

Summary
Glucagon-like peptide-1 receptor agonists (GLP-1RA) are used for the treatment of type 2 diabetes. Whether clinically important responses and adverse events (AEs) are dependent on the route of administration has not been determined. We demonstrate that nearly identical exposure-response pharmacodynamic relationships are determined by plasma semaglutide levels achieved through oral versus injectable administration for changes in HbA1c, body weight, biomarkers of cardiovascular risk, and AEs such as nausea and vomiting. At typical exposure levels for oral semaglutide, the estimated response is 1.58% (oral) versus −1.62% (subcutaneous) for HbA1c and 3.77% (oral) versus 3.48% (subcutaneous) reduction in body weight relative to baseline after 6 months. Increased body weight is the most important variable associated with reduced semaglutide exposure for both formulations. Hence, interindividual variation in GLP-1R responsivity or route of administration are not major determinants of GLP-1RA effectiveness in the clinic.
Keywords: diabetes, obesity, G protein coupled receptor, glucagon-like peptide, GLP-1, weight loss, peptide, non-alcoholic hepatosteatosis, Alzheimer’s disease
Graphical abstract
Highlights
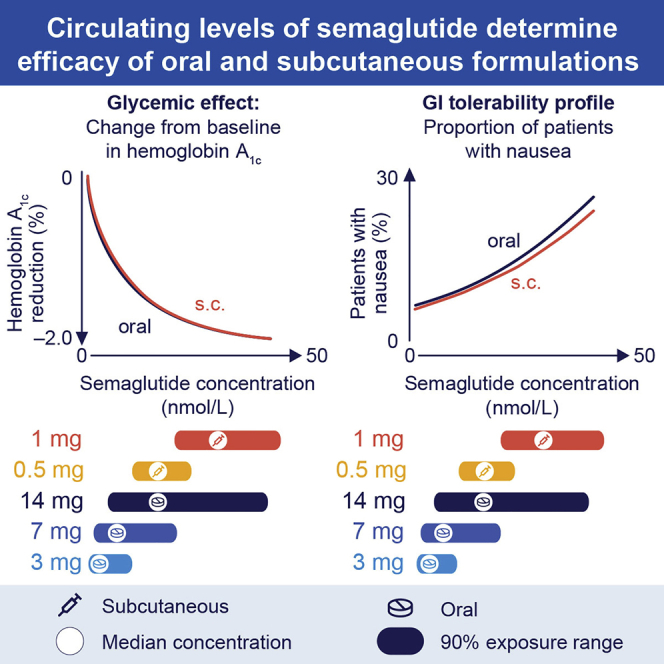
Circulating levels of semaglutide overlap with oral versus subcutaneous administration
Pharmacokinetics of semaglutide predict reductions in HbA1c and body weight
Route of semaglutide administration does not influence pharmacodynamic outcomes
Body weight influences circulating levels of semaglutide in people with diabetes
Determinants of GLP-1 action include the gut-brain axis, interindividual differences in GLP-1 receptor expression, differential drug access to key tissues, and circulating levels of bioactive peptide. Overgaard et al. analyze oral versus parenteral routes of administration, revealing that circulating levels of semaglutide predict outcomes in people with type 2 diabetes.
Introduction
The treatment of type 2 diabetes (T2D) is centered around glycemic control, based on strong relationships between blood glucose levels and the development of micro- and macrovascular complications.1The results of cardiovascular outcome studies in people with T2D have refined the approach to the selection of therapeutic agents, demonstrating the safety of three classes of glucose-lowering agents: the DPP-4 inhibitors, SGLT-2 inhibitors, and the glucagon-like peptide-1 receptor agonists (GLP-1RAs).2 Moreover, the last two classes of drugs reduced the development of several complications of T2D, enabling strategies directed at the reduction of cardiovascular disease and progression of chronic kidney disease.3
GLP-1RAs exhibit highly different molecular structures and unique pharmacokinetics. These molecules encompass small non-modified naturally occurring peptide agonists such as exenatide administered twice daily, and lixisenatide, an exendin-4 derivative modified at the carboxyl terminus and developed as a once-daily injection.4 Exenatide has also been developed as a long-acting microsphere preparation suitable for once-weekly administration.5 Alternatively, acylated human GLP-1RAs such as liraglutide and semaglutide exhibit a longer circulating t1/2 via non-covalent binding to serum albumin.6 Moreover, much larger molecules exemplified by dulaglutide, a once-weekly GLP-1RA containing two GLP-1 peptides coupled to an immunoglobulin (IgG4) Fc fragment, have been introduced for clinical use.4 These substantial differences in structure and pharmacokinetics necessitate scrutiny of the individual properties of and outcomes achieved with GLP-1RAs in the clinical setting.
An oral formulation of semaglutide was recently developed for clinical use, based on co-formulation with SNAC (sodium N-[8-(2-hydroxybenzoyl)amino] caprylate).7 Historically, peptide-based drugs exhibited limited oral bioavailability because of their chemical nature and multiple physicochemical challenges in absorption via the gastrointestinal (GI) tract.8 Oral semaglutide, administered once daily, in the morning on an empty stomach, was studied at 3 different doses, 3, 7, and 14 mg in the PIONEER (peptide innovation for early diabetes treatment) clinical trial program. Remarkably, the oral formulation of semaglutide achieved clinically relevant reductions of hemoglobin A1c (HbA1c) and body weight, in people with T2D, either as monotherapy in combination with other oral glucose-lowering agents or when added to insulin regimens. The results of the PIONEER program enabled the approval of the first oral GLP-1-based therapy for the treatment of T2D.9, 10, 11, 12, 13
The development of the field of GLP-1 therapeutics has prompted a number of unresolved mechanistic questions. For example, is genetic variation in the GLP-1 receptor (GLP-1R) or GLP-1 resistance, as described in preclinical and clinical studies,14,15 an important determinant of interindividual drug responses? Does the activity of local GI or neural GLP-1Rs preferentially activated by GLP-1RAs absorbed from the stomach contribute to differential efficacy of these oral agents?14 Is the reporting of nausea and vomiting across trials reflective of interindividual sensitivity, modified by local GI concentrations of drug, or largely attributable to actual circulating levels of GLP-1R agonists, whether achieved through enteral or parenteral administration?
