

Abstract
Fragile X syndrome (FXS) is caused by the loss of fragile X mental retardation protein (FMRP), an RNA-binding protein that can regulate the translation of specific mRNAs. Here we have developed an FXS human forebrain organoid model and observed the loss of FMRP led to dysregulated neurogenesis, neuronal maturation, and neuronal excitability. Bulk and single-cell gene expression analyses of FXS forebrain organoids revealed that the loss of FMRP altered gene expression in a cell type-specific manner. The developmental deficits in FXS forebrain organoids could be rescued by inhibiting the phosphoinositide 3-kinase pathway, but not the metabotropic glutamate pathway disrupted in the FXS mouse model. We identified a large number of human-specific mRNAs bound by FMRP. One of these human-specific FMRP targets, CHD2, contributed to the altered gene expression in FXS organoids. Collectively, our study revealed molecular, cellular, and electrophysiological abnormalities associated with the loss of FMRP during human brain development.
INTRODUCTION
Fragile X syndrome (FXS, OMIM#300624) is the most common inherited form of intellectual disability and a leading genetic cause of autism spectrum disorder (ASD)1, 2. CGG trinucleotide repeat expansion within the 5’-untranslated region of the FMR1 gene has been identified as the most prevalent cause of FXS3. This CGG expansion beyond 200 repeats, referred to as the full mutation, leads to hypermethylation of the FMR1 promoter, thereby preventing FMR1 expression. FXS is caused by the loss of functional fragile X mental retardation protein (FMRP).
Over the course of nearly three decades of research, a great deal has been learned about the function of FMRP and the consequence of its absence in FXS, primarily using mouse and fruit fly model systems. FMRP is a selective RNA-binding protein associated with translating polyribosomes that appears to be involved in the regulation of local protein synthesis at synapses3. The loss of FMRP leads to abnormal translation of selective mRNAs, many of which are autism-linked genes3. Substantial progress in characterizing the underlying disease mechanisms in animal models has led to highly successful preclinical studies of drugs modulating metabotropic glutamate and GABA receptors4. However, follow-up clinical trials in humans have been largely unsuccessful, suggesting that mouse models may not fully recapitulate human disease, and highlighting the need for human cell-based models.
Recent technical advances have enabled longer neuroepithelium formation and support growth of large, complex three dimensional (3D) organoids to recapitulate key features of human brain development such as progenitor zone organization, neurogenesis, gene expression profile, and, notably, a distinct human-specific outer radial glia cell layer5–7. Thus, the human brain organoid model provides a new platform to investigate human brain development in a dish, otherwise inaccessible to experimentation.
In this study, we developed FXS forebrain organoids from patient-derived iPSCs and observed reduced proliferation of neural progenitor cells, dyregulated neural differentiation, increased synapse formation and neuronal hyperexcitability, and a deficit in the production of GABAergic neurons. Bulk and single-cell transcriptome analyses revealed that loss of FMRP could lead to pervasive gene expression alterations in human brain development and an altered developmental trajectory of forebrain organoids in a cell type-specific manner. Furthermore, mechanism-guided pharmacological inhibition of the PI3K pathway, but not the mGluR5 pathway that was previously identified in mouse models, rescued the defects in neurodevelopment and synapse formation of FXS forebrain organoids. To identify the mRNAs bound by FMRP, we performed enhanced crosslinking and FMRP immunoprecipitation followed by high-throughput sequencing using human forebrain organoids and mouse embryonic forebrain at similar developmental stages. Our comparative analyses identified a large number of mRNAs that were bound by FMRP in a human-specific manner. Furthermore, many FMRP mRNA targets involved in neurogenesis and synaptic plasticity were found to be upregulated at the protein level. We showed that one of the human-specific FMRP targets, CHD2, contributes to the altered gene expression in FXS organoids. Our analyses revealed molecular, cellular and electrophysiological abnormalities associated with the loss of FMRP during human brain development. Our study has identified human-specific mRNA targets of FMRP, which have the potential to serve as human-specific druggable targets for FXS and autism in general.
RESULTS
FMRP regulates neurogenesis and neuronal differentiation in human forebrain organoids
To model FXS and study the consequence of FMRP deficiency in the context of neurodevelopment, we generated induced pluripotent stem cell (iPSC) lines from dermal fibroblasts of three FXS male patients and three age- and sex- matched healthy and unaffected controls (Supplementary Table 1). We also utilized the previously generated isogenic correction line by excising the expanded CGG-repeat in one of the FXS iPSC line (FXS1) through the CRISPR/Cas9 gene editing8 (Supplementary Table 1). All iPSC lines exhibited normal karyotypes (Supplementary Table 1) and were positive for pluripotency markers Tra-1–60 and NANOG (Supplementary Fig. 1a). Transcriptome analysis demonstrated that these iPSC lines express pluripotency-associated genes, including NANOG, KLF4, OCT4, SOX1, SOX2, DPPA3, and PODXL, at similar levels, further confirming their pluripotency (Supplementary Fig. 1d). To determine whether loss of FMRP dysregulates human cortical development, we derived three-dimensional (3D) forebrain organoids from the iPSCs using a recently established protocol with miniature bioreactors7 (Supplementary Fig. 1b). We focused on day 56 (D56) forebrain organoids, which strongly correlate with fetal human brain development during mid fetal period7, a critical period for human cortical neurogenesis9 and FMRP depletion in FXS patients10, 11. We confirmed that FMRP was absent in iPSCs, embryoid bodies (EB), as well as D28 and D56 forebrain organoids derived from FXS patient iPSC lines (Supplementary Fig. 1c).
To examine the impact of FMRP loss on neural progenitor cell (NPC) proliferation, we co-immunostained Ki67, a proliferation marker, with NPC markers SOX2 or PAX65, 7 (Fig. 1a and Extended Data Fig. 1b). Although the loss of FMRP didn’t affect the overall size of forebrain organoids in general (Extended Data Fig. 1a), we found a significant reduction of Ki67+ cells among all SOX2+ or PAX6+ cells (Fig. 1a, b and Extended Data Fig. 1b, c), indicating a reduction in NPC proliferation at Day 56. In addition, we pulsed D56 forebrain organoids with 5-ethynyl-2’-deoxyuridine (EdU; 10 μM) for 2 hr to label proliferating cells in S phase (Extended Data Fig. 1d). Quantitative analysis showed that loss of FMRP in FXS forebrain organoids led to reductions in the percentage of EdU+ cells among all SOX2+ cells compared to control organoids (Extended Data Fig. 1e), confirming deficits in NPC proliferation in FXS organoids. Interestingly, we observed increased Ki67+ cells among all SOX2+ cells in FXS forebrain organoids at an earlier time point (Day 28)12, suggesting that the alteration of NPC proliferation rate in FXS organoids could be developmental stage-specific, and loss of functional FMRP may accelerate neural differentiation overall. To further confirm the impact of FMRP loss on neurogenesis, we exposed control and FXS organoids to EdU for 24 hours and then measured the cell cycle exit by assessing the colocalization of EdU with Ki6713. Ki67−EdU+ indicates the cells that had exited the cell cycle within 24 hours of EdU exposure. We found that the number of Ki67−EdU+ cells was significantly increased in FXS organoids compared to the controls at D56 (Fig. 1c, d), suggesting an accelerated neural progenitor cell cycle exit in FXS forebrain organoids. Moreover, to examine whether FMRP deficiency alters neuronal differentiation, we expressed green fluorescent protein (GFP) specifically in proliferating NPCs of forebrain organoids at day 49 using a retroviral strategy, which provides adequate time resolution for birth-dating14. One week after the retroviral GFP infection, we examined the location of GFP+ cells in control and FXS organoids. Intriguingly, we found that while most of GFP+ cells in the control organoids co-expressed SOX2 and maintained as NPCs, many GFP+ cells in the FXS organoids had differentiated into MAP2+ neurons (Fig. 1e, f), suggesting an altered neuronal differentiation in FXS organoids.
Figure 1. FMRP regulates cortical neurogenesis in a human forebrain organoid model.
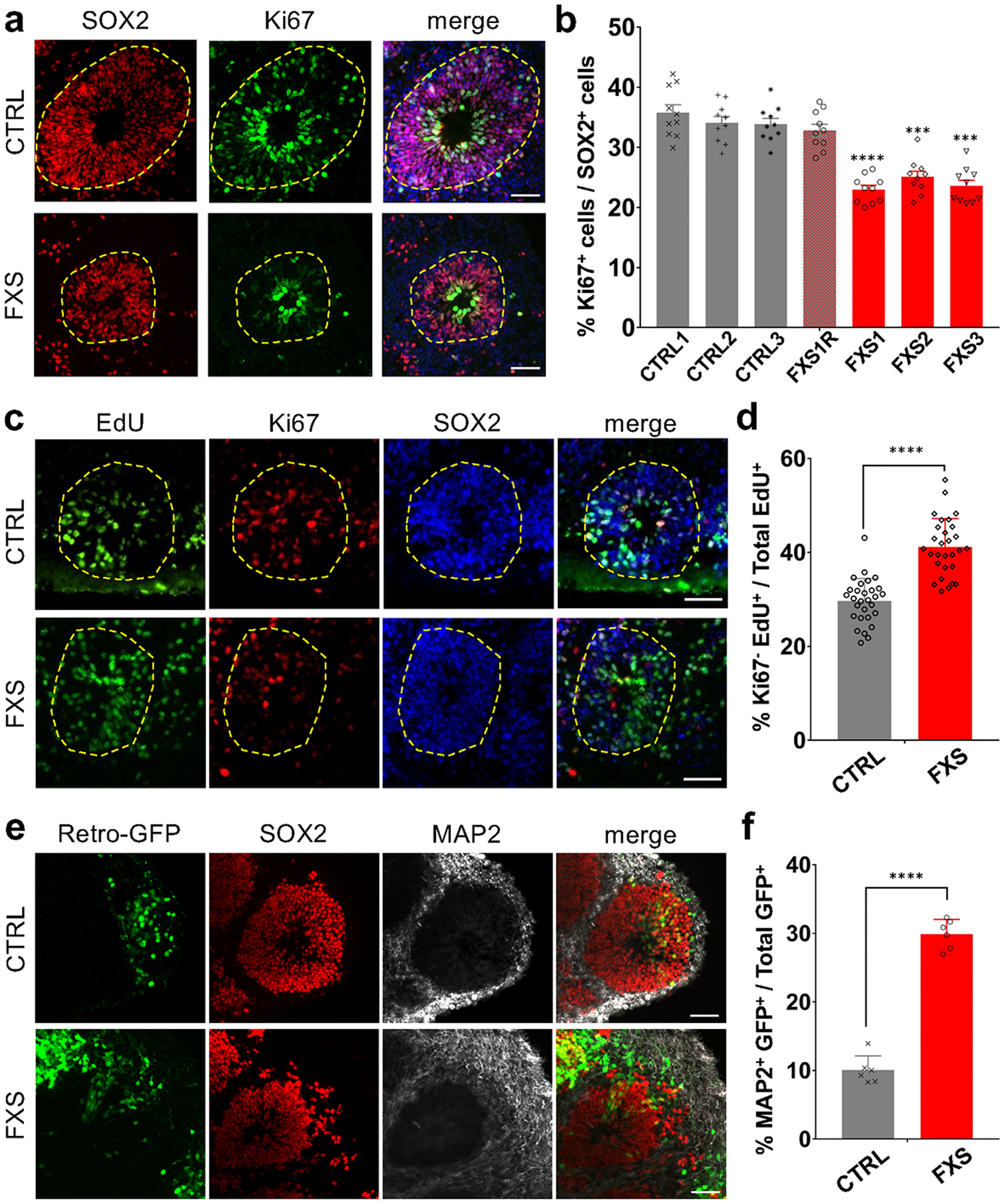
(a-b) Loss of FMRP reduces NPC proliferation. Shown are representative images (a) and stereological quantification (b) of the proportion of Ki67+ proliferating neuronal progenitor cells in both control and FXS-derived forebrain organoids at day 56. Yellow dashed lines indicate the borders of VZ-like structures. Data are presented as mean ± s.e.m. (n = 10 organoids from each line with 15–20 cortical structures analyzed per organoid; ***P = 0.0002 (FXS2 v.s. CTRL1) or 0.0001 (FXS3 v.s. CTRL1), ****P < 0.0001 (FXS1 v.s. CTRL1), one-way ANOVA). Scale bars: 50 μm. (c-d) FMRP deficiency accelerates NPC cell cycle exit during the 24-hour EdU exposure in FXS forebrain organoids. Shown are representative images (c) and quantification (d) of the proportion of Ki67− NPCs in total EdU+ cells in both control and FXS-derived forebrain organoids after 24-hour EdU exposure. Yellow dashed lines indicate the borders of VZ-like structures. Data are presented as mean ± s.e.m. (n = 29 cortical structures from at least ten organoids each condition; ****P < 0.0001, one sided student’s t-test). Scale bars: 50 μm. (e-f) Loss of FMRP accelerates neural differentiation. Day 49 control and FXS forebrain organoids were infected with retroviruses expressing GFP and analyzed 7 days later (day 56). Shown are sample images for immunostaining for GFP, SOX2 and MAP2 (e) and quantifications of percentages of MAP2+GFP+ cells among all GFP+ cells (f). Values represent mean ± s.e.m. (n = 6 organoids from control or FXS lines each with 10–12 cortical structures analyzed per organoid; ****p < 0.0001; one sided student’s t test). Scale bars: 50 μm.
NPCs give rise to TBR2+ intermediate neural progenitor cells (IPCs), which differentiate into neurons15. We found that there were more TBR2+ IPCs in FXS organoids compared to the controls at D56 (Extended Data Fig. 2a). Interestingly, while most TBR2+ IPCs were located at the edge of SOX2+CTIP2− or SOX2+MAP2− ventricular zone (VZ)-like structures in the control forebrain organoids, many of TBR2+ IPCs were located in the CTIP2+ or MAP2+ cortical plate-like (CP) layer in the FXS organoids (Fig. 2a, b and Extended Data Fig. 2b, c), further suggesting an altered neural differentiation caused by loss of FMRP. Doublecortin (DCX), a marker of neuronal precursor and immature neurons, is involved in neuronal migration via stabilizing microtubules16. The DCX expression is abnormally stronger in PAX6+ NPCs in FXS organoids compared to controls (Fig. 2c, d), further confirming the dysregulated differentiation of FXS forebrain organoids.
Figure 2. Loss of FMRP impairs cortical neurodevelopment.
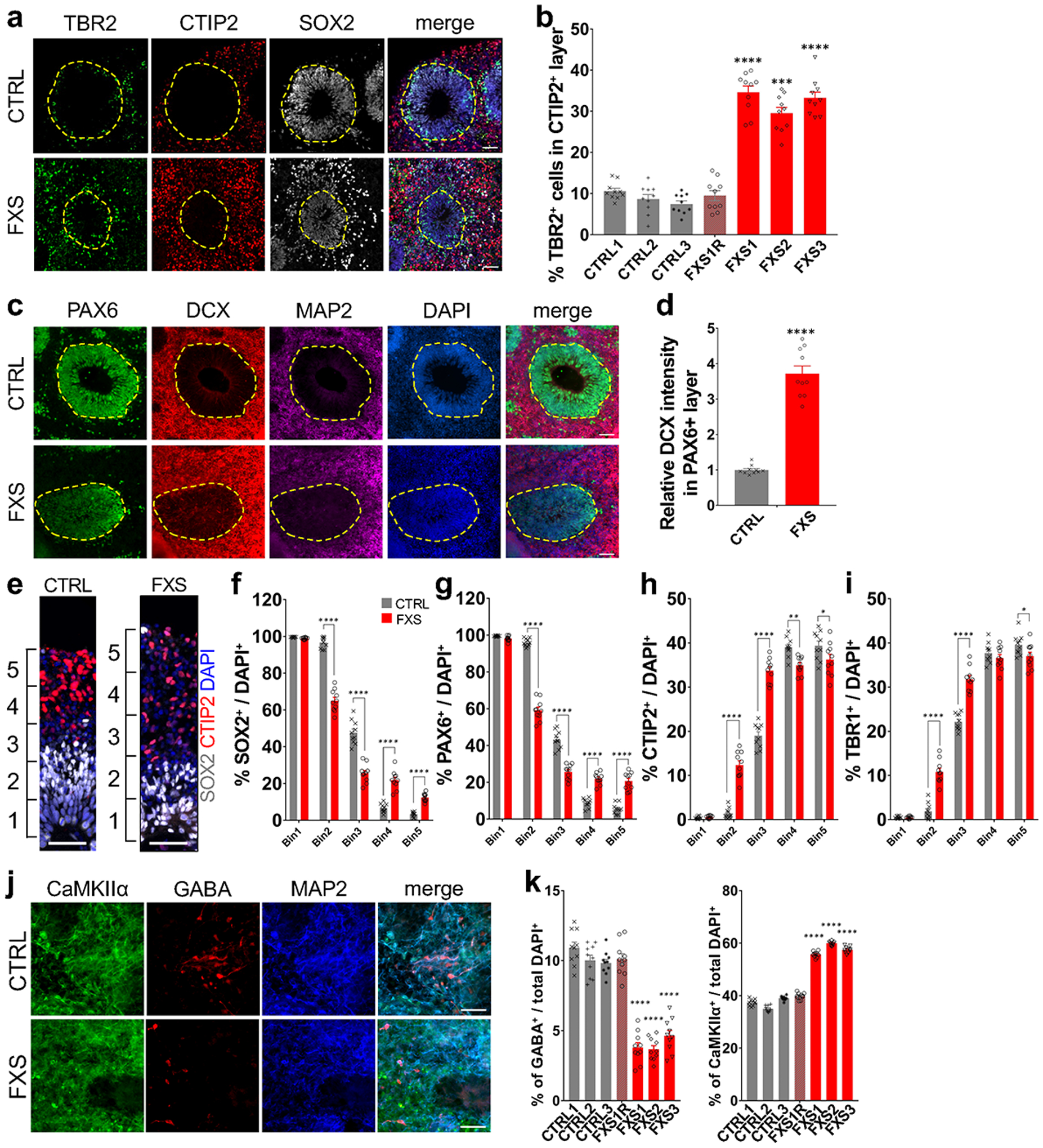
(a-b) Loss of FMRP dysregulates distribution of TBR2+ intermediate neural progenitor cells. Shown are representative images (a) and quantification (b) of the proportion of TBR2+ IPCs in CTIP2+ layer of both control and FXS-derived forebrain organoids. Yellow dashed lines indicate the borders of VZ-like structures. Data are presented as mean ± s.e.m. (n = 10 organoids from each line with 15–20 cortical structures analyzed per organoid; *** P = 0.0001, ****P < 0.0001, one-way ANOVA). Scale bars: 50 μm. (c-d) FMRP deficiency induces premature neural differentiation. Shown are sample images for immunostaining for PAX6, DCX, and MAP2 (c) and quantifications of relative expression level of DCX in PAX6+ NPCs in the VZ-like structures (d). Yellow dashed lines indicate the borders of VZ-like structures. Values represent mean ± s.e.m. (n = 10 organoids from control or FXS lines each with 15–20 cortical structures analyzed per organoid; ****p < 0.0001; one sided student’s t test). Scale bars: 50 μm. (e-i) Loss of FMRP alters cortical layer formation. Shown in (e) are sample images of the expanded neuroepithelium in D56 control and FXS organoids stained with SOX2 and CTIP2. Scale bars: 50 μm. Shown in (f-i) are quantifications of distributions of SOX2 (f), PAX6 (g), CTIP2 (h), and TBR1 (i) in the entire span of the neuroepithelium which was divided into five equal portions (bins). Data are presented as mean ± s.e.m. (n = 10 organoids from control or FXS lines each with 15–20 cortical structures analyzed per organoid; *p = 0.0240 (h) or 0.0488 (i), **p = 0.0014 (h), ****p < 0.0001; one-way ANOVA). (j-k) Loss of FMRP prevents differentiation of GABAergic interneurons. Shown are sample images for immunostaining for CaMKIIα, GABA, and MAP2 (g) and quantification (h) of percentages of GABA+ inhibitory neurons (left) and CaMKIIα+ excitatory neurons (right) in total DAPI+ cells in both control and FXS-derived forebrain organoids. Data are presented as mean ± s.e.m. (n = 10 sections from 10 organoids each line; ****P < 0.0001, one-way ANOVA). Scale bars: 50 μm.
