

Abstract
The sphingomyelin pathway is important in cell regulation and determining cellular fate. Inhibition of sphingosine kinase isoform 1 (SK1) within this pathway, leads to a buildup of sphingosine and ceramide, two molecules directly linked to cell apoptosis, while decreasing the intracellular concentration of sphingosine-1-phosphate (S1P), a molecule linked to cellular proliferation. Recently, an inhibitor capable of inhibiting SK1 in vitro was identified, but also shown to be ineffective in vivo. A set of compounds designed to assess the impact of synthetic modifications to the hydroxynaphthalene ring region of the template inhibitor with SK1 to obtain a compound with increased efficacy in vivo. Of these fifteen compounds, 4A was shown to have an IC50 = 6.55 μM with improved solubility and in vivo potential.
Graphical Abstract

Targeted therapy is a developing form of cancer treatment capable of interfering with specific molecular entities involved in cell growth and survival.1 Traditionally, chemotherapeutic agents attack all actively dividing cells, whether they are cancerous or not. Targeted therapies have the ability to attack cancer cells and specific pathways within them, doing little to no harm to the surrounding healthy cells.2 The research described herein focuses on the sphingomyelin pathway (Figure 1), which is important in cell regulation, signaling, and cell fate.3
Figure 1:

Stress induced degradation of sphingomyelin, a component of the cell membrane, to form proapoptotic ceramide and sphingosine as well as proliferative sphingosine-1-phosphate.
One of the therapeutic targets of interest in this pathway is the phosphorylation of sphingosine into sphingosine-1-phosphate (S1P), catalyzed by sphingosine kinase isoform 1 (SK1). Inhibition of SK1 is important because S1P has been shown to promote cell proliferation, angiogenesis, and inflammation. SK1 has also been shown to be overexpressed in cancer cells.4 Inhibition of SK1 should result in a decreased cellular concentration of S1P and a buildup of ceramide and sphingosine, two molecules linked to apoptosis.5 Because of this, SK1 provides a potential target for new anticancer targeted therapeutics.6,7
In this study, modifications to a known SK1 inhibitor are being investigated (Fig. 2). Initial research conducted by Smith et al.4 led to the discovery of a template compound, sphingosine kinase inhibitor-I (SKI-I), which showed success against SK1 in vitro, but, in a follow-up study, not in vivo3, presumably due to its low solubility. The objective of this work focuses on the design and synthesis of potential inhibitors that assess modifications to the hydroxynaphthalene ring (Figure 2) that may improve the in vivo effectiveness as SK1 inhibitors.
Figure 2:

The template inhibitor (SKI-I) compound by Smith et al.4 The area of modification in the current work is shown in the brackets.
Due to the complexity of the SKI-I template structure, several potential areas of chemical modification are available, and we have focused our initial efforts on making synthetic modifications to the hydroxynaphthalene ring (Figure 2), with a specific goal of decreasing the theoretical log P value of the compound thereby increasing its aqueous solubility and in vivo activity. Log P values are good predictors of a compound’s ability to penetrate through the lipid bilayer, a component of the cellular membrane which acts as a barrier for a cell.10–12 SKI-I has a calculated log P value of 5.613, which is too high to be effectively solubilized and circulated through the serum. Candidate modifications made to the SKI-I hydroxynaphthalene ring template were designed to improve the hydrophilic nature of the structure by as much as four times relative to SKI-1 which would give a calculated log P value of at least 5.0.
We designed a library of candidate compounds and obtained initial estimates of their efficacy via in silico docking experiments using the proposed derivatives and the known crystal structure of an inhibitor bound form of the enzyme (PDB ID: 3VZB).9 The inhibitor compounds were prepared following a modified method8 using microwave synthesis to improve time and overall yield. These purified derivatives were then submitted for bioassay testing to assess whether improvements were made in lipid bilayer absorption and penetration and ultimately increased inhibition success rates in vivo.