The availability of pharmacokinetic data from people treated with oral versus injectable semaglutide provided the opportunity to compare clinically meaningful outcomes achieved with different routes of drug administration. Here, we compared the extent of HbA1c and body weight reduction, changes in cardiovascular biomarkers, and commonly reported AEs such as nausea and vomiting, in people with T2D treated with oral or subcutaneous (s.c.) semaglutide. The data reveal that systemic exposure to circulating semaglutide, irrespective of the mode of administration, is the major predictor of semaglutide outcomes in people with T2D. These findings indirectly suggest that preferential activation of portal or enteric neuronal GLP-1Rs, or interindividual differences in GLP-1R signaling, are not likely to be substantial determinants of common clinical outcomes in people with T2D treated with oral GLP-1R agonists.
Results
Baseline parameters for the study populations enrolled in the PIONEER and SUSTAIN clinical trial programs are shown in Table 1. To compare results with oral versus injectable semaglutide, we examined whether oral dosing of semaglutide in the PIONEER program was affected by variability in common clinical parameters. Based on population pharmacokinetic (PK) modeling described in STAR Methods and Methods S1 and S2, body weight was the covariate with the greatest impact on semaglutide exposure as illustrated for the model including all covariate factors (Figure S1) and via the parameter estimates for the final model (Table S1). Average semaglutide concentration levels at steady state were obtained for each subject planned for PK assessment in the PIONEER program. These exposure levels were subsequently used to derive exposure-response relationships as described below. Exposure-response relationships were evaluated after 26 weeks of treatment with once-daily oral semaglutide, which represents the primary evaluation time point in the 6 PIONEER trials.
Table 1.
Demographic characteristics of data from matched and unmatched subjects from SUSTAIN and PIONEER phase 3a trials
| Category | PIONEER |
SUSTAIN (N = 1,551) | |
|---|---|---|---|
| Unmatched (N = 3,003) | Matched (N = 1,551) | ||
| Age, y | 59.4 ± 10.9 | 57.3 ± 10∙5 | 56.0 ± 10.6 |
| Female | 1,345 (44.8) | 653 (42.1) | 658 (42.4) |
| HbA1c, % | 8.1 ± 0.8 | 8.1 ± 0.9 | 8.1 ± 0.9 |
| Body weight, kg | 88.2 ± 21.9 | 87.2 ± 22.4 | 86.3 ± 22.7 |
| Diabetes duration, y | 9.5 ± 7.9 | 7.3 ± 6.3 | 7.2 ± 6.0 |
| Race | |||
| White, other | 2,034 (67.7) | 971 (62.6) | 834 (53.8) |
| Asian | 779 (25.9) | 487 (31.4) | 647 (41.7) |
| Black or African American | 190 (6.3) | 93 (6.0) | 70 (4.5) |
| Renal function | |||
| Normal | 1,809 (60.2) | 1,099 (70.9) | 1,024 (66.0) |
| Mild impairment | 865 (28.8) | 415 (26.8) | 502 (32.4) |
| Moderate impairment | 329 (11.0) | 37 (2.4) | 25 (1.6) |
| Background therapy | |||
| 1–2 OADs | 1,335 (44.5) | 994 (64.1) | 1,077 (69.4) |
| Monotherapy | 703 (23.4) | 431 (27.8) | 345 (22.2) |
| Insulin | 845 (28.1) | 12 (0.8) | – |
| Diet and exercise | 120 (4.0) | 114 (7.4) | 129 (8.3) |
| Maintenance dose | |||
| Placebo | 572 (19.0) | 191 (12.3) | 129 (8.3) |
| 0.5 mg s.c. | – | – | 556 (35.8) |
| 1.0 mg s.c. | – | – | 866 (55.8) |
| 3 mg oral | 629 (20.9) | 345 (22.2) | – |
| 7 mg oral | 620 (20.6) | 331 (21.3) | – |
| 14 mg oral | 1,182 (39.4) | 684 (44.1) | – |
Data are n (%) or means ± SDs. OAD, oral antidiabetic drug; s.c., subcutaneous; SD, standard deviation.
Oral semaglutide exposure-response relationships determine HbA1c reduction
The change in HbA1c from baseline until week 26 with once-daily oral semaglutide was exposure dependent and started to level off at higher exposures (Figure S2A). A reduction in HbA1c was observed across the concentration range evident with oral semaglutide doses of 7 and 14 mg, with a 90% exposure range from 2 to 41 nmol/L. We next explored the exposure-response relationship for HbA1c for various covariate effects in the PIONEER trials. The exposure-response relationship was primarily dependent on baseline HbA1c (Figure S2B). Subjects with higher baseline HbA1c had greater HbA1C reductions compared to subjects with lower HbA1c and did not reach the same absolute HbA1c level compared to subjects with lower HbA1c (Figure S2C). At high exposure levels of semaglutide, subjects with the highest baseline HbA1c (≥9.1%) had a ∼3% decrease in HbA1c compared to a ∼1% reduction in subjects with a lower baseline HbA1c (<7.5%). Other covariates such as baseline body weight, BMI, diabetes duration, gender, age group, race, ethnicity, and presence of upper GI disease were of no or only minor importance and did not affect the exposure response relationships for oral semaglutide (Figure S1).
