

Abstract
Behaviors and disorders related to self-regulation, such as substance use, antisocial behavior, and ADHD, are collectively referred to as externalizing and have shared genetic liability. We applied a multivariate approach that leverages genetic correlations among externalizing traits for genome-wide association analyses. By pooling data from ~1.5 million people, our approach is statistically more powerful than single-trait analyses and identifies more than 500 genetic loci. The loci were enriched for genes expressed in the brain and related to nervous system development. A polygenic score constructed from our results predicts a range of behavioral and medical outcomes that were not part of genome-wide analyses, including traits that until now lacked well-performing polygenic scores, such as opioid use disorder, suicide, HIV infections, criminal convictions, and unemployment. Our findings are consistent with the idea that persistent difficulties in self-regulation can be conceptualized as a neurodevelopmental trait with complex and far-reaching social and health correlates.
Introduction
Behaviors related to self-regulation, such as substance use disorders or antisocial behaviors, have far-reaching consequences for affected individuals, their families, communities, and society at large1,2. Collectively, this group of correlated traits are classified as externalizing3. Twin studies have demonstrated that externalizing liability is highly heritable (~80%)4,5. To date, however, no large-scale molecular genetic studies have utilized the extensive degree of genetic overlap among externalizing traits to aid gene discovery, as most studies have focused on individual disorders6. For many high-cost, high-risk behaviors with an externalizing component – opioid use disorder and suicide attempts7 being salient examples – there are limited genotyped cases available for gene discovery8,9.
A complementary strategy to the single-disease approach is to study the shared genetic architecture across traits in multivariate analyses, which boosts statistical power by pooling data across genetically correlated traits10. Multivariate approaches can use summary statistics from genome-wide association studies (GWAS) to discover connections between phenotypes not typically studied together because they span different domains, fields of study, or life stages. Novel statistical methods can increase the effective sample size by adjusting for sample overlap. Elucidating the shared genetic basis of externalizing liability can advance our understanding of the developmental etiology of self-regulation and enables mapping the pathways by which genetic risk and socio-environmental factors contribute to the development of externalizing outcomes.
We applied genomic structural equation modeling (Genomic SEM) to summary statistics from GWAS on multiple forms of externalizing for which large samples were available10. We posited that applying this multivariate approach would lead to identification of genetic variants associated with a broad array of externalizing phenotypes, and with related behavioral, social, and medical outcomes that were not directly included in our GWAS. This approach was grounded in the literature showing shared genetic liability across numerous externalizing disorders and with non-psychiatric variation in externalizing behavior5,11.
Results
Genomic SEM of externalizing liability
Following our preregistered analysis plan (https://doi.org/10.17605/OSF.IO/XKV36), we collated summary statistics from GWAS on externalizing-related traits (Supplementary Information section 1). For an exhaustive description of the phenotype selection procedure and GWAS protocol, see Supplementary Information section 2. All phenotypes considered for inclusion are listed in Supplementary Table 1. We first applied quality control (Supplementary Table 2) and excluded summary statistics based on power considerations (i.e., < 0.05, or mean χ2 < 1.05)12. After applying these filters, 11 externalizing phenotypes remained, with sample sizes >50,000 (N = 53,293–1,251,809) (Supplementary Table 3). All samples were of European ancestry. The following seven phenotypes made it to the final multivariate model specification (Table 1 & Supplementary Table 4): (1) attention-deficit/hyperactivity disorder (ADHD)13, (2) problematic alcohol use (ALCP; a meta-analysis of alcohol dependence and AUDIT-P)14,15, (3) lifetime cannabis use (CANN)16, (4) reverse-coded age at first sexual intercourse (FSEX)17, (5) number of sexual partners (NSEX)17, (6) general risk tolerance (RISK)17, and (7) lifetime smoking initiation (SMOK)18.
Table 1.
Summary of seven externalizing-related disorders and behaviors with GWAS summary statistics
| Phenotype (abbreviation) | N | h2 (SE) | λGC | Mean χ2 | Intercept | Ratio | Reference |
|---|---|---|---|---|---|---|---|
| Attention-deficit/hyperactivity disorder (ADHD) | 53,293 | 0.235 (0.015) | 1.253 | 1.297 | 1.034 | 0.113 | 13 |
| Problematic alcohol use (ALCP) | 164,121 | 0.055 (0.004) | 1.149 | 1.174 | 1.013 | 0.073 | 14,15 |
| Lifetime cannabis use (CANN) | 186,875 | 0.066 (0.004) | 1.230 | 1.267 | 1.026 | 0.098 | 16 |
| Age at first sexual intercourse (FSEX)* | 357,187 | 0.115 (0.004) | 1.623 | 1.869 | 1.036 | 0.041 | 17 |
| Number of sexual partners (NSEX) | 336,121 | 0.097 (0.004) | 1.492 | 1.682 | 1.027 | 0.041 | 17 |
| General risk tolerance (RISK) | 426,379 | 0.053 (0.002) | 1.372 | 1.461 | 1.019 | 0.041 | 17 |
| Lifetime smoking initiation (SMOK) | 1,251,809 | 0.078 (0.002) | 2.328 | 3.152 | 1.126 | 0.058 | 18 |
|
| |||||||
Notes: The statistics reported in this table were all estimated with LD Score regression12. Heritability (h2) is on the observed scale12. λGC is the median χ2 statistic divided by the expected median of the χ2 distribution with 1 degree of freedom95. Mean χ2 is the average χ2 statistic. Intercept is the estimated LD Score regression intercept. Ratio measures stratification bias, defined as (Intercept – 1) / (Mean χ2 – 1)12.
Reverse-coded (see Online Methods).
For a complete description of the model selection procedure, see Supplementary Information section 3. In summary, prior to Genomic SEM, we first applied hierarchical clustering to a matrix of LD Score genetic correlations, which identified three (k) clusters (Supplementary Table 5). An exploratory factor analysis benchmarked four factor models, specifying one to four (k + 1) latent factors, with the aim to best explain the genetic correlations amongst the 11 phenotypes (Supplementary Table 6). The three-factor solution was determined to be the best-fitting exploratory model, which aligned with the hierarchical clustering.
We proceeded with confirmatory factor analysis to formally model genetic covariances with Genomic SEM, which is unbiased by sample overlap and sample-size imbalances10,19. As indicated by its model fit indices: χ2(44) = 8007.35; Akaike information criterion (AIC) = 8051.35; comparative fit index (CFI) = 0.662; standardized root-mean-square residual (SRMR) = 0.161, we found that a common factor model with 11 phenotypes did not satisfy our preregistered criteria (i.e., CFI and SRMR >0.9 and <0.08, respectively). Two more complicated specifications were tested, a correlated three-factor model (i.e., akin to the best-fitting exploratory model) and a bifactor model (Supplementary Table 7), but neither of these two models met the criteria or provided a parsimonious interpretation. Finally, we estimated a revised and less complex common factor model with the seven phenotypes (Table 1 and Figure 1A) that displayed moderate-to-large (i.e., ≥ 0.5) loadings on the single factor estimated in the first common factor model with 11 phenotypes. The revised common factor model with seven externalizing phenotypes provided the best fit across all specifications, and it closely approximated the observed genetic covariance matrix (i.e., χ2(12) = 390.234, AIC = 422.23, CFI = 0.957, SRMR = 0.079). This model was selected as our final factor model because it identified a genetic factor of externalizing that was suitable for genome-wide association analysis, offered an easily interpretable factor solution, and satisfied the model fit criteria. We hereafter refer to it as “the externalizing factor” (EXT).
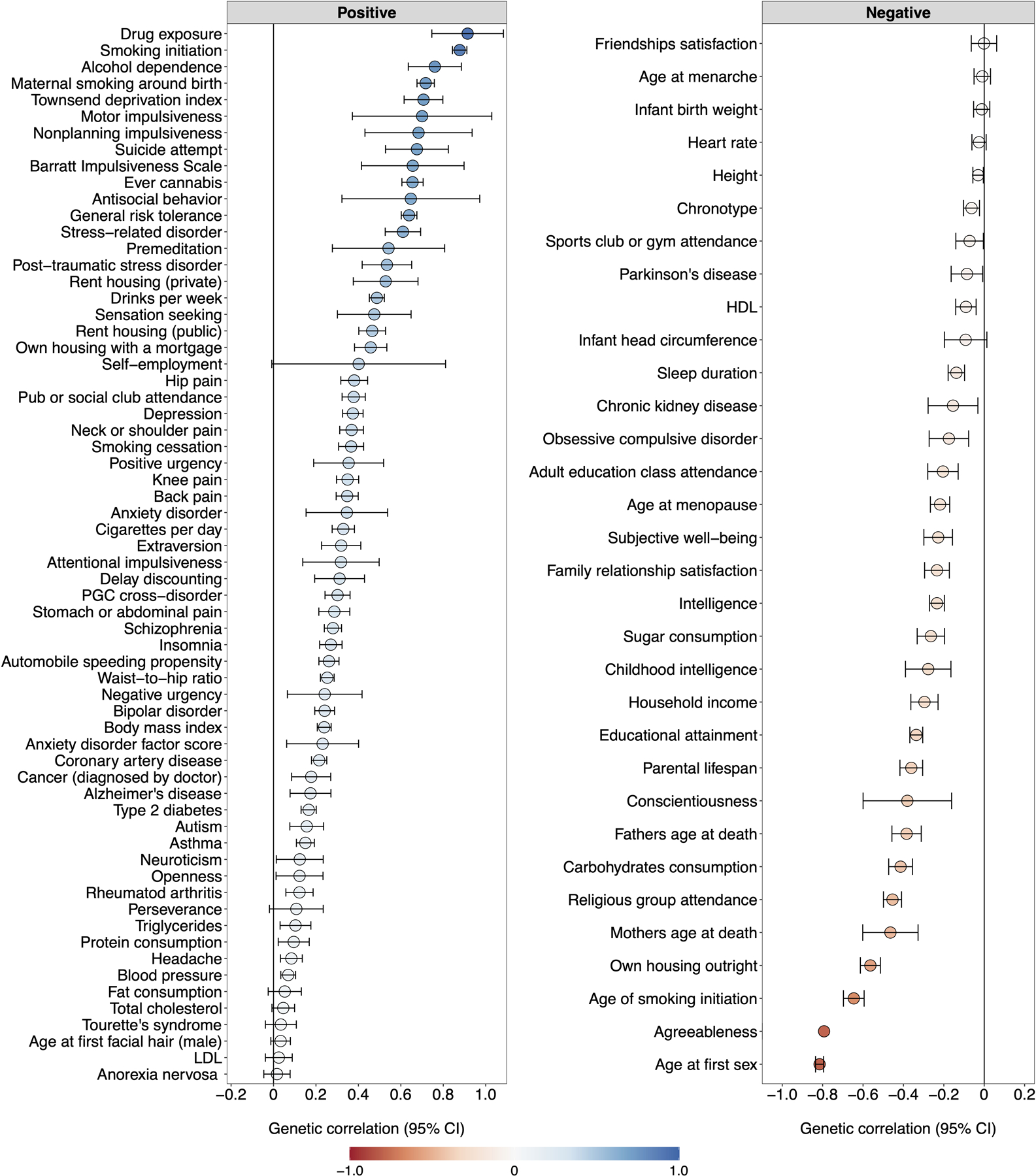
Figure 1 |. Genetic correlations and structural equation modeling with Genomic SEM.
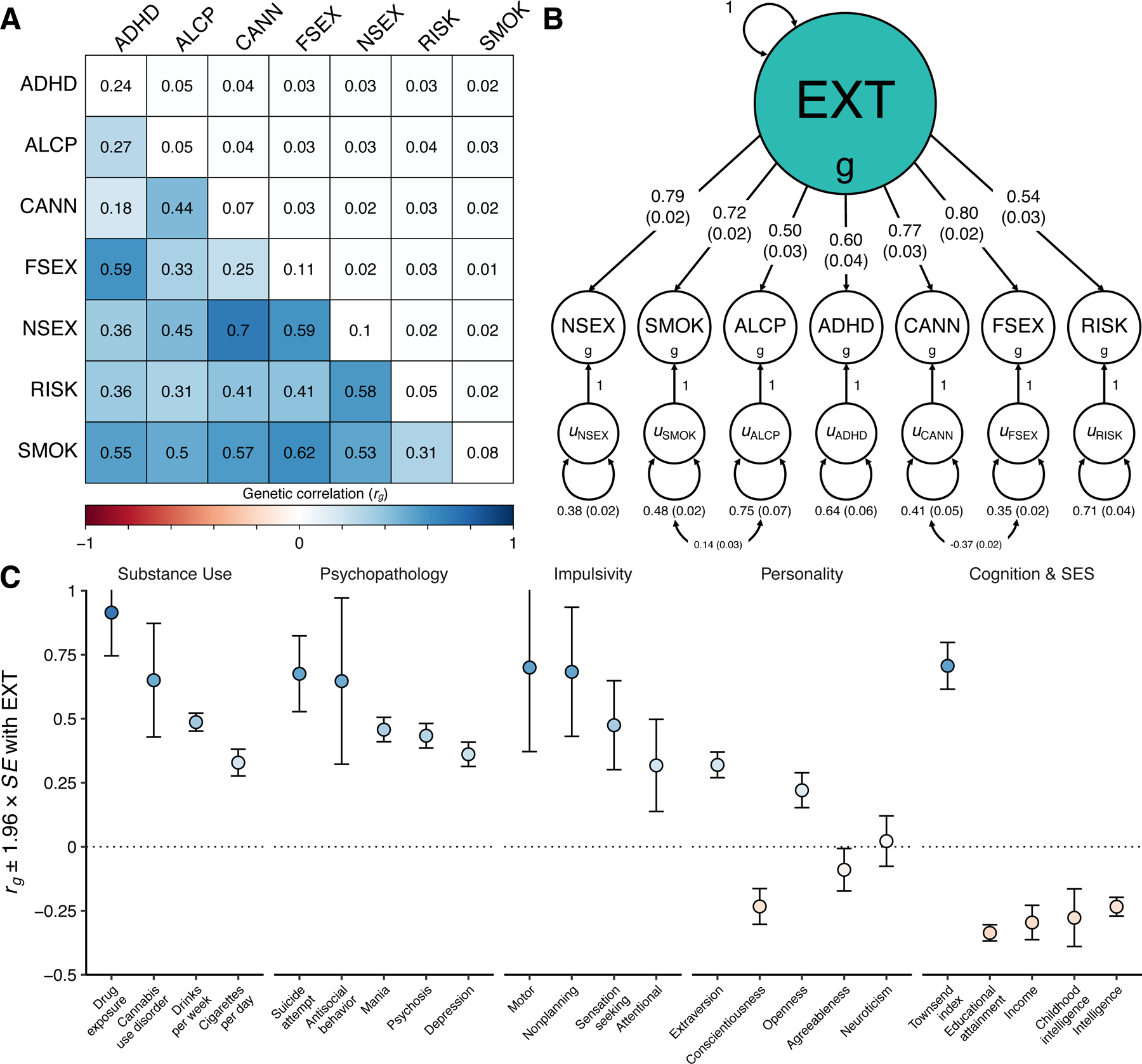
(A) The lower and upper triangles display pair-wise LD Score genetic correlations (rg) and their standard errors, respectively, among the final seven discovery phenotypes (Table 1), and the diagonal displays observed-scale SNP heritabilities (h2) (see Table 1 for standard errors). (B) Path diagram of the final revised common factor model estimated with Genomic SEM. The factor loadings were standardized, and standard errors are presented in parentheses. (C) Genetic correlations (rg) between the genetic externalizing factor (EXT, N = 1,492,085) and a subset of phenotypes selected to establish convergent and discriminant validity (Supplementary Table 8 reports all 91 estimated genetic correlations together with the exact number of independent samples used to derive each estimate), where blue and red bars represent positive and negative genetic correlations, respectively, using the same color scale as in panel A. Error bars represent 95% confidence intervals centered on the rg estimate, computed as 1.96 times the standard error. ADHD is attention deficit hyperactivity disorder (N = 53,293), ALCP is problematic alcohol use (N = 164,864), CANN is lifetime cannabis use (N = 186,875), EXT is externalizing, FSEX is reverse-coded age at first sex (N = 357,187), NSEX is number of sexual partners (N = 336,121), RISK is general risk tolerance (N = 426,379), and SMOK is lifetime smoking initiation (N = 1,251,809).