To further assess the neuronal differentiation, we divided the entire span of the neuroepithelium into five equal portions (bins) and measured the distributions of specific cell types in each bin as described previously17. We observed the distribution of CTIP2+ or TBR1+ cortical neurons was altered in the neuroepithelium of FXS organoids (Fig. 2e, h, i), suggesting dysregulated neuronal differentiation and cortical layer formation in FXS organoids. Interestingly, we also noticed that significantly more SOX2+ or PAX6+ NPCs detached from the apical VZ (SOX2+CTIP2− or PAX6+TBR1−)-like layer and were dispersed into the expanded neuroepithelium in FXS organoids (Fig. 2e, f, g), suggesting that the structure of FXS organoids is less organized. We evaluated the neuronal differentiation by quantifying the relative thickness of the VZ (SOX2+CTIP2−, or SOX2+MAP2−, or SOX2+TBR1−) layer and CP (CTIP2+ or MAP2+ or TBR1+) layer between apical and basal surfaces at D56 (Extended Data Fig. 3a–f). Quantitative analyses showed dramatically reduced VZ thickness while the CP thickness was increased in FXS organoids (Extended Data Fig. 3a–f), likely due to the dysregulated differentiation of FXS NPCs. In addition, the percentages of CTIP2+ or TBR1+ cortical neurons in total cells were significantly increased (Extended Data Fig. 2a and 3g). We also analyzed the distribution of the SOX2+, PAX6+, CTIP2+, and TBR1+ cell populations in D56 forebrain organoids (Extended Data Fig. 3i–l). The distributions of SOX2 and PAX6 were significantly reduced in the VZ layer while increased in the CP layer of the FXS organoids, indicating the dysregulated neuronal differentiation caused by the loss of FMRP. We further assessed the expression of marker genes for each cortical layer (I:RELN, II:CUX1, III:POU3F2, IV:SATB2, V:BCL11B [also known as CTIP2], VI:TBR1). The markers for deeper layers (POU3F2, SATB2, and BCL11B) were increased in FXS (Supplementary Table 2), suggesting a change in layer predominance toward deeper layers in FXS organoids. To confirm the neuronal fates after differentiation, we quantified the proportion of glutamatergic and GABAergic neurons by co-immunostaining with CaMKIIα, indicative of glutamatergic neuronal differentiation, and the GABAergic interneuron marker GABA (Fig. 2j). Intriguingly, we found that the percentage and number of GABAergic neurons was dramatically decreased in FXS organoids compared to the controls (Fig. 2k and Extended Data Fig. 4a, b), indicating a reduction of GABAergic neuronal fate. Furthermore, RNA in situ hybridization for markers of ganglionic eminence (GE) NPCs (DLX2), dorsal telencephalic NPCs (PAX6), and pan-NPCs (SOX2) revealed a reduction of DLX2+ ventral NPCs, which give rise to GABAergic interneurons, in both D28 and D56 FXS organoids (Extended Data Fig. 4c, d). This suggests that the loss of FMRP may deplete the pool of GE NPCs at an early stage, which further leads to reduction of GABAergic interneurons. Together, these results revealed that loss of FMRP reduces NPC proliferation rate, leading to dysregulated neuronal differentiation with reduction of GABAergic neuronal fate.
Loss of FMRP increases synapse formation and enhances neuronal excitability
We next characterized the synapse formation and neuronal function in FXS forebrain organoids. To investigate the impact of FMRP loss on synapse formation, we quantified the density of synaptic puncta by immunocytochemistry of presynaptic vesicle protein SYNAPSIN1 (SYN1), postsynaptic protein PSD95, and dendrite marker MAP2 (Fig. 3a). The density of SYN1+/PSD95+ synaptic boutons was significantly increased in FXS neurons compared to control neurons in D56 forebrain organoids (Fig. 3b), suggesting dysregulated formation of morphological excitatory synapses in FXS neurons.
Figure 3. Loss of FMRP alters synapse formation and enhances neuronal excitability.
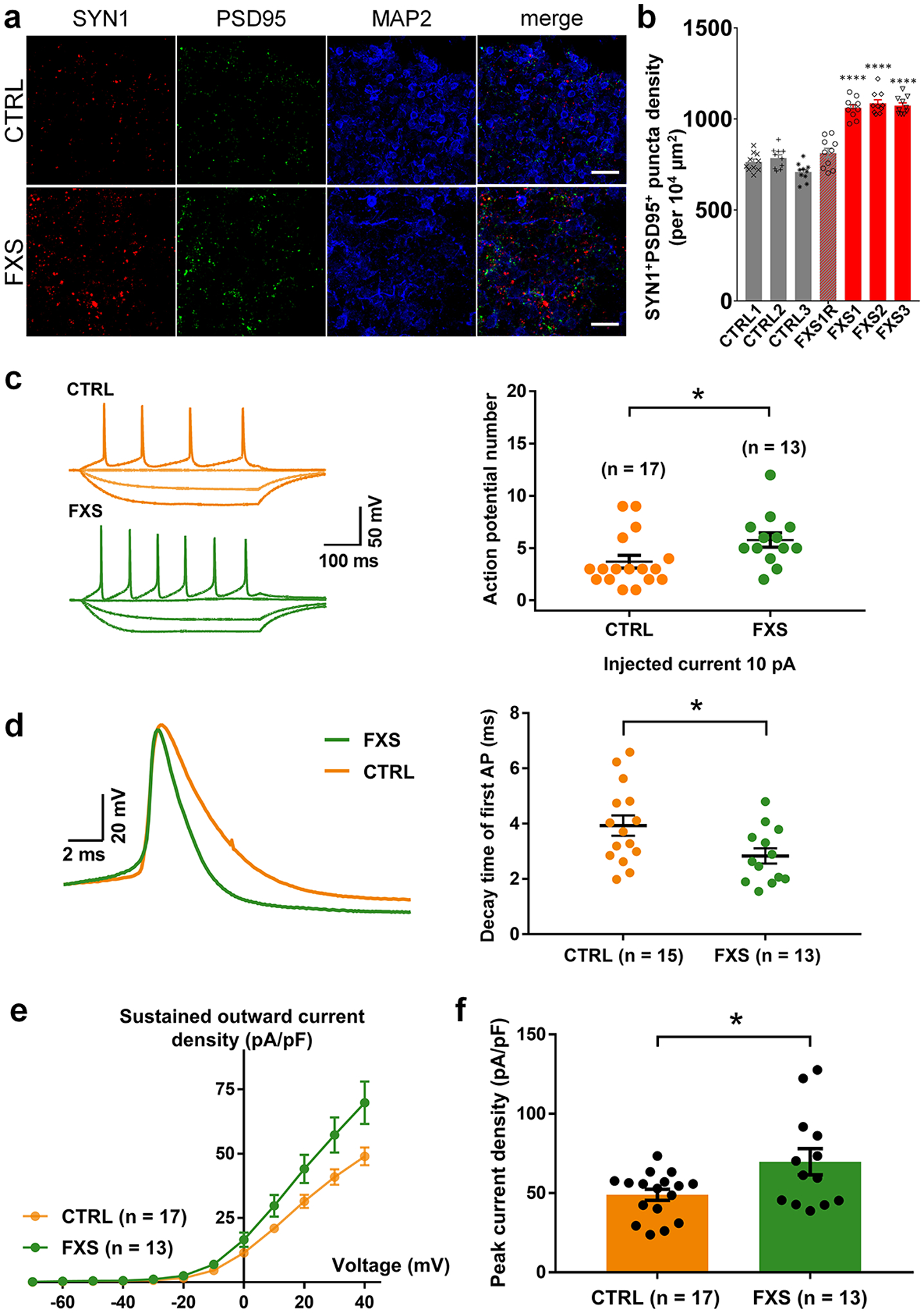
(a-b) Loss of FMRP accelerates synapse formation. Shown are sample images (a) and quantification (b) of SYN1+PSD95+ puncta density in both control and FXS-derived forebrain organoids at day 56. Data are presented as mean ± s.e.m. (n = 10 organoids each line; ****P < 0.0001, one-way ANOVA). Scale bars: 50 μm. (c-f) FXS forebrain organoids exhibit hyperexcitability. Shown in (c) are sample tracings of action potentials (left) and quantification of action potential frequency (right); (d) are sample tracing of the first action potential (left) and quantification of the decay time of first action potential (right); (e,f) are quantification of current-voltage curve (e) and peak current density (f). Cell number (n) recorded and analyzed in each condition is indicated. Data are presented as mean ± s.e.m. (*P = 0.0336 (c), 0.0421 (d), 0.0173 (f), two-tailed unpaired t test or one-way ANOVA).
To examine the physiological properties of these FXS neurons, we performed electrophysiological whole-cell recordings in slices acutely sectioned from organoids. Only CTIP2+ cortical glutamatergic neurons were analyzed, the identity of which was confirmed by injection of Alexa Fluor-594 dye into neurons after the recording and co-immunostaining of CTIP2 (Extended Data Fig. 5a). The passive membrane properties, including the resting membrane potential (RMP), membrane capacitance (Cm) and input resistance (RIN), were not altered by the loss of FMRP expression (Extended Data Fig. 5b–d). We next examined the neuronal excitability under current-clamp mode with steps of current injection. Interestingly, we observed a significant increase in action potential firing frequency (Fig. 3c), suggesting the hyperexcitability of FXS neurons. We further characterized the detailed properties of the first action potentials. Quantitative analyses showed significant reduction of decay time of the first action potentials in FXS neurons (Fig. 3d), while the amplitude, threshold, half-width, and the rise time of the first action potentials were comparable between control and FXS neurons (Extended Data Fig. 5e–h). Furthermore, FXS neurons also displayed larger voltage-activated potassium (K+) currents with normal voltage-activated sodium (Na+) currents (Fig. 3e–f and Extended Data Fig. 5i–k), suggesting an increased expression of potassium channels in FXS neurons. Indeed, the expression of voltage-gated potassium channel Kv4.2 was increased in FXS forebrain organoids (Extended Data Fig. 6). These results demonstrate that loss of FMRP may lead to neuronal hyperexcitability, potentially caused by the increase of potassium channels in FXS neurons.
Loss of FMRP leads to pervasive gene expression alterations in human brain development
Given the observed cellular and electrophysiological phenotypic changes in FXS organoids, we determined how the loss of FMRP could impact gene expression during human brain development. We performed bulk RNA-seq using both control and FXS organoids at different developmental stages, D28, D56 and D84, and identified a number of differentially expressed genes (DEG) in the absence of FMRP in a developmental stage-dependent manner (Fig. 4a, Extended Data Fig. 7 and Supplementary Table 3). The relative consistency across each line of the control and FXS organoids is shown in the heatmap of DEGs at D56 showing reasonable reproducibility across the organoids within the same group (Supplementary Fig. 2). To determine whether the changes that we observed also occurred in human fetal brain, we generated RNA-seq data using two human fragile X fetal brain cortex tissues that we obtained previously, one full mutation male (23 weeks after conception) and one full mutation female (24 weeks after conception) (Supplementary Table 4). We identified 2723 DEGs from the fetal brain data and 810, 1194 and 3839 DEGs in D28, D56 and D84 organoids, respectively. Comparing the RNA-seq data of FXS fetal brain tissues and forebrain organoids, we discovered an overlap between fragile X fetal brain tissue DEGs and FXS organoid DEGs in the same direction at different stages (p< 2.2e-16), in particular the D84 forebrain organoids which is estimated to be at a similar stage of human fetal brain at ~18–23 weeks after conception7 (Fig. 4b and Supplementary Fig. 3). Thus, our data indicate that FXS organoids could potentially mimic the developmental alterations in human fragile X fetal brain.
Figure 4. Loss of FMRP causes a more pervasive gene expression alteration in forebrain organoids than in mouse brain.
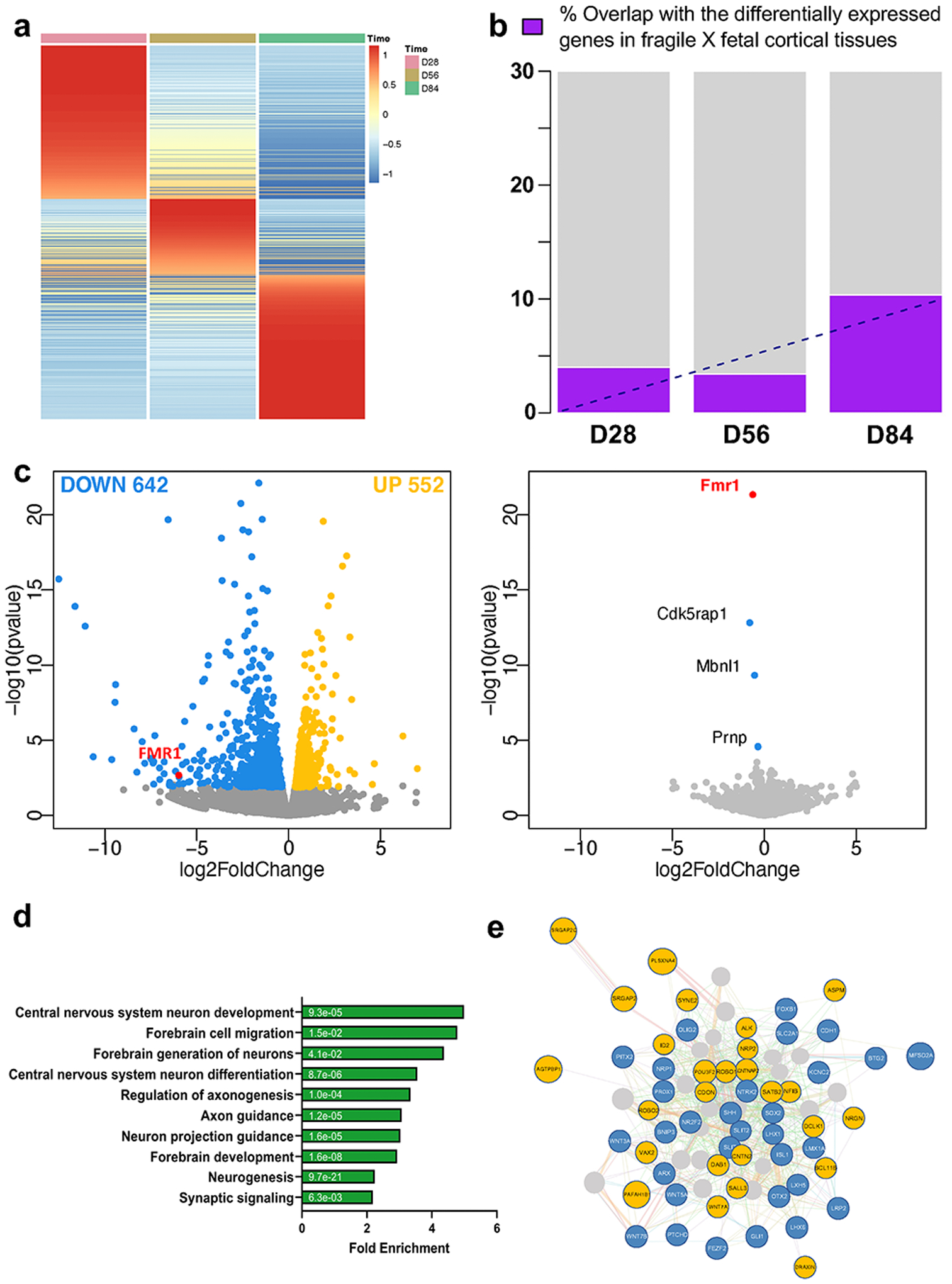
(a) Heatmap of the ratios of mRNA expression in FXS forebrain over control organoids at day 28, 56, and 84 is presented. Row normalization and ordering were applied for visualization purpose. (b) Bar plot of overlap of differentially expressed genes (DEG) in fragile X human fetal cortical tissue and DEG of forebrain organoids at day 28, 56, and 84. We only considered the DEG from human fetal tissue and DEG from forebrain organoids that showed the same directional change. (c) Shown is a volcano plot of gene expression change at day 56 human forebrain organoids (left panel) and in mouse fetal brain (right panel) upon lack of FMRP. Colored dots indicate statistically significant genes with false discovery rate < 0.20. Blue dots are down-regulated genes (log fold change < 0) and yellow dots are up-regulated genes (log fold change > 0) in FXS compared to controls. (d) GO Ontology overrepresentation tests using PANTHER (http://www.pantherdb.org/) for DEG identified from day 56 forebrain organoid RNA-seq is shown and the numbers on the bars are two-sided p-values by Fisher’s exact test. P values have been adjusted for multiple testing by Bonferroni correction. The overlapping analyses with the up and down-regulated genes are presented in Supplementary Fig. 5. (e) Interactome plot of forebrain development (GO: 0030900) is presented. Significant DE genes found in day 56 forebrain organoid RNA-seq results are shown and highlighted. Yellow represents up-regulated genes and blue represents down-regulated genes.
Given the significant number of genes with altered expression in FXS organoids, we performed similar RNA-seq analyses using mouse brain at embryonic day 13.5, which is a similar developmental stage to human D56 organoids18. Interestingly, we observed only three genes were differentially expressed in the absence of Fmrp in mouse, one of which is the Fmr1 gene, while we found a markedly greater number of DEGs in human forebrain organoids. Genes with false discovery rate <0.20 were defined as differentially expressed genes and Volcano plot was drawn using the same criteria (Fig. 4c, Supplementary Table 5). We further examined the genes that displayed human-specific alteration in the absence of FMRP in forebrain organoids. The differentially expressed genes are associated with neurodevelopment, many of which have been implicated in neuronal migration, axonogenesis, neurogenesis, regulation of membrane potential and neuron differentiation (Fig. 4d, e and Supplementary Fig. 4, 5). We did observe that both GE NPC markers DLX1 and DLX2 were decreased in FXS organoids (Supplementary Fig. 6), which is consistent with the reduction of GE NPCs and GABAergic neurons in FXS organoids (Fig. 2g, h, and Extended Data Fig. 4). Together, these data suggest that the loss of FMRP in human causes a more pervasive gene expression alteration during brain development compared to mouse.