Analysis of the proportion of subjects in the PIONEER trials achieving HbA1c levels <7.0% or ≤6.5% is shown in Figure S3. Subjects with moderate renal impairment and those receiving insulin treatment had moderately lower HbA1c responses compared to subjects with normal renal function or those treated with multiple oral antidiabetic drugs (OADs) (Figures S2D and S3E). Notably, a consistent relationship was observed between plasma levels of semaglutide achieved with oral semaglutide administration and HbA1c targets, when analyzed across a range of PIONEER trials and starting levels of HbA1c (Figures S3A, S3C, S3D, and S3F). As expected, the proportion of individuals achieving glycemic targets was inversely proportional to the starting HbA1c. Some differences in exposure response across age groups were seen, but these differences appeared to be well described by other covariate factors, and age was not identified as a covariate in the model-based analysis (Figures S1 and S4).
Oral semaglutide exposure-response relationships for body weight are modulated by gender and baseline HbA1c
Body weight loss after 26 weeks of treatment was observed across the entire concentration range investigated with oral semaglutide 7 and 14 mg, and the slope of the weight loss effect did not appear to level off at the highest exposures obtained (Figure S5A). Greater weight loss was seen in females compared to males (Figure S5B); subjects with higher baseline HbA1c appeared to have less weight loss (Figure S5C). Background medication did not affect weight loss responses by exposure (Figure S5D). Other demographic covariates had little or no effect on weight loss (Figures S5D and S5E).
Oral semaglutide exposure-response relationships predict rates of GI AEs
Exposure-response relationships were investigated for nausea and vomiting events reported during the first 26 weeks of treatment with oral semaglutide. The proportions of subjects reporting nausea or vomiting increased with greater semaglutide exposure (Figures S4C and S4D).
Larger proportions of female subjects reported GI side effects across the exposure range for both nausea and vomiting. The remaining covariate factors, including moderate renal impairment, background medication for T2D, ethnicity, upper GI disease, and race had no or little influence on reports of nausea or vomiting that remained proportional to circulating semaglutide concentrations (Figures S5F and S5G).
Overlapping plasma levels of semaglutide with oral versus injectable administration
Based on the PK data from the PIONEER trials (Figures 1 and S2A), the variability of oral semaglutide was assessed both between and within subjects and compared to different GLP-1RAs (Figure 2). Across subjects with similar demographic profiles (Table 1), the coefficient of variation (CV) between subjects was estimated to be 84%, with lower variability in plasma concentrations observed within subjects (45% CV) (Figure 2). PK variability was greater for oral semaglutide versus injectable semaglutide, liraglutide, or dulaglutide (Figure 2).
Figure 1.

Semaglutide exposure is dose proportionate in the PIONEER and SUSTAIN trials
Data are individual Cavg values (open symbols) and geometric means with 90% ranges (closed symbols with error bars). Data from PIONEER 1, 2, 3, 5, 8, and 9; SUSTAIN 1, 2, 3, and 6; and SUSTAIN Japan OAD. Cavg, average concentration; OAD, oral antidiabetic drugs.
Figure 2.

Variability estimates in exposure when adjusted for time since dose and covariate factors, assessed based on population PK analysis
PIONEER estimates are based on the model presented in the text. Estimates from SUSTAIN, SCALE, and AWARD were based on similar population PK methodology and previously published.16,17,18 PK, pharmacokinetic; CV, coefficient of variation; s.c., subcutaneous.
To compare circulating peptide levels obtained with oral versus s.c. semaglutide, the individual model derived steady-state exposure levels for subjects on oral semaglutide in the PIONEER program were compared to the individual estimates for subjects taking s.c. semaglutide in the SUSTAIN program.16 The range of exposure levels for the two routes of administration overlapped considerably (Figure 1), but variability was smaller following s.c. versus oral semaglutide. The geometric mean exposure for oral semaglutide 14 mg was similar to that of 0.5 mg s.c. semaglutide, whereas the upper exposure ranges of 14 mg oral and 1.0 mg s.c. semaglutide doses overlapped, but the mean exposure levels were nominally higher with 1 mg s.c. semaglutide (Figure 1). Likewise, the upper exposure ranges for 7 mg oral and 0.5 mg s.c. doses were similar, although the geometric mean exposure for the 7 mg oral dose was lower than for 0.5 mg s.c. (Figure 1).
Previous semaglutide population PK analysis show that individual semaglutide exposure levels following s.c. administration in a Phase III diabetes program can be reasonably simulated based on baseline demographic data.16 Simulated data for s.c. semaglutide was compared to the observed exposure levels following oral administration to estimate the relative differences in plasma semaglutide levels upon switching from oral to s.c semaglutide, or vice versa. This would mean, for subjects switching from 14 mg oral semaglutide to 0.5 mg s.c. semaglutide, 88% would have a <2-fold increase in exposure, which corresponds to the exposure increase seen during normal dose escalation (Figure 3). These findings may provide guidance for anticipating changes in semaglutide levels in individuals switching between oral and s.c. semaglutide formulations.
Figure 3.

Comparative exposures with oral versus injectable semaglutide
Data are mean relative exposures with 90% ranges obtained using the individual exposure estimates from the final PK model for subjects in PIONEER 1, 2, and 8 populations. For each individual, s.c. exposure estimates were predicted based on a dose and demographic factors using a reduced population PK model developed based on s.c. semaglutide data.16
Plasma exposure-response relationships are identical with oral versus s.c. semaglutide
We next compared a range of clinically meaningful GLP-1-responsive therapeutic and safety endpoints across a range of semaglutide exposure levels obtained with oral versus s.c. semaglutide administration. Remarkably, exposure-response analyses showed highly similar relationships reflecting plasma levels of semaglutide obtained with oral versus s.c. semaglutide (Figure 1) and subsequent reductions in HbA1c and body weight (Figures 4A and 4B). Consistent with these findings, the proportions of subjects reporting nausea or vomiting were similar for oral and s.c. semaglutide administration over a range of plasma concentrations (Figures 4C and 4D). Differential effects for oral versus s.c. administration at 14.6 nM, corresponding to 14 mg, were computed based on a sensitivity analysis using the propensity-matched dataset. The predicted change in HbA1c was −1.58% (oral) versus −1.62% (s.c.) and the body weight change was 3.77% (oral) versus 3.48% (s.c.). Nausea was predicted to occur in 11.1% (oral) versus 11.8% (s.c.) of subjects and vomiting in 4.2% (oral) versus 4.2% (s.c.). Hence, the data illustrate the importance of plasma levels, irrespective of the route of dosing, or theoretical differences in GLP-1R sensitivity, for determining the clinical pharmacodynamic consequences of semaglutide administration.