The common factor captures a shared genetic liability to the final seven externalizing traits (Figure 1B), and genetic variants associated with EXT predict central externalizing disorders and a range of behavioral and medical outcomes that were not in the model (see below). We performed a leave-one-phenotype-out Genomic SEM analysis to ensure that no single phenotype, e.g., the phenotype with the largest N, was unduly influencing the genetic architecture estimated for EXT (Supplementary Information section 3.5.3). We found that the genetic correlations between EXT and each of seven leave-one-phenotype-out models were not distinguishable from unity (rg ~ 0.984–0.999, SE ~ 0.028–0.035), which suggests that none of the phenotypes is driving the genetic architecture of EXT.
We extended Genomic SEM to estimate genetic correlations between EXT and 91 preregistered phenotypes with GWAS summary statistics that were not among the seven discovery phenotypes (Extended Data Fig. 1 and Supplementary Table 8). The genetic correlations indicate convergent and discriminant validity of the common EXT factor (Figure 1C): As anticipated, EXT showed strong positive genetic correlations with drug exposure (rg = 0.91, SE = 0.09), antisocial behavior (rg = 0.65, SE = 0.17), and impulsivity measures, including motor impulsivity (rg = 0.70, SE = 0.17) and failures to plan (rg = 0.68, SE = 0.13). We estimated similar genetic correlations with personality domains (based on 23andMe20) as to those reported in twin studies, i.e., positive correlation with extraversion (rg = 0.32, SE = 0.03), and negative with conscientiousness (rg = −0.23, SE = 0.04) and agreeableness (rg = −0.09, SE = 0.04)11,21. However, prior work has found neuroticism but not openness to be correlated with externalizing21, while we found a positive correlation with openness (rg = 0.22, SE = 0.04) but not with neuroticism (rg = 0.02, SE = 0.05). Notably, EXT was also correlated with suicide attempts (rg = 0.68, SE = 0.08) and post-traumatic stress disorder (rg = 0.53, SE = 0.06). EXT showed more modest inverse correlations with educational attainment (rg = −0.32, SE = 0.02) and intelligence (rg = −0.23, SE = 0.02), indicating that EXT is not simply reflecting genetic influences on cognitive ability. Finally, there was a significant correlation with the Townsend index (rg = 0.71, SE = 0.05), a measure of neighborhood deprivation that reflects high concentrations of unemployment, household overcrowding, and lower home- and car-ownership22. Genetic correlations can reflect correlated social processes or variables that are nonrandomly distributed with respect to genotypes, such as genetic nurture or neighborhood conditions, and we return to this topic in within-family analyses below.
Multivariate GWAS of externalizing liability
We next used Genomic SEM10 to conduct a GWAS on the shared genetic liability EXT (Figure 2 and Extended Data Fig. 2). This analysis estimated single-nucleotide polymorphism (SNP) associations directly with EXT, with an effective sample size of N = 1,492,085 individuals (Supplementary Information section 3.4). These analyses are different in their approach and substantially increase sample size, statistical power, and the range of findings compared to previous work (Supplementary Information section 2.2.1). After applying conditional and joint multiple-SNP analysis (COJO) on a set of near-independent, genome-wide significant (two-sided P < 5×10−8) lead SNPs23, we identified 579 conditionally and jointly associated “EXT SNPs” (Supplementary Tables 9–9B), meaning they were significantly associated with EXT even after statistically adjusting for each other and other lead SNPs. Of the 579 EXT SNPs and their correlates within linkage disequilibrium (LD) regions (r2 > 0.1), 121 (21%) were new loci, not previously associated with any of the seven externalizing behaviors/disorders that went into the Genomic SEM model, and 41 (7%) can be classified as entirely novel, as they have not been reported previously for any trait in the GWAS literature (i.e., neither of these 41 SNPs nor any SNPs in LD (r2 > 0.1) were reported for any traits at two-sided P < 1×10−5 in the NHGRI-EBI GWAS Catalog6, version e96 2019-05-03) (Supplementary Table 10).
Figure 2 |. Multivariate genome-wide association analysis of EXT with Genomic SEM.
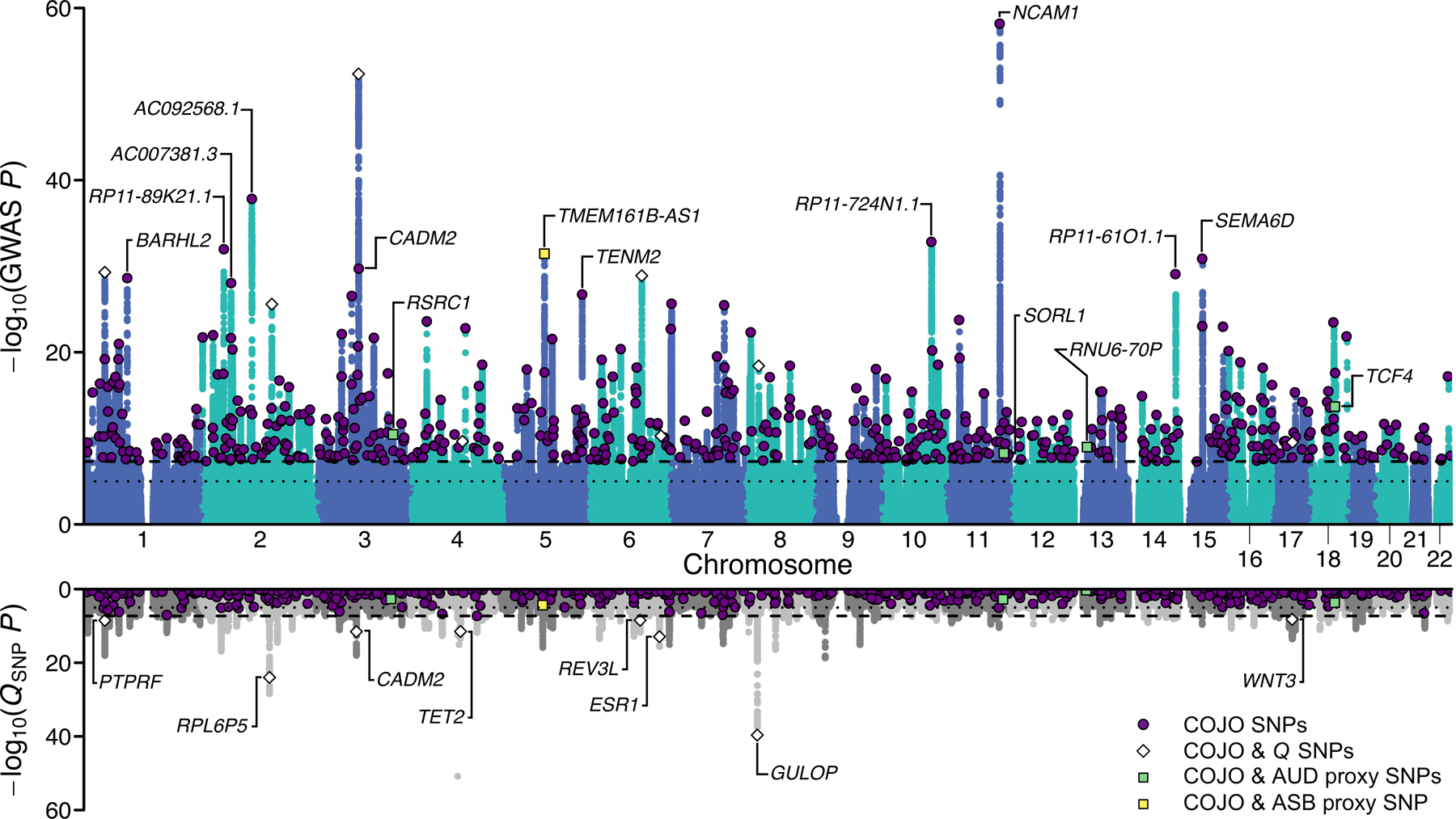
Scatterplot of −log10(P value for two-sided Z-test) for weighted least squares regression to estimate GWAS associations (top panel) and −log10(P value for one-sided χ2 test with 7–1 degrees of freedom) for QSNP tests of heterogeneity (bottom panel) for EXT. Purple dots represent the 579 EXT SNPs that are conditionally and jointly associated (COJO) at genome-wide significance (two-sided P < 5×10−8) (Supplementary Table 9). White diamonds represent eight of the 579 SNPs that also show significant QSNP heterogeneity. Four green and one yellow squares represent five out of the 579 SNPs that also were Bonferroni-significant proxy-phenotype associations with alcohol use disorder (AUD) and antisocial behavior (ASB), respectively (Supplementary Table 11–12). Gene names refer to the closest gene based on genomic location, displayed for a selection of the findings (Supplementary Table 9 reports the nearest gene for all 579 EXT SNPs).
Genomic SEM was used to perform SNP-level tests of heterogeneity (QSNP; Supplementary Information section 3.5.1; Supplementary Data 1–2) to investigate whether each SNP had consistent, pleiotropic effects on the seven input phenotypes that effectively only operate via EXT. If the EXT loci really index a shared genetic externalizing liability, we would expect to identify heterogeneity mostly in regions of the genome not associated with EXT. In the absence of heterogeneity, it is expected that a given SNP’s GWAS effects on the input phenotypes will scale proportionally to the factor loadings24 (see Supplementary Information section 3.5.2). The genome-wide QSNP analysis was adequately powered (mean = 1.864; Extended Data Fig. 2), and at one-sided QSNP P < 5×10−8, we identified 160 QSNP loci (Supplementary Information section 3.5.1). Importantly, only eight of these 160 loci overlapped with EXT loci (~1% = 8/579) (Figure 2; Supplementary Table 9). Reassuringly, we identified 3.6 times more EXT loci than QSNP loci (579/160). Using a less stringent significance threshold by focusing specifically on the 579 EXT loci, only 7% (41/579) were significant for QSNP (one-sided QSNP P < 0.05/579). The observation that a small minority of the EXT loci were heterogeneous at either significance threshold, and that the vast majority of the 160 QSNP loci were found outside of EXT loci, provide evidence that the EXT loci primarily index a unitary dimension of genetic liability rather than representing an amalgamation of variants with divergent associations across the discovery phenotypes. Notably, the strongest QSNP and most salient example of a heterogeneous, trait-specific association is SNP rs1229984 (one-sided QSNP P = 1.67×10−51; Supplementary Data 1A). This particular SNP, located in the gene ADH1B, is a missense variant with a well-established role in alcohol metabolism25, and it was not associated with EXT (two-sided P = 0.022) but only with problematic alcohol use (two-sided P = 6.43×10−57). Additionally, for each of the 579 EXT SNPs, we investigated the concordance in direction of SNP effects (i.e., the sign) on the seven phenotypes (Supplementary Information section 3.5.4). For 317 of the 579 EXT SNPs (54.7%), the concordance was perfect (i.e., the same direction of effect on all seven phenotypes), and for 203 (35.1%), 47 (8.1%), and 12 (2.1%) we observed either six, five, or four concordant effects, respectively. Thus, the analysis of sign concordance lends further support to our interpretation that the EXT loci primarily index a shared genetic liability to externalizing.
Quasi-replication analyses
Because the discovery stage effectively exhausted large study cohorts available for replication, we performed a series of preregistered quasi-replication analyses (Supplementary Tables 11–12). As quasi-replication analyses of the 579 SNPs (Supplementary Information section 4), a three-step method tested their association with two independent, GWAS meta-analyses on externalizing phenotypes: (1) alcohol use disorder (rg with EXT = 0.52; N = 202,004), and (2) antisocial behavior (rg with EXT = 0.69; N = 32,574). We had preregistered to hold out antisocial behavior from the externalizing GWAS to enable quasi-replication with a central externalizing trait that was not included in the model. First, we tested whether the 579 SNPs (or an LD proxy for missing SNPs, r2 > 0.8) showed sign concordance, i.e., the same direction of effect between EXT and alcohol use disorder or antisocial behavior: 75.4% of SNPs showed sign concordance with alcohol use disorder (two-sided test P = 6.84×10−36) and 66.9% with antisocial behavior (two-sided test P = 1.39×10−15) (Extended Data Fig. 3). For the second and third tests, we generated empirical null distributions for the two phenotypes by randomly selecting 250 near-independent (r2 < 0.1) SNPs per each of the 579 SNPs, matched on allele frequency. In the second test, a greater proportion of the 579 SNPs were nominally associated (P < 0.05) with the two phenotypes compared to their empirical null distributions: 124 (21.4% vs. 6.6%) with alcohol use disorder (two-sided P = 1.87×10−31) and 58 (10.5% vs. 4.7%) with antisocial behavior (P = 1.64×10−8). In the third test, the 579 SNPs were jointly more strongly enriched for association with alcohol use disorder (one-sided Mann-Whitney test P = 5.89×10−26) and antisocial behavior (P = 1.10×10−5) compared to their empirical null distributions. Overall, the three exercises consistently suggested that the GWAS of EXT is not spurious overall, and that it is enriched for genetic signal with two phenotypes of central importance to the literature on externalizing. Below, we perform further quasi-replication of the 579 EXT SNPs in an auxiliary polygenic score analyses (also in within-family models).
Bioinformatic analyses highlight relevant neurobiology
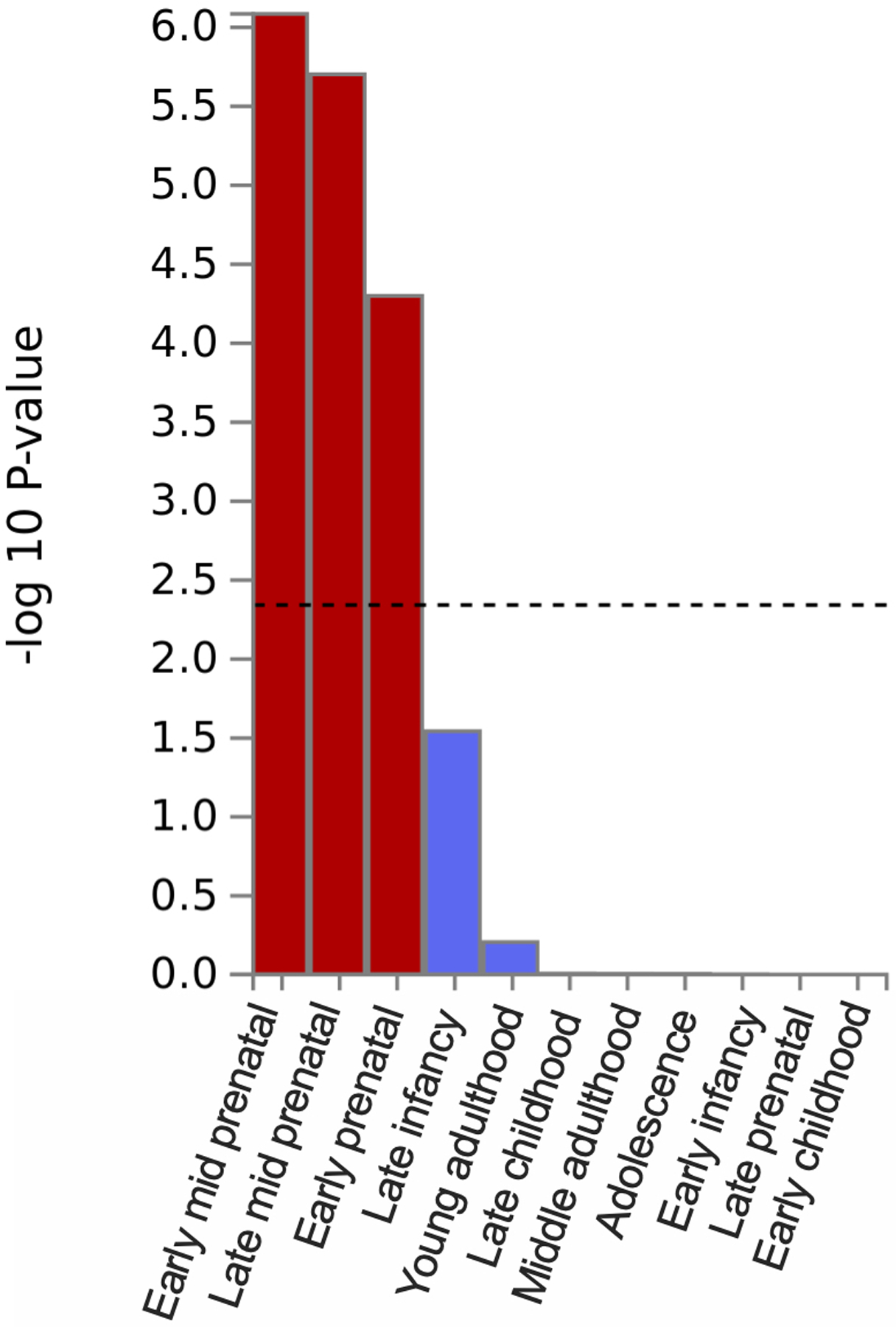
We performed bioinformatic analyses to explore biological processes underlying EXT (Supplementary Information section 6, Supplementary Tables 9–10, and 13–26; Extended Data Figs. 4–8). MAGMA gene-property analyses and gene network analysis with PCNet suggested an abundance of enrichment in genes expressed in brain tissues, particularly during prenatal developmental stages (Extended Data Figs. 6 and 8), with the strongest enrichment seen in the cerebellum, followed by frontal cortex, limbic system tissues, and pituitary gland tissues (Extended Data Fig. 5). Furthermore, MAGMA gene-set analysis and PCNet network analysis identified gene sets related to neurogenesis, nervous system development, and synaptic plasticity, among other gene-sets related to neuronal function and structure.