Loss of FMRP leads to altered developmental trajectory in forebrain organoids
Bulk-RNAseq is unable to provide detailed insights such as the distinct cell population alteration, the presence of specific cell types and developmental variation. To more comprehensively understand the specificity and complexity of the impact of loss of FMRP on human brain development, we performed single cell RNA-seq (scRNA-seq) on 3 pairs of control and FXS forebrain organoids including the isogenic line. After quality control and filtering, a total of 30,550 single cells were analyzed, and ontology analyses on DE genes between control and FXS revealed specific enrichment in selective pathways (Fig. 5a)19. Among these pathways, the PANTHER analyses on down-regulated DE genes revealed the enrichment of the genes involved in neuronal development such as neurogenesis, differentiation and neuron projection morphogenesis, while up-regulated DE genes were enriched in pathways of protein translation and targeting to the membrane and oxidative phosphorylation, which is required for the function of a neuron. Uniform Manifold Approximation and Projection (UMAP) plots were visually adopted to identify 14 clusters, which were systematically classified as representing neural lineages based on the cell type specific molecular signatures in combination with gene ontology (GO) analysis20 (Figure 5b, Extended Data Fig. 8a–c and Supplementary Table 6–8). After the assessment of the overlap between human fetal brain DEGs and cell type specific DEGs, C7 overlap was highest (Extended Data Fig. 9). The overlapped genes are highly enriched in neuron generation and development as seen in the cellular phenotype change (Extended Data Fig. 9b, Fig. 1–3). The neural stem cell/radial glia (C3, C5 and C6) are expanded in FXS organoids, which could be the consequence of increased proliferation earlier in D28 organoids12. Cyclin D2 (CCND2), a cortical progenitor proliferation promoter and differentiation blocker21, decreased in clusters of neural stem and progenitors and inhibitory neurons in FXS organoids (Supplementary Table 9), supporting the notion of the reduced NPC proliferation in D56 FXS organoids. In FXS organoids, marker genes specific for certain developmental stage are ambiguously expressed. Some undifferentiated cells express mature markers too early and some differentiated cells express abnormally stronger immature markers, but less or no expression of some mature markers. Especially, C7, inhibitory neurons are digressing from the proper maturation toward functional inhibitory neurons. (Fig. 5 and Extended Data Fig. 8). We found that several important molecules in GABAergic synapse development and function are decreased in FXS C7 compared to control (Supplementary Fig. 7). The gene expression profile in Seurat clusters of FXS organoids clearly suggests that they undergo distinct developmental progression from control, and the development and differentiation of FXS organoids are not properly orchestrated and are perturbed especially in inhibitory neurons.
Figure 5. Lack of FMRP causes altered neural differentiation and aberrant developmental trajectory in forebrain organoids.
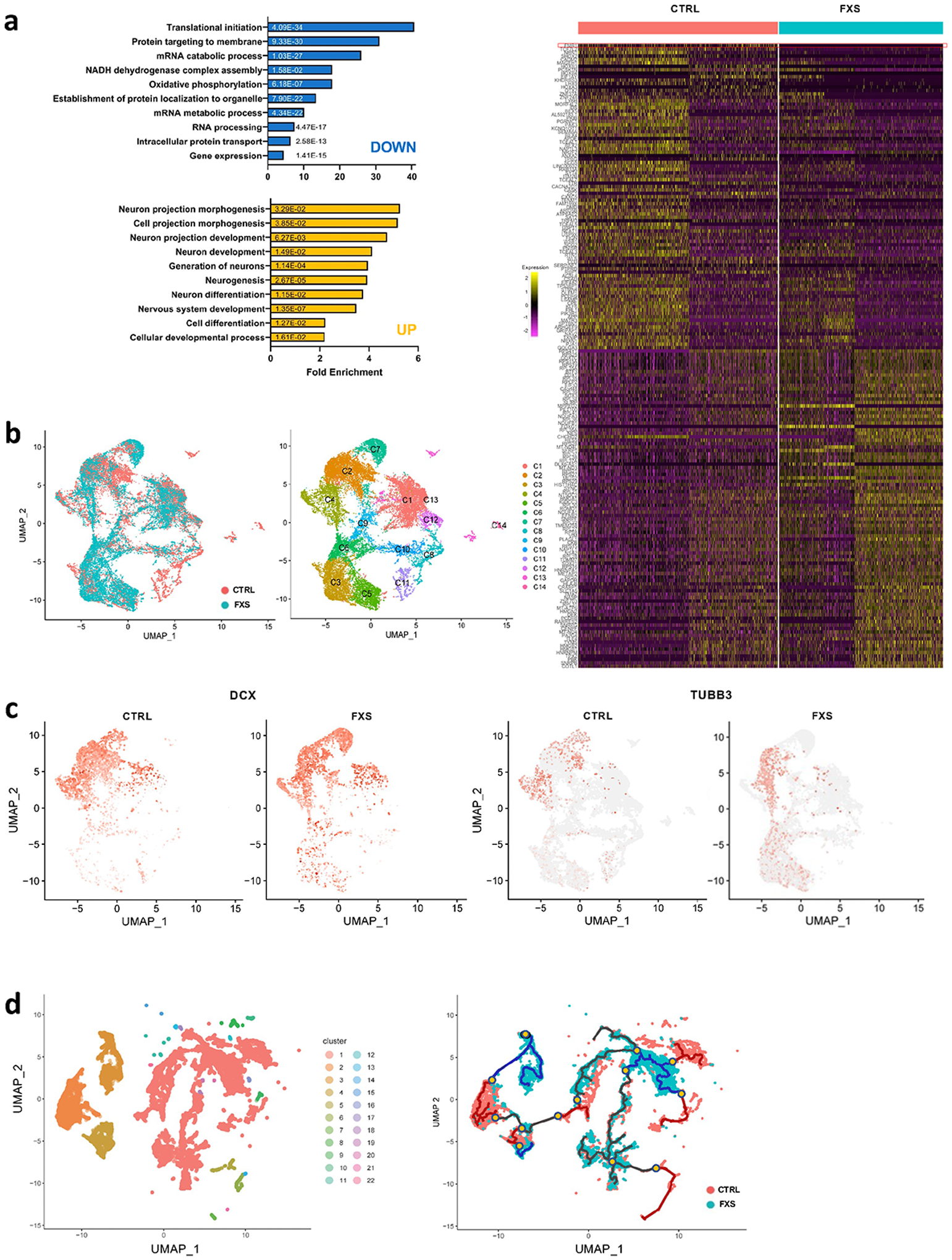
(a) Shown in the right panel is a heat map showing the normalized and scaled gene expression level in forebrain organoids at day 56 and bar plots of gene ontology analyses using PANTHER overrepresentation test on 100 upregulated genes (yellow bars) and 100 downregulated genes (blue bars) in left panel. FMR1 is shown on the top row. The numbers on the bars are two-sided p-values by Fisher’s exact test. P values have been adjusted for multiple testing by Bonferroni correction. (b) UMAP plots of control and FXS show the differential change in the population of specific cell types in FXS compared to control (left). Individual clusters were defined and annotated to distinct neural cell types (right). (c) UMAP plots of DCX, an immature neuron marker, and TUBB3, a mature neuron marker, reveal developmental progression alterations in FXS compared to control. (d) Twenty-two Monocle 3 clusters were identified and cluster-specific marker genes were determined (left). The overall pseudo-time trajectory of cells was obtained. The developmental trajectory indicated as a blue line for the FXS trajectory is quite distinct from the control organoid time trajectory in red (right). Branches are indicated in yellow dots. The progression direction is indicated by black arrowheads.
To investigate the developmental progression alteration caused by the loss of FMRP, we utilized statistical methods based on minimal spanning tree to obtain pseudo-time trajectory to investigate the developmental stages of cell populations. Twenty-two pseudo-time trajectory clusters were identified (Fig. 5d, left)22, 23. We located branches, bypasses and break points that were distinct in the FXS trajectory (blue line) compared to the control organoid time trajectory (red line) (Fig. 5d, right). The shared path between control and FXS are shown in black. Cells of these subpopulations with altered neuronal states are strongly enriched in FXS. The characteristic transcriptional features of each branch cluster (yellow marks in Fig. 5d, right) define the differential dynamics of gene regulation in the developmental progression along the distinguishing developmental trajectory (Extended Data Fig. 8d and Supplementary Table 10). The single cell RNAseq and time trajectory analyses suggest that the loss of FMRP leads to alternative and differential developmental progression, possibly related to aberrant neuronal differentiation.
PI3K inhibitors, not mGluR5 antagonists, rescue neurodevelopmental defects in FXS forebrain organoids.
Given the developmental and synaptic deficits we observed in FXS forebrain organoids, we explored pharmacological approaches to rescue these phenotypes. Previously, the group 1 metabotropic glutamate receptors (mGluRs) theory of FXS has postulated that excessive mGluR signal causes synaptic defects of FXS affecting translation at the synapse24, 25, which was supported in animal models where genetic or pharmacological inhibition of mGluR5 could rescue both behavioral and synaptic abnormalities associated with the loss of Fmrp26, 27. To test whether inhibition of the mGluR5 pathway could rescue the developmental phenotypes in FXS forebrain organoids, we treated the organoids with 2-methyl-6-(phenylethynyl)-pyridine (MPEP; 10 μM), an mGluR5 antagonist, from day 42 to 56. No significant differences in proportion of Ki67+ NPCs (Fig. 6a, b) and SYN1+PSD95+ synaptic bouton density (Fig. 6c–d) between the MPEP-treated and vehicle-treated FXS organoids were seen, suggesting that MPEP fails to rescue the developmental defects in FXS forebrain organoids. These results are consistent with recent clinical trials with several mGluR5 antagonists in humans that showed a lack of efficacy4, 28.
Figure 6. PI3K inhibitors, not an mGluR5 antagonist, rescue neurodevelopmental defects in FXS organoids.
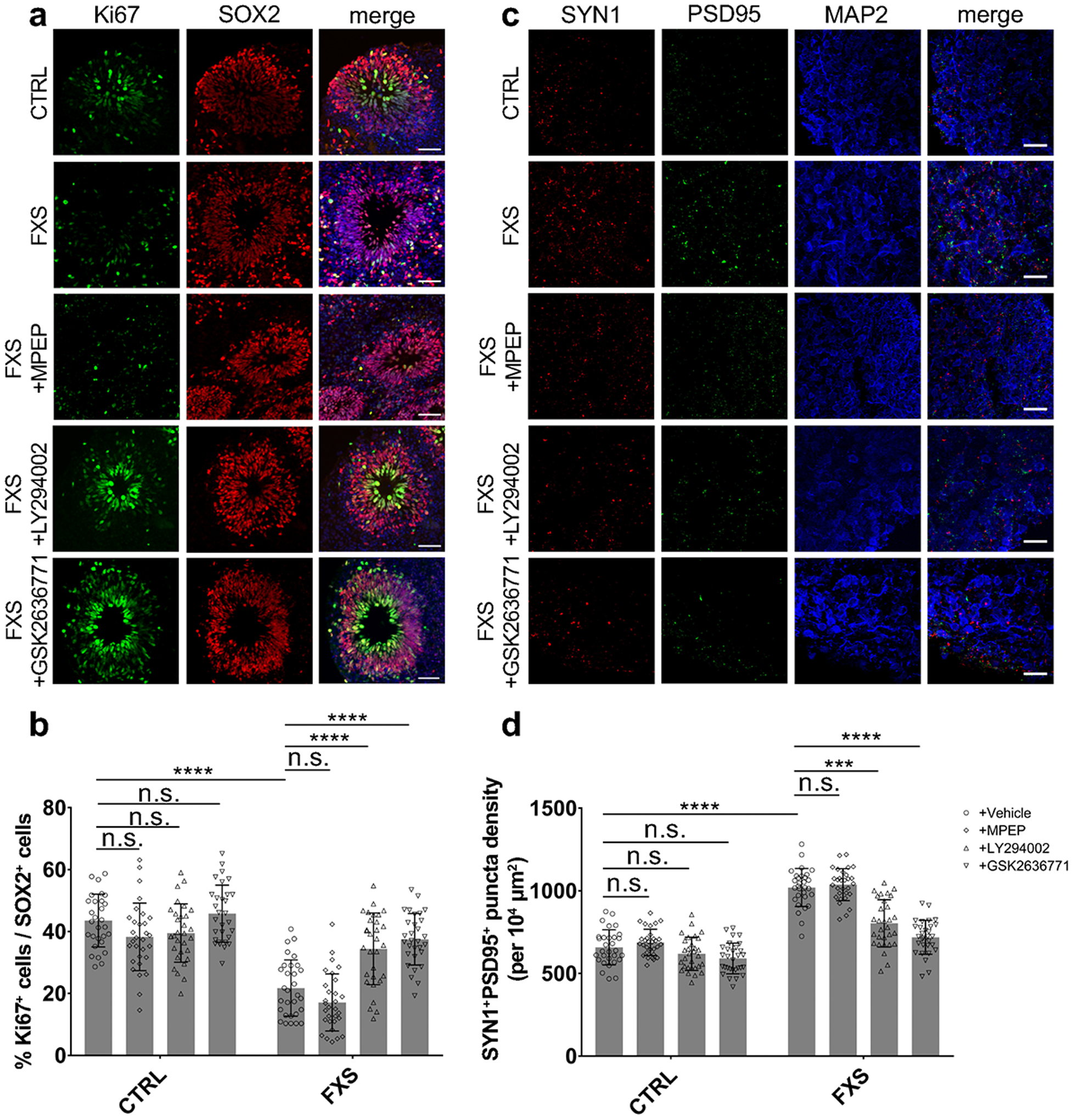
Both control and FXS-derived forebrain organoids were treated by pan-PI3K inhibitor LY294002 (10 μM), selective PI3Kβ inhibitor GSK2636771 (1 μM), mGluR5 antagonist MPEP (10 μM), or DMSO (vehicle) from day 42 to day 56 for two weeks. (a-b) Inhibition of PI3K increases NPC proliferation in FXS organoids. Representative images (a) and quantification (b) of the proportion of Ki67+ neuronal progenitor cells under different treatment conditions in both control and FXS-derived forebrain organoids at day 56. Data are presented as mean ± s.d. (n = 30 sections from at least from 10 organoids each condition; ****P < 0.0001, two-way ANOVA). Scale bars: 50 μm. (c-d) Inhibition of PI3K recues deficit of synaptic formation in FXS organoids. Shown are sample images (c) and quantification (d) of SYN1+PSD95+ puncta density under different treatment conditions in both control and FXS-derived forebrain organoids. Data are presented as mean ± s.d. (n = 30 sections from at least from 10 organoids each condition; ***P = 0.0002, ****P < 0.0001, two-way ANOVA). Scale bars: 50 μm.
In addition to mGluR5, PI3K, phosphoinositide 3-kinase, has also drawn attention due to its specific dysregulation in FXS. Intriguingly, previous studies reported elevated PI3K signaling in FXS models, and genetic reduction or pharmacological inhibition of PI3K has been shown to ameliorate FXS-associated phenotypes in animal models29–31. Therefore, we treated FXS forebrain organoids with a pan-PI3K inhibitor, LY294002 (10 μM), or a selective inhibitor of PI3Kβ, GSK2636771 (1 μM) from day 42 to 56. Quantitative analyses showed that the 2-week treatment of LY294002 (10 μM) or GSK2636771 (1 μM) largely normalized the proportion of Ki67+ NPCs (Fig. 6a–b) and SYN1+PSD95+ synaptic bouton densities (Fig. 6c, d) to similar levels as seen in control. Our results support that PI3K, but not mGluR5, could be a potential therapeutic target for intervention in human.
Identification of human FMRP mRNA targets
Given the specific neurodevelopmental deficits associated with the loss of FMRP and more pervasive gene expression alteration in fragile X organoids compared to mouse, we performed enhanced crosslinking and immunoprecipitation followed by high-throughput sequencing (eCLIP-seq) to identify the specific mRNAs that could be bound by FMRP in human forebrain organoids. As a comparison, cerebral cortices of mouse embryonic brain E13.5 were analyzed in parallel. We conducted eCLIP-seq using an FMRP antibody that was used previously for FMRP eCLIP-seq in non-neuronal cells (Fig. 7a). For all eCLIP-seq experiments, we identified significant peaks by comparing read density between eCLIP and input, excluding PCR duplications and considering only uniquely mapped reads, as previously described32. We used a stringent peak threshold of at least 8-fold enrichment in IP over the input and identified the consistent peaks across the biological replicates.
Figure 7. Identification of human FMRP mRNA targets.
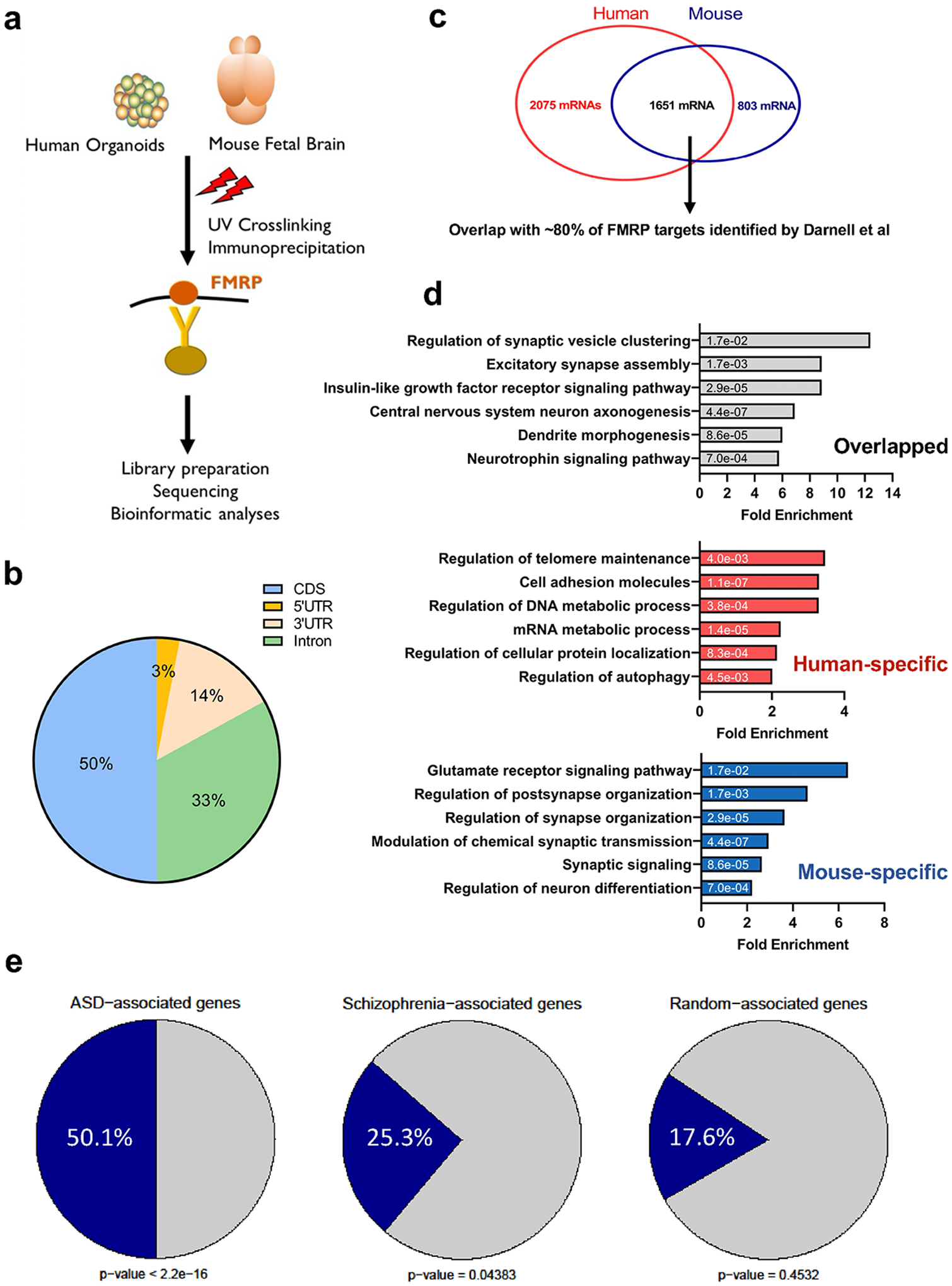
(a) A schematic plot demonstrates the experimental procedure for eCLIP using human forebrain organoids and mouse fetal brains. (b) Pie charts display the genomic location of all human FMRP-binding sites identified from eCLIP using human forebrain organoids, mostly in CDS. (c) The number of FMRP targets and the overlap between human and mouse eCLIP genes are shown in the Venn diagram. The shared eCLIP mRNAs overlap with more than 80 percent of FMRP targets previously identified by Darnell et al. (p < 2.2e-16 using Person’s Chi-square test). (d) Shown are bar plots of gene ontology analyses using PANTHER overrepresentation test on the human-specific, the mouse-specific, and the shared FMRP targets. Numbers on the bars indicate the two-sided p-values by Fisher’s exact test and the x-axis represents fold enrichment. P values have been adjusted for multiple testing by Bonferroni correction. (e) A significant overlap between autism spectrum disorders (ASD)-related risk genes, schizophrenia (SCZ)-related risk genes and FMRP binding genes are indicated. The genes that are randomly selected did not show significant enrichment. Statistical significance was calculated by Pearson’s χ2 tests, and p-values are indicated.
