhibitor against the SARS-CoV-2, therefore, they were used as reference compounds in computational DR studies. The docking study revealed 13 best compounds based on their highest binding affinity, binding energy, and dock score concerning the other screened compounds. Out of 13, only 4 compounds were further shortlisted based on their binding energy and best ADME properties. The hierarchical virtual screening yielded the best 04 drugs, DB07042 (compound 2), DB13035 (compound 3), DB13604 (compound 5) and DB08253 (compound 6), with commendable binding energies in kcal/mol, i.e. −65.45, −62.01, −52.09 and −51.70 respectively. Further, Molecular dynamics simulation with 04 best-retrieved hits has confirmed stable trajectories in protein in terms of root mean square deviation and root mean square fluctuation. During 30 ns simulation, the interactions were also found similar to the docking-based studies. However, clinical studies are necessary to investigate their therapeutic use against this outbreak.
Keywords: Binding energy, COVID, Docking, Drug repurposing, Virtual screening
Abbreviations: ACE, Angiotensin-Converting Enzyme; ADME, Absorption Distribution Metabolism Excretion; CoV, Corona Virus; CDR, Computational Drug Repurposing; HTVS, High-throughput virtual screening; MMGBSA, Molecular mechanics generalized born surface area; OPLS, Optimized Potentials for Liquid Simulations; PDB, Protein data bank; SARS, Severe Acute Respiratory Syndrome; SP, Standard Precision; XP, Extra precision
Graphical abstract

1. Introduction
The recent COVID-19 health emergency caused by the new SARS-CoV-2 has developed an immediate need to identify therapeutic agents against the SARS-CoV-2 and thereby, could put halt to its spread across the world. Coronavirus (CoVs) are enveloped viruses with a positive RNA genome, belonging to the Coronaviridae family of the order Nidovirales, which are divided into four genera (α, β, γ, and δ) (Guarner, 2020; Li et al., 2020; X. Xu et al., 2020). The SARS-CoV-2 belongs to the β genus. CoVs mainly consist of four structural proteins: spike (S) protein, envelope (E) protein, membrane (M) protein, and nucleocapsid (N) protein (El-Demerdash et al., 2021; Ibrahim et al., 2021; Mhatre et al., 2021; Hoffmann et al., 2020). Among them, spikes promote host attachment and virus-cell membrane fusion during viral infection. In addition, perpetually, viruses bind to target proteins present on the cellular surface. Similarly, the SARS virus binds to the human host receptors i.e. angiotensin-converting enzyme 2 (ACE2) receptor for entry of viruses to the human cells (J Alsaadi and Jones, 2019; F. Li, 2016; McBride et al., 2014; W. Li et al., 2005; Prabakaran et al., 2004). The structure-based drug discovery could be utilized to identify the promising compounds for the blockade of formation of SARS-CoV-2 S-protein: human ACE2 complex that facilitates this virus entry in the human body (Mhatre et al., 2021; Hoffmann et al., 2020). Currently, no specific therapies for COVID-19 are available and investigations regarding the treatment of COVID-19 are lacking (Neupane et al., 2021; X. Xu et al., 2020). However, the measures that have been implemented remain limited to preventive and supportive therapies. Common symptoms of a person infected with coronavirus include respiratory symptoms, fever, cough, shortness of breath, and dyspnea. In more severe cases, the infection can cause pneumonia, severe acute respiratory syndrome, kidney failure, and even death (Catharine I. Paules, HilaryD. Marston, 2020).
In this pandemic situation where novel therapeutics is required immediately, the most efficient way to investigate the treatment options seems to be the re-purposing of therapeutic drugs. Several drugs have already been tested, among which hydroxychloroquine and Nelfinavir, are among the promising drugs which have shown efficacy in the treatment of COVID-19 associated respiratory problems in recent clinical studies (Baildya et al., 2020; Colson et al., 2020; Gautret et al., 2020; Musarrat et al., 2020; Nicola Principi, 2020; Sahraei et al., 2020; Shah et al., 2020; Z. Xu et al., 2020; Yamamoto et al., 2004; Colson et al., 2020; X. Xu et al., 2020; Z. Xu et al., 2020; Baildya et al., 2020; Colson et al., 2020; Gautret et al., 2020).
