

Abstract
Objective
The goal of this article is to discuss the importance of differentiating hereditary angioedema (HAE) from other types of angioedema, describe advances in HAE management, especially long-term prophylaxis (LTP), and offer practical recommendations for dermatologists.
Commentary
While HAE is rare, dermatologists are likely to encounter patients with this condition at some point over the course of their clinical practice due to the fact that HAE episodes typically involve subcutaneous swelling and sometimes erythema marginatum. HAE is characterized by recurrent episodes of painful and/or disabling bradykinin-mediated angioedema. Unfortunately, HAE is commonly mistaken for other conditions such as allergic and other mast cell-mediated angioedema, but has very different treatment requirements. Delayed diagnosis of HAE can result in years of avoidable debilitating symptoms, inappropriate treatment, potentially unnecessary invasive intervention, and reduced quality of life, and can be life threatening. Thus, timely identification of HAE is essential to ensure appropriate clinical management. Patients with HAE have either deficiency or dysfunction of the C1 inhibitor (C1INH) protein that inhibits proteases in the contact, complement, and fibrinolytic systems. Pathway-specific HAE treatments include C1INH replacement, kallikrein inhibitors, and bradykinin receptor antagonists. Treatment options for managing acute attacks include C1INH replacement (plasma-derived or recombinant formulations), icatibant (kallikrein inhibitor), and ecallantide (bradykinin B2 receptor antagonist). In the past 5 years, several new options for LTP have been approved, including a subcutaneous plasma-derived C1INH formulation and two kallikrein inhibitors (lanadelumab; berotralstat). Optimal management of HAE entails the creation of a comprehensive management plan that addresses both acute and long-term patient needs and includes input from an HAE expert and the patient/caregivers.
Keywords: C1 inhibitor, Hereditary angioedema, Prophylaxis
Key Summary Points
| Hereditary angioedema (HAE), characterized by recurrent episodes of painful and/or disabling angioedema, is commonly mistaken for other conditions such as allergic (mast cell-mediated) angioedema, but has very different treatment requirements. |
| Although rare, dermatologists are likely to encounter patients with HAE at some point over the course of their clinical practice due to the fact that HAE episodes typically involve subcutaneous swelling and sometimes erythema marginatum. |
| The key distinguishing diagnostic aspects of HAE-related angioedema include an absence of wheals/urticaria; unresponsiveness to typical interventions (e.g., epinephrine, high-dose antihistamines, corticosteroids), and laboratory abnormalities in C1 inhibitor quantity and/or function and C4 level (depending on the subtype of HAE). |
| Multiple disease-specific treatments are available for all HAE management strategies, which may include acute treatment of attacks, short-term prophylaxis prior to potentially triggering events, and routine or long-term prophylaxis to prevent attacks on an ongoing basis. |
Introduction
Although hereditary angioedema (HAE) is a rare disease, dermatologists are likely to encounter patients with HAE at some point over the course of their clinical practice, due to the fact that HAE episodes typically manifest as subcutaneous swelling and are sometimes accompanied by erythema marginatum. As reviewed here, HAE-related angioedema is pathologically different from allergic angioedema and is managed differently; thus, differential diagnosis is critical for timely and appropriate management. A suspicion of HAE can be easily investigated through confirmatory laboratory diagnostics or referral to an expert HAE allergist/immunologist. Delays in HAE diagnosis and/or delays in initiation of effective treatment can result in years of patient suffering, unwarranted medical/surgical procedures, and even a risk of death in the event of a non- or mistreated laryngeal attack [1–3]. Thus, to avoid the negative consequences of diagnostic delay and/or misdiagnosis, it is of utmost importance that HAE be recognized early, differentiated from other causes of angioedema, and managed appropriately [1, 4]. This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors. Written informed consent was obtained from the patients for the publication of their images.