Because of the strong polygenic signal identified in the GWAS of EXT, four different gene-based analyses identified an abundance of implicated genes (>3,000): (1) functional annotation of the 579 SNPs to their nearest gene with FUMA26, which suggested 587 genes; (2) MAGMA gene-based association analysis27, which identified 928 Bonferroni-significant genes (one-sided P < 2.74×10−6); (3) H-MAGMA28, a method that assigns non-coding SNPs to cognate genes based on chromatin interactions in adult brain tissue and which identified 2,033 Bonferroni-significant genes (one-sided P < 9.84×10−7); and (4) S-PrediXcan29, which uses transcriptome-based analyses of predicted gene expression in 13 brain tissues and which identified 348 Bonferroni-significant gene-tissue pairs (two-sided P < 2.73×10−7).
We found 34 genes that were consistently identified by all four methods, while 741 overlapped across two or more methods (Supplementary Table 22; Extended Data Fig. 7). Several of the 34 implicated genes are novel discoveries for the psychiatric/behavioral literature and have previously been identified only in relation to non-psychiatric biomedical diseases. Such discoveries include ALMS1 (previously associated with kidney function and urinary metabolites30), and ERAP2 (blood protein levels and autoimmune disease31,32). Other genes among the 34 have previously been identified in GWAS of behavioral or psychiatric traits: Cell Adhesion Molecule 2 (CADM2, previously identified in GWAS related to self-regulation, including drug use and risk tolerance17,33), Zic Family Member 4 (ZIC4, associated with brain volume34), Gamma-Aminobutyric Acid Type A Receptor Subunit Alpha 2 (GABRA2; the site of action for alcohol and benzodiazepines, extensively studied in relation to alcohol dependence35,36, and candidate gene for psychiatric disorders37,38), NEGR1 (neuronal growth regulator 1, associated with intelligence and educational attainment39,40), and Paired Basic Amino Acid Cleaving Enzyme (FURIN, associated with schizophrenia, risk tolerance, and vulnerability to psychiatric disorders19,41).
Polygenic score analyses
We created genome-wide polygenic scores for EXT with ~1 million SNPs, adjusted for LD with PRS-CS42 (Supplementary Information section 5), among subjects from two hold-out samples selected for their detailed phenotypes related to externalizing and substance use (Supplementary Information section 5): (1) the National Longitudinal Study of Adolescent to Adult Health (Add Health; N = 5,107), a U.S.-based study of adolescents recruited from secondary schools in the mid-1990s; (2) the Collaborative Study on the Genetics of Alcoholism (COGA; N = 7,594), a U.S.-based study on genetic contributions to alcohol use disorders.
To investigate the validity of EXT, in each of these two samples, we generated a phenotypic externalizing factor by fitting a factor model to phenotypic data corresponding to the seven discovery phenotypes (Extended Data Fig. 9 and Supplementary Table 27). Controlling for age, sex, and ten genetic principal components, the genome-wide polygenic score was associated with the phenotypic factor in both data sets (βAdd Health = 0.33, 95% CI: 0.30–0.36, ΔR2 = 10.5%; βCOGA = 0.30, 95% CI: 0.27–0.34, ΔR2 = 8.9%; Figure 3A and Supplementary Table 28). The variance explained by the EXT polygenic score (ΔR2 ~ 8.9–10.5%) is commensurate with many conventional variables used in social science research, including parental socioeconomic status, family income or structure, and neighborhood disadvantage/disorder43–45. Next, as further quasi-replication, we created a polygenic score using only the 579 EXT SNPs (this score was only used for this quasi-replication exercise), and also this polygenic score was found associated with the phenotypic externalizing factor, explaining ~3–4% of the variance (βAdd Health = 0.20, 95% CI: 0.17–0.23, ΔR2 = 4.1%; βCOGA = 0.17, 95% CI: 0.13–0.20, ΔR2 = 3.0%).
Figure 3 |. Genome-wide EXT polygenic score associations with behavioral, psychiatric, and social outcomes in the independent Add Health (N = 5,107) and COGA (N = 7,594) datasets.
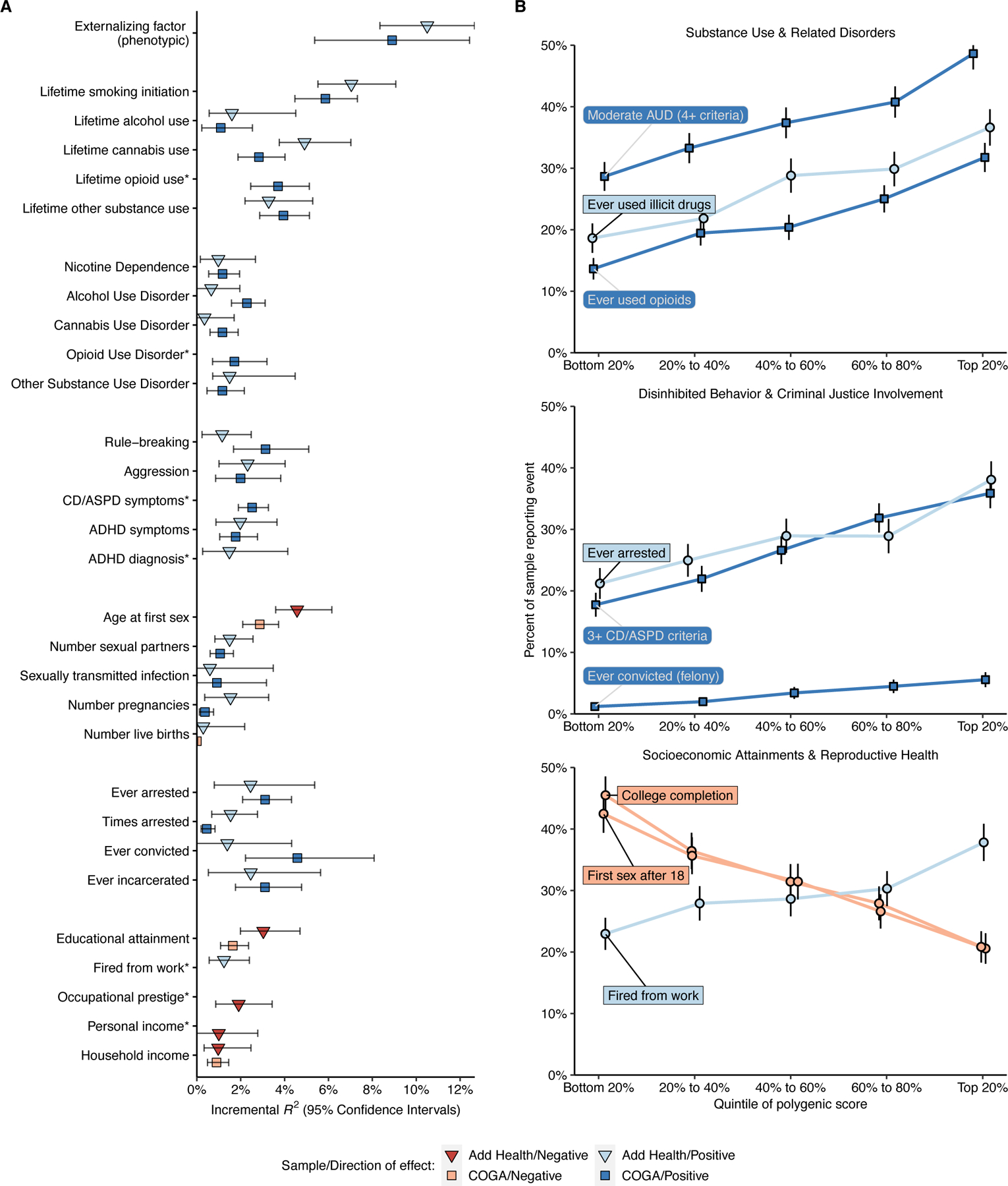
(A) Scatter plots illustrating the incremental proportion of variance (incremental R2, or ΔR2) explained by the genome-wide PRS-CS polygenic score. Light and dark hue indicates the Add Health and COGA cohort, respectively. Blue and red bars indicate positive and negative associations, respectively. The error bars represent 95% confidence intervals centered on ΔR2, computed as 1.96 times the standard error (estimated using percentile method bootstrapping over 1000 bootstrap samples). (B) Line charts illustrating the relative risks across quintiles of the polygenic score for eight (binary or dichotomized) illustrative outcomes: (1) meeting 4 or more criteria for alcohol use disorder (AUD), (2) lifetime use of an illicit substance other than cannabis, (3) lifetime opioid use, (4) ever being arrested, (5) meeting 3 or more criteria for conduct disorder (CD) or antisocial personality disorder (ASPD), (6) ever being convicted of a felony, (7) completing college, and (8) first sexual intercourse at the age of 18 or older. The error bars represent 95% confidence intervals centered on the per-quintile prevalence, computed as 1.96 times the analytical standard error.
In Add Health, COGA, and the Philadelphia Neurodevelopmental Cohort (PNC), we next explored to what extent genome-wide polygenic scores for EXT were associated with childhood externalizing disorders and a variety of phenotypes that reflect difficulty with self-regulation or its consequences (Figure 3B and Supplementary Tables 29–31, see the tables for standard errors per hold-out sample). Polygenic scores for EXT explained significant variance (ΔR2) in criteria counts of ADHD (mean ΔR2 = 1.65%), conduct disorder (CD; mean ΔR2 = 3.1%), and oppositional defiant disorder (ODD; ΔR2 = 1.96%), as well as in the categories substance use initiation (mean ΔR2 = 1.3–6.5%), substance use disorders (mean ΔR2 = 0.8–1.7%), disinhibited behaviors (mean ΔR2 = 1.5–2.5%), criminal justice system involvement (mean ΔR2 = 1.0–3.0%), reproductive health (mean ΔR2 = 0.3–3.7%), and socioeconomic attainment (mean ΔR2 = 0.1–2.3%). Many of the phenotypes – such as opioid use disorder criteria count, conduct disorder and antisocial personality disorder criteria count, lifetime history of arrest or incarceration, and lifetime history of being fired from work – were not included in our Genomic SEM analyses. The associations between the EXT polygenic score and this broad range of phenotypes represents an affirmative test of the hypothesis that genetic variants associated with externalizing liability generalize to a variety of behavioral and social outcomes related to self-regulation.
PheWAS with externalizing polygenic score
To evaluate medical outcomes associated with EXT, we conducted a phenome-wide association study (PheWAS) in 66,915 genotyped individuals of European-ancestry in the BioVU biorepository, a U.S.-based biobank of electronic health records from the Vanderbilt University Medical Center46. A logistic regression was fit to 1,335 case/control disease phenotypes. 255 disease phenotypes were associated with the EXT polygenic score at false discovery rate <0.05, with odds ratios ranging from 0.8 to 1.4 per standard deviation increase in the score (Figure 4 and Supplementary Table 32). The most abundant associations were with mental and behavioral disorders, such as substance use, mood disorders, suicidal ideation, and attempted suicide. Individuals with higher EXT polygenic scores also showed worse health in nearly every bodily system. They were more likely to suffer, for example, from ischemic heart disease, viral hepatitis C and HIV infection, type 2 diabetes and obesity, cirrhosis of liver, sepsis, and lung cancer. Behaviors related to self-regulation, e.g., smoking, drinking, drug use, condomless sex, and overeating, contribute to many of these medical outcomes.
Figure 4 |. Phenome-wide association study in the BioVU biorepository.
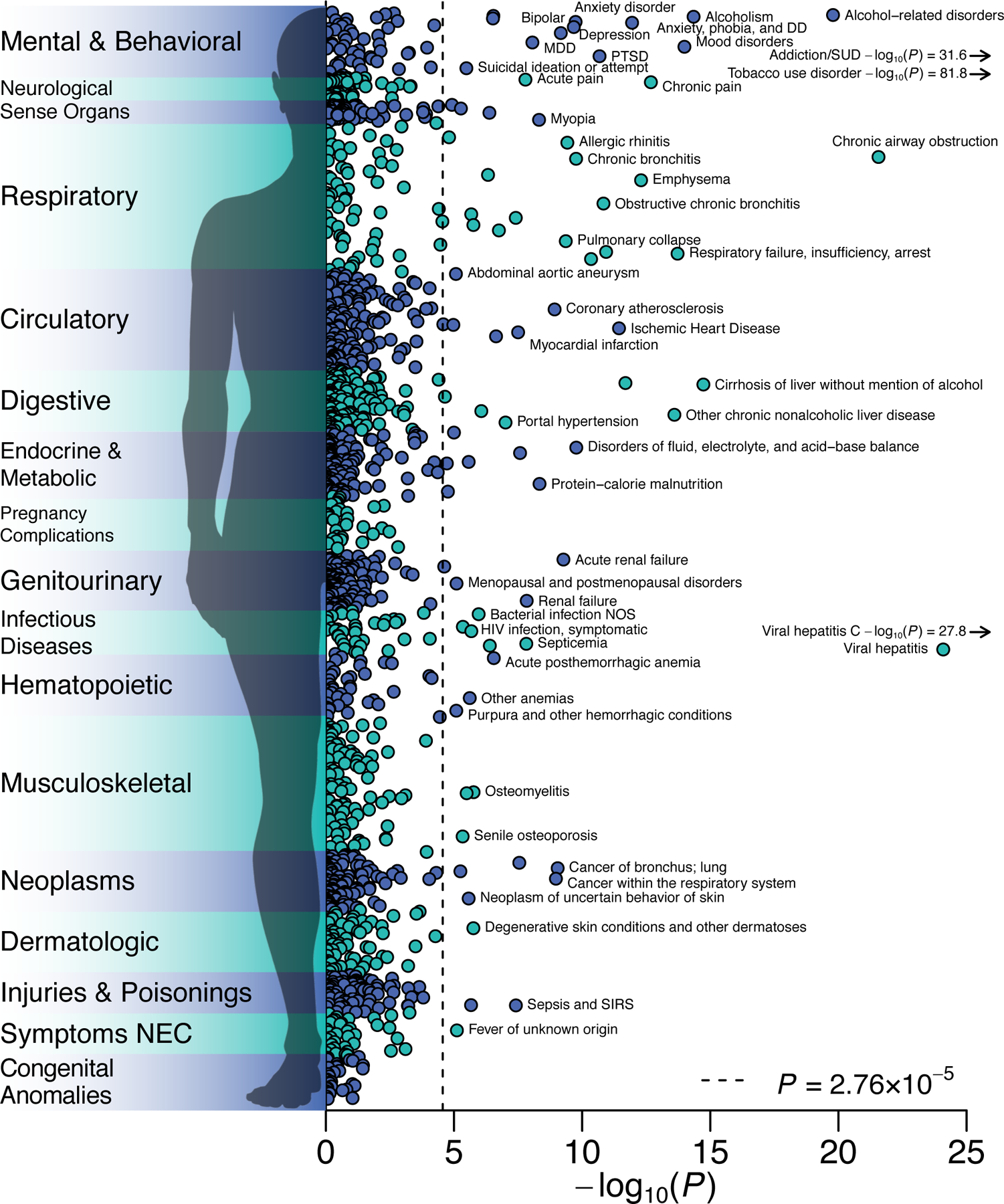
−log10 P values for two-sided Z-test of the log of the odds ratio for the genome-wide PRS-CS polygenic score for EXT with 1,335 medical outcomes, estimated with logistic regression in up to 66,915 patients, adjusted for sex, median age in the EHR data, and the first 10 genetic PCs. The dashed line is the Bonferroni-corrected significance threshold; adjusted for the number of tested medical conditions. 84 medical conditions were Bonferroni-significant, while 255 conditions were significant at a false discovery rate less than 0.05. The labels for some conditions were omitted. The complete results, including case-control counts, effect sizes, and standard errors, are reported in Supplementary Table 20.