In the present study, drug repurposing using in silico studies has been used, in which all drugs of drug bank were subjected to the ensemble docking campaign and binding energy prediction to identify hit candidate that could bind at the active site of SARS-CoV-2 protein (6LU7) and may cause inhibition of SARS-CoV-2 (Amaro et al., 2018; Van Norman, 2016; Choudhary and Silakari, 2020; Amaro et al., 2018; Van Norman, 2016). Further, the present studies were able to identify thirteen top-ranked compounds, which are currently available and have had either regulatory approval as drugs or had multiple prior studies that indicate high potential for therapeutic use. Out of the 13 drugs selected, based on their binding affinity and interactions with SARS-CoV-2, 04 best drug hits were shortlisted that were also compared with currently available standard drugs against COVID-19 i.e. Hydroxychloroquine and Nelfinavir. These four drugs have a similar type of protein-ligand interaction in the binding pocket of SARS-CoV-2 (6LU7) protein as standard drugs (Hydroxychloroquine and Nelfinavir) and thus, could serve as appropriate candidates for immediate experimental studies.
2. Material and methodology
The X-Ray crystal structure of SARS-CoV-2 which causes COVID-19 was chosen from protein databank (PDB ID 6LU7) as docking template because it was found to have good resolution of 2.1 Å (Jin et al., 2020a, 2020b, 2020b). The protein preparation wizard was used to correct the addition of hydrogens, disulfide bonds establishment, assigning bond orders, and deletion of water molecules beyond 5 Å. Further, steric hindrances in the target site were removed by optimizing and minimizing the hydrogen bonds. Moreover, the missing side chains and loops were used for filling through the Prime module (Roos et al., 2019; Genheden and Ryde, 2015). Docking studies were performed on a prepared X-Ray crystal structure of SARS-CoV-2 which was chosen from protein data bank (PDB ID 6LU7).
2.1. Grid generation
3D grid was generated at the N3 site which is a Michael acceptor inhibitor having (cytotoxicity concentration) CC50 > 133 μM and the substrate-binding site is situated between domain I and II as reported in previous works of literature (Jin et al., 2020a, 2020b, 2020b). The generation of the 3D grid was further done by using SiteMap analysis, which provides information about the size, hydrophilic and hydrophobic areas of the binding site. Hydrogen bond acceptors, as well as donor areas on ligand or receptor, are also evaluated by SiteMap analysis (Halgren, 2007).
2.2. Computational screening
The chemical structures downloaded from drug bank (https://www.drugbank.ca/releases/latest#structures), consists of 8,135 numbers of compounds which included all approved drugs along with hydroxychloroquine and nelfinavir. The structures were prepared using the Ligprep module of the Schrodinger package for desalting, cleaning, and adding up hydrogen atoms. Subsequently, the generation of various stereoisomers, tautomers, and ionization states at pH 7 ± 2 was done. The out.mae file of prepared drug compounds was then subjected for computational drug repurposing. Most of the looming drugs were recognized from docking-based virtual screening which may act against SARS-CoV-2. These studies were performed on the grid of 6LU7 using virtual hierarchical screening workflow of GLIDE module version 8.2 (ADME, Lipinski filter, HTVS, SP, XP) for identification of compounds based on protein-ligand interactions and ligand binding affinity in energy terms. The hierarchal virtual screening was set at 20% out ligands from HTVS, 20% out from SP, and 10% out ligands from XP mode of docking. Calculation of the Epik-based state penalties which generate the possible states of ligands was set in docking-based virtual screening. The count of penalties in HTVS was lowest whereas, in XP mode of docking, the count of penalties was higher. In docking-based virtual screening protocol, the dock flexibility was set as default i.e. penalizing non planar confirmations for amide bonds.
2.3. Molecular Mechanics studies
Retrieved drug compounds as hits against SARS-CoV-2 (6LU7) were taken for relative binding energy prediction from protein-ligand complexes using the prime module of Schrodinger software 2019(1). The physics-centered technique that drives the force field in an implicit solvent of the bound and unbound candidates participating in the binding process is well known as MM-GBSA (Molecular Mechanics, the Generalized Born model, and Solvent Accessibility). The MMGBSA binding energy consists of terms such as protein-ligand van Der Waals associates, electrostatic contacts, ligand desolvation, and internal strain (ligand and protein) energies. Determination of binding energy was done by VSGB2.0 implicit solvent model in prime with the help of force field (OPLS3e) (Halgren, 2007; Genheden and Ryde, 2015). The collective hits achieved from the docking-based screening were further studied to the MM-GBSA exploration. In the course of these calculations, the active site of protein was established to amend itself up to a distance of 5 Å for ligand accordingly. This procedure imported the pose viewer file of XP docked mode of the protein-ligand complex in docking, which resulted in the ligands ranking based on predicted binding energies (Humphrey et al., 1996; Malik et al., 2016).
