

Abstract
SET (TAF-1β/I2PP2A) is a ubiquitously expressed, multifunctional protein that plays a role in regulating diverse cellular processes, including cell cycle progression, migration, apoptosis, transcription, and DNA repair. SET expression is ubiquitous across all cell types. However, it is overexpressed or post-translationally modified in several solid tumors and blood cancers, where expression levels are correlated with worsening clinical outcomes. SET exerts its oncogenic effects primarily through the formation of antagonistic protein complexes with the tumor suppressor, protein phosphatase 2A (PP2A), and the well-known metastasis suppressor, nm23-H1. PP2A inhibition is often observed as a secondary driver of tumorigenesis and metastasis in human cancers. Preclinical studies have shown that the pharmacological reactivation of PP2A combined with potent inhibitors of the primary driver oncogene produces synergistic cell death and decreased drug resistance. Therefore, the development of novel inhibitors of the SET-PP2A interaction presents an attractive approach to reactivation of PP2A, and thereby, tumor suppression. NMR provides a unique platform to investigate protein targets in their natively folded state to identify protein and small-molecule ligands and report on the protein internal dynamics. The backbone 1HN, 13C, and 15N NMR resonance assignments were completed for the 204 amino acid nucleosome assembly protein-1 (NAP-1) domain of the human SET oncoprotein (residues 23-225). These assignments provide a vital first step toward the development of novel PP2A reactivators via SET-selective inhibition.
Keywords: SET (Su(Var)3-9, Enhancer-of-zeste, Trithorax); TAF-1β (Template-Activating Factor-I (β subunit); I2PP2A (Inhibitor 2 of Protein Phosphatase 2A); NAP (Nucleosome Assembly Protein); Proto-oncogene
Biological context
Protein SET (su(var)3-9, enhancer-of-zeste, trithorax), also known as I2PP2A (inhibitor 2 of protein phosphatase 2A), TAF-1β (template-activating factor-I (β subunit), and INHAT (inhibitor of histone acetyltransferases) is a 290 amino acid (~32 kDa) multifunctional protein involved in several cellular processes, including cell cycle progression, cell migration, apoptosis, transcription, and DNA repair (reviewed in Bayarkhangai et al. 2018). The first crystal structure of SET (PDB ID: 2E50) revealed a typical nucleosome assembly protein (NAP) core; a headphone-shaped homodimer with extended N-terminal helices that form the coiled-coil domain (23-79) and globular “earmuff” domains (80-225) which are essential for core histone and DNA binding (Muto et al. 2007). Not present in the crystal structure is an acidic C-terminal tail (226-277) that mediates the inhibition of histone acetyltransferase. SET was first discovered as a chimera fused with the nucleoporin NU214 (CAN) in an acute undifferentiated non-lymphocytic leukemia patient and has since been implicated in various cancer types, including solid tumors of the head and neck, breast, brain, kidney, testes, colon, liver, lung and prostate. SET overexpression is prevalent in chronic myeloid leukemia (CML), chronic leukocytic leukemia (CLL), acute myeloid leukemia (AML), and non-Hodgkin’s lymphoma (NHL), where expression levels are correlated with poor outcomes (Bayarkhangai et al. 2018). It exerts its tumorigenic and metastatic effects by forming inhibitory complexes with key suppressors of cancer progression, pro-apoptotic protein phosphatase 2A (PP2A), and the well-known tumor metastasis suppressor nm23-H1. PP2A is an endogenous regulator of cell death and cell division, acting as a counterbalance to tumorigenic pathways mediated by protein kinases. Inhibition of PP2A, whether by modification of its subunits or by direct interaction with antagonistic binding partners, is observed in non-neoplastic disease (Alzheimer’s disease) as well as solid cancers and leukemias (reviewed in Perrotti and Neviani 2013). Therefore, pharmacological activation of PP2A is an attractive strategy for cancer prevention and treatment, either as a monotherapy or in combination with specific inhibitors of driver oncogenes. For example, synergistic cell death has been reported in preclinical studies in which SET antagonist-mediated PP2A reactivation is coupled with potent inhibitors of proliferative pathways (reviewed in Mazhar et al., 2019). These studies highlight the capacity for improved efficacy of front-line treatments for malignancies with elevated SET expression. To gain structural information that may facilitate our understanding of SET regulation, inhibition, and tumor-promoting interactions, we have initiated solution NMR spectroscopy studies and report here the sequence-specific backbone chemical shift assignments of the SET oncoprotein NAP domain (residues 23-225).