The majority of FMRP binding sites are located in the coding sequence (CDS) and introns in both human and mouse analyses (Fig. 7b and Supplementary Fig. 8). Overall, our analyses identified more than 3700 mRNAs bound by FMRP in human organoids (Fig. 7c). Of the 3726 mRNAs, 1651 overlap with the mRNAs bound by Fmrp in mouse embryonic cortex. Approximately, 80% of the mRNAs bound by both human and mouse FMRP overlap with the previously identified Fmrp mRNA targets in mouse adult brain (Supplementary Table 11)33. Gene Ontology (GO) analyses of different groups of FMRP targets revealed different terms. The mRNA targets that overlap between human and mouse are enriched for genes involved in neurodevelopment, neurogenesis and synaptic plasticity. The human-specific targets are more involved in synaptic signaling and the Wnt pathway. Intriguingly, the most enriched pathway among mouse-specific mRNA targets is glutamate receptor signaling pathway (Fig. 7d). Among 1197 D56 DEGs, we found 170 genes overlapped with human-specific targets while 78 genes overlapped with shared targets. We next assessed whether FMRP targets are enriched for neurodevelopmental disease genes (Fig. 7e). We obtained the genes associated with autism spectrum disorder (ASD), schizophrenia (SCZ), depression, aortic lesion, and obesity, and overlapped the disease related genes with FMRP targets of both human and mouse34–37. FMRP mRNA targets, particularly those from human, are significantly enriched among ASD-associated genes (Fig 7e and Extended Data Fig. 10). These data are consistent with previous studies showing overlap between the genes implicated in autism and FMRP mRNA targets, and further expand the FMRP targets with the relevance to the developing human brain.
Given the defects of neurogenesis and synaptic plasticity associated with FXS organoids, we further examined the protein level of selective mRNA targets identified above. Indeed, most also displayed augmented protein levels, which suggests that FMRP could suppress the translation of these mRNA targets (Fig. 8a).
Figure 8. CHD2 is a human-specific mRNA target of FMRP.
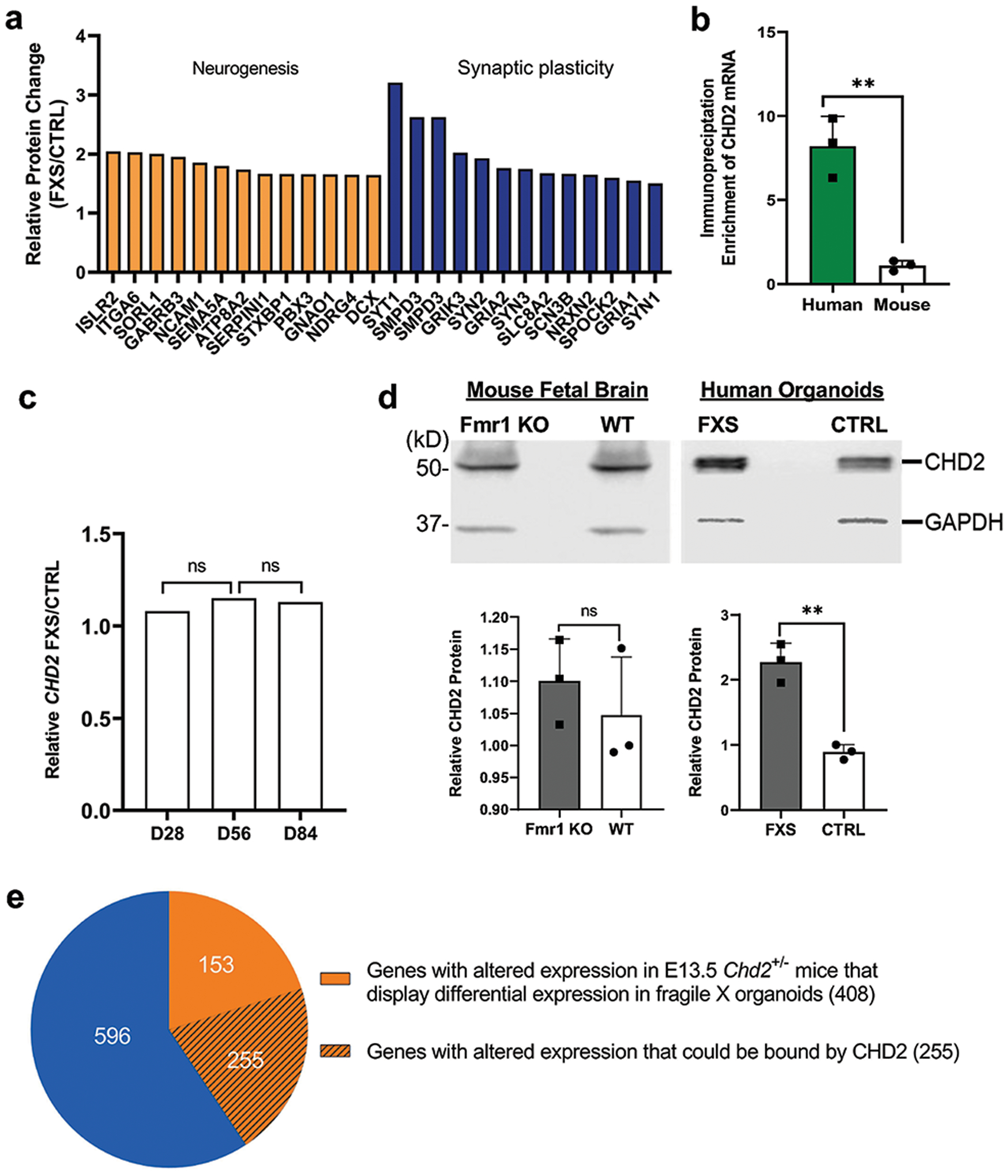
(a) FMRP target genes identified by eCLIP and related to neurogenesis and synaptic plasticity showed alteration at the protein expression level. (b) Shown is the fold enrichment of CHD2 mRNA by FMRP immunoprecipitation followed by qRT-PCR in human forebrain organoids and mouse brain. Data are presented as mean ± s.e.m. (N=3, ** P=0.0024, two-tailed t test). (c) CHD2 mRNA level did not display significant alteration in D28, D56, and D84 fragile X forebrain organoids based on chi-square test. “ns” indicates that it is not statistically significant (p-value > 0.05). (d) Western blots are presented for comparing the CHD2 protein level in human organoids (Control vs FXS) and mouse embryonic brains (WT vs. Fmr1 KO) using GAPDH as loading control. Upper panel shows Western blot of CHD2 and GAPDH and the lower panel is quantification of the Western blot in human (lower left, N=3, ** P=0.0016, two-tailed t test) and mouse (lower right, N=3, NS P>0.05, two-tailed t-test). Data are presented as mean ± s.e.m. (e) Shown is overlap of the DE genes between FXS organoids and E13.5 Chd2+/− mice. Approximately half of the common DE genes (408) between human forebrain organoids and Chd2+/− mice were also verified to be bound by CHD2 in human cell (255).
CHD2 is a Human-specific FMRP mRNA target
One of the human-specific mRNA targets of FMRP is CHD2, Chromodomain-helicase-DNA-binding protein 2 (CHD2), which belongs to the CHD family, characterized by presence of chromatin organization modifier domains (chromo domain) and SNF2-related helicase/ATPase domains38. CHD2 mutations have been associated with intellectual disability and ASD39. CHD2 could play critical roles in neurogenesis and the generation of GABAergic neurons. To further validate CHD2 mRNA as a human-specific FMRP target, we performed parallel FMRP immunoprecipitation followed by qRT-PCR and confirmed that only human FMRP could bind to CHD2 mRNA (Fig. 8b). Furthermore, we examined the expression of CHD2 at both mRNA and protein levels. CHD2 mRNA levels are similar without a significant difference in either mouse embryonic cortex or human forebrain organoids in the presence or absence of FMRP (Fig. 8c and Supplementary Fig. 9). However, at the protein level, while no significant change was detected in Fmr1 KO mouse embryonic cortex, a significant increase of CHD2 protein was observed in FXS forebrain organoids compared to unaffected control (Fig. 8d). These results suggest that human FMRP, but not mouse Fmrp, could regulate the expression of CHD2 post-transcriptionally.
A previous study has shown that Chd2 is necessary for neural circuit development and long-term memory in mice, and the differentially expressed genes in embryonic cortex of E13.5 were also identified40. To determine whether the misregulation of CHD2 in FXS forebrain organoids could have any functional consequence, we compared the differentially expressed genes between FXS organoids and E13.5 Chd2+/− mice, and observed significant overlap (408 out of 1004 genes were altered in both FXS organoids and E13.5 Chd2+/− mice, p value < 2.2e-16) (Fig. 8e and Supplementary Table. 12). Furthermore, utilizing the available ChIP-seq data of CHD2 in human cells generated by ENCODE, we found that 255 out of 408 genes could be bound by CHD2 (p value = 2.97e-05, Fig. 8e and Supplementary Table. 12). These analyses suggest that the loss of FMRP could lead to the increased expression of CHD2 protein, which in turn alters the expression of its downstream targets. Thus, the identification of these human-specific FMRP mRNA targets will provide new insights into the molecular pathogenesis of fragile X syndrome in a human context. human context.
DISCUSSION
Here we developed FXS forebrain organoids from patient-derived induced pluripotent stem cells and observed dysregulated neurodevelopment in these FXS organoids. Transcriptomic analyses revealed that loss of FMRP led to more pervasive gene expression alterations in human brain development and an altered developmental trajectory in forebrain organoids in a cell type-specific manner. Mechanistically, we demonstrated that FMRP could bind to a number of human-specific mRNA targets, which are significantly enriched in ASD-associated genes. Furthermore, we have shown that pharmacological inhibition of the PI3K pathway, but not the mGluR5 pathway, could largely rescue the defects associated FXS forebrain organoids. These data together provide new insights into mechanisms underlying FXS in human context and identify potential human-specific druggable targets for FXS and autism in general.
Major progress into the understanding of the molecular mechanisms underlying FXS in animal models led to several clinical trials, such as modulating metabotropic glutamate and GABA receptors; however, these clinical trials have been unsuccessful so far. One potential explanation for the lack of successful clinical trials could be the fundamental developmental, biochemical and physiological differences between animal models and humans, which motivated us in the current study. Previous studies using 2D culture of FXS human neurons have shown impaired differentiation and hyperexcitability41–45. Recently, 3D organoids have been generated from human iPSCs via 3D culturing methods46, 47. Human organoids resemble critical organ and tissue-specific features of cell assembly, integration, and organization, and they exhibit unique properties not observed in animal models. Thus, 3D organoids provide a unique opportunity to model human organ structure and function under healthy and disease conditions. Studies have shown that human organoids derived from patient-specific iPSCs, such as patients with autism and other diseases, recapitulate specific disease phenotypes, respond to drug treatments, and are valuable in mechanistic studies48–57.
In the present study we observed dysregulated neurodevelopment in FXS forebrain organoids, much of which was not observed in the FXS mouse model. Furthermore, our transcriptome analyses revealed a large number of overlapped genes differentially expressed in both FXS forebrain organoids and fetal brain tissues, with only a few genes dysregulated in FXS mouse brain, suggesting that human brain organoids could better mimic the FXS-related phenotypes in a human context. Our comparative analyses have led to the identification of a large number of mRNAs that were bound by FMRP in human but not mouse. Interestingly, manipulating mGluR5 signaling, one well-studied pathway that is dysregulated in FXS animal models leading to impairments in protein synthesis, synaptic structure and function25, showed no effect on rescuing neurodevelopmental defects in FXS forebrain organoids. However, we should note here that the mGlu5R clinical trial was done in adolescents and adults, not during early development. Dysregulated PI3K signaling has also been linked to FXS29, 58–64, and our data presented here suggest that pharmacological inhibition of the PI3K pathway can rescue some of the defects in neurodevelopment and synapse formation in FXS forebrain organoids. Future comprehensive evaluation of the effect of PI3K inhibitors on fragile X organoids are certainly warranted. Our findings also indicate that human brain organoids may provide a human-specific preclinical model for studying disease mechanisms underlying FXS and identifying human-specific therapeutic targets.
FMRP is widely expressed throughout the embryonic brain development and its expression increases during neuronal differentiation, indicating the importance of FMRP for neurogenesis65–67. Importantly, there is evidence that in chorionic villi samples taken from FXS fetuses, FMRP is absent at the early fetal period (12 PCW)68, a critical period for neurogenesis15. Indeed, examination of the postmortem brain tissues from FXS patients has revealed an increased density of long and tortuous dendritic spines67. In addition, previous studies have shown neuroanatomical abnormalities in children with FXS, which are likely due to altered neurogenesis during early development9. Consistent with previous analyses, we observed dysregulated neurodevelopment in FXS forebrain organoids at both the molecular and cellular level. Furthermore, our transcriptome analyses on FXS forebrain organoids revealed human-specific molecular signatures associated with FXS, which largely overlapped with signatures of FXS fetal brain tissues but not with mouse brains. These results highlight the unique impact of loss of functional FMRP on human brain development and provide mechanistic insights into the molecular pathogenesis of FXS. One potential mechanism underlying the striking difference of FMRP-dependent gene expression changes in human organoids and mouse embryonic brains could be due to the differential m6A RNA modification. Earlier works have shown that FMRP could utilize the m6A pathway to regulate gene expression, and that the m6A modification is more prevalent in human brain tissues than mouse13, 69.
Emerging evidence suggests that FMRP plays a critical role during earlier stages of neural development. Our data suggest that absence of FMRP leads to fewer actively proliferating (Ki67+) NPCs, coinciding with an increase in TBR2+ intermediate progenitors/neuroblasts and accelerated cortical layer formation, which is consistent with previous reports in mouse and human models70–73. Intriguingly other groups and our group have also shown that loss of FMRP increases the proliferation of adult neural stem cells in mouse hippocampus74 and early stage of human iPSC-derived neural stem cells12. Thus, the effect of loss of FMRP could be developmental stage-specific: loss of FMRP increases NPC proliferation at early stages while it decreases NPC proliferation and promote neural differentiation at the later stages, and its overall effect is to accelerate premature neural differentiation.
The GABAergic inhibitory neurons are ~10% of total cells in our control forebrain organoids, which is similar to organoids generated by other groups75. Interestingly, while they found that GABAergic inhibitory neurons were overproduced in organoids from patients with idiopathic autism, we observed significant reduction of GABAergic inhibitory neurons in FXS organoids. To further investigate how loss of FMRP affects the development of GABAergic neurons, a model system with enrichment of GABAergic neurons, such as the ventral forebrain organoids or the forebrain assembloids76 will be needed in future studies.
To identify the druggable targets for FXS therapeutic interventions, it is important to identify the mRNA targets regulated by FMRP in human brain. Given the complexity of the CLIP assay itself, it was technically challenging to perform the CLIP assay using postmortem tissues. Accordingly, development of human brain organoids has provided us the first opportunity to identify the FMRP mRNA targets during human brain development. By performing eCLIP-seq on both human forebrain organoids and mouse brains, we identified a large number of mRNAs that were bound by FMRP only in human, many of which are ASD-associated genes. The identification of these human-specific FMRP mRNA targets may provide new insights into the molecular pathogenesis of FXS in human context and reveal potential therapeutic targets for interventions. Intriguingly, one of the human-specific targets of FMRP is CHD2, which is a well-known risk factor for intellectual disability and ASD that plays critical roles in neurogenesis and generation of GABAergic neurons. Increased expression of CHD2 in FXS forebrain organoids not only provides a potential mechanistic link to the neurodevelopmental deficits associated with loss of functional FMRP, but also suggests that CHD2 could serve as a converging point of ASD and a potential druggable target for FXS and autism, in general.
In summary, we have developed a human forebrain organoid model of fragile X syndrome and shown that that loss of functional FMRP could lead to neurodevelopmental defects and alter the developmental trajectory. The identification of human FMRP mRNA targets could provide potential druggable targets for the therapeutic development of fragile X syndrome. The human forebrain organoid model of fragile X syndrome presented here could serve as a human specific preclinical model and a resource for further studying the molecular pathogenesis of FXS.
METHODS
Human forebrain-specific organoid cultures
The human iPSC lines were previously generated from skin biopsy samples of three FXS male patients and three age-matched healthy males at Emory University. The isogenic line was generated by excising the expanded CGG-repeat in one of the FXS iPSC line (FXS1) using the CRISPR/Cas9 gene editing. All iPSC lines have been fully characterized and cultured on irradiated MEFs in human iPSC medium consisting of D-MEM/F12 (Invitrogen), 20% Knockout Serum Replacement (KSR, Invitrogen), 1X Glutamax (Invitrogen), 1X MEM Non-essential Amino Acids (Invitrogen), 100 μM β-Mercaptoenthanol (Invitrogen), and 10 ng/ml human basic FGF (PeproTech) as described77. Forebrain-specific organoids were generated using established protocols as previously described7. Briefly, human iPSC colonies were detached from the feeder layer with 1 mg/ml collagenase treatment for 1 hour and suspended in embryonic body (EB) medium, consisting of FGF-2-free iPSC medium supplemented with 2 μM Dorsomorphin and 2 μM A-83 in non-treated polystyrene plates for 4 days with a daily medium change. On days 5–6, half of the medium was replaced with induction medium consisting of DMEM/F12, 1X N2 Supplement (Invitrogen), 10 μg/ml Heparin (Sigma), 1X Penicillin/Streptomycin, 1X Non-essential Amino Acids, 1X Glutamax, 4 ng/ml WNT-3A (R&D Systems), 1 μM CHIR99021 (Tocris), and 1 μM SB-431542 (Tocris). On day 7, organoids were embedded in Matrigel (BD Biosciences) and continued to grow in induction medium for 6 more days. On day 14, embedded organoids were mechanically dissociated from Matrigel by pipetting up and down onto the plate with a 5 ml pipette tip. Typically, 10–20 organoids were transferred to each well of a 12-well spinning bioreactor (SpinΩ) containing differentiation medium, consisting of DMEM/F12, 1X N2 and B27 Supplements (Invitrogen), 1X Penicillin/Streptomycin, 100 μM β-Mercaptoenthanol (Invitrogen), 1X MEM NEAA, 2.5 μg/ml Insulin (Sigma). At day 71, differentiation medium was exchanged with maturation medium, consisting of Neurobasal (Gibco), 1X B27 Supplement, 1X Penicillin/Streptomycin, 1X β-Mercaptoenthanol, 0.2 mM Ascorbic Acid, 20 ng/ml BDNF (Peprotech), 20 ng/ml GDNF (Peprotech), 1 ng/ml TGFβ (Peprotech), and 0.5 mM cAMP (Sigma). All media was changed every other day. LY294002 and MPEP were purchased from Millipore Sigma, and GSK2636771 was purchased from Cayman Chemical.