2.4. ADME predictions
Many of the drugs are rejected due to their abnormal pharmacokinetic features that may include alarming absorption, distribution, metabolism, or excretion (ADME) parameters. Therefore, the pharmacokinetic features of the best-obtained candidates were assessed using the QikProp module version5.9 by conforming to the rule of five and three to evaluate some vital properties such as blood-brain barrier permeability, polar surface area, human oral absorption percentage, and molecular weight (Chatterjee et al., 2014, Dzierba et al., 2007).
2.5. Molecular dynamics simulation study
Molecular Dynamics simulation study was performed using Desmond v3.8, the 04 retrieved protein-ligand complexes were first solvated into explicit SPC (simple point charge) water model in an orthorhombic box. Counter ions were neutralizing the system, further, minimization of the complex system of 6LU7 with DB07042, DB08253, DB13604, and DB13035 was done which consist of 36914, 36866, 36905, and 36920 atoms respectively. The MD has performed in the NPT (Normal Pressure Temperature) ensemble for 30 ns with recording intervals of 1.2 ps for energy and 4.8 ps for trajectory. Simulations were run under the OPLS-2005 force field. The workflow of exhaustive virtual screening and Simulation was depicted in Fig. 1 .
Fig. 1.

Methodology of Structure-based virtual screening.
3. Results and discussion
The intent of the current research was to implement target-based virtual screening of drugbank database on SARS-CoV-2 (PDB ID 6LU7) to obtain promising hit drugs. The active site of 6LU7 i.e. N3 site and was validated based on site score Further, based on the predicted binding energy, insilico ADME along with the protein-ligand interactions, 04 hit drugs was achieved which may act against the novel coronavirus.
3.1. Validation of site of SARS-CoV-2 (6LU7)
The grid at the N3 site of 6LU7 was successfully generated which was further validated by using SiteMap and revealing vital information from this validation study. The results of the SiteMap study were signified in the arrangement of site points which seem like the white grey translucent area within the receptor. The yellow mesh showed the hydrophobic nature and the green mesh shows a hydrophilic map. The hydrogen-bond donor points were shown in blue mesh whereas the hydrogen-bond acceptors were presented by red mesh as depicted in Fig. 2 a and b.
Fig. 2.

SiteMap analysis a) Illustration of hydrogen donor and acceptor points; b) Illustration of Hydrophilic and hydrophobic meshes.
3.2. Structure-based screening
The docking-based approach of virtual screening was used in a hierarchical mode of elimination technique, i.e. ADME, Lipinski filter, high throughput virtual screening (HTVS) followed by Standard precision (SP) and the extra precision (XP). PDB ID 6LU7 was chosen for targeting COVID-19 in structure-based virtual screening, and it indicated that the 8135 drugs from different pharmacological categories were able to form bonds that interact with different amino acids in the active site of 6LU7 for the computational model of drug repurposing. Hydroxychloroquine (anti-malarial) and Nelfinavir (HIV-1 protease inhibitor) were found to act against COVID-19. Therefore, they were XP docked and prediction of binding energy was done for the finding of essential interacting residues, Hydroxychloroquine showed interaction with Glu290, Asp289, Lys137, Tyr126, Gln127 residues while Nelfinavir showed Tyr154, Asp153, Asn151, Ile152, Gln110, Phe294 interactions as showed in Fig. 3, Fig. 4 . Structure-based virtual screening and binding energy prediction gave 13 drug compounds with structural diversity.
Fig. 3.

2D and3D interaction of Nelfinavir with 6LU7.
Fig. 4.

2D and 3D interaction of Hydroxychloroquine with 6LU7.
3.3. Interpretation of hierarchical structure-based screening
The scrutinizing process included protein-ligand interaction study and predicted binding energy. The docking-based screening of 8,135 compounds resulted in the identification of 13 drugs, out of which 04 best drugs were selected based on interactions and binding affinity as listed in Table 1, Table 2 . These 04 drug hits may act against COVID-19 as they possess similar protein-ligand interaction in the binding pocket as Hydroxychloroquine and Nelfinavir with 6LU7.
Table 1.
Data of hit drugs with their protein-ligand interactions.
| Sr.No. | Name of Drug/Drug Bank ID | Dock Score (kcal/mol) | Binding energy (kcal/mol) | Protein-Ligand Interactions |
|---|---|---|---|---|
| 1 | DB07212 | −6.150 | −60.397 | Gln127, Glu209, Lys137, Gly170, Gly138 |
| 2 | DB07908 | −4.755 | −57.268 | Asp153, Phe294, Thr111 |
| 3 | DB13779 | −4.894 | −48.362 | Gln127, Glu290, Lys137 |
| 4 | DB07206 | −5.429 | −45.706 | Asp153, Phe294 |
| 5 | DB13211 | −4.735 | −43.825 | Gln127, Glu290, Lys137 |
| 6 | DB12890 | −6.594 | −43.703 | Gln110, Thr111, Phe294, Lys102, Asp153 |
| 7 | DB06753 | −4.648 | −37.126 | Glu290, Lys137, Lys5 |
| 8 | DB08178 | −4.814 | −24.803 | Lys5, Lys137 |
| 9 | DB01140 | −4.644 | −22.589 | Gln127, Glu290, Lys137 |
Table 2.