Methods and experiments
Expression and purification of (2H, 13C, 15N)-SETNAP
We have generated a ~24 kDa truncated version of the human SET/TAF-1β/I2PP2A spanning residues 23-225 (hereafter referred to as SETNAP). The protein sequence was codon-optimized for E. coli expression and cloned into the pET28a (+) vector in frame with the Tobacco Etch Virus (TEV) protease recognition site ENLYFQG located directly upstream. The expression plasmid was transformed into Escherichia coli BL21(DE3), and a single colony was selected for growth in minimal media. For deuterated, doubly labeled samples, cells were adapted to growth in 99.9% D2O by successive passages into (15N, 13C)-enriched M9 minimal media containing 90, 95, 99, and 99.9% D2O and utilizing 15NH4Cl (1.4 g/L) and 13C6-glucose (4.0 g/L) as the sole nitrogen and carbon sources, respectively. Cells were incubated at 37 °C and grown to an optical density of A595 = 0.8-1.0, the temperature was decreased to 18 °C, and protein expression was induced by the addition of 0.3 M IPTG overnight. Cells were pelleted by centrifugation at 10,500 x g for 20 min at 4 °C and resuspended in Lysis Buffer containing 20 mM Tris pH 8.0, 500 mM NaCl, 20 mM Imidazole, 1mM PMSF, and 1x protease inhibitor cocktail (Thermo Scientific #A32963). Cells were lysed by sonication and pelleted at 15,000 x g for 30 min at 4 °C. The clarified lysate was applied to a 5 ml HisTrap FF column (Cytiva #17528601) pre-equilibrated with Binding/Wash Buffer (20 mM Tris pH 7.4, 500 mM NaCl, 20 mM Imidazole), and protein was eluted in buffer containing 20 mM Tris pH 7.4, 500 mM NaCl, and 500 mM Imidazole. Fractions containing His6-TEV-SETNAP were pooled (~ 30 ml), and the N-terminal His6 tag was cleaved with the addition of 3 mg TEV protease. Simultaneously, the protein was back-exchanged against 4 L Binding Buffer by dialysis at 4 °C overnight. Cleaved SETNAP was applied to a pre-equilibrated 5 ml HisTrap HP column (Cytiva #17524801). The flow-through containing purified protein was collected, and the sample volume was reduced to ~18 ml using a centrifugal concentrator (Millipore Sigma #UFC9010). The purified sample was buffer exchanged and further purified by isocratic elution in NMR buffer (20 mM Tris pH 8.0, 50 μM EDTA, 3 mM NaN3) over a preparation-scale size exclusion column (Cytiva #GE28-9893-34) and concentrated to ~1.5 mM using a centrifugal concentrator. Approximately 75 mg of >95% pure recombinant SETNAP was produced per liter of isotopically enriched M9 media.
NMR experiments
All NMR experiments were acquired at 310 K on a Bruker Avance III 950 MHz spectrometer equipped with a z-axis gradient cryogenic probe. A 2D [1H–15N]–TROSY–HSQC, shown in Figure 1, was used as the root spectrum to assign backbone resonances via pairwise comparison of inter/intra-residue 13Cα, 13Cβ, and 13C′ chemical shifts. Even at relatively low sample concentrations (<200 μM), we observe protein dimerization in solution, confirmed by size exclusion chromatography (unpublished) and consistent with our crystal structure of the same construct (PDB ID: 7MTO). Therefore, to overcome signal-to-noise limitations observed with fully-protonated samples, we exploited TROSY-based pulse sequences to enhance sensitivity and resolution. Triple resonance trHNCO, trHN(CA)CO, trHNCA, trHN(CO)CA, trHNCACB and trHN(CO)CACB experiments were collected on a triply labeled 1.5 mM [2H,15N,13C]-SETNAP sample back-exchanged into 95% H2O/5% D2O NMR buffer (20 mM Tris pH 8.0, 50 μM EDTA, and 3 mM NaN3) at 37 °C. All 3D datasets were acquired by non-uniform sampling (NUS) of 10 % of the linear points using the default sinusoidal-weighted Poisson-gap scheduler included with Bruker’s Topspin 3.2.6 software package. Reconstruction of sparse data was achieved with SMILE (Sparse Multidimensional Iterative Lineshape-Enhanced reconstruction; Ying et al. 2017) using default settings. 15N–{1H} heteronuclear NOE experiments were acquired at 950 MHz by fully interleaving NOE and reference spectra and were recorded as a series of three experiments with relaxation delays of 3-, 4-, and 5-s to ensure the steady-state NOE was achieved. Data were processed using NMRPipe (Delaglio et al. 1995) and analyzed with CcpNmr Analysis v2.5.0 (Vranken et al. 2005). 1H chemical shifts were referenced to internal trimethylsilyl propanoic acid (TSP), while 15N and 13C chemical shifts were indirectly calibrated. Talos+ (Shen et al. 2009) was used to generate secondary structure predictions based on experimentally derived HN, N, C′, Cα, and Cβ chemical shifts (Fig. 2).