Previously, the synthetic scheme for the SKI-I template and its derivatives followed a traditional three step process, however this work performed all three synthetic steps in a microwave reactor. These modified inhibitors are synthesized using microwave-assisted organic synthesis (MAOS) to improve yields and promote cleaner reactions in a timely manner which have become more popular as safe alternatives to traditional synthetic strategies with little to no environmental impact and greater efficiency of the reactions.14–17 Utilizing MAOS to synthesize inhibitors with improved hydrophilicity decreasing the overall reaction time while simultaneously increasing purity and product yields. Using MAOS, a total of fifteen SK1 inhibitors with modifications to the SKI-I hydroxynaphthalene ring moiety have been synthesized successfully and are described herein.
In silico docking:
The x-ray crystallographic structure of sphingosine kinase isoform 1 in complex with sphingosine (PDB ID: 3VZB) was used as a base structure for all docking runs. SKI-I and design analogues were built in MarvinSketch.18 Proteins and ligands were converted to PDBQT files using the DockPrep module of Chimera and were submitted for molecular docking with Autodock Vina using the dimensions of the sphingosine binding pocket of SK1 as the receptor. The results of the docking calculations were subsequently viewed in the ViewDock module of Chimera.
In vitro studies:
The inhibitors were tested in biochemical kinase activity assays using the Invitrogen SelectScreen Kinase Profiling Service (LifeTechnologies Corporation, Madison, WI) to determine IC50 values.17 The compounds were assayed at a starting concentration of 10μM in 1% DMSO with threefold serial dilutions at [Km,app] ATP, which was conducted at 100μM ATP. These inhibitors were evaluated against a known inhibitor of sphingosine kinase 1, PF-54319, which has a published Ki of 4.3 nM.
The bioavailability of SKI-1 is a major concern, likely preventing an effective dosage from accumulating in the blood stream and reaching the target sites, and may be responsible for its lack of success in vivo.10 With increased hydrophilicity and improved enzyme binding, we hypothesize that these inhibitors will be more readily bioavailable and potent in vivo. The binding of each new inhibitor was assessed comparatively to the binding of SKI-I. These results are provided in Table 1 along with the synthetic yield, in silico docking energies and calculated log P values.
Table 1.
Final reaction aldehyde derivatives, percent yieldsa and Log P values.

| ||||||
|---|---|---|---|---|---|---|
| Inhibitor | Aldehyde Derivative (R) | Yield (%) | Binding Energy with SK1 (kcal/mol)b | Mean SinglePoint(% inhibition)c [Std. Dev (n = 4)] | IC50 (μM)c | Log P valued |
|
| ||||||
| 4A | 3,4-dimethoxybenzaldehyde | 99 | −11.2 | 57 [9.9] | 6.55 | 4.222 |
| 4B | 1,4-benzodioxan-6-carboxaldehyde | 68 | −11.9 | 1 [7.9] | >10 | 4.466 |
| 4C | p-methoxybenzaldehyde | 80 | −11.2 | −2 [5.4] | >10 | 4.632 |
| 4D | o-methoxybenzaldehyde | 72 | −11.1 | 4 [4.9] | >10 | 4.584 |
| 4E | m-methoxybenzaldehyde | 58 | −10.8 | −3 [7.5] | >10 | 4.608 |
| 4F | 2-methoxynaphthaldehyde | 64 | −12.5 | 6 [4.2] | >10 | 5.740 |
| 4G | p-hydroxybenzaldehyde | 92 | −11.2 | 17 [7.9] | >10 | 4.072 |
| 4H | m-hydroxybenzaldehyde | 85 | −11.2 | 10 [3.0] | >10 | 4.072 |
| 4I | o-hydroxybenzaldehyde | 69 | −10.5 | −6 [6.2] | >10 | 4.516 |
| 4 J | cyclohexane carboxaldehyde | 47 | −11.0 | −9 [12.7] | >10 | 4.736 |
| 4 K | 1 -naphthaldehyde | 62 | −12.3 | −7 [8.6] | >10 | 5.735 |
| 4L | benzaldehyde | 47 | −11.0 | 10 [3.5] | >10 | 4.580 |
| 4 M | 3-pyridinecarboxaldehyde | 98 | −10.9 | 18 [5.0] | >10 | 3.340 |
| 4 N | p-chlorobenzaldehyde | 91 | −11.4 | −15 [8.3] | >10 | 5.253 |
| 4O | m-nitrobenzaldehyde | 92 | −11.4 | −2 [3.8] | >10 | 4.510 |
| SKI-1 | 2-hydroxynapthaldehyde | 88 | −12.4 | 3 [2.2] | >10 | 5.675 |
Reagents and conditions: Pyrazole/hydrazide (1 mmol), aldehyde (1 mmol), ethanol (3 ml), 12 M HCl (1 drop).