Overview of HAE
Pathophysiology of HAE
HAE is a chronic nonallergic condition, characterized by episodes of non-pitting, non-urticarial, painful and/or disabling subcutaneous or submucosal swelling [5]. HAE-related swelling is mediated by bradykinin, a byproduct of the contact system regulated in large part by C1 inhibitor (C1INH) [6, 7]; this is a distinct pathway from mast cell-mediated allergic angioedema, which is far more common [8]. The main phenotypic variants are HAE type 1 (~ 85% of patients), which is characterized by a quantitative deficiency of C1INH, and HAE type 2 (~ 15% of patients), characterized by normal levels of dysfunctional C1INH (“functional” deficiency) [5, 9]. These most prevalent types of HAE are consequences of mutations in the SERPING1 gene that codes for the C1INH protein. A third and very rare subtype of the disease is HAE with normal C1INH levels; this type appears to result from a number of different genetic mutations that continue to be identified [10], including the gene coding for Factor XII [11].
Figure 1 illustrates that C1INH is an important endogenous protein that inhibits a number of steps along the pathway of bradykinin formation [11]. Quantitative or functional deficiency of C1INH results in dysregulation of these pathways. HAE is inherited in about 75% of cases; however, de novo genetic mutations (i.e., no family history) are responsible in the remaining 25% of cases [13]. Acquired forms of bradykinin-mediated angioedema can be the result of a complement consumptive process (e.g., lymphoproliferative disorder and/or antibodies directed against C1INH) or be drug induced (i.e., caused by angiotensin-converting enzyme inhibitors) [8].
Fig. 1.

Simple schematic comparing the different pathophysiologic pathways underlying A mast cell-mediated (allergic) angioedema and B bradykinin-mediated angioedema, with sites of C1INH regulation [7, 8, 12]. C1INH C1 inhibitor, HMW high molecular weight, IgE Immunoglobulin E, NSAIDs nonsteroidal anti-inflammatory drug. *E.g., tryptase, chymase, heparin, prostaglandins, cysteinyl leukotrienes, and anaphylatoxins
Clinical Characteristics of HAE
HAE typically manifests during childhood or young adulthood, with symptoms often worsening around the time of puberty [9]. Attacks are recurrent and generally unpredictable, with severity and frequency variable between patients and also within individual patients at different stages of life [14]. Common sites of HAE swelling include the face (Fig. 2A), extremities (Fig. 2B), the gastrointestinal tract (abdominal swelling), and genitalia. Attack frequency can range from almost never to multiple attacks per week, with a frequency of 1 to 3 attacks per month being the average in the absence of prophylaxis [9]. Many HAE attacks happen spontaneously, but about half of attacks are triggered by factors such as hormones (e.g., during menstrual cycle, pregnancy, or birth control use), emotional stress, and physical trauma, including medical and dental surgeries and procedures [9, 14]. Some attacks are preceded by prodromal symptoms such as a transient reticular rash [erythema marginatum (Fig. 3)], skin tingling, or vague complaints such as nausea, anxiety, fatigue, or flu-like symptoms [14].
Fig. 2.

Representative photographs of hereditary angioedema attacks affecting A the face (baseline and during) and B hands
Fig. 3.
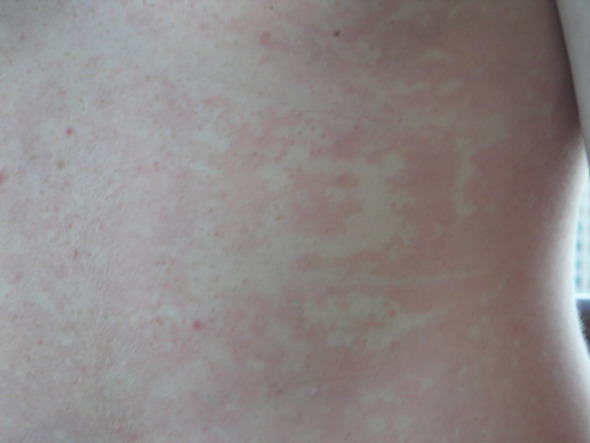
Representative photograph of erythema marginatum, a transient reticular rash that sometimes appears as a prodrome to hereditary angioedema attacks
If untreated, a typical HAE attack escalates gradually over several hours, peaks in intensity between 12 and 36 h, then resolves slowly after 2 to 5 days. Abdominal and laryngeal attacks may have more rapid onset [8, 14]. Abdominal attacks can be excruciatingly painful and can result in unfortunate consequences such as narcotic dependency or unnecessary surgery [9, 15]. Laryngeal attacks are potentially fatal and are the most feared site of involvement [2].