Figure 4.

Efficacy and tolerability by semaglutide exposure in the PIONEER and SUSTAIN trials
Data are means and 95% CIs at week 26 for PIONEER and week 30 for SUSTAIN. Exposure is presented as quantiles of Cavg for semaglutide and 1 quantile for placebo (at Cavg of 0 nmol/L). The fitted solid line represents model-derived relations for each clinical program. The horizontal lines along the x axes represent medians and 90% exposure ranges, with the median exposure represented by a diamond. Data from SUSTAIN 1, 2, and 3; SUSTAIN-Japan; and PIONEER 1, 2, 3, 5, 8, and 9. The PIONEER and SUSTAIN populations differed somewhat with respect to demographic composition, and datasets were propensity matched. CI, confidence interval.
The exposure-response relationships for changes in blood pressure (BP), C-reactive protein (CRP), and fasting levels of triglycerides, indirect biomarkers of cardiovascular risk, are shown in Figure 5. Progressive reductions in CRP and triglycerides (Figures 5A and 5B) were associated with increasing levels of semaglutide exposure, whether obtained with oral or s.c. administration. Changes in diastolic BP were modest over a range of semaglutide concentrations; however, a clear exposure-response relationship was observed for systolic BP (Figures 5C and 5D). Collectively, these findings highlight the dominant importance of circulating levels of semaglutide, irrespective of the route of administration, for the determination of a broad range of clinically meaningful endpoints in people with T2D.
Figure 5.

Change in CRP, triglycerides, and blood pressure reflect semaglutide exposure in the PIONEER and SUSTAIN trials
Data are means and 95% CIs. Exposure is presented as quantiles of Cavg for semaglutide and 1 quantile for placebo (at Cavg of 0 nmol/L). The fitted solid line represents model-derived relations not adjusted for covariate factors, based on the PIONEER program. Triglycerides and systolic and diastolic BP: data from PIONEER 1, 2, 3, 5, 8, and 9 at week 26 and SUSTAIN 1, 2, and 3 and SUSTAIN Japan OAD at week 30. CRP: PIONEER 1, 2, and 5 data at week 26 and SUSTAIN 3 data at week 56. BP, blood pressure; CRP, C-reactive protein.
Discussion
Although GLP-1Ras have been used for the treatment of T2D for ∼16 years,19 the key determinants of clinical outcomes and their relationship to circulating drug levels in people with T2D have not been systematically compared using different routes of peptide administration. Here, we demonstrate that the plasma levels of semaglutide, independent of a wide range of co-variables, or theoretical interindividual differences in GLP-1R expression and sensitivity, are the major determinants of efficacy, as judged by reductions in HbA1c and body weight in people treated with oral or s.c. semaglutide. Not surprisingly, semaglutide plasma exposure also correlated with reports of nausea and vomiting in the same individuals, and the dose-response relationships for these endpoints were not different in the PIONEER versus SUSTAIN clinical trial programs.
Previous studies have identified neural circuits triggered by endogenous GLP-1 released from the gut that communicate with the autonomic and central nervous systems to mediate multiple actions of GLP-1, including control of satiety, glycemia, and gastric emptying.20,21 Notably, oral semaglutide is administered before food ingestion in the morning, whereas s.c. semaglutide may be administered at any time throughout the day. Despite an extensive preclinical literature highlighting the existence of a GLP-1R-dependent gut-brain axis triggered by oral nutrient ingestion and linked to the control of glucose homeostasis,22, 23, 24 the importance of this axis has been questioned,25 and its relevance to human physiology or the treatment of T2D remains uncertain. Critically, we did not detect evidence for preferential or detrimental actions of oral (relative to s.c. administered) semaglutide across a broad range of semaglutide plasma concentrations.
Notably, the venous drainage of the stomach via the splenic vein allows rapid access of semaglutide, administered orally, to the GLP-1Rs within the portal vein.24 Nevertheless, in most species, the majority of L cells are located in the distal gut, and the quantitative contributions of proximal gut L cells to the control of glucose homeostasis and circulating levels of GLP-1 throughout the day remains questionable.26 Our current observations with oral semaglutide absorbed through the stomach do not rule out the potential existence and importance of an endogenous GLP-1R-dependent gut-brain axis in humans, with physiological circuits potentially activated transiently by food ingestion triggering GLP-1 secretion from L cells within the distal gut enteroendocrine system.
Examining the clinical importance of PK variability reveals insights for oral peptide delivery. The variability estimates for oral semaglutide were compared to results obtained with similar methodology for s.c. semaglutide16 and dulaglutide.17 Interindividual variability of oral semaglutide was higher than for other GLP-1RAs dosed s.c., whereas intra-individual variability was just marginally higher, confirming previous findings from clinical pharmacology trials.16 Importantly, the higher day-to-day variability was mitigated by frequent dosing and a long half-life of ∼1 week for semaglutide, thereby leading to similar effects on metabolic outcomes and body weight.
Similar exposure-response relationships with s.c. versus oral semaglutide for GI side effects further support the limited clinical impact of intra-individual variability in plasma concentrations for oral semaglutide. The starting dose of 3 mg semaglutide administered orally generally provides lower exposure than 0.25 mg s.c., which presumably helps to ensure tolerability across the exposure range and comparable tolerability for oral and s.c. semaglutide. Nevertheless, compared to s.c. semaglutide, more subjects achieved lower plasma exposures, leading to suboptimal pharmacodynamics effects. At 14 mg, a substantial proportion of the subjects achieved exposure and reductions in HbA1c and body weight similar to that achieved with s.c. semaglutide 1 mg.