Tissue infection and immunocytochemistry
For retroviral infection, retroviruses expressing GFP were prepared by Emory Viral Core and forebrain organoids were infected at D49. For immunocytochemistry, forebrain organoids were processed at day 56 as previously described7. Briefly, whole organoids were fixed in 4% Paraformaldehyde in Phosphate Buffered Saline (BPS) for 30–60 min at room temperature. Organoids were washed 3 times with PBS and then incubated in 30% sucrose solution overnight. Organoids were embedded in tissue freezing medium (General Data) and sectioned with a cryostat (Leica). For immunostaining, freezing medium was washed with PBS before permeabilization with 0.2% Triton-X in PBS for 1 hr. Tissues were then blocked with blocking medium consisting of 10% donkey serum in PBS with 0.1% Tween-20 (PBST) for 30 min. The following primary antibodies were used: anti-Ki67 (mouse; 1:500; BD-Pharmingen), anti-SOX2 (goat; 1:500; R&D Systems), anti-PAX6 (rabbit; 1:500; Thermofisher), anti-CTIP2 (rat; 1:400; Abcam), anti-TBR1 (rabbit; 1:500; Abcam), anti-TBR2 (rabbit; 1:500; Abcam), anti-CaMKIIα (mouse; 1:800; Cell Signaling), anti-GABA (rabbit, 1:800; Sigma), anti-SYN1 (mouse; 1:500; Synaptic System), anti-PSD95 (rabbit; 1:500; Thermofisher), anti-DCX (rabbit; 1:500; Cell Signaling), anti-MAP2 (chicken; 1:500; Novus), anti-Tra-1–60 (mouse; 1:500; Millipore), anti-NANOG (goat; 1:500; R&D Systems). Primary antibodies diluted in blocking solution were applied to the sections overnight at 4°C. After washing with PBST, secondary antibodies diluted in blocking solution were applied to the sections for 1 hr at room temperature. Finally, sections were washed with PBST and stained with DAPI. For EdU labeling, forebrain organoids were pulsed with 10 μM EdU for 2 hrs or 24 hrs on day 56, followed by fixation and immunostaining. All images were captured by Nikon Eclipse Ti-E microscope. Quantitative analyses were conducted on randomly selected cortical structures in a blind fashion. The numbers of cells positive for each marker were measured using ImageJ software (NIH). For analysis of synaptic bouton density, five regions of interest (ROIs) in each organoid section were randomly selected in cortical-like layer, and the numbers of SYN1+PSD95+were counted using ImageJ software (NIH). To quantify the layer thickness, the ventricular-like zone (VZ) was defined by SOX2+ (or PAX6+)/CTIP2− (or TBR1−, or MAP2−), and the cortical plate (CP) was defined by CTIP2, or TBR1, or MAP2 immunoreactivity. For each cortical structure, three measurements were performed forming a right angle fan area pointing to the nearest pial surface, at 0, 45 and 90 degrees. The length for VZ and CP layers was measured in ImageJ software. Relative thicknesses for VZ and CP were calculated by the ratio to total thickness from ventricular to pial surface. To quantify the distribution of specific cell types in the entire span of the neuroepithelium, the entire span of the neuroepithelium was divided into five equal portions (bins) and measured the distributions of specific cell types by calculating the percentage of each marker in the total DAPI+ cells in each bin.
Electrophysiology
Organoid slices were prepared by embedding organoids in 4% low melting point agarose cooled to approximately 32°C. Slices (225 μm) were sectioned using a vibratome (Leica VT1000S) and stored at room temperature, oxygenated (95% 02, 5% CO2) ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM): 125 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 3 KCl, 25 dextrose, 1 MgCl2, and 2 CaCl2. Slices were immediately ready for recording. ACSF was oxygenated (95% O2, 5% CO2) and bath temperature was approximately 31°C. Patch pipettes were fabricated from borosilicate glass (1.5 mm OD / 0.86 mm ID, Sutter Instrument) to a resistance of 5–6 MΩ. For current- and voltage-clamp measurements, pipettes were filled with (in mM): 130 potassium gluconate, 8 KCl, 10 HEPES, 1 EGTA, 8 Na2-phosphocreatine, 4 Mg-ATP, 0.3 Na2-GTP; pH 7.3 adjusted with KOH. Signals were recorded with a Multiclamp 700B amplifier (Molecular Devices). Signals were filtered at 2 kHz and sampled at 20 kHz via a Digidata 1550B digitizer (Molecular Devices) controlled by pCLAMP software. To record whole cell ionic currents, cells were voltage clamped at −60 mV and depolarized to voltages stepped from −90 to 50 mV with a 10 mV increment for 500 ms. Membrane resting potential depolarized more than −45 mV was discarded. The spontaneous PSCs recording were held at −60 mV and the amplitudes less than 10 pA were excluded from analysis. The evoked action potential measurements were conducted in current clamp recording. Resting membrane potential was adjusted to −60 mV by injecting small positive currents usually less than 30 pA. Cells were stimulated with current steps from −20 to 100 pA with a 10 pA increment for 500 ms to measure the firing number and pattern of action potential. Action potential parameters were characterized from the first evoked action potential for which the maximum rising slope exceeds than 20 mV/ms.
RNA in situ hybridization
RNA in situ hybridization was performed using the RNAscope Fluorescent Multiplex Reagent Kit (Advanced Cell Diagnostics). The samples were prepared according to the manufacturer’s instructions. Briefly, forebrain organoids were collected and embedded in OCT. The fresh frozen sample were equilibrated to −20°C in a cryostat for 1 h. 15um FFPE tissue sections were mounted on SuperFrost Plus slides, fixed in 4% PFA for 15 min at 4°C, dehydrated using 50%, 70% and 100% ethanol, and treated with RNAscope protease IV (Advanced Cell Diagnostics) for 15 min at room temperature. Probes were mixed at a 50:1:1 ratio (C1: C2:C3) and hybridized for 2 h at 40°C in the HybEZ Oven (Advanced Cell Diagnostics). The following probes from Advanced Cell Diagnostics were used: Hs-SOX2 (catalog #400871-C1), Hs-DLX2-C2 (catalog #483311-C2), and Hs-PAX6-C3 (catalog #588881-C3). Amplification and detection were performed according to the manufacturer’s instructions. Slides were applied to DAPI for 30s at room temperature, added fluorescent mounting medium and covered with coverslips. All images were captured by Nikon Eclipse Ti-E microscope. Quantitative analyses were conducted randomly on 5 regions per section in a blind fashion. The area positive for each probe was measured using ImageJ software (NIH).
RNA isolation, quantitative RT-PCR, and RNA-seq
The study of human fetal tissue was approved by Central South University Medical Genetics National Laboratory Ethics Review Committee (2013051202) and consented by the patients and their families. After the prenatal diagnosis confirmed that the fetus was fragile X full mutation, the patients decided to terminate the pregnancy and donate the fetal tissues for research. The age-matched control cortex tissues were obtained from local brain banks. All protocols for mouse experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Emory University. Human fetal brain cortex tissues, human organoids and mouse embryonic whole forebrain were homogenized in Trizol (Invitrogen) and processed according to the manufacturer’s instructions. RNA was then reverse-transcribed using a SuperScript® III First-Strand Synthesis System (Invitrogen). qPCR quantified cDNA via TaqMan assay (Invitrogen). Each reaction was run in triplicate and analyzed following the ΔΔCt method using glyceraldehyde-3-phosphate dehydrogenase (Gapdh) as a normalization control.
RNA-seq library construction was performed following the instructions of Illumina mRNA sample prep kit (cat# RS-100–0801). In brief, the poly-A–containing mRNA was purified using poly-T oligo-attached magnetic beads. The mRNA was then fragmented into small pieces using divalent cations under elevated temperature. The cleaved RNA fragments were copied into first strand cDNA using reverse transcriptase and random primers. This was followed by second strand cDNA synthesis using DNA Polymerase I and RNase H. These cDNA fragments went through an end repair process, the addition of a single ‘A’ base, and then ligation of the adapters. These products were gel purified and enriched with PCR to create the final cDNA libraries. The library constructs were run on the bioanalyzer to verify the size and concentration before sequencing on the Illumina HiSeq2500 machine. Raw reads were examined for quality issues to ensure library generation and sequencing were suitable for further analysis. As necessary, adapter sequences, other contaminant sequences such as polyA tails and low-quality sequences with PHRED quality scores less than five were trimmed from reads using cutadapt. RNA-seq reads were aligned using STAR v2.7, reference genome hg19 for human samples and mm9 for mouse samples. and differential expression values were extracted using DESeq2 v1.26.078. Genes with false discovery rate < 0.20 are defined as differentially expression genes. All downstream analysis including plotting and statistical tests were conducted using R 3.6.2.
Single cell RNA sequencing (scRNA-seq)
Three batches of human D56 forebrain organoids generated from three sets of unaffected and FXS iPSCs including an isogenic line were dissociated with Accutase solution (Sigma-Aldrich) and the single cell suspension was subjected to Chromium 10x Genomics 10X Genomics Chromium Controller (Emory Integrated Genomics Core) with 5000 target cell number and HiSeq4000 sequencing (Novogene Inc.). Briefly, the captured cells were processed for barcoding and generation of a single cell library with Chromium Next GEM Single Cell 3′ Library kit, and the resulting single cell libraries were sequenced with HiSeq 4000. Using Cell Ranger 3.0.2 from 10X Genomics, raw Chromium scRNA-seq output was processed to align reads, generate feature-barcode matrices and perform clustering and gene expression analysis.
The CRCh38 human genome assembly was adopted to map the single-cell sequence reads. The median number of genes detected per cell is 1,995, and the fraction reads in cells is 76.3%. The median Unique Molecular Identifier (UMI) counts per cell is 4,617. We kept the genes with expression in at least 10 cells and cells that had expression of at least 200 genes. We filtered out cells expressing high level of genes mapped to the mitochondrial genome. To preserve high-quality cells, we removed cells with greater than 30% of mitochondrial sequencing reads. After filtering, normalization and scaling were performed. We analyzed 30,550 single cells with 1,528,965,000 unique mRNAs reads in downstream analysis with Seurat 3.1 and Monocle 3 packages in R.
scRNA-seq data analysis
Using all 29,025 genes, we identified anchor genes to integrated FXS and control samples for comparison in Seurat. In order to acquire the integrated object, we created an object list containing both the control and FXS Seurat object and performed normalization as well as variable feature selection. The objects were normalized using “LogNormalize” and the scale factor was set as 10,000. We identified a set of features exhibiting high cell-to-cell variability, using Variance-Stabilizing Transformation (VST). The integration anchors were found using the two objects and employed 1:20 dimensions using Canonical Correlation Analysis (CCA). After data integration, we performed principal components analysis (PCA) using the previously identified variable genes. Fourteen cell types (clusters) were identified using the Uniform Manifold Approximation and Projection (UMAP) dimensional reduction technique. We utilized the top 20 principal components to retain sufficient information for dimension reduction. Using known marker genes in combination with gene ontology analysis, we were able to identify each cell type representing neural lineages, such as neural progenitors, astrocytes, immature neurons, inhibitory neurons and excitatory neurons.
We used the “FindMarkers” function in Seurat to identify the Differential Expressed (DE) genes comparing FXS with control in each cluster using Wilcoxon Rank Sum test. Analyses were controlled for multiple testing. Adjusted p-value and False discovery rate (FDR) was calculated. FDR < 0.05 was defined as statistical significance.
Cell types were assigned to the clusters as modified from previous studies with scRNA-seq20, 79, 80. Briefly, all genes expressed in each cell are ranked by relative expression to the average of all cells and the clusters were first annotated by collectively weighing the expression of markers of each specific cell type such as NPC markers (SOX2, NES, NOTCH1, HES1, CDH1), radial glia markers (HES5, PAX6, VIM, FABP5, FABP7, CHD2, LIFR, FAM107A, PTPRZ1, BMP7, HOPX), glial cell/ oligodendrocyte and mature astrocytes markers (SLC1A2, SLC1A3, SLC1A4, GLUD1, OLIG1, OLIG2, GFAP, and S100B), intermediate progenitor markers (NEUROG2, DLX1, ASCL1), migrating neuronal committed cell marker (DCX, GAP43), cortical neuronal marker (TBR1), immature neuron markers (STMN1, STMN2, NEUROD1, NEUROD2, NEUROD4, NEUROD6, TUBB3.1), mature neuron markers (RBFOX3, MAP2, SYP, SYN1, NEFM), inhibitory neuron markers (GAD1, GAD2, GABBR1, GABBR2, SLC32A1), excitatory neuron markers (SLC17A6, SLC17A7, GRIN2B, GLS, GLUL), mesoderm markers (MYL1, MYH3). First, any mesodermal cell cluster was identified with myosin and myofibril (MYL1 and MYH3). Then, the annotation term of each cluster was verified with GO term analysis to correlate the enriched GO term to the identity of each cluster.
Time-Trajectory analysis in Monocle 3
We first created a new Seurat object containing all the samples using their indexes from the original object. It should be noted that the quality control and pre-processing had already been performed on the original Seurat object. In Monocle 3, the cell dataset (CDS) object was created using the expression matrix, cell metadata, and gene metadata generated previously. We retained the first 50 principle components (PCs) for downstream analysis. We performed the dimension reduction using the “reduce_dimension” function using UMAP. Then, the clusters were identified through “cluster_cells” with a resolution of 10−4. To obtain the marker genes expressed by each cluster for CDS objects, the marker genes were identified using the function “top_markers”. Then, we grouped the cells by cluster to find the cluster-specific marker genes. Next, we used the “learn_graph” function to investigate the overall pseudo-time trajectory of cells and labeled them in the trajectory path.
Enhanced crosslinking and immunoprecipitation followed by high-throughput sequencing (eCLIP-seq)
To fix the interactions between the RNA binding proteins and their related RNA permanently, UV crosslinking was conducted at 254 nm, 400 mJ/cm2 three times on an iced waterbed using the D56 organoids of both unaffected and fragile X as well as the mouse embryonic brains of E13.5.. The UV crosslinked samples were lysed in lysis buffer (50 mM Tris-HCl pH7.5, 100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate and protease inhibitor) on ice for 10–30 min followed by digestion with RNase I and DNase (Invitrogen) at 37 °C for 5 min. After digestion, RNase inhibitor was added immediately to avoid further degradation of RNA, and centrifugation was carried out at 14,000 rpm for 10 min at 4°C. The supernatant was used for immunoprecipitation of FMRP–RNA complexes with specific anti-FMRP primary antibody (Abcam) and normal IgG as negative IP control using Dynbeads Protein G (Invitrogen).
The IPed beads were washed stringently and sequentially with cold high salt buffer, cold wash buffer, and finally with Fast AP buffer. The washed beads were treated with FastAP (ThermoFisher) and followed by PNK (NEB) for dephosphorylation using 37°C Thermomixer at 1200 rpm to avoid bead precipitation followed by washing with the ice-cold wash buffer, the cold high salt buffer, and the 1X ligase buffer, respectively. To enhance the ligation efficiency and to wash away unincorporated adapters, the on-beads ligation was carried out to ligate the RNA adapter RiL19 to the 3′ end of the bound RNA by FMRP using T4 RNA Ligase (NEB), PEG8000 (NEB), and DMSO (Sigma). After stringent washes, the washed/ligated beads for each sample were resuspended in 100 ul of 1XPBS buffer followed by dividing the beads into two tubes: one for 20% and the other for 80%, respectively. After the PBS buffer was completely removed from both tubes for each sample, the FMRP/RNA complexes were eluted with 4X NuPAGE buffer (Invitrogen) at 70°C for 10 min at 1200 rpm. The eluted FMRP-RNA complex samples were then separated on NuPAGE gels and transferred to nitrocellulose membranes. The membrane containing the elution from 20% of the beads was applied for detection of efficiency for both IP and crosslink via Western blot, while the membrane for 80% of the eluted was used for recovery of the FMRP/RNA complexes.
A region starting from the FMRP protein size (RNA fragment free FMRP) to above about 75 kDa (FMRP bound with around 220 nt of RNA fragments) on the membrane was recovered. The recovered region of the membrane was digested with proteinase K (NEB) and the released RNA fragments were purified by Zymo column (Zymo Research). Using AR17 as the primer, the recovered RNA fragments was reverse transcribed with AffinityScript (Agilent). The first strand cDNA was purified by treatment with ExoSAP-IT (Affymetrix) and binding to MyONE Silane beads (ThermoFisher). After the MyONE Silane beads were stringently washed to remove the excess AR17 oligos, ligation was carried out on beads to ligate the DNA oligo rand3Tr3 adapter to the 3’end of the cDNA at room temperature overnight using the same RNA ligase as mentioned above, followed by a stringent wash to get rid of the primer dimers. After purification, both 3’- and 5’-ligated cDNAs were first applied as templates for q-PCR to determine the proper number of final PCR cycles for enrichment of the eCLIP-seq library using Q5 high fidelity DNA polymerase and i5/i7 primer pairs. Finally, the amplified eCLIP library size range from 175–300bp was selected by PAGE purification, followed by elution and precipitation with glycogen (AMBION), 3M sodium acetate (pH5.2), and ethanol. The library constructs were run on the bioanalyzer to verify the size and concentration before sequencing on the Illumina HiSeq2500 machine.
eCLIP-seq Bioinformatic analyses
After standard HiSeq demultiplexing, reads were aligned using STAR 2.7 with human genome (hg19) for human forebrain organoids and mouse genome (mm9) for mouse fetal brain. Candidate binding sites were identified using CLIPper81. To normalize peak-level read counts, Input and IgG samples were processed identically to eCLIP samples. The number of reads overlapping with CLIPper-identified peaks for eCLIP samples, Input and IgG samples were counted. Log fold change of eCLIP versus Input and IgG versus Input were computed separately. Enrichment p-values were calculated by Yates’ Chi-square test. The top 1 percent quantile candidate peaks over-expressed in IgG compared over Input were removed from further analysis. Among the rest of the peaks, the ones with log fold change >1.5 and p value < 1e-10 in eCLIP samples over Inputs were selected as significant FMRP binding peaks. Regions were then mapped back to genome data to identify significant FMRP binding genes in both human and mouse data. Homer (v4.10) was used to annotate FMRP binding peaks into functional genomic regions. The gene interactome plot was obtained using GENEMANIA (genemania.org)82. Gene ontology (GO) analyses were performed using the Panther overrepresentation test and GO database annotation with Fisher test and Bonferroni correction.
Western blotting
Protein samples were separated on SDS-PAGE gels and then transferred to polyvinylidene fluoride membranes (Millipore). Membranes were processed according to the ECL Western Blotting Protocol (GE Healthcare). The membranes were probed with anti-FMRP, anti-CHD2, and anti-GAPDH, respectively, at the dilution of 1:1,000 for anti-FMRP, 1:2000 for anti-CHD2, and 1:100000 for GAPDH. HRP-labeled secondary antibodies were obtained from Cell Signaling Technology (7074S & 7076S) and were used at a dilution of 1:5,000. The antibodies against GAPDH (AM4300; Thermo Fisher) were used for loading controls. All Western blot quantifications were performed using ImageJ software.
Extended Data
Extended Data Fig. 1. FMRP regulates cortical neurogenesis in a human forebrain organoid model.