Binding energy values recorded from Auto Dock Vina with 3vzb model.
Data obtained from Thermo Fisher Scientific SelectScreen™ Profiling Service, SSBK-Adapta, Madison, WI.
Log P values calculated using www.molinspiration.com.13
The synthesis of SK1 inhibitors was accomplished in three steps (Schemes 1 – 3), adapted from Sharma et. al.8 The synthesis of 2 was done via a Claisen-like reaction in the microwave reactor with 1 (1 equiv), sodium ethoxide in ethanol (21%) (1.5 equiv) and diethyl oxalate (1.2 equiv) (Scheme 1), reacting to a very high yield of 96%.20 Previous efforts involved running this reaction overnight while the method described has reduced that time down to one hour, dramatically improving the reaction efficiency while consistently maintaining very high yields, greater than 90%, which is a significant improvement over previous methodology.8
Scheme 1:

Claisen-like reaction to produce compound 2.
Scheme 3:
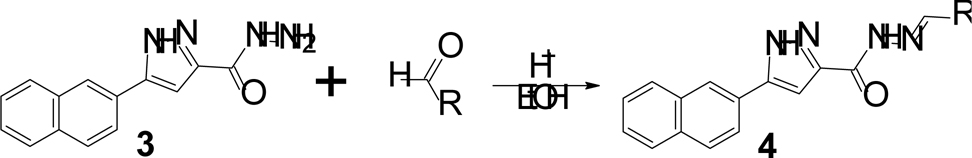
Acid catalyzed imine formation to yield the final derivatives.
Synthesis of 3 was adapted from Sharma et al8 using a 50% hydrazine hydrate solution (Scheme 2). This reaction is traditionally performed under reflux for 6 hours. Substituting the microwave in this step resulted in a 99% yield21 after just one hour, which is an improvement over previous work based on yield alone with a substantial reduction in reaction time.
Scheme 2:

Pyrazole and hydrazide formation of compound 3
The final step was prepared by reacting 3 with an appropriate aldehyde derivative via an imine formation (Scheme 3)22. The aldehyde derivatives are responsible for the modifications and were chosen in an attempt to assess improvements in the hydrophilicity of the final inhibitors and potentially the in vivo potency. Log P values for the inhibitors are mostly lower than SKI-1 (Table 1) due to these modifications with improved bioavailability.
A total of fifteen potential SK1 inhibitors have been synthesized using this process. These derivatives were all formed in good to high yields with high purity. While the yields are an improvement, the reduction in overall reaction time from two overnight reactions plus a 6 hour reflux to approximately 4 reaction hours overall is perhaps one of the most significant aspects of this research. Percent yields for the final reactions, aldehyde derivatives, and calculated log P values are included (Table 1). Table 1 also includes the in silico binding energy values calculated with AutoDock Vina and Thermo Fisher Scientific’s SelectScreenServices™ assay results compared to PF-543.
Observations of the crystal structure of human sphingosine kinase in complex with sphingosine (PDB ID: 3VZB), Figure 3A, Figure 3B, reveal a distinctly cylindrical binding pocket bounded by Phe288 and His311 at one end, the hydrophobic main body of the pocket defined by amino acids Ile174, Val177, Thr196, Leu 268 and Met272 and the end closest to the ATP binding pocket by Ser168. The C-2 amino group of the bound sphingosine substrate forms a hydrogen bond with the hydroxyl oxygen of Ser168, serving to orient the C-1 hydroxyl towards the ATP binding site of the enzyme. Using the dimensions of this sphingosine binding site as the boundaries for the in silico studies, docking of the SKI-2 molecule into the crystal structure of hSK1 resulted in a less than 0.5 angstrom RMSD between the location of the bound, crystallized SKI-2 molecule and the placement of the same molecule in the binding site by Autodock Vina, thereby validating the method.9
Fig. 3.