Differentiating HAE from Allergic Angioedema
Given that bradykinin-mediated angioedema is nonallergic and does not involve mast cell activation (Fig. 1), it cannot be managed by standard treatments for allergic angioedema such as antihistamines, corticosteroids, and epinephrine [8]. Thus, differential diagnosis of HAE is crucial (Table 1).
Table 1.
Differential diagnostic components for workup of patients with unexplained recurrent angioedema [16]
| Urticaria | Family history | C1INH, C4 | Responsive to antihistamines | Onset > 40, low C1q | FXII genetic mutation | Diagnosis |
|---|---|---|---|---|---|---|
| Yes | Mast cell-mediated or idiopathic angioedema | |||||
| No | Yes (75% of cases) | Lowa | No | HAE-C1INH | ||
| No | Yes or No | Normalb | Yes | Idiopathic histaminergic angioedema | ||
| No | No | Normalb | No | Idiopathic non-histaminergic angioedema | ||
| No | Yes | Normalb | No | Yes | HAE-FXII | |
| No | Yes | Normalb | No | No | HAE-U | |
| No | No | Lowa | No | Yes | Acquired C1INH deficiency |
C1INH C1 inhibitor, HAE hereditary angioedema, HAE-C1INH hereditary angioedema due to C1 inhibitor deficiency, HAE-FXII hereditary angioedema with F12 mutation, HAE-U hereditary angioedema, unknown
aLow (< 50% of normal) C1INH antigenic levels and/or low (< 50% of normal) functional C1INH activity (low C4 levels and normal antigenic C1INH levels should prompt functional C1INH activity assessment)
bNormal C1INH antigenic levels and C1INH functional activity
A history of recurrent and unexplained cutaneous swelling without wheals/urticaria is strongly suggestive of HAE [1, 5]. Additional factors associated with HAE include family history (although 25% of cases may not) [8], onset in childhood/adolescence, prodromal signs/symptoms preceding the swellings, recurrent and painful abdominal symptoms (due to gastrointestinal swelling), upper airway edema, and lack of response to conventional anti-allergy treatments. Clinically suspected HAE (e.g., individuals with more than one episode of non-urticarial angioedema and with no apparent allergic cause) should promptly be investigated by measuring blood levels of complement C4, C1INH protein, and C1INH function. Low C4 levels are generally ubiquitous and considered diagnostic for HAE types 1 and 2, although in rare cases, C4 levels may be normal in between attacks [17, 18]. Regardless of type 1 or 2, C4 will always be low during an HAE attack [5]. Combined with a low C4 level, low (< 50% of normal) C1INH protein (antigenic) levels suggest a diagnosis of HAE type 1, while low C1INH function in the presence of normal C1INH antigenic levels is indicative of HAE type 2 [1, 5]. As noted above, there are very rare forms of HAE in which C1INH levels are normal.
Treatment of HAE
It is recommended that a detailed and individualized treatment plan be developed for each patient with HAE, ideally in consultation with an HAE specialist. Such plans should factor in access to effective acute (“on-demand”) treatment for attacks, consideration of long-term prophylaxis (LTP) to prevent attacks, and implementation of short-term prophylaxis (STP) prior to situations that carry a high risk of precipitating an attack.
On-Demand Treatments
Acute treatment of HAE attacks is important to limit the morbidity and mortality of the disease. Current US and international HAE clinical guidelines recommend that all patients with HAE should have access to on-demand treatment to be used for acute attacks. Further, all patients should have sufficient medication for treatment of two attacks and should carry on-demand medication at all times, even if using LTP [1, 5].