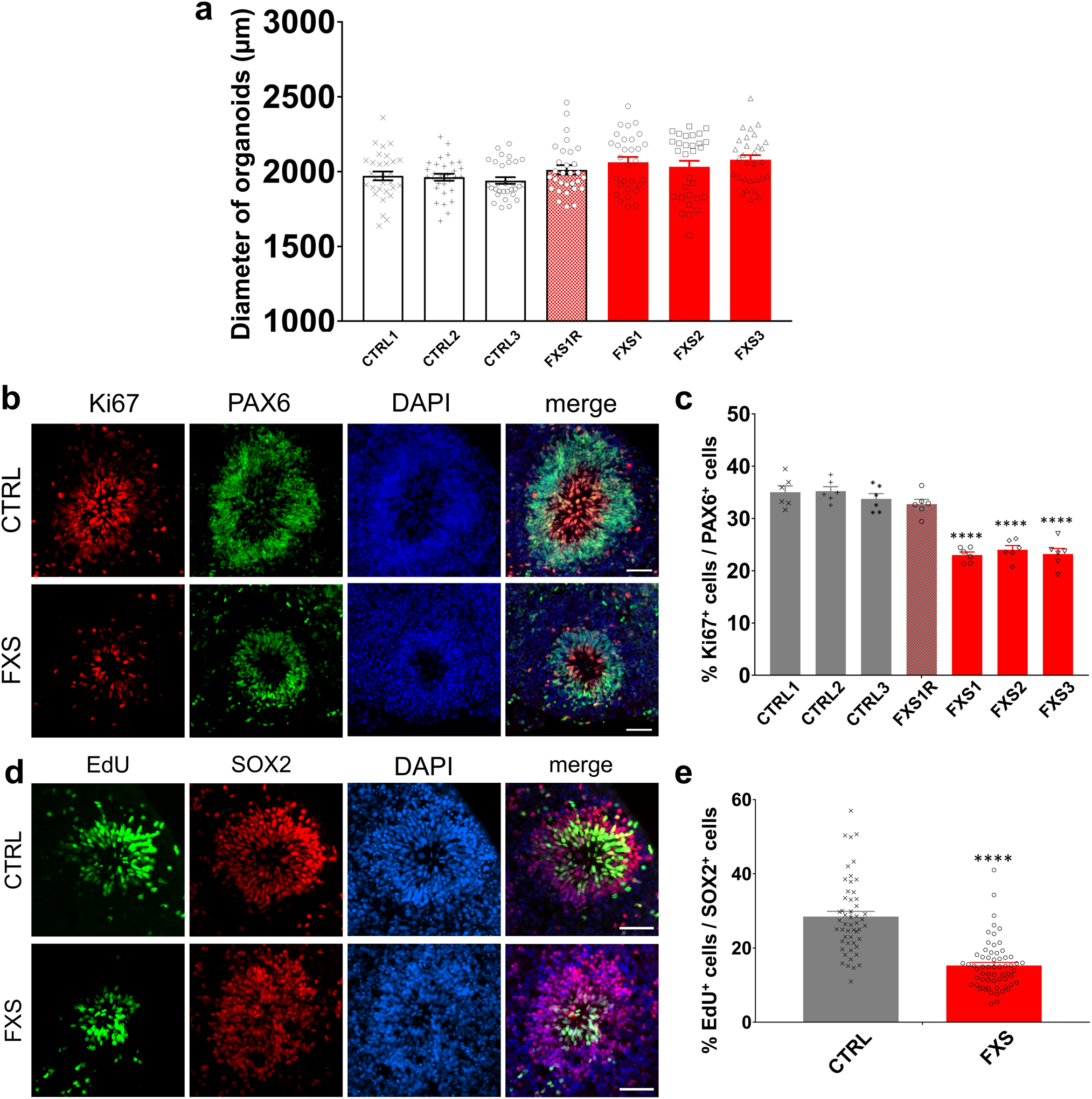
(a) Quantification of the size of control and FXS forebrain organoids. Data are presented as mean ± s.e.m. (n = 30 organoids from each line; one-way ANOVA). (b-c) Loss of FMRP reduces NPC proliferation. Shown are representative images (b) and quantification (c) of the proportion of Ki67+ proliferating neuronal progenitor cells in total PAX6+ dorsal forebrain neuronal progenitor cells of both control and FXS-derived forebrain organoids at day 56. Data are presented as mean ± s.e.m. (n = 6 organoids from each line with 15–20 cortical structures analyzed per organoid; ****P < 0.0001, one-way ANOVA). Scale bars: 50 μm. (d-e) D56 forebrain organoids were pulsed with EdU (10 μM) for 2 hr. Shown are representative images (d) and quantification (e) of the proportion of EdU+ proliferating cells in total SOX2+ NPCs in both control and FXS-derived forebrain organoids at day 56. Data are presented as mean ± s.e.m. (n = 62 cortical structures from at least ten organoids each condition; ****P < 0.0001, one-way ANOVA). Scale bars: 50 μm.
Extended Data Fig. 2. Loss of FMRP impairs cortical neurodevelopment.
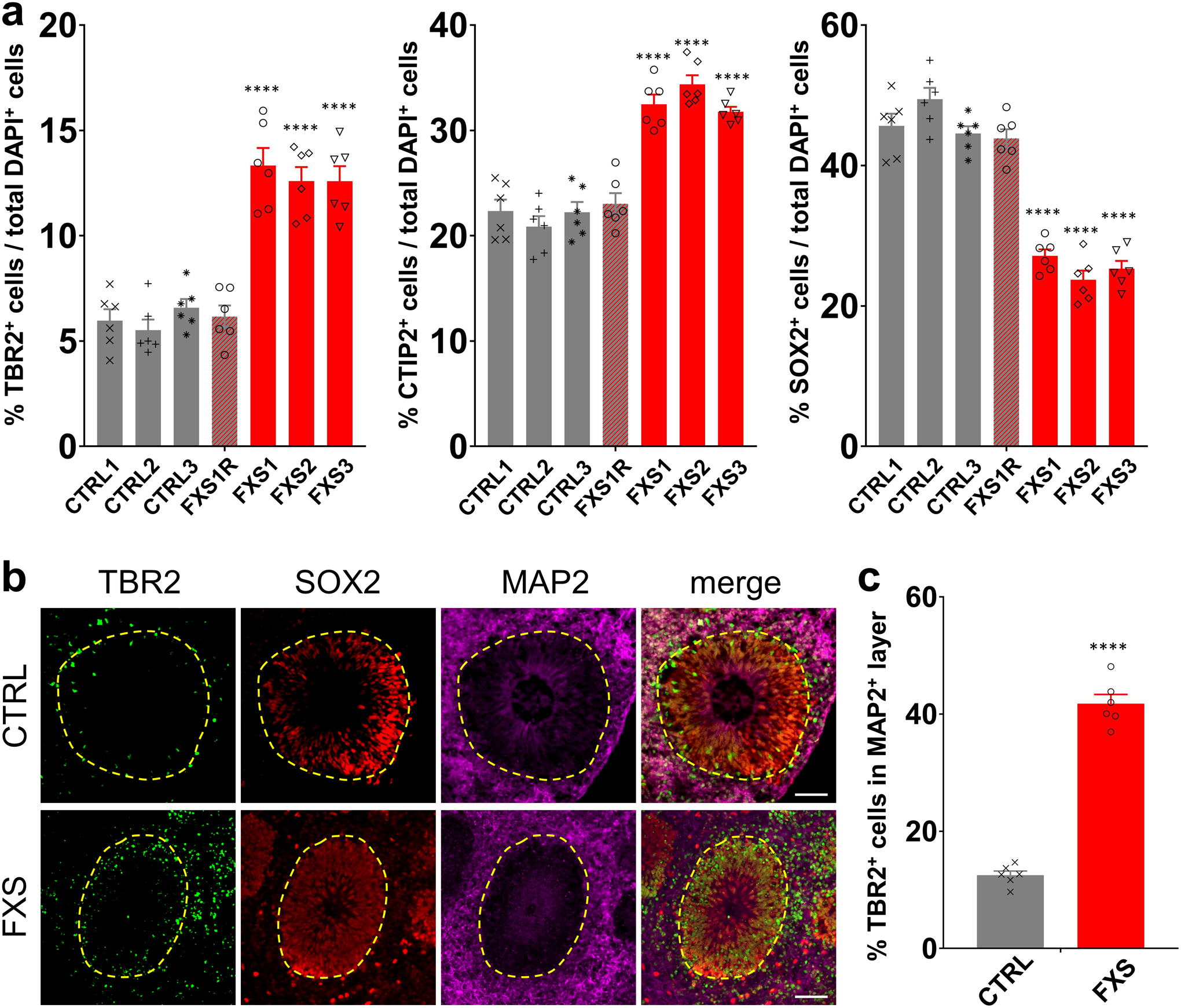
(a) Quantification of the proportions of TBR2+ IPCs, CTIP2+ cortical neurons, and SOX2+ NPCs in total DAPI+ cells in control and FXS-derived forebrain organoids at day 56. Data are presented as mean ± s.e.m. (n = 6 organoids from each line with 15–20 cortical structures analyzed per organoid; ****P < 0.0001, one-way ANOVA). (b-c) Loss of FMRP dysregulates distribution of TBR2+ intermediate neural progenitor cells. Shown are representative images (b) and quantification (c) of the proportion of TBR2+ IPCs in MAP2+ layer of both control and FXS-derived forebrain organoids. Yellow dashed lines indicate the borders of VZ-like structures. Data are presented as mean ± s.e.m. (n = 6 organoids from each condition with 15–20 cortical structures analyzed per organoid; ****P < 0.0001, one-way ANOVA). Scale bars: 50 μm.
Extended Data Fig. 3. Loss of FMRP accelerates cortical layer formation.
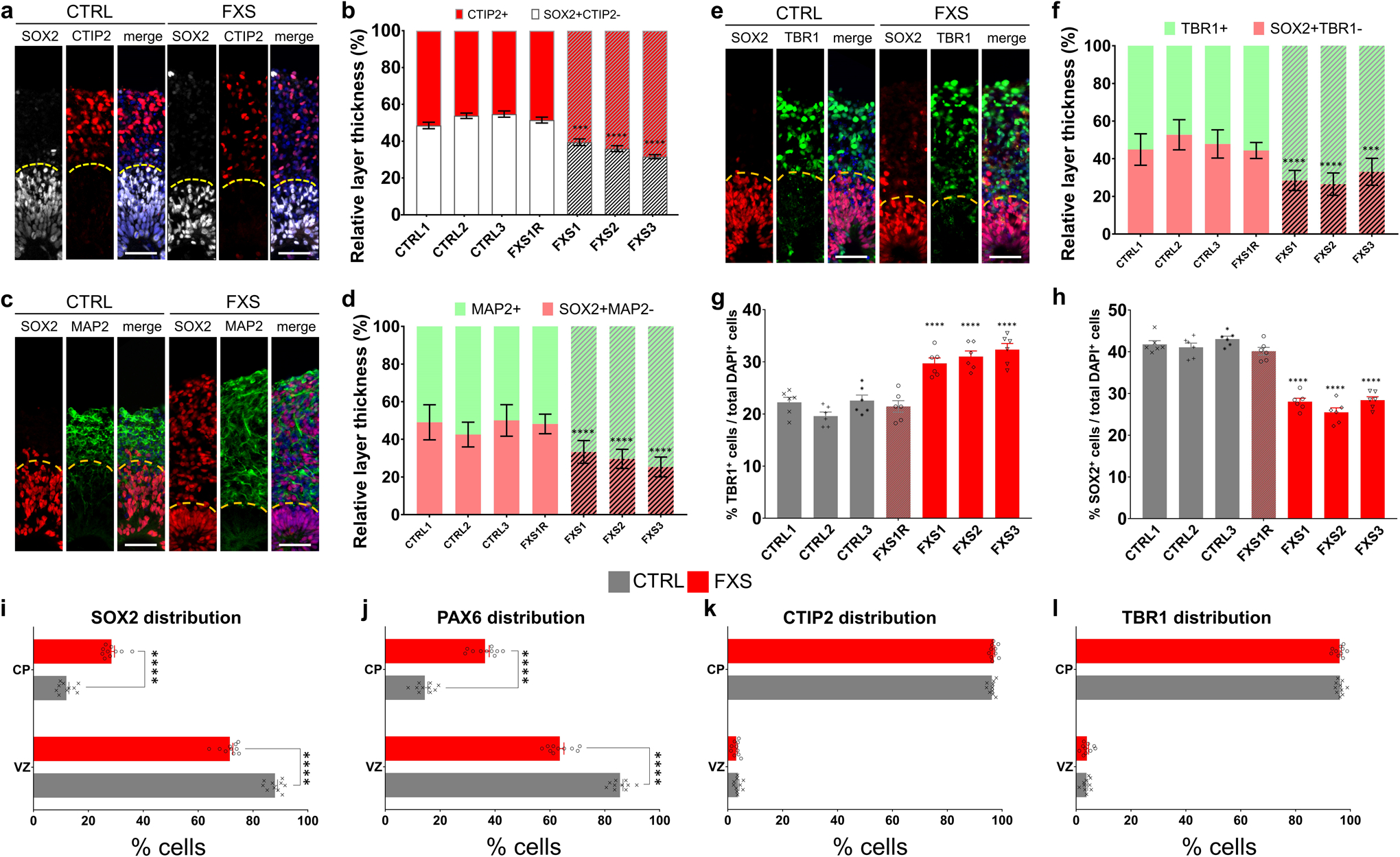
(a-b) Shown are sample images (a) and quantification (b) of relative thickness of SOX2+CTIP2−VZ layer and CTIP2+ CP layer in day 56 forebrain organoids. Yellow dashed lines indicate the borders between VZ and CP layers. Data are presented as mean ± s.e.m. (n = 15 cortical structures per organoid from at least 12 organoids each line; ***P = 0.0005, ****P < 0.0001, one-way ANOVA). Scale bars: 50 μm. (c-d) Shown are sample images (c) and quantification (d) of relative thickness of SOX2+MAP2− VZ layer and MAP2+ CP layer in day 56 forebrain organoids. Yellow dashed lines indicate the borders between VZ and CP layers. Data are presented as mean ± s.e.m. (n = 15 cortical structures per organoid from at least 12 organoids each line; ****P < 0.0001, two-way ANOVA). Scale bars: 50 μm. (e-f) Shown are sample images (e) and quantification (f) of relative thickness of SOX2+TBR1− VZ layer and TBR1+ CP layer in day 56 forebrain organoids. Yellow dashed lines indicate the borders between VZ and CP layers. Data are presented as mean ± s.e.m. (n = 15 cortical structures per organoid from at least 12 organoids each line; ***P = 0.0009, ****P < 0.0001, one-way ANOVA). Scale bars: 50 μm. (g-h) Quantification of the proportions of TBR1+ cortical neurons (c) and SOX2+ NPCs (d) in total DAPI+ cells in control and FXS-derived forebrain organoids at day 56. Data are presented as mean ± s.e.m. (n = 6 organoids from each line with 15–20 cortical structures analyzed per organoid; ****P < 0.0001, one-way ANOVA). (i-l) Analysis of marker distribution across the VZ/CP layers. Data are presented as mean ± s.e.m. (n = 10 organoids from control or FXS lines each with 15–20 cortical structures analyzed per organoid; ****p < 0.0001; one-way ANOVA).
Extended Data Fig. 4. Loss of FMRP prevents differentiation of GABAergic interneurons.
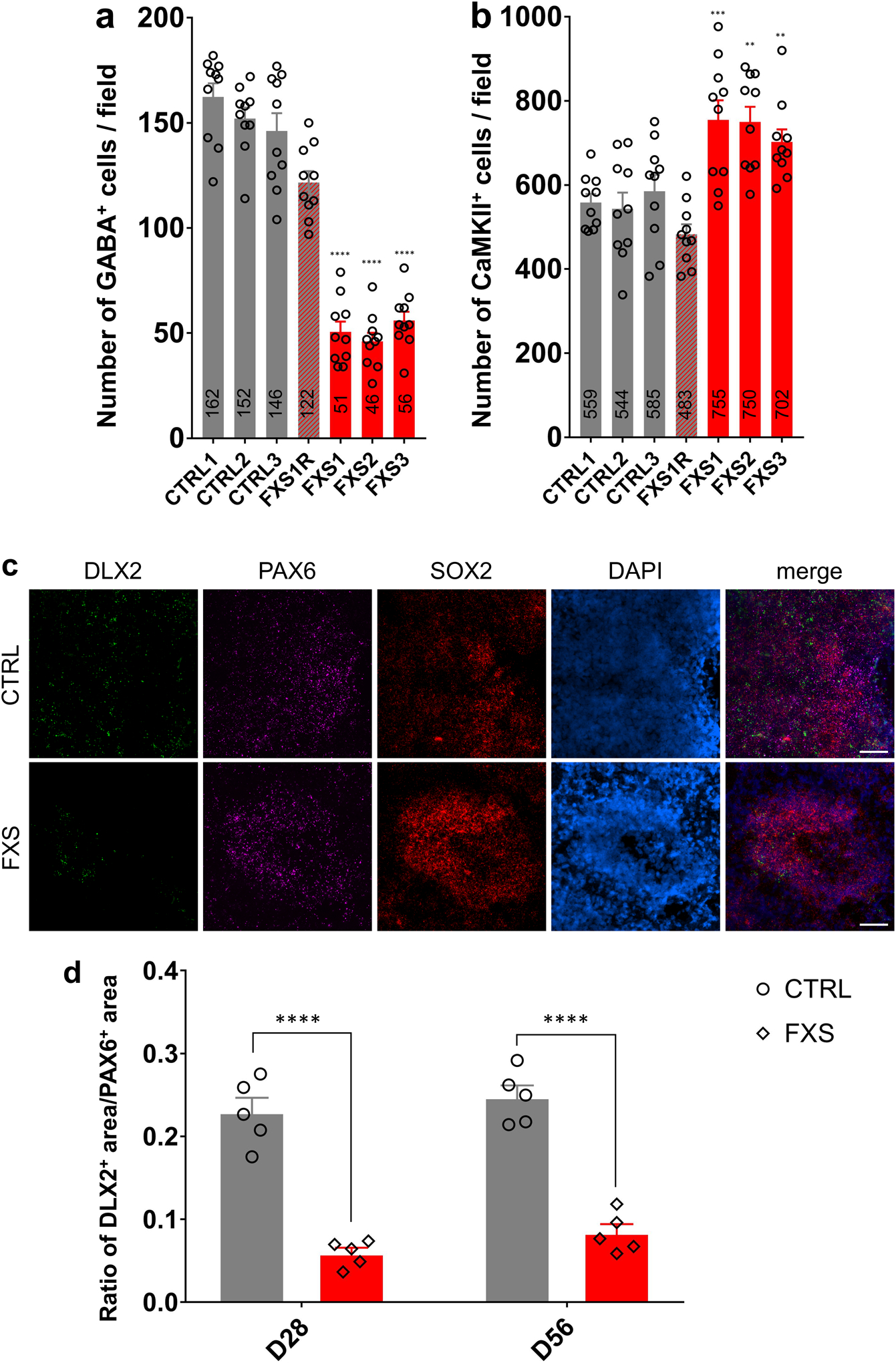
(a-b) Quantifications of the numbers of GABA+ inhibitory neurons (a) and CaMKIIα+ excitatory neurons (b) in a field of 588 μm X 588 μm in both control and FXS-derived forebrain organoids. Data are presented as mean ± s.e.m. (n = 10 sections from 10 organoids each line; **p = 0.0012 (FXS2 v.s. CTRL1 in b) or 0.0097 (FXS3 v.s. CTRL1 in b), ***p = 0.0008 (b), ****P < 0.0001, one-way ANOVA). (c) Sample images of RNA expression of DLX2, PAX6 and SOX2 by RNAscope in control and FXS forebrain organoids at day 56. Blue staining represents DAPI. Scale bars: 50 μm. (d) Quantification of ratio of DLX2+ MGE-like NPC area v.s. PAX6+ dorsal forebrain NPC area in D28 and D56 control and FXS-derived forebrain organoids. Data are presented as mean ± s.e.m. (n = 5 organoids from each condition with 15–20 sections analyzed per organoid; ****P < 0.0001, one-way ANOVA).
Extended Data Fig. 5. Basic electrophysiological characterization of FXS neurons in forebrain organoids.