Ligands occupying the binding pocket of SK1. A) Crystallographic structure of sphingosine bound to the enzyme surrounded by hydrophobic side chains; B) Crystallographic structure of SKI-1 bound to the enzyme. C) Crystallographic structure of SKI-2 bound to the enzyme; D) Best conformer of compound 4A in the active site of SK1 as determined by Autodock Vina. E) Best conformer of compound 4G in the active site of SK1 as determined by Autodock Vina.
Of the eight interactions observed between sphingosine and SK1 in the crystal structure, seven are hydrophobic in nature, Figure 3A, an observation that complicates the design of targeted inhibitors with increased hydrophilicity. Inhibitors 4F and 4K have log P values matching that of the template inhibitor and were synthesized to determine the role of the hydroxyl group on the naphthyl ring interacting with the enzyme. Despite the increased hydrophobic character of the two compounds, both showed improved interactions with SK1 based upon the docking studies, but did not inhibit sphingosine kinase 1 significantly better than SKI-1 in the in vitro kinase assay (Table 1). Closer examination of these compounds, in silico, shows a preference for the naphthalene in the modified area remaining in the hydrophobic pocket, while in the other examples tested removal of this naphthalene ring flips the inhibitor within the binding site placing the “zone 1” naphthalene ring in this region (Figures 3B and 3D). This results in a new orientation of the structure, where the modified ring is in close contact to Ser168, an amino acid integral to sphingosine binding in the active site. This flip may explain the lower binding energy values for the inhibitors without the retained naphthalene ring.
Within the enzyme binding pocket, there are eight amino acids that can interact with these new inhibitors. Compared to SKI-I, five new inhibitors scored better in the single point inhibition assay and in each of these structures the calculated log P values are 4.58 or below. This indicates that the drugs may have reoriented in the binding site, as predicted by the docking study leading to a higher efficacy of inhibition during in vivo testing as they have in these in vitro studies.
The five best inhibitors based on the single point titration assay data (4A, 4G, 4M, 4L and 4H) show strong interactions within the binding pocket with the primary improvement being the interaction with the polar side chain, Ser168, as well as the backbone interactions with Thr196. The best inhibitor using both the single point and IC50value, 4A, has the methoxy group in the meta position which places it in close proximity (2.540Å) to Ser168 for a more stabilizing binding interaction with SK1 (Figure 3D). There are also two potential interactions between the Thr196 and the vinyl pyrazole nitrogen/carbonyl oxygen. For the 4G derivative, the meta-hydroxy group interacts with Asp81 (2.646Å) along with interactions between the pyrazole nitrogens and the Thr198 (Figure 3E). 4M, 4L and 4H also show comparable interactions with Ser168 and Thr198 as an explanation to their improved inhibition assay results.
To validate whether or not the correct products were produced, proton and carbon NMR were performed along with IR and high-resonance mass spectrometry. Carbon NMR spectra were extremely complex due to the number of peaks in the aromatic region of the spectra so attached proton test (APT) carbon NMR was performed to provide greater spectral clarity. One interesting result that was noted for a number of derivatives in the NMR spectra was the potential for geometric isomers to be formed during the final step. This was especially true for the methoxy derivatives (4C, 4D, 4E and 4F) where two peaks appeared in the APT spectra in the methyl region (55ppm) identifying geometric isomerization.
Future testing will involve modification to this template compound in other areas of the structure, specifically in the other naphthalene group or pyrazole ring to evaluate the interaction of the inhibitors with crystallized human SK1, using in silico docking and single point inhibition assays. Combining the best inhibitor modifications from this study in these synthetic approaches will also assess the combined improvements within the binding site of SK1. The results will be compared to SKI-1 to see the relative efficiency of the compounds at inhibiting SK1. In vivo studies will be pursued to definitively determine the modifications made in these inhibitor structures and provide direction of future synthetic efforts.