Within-family analyses demonstrate robustness to confounding
Genetic associations detected in GWAS can be due to direct genetic effects, but can also be confounded by population stratification, indirect genetic effects from e.g., parental environment, and assortative mating47,48. While reducing statistical power, sibling comparisons overcome these methodological challenges, because meiosis randomizes genotypes to siblings47,49. We therefore conducted within-family analyses of polygenic score associations in the sibling sub-samples of Add Health (N = 994 siblings from 492 families) and COGA (N = 1,353 siblings from 621 families), and a sibling sample from the UKB (N = 39,640), which were held-out from the discovery stage (Supplementary Information section 2.3.2).
In Add Health and COGA, the phenotypic externalizing factor corresponding to the seven discovery phenotypes (see above) was regressed on the genome-wide EXT polygenic scores in a within-family model (Supplementary Table 33). Parameter estimates from the within-family model (βWF Add Health = 0.12, 95% CI: 0.04–0.20; βWF COGA = 0.14, 95% CI: 0.08–0.20) were smaller compared to OLS models without family-specific intercepts (βAdd Health = 0.20, 95% CI, 0.16 to 0.24; βCOGA = 0.16, 95% CI, 0.12 to 0.20), but remained statistically significant (Add Health two-sided P = 4.89×10−3; COGA two-sided test P = 1.87×10−6). As a formal test of attenuation, we evaluated the standardized difference between βWF and β (i.e., a Z-statistic assumed to be normally distributed, see Supplementary Information section 5.2.6) and found that it was −1.988 (two-sided P = 0.047) and −0.704 (two-sided P = 0.481) for the PRS-CS polygenic score in Add Health and COGA, respectively. Thus, we conclude that there was some, but not extreme, attenuation when predicting the phenotypic externalizing factor within families. Also, the association of the quasi-replication polygenic score constructed with the 579 EXT SNPs remained significant and basically did not attenuate in within-family models (for this score, the standardized difference between βWF and β was −0.338 (two-sided P = 0.735) and 0.07 (two-sided P = 0.944) in Add Health and COGA, respectively).
In the UKB sibling hold-out sample, we conducted analyses of the genome-wide EXT polygenic scores with 37 phenotypes from the domains of (a) risky behavior, (b) overall and reproductive health, (c) cognitive ability, (d) personality, and (e) socioeconomic status (Supplementary Information section 5.2.5; Supplementary Table 34). We evaluated the per-category mean of the standardized difference between βWF and β, and found that within-family estimates were, on average, the same for the risky behavior category (mean attenuation = 0.08; 95% CI: −1.67 to 1.83), and only attenuated modestly for personality (mean attenuation = −0.35; 95% CI: −1.06 to 0.36). However, the within-family estimates attenuated more for cognitive ability (mean attenuation −6.55; 95% CI: −9.93 to −3.17), socioeconomic status (mean attenuation −2.43; 95% CI: −4.39 to −0.48), and overall and reproductive health (mean attenuation −2.20; 95% CI: −4.18 to −0.21). Nonetheless, the EXT polygenic score remained nominally significant (two-sided P < 0.05) with 24 outcomes across the five categories, showing that the externalizing GWAS captures genetic effects that are not solely a consequence of uncontrolled population stratification, indirect genetic effects, or other forms of environmental confounding.
Discussion
Externalizing disorders and behaviors are a widely prevalent cause of human suffering, but understanding of the molecular genetic underpinnings of externalizing has lagged behind progress made in other areas of medical and psychiatric genetics. For example, dozens of genetic loci have been discovered for schizophrenia (>100 loci)50, bipolar disorder (30 loci)51, and major depressive disorders (44 loci)52, whereas for antisocial behavior53, alcohol use disorders54, and opioid use disorders8, only a very small number of loci have been discovered. We used multivariate genomic analyses to accelerate genetic discovery, identifying 579 genome-wide significant loci associated with a liability toward externalizing outcomes, 121 of which are entirely novel discoveries for any of the seven phenotypes analyzed. Follow-up bioinformatic analyses suggest the implicated genes have early neurodevelopmental effects, which are then associated with behavioral patterns that have repercussions across the lifespan.
Our results demonstrate that moving beyond traditional disease classification categories can enhance gene discovery, improve polygenic scores, and provide information about the underlying pathways by which genetic variants impact clinical outcomes. GWAS efforts find almost ubiquitous genetic correlations across psychiatric disorders55,56; new analytic methods now allow us to capitalize on these genetic correlations. Pragmatically, non-disease phenotypes such as the ones we use here (e.g., self-reported age at first sex) are often easier to measure in the general population than diagnostic status, making it easier to achieve large sample sizes. Expanding beyond individual diagnoses increases our ability to detect genes underlying human behavioral and medical outcomes of consequence. Our polygenic score for externalizing has one of the largest effect sizes of any polygenic score in psychiatric and behavioral genetics, accounting for ~10% of the variance in a phenotypic externalizing factor. These effect sizes rival the associations observed with “traditional” covariates used in social science research.
Polygenic scores created using our GWAS results were associated not just with psychiatric and substance use disorders, but also with correlated social outcomes, such as lower employment and greater criminal justice system involvement, as well as with biomedical conditions affecting nearly every system in the body. These results highlight again that there is no distinct line between the genetic study of biomedical conditions and the genetic study of social and behavioral traits57. Linking biology with socially-valued behavioral outcomes can be politically sensitive58. Modern genetics research is routinely appropriated by white supremacist movements to argue that racialized disparities in health, employment, and criminal justice system involvement are due to the genetic inferiority of people of color rather than environmental and historical disadvantages59. At the same time, failing to understand how individual genetic differences contribute to vulnerability to externalizing can increase stigma and blame for these behaviors60. Given the horrific legacy of eugenics, the ongoing reality of racism in the medical and criminal justice systems, and the importance of combatting stigma in psychiatric disorders, the scientific results we report here (which are, for technical reasons, limited to European-ancestry individuals) must be interpreted with great care. Our results are not evidence that some people are genetically determined to experience certain life outcomes or are “innately” antisocial. Genetic differences are probabilistically associated with psychiatric, medical, and social outcomes, in part via environmental mechanisms that might differ across historical, political, and economic contexts61. Please see our Frequently Asked Questions (FAQ) and supporting materials at www.externalizing.org.
In conclusion, our analyses demonstrate the far-reaching toll of human suffering borne by people with high genetic liabilities to externalizing. Future work will be needed to tease apart the pathways by which biological and social risks unfold within and across generations, and our findings can contribute to that effort.
Online methods
The article is accompanied by Supplementary Information. The study followed a preregistered analysis plan (https://doi.org/10.17605/OSF.IO/XKV36), which specified that we would generate new, or collect existing, single-phenotype genome-wide association study (GWAS) summary statistics on externalizing phenotypes (Supplementary Information section 1). Summary statistics were to be analyzed with Genomic SEM to (a) estimate a genetic factor structure underlying externalizing liability, (b) identify single-nucleotide polymorphisms (SNPs) and genes involved in a shared genetic liability to externalizing rather than individual traits, and (c) increase the accuracy of polygenic scores for specific externalizing phenotypes that are difficult or intractable to study in large samples. To ensure satisfying statistical power, we preregistered a minimum sample-size of N > 15,000, and that additional exclusions would be based on negligible SNP-based heritability or GWAS signal. All considered traits are discussed in the preregistration and listed in Supplementary Table 1, while the following sections focus on 11 phenotypes that were not excluded due to negligible SNP-based heritability or GWAS signal. The study did not manipulate an experimental condition or collect any new individual-level data, and thus, was neither randomized nor blinded.
Collecting single-phenotype GWAS on externalizing phenotypes
A detailed definition of “externalizing phenotypes” was preregistered to delimit the collection of single-phenotype summary statistics (Supplementary Information section 2.1). Summary statistics from existing studies were provided by, or downloaded from the public repositories of, 23andMe, the Psychiatric Genomics Consortium (PGC), the Million Veterans Program (MVP), the International Cannabis Consortium (ICC), the GWAS & Sequencing Consortium of Alcohol and Nicotine Use (GSCAN), the Social Science Genetics Association Consortium (SSGAC), the Genetics of Personality Consortium (GPC), and the Broad Antisocial Behavior Consortium (Broad ABC) (Supplementary Information section 2.2). All considered GWAS are listed in Supplementary Table 1, and Supplementary Table 4 reports the 67 underlying cohorts of the summary statistics in the final Genomic SEM specification (see below).
GWAS in UK Biobank (UKB)
For the Genomic SEM analyses, we conducted a total of 10 GWAS in UKB (listed in Supplementary Table 1). These GWAS were conducted for two reasons: (1) to generate summary statistics for phenotypes that had not yet been studied in the full genetic data release, or (2) to generate hold-out summary statistics that excluded participants for follow-up analyses. The hold-out summary statistics were used to replace, in our Genomic SEM analyses, summary statistics from existing studies that had included UKB. With respect to (1), summary statistics for “age at first sexual intercourse” and “Alcohol Use Disorder Identification Test problem items” (AUDIT-P) were later included in the final Genomic SEM specification (the latter as a meta-analysis with a GWAS on alcohol dependence by the PGC). With respect to (2), the final specification included replacement summary statistics on “lifetime cannabis use”, “general risk tolerance”, and “lifetime smoking initiation”, and “number of sexual partners”. The seventh phenotype in the final specification—ADHD by the PGC—did not include analyses from UKB. For a detailed description, see Supplementary Information section 2.2–2.3.
The GWAS in UKB were conducted with linear mixed models (BOLT-LMM version 2.3.2) and were adjusted for sex, birth year, sex-specific birth-year dummies, genotyping array and batch, and 40 genetic principal components (PCs) estimated with FlashPCA2 (version 2.0). Two partly overlapping hold-out subsamples of UKB participants were excluded from all single-phenotype GWAS summary statistics that included UKB data, and the participants were instead retained as a hold-out sample for polygenic score analyses (Supplementary Information section 2.3.2). Genetic relatives (pairwise KING coefficient ≥ 0.0442, version 2.1.5) of the held-out individuals were excluded from the study altogether to ensure independence between the discovery and follow-up analyses. In summary, whenever an existing GWAS (or GWAS meta-analysis) was based on UKB, we re-conducted it using the same phenotype definition to generate summary statistics that excluded the hold-out sample and their genetic relatives.
GWAS inclusion criteria, quality control, and meta-analysis
All GWAS were conducted among individuals that (a) were of European ancestry, (b) were not missing any relevant covariates, (c) were successfully genotyped and passed standardized sample-level quality control (according to study-specific protocols13–16,18), and (d) were unrelated (unless a particular GWAS was conducted with linear mixed models). Genotypes were imputed with reference data from either the 1000 Genomes Consortium62, the Haplotype Reference Consortium63, the UK10K Consortium64, or a combination thereof. We performed quality control with EasyQC (version 9.1)65. For that purpose, we used a whole-genome sequenced reference panel, assembled from 1000 Genomes Consortium62 and UK10K Consortium64 data by using BCFtools (version 1.8; Supplementary Information section 2.4.1). Our quality-control procedure applied recommended65 SNP-filtering to (i) remove rare SNPs (minor allele frequency < 0.005), (ii) SNPs with an IMPUTE imputation quality (INFO) score less than 0.9, (iii) SNPs that could not be mapped to or had discrepant alleles with the reference panel, and (iv) otherwise low-quality variants (Supplementary Table 2). For a complete description of the quality-control, see Supplementary Information section 2.4.
We performed sample-size weighted meta-analysis with METAL (versions 2011-03-25 and 2020-05-05)66, while ensuring absence of sample overlap (Supplementary Information section 2.5.1). We excluded any summary statistics with negligible SNP-based heritability (h2 < 0.05) or GWAS signal ( < 1.05), estimated with LD Score regression (version 1.0.0)12,55. At this stage, we had collected or generated well-powered summary statistics for 11 phenotype-specific GWAS (or meta-analysis) that satisfied our inclusion criteria (Supplementary Table 3): (1) attention-deficit/hyperactivity disorder (ADHD, N = 53,293), (2) reverse-coded age at first sexual intercourse (FSEX, N = 357,187), (3) problematic alcohol use (ALCP, N = 164,684), (4) automobile speeding propensity (DRIV, N = 367,151), (5) alcoholic drinks per week (DRIN, N = 375,768), (6) reverse-coded educational attainment (EDUC, N = 725,186), (7) lifetime cannabis use (CANN, N = 186,875), (9) lifetime smoking initiation (SMOK, N = 1,251,809), (9) general risk tolerance (RISK, N = 426,379), (10) irritability (IRRT, N = 388,248), and (11) number of sexual partners (NSEX, N = 336,121) (Supplementary Table 4). The GWAS effect-sizes of age at first sexual intercourse and educational attainment were reversed to anticipate positive correlations with externalizing liability.
Exploratory factor analysis
As an initial analysis to guide the multivariate analyses, we performed hierarchical clustering of a matrix with pair-wise LD Score (version 1.0.0) genetic correlations (rg) (Supplementary Information section 3). The 11 phenotypes displayed appreciable genetic overlap with at least one other phenotype (max |rg| = 0.245–0.773) (Supplementary Table 5). Three (k) clusters were identified: (1) attention deficit/hyperactivity disorder (ADHD), educational attainment (EDUC), age at first sexual intercourse (FSEX), irritability (IRRT), and smoking initiation (SMOK); (2) problematic alcohol use (ALCP), drinks per week (DRIN); and (3) lifetime cannabis use (CANN), automobile speeding propensity (DRIV), number of sexual partners (NSEX), general risk tolerance (RISK).
Exploratory factor analysis tested four factor solutions, specifying 1 to k + 1 factors with the factanal function of R (“stats” package version 3.5.1) (Supplementary Information section 3.2), where k is the number of clusters identified in the genetic correlation matrix, while retaining factors that explained ≥15% variance (preregistered threshold). The fourth factor explained only 12.5% variance, and thus, the three-factor solution was considered the best-fitting exploratory model (Supplementary Table 6). The factor loadings were consistent with the hierarchical clustering. However, as detailed in Supplementary Information section 3.2, the second and third factor accounted for complex residual variation and divergent residual cross-trait correlations among the subset of phenotypes that had the weakest loadings on the single common factor. Thus, we learned from exploratory analysis that some of the 11 indicators may not be optimal for identifying a common genetic liability to externalizing, and that a less complex model with fewer indicators may perform better in subsequent confirmatory analyses.