In the USA, the following HAE pathway-specific on-demand treatments are approved and recommended for the first-line treatment of acute HAE attacks: intravenous (IV) plasma-derived C1INH (BERINERT), IV recombinant C1INH (RUCONEST), the subcutaneous (SC) kallikrein inhibitor ecallantide (KALBITOR), or the SC bradykinin receptor antagonist, icatibant (FIRAZYR) [5]. Early treatment is important to minimize progression of an attack, and each of these therapies can be self-administered by the patient or a caregiver upon attack onset, with the exception of ecallantide, which needs to be administered by a qualified health care provider because of a small risk of anaphylaxis [19]. Onset of effect is usually within about 60 min [5]; in contrast, untreated attack symptoms can persist for days. Patients experiencing attacks involving the airway should seek medical attention even if self-administering on-demand treatment. Current guidelines encourage consideration of treatment of all attacks, regardless of location, as any attack has the potential for affecting quality of life and causing some degree of disability or distress.
Long-Term Prophylaxis
The most recent US guidelines emphasize that the decision to use LTP should “reflect the needs of the individual patient” [5]. Factors for consideration can include frequency and severity of HAE attacks, history of laryngeal attacks, excessive loss of work or school related to HAE, significant anxiety, impaired quality of life, and/or poor disease control with on-demand therapy. Ideally, patients should be evaluated for LTP routinely, and at least annually [1, 20]. In the USA, the following are currently recommended as first-line options for LTP: C1INH replacement with either IV C1INH (CINRYZE) or SC C1INH (HAEGARDA), or kallikrein inhibition with the SC monoclonal antibody lanadelumab-flyo (TAKHZYRO) [5].
Plasma-derived C1INH(IV) (CINRYZE) was the first HAE pathway-specific product approved for routine prophylaxis of HAE and is indicated in patients aged 6 years and older [21]. In the pivotal C1INH(IV) prophylaxis study, which was a crossover design, the average normalized 12-week attack rates were 6.26 attacks during treatment with C1INH(IV) 1000 units every 3–4 days and 12.73 attacks during placebo treatment. The severity and duration of attacks were both lower during C1INH(IV) treatment compared with placebo treatment [22]. While effective in reducing attack frequency, limitations of C1INH(IV) include breakthrough attacks, IV access-related issues, indwelling port-related risks of thrombosis and infection, and inconvenience [23–25].
C1INH(SC) (HAEGARDA) is indicated for the routine prevention of HAE attacks in patients age 6 years and older [26]. In contrast to the C1INH(IV) formulation, which resulted in peaks and troughs of functional C1INH activity below the 40% threshold considered protective against attacks, the SC formulation’s pharmacokinetic profile demonstrated smoother steady-state levels of C1INH activity above the 40% threshold [27, 28]. The pivotal phase 3 study with C1INH(SC) (COMPACT; NCT01912456) [29] included patients ≥ 12 years old who had HAE type 1 or 2 and ≥ 4 attacks in 2 consecutive months over a 3-month period prior to screening. In the crossover-designed study, patients had a significantly lower rate of mean (95% CI) time-normalized attacks per month while using the FDA-approved dose of 60 IU/kg twice weekly (0.52 [0.00, 1.04] attacks/month) than while using placebo (4.03 [3.51, 4.55] attacks/month) (within-patient difference, −3.51 [−4.21, −2.81]; P < 0.001). Attack reductions of ≥ 50%, ≥ 70%, and ≥ 90% versus placebo were demonstrated among 90%, 83%, and 58% of patients, respectively, with no laryngeal attacks reported while using C1INH (25 laryngeal attacks occurred during placebo use). The reductions in HAE attacks with C1INH(SC) were sustained in the 1-year open-label extension study (COMPACT OLE; NCT02316353) with a median annualized attack rate of 1 attack/year and no unexpected safety or tolerability issues [30]. In a post-hoc analysis, it was shown that switching to C1INH(SC) for LTP reduced HAE attacks by approximately 50% among patients who had previously used C1INH(IV) [31].