Scores of reference drugs and best hit drugs with their structures and protein-ligand interactions.
| Name of Drug/Drug Bank ID | Structures of Drugs | Dock Score (kcal/mol) | Binding energy (kcal/mol) | Protein-Ligand Interactions |
|---|---|---|---|---|
| Nelfinavir | 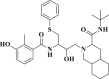 |
−6.139 | −77.128 | Asn151, Asp153, Tyr154 |
| DB07042 | 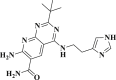 |
−4.647 | −65.453 | Asn151, Phe294, Gln110, Ile152, Asp153, Ser158 |
| DB13035 | 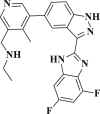 |
−5.138 | −62.015 | Asp153, Phe8, Tyr154, Phe294 |
| Hydroxychloroquine | 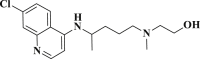 |
−3.612 | −50.052 | Glu290, Asp289, Lys137, Tyr126 |
| DB13604 | 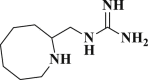 |
−5.129 | −52.096 | Lys137, Glu290, Gln127, Tyr126 |
| DB08253 | 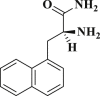 |
−4.625 | −51.708 | Lys137, Gln127, Glu290, Tyr126 |
DB07042 (compound 2) and DB13035 (compound 3) showed a similar type of interactions with 6LU7 protein as Nelfinavir whereas, DB13604 (compound 5) and DB08253 (compound 6) showed Hydroxychloroquine type of interactions with the protein as showed in Fig. 1, Fig. 2. Identical protein-ligand interactions of DB07042 (compound 2) and DB13035 (compound 3) were matched with Nelfinavir while DB13604 (compound 5) and DB08253 (compound 6) were matched with Hydroxychloroquine with 6LU7. These four drugs have comparable scores and admissible ADME features with 2D structural similarity as the reference drug compounds. Compound 2 showed the formation of hydrogen bonding with Asn151, Gln110, Ile152, Asp153, Ser158 while Phe294 was found to be involved in pi-pi bonding. Compound 3 had established hydrogen bonding with Asn151, Asp153, Tyr154 while Phe8, Phe294 showed pi-pi interactions with the protein. Compound 5 formed hydrogen bonding interactions with Lys137, Glu290, Gln127 while Tyr126 showed pi-pi bonding whereas compound 6 was involved in hydrogen bonding interactions with Lys137, Gln127, Glu290 and Tyr126 showed pi-pi interaction. These hit drug candidates and may serve as effective and potential drugs against the novel CORONA virus as depicted from Fig. 5, Fig. 6, Fig. 7, Fig. 8 . Their dock scores range from −4.625 to −5.138 kcal/mol and the dGBind score ranges between −51.708 and −65.453 kcal/mol, which was very nearby to the scores of hydroxychloroquine and nelfinavir and listed in Table 2. Therefore, may also be helpful in the new drug development process and thus, restrict this epidemic condition. Four major pharmacokinetic concerns (ADME) persuade the kinetics of drug experience to the tissues and thus influence the potential of the drug molecule. In silico pharmacokinetic properties of the hits were predicted using Qikprop software and tabulated in Table 3 . Oral administration and bioavailability depend on the aqueous solubility which was recorded in all retrieved hit drugs in the range i.e. −6.5 to 0.5 mol dm-3. Due to admissible predicted solubility, the Percentage of human oral absorption was also good and scaled from 45% to 90%. The predicted gut blood barrier which is non-active transport and these four hit drugs seems in the admissible range i.e. 25 nm/s to 500 nm/s. These hit drug candidates were reported as experimental or investigational drugs in the drugbank database and may help against the novel CORONA virus. DB13604, which is also a lead compound and known as AG-24322; an investigational drug that was tested against the cancer cells as a cell cycle inhibitor whereas, all other three hit drugs were in the experimental category and their pharmacological action was unrevealed in the database.
Fig. 5.

2D and 3D interaction of DB07042 with 6LU7.
Fig. 6.