Confirmatory factor analyses with Genomic SEM
We formally modelled genetic covariances (rather than rg) in confirmatory factor analyses using genomic structural equation modeling (Genomic SEM, versions 0.0.2a-c)10 (Supplementary Information section 3.3). Genomic SEM is unbiased by sample overlap and imbalanced sample size, and by applying to summary statistics allows for genetic analyses of latent factors with more observations than is typically possible with individual-level data10. We estimated and benchmarked four models: (1) a common factor model with the 11 phenotypes, (2) a correlated three-factors model with the 11 phenotypes (with and without cross-loadings), (3) a bifactor model with the 11 phenotypes, and finally, (4) a revised common factor model that only included seven of the phenotypes that satisfied moderate-to-large (i.e., ≥ 0.50) loadings on the single latent factor in model (1) (Supplementary Table 7). We found that model (4) was the only model that closely approximated the observed genetic covariance matrix (χ2(12) = 390.234, AIC = 422.234, CFI = 0.957, SRMR = 0.079), fulfilled our preregistered model fit criteria, and coalesced with theoretical expectations of a common genetic liability to externalizing. This model was selected as final specification, and is hereafter referred to as “the externalizing factor” (EXT). To explore the convergent and discriminant validity of the externalizing factor, we estimated its genetic correlation with 91 traits from various domains (Supplementary Table 8).
Multivariate GWAS analyses with Genomic SEM
Using Genomic SEM, we performed multivariate genome-wide association analysis by estimating SNP associations with EXT, which is our main discovery analysis (Supplementary Information section 3.4). The estimated effective sample size of the “externalizing GWAS” is Neff = 1,492,085, and the mean χ2 and genomic inflation factor (λGC) are 3.114 and 2.337, respectively. LD Score regression suggested polygenicity rather than bias from population stratification10,12, as the (default settings) LD Score intercept and attenuation ratio were estimated to be 1.115 (SE = 0.019) and 0.054 (SE = 0.009), respectively.
Conventional “clumping” was applied with PLINK (v1.90b6.13)67 to identify near-independent genome-wide significant lead SNPs, with the primary (two-sided) P-value threshold of 5×10−8, the secondary P-value threshold (for computational efficiency) of 1×10−4, and an r2 threshold of 0.1 together with a wide SNP window (1,000,000 kb). Before counting the number of hits to report, we first subjected the 855 lead SNPs to “multi-SNP-based conditional & joint association analysis using GWAS summary data” (GCTA-COJO, version 1.93.1beta)23,68 (Supplementary Information section 3.4.2). This procedure identified 579 lead SNPs that were conditionally and jointly associated with EXT (Supplementary Table 9). We performed lookups of these “579 EXT SNPs”, and any correlated SNPs (r2 > 0.1), in the NHGRI-EBI GWAS Catalog6 (e96 2019-05-03) to investigate whether the loci have previously reported with other traits (at two-sided P < 1×10−5) (Supplementary Table 10). To evaluate whether each SNP acted through the externalizing factor, we estimated QSNP heterogeneity statistics genome-wide with Genomic SEM (Supplementary Information section 3.5.1). The null hypothesis of the QSNP test is that SNP effects on the constituent phenotypes operate (i.e., are statistically mediated) via the EXT factor, so a significant QSNP test indicates that the SNP effects are better explained by pathways independent of EXT. The QSNP analysis identified substantial heterogeneity (160 near-independent genome-wide significant QSNP loci), but reassuringly, did not identify heterogeneity among 99% (571/579) of the EXT SNPs (Supplementary Table 10). An analysis of sign concordance further supported homogeneity among the EXT SNPs (Supplementary Information section 3.5.4).
Proxy-phenotype and quasi-replication analysis
We conducted proxy-phenotype69 and quasi-replication70 analyses by investigating the 579 EXT SNPs for association in two independent, second-stage GWAS on (1) alcohol use disorder (N = 202,004, rg = 0.52) and (2) antisocial behavior (N = 32,574, rg = 0.69) (Supplementary Information section 4) (Supplementary Tables 11–12). For SNPs missing from the second-stage GWAS, we analyzed proxy SNPs (r2 > 0.8). Significant proxy-phenotype associations were evaluated for Bonferroni-corrected significance (two-sided test P < 0.05/579). For the quasi-replication, we generated empirical null distributions for the second-stage GWAS by randomly selecting 250 near-independent (r2 < 0.1) SNPs matched on MAF (± 1 percentage point) for each of the 579 SNPs. The quasi-replication included three steps: (1) a binomial test of sign concordance to test whether the direction of effect of the SNPs were in greater concordance between the externalizing GWAS and each of the second-stage GWAS compared to what would be expected by chance (H0 = 0.5); (2) a binomial test of whether a greater proportion of the SNPs were nominally significant (two-sided P < 0.05) in the second-stage GWAS compared to the empirical null distribution; (3) a test of joint enrichment, using a non-parametric (one-sided) Mann-Whitney test of the null hypothesis that the P values of the SNPs are derived from the empirical null distribution. We strongly rejected the null hypotheses in all quasi-replication tests, suggesting that the externalizing GWAS is not spurious overall and that it was more enriched for association with the second-stage phenotypes than their respective polygenic background GWAS signal.
Polygenic score analyses
We generated polygenic scores by summing genotypes weighed by the effect sizes estimated in the externalizing GWAS, among individuals of European ancestry in five hold-out cohorts: (1) Add Health71,72, (2) COGA73–75, (3) PNC76,77, (4) the UKB siblings hold-out cohort78, and (5) BioVU46 (Supplementary Information section 5). In each dataset, we generated three scores, of which two were adjusted for linkage disequilibrium (LD): (1) PRS-CS (version October 20, 2019; default Bayesian gamma-gamma prior of 1 and 0.5, and 1,000 Monte Carlo iterations with 500 burn-in iterations)79, (2) LDpred (version 0.9.09; infinitesimal Bayesian prior)80, and (3) unadjusted scores81, while using SNPs that overlapped the HapMap 3 Consortium consensus set82 (for comparability across methods and with previous work, and because PRS-CS imposes that restriction). We evaluated the incremental R2/pseudo-R2 (ΔR2) attained by adding the polygenic score to a regression model with baseline covariates, as in previous efforts17. The baseline model included covariates for sex, age, and genetic principal components (PCs), and genotyping array and batch. The choice of statistical model (e.g., least squares vs. logit) and adjustment of standard errors depended on (1) the phenotype distribution and (2) the cohort data structure (independent vs. clustered observations), see Supplementary Information section 5.2.4. We estimated 95% confidence intervals for ΔR2 using percentile method bootstrapping (1000 iterations).
In Add Health and COGA, we performed out-of-sample validation of EXT by modeling a latent phenotypic externalizing factor corresponding to the seven Genomic SEM phenotypes (Supplementary Information section 5.2.3) (Supplementary Tables 27–28). In Add Health, COGA, PNC, and the UKB siblings hold-out cohort, we performed exploratory analyses with a range of preregistered phenotypes from various domains (Supplementary Tables 29–31, 34). In BioVU, we performed a phenome-wide association study (PheWAS) of medical outcomes by fitting a logistic regression to 1,335 case/control disease “phecodes”83 (N = 66,915) (Supplementary Table 32), adjusted for sex, median age in the EHR data, and the first 10 genetic PCs.
We performed within-family analyses among full siblings in Add Health, COGA, and the UKB siblings hold-out cohort (Supplementary Information section 5.2.5). We analyzed 492 families in Add Health (Nsiblings = 994), 621 families in COGA (Nsiblings = 1,353), and 19,252 families in the UKB (Nsiblings = 39,640). In Add Health and COGA, we applied least squares regression on a single outcome: the factor scores of the phenotypic externalizing factor (a continuous variable), while adjusting for family-specific dummy variables (Supplementary Table 33), and calculated the standardized difference (i.e., a Z-statistic) between the within-family coefficient to the coefficient from a model without family dummies (Supplementary Information section 5.2.6). In the UKB siblings hold-out cohort, we performed an analogous analysis of exploratory phenotypes (Supplementary Table 34). We analyzed heteroskedasticity-consistent and cluster-robust standard errors, clustered at the family level.
Bioannotation
We conducted bioannotation and bioinformatic analyses (Supplementary Information section 6). The method FUMA (version 1.3.5e)26 was applied to explore the functional consequences of the 579 SNPs (Supplementary Table 9), which included ANNOVAR categories (i.e., the functional consequence of SNPs on genes), Combined Annotation Dependent Depletion (CADD) scores, RegulomeDB scores, expression quantitative trait loci (eQTLs), and chromatin states. The default external reference data for FUMA is described elsewhere26.
Gene-based analyses was performed with “multi-marker analysis of genomic annotation” (MAGMA, version 1.08)27 (Supplementary Information sections 6.1.2). Genome-wide SNPs were first mapped to 18,235 protein-coding genes from Ensembl (build 85)84, and SNPs within each gene were jointly tested for association with EXT. We evaluated Bonferroni-corrected significance, adjusted for the number of genes (one-sided P < 2.74×10−6) (Supplementary Table 13). Next, MAGMA gene-set analysis was performed using 15,481 curated gene sets and Gene Ontology (GO)85 terms obtained from the Molecular Signatures Database (MsigDB version 7.0)86. We evaluated Bonferroni-corrected significance, adjusted for the number of gene sets (one-sided P < 3.23×10−6) (Supplementary Table 14). A gene property analysis tested the relationships between 54 tissue-specific gene expression profiles and gene associations, while adjusting for the average expression of genes per tissue type as a covariate (Supplementary Table 15), and between brain gene expression profiles and gene associations across 11 brain tissues from BrainSpan87 (Supplementary Table 16). Gene expression values were log2 transformed average Reads Per Kilobase Million (RPKM) per tissue type (after replacing RPKM > 50 with 50) based on GTEx RNA-seq data (version 8.0)88. We evaluated Bonferroni-corrected significance, adjusted for the number of tested profiles (one-sided P < 9.26×10−4).
We used an extension of MAGMA: “Hi-C coupled MAGMA” or “H-MAGMA” (based on MAGMA version 1.08)28, to assign non-coding (intergenic and intronic) SNPs to cognate genes based on their chromatin interactions. Exonic and promoter SNPs were assigned to genes based on physical position. We used four Hi-C datasets provided with the software89–91. We evaluated Bonferroni-corrected significance, adjusted for the number of tests within each of the four Hi-C datasets (one-sided P < 9.83–9.86×10−7) (Supplementary Tables 17–20).
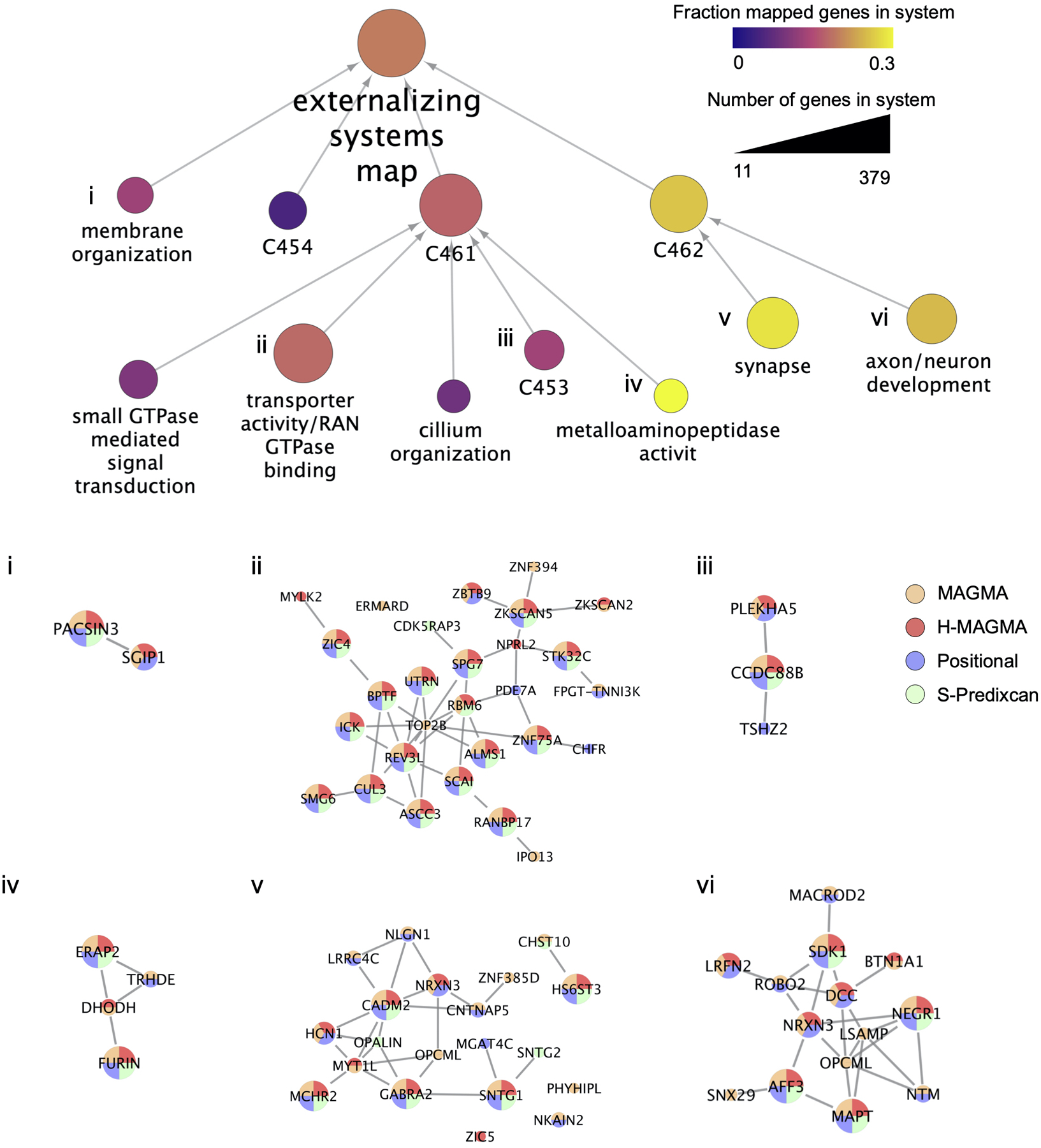
The method S-PrediXcan (version 0.6.2)92 tested the association of EXT with gene expression in brain tissues. We used pre-computed tissue weights from the Genotype-Tissue Expression (GTEx, version 8.0) database as the reference dataset88. As inputs, we used the EXT summary statistics, LD matrices of the SNPs (available at the PredictDB Data Repository, http://predictdb.org, no version number), and transcriptome-tissue data related to 13 brain tissues: anterior cingulate cortex, amygdala, caudate basal ganglia, cerebellar hemisphere, cerebellum, cortex, frontal cortex, hippocampus, hypothalamus, nucleus accumbens basal ganglia, putamen basal ganglia, spinal cord and substantia nigra. We evaluated transcriptome-wide significance at the two-sided P < 2.77×10−7, which is Bonferroni-corrected adjusted for 13 tissues times 13,876 tested genes (180,388 gene-tissue pairs) (Supplementary Table 21). In Supplementary Table 22 we summarize the gene findings. Finally, we followed-up on the subset of gene findings that were consistently implicated in all gene-based methods, by generating an “externalizing gene network” as a parsimonious composite network (PCNet) and an “externalizing systems map” with Cytoscape (version 3.8.2)93,94, and applied Tissue Specific Expression Analysis (TSEA, version 1.0) and Specific Expression Analysis (SEA, version 1.1) to explore tissue and brain region specificity (Supplementary Tables 23–26).