An SC recombinant, human monoclonal antibody long-acting inhibitor of kallikrein, lanadelumab-flyo (TAKHZYRO), is indicated for prophylaxis to prevent HAE attacks [32, 33]. The phase 3 study (HELP; NCT02586805) included patients ≥ 12 years old who had HAE type 1 or 2 and experienced ≥ 1 attacks/month during run-in [34]. Lanadelumab 300 mg every 2 weeks (FDA-approved dose) reduced the monthly HAE attack rate by 87% versus placebo. The model-based mean number of attacks per month (95% CI) was 1.97 (1.64–2.36) with placebo compared with 0.26 (0.14–0.46) with lanadelumab. In this dose group, attack reductions of ≥ 50%, ≥ 70%, and ≥ 90% from baseline were demonstrated among 100.00%, 88.89%, and 66.67% of patients, respectively. The most commonly reported adverse events in patients receiving lanadelumab that were considered treatment-related included injection site pain (42.86%), upper respiratory tract infection (23.81%), and headache (20.24%). In a long-term open-label extension of the HELP study (mean duration of treatment, 29.6 months), mean HAE attack rate reduction from baseline among patients using lanadelumab was 87.4% overall [35]. Injection site pain was reported in 47.17% of patients, and upper respiratory tract infection was reported in 41.98%.
An oral plasma kallikrein inhibitor, berotralstat (ORLADEYO) was approved in 2020 as prophylaxis to prevent attacks of HAE in adults and pediatric patients aged ≥ 12 years [36]. In the pivotal phase 3 study (APeX-2; NCT03485911) involving 120 patients from the USA and Canada (≥ 12 years of age) or Europe (≥ 18 years of age) with HAE type 1 or 2 and who had two or more clinically relevant attacks during run-in [37], berotralstat 150 mg once daily (FDA-approved dose) significantly reduced attacks versus placebo over a 24-week treatment period (1.31 versus 2.35 attacks/month [P < 0.001]). In this dose group, attack reductions of ≥ 50%, ≥ 70%, and ≥ 90% from baseline were demonstrated among 58%, 50%, and 23% of patients, respectively. The most commonly reported adverse events in patients taking berotralstat 150 mg once daily were upper respiratory tract infection (30.00%), abdominal pain (22.50%), diarrhea (15.00%), nausea (15.00%), and vomiting (15.00%), and, overall, gastrointestinal events were reported in 50% of patients in this dose group.
Prior to the availability of these disease-specific therapies, oral anabolic steroids (e.g., danazol) were widely used for HAE LTP, but are currently recommended only as second-line therapy. These agents have largely fallen out of favor for routine use given concerns over side effects and long-term hepatotoxicity [5, 38].
Short-Term Prophylaxis
Even though many HAE attacks appear spontaneously, certain situations present risks for triggering an attack, specifically physical trauma (including medical and dental procedures), and stressful life events. Current US guidelines recommend a single dose of C1INH(IV) 1 to 12 h prior to the stressor event or a course of anabolic androgens starting 5 to 7 days prior and continuing for 2 to 5 days after the event. Even if STP is given prior to a stressor event, it is critical that a supply of on-demand medication be available [5].
Clinical Practice Recommendations/Call to Action
As reviewed earlier, certain clinical and historical findings should raise suspicion of bradykinin-mediated angioedema and prompt appropriate diagnostic laboratory testing, including failure to respond to high-dose antihistamines/steroids/epinephrine (current or past attacks), absence of urticaria, abdominal symptoms, and laryngeal edema. Appropriate diagnostic laboratory evaluations should be obtained. Enlisting the help of a HAE expert will aid in the diagnosis and development of a treatment plan.
After an HAE diagnosis is confirmed, patient education should include clarification regarding genetics and screening family members, as many patients may not be familiar with the genetic component of HAE. Proactive screening for C1INH function, C1INH protein, and C4 plasma levels should be implemented for family members of patients with HAE type 1 or type 2, including parents and grandparents, as well as for siblings, children, and grandchildren aged > 1 year [1, 5]. Although antigenic levels of C4 and C1INH are quite variable in younger children aged < 1 year, functional testing may have adequate sensitivity/specificity to be considered, if needed.