Supplementary Material
Acknowledgments
We would like to acknowledge the funding and support provided by an NIH-INBRE grant from the National Center for Research Resources and the National Institute for General Medical Sciences (8 P20 GM103499), Winthrop University Research Council grants (529230, 529231, SC 10011, 381118-2555-200) and the Winthrop University Department of Chemistry, Physics and Geology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Sawyers C Targeted Cancer Therapy. Nature. 2004;432:294–297. 10.1038/nature03095. [DOI] [PubMed] [Google Scholar]
- [2].Wu H, Chang DK, Huang CT. Targeted Therapy for Cancer. J. Cancer Mol. 2006;2:57–66. [Google Scholar]
- [3].French KJ, Upson JJ, Keller SN, Zhuang Y, Yun JK, Smith CD. Antitumor Activity of Sphingosine Kinase Inhibitors. Pharmacol. Ther. 2006;318:596–603. 10.1124/jpet.106.101345. [DOI] [PubMed] [Google Scholar]
- [4].French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD. Discovery and Evaluation of Inhibitors of Human Sphingosine Kinase. Cancer Res. 2003;63:5962–5969. [PubMed] [Google Scholar]
- [5].Maceyka M, Payne SG, Milstien S, Spiegel S. Sphingosine Kinase, Sphingosine-1-phosphate, and Apoptosis. Biochim. et Biophys. Acta. 2000;13:193–201. 10.1016/s1388-1981(02)00341-4. [DOI] [PubMed] [Google Scholar]
- [6].Gustin DJ, Li Y, Brown ML, Min X, Schmitt MJ, Wanska M, Wang X, Connors R, Johnstone S, Cardozo M, Cheng AC, Jefferies S, Franks B, Li S, Shen S, Wong M, Wesche H, Xu G, Carlson TJ, Plant M, Morgenstern K, Rex K, Schmitt J, Coxon A, Walker N, Kayser F, Wang Z. Structure Guided Design of a Series of Sphingosine Kinase (SphK) Inhibitors. Bioorg. Med. Chem. Lett. 2013;23:4608–4616. 10.1016/j.bmcl.2013.06.030. [DOI] [PubMed] [Google Scholar]
- [7].Li P, Wu J, Zheng J, Pei D. A Sphingosine Kinase-1-Inhibitor, SKI-ll, Induces Growth Inhibition and Apoptosis in Human Gastric Cancer Cells. Asian Pac. Cancer Prev. 2014;15:10381–10385. 10.7314/apjcp.2014.15.23.10381. [DOI] [PubMed] [Google Scholar]
- [8].a)Sharma AK, Su HU, Gimbor MA, Hengst JA, Wang X, Yun J, Amin S. Synthesis and Bioactivity of Sphingosine Kinase Inhibitors and their Novel Aspirinyl Conjugated Analogs. Eur. J. Med. Chem. 2010;45;4149–4156. 10.1016/j.ejmech.2010.06.005. [DOI] [PubMed] [Google Scholar]; b)Hengst JA, Wang X, Sk UH, Sharma AK, Amin S, Yun JK. Development of a sphingosine kinase 1 specific small-molecule inhibitor. Bioorg. Med. Chem. Lett., 2010;20:7498–7502. 10.1016/j.bmcl.2010.10.005. [DOI] [PubMed] [Google Scholar]
- [9].Wang Z, Min X, Xiao S-H, Johnstone S, Romanow W, Meininger D, Xu H, Liu J, Dai J, An S, Thibault S, Walker N. Molecular Basis of Sphingosine Kinase 1 Substrate Recognition and Catalysis. Structure 2013;21;789–809. 10.1016/j.str.2013.02.025. [DOI] [PubMed] [Google Scholar]
- [10].Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple DK. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002;45:2615–2623. 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- [11].Walters PW, Murcko MA. Prediction of ‘Drug-likness’. Adv. Drug Deliv. Rev. 2002;54: 255–271. 10.1016/s0169-409x(02)00003-0. [DOI] [PubMed] [Google Scholar]
- [12].Levine YK, Wilkins MHF. Structure of Oriented Lipid Bilayers. Nat. Biol. 1971;230: 69–72. 10.1038/newbio230069a0. [DOI] [PubMed] [Google Scholar]
- [13].Molinspiration (2015) Calculation of Molecular Properties and Bioactivity Score. http://www.molinspiration.com/cgi-bin/properties