2D and 3D interaction of DB13035 with 6LU7.
Fig. 7.

2D and 3D interaction of DB13604 with 6LU7.
Fig. 8.

2D and3D interaction of 08253 with 6LU7.
Table 3.
Predicted ADME Scores of selected hit drugs.
| Drug Bank ID | Mol.Wt. | logPo/w | logS | logHERG | PCaco | logBB | PMDCK | PercentHumanOralAbsorption | logKp |
|---|---|---|---|---|---|---|---|---|---|
| DB07042 | 354.414 | 1.093 | −3.802 | −5.074 | 45.02 | −2.287 | 17.332 | 49.979 | −4.894 |
| DB13035 | 418.448 | 3.853 | −5.506 | −7.053 | 145.67 | −0.436 | 205.849 | 88.228 | −4.51 |
| DB13604 | 184.284 | −0.044 | −0.509 | −3.796 | 105.815 | −0.393 | 48.293 | 62.924 | −8.016 |
| DB08253 | 214.266 | 0.057 | 0.246 | −3.674 | 66.012 | −0.427 | 53.321 | 59.846 | −4.892 |
logP o/w: Predicted octanol/water partition coefficient (−2.0–6.5).
logS: Predicted aq.solubility (−6.5–0.5).
logHERG: Predicted IC50 value for blockage of HERG K + channel (below −5).
QPPCaco: Caco-2 cell permeability in nm/s. Caco-2 cells are a model for the gut–blood barrier (<25 poor, >500 great).
logBB: Predicted brain/blood partition coefficient (−3.0–1.2).
Percent human oral absorption predicted human oral absorption on 0–100% scale (>80% high, <25% poor).
MDCK predicted apparent MDCK cell permeability in nm/sc. MDCK cells are considered to be a good mimic for the blood–brain barrier <25 poor, >500 great
logKp: Predicted skin permeability (−8.0–1.0)
3.4. Molecular dynamics simulations studies
Different facets were assessed in these studies, which incorporate the stability of protein-ligand complex by studying root mean square deviation (RMSD) and root mean square fluctuation (RMSF) of backbone. 30 ns Molecular dynamics simulation studies of 6LU7 with 04 hit candidates were conducted to analyze the protein-ligand interactions as obtained from extensive computational screening.
The trajectory analysis revealed the RMSD plot which was found stable throughout the simulation with all hits. RMSD and RMSF were recorded differently in individual P-L complexes which were found below 2.4 Å and approximately 2.0 Å respectively for all 04 hits. The results of MD simulations are shown comprehensively in Table 4 and Fig. 9, Fig. 10, Fig. 11 .
Table 4.
Comprehensive MD simulation results of complex of retrieved hits with 6LU7.
| 6LU7 Complexes with Hits | Counter Ions | RMSD (Å) | RMSF (Å) | P-L Contacts & Percentage Occupancy |
|---|---|---|---|---|
| DB07042 | +32 Na, −30Cl | 2.4 | <1.5 | Cys160(53%), Thr111(86%), Ser158(60%), Gln110(80%), Phe294(50%), Asp248(50%), Ile106(40%), Asn151(19%), Ile152(8%), Asp153(10%) |
| DB13035 | +32 Na, −30Cl | 2.4 | <2.4 | Asp153(79%), Tyr154(65%), Phe294(38%), Thr111(40%), Asn151(39%), Phe8(45%), Gln110(10%), Ile152(15%), Ser158(10%) |
| DB13604 | +31 Na, −30Cl | 1.8 | <1.5 | Gln288(>100%), Gln290(80%), Asp289(98%), Lys137(35%), Gln127(10%), Tyr126(2%) |
| DB08253 | +32 Na, −30Cl | 1.6 | <1.5 | Gln288(>100%), Gln290(>100%), Gln127(60%), Lys5(99%) Lys137(21%), Tyr126(30%), Asp(2%) |
Fig. 9.

Root mean square deviation plots of protein-ligand complexes.
a) 6LU7-DB07042; b) 6LU7-DB13035; c) 6LU7-DB13604; d) 6LU7-DB08253.
Fig. 10.

Root mean square fluctuation plots of protein-ligand complexes
a) 6LU7-DB07042; b) 6LU7-DB13035; c) 6LU7-DB13604; d) 6LU7-DB08253.
Fig. 11.

Protein-ligand interactions and their percentage occupancy.
a) 6LU7-DB07042; b) 6LU7-DB13035; c) 6LU7-DB13604; d) 6LU7-DB08253.