Data availability
All data sources are described in the Supplementary Information and are listed in the Reporting Summary. No new data was collected. Only data from existing studies or study cohorts were analyzed, some of which are restricted access to protect the privacy of the study participants (see Reporting Summary for accession codes or URLs). The minimum data set necessary to interpret, verify, and extend the research, i.e., the GWAS summary statistics for the externalizing (EXT) GWAS (our main discovery analysis), can be obtained by following the procedures detailed at https://externalizing.org/request-data/. In brief, summary statistics are derived from analyses based in part on 23andMe data, for which we are restricted to only publicly report results for up to 10,000 SNPs. The full set of externalizing GWAS summary statistics can be made available to qualified investigators who enter into an agreement with 23andMe that protects participant confidentiality. Once the request has been approved by 23andMe, a representative of the Externalizing Consortium can share the full GWAS summary statistics. No source data is published alongside the paper.
Code availability
No custom algorithms or software was developed in this study. The Reporting Summary lists all software, code, and webtools that were used for the genetic and bioinformatic analyses we report.
Extended Data
Extended Data Fig. 1. Genetic correlations with the genetic externalizing factor (EXT).
Dot plot of genetic correlations (rg) estimated with Genomic SEM between the genetic externalizing factor (EXT) with 91 other complex traits (Supplementary Information section 3). Error bars are 95% confidence intervals, calculated as 1.96×SE, centered on the rg estimate (omitted for Agreeableness). The estimates are also reported in Supplementary Table 8, together with the exact number of independent samples used to derive each estimate. This figure displays genetic correlations with personality measures based on GWAS summary statistics from the Genomics of Personality Consortium, while Figure 1 instead reports genetic correlations with personality measures based on more recent and substantially larger GWAS provided by 23andMe.
Extended Data Fig. 2. Quantile-quantile (Q-Q) plots of the externalizing GWAS and QSNP results.
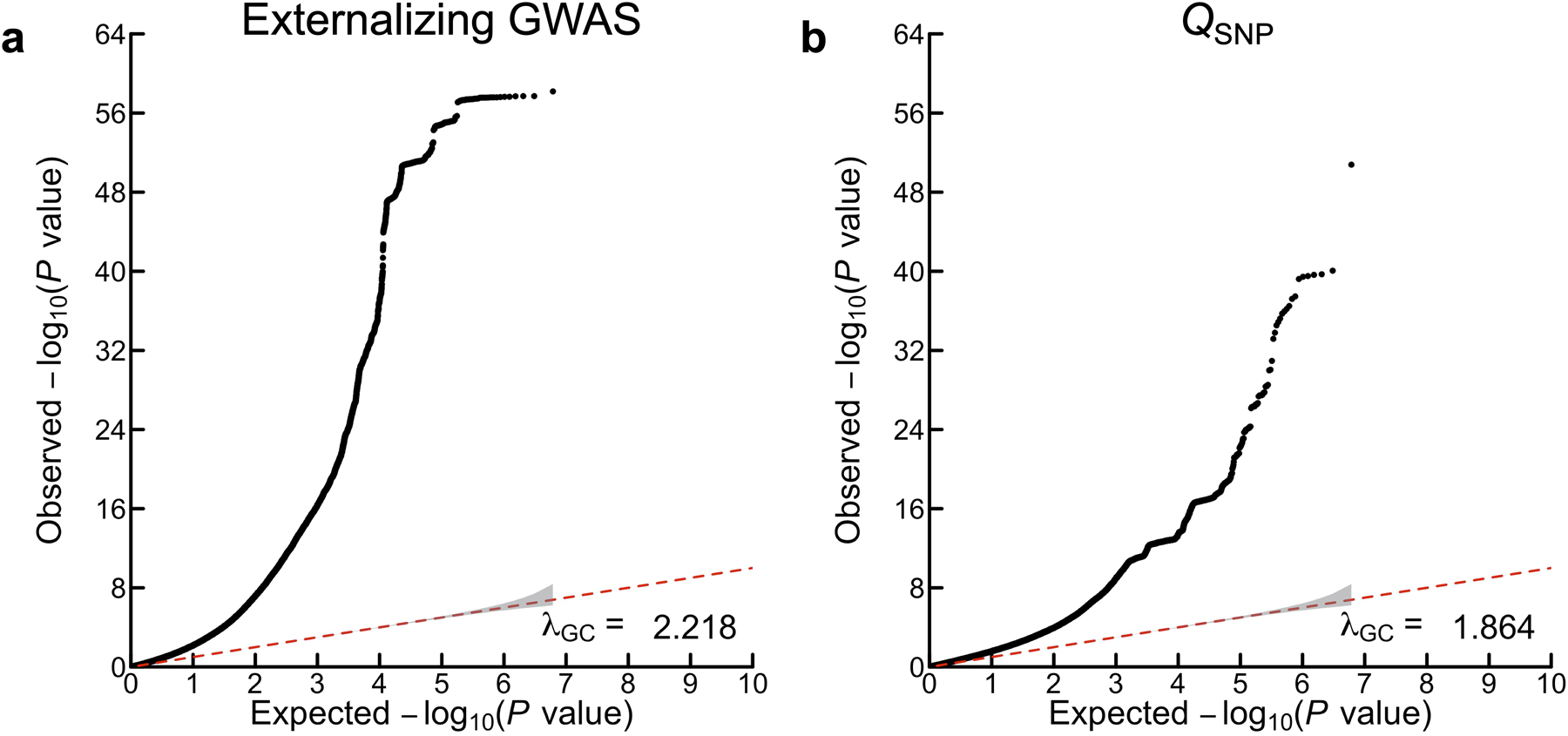
The panels display Q-Q plots for (a) the externalizing GWAS (Neff = 1,492,085), and (b) SNP-level tests of heterogeneity (QSNP) with respect to the SNP-effects estimated in the externalizing GWAS (for more details see Supplementary Information section 3). The y-axis is the observed association P value on the −log10 scale (based on a two-sided Z-test in panel a, and based on a one-sided χ2 test scaled to 1 degree of freedom in panel b). The gray shaded areas represent 95% confidence intervals centered on the expected −log10(P) of the null distribution. The genomic inflation factors displayed here, λGC, is defined as the median χ2 association test statistic divided by the expected median of the χ2 distribution with 1 degree of freedom, and were calculated with 6,132,068 and 6,107,583 SNPs for (a) and (b), respectively. Although there is a noticeable early “lift-off”, the estimated LD Score regression intercepts of (a) 1.115 (SE = 0.019) and (b) 0.9556 (SE = 0.013) suggest that most of the inflation of the test statistics is attributable to polygenicity rather than bias from population stratification
Extended Data Fig. 3. Quantile-quantile (Q-Q) plots of the proxy-phenotypes analyses.
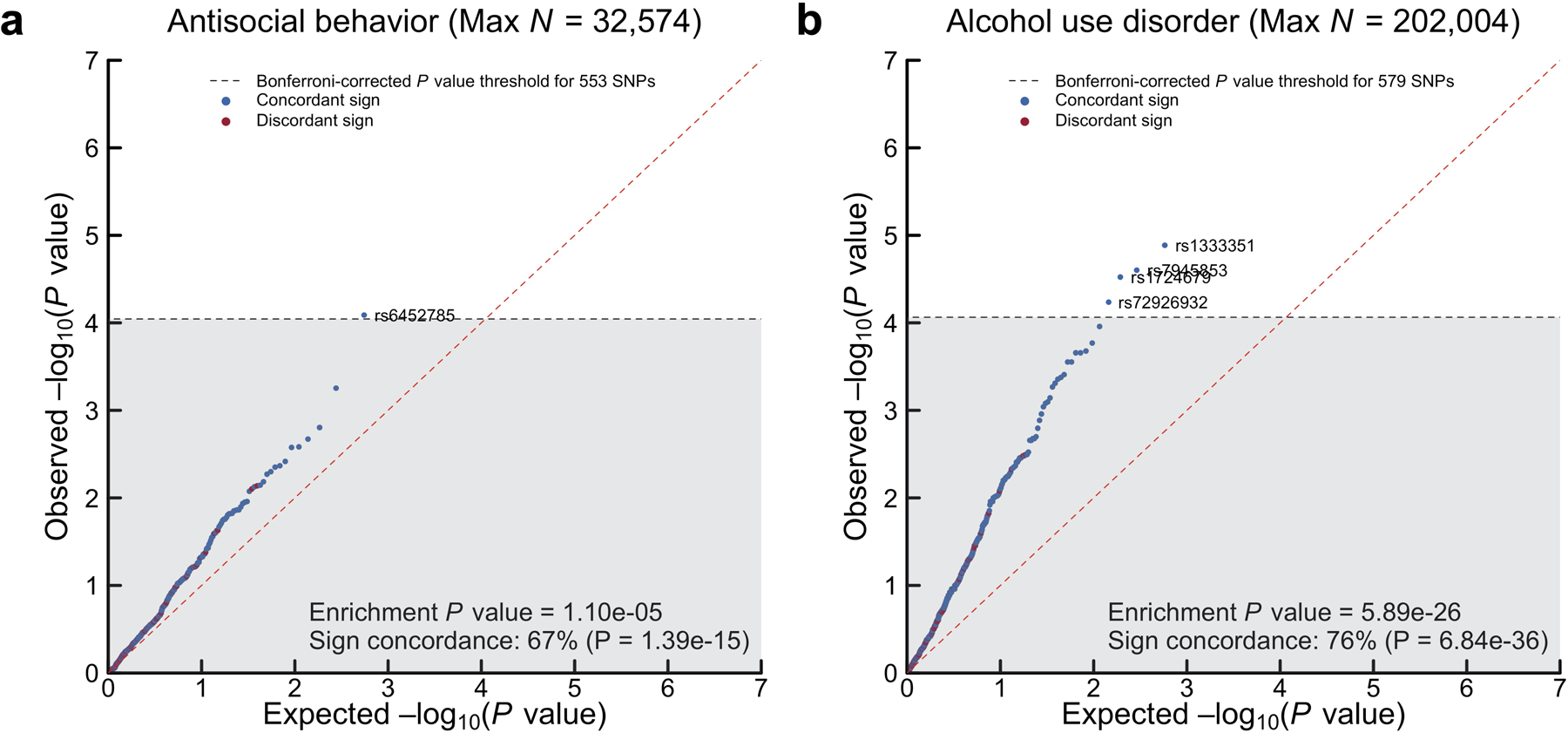
Panels (a–b) show −log10(P values from a two-sided Z-test) for linear regression of the 553 and 579 EXT SNPs (or such SNPs that could be proxied in case of missingness, r2 > 0.8) that were looked up in independent, second-stage GWAS samples on (1) antisocial behavior (N = 32,574) and (2) alcohol use disorder (N = 202,400), respectively (Supplementary Information section 4). Dashed line denotes experiment-wide significance at P < 0.05/553 and 0.05/579 for (1) and (2), respectively. Enrichment P value is the result of a one-sided test of joint enrichment with the non-parametric Mann-Whitney test against an empirical null distribution of 138,250 and 144,750 near-independent (r2 < 0.1) SNPs, matched on MAF, that were randomly selected from the GWAS on (1) and (2), respectively. Sign concordance is the proportion of looked-up SNPs with concordant direction of effect sizes across the externalizing GWAS and the second-stage GWAS, and the sign concordance P value is from a one-sided binomial tests of the sign concordance for the 579 SNPs (against the null hypothesis of 50% concordance that is expected by chance).
Extended Data Fig. 4. MAGMA gene-based association analysis.
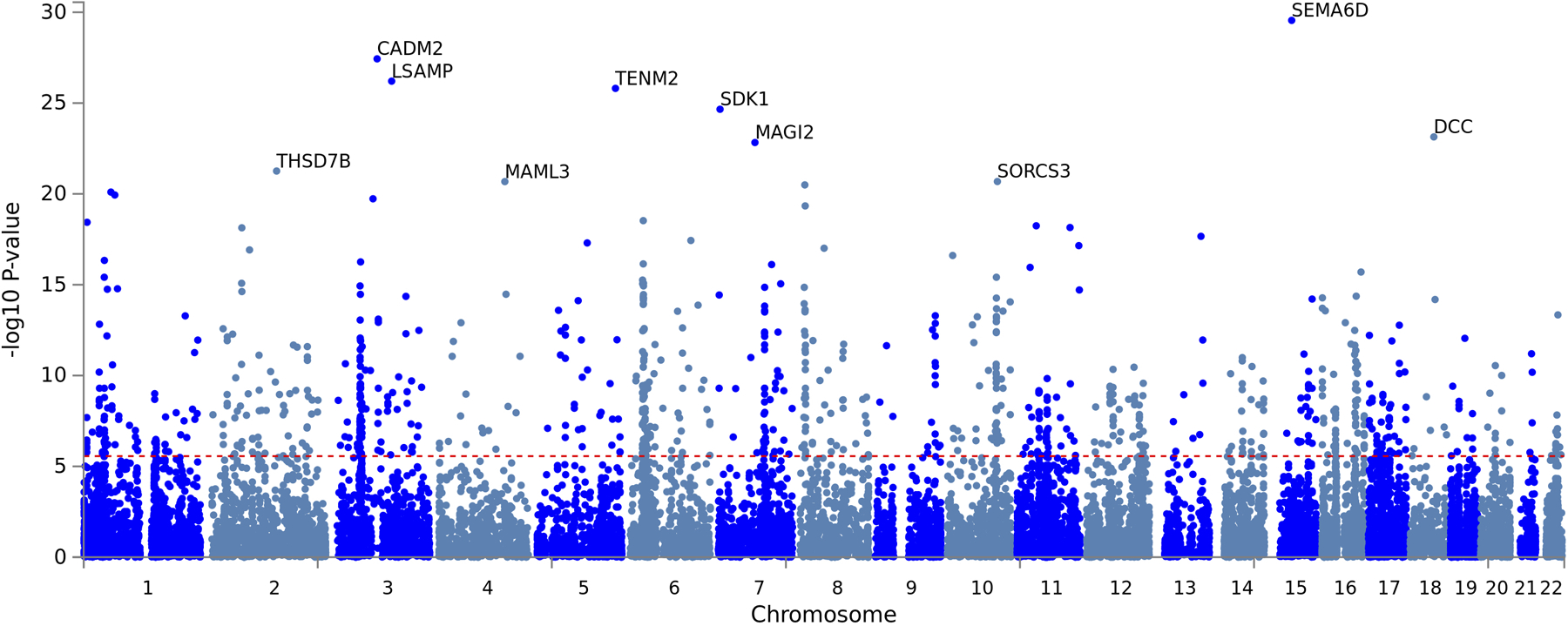
Manhattan plot of the −log10(P from a one-sided Z-test) of 18,093 genes that were tested for association in the MAGMA (v.1.08) gene-based association analysis (Supplementary Information section 6). The 10 most significant genes are labeled with gene names. Red dashed line represents Bonferroni-significance, adjusted for the number of tested genes (one-sided P = 2.74×10−6). 928 genes were found to be significant, of which 244 have one or more genome-wide significant SNPs from the externalizing GWAS within their gene breakpoints. The results are also report in Supplementary Table 13.
Extended Data Fig. 5. MAGMA gene-property analysis.