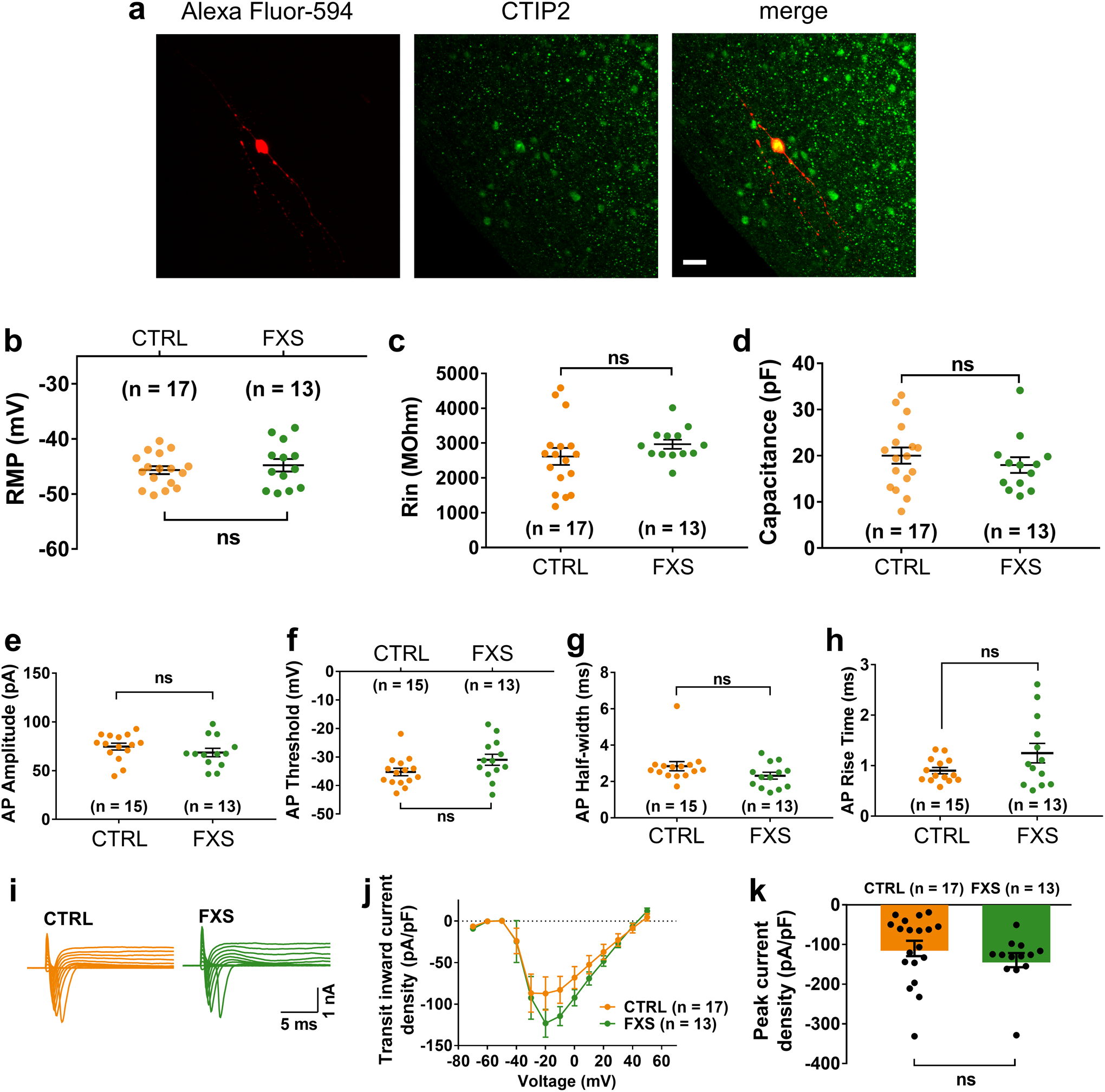
(a) Shown are sample images of a CTIP2+ cortical neurons that was filled with Alexa Fluor-594 dye after the electrophysiological recording. Scale bars: 20 μm. Experiment was repeated at least 13 times independently for each condition with similar results. (b-d) Characterization of passive membrane properties, including the resting membrane potential (RMP; b), input resistance (RIN; c), and membrane capacitance (d). Data are presented as mean ± s.e.m. (two-tailed unpaired t test or one-way ANOVA). (e-h) Basic properties of action potentials, including the amplitude (e), threshold (f), half-width (g), and the rise time (h) of the first action potentials. Data are presented as mean ± s.e.m. (two-tailed unpaired t test or one-way ANOVA). (i-k) Characterization of transient inward currents and sustained outward currents of FXS neurons. Shown are sample tracings of transient inward and sustained outward currents (i), quantification of transient inward current-voltage curve (j) and peak density of transient inward currents (k). Data are presented as mean ± s.e.m. (two-tailed unpaired t test or one-way ANOVA). Cell number (n) recorded and analyzed in each condition is indicated.
Extended Data Fig. 6. Expression of Kv4.2 voltage-gated potassium channel in human forebrain organoids.
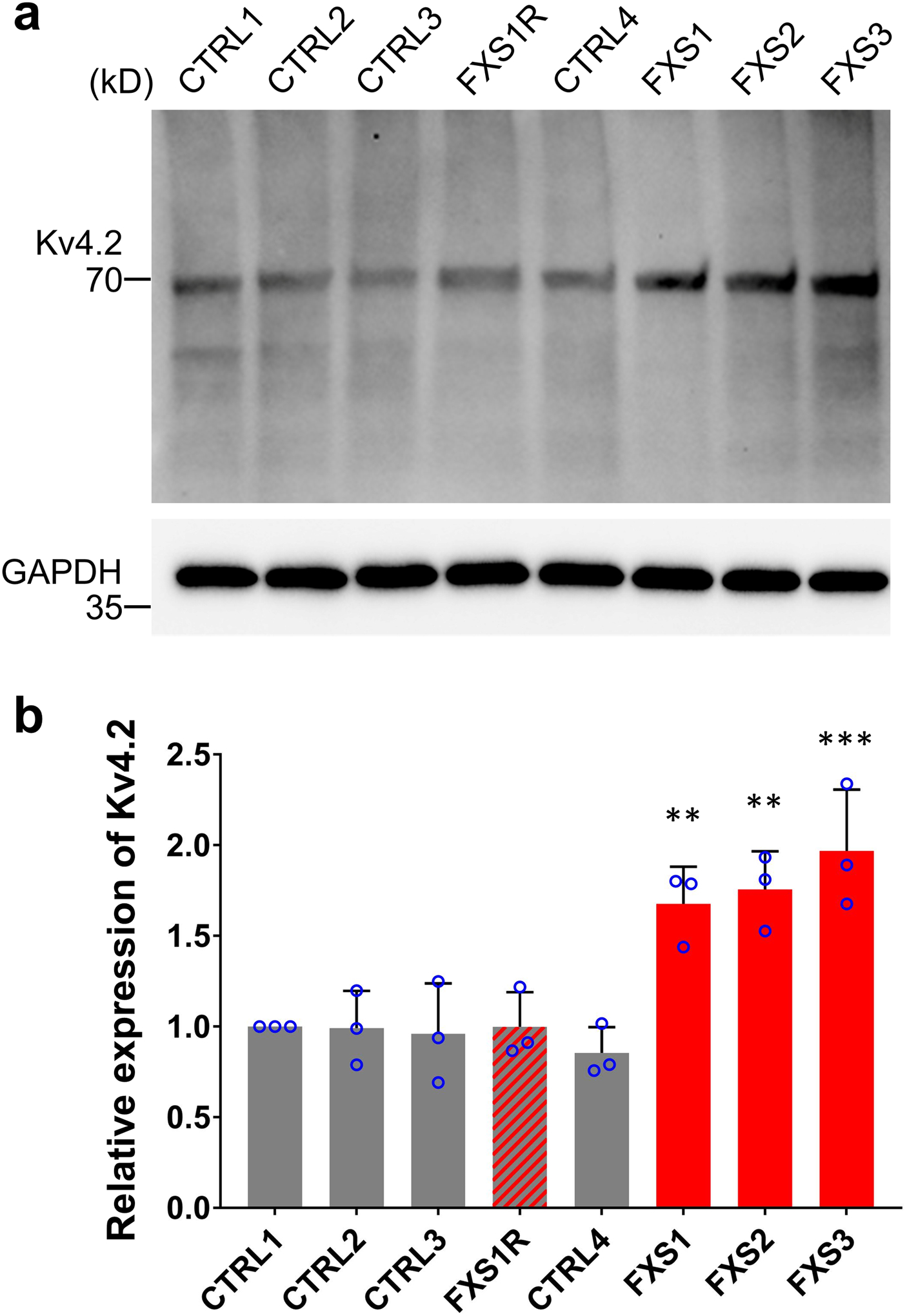
Sample images (a) and quantification (b) of Western blots are presented for comparing Kv4.2 protein level in D56 control and FXS forebrain organoids using GAPDH as loading control. Data are presented as mean ± s.d. (n = 3 cultures; **P = 0.0085 (FXS1 v.s. CTRL1) or 0.0033 (FXS2 v.s. CTRL1), ***P = 0.0003, one-way ANOVA).
Extended Data Fig. 7. The PANTHER overrepresentation test on the upregulated genes in FXS organoid at each stage show enrichment in distinct pathways.
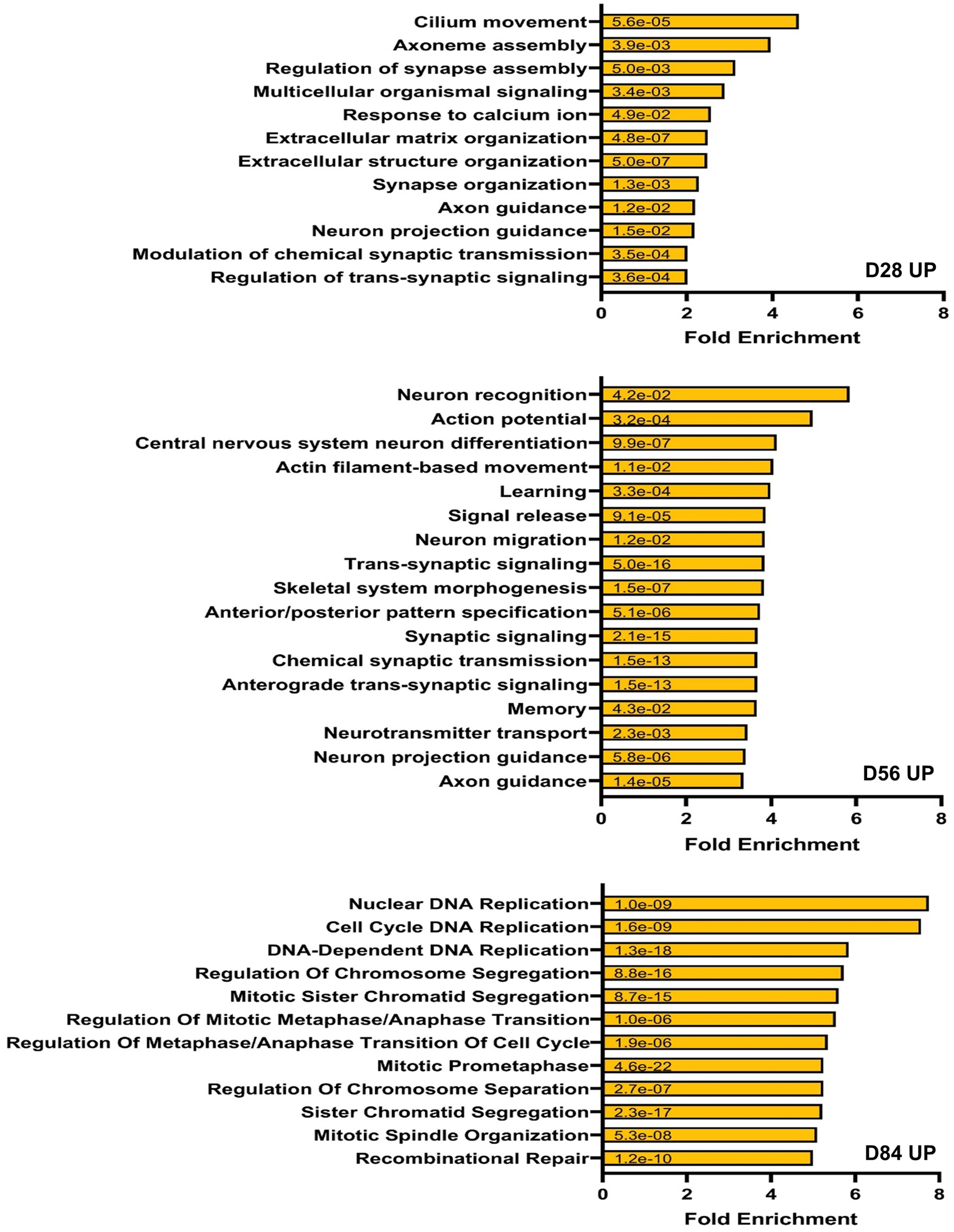
The upregulated genes in FXS at a given developmental stage, D28, D56, or D84 show specific pathway enrichment. The upregulated genes at D28 in FXS organoids are enriched in ciliary locomotion of neuron, axoneme assembly, and other synaptogenesis related pathways while the up-regulated genes at D56 in FXS organoids show more relevance to the pathways associated with synaptic function. Interestingly, genes with higher expression in FXS than in control organoids at the more developed D84 are concentrated in DNA replication, cell division and cell cycle pathways. This suggests aberrant developmental manifestation in FXS organoids. The numbers on the bars indicate the two-sided p values by Fisher’s exact test. The p values have been adjusted for multiple testing using Bonferroni correction.
Extended Data Fig. 8. Lack of FMRP causes altered neural differentiation and aberrant developmental trajectory in forebrain organoids.
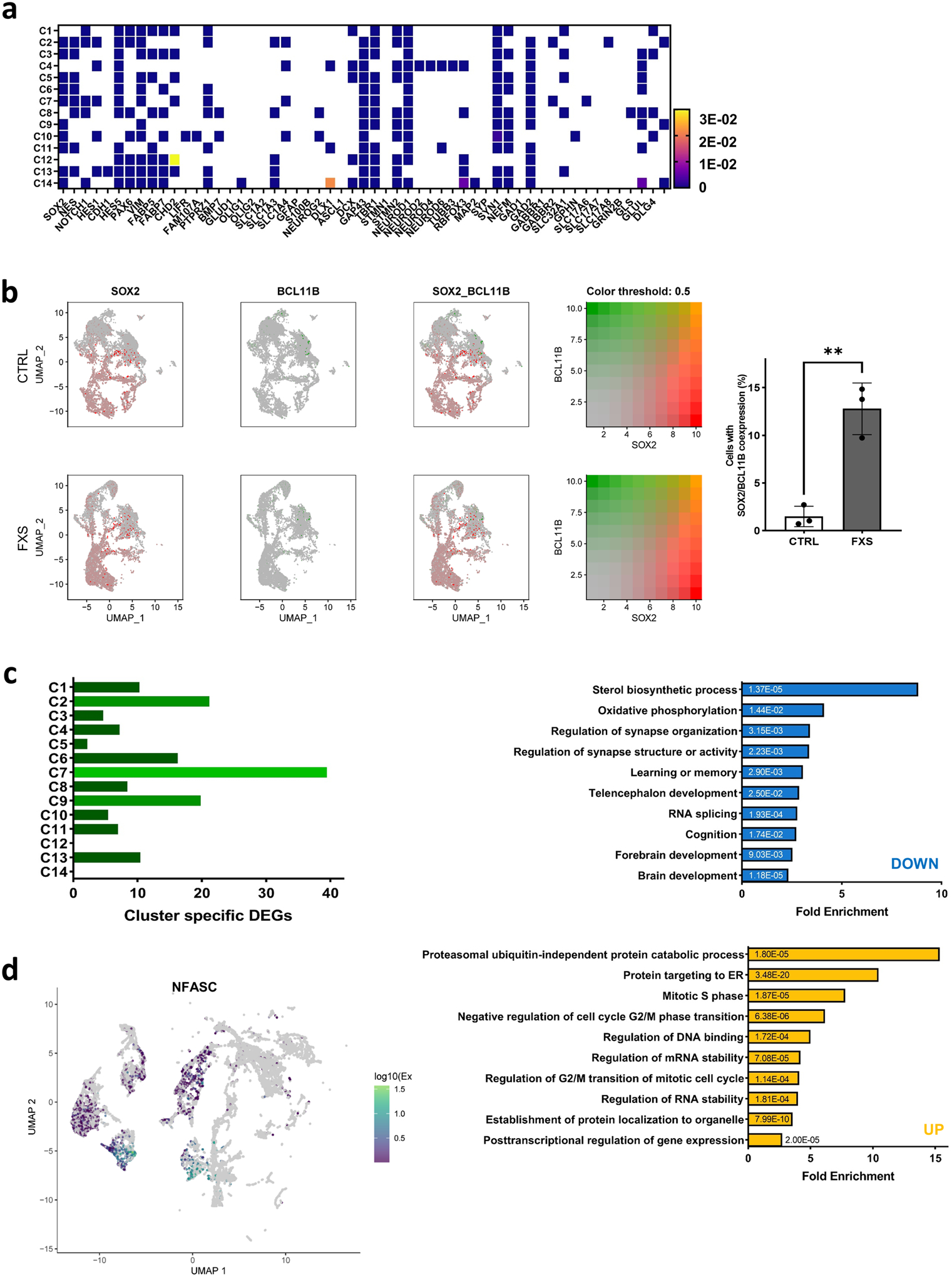
(a) A heat map of expression of annotation reference genes in 14 cell type specific clusters present during human forebrain organoids shows the differential expression of various marker genes for specific cell types in each cluster. (C1: fate determining stage neurons toward excitatory neuron, C2: excitatory neuron, C3: neural stem cell /radial glia2, C4: immature neuron, C5: neural stem cell /radial glia1 cell, C6: glial progenitor, C7: inhibitory neuron, C8: astrocyte, C9: radial glia, C10: astrocyte, C11: immature neuron very early stage, C12: oligodendrocyte, C13: ectodermal origin non-neuronal cells, C14: non-neuronal cells) (b) The expression of neural stem cell/progenitor marker, SOX2 (red) and differentiated cortical plate neuron marker, BCL11B (CTIP2, green) were presented simultaneously in the UMAP plot. Compared to control, cells in FXS organoids expressing BCL11B/CTIP2 at low level were increased and widely distributed spanning various cell types regardless of differentiation status and cell function. Many of these are accompanied by the expression of SOX2. Significantly high co-expression rate of the NPC marker, SOX2, and cortical plate marker, BCL11B in the C7, young inhibitory neuron cluster (19% in FXS forebrain organoids compared to 0% in control forebrain organoids), suggest that the spatiotemporal regulation of SOX2 and BCL11B expression critical for proper specification and lamination of neurons is severely perturbed in FXS organoids. Data are presented as mean ± s.e.m. (n=3 single cell RNAseq of 3 independent culture sets, **P=0.0025, two-tailed unpaired t test) (c) Among the 14 clusters, the highest number of DEGs were detected in the young inhibitory neuron cluster, C7. PANTHER analyses show high relevance to regulation of synapse organization, learning and memory, and forebrain development with down-regulated DEGs and protein targeting. mRNA stability and regulation of cell cycle. Yellow represents up-regulated genes and blue represents down-regulated genes. The numbers on the bars are the associated two-sided p-values by Fisher’s Exact test. The p values have been adjusted for multiple testing using Bonferroni correction. (d) Transcriptional features of the cluster 6 at the developmental break point between FXS and control (arrow in red) in the time trajectory was assessed. The Monocle cluster 4, one of the major break point in the time trajectory, has marker genes associated with cell proliferation and regulation of DNA methylation, (e.g., KMT2A), neuron migration and regulation of neuron projection development (ACAP3), synapse organization and axon guidance (NFASC).
Extended Data Fig. 9. The overlap between human fetal brain DEGs and cell type specific DEGs.