The fraction pattern in contrast to timeline representation was found favorable throughout the simulation. The individual docking interactions of 6LU7 with DB07042 and DB13035 were validated and found similar in the 30ns MD simulation study. Significant interactions of amino acid residues on active site of 6LU7 with DB07042 through different bonding patterns such as Ser158, Cys160, Asp248, Gln110, Thr111 formed direct hydrogen bonding and through water bridging, while, hydrophobic interactions were seen with Phe294, Ile106, and Ile249. DB13035 showed Thr111, Asp153, Asn151, Ser301, and Arg298 take part in hydrogen bond formation and water bridging, whereas, Phe8, Tyr154, Phe294 formed hydrophobic contacts. Similarly, DB13604 and DB08253 resulted in significant interactions having various types of bond with amino acids such as Gln288, Gln290, Gln127, and Lys137. These amino acids majorly interact and formed hydrogen bonds along with the water bridging in which ionic exchange was also found in Gn288 and Gln290 for a very short duration.
3.5. Structural integrity and chemical template analysis
Two-dimensional structural features of Nelfinavir and Hydroxychloroquine were matched with obtained hit drugs and it was observed that these compounds have shape similarity with identical types of chemical features and thus, they have been depicted in the same colors as that of reference drugs as depicted in Fig. 12 . In the case of Nelfinavir, the pink region showed the substituted heterocyclic moiety, orange-colored region indicated branching with heteroatom which may be replaceable with chain shortening or heterocyclic ring as linker while the blue and green colored region is useful for hydrophobic interactions. Similarly, in the case of Hydroxychloroquine, the yellow-colored region shows the necessity of cyclic moiety (quinoline, naphthalene, azocane), blue region acts as a linker between cyclic moiety and groups responsible for hydrogen bond formation as depicted in light green color, the linker may have a longer or shorter chain of carbon with an amino group. Additionally, differences in the 3D orientation but the fitness of Nelfinavir (grey) in the binding pocket with compound 2 (orange) and compound 3 (green) were noticeably seen in surface view of pictorial representation of binding cavity, whereas, Hydroxychloroquine was showed in the grey colored binding pocket and was well overlapped with compound 5 (red) and compound 6 (purple) as showed in Fig. 13 .
Fig. 12.

Shape similarity with identical chemical features of Nelfinavir and Hydroxychloroquine with obtained hits.
Fig. 13.

Different 3D-orientation of drugs with their fitness in the binding pocket of 6LU7; (a) Hydroxychloroquine with DB13604 and DB08253 (b) Nelfinavir with DB07042 and DB13035.
4. Conclusion
The worldwide novel coronavirus health emergency has emerged as a potential threat to global health. Moreover, the major problem lies in the unavailability of any approved drug against SARS-CoV-2 infection. The in silico drug discovery through drug repurposing could prove a fast and most appropriate option to find a potential therapeutic solution for the identification of drugs against SARS-CoV-2. In the present study, the site of SARS-CoV-2 protein (6LU7) i.e. N3 site was validated from sitemap analysis and based on site score selected for virtual screening. Hydrophobic, hydrophilic regions of the active site along with the hydrogen bond donor and acceptor regions were also analyzed using sitemap which will be useful for the designing of highly efficacious novel selective inhibitors. Disparate docking module (HTVS, SP, XP) based screening of 8,135 drug compounds was performed and retrieve 13 hits which were further passed from scrutinizing process (binding energy and P-L interactions) hence, top four best hit drugs were selected that may bind to and inhibit SARS-CoV-2 activity. Insight the course of study, data of molecular dynamics simulation studies revealed the stable complex formation of 04 best-retrieved hit drugs with the target protein, with RMSD and RMSF below 2.4 and 2.0, respectively. Besides these, the interactions were maintained and found similar as in docking studies during 30 ns simulation. Present work indicates that the four identified compounds may have a reasonable positive preliminary effect in limiting SARS-CoV-2 prevalence. However, these four hit candidates were in different phases of biological studies as they are under experimental or investigational class thus, pre-clinical and clinical experimental studies are required to validate these potential inhibitors for use as a potential clinical treatment. The Computational Drug Repurposing (CDR) anticipates and may prove valuable for exploring and developing novel anti-COVID-19 therapeutic agents in the future.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Virendra Nath: Conceptualization, Methodology, Research Work, Writing – original draft. A. Rohini: Support in scientific writing. Vipin Kumar: Supervision.