Examining the subset of individuals on oral semaglutide 14 mg (19%) that achieved exposure levels below those obtained with 0.5–1.0 mg s.c. semaglutide, revealed that these subjects still achieved an ∼1% reduction in HbA1c, which is considered a clinically relevant treatment outcome. Only a small fraction (3%) of subjects obtained exposure levels considered below the therapeutic range, with an expected HbA1c reduction <0.5%. Whether these observations reflect real problems with drug absorption and/or compliance remains uncertain. These findings demonstrate that the variability associated with the oral administration of semaglutide has limited consequences for clinical responses in the majority of treated subjects.
It is widely appreciated that the extent of insulin resistance, and not just circulating levels of insulin, influences the biological response to insulin therapy. In contrast, despite the growing clinical use of GLP-1RAs, the potential importance of various factors modifying therapeutic responses to GLP-1RAs remains poorly defined. Notably, relative GLP-1 resistance and impairment of the gut-brain GLP-1 axis has been described in preclinical studies.14 Clinically, genetic variation in the GLP1R has been associated with changes in receptor signaling and glycemia27 and differential responses to GLP-1RA in some human studies.28,29 Variables such as local tissue expression of the GLP-1R, interindividual differences in GLP-1 receptor binding, receptor structure, or downstream signaling activity are difficult to measure in the context of large clinical trials. Here, we determined that when corrected for plasma level of semaglutide achieved, irrespective of the mode of administration, factors such as body weight or BMI, race, and ethnicity did not meaningfully influence the exposure-response relationships for key determinants such as HbA1c and weight loss. In contrast, individuals on insulin, those with modest degrees of renal impairment, and subjects with lower starting baseline HbA1c exhibited diminution in exposure-response relationships, relative to other analyzed groups. The characteristics of the first two subgroups are related in part to a more prolonged duration of disease and so their diminished responses are in line with general experience surrounding the relative efficacy of glucose-lowering agents and the extent and duration of preexisting T2D.
The observations that multiple long-acting GLP-1RAs reduced the rates of CV events and all-cause mortality across a broad range of cardiovascular safety trials30 have sparked tremendous interest in understanding mechanisms linking GLP-1R activation to cardioprotection. Notably, our analyses demonstrate a consistent exposure-response relationship for biomarkers and physiological variables indirectly related to CV risk, such as CRP, BP, and plasma triglycerides, with both oral and s.c. semaglutide. These findings align with the results of the SUSTAIN-6 and PIONEER-6 trials, showing a 26% and non-significant 21% reduction, respectively, in the hazard ratio of major adverse cardiovascular events (MACE) in T2D populations with high CV risk.31,32 Hence, the effects of semaglutide in modifying cardiovascular risk factors are not dissociated from plasma levels of semaglutide achieved using different modes of drug administration.
The finding that increasing semaglutide exposure is linked to the extent of HbA1c reduction and weight loss provides further guidance for the ongoing development of GLP-1RAs for the treatment of obesity. These observations are consistent with more limited analysis of the pharmacodynamic differences obtained for 0.5 versus 1.0 mg of injectable semaglutide,16,33 as well as the greater weight loss achieved with 3.0 versus 1.8 mg liraglutide34, 35 and collectively support the evaluation of higher doses of oral semaglutide, and the higher dose of 2.4 mg semaglutide once weekly investigated in the STEP program. These findings may also inform the dose selection of semaglutide in some subjects with monogenic obesity, an important population that may benefit from therapy with GLP-1Ras.36 Interestingly, an upper dose and plasma concentration range for semaglutide, or for that matter, any other GLP-1RA, that is not associated with additional incremental weight loss, has not yet been defined. These findings highlight the importance of progressive dose escalation, while balancing tolerability, for the development of new GLP-1RAs with improved weight loss profiles.
Translational relevance
Importantly, body weight was a relevant modifier of the plasma levels of semaglutide achieved across a range of semaglutide dosing regimens, although its impact on semaglutide plasma levels does not substantially modify clinical outcomes and does not imply dose adjustments based on baseline body weight. Nevertheless, the body weight-exposure relationship may influence the option of increasing or decreasing dose levels on an individual basis. Individuals with greater degrees of obesity not achieving glycemic and weight goals on oral semaglutide may consider switching to injectable semaglutide. These relationships may also explain in part the frequency of AEs in people across a range of BMIs.
The presence of upper GI disorders may modify the absorption of oral semaglutide37 in some individuals; however, 85% of subjects with GI disorders studied at 14 mg (81% overall) achieved circulating levels of semaglutide comparable to that achieved with s.c. administration. Higher exposure with GI disease was driven by gastroesophageal reflux disease, which constituted the majority of reported GI diseases (412 subjects). Gastric ulcer was less common in 34 patients; a non-significant 11% lower exposure was seen in these subjects compared to subjects not reporting GI disease. No effect differences were seen in 174 subjects with chronic gastritis. For subjects in whom switching between oral versus s.c semaglutide is relevant, our data provide guidance on how to avoid exposure increases leading to tolerability issues. The interindividual and, to a lesser extent, day-to-day intra-individual variability, in plasma semaglutide levels was greater with oral versus s.c. semaglutide administration, likely reflecting the greater reproducibility of semaglutide delivery with parenteral administration.
Our comparative findings simplify the interpretation of outcomes in thousands of people studied in the PIONEER and SUSTAIN clinical trials. The predictability of the dose-response relationships for oral semaglutide support ongoing trials that include higher dose levels of oral semaglutide in people with T2D and obesity, to maximize efficacy by achieving levels of circulating semaglutide with oral dosing that more approximates levels achieved with higher dose of s.c. semaglutide.