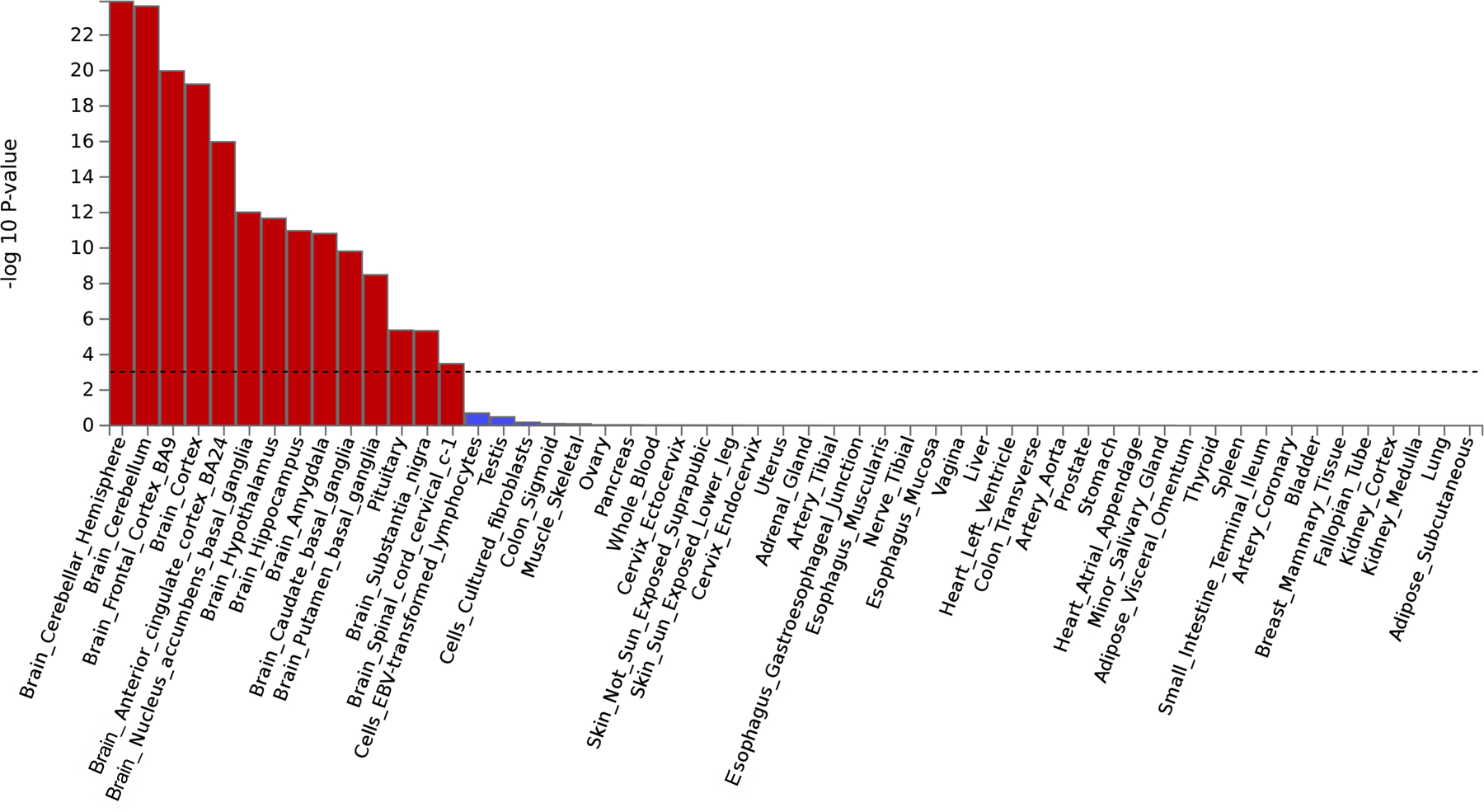
Bar plot of the −log10(P from one-sided Z-tests) of the point estimate from a generalized least squares regression. The analysis identified that the externalizing GWAS is significantly enriched in brain and pituitary gland tissues (Supplementary Information section 6). Dashed line denotes Bonferroni-corrected significance, adjusted for testing 54 tissues (one-sided P < 9.26×10−4). 14 tissues were significantly associated with the externalizing GWAS, including 13 brain related tissues and the pituitary tissue. The results are also report in Supplementary Table 15.
Extended Data Fig. 6. MAGMA gene-property analysis of enrichment in brain tissues across 11 developmental stages (BrainSpan).
Bar plot of the −log10(P from one-sided Z-tests) of the point estimate from a generalized least squares regression. The analysis identified that the externalizing GWAS is significantly enriched during prenatal developmental stages (Supplementary Information section 6). Dashed line denotes Bonferroni-corrected significance, adjusted for testing 54 tissues (one-sided P < 9.26×10−4). The results are also report in Supplementary Table 16.
Extended Data Fig. 7. Gene overlap across multiple gene-association methods.
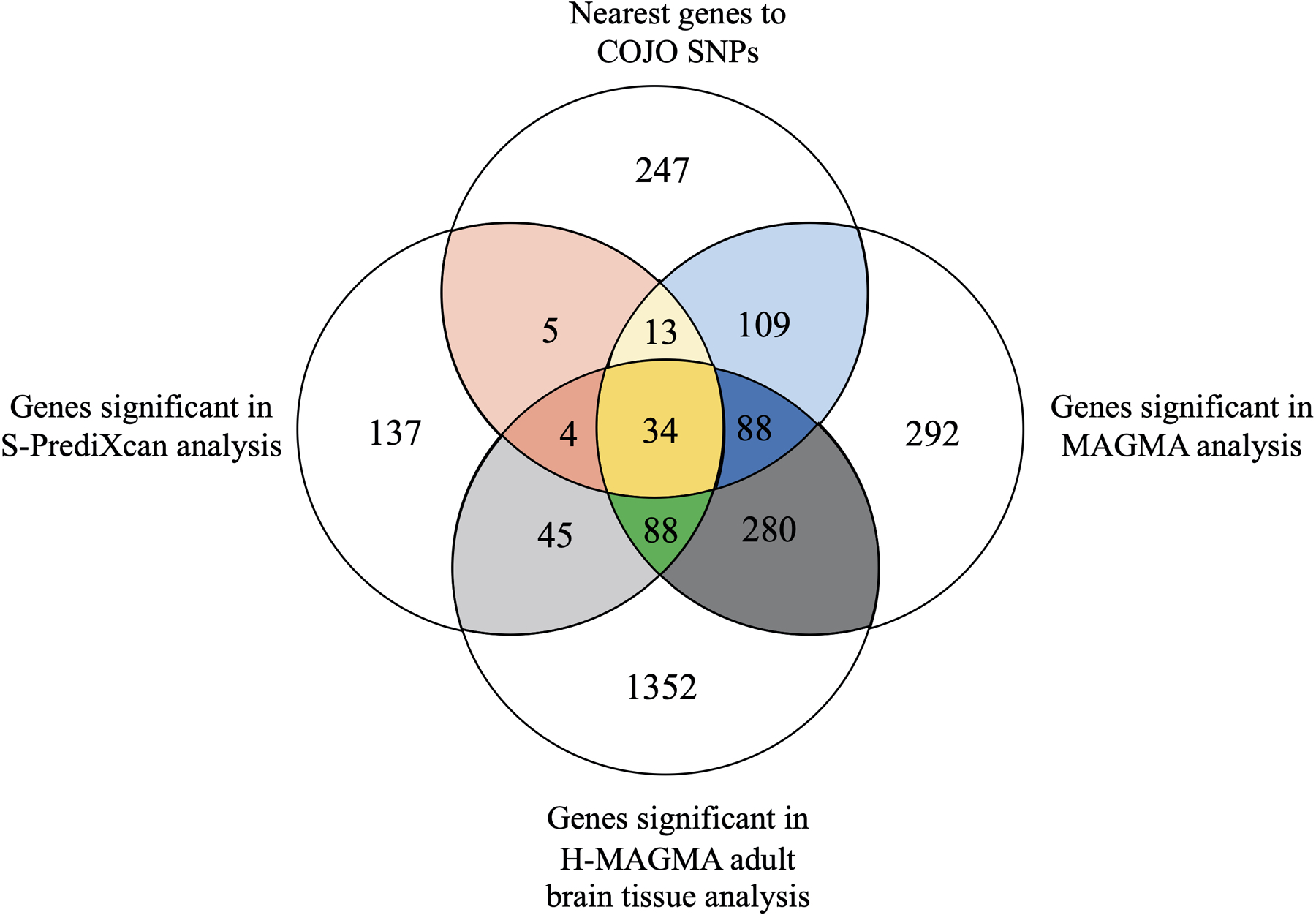
Venn diagram illustrating the overlap between (1) the nearest genes to the 579 jointly associated lead SNPs (denoted as the COJO EXT SNPs, see Supplementary Table 9), (2) the genes significant in the MAGMA gene-based analysis (Supplementary Table 13), (3) the genes significant in the H-MAGMA adult brain tissue analysis (Supplementary Table 17), and (4) the genes significant in the S-PrediXcan analysis (Supplementary Table 21). Across these four approaches, 34 genes were consistently implicated; these genes include CADM2, PACSIN3, ZIC4, MAPT, and GABRA2. Colored regions of this diagram correspond to the coloring shown in Supplementary Table 22, which lists all identified genes. No new statistical test was performed to generate this figure, and the statistical test used in each gene-based approach is reported in the notes of Supplementary Tables 9, 13, 17, and 21.
Extended Data Fig. 8. Externalizing systems map estimated with the Order Statistics Local Optimization Method (OSLOM) algorithm.
Representation of the externalizing network neighborhood estimated with PCNet as modular gene systems. In the top panel, circles represent distinct systems, with size indicating the number of genes belonging to each system (min 11 for “cilium organization”, and max 379 for the “externalizing systems map”). System color indicates the fraction of genes in each system that have been mapped to the externalizing phenotype by at least one of the four gene mapping methods (positional, MAGMA, H-MAGMA, and S-PrediXcan). Systems have been annotated with significantly enriched gene ontology terms. Systems without significant enrichment of biological pathways are labeled with a unique system ID (C454, C461, C453, C462), and may represent novel pathways. (i-vi) Visualization of genes within selected systems that have been mapped to the externalizing phenotype by one or more gene mapping methods, and their molecular interactions. In the bottom panel, the gene size is mapped to the number of methods in which the gene was found associated with externalizing (with the largest genes indicating the gene was identified by all 4 methods), and gene color(s) indicates which method(s) have mapped the gene.
Extended Data Fig. 9. Confirmatory factor analysis of phenotypic externalizing factor in Add Health and COGA.
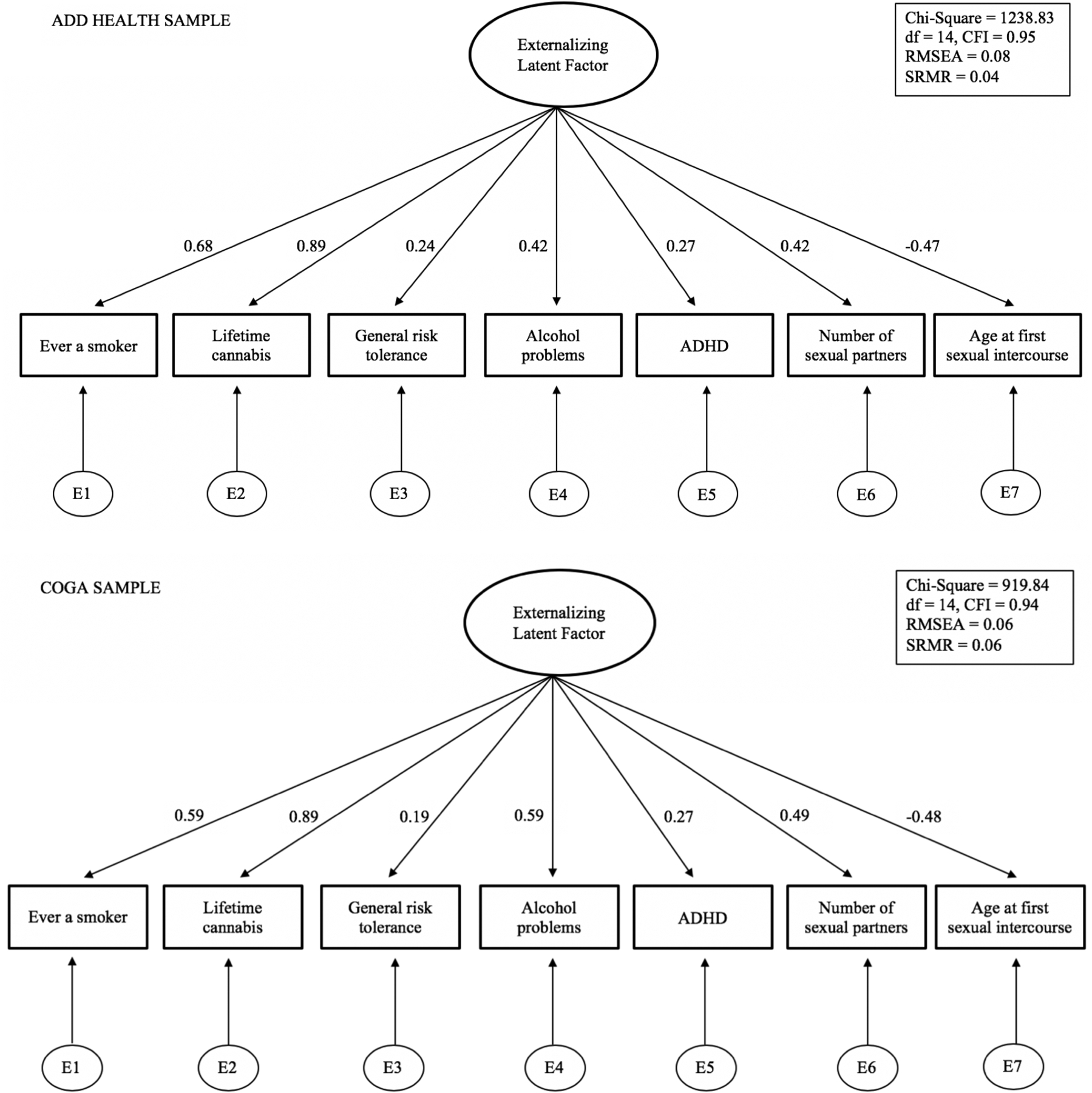
Path diagram of confirmatory factor analysis (CFA) models in (top panel) Add Health (N = 15,107) and (bottom panel) COGA (N = 16,857) (Supplementary Information section 5). The reported model fit statistics and fit indices are degrees of freedom (df), comparative fit index (CFI), root mean square error (RMSEA), standardized root mean squared residual (SRMR). Standardized factor loadings presented as numbers on the paths.
Supplementary Material
Acknowledgements
This research was carried out under the auspices of the Externalizing Consortium. The study was classified as secondary research of de-identified subjects, and the study was awarded ethical approval by the internal review board (IRB) of Virginia Commonwealth University (VCU), with reference number HM20019386. These analyses were made possible by the generous public sharing of summary statistics from published GWAS from the Psychiatric Genomics Consortium (PGC), the Million Veterans Program (MVP), the International Cannabis Consortium (ICC), the GWAS & Sequencing Consortium of Alcohol and Nicotine use (GSCAN), the Social Science Genetics Association Consortium (SSGAC), the Genetics of Personality Consortium (GPC), and the Broad Antisocial Behavior Consortium (BroadABC). We would like to thank the many studies that made these consortia possible, the researchers involved, and the participants in those studies, without whom this effort would not be possible. We would also like to thank the research participants and employees of 23andMe for making this work possible. This research was conducted in part using the UK Biobank Resource under applications 40830 and 11425. We thank all UKB cohort participants for making this study possible. We thank Lea K. Davis for providing access to BioVU. Finally, we thank the Collaborative Study on the Genetics of Alcoholism (COGA), Principal Investigators B. Porjesz, V. Hesselbrock, H. Edenberg, L. Bierut, includes eleven different centers: University of Connecticut (V. Hesselbrock); Indiana University (H.J. Edenberg, J. Nurnberger Jr., T. Foroud; Y. Liu); University of Iowa (S. Kuperman, J. Kramer); SUNY Downstate (B. Porjesz); Washington University in St. Louis (L. Bierut, J. Rice, K. Bucholz, A. Agrawal); University of California at San Diego (M. Schuckit); Rutgers University (J. Tischfield, A. Brooks); Department of Biomedical and Health Informatics, The Children’s Hospital of Philadelphia; Department of Genetics, Perelman School of Medicine, University of Pennsylvania, Philadelphia PA (L. Almasy), Virginia Commonwealth University (D. Dick), Icahn School of Medicine at Mount Sinai (A. Goate), and Howard University (R. Taylor). Other COGA collaborators include: L. Bauer (University of Connecticut); J. McClintick, L. Wetherill, X. Xuei, D. Lai, S. O’Connor, M. Plawecki, S. Lourens (Indiana University); G. Chan (University of Iowa; University of Connecticut); J. Meyers, D. Chorlian, C. Kamarajan, A. Pandey, J. Zhang (SUNY Downstate); J.C. Wang, M. Kapoor, S. Bertelsen (Icahn School of Medicine at Mount Sinai); A. Anokhin, V. McCutcheon, S. Saccone (Washington University); J. Salvatore, F. Aliev, B. Cho (Virginia Commonwealth University); and Mark Kos (University of Texas Rio Grande Valley). A. Parsian and H. Chen are the NIAAA Staff Collaborators. All studies included in the externalizing GWAS are listed in the Supplementary Information.