References
- Amaro R.E., Baudry J., Chodera J., Demir Ö., McCammon J.A., Miao Y., Smith J.C. Ensemble docking in drug discovery. Biophys. J. 2018;114:2271–2278. doi: 10.1016/j.bpj.2018.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baildya N., Ghosh N.N., Chattopadhyay A.P. Inhibitory activity of hydroxychloroquine on COVID-19 main protease: an insight from MD-simulation studies. J. Mol. Struct. 2020;1219:128595. doi: 10.1016/j.molstruc.2020.128595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C S., S D.K., Ragunathan V., Tiwari P., A S., Brindha Devi B.D. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J. Biomol. Struct. Dyn. 2020:1–27. doi: 10.1080/07391102.2020.1815584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A., Cutler S.J., Doerksen R.J., Khan I.A., Williamson J.S. Discovery of thienoquinolone derivatives as selective and ATP non-competitive CDK5/p25 inhibitors by structure-based virtual screening. Bioorg. Med. Chem. 2014;22:6409–6421. doi: 10.1016/j.bmc.2014.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary S., Silakari O. Scaffold morphing of arbidol (umifenovir) in search of multi-targeting therapy halting the interaction of SARS-CoV-2 with ACE2 and other proteases involved in COVID-19. Virus Res. 2020;289:198146. doi: 10.1016/j.virusres.2020.198146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colson P., Rolain J.M., Lagier J.C., Brouqui P., Raoult D. Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. Int. J. Antimicrob. Agents. 2020 doi: 10.1016/j.ijantimicag.2020.105932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzierba C.D., Tebben A.J., Wilde R.G., Takvorian A.G., Rafalski M., Kasireddy-Polam P., Klaczkiewicz J.D., Pechulis A.D., Davis A.L., Sweet M.P., Woo A.M., Yang Z., Ebeltoft S.M., Molski T.F., Zhang G., Zaczek R.C., Trainor G.L., Combs A.P., Gilligan P.J. Dihydropyridopyrazinones and dihydropteridinones as corticotropin-releasing factor-1 receptor antagonists: structure-activity relationships and computational modeling. J. Med. Chem. 2007;50:2269–2272. doi: 10.1021/jm0611410. [DOI] [PubMed] [Google Scholar]
- El-Demerdash A., Metwaly A.M., Hassan A., El-Aziz T.M.A., Elkaeed E.B., Eissa I.H., Arafa R.K., Stockand J.D. Comprehensive virtual screening of the antiviral potentialities of marine polycyclic guanidine alkaloids against sars-cov-2 (Covid-19) Biomolecules. 2021;11:1–25. doi: 10.3390/biom11030460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautret P., Lagier J.C., Parola P., Hoang V.T., Meddeb L., Mailhe M., Doudier B., Courjon J., Giordanengo V., Vieira V.E., Tissot Dupont H., Honoré S., Colson P., Chabrière E., La Scola B., Rolain J.M., Brouqui P., Raoult D. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents. 2020;56:105949. doi: 10.1016/j.ijantimicag.2020.105949. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Genheden S., Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expet Opin. Drug Discov. 2015;10:449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarner J. Three emerging coronaviruses in Two decades: the story of SARS, MERS, and now COVID-19. Am. J. Clin. Pathol. 2020;153:420–421. doi: 10.1093/ajcp/aqaa029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007;69:146–148. doi: 10.1111/j.1747-0285.2007.00483.x. [DOI] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Krueger N., Mueller M.A., Drosten C., Poehlmann S. The novel coronavirus 2019 (2019-nCoV) uses the SARS-coronavirus receptor ACE2 and the cellular protease TMPRSS2 for entry into target cells. bioRxiv. 2020 doi: 10.1101/2020.01.31.929042. 2020.01.31.929042. [DOI] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VDM: visual molecular dynamics. J. Mol. Graph. 1996;14(33–8):27–28. doi: 10.1016/0263-7855(96)00018-5. plates. [DOI] [PubMed] [Google Scholar]
- Ibrahim M.A.A., Abdelrahman A.H.M., Allemailem K.S., Almatroudi A., Moustafa M.F., Hegazy M.E.F. In silico evaluation of prospective anti-COVID-19 drug candidates as potential SARS-CoV-2 main protease inhibitors. Protein J. 2021 doi: 10.1007/s10930-020-09945-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- J Alsaadi E.A., Jones I.M. Membrane binding proteins of coronaviruses. Future Virol. 2019;14:275–286. doi: 10.2217/fvl-2018-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang Xinglou, You T., Liu Xiaoce, Yang Xiuna, Bai F., Liu H., Liu Xiang, Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. 2020 doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang Xinglou, You T., Liu Xiaoce, Yang Xiuna, Bai F., Liu H., Liu Xiang, Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. bioRxiv. 2020 doi: 10.1101/2020.02.26.964882. 2020.02.26.964882. [DOI] [PubMed] [Google Scholar]