The complexity of interpreting dose-response relationships for clinical insulin action requires consideration that insulin bioactivity is greatly influenced by interindividual differences in insulin resistance, and potentially through differential routes of administration such as selective preferential targeting of hepatic versus adipose tissue insulin receptors.38 In contrast, the present data highlight the predominant importance of plasma semaglutide levels, generally independent of a range of potential co-variables, as critical for determining the efficacy of GLP-1RA therapy for the management of T2D.
Conclusions
The present data advance our understanding of key variables potentially modifying the efficacy of oral and injectable semaglutide for the therapy of T2D. Neither gender nor age was found to be an important predictor of semaglutide action; however, plasma levels trended higher, and weight loss achieved was somewhat greater in female subjects at a given exposure level. Intra-individual variability did not appear to have a clinical impact, and plasma levels of semaglutide achieved with oral semaglutide overlapped substantially with those detected after s.c. semaglutide administration. Neither renal impairment nor hemodialysis meaningfully affected dose-response relationships. Moreover, plasma semaglutide levels increased in a dose-proportional manner with both modes of administration and represented the major determinant of the magnitude of clinical benefit.
Limitations of the study
Exposure-response data following oral and s.c. administration were obtained in separate large clinical programs with potential differences in trial population and conduct; differences were corrected for in part by propensity matching. Additional limitations of the present analysis include the lack of measurements of insulin, glucagon, and gastric emptying or the assessment of GLP1R expression in tissues from study subjects, and the absence of genotyping for individual variation within the GLP1R. We were unable to measure free semaglutide; hence, the measurement of total semaglutide concentrations is an indirect surrogate of true semaglutide bioavailability in vivo. Nevertheless, the present data solidify the importance of plasma exposure, independent of the route of administration, as a key determinant of semaglutide action in the therapy of T2D.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Software and algorithms | ||
| NONMEM version 7.3 | ICON Development Solutions | https://www.iconplc.com/innovation/nonmem/ |
| R version 3.2.3 | R-Project | https://www.r-project.org/ |
| STAR Methods. Oral semaglutide population PK model: Parameterization and NONMEM settings for parameter estimation | This paper Supplemental Methods S1 | N/A |
| STAR Methods. Subcutaneous semaglutide population PK model: Parameterization and NONMEM settings for parameter estimation | This paper Supplemental Methods S2 | N/A |
| Other clinical trial pharmacokinetic data | ||
| SUSTAIN clinical trial program | Published analyses | PMID 29907893 |
| PIONEER clinical trial program | Blood samples analyzed from PIONEER trials | PMID: 31186300 |
| PIONEER 1,2,3,5,8, and 9 trials | PMID: 31530666 | |
| PMID: 30903796 | ||
| PMID: 31189517 | ||
| PMID: 31530667 | ||
| PMID: 32333875 | ||
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Daniel Drucker (danieljdrucker@gmail.com or drucker@lunenfeld.ca).
Materials availability
No new reagents or research materials were generated in this study.
Experimental model and subject details
This study presents exposure data and exposure-response analyses for oral semaglutide in male and female subjects with type 2 diabetes and furthermore, compares with previously published data on s.c. semaglutide for exposure16 and exposure-response.33 The pharmacokinetic analyses presented here are consistent with the informed consent forms approved by participating institutions and signed by participants enrolling in the PIONEER and SUSTAIN clinical trial programs.
Clinical data
Data from six phase 3 trials of oral semaglutide was used for assessment of semaglutide PK and exposure-response analysis (global trials PIONEER 1, 2, 3, 5, 8 and Japanese trial PIONEER 9).9, 10, 11, 12, 13,32,39,40 Each trial was powered for efficacy versus comparator or placebo, and not for population PK or exposure-response analyses. Randomization across the dose levels were 1:1:1 for PIONEER 1, 3, 8, and 9, whereas PIONEER 2 and 5 included only the 14 mg dose. The current exposure-pharmacodynamic relationships for oral semaglutide in the PIONEER trials were compared to those obtained from the phase 3a program of injectable semaglutide in subjects with T2D (SUSTAIN trials). Overall, the PIONEER and SUSTAIN exposure-response populations were similar, with the main difference being the inclusion of a dedicated study of patients with moderate renal impairment and a trial with concomitant insulin treatment in the PIONEER program. Propensity score matching was used to analyze the effect of balancing the differences between SUSTAIN and PIONEER populations. Patient characteristics are summarized in Table S1.
Oral semaglutide dosing was administered with up to 120 mL of water, at least 30 min before the first meal of the day and any other oral medication. The PK population was defined as all subjects from the full analysis sets who were randomized to oral semaglutide, except for PIONEER 3 which had PK sampling scheduled for subjects recruited in USA, Germany and Japan, constituting approximately 50% of the subjects in PIONEER 3. Only PK data obtained during treatment was included, thus excluding data obtained at baseline and following premature discontinuation of treatment. Blood samples for estimation of semaglutide concentrations in plasma were obtained at week 4, 8, 14 and 26 (PIONEER 1), week 4, 26 and 52 (PIONEER 2), week 4, 8, 14, 26, 38, 52 and 78 (PIONEER 3), week 4, 8, 14, 26 (PIONEER 5), week 4, 14, 26, 38, 52 (PIONEER 8) and week 4, 14, 26, 38 and 52 (PIONEER 9). A single blood sample was obtained at each occasion, except for PIONEER 9 which had two samples obtained at week 26 (one pre-dose and one 60-90 min post-dose). Timing of blood sampling was at the investigator’s discretion except in PIONEER 2 which aimed at sampling 25 min post-dose to approximately capture Cmax of SNAC. Actual sampling times as well as dosing times for the last dose prior to sampling were recorded. Total protein bound semaglutide concentrations were estimated in plasma by a validated liquid chromatography-mass spectrometry assay with a lower limit of quantification (LLOQ) of 0.729 nmol/L.41
Method details
Population PK assessment of oral semaglutide
In total, 10,527 PK observations from 2,345 subjects during treatment with oral semaglutide from the PIONEER 1,2,3,5,8, and 9 trials were included in the PK dataset. Of these, 914 observations were below the LLOQ and were assigned the value LLOQ/2. 154 PK observations (1.5%) were excluded due to inadequate dosing history.