Fig. 1.
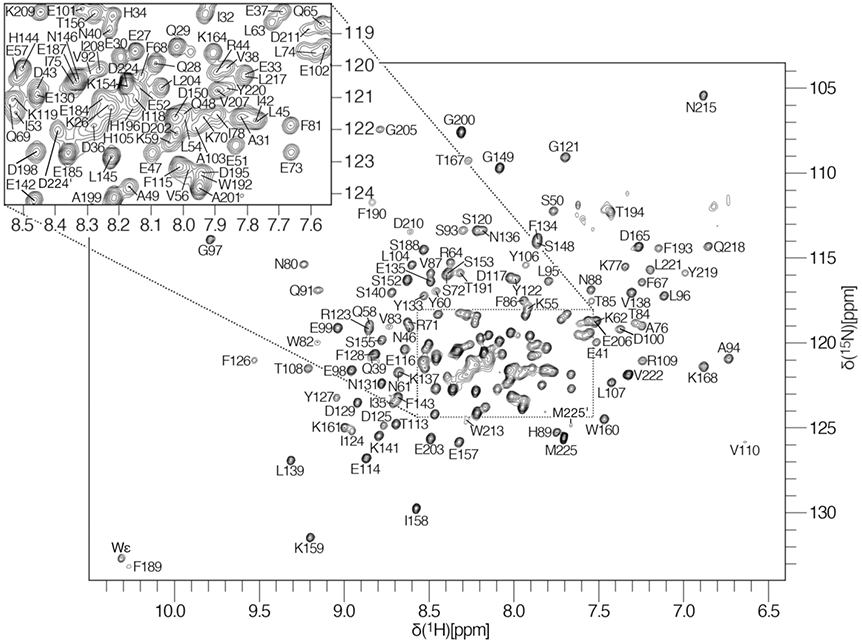
2D [1H-15N]-TROSY-HSQC of residues 23-225 of the human proto-oncogene SET/TAF-1β/I2PP2A (SETNAP) recorded on a Bruker Avance III 950 MHz spectrometer at pH 8.0 and 310 K. An enlarged view of the most crowded region of the spectrum is shown in the upper-left corner. Backbone amide N-H correlations are labeled with the singleletter amino acid code and residue number of the mature native protein. The prime (′) notation represents correlations in the C-terminus (residues 224 and 225) from a minor secondary conformation. An ambiguous tryptophan sidechain indole resonance is marked as Wε.
Fig. 2.

Characterization of the NAP domain (residues 23-225) of human SET/TAF-1β/I2PP2A based on NMR chemical shifts. a. Raw data depicting chemical shift deviations of Cα and Cβ carbons with respect to random coil values (Δδ(Cβ)- Δδ(Cα)) are plotted against residue numbers. Positive and negative values indicate β-strand and α-helix characters, respectively. b. The per-residue probability of secondary structure formation as predicted by Talos+, with blue bars representing α-helices and red bars indicating β-strands. Heteronuclear NOE relaxation parameters are shown as open diamonds. These data represent the average of three experiments collected with 3, 4, and 5-s delays acquired at 950 MHz, with error bars indicating one standard deviation above and below the average. c. NMR-based secondary structure predictions show good agreement with a high-resolution crystal structure of the same construct (boxes labeled α2-8 and β1-4).