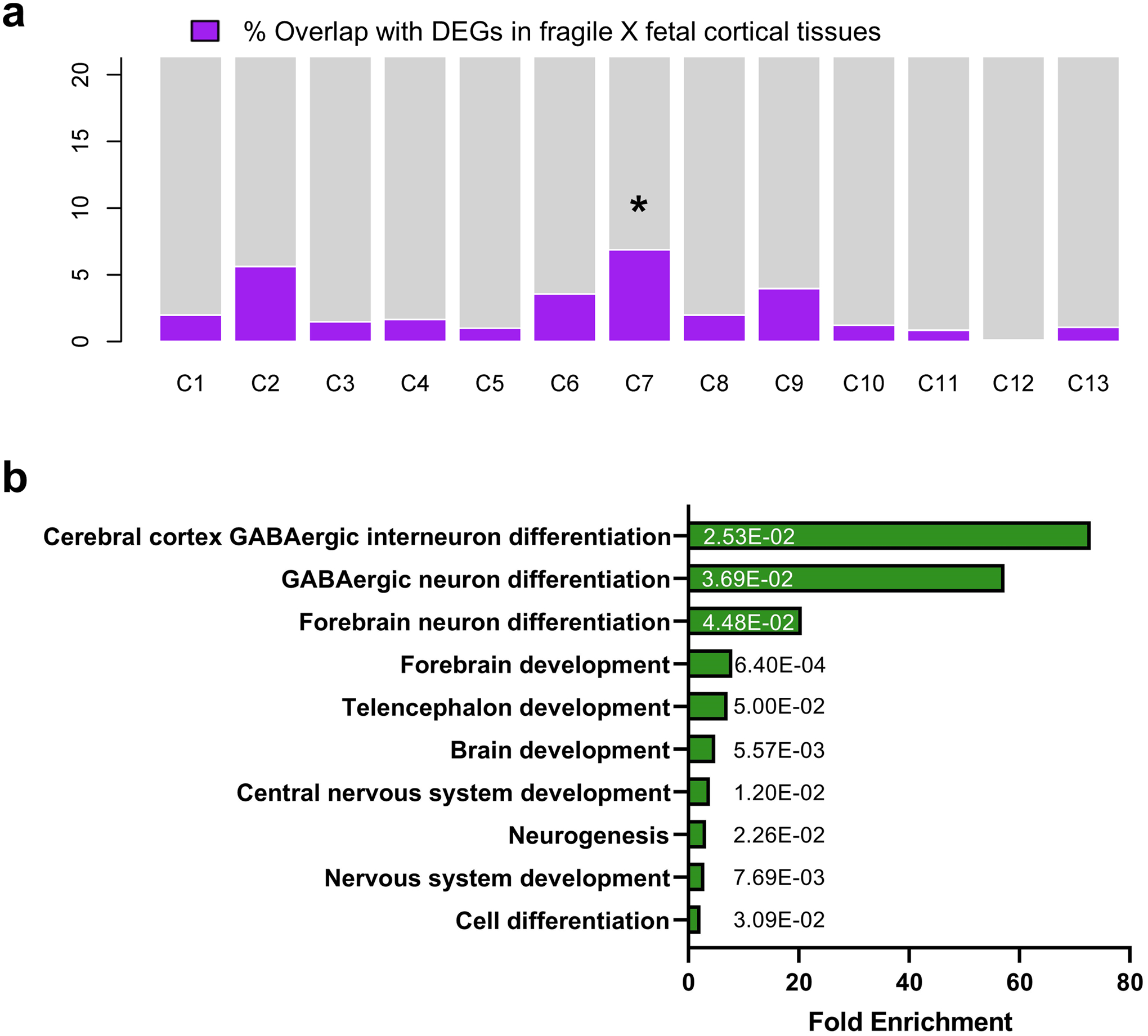
(a) all single cell cluster specific DEGs were compared with human fragile X fetal brain RNAseq DEGs. The highest overlap is marked with an asterisk above the bar. (b) PANTHER gene ontology revealed that theyare involved in GABAergic neuron differentiation, forebrain neuron generation and differentiation. Downregulated genes are enriched in regulation of neural precursor cell, neurogenesis and proliferation, cerebral cortex and forebrain development, gliogenesis, and cell differentiation. The numbers on the bars are two-sdied p-values by Fisher’s exact test. The p values have been adjusted for multiple testing using Bonferroni correction.
Extended Data Fig. 10. An overlap between disease risk genes and the subset of human and mouse FMRP binding genes are shown.
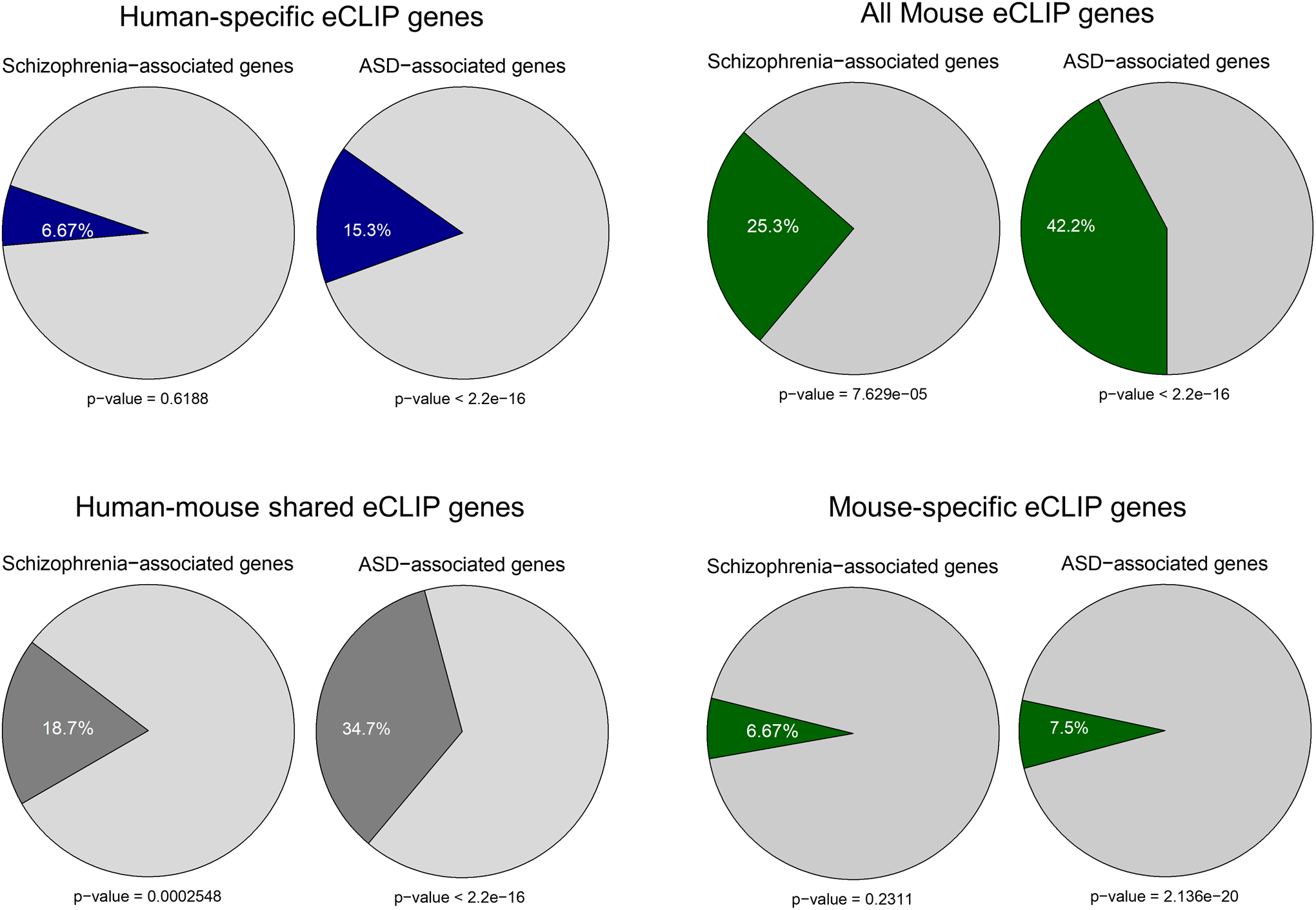
The percentage of overlap between Schizophrenia and ASD risk genes and human-specific, mouse-specific or human-mouse shared FMRP binding genes are indicated. Statistical significance was calculated by Pearson’s χ2 tests, and p-values are indicated.
Supplementary Material
{kind=link}
ACKNOWLEDGEMENTS
This work is dedicated to late Steve Warren, and was supported in part by National Institutes of Health (NS091859 to S.T.W. and P.J., HD104458 to S.T.W., P.J., G.B., and Z.W., HD082013 to G.B., AI131130 to Z.W. and P.J., MH123711 and MH121102 to Z.W., NS051630 and NS111602 to P.J.), Department of Defense (W81XWH1910068 to E.G.A., W81XWH1910353 to Z.W.), Edward Mallinckrodt, Jr. Foundation (Z.W.). and FRAXA Research Foundation (Y.K.). We would like to thank Dr. Steven Sloan at Emory University for the help with scRNA-seq analyses. This study was supported in part by the Emory Integrated Genomics Core (EIGC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. Additional support was provided by the Georgia Clinical & Translational Science Alliance of the National Institutes of Health under Award Number UL1TR002378. This work was performed with the support of the Georgia Genomics and Bioinformatics Core at the University of Georgia. The scRNAseq work was performed at the Georgia Genomics and Bioinformatics Core (GGBC) at the University of Georgia, Athens. We thank Dr. Magdy Alabady and his team at GGBC for their support and contribution to this work.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing interests.
Code Availability
RNA-seq data were analyzed following the standard pipeline with software STAR 2.7 (https://hbctraining.github.io/Intro-to-rnaseq-hpc-O2/lessons/03_alignment.html) and DESeq2 (https://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html). CLIP-seq data were analyzed following the pipeline with software STAR 2.7 and CLIPper (https://github.com/YeoLab/clipper). Single cell RNA-seq data were anlyzed following the pipeline with software CellRanger 3.0.2 (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/tutorial_ov), Seurat 3.1 (https://satijalab.org/seurat/articles/pbmc3k_tutorial.html), Monocle 3 (https://cole-trapnell-lab.github.io/monocle3/docs/starting/). Analysis code is available upon reasonable request.
Data Availability
We have deposited the scRNA-seq, RNA-seq, eCLIP-seq data into the Gene Expression Omnibus (GEO) at https://www.ncbi.nlm.nih.gov/geo/. The accession number is GSE146878.
REFERENCES
- 1.Santoro MR, Bray SM & Warren ST Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annual review of pathology 7, 219–245 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Richter JD, Bassell GJ & Klann E Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat Rev Neurosci 16, 595–605 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson DL, Orr HT & Warren ST The unstable repeats--three evolving faces of neurological disease. Neuron 77, 825–843 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hagerman RJ, et al. Fragile X syndrome. Nat Rev Dis Primers 3, 17065 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Lancaster MA, et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasca AM, et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods 12, 671–678 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qian X, et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 165, 1238–1254 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie N, et al. Reactivation of FMR1 by CRISPR/Cas9-Mediated Deletion of the Expanded CGG-Repeat of the Fragile X Chromosome. PLoS One 11, e0165499 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kostovic I, Sedmak G & Judas M Neural histology and neurogenesis of the human fetal and infant brain. Neuroimage 188, 743–773 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Agulhon C, et al. Expression of FMR1, FXR1, and FXR2 genes in human prenatal tissues. J Neuropathol Exp Neurol 58, 867–880 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Tamanini F, et al. Differential expression of FMR1, FXR1 and FXR2 proteins in human brain and testis. Hum Mol Genet 6, 1315–1322 (1997). [DOI] [PubMed] [Google Scholar]
- 12.Raj N, et al. Cell-type-specific profiling of human cellular models of fragile X syndrome reveal PI3K-dependent defects in translation and neurogenesis. Cell reports 35, 108991 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoon KJ, et al. Temporal Control of Mammalian Cortical Neurogenesis by m(6)A Methylation. Cell 171, 877–889 e817 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JY, et al. DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron 63, 761–773 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gotz M & Huttner WB The cell biology of neurogenesis. Nat Rev Mol Cell Biol 6, 777–788 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Gleeson JG, et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92, 63–72 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Qian X, et al. Sliced Human Cortical Organoids for Modeling Distinct Cortical Layer Formation. Cell Stem Cell 26, 766–781 e769 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clancy B, et al. Web-based method for translating neurodevelopment from laboratory species to humans. Neuroinformatics 5, 79–94 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Darmanis S, et al. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci U S A 112, 7285–7290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsunekawa Y, et al. Cyclin D2 in the basal process of neural progenitors is linked to non-equivalent cell fates. EMBO J 31, 1879–1892 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qiu X, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 14, 979–982 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trapnell C, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huber KM, Gallagher SM, Warren ST & Bear MF Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A 99, 7746–7750 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bear MF, Huber KM & Warren ST The mGluR theory of fragile X mental retardation. Trends Neurosci 27, 370–377 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Michalon A, et al. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dolen G, et al. Correction of fragile X syndrome in mice. Neuron 56, 955–962 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Youssef EA, et al. Effect of the mGluR5-NAM Basimglurant on Behavior in Adolescents and Adults with Fragile X Syndrome in a Randomized, Double-Blind, Placebo-Controlled Trial: FragXis Phase 2 Results. Neuropsychopharmacology 43, 503–512 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gross C, et al. Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in fragile x syndrome. Cell Reports 11, 1–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gross C, et al. Selective Role of the Catalytic PI3K Subunit p110β in Impaired Higher Order Cognition in Fragile X Syndrome. Cell reports 11, 681–688 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gross C, et al. Isoform-selective phosphoinositide 3-kinase inhibition ameliorates a broad range of fragile X syndrome-associated deficits in a mouse model. Neuropsychopharmacology 44, 324–333 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Nostrand EL, et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods 13, 508–514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Darnell JC, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basu SN, Kollu R & Banerjee-Basu S AutDB: a gene reference resource for autism research. Nucleic Acids Res 37, D832–836 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jia P, Sun J, Guo AY & Zhao Z SZGR: a comprehensive schizophrenia gene resource. Mol Psychiatry 15, 453–462 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hyde CL, et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet 48, 1031–1036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Flint J & Eskin E Genome-wide association studies in mice. Nat Rev Genet 13, 807–817 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tajul-Arifin K, et al. Identification and analysis of chromodomain-containing proteins encoded in the mouse transcriptome. Genome Res 13, 1416–1429 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chenier S, et al. CHD2 haploinsufficiency is associated with developmental delay, intellectual disability, epilepsy and neurobehavioural problems. J Neurodev Disord 6, 9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim YJ, et al. Chd2 Is Necessary for Neural Circuit Development and Long-Term Memory. Neuron 100, 1180–1193 e1186 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu XS, et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 172, 979–992 e976 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Graef JD, et al. Partial FMRP expression is sufficient to normalize neuronal hyperactivity in Fragile X neurons. Eur J Neurosci 51, 2143–2157 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Telias M, Segal M & Ben-Yosef D Neural differentiation of Fragile X human Embryonic Stem Cells reveals abnormal patterns of development despite successful neurogenesis. Dev Biol 374, 32–45 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Doers ME, et al. iPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev 23, 1777–1787 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Halevy T, Czech C & Benvenisty N Molecular mechanisms regulating the defects in fragile X syndrome neurons derived from human pluripotent stem cells. Stem cell reports 4, 37–46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lancaster MA & Knoblich JA Organogenesis in a dish: modeling development and disease using organoid technologies. Science 345, 1247125 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Sasai Y Next-generation regenerative medicine: organogenesis from stem cells in 3D culture. Cell stem cell 12, 520–530 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Gonzalez C, et al. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol Psychiatry (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin YT, et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 98, 1141–1154 e1147 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seo J, et al. Inhibition of p25/Cdk5 Attenuates Tauopathy in Mouse and iPSC Models of Frontotemporal Dementia. J Neurosci 37, 9917–9924 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Raja WK, et al. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS One 11, e0161969 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee HK, et al. Three Dimensional Human Neuro-Spheroid Model of Alzheimer’s Disease Based on Differentiated Induced Pluripotent Stem Cells. PLoS One 11, e0163072 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shang L, et al. beta-cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes 63, 923–933 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Teo AK, et al. Early Developmental Perturbations in a Human Stem Cell Model of MODY5/HNF1B Pancreatic Hypoplasia. Stem Cell Reports 6, 357–367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nishita M, et al. Ror2 signaling regulates Golgi structure and transport through IFT20 for tumor invasiveness. Sci Rep 7, 1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hosokawa Y, et al. Insulin-producing cells derived from ‘induced pluripotent stem cells’ of patients with fulminant type 1 diabetes: Vulnerability to cytokine insults and increased expression of apoptosis-related genes. J Diabetes Investig (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zeng H, et al. An Isogenic Human ESC Platform for Functional Evaluation of Genome-wide-Association-Study-Identified Diabetes Genes and Drug Discovery. Cell stem cell 19, 326–340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bhattacharya A, et al. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron 76, 325–337 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bhattacharya A, et al. Targeting Translation Control with p70 S6 Kinase 1 Inhibitors to Reverse Phenotypes in Fragile X Syndrome Mice. Neuropsychopharmacology (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gross C & Bassell GJ Excess protein synthesis in FXS patient lymphoblastoid cells can be rescued with a p110beta-selective inhibitor. Mol Med 18, 336–345 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gross C, et al. Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J Neurosci 30, 10624–10638 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoeffer CA, et al. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav 11, 332–341 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kumari D, et al. Identification of Fragile X Syndrome-Specific Molecular Markers in Human Fibroblasts: A Useful Model to Test the Efficacy of Therapeutic Drugs. Human mutation (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sharma A, et al. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci 30, 694–702 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dockendorff TC & Labrador M The Fragile X Protein and Genome Function. Mol Neurobiol 56, 711–721 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Bardoni B, Mandel JL & Fisch GS FMR1 gene and fragile X syndrome. Am J Med Genet 97, 153–163 (2000). [DOI] [PubMed] [Google Scholar]
- 67.Castren ML Cortical neurogenesis in fragile X syndrome. Front Biosci (Schol Ed) 8, 160–168 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Willemsen R, Bontekoe CJ, Severijnen LA & Oostra BA Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum Genet 110, 601–605 (2002). [DOI] [PubMed] [Google Scholar]
- 69.Zhang F, et al. Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum Mol Genet 27, 3936–3950 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sourial M & Doering LC Abnormal neural precursor cell regulation in the early postnatal Fragile X mouse hippocampus. Brain Res 1666, 58–69 (2017). [DOI] [PubMed] [Google Scholar]
- 71.Tervonen TA, et al. Aberrant differentiation of glutamatergic cells in neocortex of mouse model for fragile X syndrome. Neurobiol Dis 33, 250–259 (2009). [DOI] [PubMed] [Google Scholar]
- 72.Castren M, et al. Altered differentiation of neural stem cells in fragile X syndrome. Proc Natl Acad Sci U S A 102, 17834–17839 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khalfallah O, et al. Depletion of the Fragile X Mental Retardation Protein in Embryonic Stem Cells Alters the Kinetics of Neurogenesis. Stem Cells 35, 374–385 (2017). [DOI] [PubMed] [Google Scholar]
- 74.Luo Y, et al. Fragile x mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet 6, e1000898 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mariani J, et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 162, 375–390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Birey F, et al. Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wen Z, et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 515, 414–418 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xiang Y, et al. Fusion of Regionally Specified hPSC-Derived Organoids Models Human Brain Development and Interneuron Migration. Cell stem cell 21, 383–398 e387 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xiang Y, et al. hESC-Derived Thalamic Organoids Form Reciprocal Projections When Fused with Cortical Organoids. Cell stem cell 24, 487–497 e487 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lovci MT, et al. Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat Struct Mol Biol 20, 1434–1442 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Warde-Farley D, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 38, W214–220 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We have deposited the scRNA-seq, RNA-seq, eCLIP-seq data into the Gene Expression Omnibus (GEO) at https://www.ncbi.nlm.nih.gov/geo/. The accession number is GSE146878.