The population PK model for oral semaglutide was pre-specified based on the model obtained from data across clinical pharmacology trials of oral, s.c. and intravenous administration of semaglutide.42 This was a two compartment model with separate first order absorption processes for oral and s.c. administration, and with the same distribution and elimination processes across all routes of administration. All parameter values in the oral PK model were based on these previously established results, except for bioavailability, which was estimated on PIONEER PK data. The PK model was used to characterize variability in exposure (within and between subjects), demographic factors influencing exposure, as well as individual average steady state exposure levels (Cavg) used for exposure-response. A single Cavg value was computed for each subject based on the individual parameter estimates, as the average concentration in a dosing interval at steady state at the target dose level. Several sensitivity analyses with different structural PK parameters and different procedures for LLOQ handling were conducted, all demonstrating robust Cavg values, with above 0.99 correlation to the final estimates.
A confirmatory approach with estimation of a base model without covariates, a full model including all investigated covariates and a final model including only significant covariates, was applied.43 The full model used for covariate analysis included the following covariates on exposure: gender (male, female), age group (18-64, 65-74, 75-92 years), race (White, Black or African American, Asian), ethnicity (Non-Hispanic or Latino, Hispanic or Latino), body weight as continuous covariate, renal function (normal, mild and moderate impairment), semaglutide dose (3, 7, 14 mg), subjects with/without upper GI disease and trial effects for PIONEER 2, 3, 5 and 8. Presence of upper GI disease was defined as having ticked ‘yes’ to an ongoing diagnosis of gastresophageal reflux disease, chronic gastritis, gastric ulcer and/or gastroparesis on the GI disease history form filled in by the investigator at the baseline visit. In the final model, the following covariates were retained with minor impact: gender, race (Asian and Whites), body weight, upper GI disease and trial effects for PIONEER 3 and 5. The latter corrected for overall lower exposures in PIONEER 3 and 5 compared to PIONEER 1, 2, 8 and 9. The final model included log-normal inter-individual variability model for bioavailability (F), and a proportional residual error model. Details on the parameterization and software settings for parameter estimation is given in the Supplemental information section.
Population PK assessment of s.c. semaglutide
Assessment of s.c. semaglutide exposure levels in type 2 diabetes from SUSTAIN trials has previously been reported.16 Due to the relatively slow absorption of s.c. semaglutide, a one-compartment model was applied as this was the simplest model to adequately describe steady PK for s.c. semaglutide.16 The one-compartment model and the two-compartment model used for oral semaglutide were assessed to provide almost identical Cavg values for s.c. semaglutide across individual subjects,42 thus reassuring that Cavg levels can be compared across SUSTAIN and PIONEER. The final model included the same demographic covariates as for oral semaglutide described above, with the exception of GI disease and the addition of injection site. log-normal inter-individual variability model for the apparent clearance (CL/F) and the apparent volume of distribution (V/F), and a proportional residual error model.
Quantification and statistical analysis
Exposure-response analyses aim to reflect the efficacy observed for subjects adhering to treatment and not taking anti-diabetic rescue medication.
Subjects scheduled for PK assessment, but without valid concentration data were assigned the population PK parameter value based on the subject’s covariate profile. Missing efficacy data for efficacy-response analyses were predicted using a mixed model repeated-measures (MMRM) model.
Propensity score matching based on baseline HbA1c, diabetes duration, race/ethnicity, and gender was used to obtain a dataset of subjects with matched baseline characteristics from SUSTAIN and PIONEER.
All statistical tests, e.g., used during model development for the population PK models and exposure-response models, were performed using likelihood ratio tests. Details regarding summary statistics presented is given in Figure Legends.
Software
NONMEM (ICON Development Solutions, Ellicott City, MD, USA) version 7.3 was used for the population PK analysis. Parameter uncertainty was obtained via bootstrapping via PsN version 4.60.
R version 3.2.3 was used for data file processing, exposure-response modeling and plotting. Nls was used for the non-linear regression models of HbA1c and body weight change from baseline. Gnls was used for non-linear regression models of HbA1c responder endpoints on a logit scale. Gnls was used to implement a weighted regression consistent with modeling of the probability of response. Glm was used for the more standard linear logistic regression models for proportion of subjects reporting nausea or vomiting.
Propensity matching was performed based on baseline HbA1c, trial population, diabetes duration, race, ethnicity and gender, using the MatchIt library with default settings for a logit distance and a nearest neighbor method.
Exposure-response analysis
The exposure-response population was comprised of the PK population with addition of placebo-treated subjects from PIONEER 1, 5, 8 and 9, adding up to a total of 3,003 subjects. Pre-specified exposure-response analysis for efficacy was conducted for change from baseline of HbA1c and body weight as well as for the proportions of subjects reaching the HbA1c target defined by ADA (HbA1c < 7.0%) and AACE (HbA1c ≤ 6.5%), respectively, following 26 weeks of treatment. Pre-specified exposure-response analysis for tolerability was conducted for proportions of subjects reporting nausea and vomiting, respectively, at any time and severity during 26 weeks of treatment. Stepwise model development was executed for each endpoint, ensuring that each covariate factor in the final model demonstrated an impact on the exposure-response that was statistically significant and reasonably identifiable from exposure-response figures.
HbA1c change from baseline data was described by a maximum response (Emax) model with baseline HbA1c and trial population influencing the treatment effect, and additional placebo effects of diabetes duration and baseline HbA1c. The same model structure was used to model the logit of the probability for reaching HbA1c targets.
Body weight change from baseline (%) was described by an Emax model with gender and baseline HbA1c influencing the treatment effect and additional placebo effects of diabetes duration, race and ethnicity.
For binary safety endpoints (patients reporting nausea and vomiting), linear models on the logit scale were used. For nausea, covariate factors were identified as trial population, gender, race, ethnicity and upper GI disease. For vomiting, covariate factors were identified for trial population and gender.