Assignments and data deposition
Backbone assignments were obtained for the human oncoprotein SET/TAF-1β/I2PP2A. The 204 amino acid-containing-construct described here contains a non-native TEV protease-generated residual glycine at position −1 followed by NAP domain residues 23-225 of human SET. Deleting the nuclear localization signal-containing N-terminal helix and the unstructured acidic tail was required to produce a stable, soluble protein with relatively equal peak heights and good peak dispersion. The 2D [1H-15N]-TROSY-HSQC spectrum of SETNAP is shown in Figure 1. Under the conditions used in this experiment, 100% (168/168) of observable 1H-15N correlations were assigned unambiguously. Two residues are marked with a (′), D224′ and M225′, to denote the presence of a weak secondary conformation for the penultimate and ultimate amino acids. In total, 717 of 809 (89%) backbone resonances were assigned, including 84% 1HN proton and 15N amide resonances (336/398), 91% Cα (185/204), 91% Cβ (179/197), and 91% C′ (185/204) resonances. Twenty-six residues did not provide correlations in the [1H-15N]-TROSY-HSQC spectrum, most notably the fifteen amino acids spanning R169-H183. High-confidence Phyre2 modeling (Kelley et al. 2015) suggests that this region could adopt an out-and-back loop structure containing two five-residue antiparallel beta-strands seen in the crystal structure of a functionally similar yeast homolog, NAP-1 (PDB ID: 2AYU; Park and Luger 2006). However, our laboratories' crystallographic efforts were not able to provide the experimental data to support such a model. Other missing correlations include the first four N-terminal residues G-1, T23, S24, and E25, as well as E111, V112, E147, S162, G163, S197, and I212. Per-residue mapping against the Talos+-predicted secondary structure suggests that, except for E111 and V112, all are found in flexible loop regions where they are likely to experience solvent exchange or conformational averaging on an unfavorable timescale. Along with the N-terminal stretch and the R169-H183 loop, two residues (E111 and S162) failed to provide Cα, Cβ, or C′ correlations as well. The chemical shift assignments from these experiments were all submitted to the BioMagResBank (www.bmrb.wisc.edu) under accession number 50899. The backbone Cα, Cβ, and C′ assignments presented here were used to calculate chemical shift deviations from the random coil and map the secondary structure. As shown in Figure 2, the predicted secondary structure of SETNAP contains six alpha-helices and four beta-strands, numbered in the context of full-length SET (α2:K26-A76, α3:F81-F86, α4:Q91-A94, α5:E99-H105, α6:F189-W192, α7:L204-K209, β1:L107-E114, β2:Y122-D129, β3:V138-L145, and β4:S153-K159). Heteronuclear NOE analysis reveals dynamic regions of the protein, and the secondary structure predictions provided by Cα, Cβ, and C′ chemical shifts are in agreement. In addition, the secondary structure architecture derived by solution NMR closely resembles that of the crystal structure, with only minor differences occurring at the margins of secondary structure elements. Such consistencies warrant confidence that these assignments are derived from a correctly folded protein construct suitable for NMR-based characterization of regulatory ligands as well as NMR-based screening for pharmacological inhibitors of the SET oncoprotein.
Funding
This work is supported in part by shared instrumentation grants to the UMB NMR center from the National Institutes of Health [S10 RR10441, S10 RR15741, S10 RR16812, and S10 RR23447 (D.J.W.)] and the National Science Foundation (DBI 1005795 to D.J.W.). This work was also supported via the Center for Biomolecular Therapeutics (CBT) at the University of Maryland. This work was also supported by research funding from the National Institutes of Health (CA214461, DE016572, and P01 CA203628 to BO). The core facilities utilized are supported by NIH (C06 RR015455), Hollings Cancer Center Support Grant (P30 CA138313), or Center of Biomedical Research Excellence (Cobre) in Lipidomics and Pathobiology (P30 GM103339).
Footnotes
Conflicts of interest
The authors declare that they have no conflict of interest.
Availability of data and material
The datasets generated during and/or analyzed during the current study are available in the BioMagResBank (www.bmrb.wisc.edu) under accession number 50899.
Ethical standards
All experiments comply with the current laws of the United States of America.
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- Bayarkhangai B, Noureldin S, Yu L, et al. (2018) A comprehensive and perspective view of oncoprotein SET in cancer. Cancer Med. 7:3084–3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S. Vuister GW, et al. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293 [DOI] [PubMed] [Google Scholar]
- Farrow NA, Muhandiram R, Singer AU, et al. (1994) Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33:5984–6003 [DOI] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, et al. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858. doi: 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazhar S, Taylor SE, Sangodkar J, Narla G (2019) Targeting PP2A in cancer: Combination therapies. Biochim Biophys Acta - Mol Cell Res 1866:51–63. doi: 10.1016/J.BBAMCR.2018.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto S, Senda M, Akai Y, et al. (2007) Relationship between the structure of SET/TAF-Iβ/INHAT and its histone chaperone activity. Proc Natl Acad Sci U S A 104:4285–4290. doi: 10.1073/pnas.0603762104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y-J, Luger K (2006) The structure of nucleosome assembly protein 1. Proc Natl Acad Sci U S A 103:1248–53. doi: 10.1073/pnas.0508002103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotti D, Neviani P (2013) Protein phosphatase 2A: A target for anticancer therapy. Lancet Oncol. 14:e229–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, Bax A (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR 44:213–223. doi: 10.1007/s10858-009-9333-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, et al. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59:687–696. doi: 10.1002/prot.20449 [DOI] [PubMed] [Google Scholar]
- Ying J, Delaglio F, Torchia DA, Bax A (2017) Sparse multidimensional iterative lineshape-enhanced (SMILE) reconstruction of both non-uniformly sampled and conventional NMR data. J Biomol NMR 68:101–118. doi: 10.1007/s10858-016-0072-7 [DOI] [PMC free article] [PubMed] [Google Scholar]