Pregnancy may be associated with an increased rate of HAE attacks in some patients [39]. Attacks are generally rare during labor and delivery but may develop within 48 h of delivery. C1INH is considered the preferred option in pregnant patients [5] and has been used safely for acute management as well as LTP [39]. There are currently insufficient data to support the use of other HAE pathway-specific agents in pregnant patients. Of note, although anabolic steroids have been used for prophylaxis of HAE, their use is contraindicated in pregnant women owing to the potential for adverse fetal effects, such as virilization [5].
Dermatologists who identify and/or care for patients with HAE, as well as patients (and caregivers for pediatric patients), should reach out to the HAE association (HAEA.ORG) to help connect to local HAE experts. In addition, the HAEA and HAE experts can connect providers and patients to educational resources such as webinars and podcasts. Optimal management of HAE calls for the creation of an individualized and comprehensive management plan that incorporates input from an HAE expert and the patient/caregivers. Managing patients with HAE is truly a team effort that is constantly evolving based on the changing needs of the patient.
Acknowledgements
Funding
Funding for writing and editorial support was provided by CSL Behring; CSL had no role in the development of manuscript content. Any fees associated with publication were funded by CSL Behring.
Medical Writing and Editorial Assistance
Writing and editorial assistance was provided by Kulvinder Singh, PharmD, and Sandra Westra, PharmD, Churchill Communications (Maplewood, NJ), funded by CSL Behring.
Authorship
Michael Manning meets the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, takes responsibility for the integrity of the work as a whole, and has given his approval for this version to be published.
Author Contributions
Michael Manning contributed to the concept/design and the writing and revision of this manuscript.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors. Written informed consent was obtained from the patients for the publication of their images.
Disclosures
Michael Manning is a researcher/PI and speaker/consultant at BioCryst, CSL Behring, Ionis, KalVista, Pharming, and Shire/Takeda.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
- 1.Maurer M, Magerl M, Ansotegui I, et al. The international WAO/EAACI guideline for the management of hereditary angioedema—the 2017 revision and update. Allergy. 2018;73(8):1575–1596. doi: 10.1111/all.13384. [DOI] [PubMed] [Google Scholar]
- 2.Bork K, Hardt J, Witzke G. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1-INH deficiency. J Allergy Clin Immunol. 2012;130(3):692–697. doi: 10.1016/j.jaci.2012.05.055. [DOI] [PubMed] [Google Scholar]
- 3.Hahn J, Hoess A, Friedrich DT, et al. Unnecessary abdominal interventions in patients with hereditary angioedema. J Dtsch Dermatol Ges. 2018;16(12):1443–1449. doi: 10.1111/ddg.13698. [DOI] [PubMed] [Google Scholar]
- 4.Zanichelli A, Longhurst HJ, Maurer M, et al. Misdiagnosis trends in patients with hereditary angioedema from the real-world clinical setting. Ann Allergy Asthma Immunol. 2016;117(4):394–398. doi: 10.1016/j.anai.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 5.Busse PJ, Christiansen SC, Riedl MA, et al. US HAEA Medical Advisory Board 2020 guidelines for the management of hereditary angioedema. J Allergy Clin Immunol Pract. 2021;9(1):132–50.e3. doi: 10.1016/j.jaip.2020.08.046. [DOI] [PubMed] [Google Scholar]
- 6.Fields T, Ghebrehiwet B, Kaplan AP. Kinin formation in hereditary angioedema plasma: evidence against kinin derivation from C2 and in support of "spontaneous" formation of bradykinin. J Allergy Clin Immunol. 1983;72(1):54–60. doi: 10.1016/0091-6749(83)90052-0. [DOI] [PubMed] [Google Scholar]