- [14].Herr H, Chun J. Effects of LPA and S1P on the nervous system and implications for their involvement in disease. Curr. Drug Targets. 2007;8:155–167. 10.2174/138945007779315669. [DOI] [PubMed] [Google Scholar]
- [15].Mollo MC, Orelli LR. Microwave-assisted synthesis of 2-aryl-2-oxazolines, 5, 6-dihydro-4 h-1, 3-oxazines, and 4, 5, 6, 7-tetrahydro-1, 3-oxazepines. Organic Lett, 2016;18:6116–6119. 10.1021/acs.orglett.6b03122. [DOI] [PubMed] [Google Scholar]
- [16].Chundawat TS, Sharma N, Kumari P, Bhagat S. Microwave-assisted nickel-catalyzed one-pot synthesis of 2, 4, 5-trisubstituted imidazoles. Synlett, 2016;27:404–408. 10.1055/s-0035-1560825. [DOI] [Google Scholar]
- [17].Laclef S, Harari M, Godeau J, Schmitz-Afonso I, Bischoff L, Hoarau C, Levacher V, Fruit C, Besson T. Ligand-free Pd-catalyzed and copper-assisted C-H arylation of quinazolin-4-ones with aryl iodides under microwave heating. Organic Lett, 2016;17:1700–1703. 10.1021/acs.orglett.5b00467. [DOI] [PubMed] [Google Scholar]
- [18].Marvin was used for drawing, displaying and characterizing chemical structures, substructures and reactions, Marvin sketch (18.22.0), 2018, ChemAxon; (http://www.chemaxon.com). [Google Scholar]
- [19].Wang J, Knapp S, Pyne NJ, Pyne S, Elkins JM. Crystal Structure of Sphingosine Kinase 1 with PF-543. ACS Med. Chem. Lett. 2014;5:1329–1333. 10.1021/ml5004074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].2-Acetonaphthone (0.3g, 1.76 mmol) was dissolved in ethanol (3 mL) in a 10-mL microwave tube. To the tube was added sodium ethoxide in ethanol (21%) (0.9 mL, 2.64 mmol) via syringe. Finally, diethyl oxalate (0.29 mL, 2.11 mmol) was added via syringe. The solution was then placed in the microwave reactor and heated to 80 °C at 150 W for 1 hour with stirring. Upon cooling the solution was combined with a 10% hydrochloric acid solution (20 mL) and diethyl ether (20 mL). The organic layer was collected, dried over a small amount of anhydrous sodium sulfate, and transferred to a pre-weighed round bottom flask. Using a rotary evaporator, the solvent was removed. The remaining material was dried in vacuo overnight. Yield: 96%, 0.46 g. 1H NMR (CHCl3, 400 MHz): δ 8.54 (1H, s), 7.93–8.01 (4H, m), 7.52–7.64 (2H, m), 7.25 (1H, s), 4.41 (2H, q), 1.43 (3H, t).
- [21].A 50% hydrazine hydrate solution (0.32 mL, 6.67 mmol) was combined with 2 (0.30 g, 1.11 mmol) and ethanol (3 mL) in a microwave test tube. The reactants stirred for one minute and were placed in a CEM microwave reactor for 1 hour at 80 °C. After the reaction was complete, the tube was placed in an ice bath for 15 minutes and filtered via vacuum filtration. The remaining solid was placed in vacuo overnight to remove residual solvent. Yield: 99%, 0.13 g. 1H NMR confirms the formation of 5-naphthalen-2-yl-1H-pyrazol-3-carboxylic acid hydrazide. 1H NMR (DMSO-d6, 400 MHz): δ 8.4 (1H, s), 8.03–7.93 (4H, m), 7.51–7.57 (2H, m), 7.35 (1H, s).
- [22].General procedure for the formation of inhibitors (4) Compound 3 (0.080g, 0.317mmol) was combined with 1 equiv. of an appropriate aldehyde derivative(HCO-R), 3 mL ethanol, and 1 drop of 12M hydrochloric acid, which reacted in the CEM microwave reactor for 2 hours at 80 °C. The tube was cooled in an ice bath for 15 minutes and filtered via vacuum filtration. The remaining solid was dried in vacuo overnight to remove residual solvent. 1H NMR confirms the formation of each final inhibitor structure, which is provided in the supplementary material.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.