- Li F. Structure, function, and evolution of coronavirus spike proteins. Annual Review of Virology. 2016;3:237–261. doi: 10.1146/annurev-virology-110615-042301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Zhang C., Sui J., Kuhn J.H., Moore M.J., Luo S., Wong S.K., Huang I.C., Xu K., Vasilieva N., Murakami A., He Y., Marasco W.A., Guan Y., Choe H., Farzan M. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24:1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Ma Y., Wang N., Hu Y., Research Z.L.-A.T. Undefined, 2020. New coronavirus-infected pneumonia engulfs Wuhan. Tmrjournals.Com. 2020;2:1–7. doi: 10.12032/atr2020-0202-007. [DOI] [Google Scholar]
- Malik R., Bunkar D., Choudhary B.S., Srivastava S., Mehta P., Sharma M. High throughput virtual screening and in silico ADMET analysis for rapid and efficient identification of potential PAP248-286aggregation inhibitors as anti-HIV agents. J. Mol. Struct. 2016;1122:239–246. doi: 10.1016/j.molstruc.2016.05.086. [DOI] [Google Scholar]
- McBride R., van Zyl M., Fielding B.C. The coronavirus nucleocapsid is a multifunctional protein. Viruses. 2014;6:2991–3018. doi: 10.3390/v6082991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhatre S., Naik S., Patravale V. A molecular docking study of EGCG and theaflavin digallate with the druggable targets of SARS-CoV-2. Comput. Biol. Med. 2021;129:104137. doi: 10.1016/j.compbiomed.2020.104137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musarrat F., Chouljenko V., Dahal A., Nabi R., Chouljenko T., Jois S.D., Kousoulas K.G. The anti-HIV drug nelfinavir mesylate (Viracept) is a potent inhibitor of cell fusion caused by the SARSCoV-2 spike (S) glycoprotein warranting further evaluation as an antiviral against COVID-19 infections. J. Med. Virol. 2020;92:2087–2095. doi: 10.1002/jmv.25985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupane N.P., Karn A.K., Mukeri I.H., Pathak P., Kumar P., Singh S., Qureshi I.A., Jha T., Verma A. Molecular dynamics analysis of phytochemicals from Ageratina adenophora against COVID-19 main protease (Mpro) and human angiotensin-converting enzyme 2 (ACE2) Biocatalysis and Agricultural Biotechnology. 2021;32 doi: 10.1016/j.bcab.2021.101924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabakaran P., Xiao X., Dimitrov D.S. A model of the ACE2 structure and function as a SARS-CoV receptor. Biochem. Biophys. Res. Commun. 2004;314:235–241. doi: 10.1016/j.bbrc.2003.12.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos K., Wu C., Damm W., Reboul M., Stevenson J.M., Lu C., Dahlgren M.K., Mondal S., Chen W., Wang L., Abel R., Friesner R.A., Harder E.D. OPLS3e: extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theor. Comput. 2019;15:1863–1874. doi: 10.1021/acs.jctc.8b01026. [DOI] [PubMed] [Google Scholar]
- Sahraei Z., Shabani M., Shokouhi S., Saffaei A. Aminoquinolines against coronavirus disease 2019 (COVID-19): chloroquine or hydroxychloroquine. Int. J. Antimicrob. Agents. 2020;55:105945. doi: 10.1016/j.ijantimicag.2020.105945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S., Das S., Jain A., Misra D.P., Negi V.S. A systematic review of the prophylactic role of chloroquine and hydroxychloroquine in coronavirus disease-19 (COVID-19) International Journal of Rheumatic Diseases. 2020;23:613–619. doi: 10.1111/1756-185X.13842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Norman G.A. Drugs, devices, and the FDA: Part 1: an overview of approval processes for drugs. JACC (J. Am. Coll. Cardiol.): Basic to Translational Science. 2016;1:170–179. doi: 10.1016/j.jacbts.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Chen P., Wang J., Feng J., Zhou H., Li X., Zhong W., Hao P., et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020;63:457–460. doi: 10.1007/s11427-020-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Peng C., Shi Y., Zhu Z., Mu K., Wang X., Zhu W. Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. bioRxiv. 2020;1201 doi: 10.1101/2020.01.27.921627. 2020.01.27.921627. [DOI] [Google Scholar]
- Yamamoto Norio, Yang R., Yoshinaka Y., Amari S., Nakano T., Cinatl J., Rabenau H., Doerr H.W., Hunsmann G., Otaka A., Tamamura H., Fujii N., Yamamoto Naoki. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem. Biophys. Res. Commun. 2004;318:719–725. doi: 10.1016/j.bbrc.2004.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]