- 7.Kaplan AP, Joseph K. The bradykinin-forming cascade and its role in hereditary angioedema. Ann Allergy Asthma Immunol. 2010;104(3):193–204. doi: 10.1016/j.anai.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 8.Bernstein JA, Cremonesi P, Hoffmann TK, Hollingsworth J. Angioedema in the emergency department: a practical guide to differential diagnosis and management. Int J Emerg Med. 2017;10(1):15. doi: 10.1186/s12245-017-0141-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernstein JA. HAE update: epidemiology and burden of disease. Allergy Asthma Proc. 2013;34(1):3–6. doi: 10.2500/aap.2013.34.3623. [DOI] [PubMed] [Google Scholar]
- 10.Bork K, Machnig T, Wulff K, Witzke G, Prusty S, Hardt J. Clinical features of genetically characterized types of hereditary angioedema with normal C1 inhibitor: a systematic review of qualitative evidence. Orphanet J Rare Dis. 2020;15(1):289. doi: 10.1186/s13023-020-01570-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zuraw BL, Christiansen SC. HAE pathophysiology and underlying mechanisms. Clin Rev Allergy Immunol. 2016;51(2):216–229. doi: 10.1007/s12016-016-8561-8. [DOI] [PubMed] [Google Scholar]
- 12.Maurer M, Magerl M. Differences and similarities in the mechanisms and clinical expression of bradykinin-mediated vs mast cell-mediated angioedema. Clin Rev Allergy Immunol. 2021;61(1):40–49. doi: 10.1007/s12016-021-08841-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Busse PJ, Christiansen SC. Hereditary angioedema. N Engl J Med. 2020;382(12):1136–1148. doi: 10.1056/NEJMra1808012. [DOI] [PubMed] [Google Scholar]
- 14.Bygum A. Hereditary angio-oedema for dermatologists. Dermatology. 2019;235(4):263–275. doi: 10.1159/000500196. [DOI] [PubMed] [Google Scholar]
- 15.Tilles SA, Borish L, Cohen JP. Management of hereditary angioedema in 2012: scientific and pharmacoeconomic perspectives. Ann Allergy Asthma Immunol. 2013;110(2):70–74. doi: 10.1016/j.anai.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zuraw BL, Christiansen SC. How we manage persons with hereditary angioedema. Br J Haematol. 2016;173(6):831–843. doi: 10.1111/bjh.14059. [DOI] [PubMed] [Google Scholar]
- 17.Karim Y, Griffiths H, Deacock S. Normal complement C4 values do not exclude hereditary angioedema. J Clin Pathol. 2004;57(2):213–214. doi: 10.1136/jcp.2003.12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nzeako UC, Frigas E, Tremaine WJ. Hereditary angioedema: a broad review for clinicians. Arch Intern Med. 2001;161(20):2417–2429. doi: 10.1001/archinte.161.20.2417. [DOI] [PubMed] [Google Scholar]
- 19.KALBITOR® [package insert]. Lexington, MA: Dyax Corp., a Takeda Company; 2020.
- 20.Craig T, Riedl M, Dykewicz MS, et al. When is prophylaxis for hereditary angioedema necessary? Ann Allergy Asthma Immunol. 2009;102(5):366–372. doi: 10.1016/S1081-1206(10)60506-6. [DOI] [PubMed] [Google Scholar]
- 21.CINRYZE® [package insert]. Lexington, MA: Shire ViroPharma, Inc.; 2018.
- 22.Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. 2010;363(6):513–522. doi: 10.1056/NEJMoa0805538. [DOI] [PubMed] [Google Scholar]
- 23.Zuraw BL, Banerji A, Bernstein JA, et al. US Hereditary Angioedema Association Medical Advisory Board 2013 recommendations for the management of hereditary angioedema due to C1 inhibitor deficiency. J Allergy Clin Immunol Pract. 2013;1(5):458–467. doi: 10.1016/j.jaip.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 24.Yoo SM, Khan DA. Implantable venous access device associated complications in patients with hereditary angioedema. J Allergy Clin Immunol Pract. 2013;1(5):524–525. doi: 10.1016/j.jaip.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 25.Riedl MA, Banerji A, Busse PJ, et al. Patient satisfaction and experience with intravenously administered C1-inhibitor concentrates in the United States. Ann Allergy Asthma Immunol. 2017;119(1):59–64. doi: 10.1016/j.anai.2017.05.017. [DOI] [PubMed] [Google Scholar]
- 26.HAEGARDA® [package insert]. Kankakee, IL: CSL Behring, LLC; 2020.