Funding
The Externalizing Consortium has been supported by the National Institute on Alcohol Abuse and Alcoholism through an administrative supplement to R01AA015416, and by the National Institute of Drug Abuse (R01DA050721). D.M.D. was supported through funding from the National Institute on Alcohol Abuse and Alcoholism (K02AA018755, U10AA008401, and P50AA0022527). P.D.K. was supported through a European Research Council Consolidator Grant (647648 EdGe). K.P.H. was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development: (R01HD092548 and R01HD083613) and the Jacobs Foundation. A.A.P. was supported by the National Institute on Alcohol Abuse and Alcoholism (R01AA026281) and the National Institute of Drug Abuse (P50DA037844). S.S-R. was supported through a NARSAD Young Investigator Award from the Brain and Behavior Foundation (grant number 27676). Both A.A.P. and S.S-R. were supported by funds from the California Tobacco-Related Disease Research Program (TRDRP, grant numbers 28IR-0070 and T29KT0526). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the above funding bodies. This research used data from Add Health, a program project directed by K.M.H. (principal investigator) and designed by J. R. Udry, P. S. Bearman, and K.M.H. at the University of North Carolina at Chapel Hill, and funded by grant P01HD031921 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, with cooperative funding from 23 other federal agencies and foundations. Information on how to obtain the Add Health data files is available on the Add Health website (www.cpc.unc.edu/addhealth). This research used Add Health GWAS data funded by Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) grants R01HD073342 to K.M.H. (principal investigator) and R01HD060726 to K.M.H., J. D. Boardman, and M. B. McQueen (multiple principal investigators). COGA is a national collaborative study supported by NIH Grant U10AA008401 from the National Institute on Alcohol Abuse and Alcoholism and the National Institute on Drug Abuse. Data were obtained from Vanderbilt University Medical Center’s BioVU which is supported by numerous sources: institutional funding, private agencies, and federal grants. These include the NIH funded Shared Instrumentation Grant S10RR025141; and CTSA grants UL1TR002243, UL1TR000445, and UL1RR024975. Genomic data are also supported by investigator-led projects that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962, R01HD074711; and additional funding sources listed at https://victr.vumc.org/biovu-funding/. Support for data collection for the Philadelphia Neurodevelopment Cohort (PNC), acquired through dbGaP (accession number phs000607.v3.p2), was provided by grant RC2MH089983 awarded to Raquel Gur and RC2MH089924 awarded to Hakon Hakonarson. Subjects were recruited and genotyped through the Center for Applied Genomics (CAG) at The Children’s Hospital in Philadelphia (CHOP). Phenotypic data collection occurred at the CAG/CHOP and at the Brain Behavior Laboratory, University of Pennsylvania. A full list of funding for investigator effort is listed in the supplementary material.
Footnotes
Competing interests Dr. Kranzler is a member of the American Society of Clinical Psychopharmacology’s Alcohol Clinical Trials Initiative, which was supported in the last three years by AbbVie, Alkermes, Ethypharm, Indivior, Lilly, Lundbeck, Otsuka, Pfizer, Arbor, and Amygdala Neurosciences. Drs. Kranzler and Gelernter are named as inventors on PCT patent application #15/878,640 entitled: “Genotype-guided dosing of opioid agonists,” filed January 24, 2018. Dr. Gelernter did paid editorial work for the journal Complex Psychiatry. Authors declare no other competing interests.
Additional information Supplementary Information is available for this paper. Online Content Methods, along with any additional Extended Data display items and Supplementary Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Main References:
- 1.Richmond-Rakerd LS et al. Clustering of health, crime and social-welfare inequality in 4 million citizens from two nations. Nat. Hum. Behav 4, 255–264 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Case A & Deaton A Mortality and Morbidity in the 21st Century. Brookings Pap. Econ. Act 2017, 397–476 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Achenbach TM The classification of children’s psychiatric symptoms: A factor-analytic study. Psychol. Monogr. Gen. Appl 80, 1–37 (1966). [DOI] [PubMed] [Google Scholar]
- 4.Hicks BM, Krueger RF, Iacono WG, McGue M & Patrick CJ Family transmission and heritability of externalizing disorders: a twin-family study. Arch. Gen. Psychiatry 61, 922–928 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Krueger RF et al. Etiologic connections among substance dependence, antisocial behavior and personality: Modeling the externalizing spectrum. J. Abnorm. Psychol 111, 411–424 (2002). [PubMed] [Google Scholar]
- 6.Buniello A et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47, D1005–D1012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swann AC, Lijffijt M, O’Brien B & Mathew SJ Impulsivity and Suicidal Behavior. in Recent Advances in Research on Impulsivity and Impulsive Behaviors (eds. de Wit H & Jentsch JD) 179–195 (Springer International Publishing, 2020). [DOI] [PubMed] [Google Scholar]
- 8.Zhou H et al. Association of OPRM1 Functional Coding Variant With Opioid Use Disorder: A Genome-Wide Association Study. JAMA Psychiatry (2020). doi: 10.1001/jamapsychiatry.2020.1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mullins N et al. GWAS of Suicide Attempt in Psychiatric Disorders and Association With Major Depression Polygenic Risk Scores. Am. J. Psychiatry 176, 651–660 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grotzinger AD et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat. Hum. Behav (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kendler KS & Myers J The boundaries of the internalizing and externalizing genetic spectra in men and women. Psychol. Med 44, 647–655 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Demontis D et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet 51, 63–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walters RK et al. Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat. Neurosci 21, 1656–1669 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchez-Roige S et al. Genome-Wide Association Study Meta-Analysis of the Alcohol Use Disorders Identification Test (AUDIT) in Two Population-Based Cohorts. Am. J. Psychiatry 176, 107–118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pasman JA et al. GWAS of lifetime cannabis use reveals new risk loci, genetic overlap with psychiatric traits, and a causal influence of schizophrenia. Nat. Neurosci 21, 1161–1170 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karlsson Linnér R et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat. Genet 51, 245–257 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu M et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet 51, 237–244 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee PH et al. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 179, 1469–1482 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lo M-T et al. Genome-wide analyses for personality traits identify six genomic loci and show correlations with psychiatric disorders. Nat. Genet 49, 152–156 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenström T et al. Joint factorial structure of psychopathology and personality. Psychol. Med 49, 2158–2167 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Townsend P Health and deprivation: Inequality and the North (Croom Helm, 1988). [Google Scholar]
- 23.Yang J et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet 44, 369–75, S1–3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de la Fuente J, Davies G, Grotzinger AD, Tucker-Drob EM & Deary IJ A general dimension of genetic sharing across diverse cognitive traits inferred from molecular data. Nat. Hum. Behav 5, 49–58 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hart AB & Kranzler HR Alcohol Dependence Genetics: Lessons Learned From Genomae-Wide Association Studies (GWAS) and Post-GWAS Analyses. Alcohol. Clin. Exp. Res 39, 1312–27 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe K, Taskesen E, van Bochoven A & Posthuma D Functional mapping and annotation of genetic associations with FUMA. Nat. Commun 8, 1826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Leeuw CA, Mooij JM, Heskes T & Posthuma D MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol 11, 1–19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sey NYA et al. A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. Nat. Neurosci 23, 583–593 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gamazon ER et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet 47, 1091–1098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaykumar AB et al. Role of Alström syndrome 1 in the regulation of blood pressure and renal function. JCI Insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun BB et al. Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li YR et al. Meta-analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat. Med 21, 1018–1027 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanchez-Roige S et al. Genome-wide association studies of impulsive personality traits (BIS-11 and UPPS-P) and drug experimentation in up to 22,861 adult research participants identify loci in the CACNA1I and CADM2 genes. J. Neurosci 39, 2562–2572 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao B et al. Genome-wide association analysis of 19,629 individuals identifies variants influencing regional brain volumes and refines their genetic co-architecture with cognitive and mental health traits. Nat. Genet 51, 1637–1644 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edenberg HJ et al. Variations in GABRA2, Encoding the α2 Subunit of the GABAA Receptor, Are Associated with Alcohol Dependence and with Brain Oscillations. Am. J. Hum. Genet 74, 705–714 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dick DM et al. The Role of GABRA2 in Risk for Conduct Disorder and Alcohol and Drug Dependence across Developmental Stages. Behav. Genet 36, 577–590 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Duman RS, Sanacora G & Krystal JH Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron 102, 75–90 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brambilla P, Perez J, Barale F, Schettini G & Soares JC GABAergic dysfunction in mood disorders. Mol. Psychiatry 8, 721–737 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Okbay A et al. Genome-wide association study identifies 74 loci associated with educational attainment. Nature 533, 539–542 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hill WD et al. A combined analysis of genetically correlated traits identifies 187 loci and a role for neurogenesis and myelination in intelligence. Mol. Psychiatry 24, 169–181 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schrode N et al. Synergistic effects of common schizophrenia risk variants. Nat. Genet 51, 1475–1485 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ge T, Chen C-Y, Ni Y, Feng Y-CA & Smoller JW Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun 10, 1776 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Derzon JH The correspondence of family features with problem, aggressive, criminal, and violent behavior: A meta-analysis. J. Exp. Criminol (2010). doi: 10.1007/s11292-010-9098-0 [DOI] [Google Scholar]
- 44.O’Brien DT, Farrell C & Welsh BC Broken (windows) theory: A meta-analysis of the evidence for the pathways from neighborhood disorder to resident health outcomes and behaviors. Social Science and Medicine (2019). doi: 10.1016/j.socscimed.2018.11.015 [DOI] [PubMed] [Google Scholar]
- 45.Chang LY, Wang MY & Tsai PS Neighborhood disadvantage and physical aggression in children and adolescents: A systematic review and meta-analysis of multilevel studies. Aggress. Behav (2016). doi: 10.1002/ab.21641 [DOI] [PubMed] [Google Scholar]
- 46.Davis L Psychiatric Genomics, Phenomics, and Ethics Research In A 270,000-Person Biobank (BioVU). Eur. Neuropsychopharmacol 29, S739–S740 (2019). [Google Scholar]
- 47.Young AI, Benonisdottir S, Przeworski M & Kong A Deconstructing the sources of genotype-phenotype associations in humans. Science 365, 1396–1400 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kong A et al. The nature of nurture: Effects of parental genotypes. Science 359, 424–428 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Selzam S et al. Comparing Within- and Between-Family Polygenic Score Prediction. Am. J. Hum. Genet 105, 351–363 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ripke S et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stahl EA et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet 51, 793–803 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wray NR et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet 50, 668–681 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tielbeek JJ et al. Genome-wide association studies of a broad spectrum of antisocial behavior. JAMA Psychiatry 74, 1242 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kranzler HR et al. Genome-wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nat. Commun 10, 1499 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bulik-Sullivan BK et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet 47, 1236–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anttila V et al. Analysis of shared heritability in common disorders of the brain. Science 360, eaap8757 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gage SH, Smith GD, Ware JJ, Flint J & Munafò MR G = E: What GWAS Can Tell Us about the Environment. PLOS Genet 12, e1005765 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fox D Subversive science. Penn State Law Rev 124, 153–191 (2019). [Google Scholar]
- 59.ASHG Denounces Attempts to Link Genetics and Racial Supremacy. Am. J. Hum. Genet 103, 636 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kvaale EP, Gottdiener WH & Haslam N Biogenetic explanations and stigma: A meta-analytic review of associations among laypeople. Soc. Sci. Med 96, 95–103 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Tucker-Drob EM, Briley DA & Harden KP Genetic and environmental influences on cognition across development and context. Curr. Dir. Psychol. Sci 22, 349–355 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Auton A et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCarthy S et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet 48, 1279–1283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Walter K et al. The UK10K project identifies rare variants in health and disease. Nature 526, 82–90 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Winkler TW et al. Quality control and conduct of genome-wide association meta-analyses. Nat. Protoc 9, 1192–1212 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Willer CJ, Li Y & Abecasis GR METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang CC et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang J, Lee SH, Goddard ME & Visscher PM GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet 88, 76–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rietveld CA et al. Common genetic variants associated with cognitive performance identified using the proxy-phenotype method. Proc. Natl. Acad. Sci. U. S. A 111, 13790–13794 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Okbay A et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat. Genet 48, 624–633 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harris KM, Halpern CT, Haberstick BC & Smolen A The National Longitudinal Study of Adolescent Health (Add Health) sibling pairs data. Twin Res. Hum. Genet 16, 391–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McQueen MB et al. The National Longitudinal Study of Adolescent to Adult Health (Add Health) sibling pairs genome-wide data. Behav. Genet 45, 12–23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Begleiter H The Collaborative Study on the Genetics of Alcoholism. Alcohol Health Res. World 19, 228–236 (1995). [PMC free article] [PubMed] [Google Scholar]
- 74.Edenberg HJ The collaborative study on the genetics of alcoholism: An update. Alcohol Res. Heal (2002). [PMC free article] [PubMed] [Google Scholar]
- 75.Bucholz KK et al. Comparison of Parent, Peer, Psychiatric, and Cannabis Use Influences Across Stages of Offspring Alcohol Involvement: Evidence from the COGA Prospective Study. Alcohol. Clin. Exp. Res (2017). doi: 10.1111/acer.13293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Calkins ME et al. The Philadelphia Neurodevelopmental Cohort: constructing a deep phenotyping collaborative. J Child Psychol Psychiatry 56, 1356–1369 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Satterthwaite TD et al. The Philadelphia Neurodevelopmental Cohort: a publicly available resource for the study of normal and abnormal brain development in youth. Neuroimage 124, 1115–1119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bycroft C et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ge T, Chen C-Y, Ni Y, Feng Y-CA & Smoller JW Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun 10, 1776 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vilhjálmsson BJ et al. Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. Am. J. Hum. Genet 97, 576–592 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dudbridge F Power and Predictive Accuracy of Polygenic Risk Scores. PLoS Genet 9, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Altshuler DM, Gibbs RA & Peltonen L Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–58 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wei W-Q et al. Evaluating phecodes, clinical classification software, and ICD-9-CM codes for phenome-wide association studies in the electronic health record. PLoS One 12, e0175508 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hubbard T et al. The Ensembl genome database project. Nucleic Acids Res 30, 38–41 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Consortium TGO The Gene Ontology project in 2008. Nucleic Acids Res 36, D440–D444 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liberzon A et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miller JA et al. Transcriptional landscape of the prenatal human brain. Nature 508, 199–206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lonsdale J et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet 45, 580–5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang D et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Won H et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rajarajan P et al. Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barbeira AN et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun 9, 1–20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shannon P et al. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Singhal A et al. Multiscale community detection in Cytoscape. PLOS Comput. Biol 16, e1008239 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang J et al. Genomic inflation factors under polygenic inheritance. Eur. J. Hum. Genet 19, 807–812 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data sources are described in the Supplementary Information and are listed in the Reporting Summary. No new data was collected. Only data from existing studies or study cohorts were analyzed, some of which are restricted access to protect the privacy of the study participants (see Reporting Summary for accession codes or URLs). The minimum data set necessary to interpret, verify, and extend the research, i.e., the GWAS summary statistics for the externalizing (EXT) GWAS (our main discovery analysis), can be obtained by following the procedures detailed at https://externalizing.org/request-data/. In brief, summary statistics are derived from analyses based in part on 23andMe data, for which we are restricted to only publicly report results for up to 10,000 SNPs. The full set of externalizing GWAS summary statistics can be made available to qualified investigators who enter into an agreement with 23andMe that protects participant confidentiality. Once the request has been approved by 23andMe, a representative of the Externalizing Consortium can share the full GWAS summary statistics. No source data is published alongside the paper.