- 27.Späth PJ, Wüthrich B, Bütler R. Quantification of C1-inhibitor functional activities by immunodiffusion assay in plasma of patients with hereditary angioedema–evidence of a functionally critical level of C1-inhibitor concentration. Complement. 1984;1(3):147–159. doi: 10.1159/000467830. [DOI] [PubMed] [Google Scholar]
- 28.Zuraw BL, Cicardi M, Longhurst HJ, et al. Phase II study results of a replacement therapy for hereditary angioedema with subcutaneous C1-inhibitor concentrate. Allergy. 2015;70(10):1319–1328. doi: 10.1111/all.12658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Longhurst H, Cicardi M, Craig T, COMPACT Investigators et al. Prevention of hereditary angioedema attacks with a subcutaneous C1 inhibitor. N Engl J Med. 2017;376(12):1131–1140. doi: 10.1056/NEJMoa1613627. [DOI] [PubMed] [Google Scholar]
- 30.Craig T, Zuraw B, Longhurst H, COMPACT Investigators et al. Long-term outcomes with subcutaneous C1-inhibitor replacement therapy for prevention of hereditary angioedema attacks. J Allergy Clin Immunol Pract. 2019;7(6):1793–802.e2. doi: 10.1016/j.jaip.2019.01.054. [DOI] [PubMed] [Google Scholar]
- 31.Craig T, Lumry W, Cicardi M, et al. Treatment effect of switching from intravenous to subcutaneous C1-inhibitor for prevention of hereditary angioedema attacks: COMPACT subgroup findings. J Allergy Clin Immunol Pract. 2019;7(6):2035–2038. doi: 10.1016/j.jaip.2019.01.007. [DOI] [PubMed] [Google Scholar]
- 32.Banerji A, Busse P, Shennak M, et al. Inhibiting plasma kallikrein for hereditary angioedema prophylaxis. N Engl J Med. 2017;376(8):717–728. doi: 10.1056/NEJMoa1605767. [DOI] [PubMed] [Google Scholar]
- 33.TAKHZYROTM [package insert]. Lexington, MA: Dyax Corp; 2018.
- 34.Banerji A, Riedl MA, Bernstein JA, et al. Effect of lanadelumab compared with placebo on prevention of hereditary angioedema attacks: a randomized clinical trial [published correction appears in JAMA. 2019;321(16):1636] JAMA. 2018;320(20):2108–2121. doi: 10.1001/jama.2018.16773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Banerji A, Bernstein JA, Johnston DT, et al. Long-term prevention of hereditary angioedema attacks with lanadelumab: the HELP OLE Study. Allergy. 2021 doi: 10.1111/all.15011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.ORLADEYOTM [package insert]. Durham, NC: BioCryst Pharmaceuticals, Inc.; 2020.
- 37.Zuraw B, Lumry WR, Johnston DT, et al. Oral once-daily berotralstat for the prevention of hereditary angioedema attacks: a randomized, double-blind, placebo-controlled phase 3 trial. J Allergy Clin Immunol. 2021;148(1):164–72.e9. doi: 10.1016/j.jaci.2020.10.015. [DOI] [PubMed] [Google Scholar]
- 38.Riedl MA. Critical appraisal of androgen use in hereditary angioedema: a systematic review. Ann Allergy Asthma Immunol. 2015;114(4):281–8.e7. doi: 10.1016/j.anai.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 39.Caballero T, Canabal J, Rivero-Paparoni D, Cabañas R. Management of hereditary angioedema in pregnant women: a review. Int J Womens Health. 2014;6:839–848. doi: 10.2147/IJWH.S46460. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.