Exposure-response was illustrated in quantiles of subjects based on individual steady-state exposure levels (Cavg) or placebo with up to 200 subjects per quantile (100 subjects when further subgroup stratification is used). To correct for confounding factors due to differences in demographic profiles across quantiles, mean exposure-response model predictions are provided to show estimated outcomes of changing the exposure without a simultaneous change in the distribution of subjects.44 The model lines were generated as the mean model prediction across subjects, for each exposure level.
Comparison of exposure-response for oral and s.c. data was conducted by adding previously published exposure-response data from SUSTAIN trials. Propensity matching was conducted to identify the subjects treated with oral semaglutide that best match subjects investigated with s.c. semaglutide. Previous exposure-response models for s.c. semaglutide support the relationship observed across exposure quantiles for s.c. semaglutide,33,42 and no updated exposure-response modeling of s.c. semaglutide are provided in the present report. The exposure-response model obtained for oral semaglutide was used to provide model derived exposure-response lines both for a population with baseline characteristics as in the matched PIONEER population and for a population with baseline characteristics as in the SUSTAIN population.
The covariate factors tested for inclusion in the model were: “baseline HbA1c, body weight, BMI, diabetes duration, gender, age, Hispanic ethnicity, Asian race, upper GI disease, renal impairment, Trial effects, and Trial population, i.e. “Monotherapy” corresponding to PIONEER 1 and 9, “1-2 OAD” corresponding to PIONEER 2 and 3, “Moderate renal impairment” corresponding to PIONEER 5, and “Insulin treatment” corresponding to PIONEER 8.
Model qualification
The population PK model was qualified to meet the assumptions in the prespecified model. Model qualification included 1) Model fit to median concentration profiles, overall and across covariate subgroups 2) Standard GOF plots, including investigation of observed versus predicted PK observations, conditional weighted residuals versus predictions and time, and the distribution of the conditional weighted residuals. 3) Visual predictive checks, investigating if simulations from the model could reproduce the median and 90% range in PK observations over time. 4) Sensitivity analyses were conducted, investigating sensitivity of estimated covariate effects and individual Cavg values with different fixed structural PK parameters (changing to 50% higher value) and different procedures for LLOQ handling (either excluding values below LLOQ, or handling these using a censoring technique).
The exposure-response models were qualified by model fit to mean response data overall and across covariate subgroups as included in Figures S4 and S5E–S5G.
Additional resources
The PIONEER and SUSTAIN clinical trials used to generate the pharmacokinetic data are registered and described within the following clinical trial online registration sites.
PIONEER 9: NCT03018028
PIONEER 1: NCT02906930
PIONEER 3: NCT02607865
PIONEER 5: NCT02827708
PIONEER 2: NCT02863328
PIONEER 8: NCT03021187
SUSTAIN 1: NCT02054897
SUSTAIN 2: NCT01930188
SUSTAIN 3: NCT01930188
SUSTAIN 6: NCT01720446
SUSTAIN Japan OAD: NCT02207374
Acknowledgments
The clinical trials and subsequent analyses were funded by Novo Nordisk A/S. D.J.D. is supported in part by a Banting and Best Diabetes Centre-Novo Nordisk Chair in Incretin Biology, a Sinai Health-Novo Nordisk Foundation Fund in Regulatory Peptides, and CIHR grant no. 154321. Preclinical studies in the Drucker lab are supported by grants to Mt. Sinai Hospital from Novo Nordisk and Pfizer.
Author contributions
R.V.O., C.L.H., S.H.I., and A.N. carried out the initial primary analyses and wrote and edited the paper. D.J.D. analyzed the data and wrote the paper.
Declaration of interests
R.V.O., C.L.H., S.H.I., and A.N. are full-time employees of and own stock in Novo Nordisk A/S. D.J.D. has served as an advisor or consultant or speaker within the past 12 months to Eli Lilly, Forkhead Biotherapeutics, Intarcia Therapeutics, Kallyope, Merck Research Laboratories, and Novo Nordisk, and holds non-exercised options in Kallyope.
Published: September 1, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100387.
Supplemental information
Data and code availability
-
•
Original data may be reviewed consistent with conditions within the informed consent signed by participants in the PIONEER and SUSTAIN clinical trial programs, upon contacting the authors, upon request to the lead contact, Daniel Drucker.
-
•
No new code was generated in this study. For the oral semaglutide pharmacokinetics model, parameterization and NONMEM computer program settings for parameter estimation are provided in the Methods S1 and S2.
-
•
The patient level data reported in this study cannot be deposited in a public repository because of limitations associated with the informed consent for the SUSTAIN and PIONER trials.
-
•
To request access, contact the Lead contact DanieljDrucker@gmail.com.
-
•
All of the data reported in this manuscript will be shared upon contact of the lead author Danieljdrucker@gmail.com.
-
•
Any additional information required to re-analyze the data reported in this work paper is available from the Lead Contact upon request.
References
- 1.Home P. Controversies for Glucose Control Targets in Type 2 Diabetes: Exposing the Common Ground. Diabetes Care. 2019;42:1615–1623. doi: 10.2337/dci19-0002. [DOI] [PubMed] [Google Scholar]
- 2.Khunti K., Davies M.J., Seidu S. Cardiovascular outcome trials of glucose-lowering therapies. Expert Rev. Pharmacoecon. Outcomes Res. 2020;20:237–249. doi: 10.1080/14737167.2020.1763796. [DOI] [PubMed] [Google Scholar]
- 3.Buse J.B., Wexler D.J., Tsapas A., Rossing P., Mingrone G., Mathieu C., D’Alessio D.A., Davies M.J. 2019 Update to: Management of Hyperglycemia in Type 2 Diabetes, 2018. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) Diabetes Care. 2020;43:487–493. doi: 10.2337/dci19-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Müller T.D., Finan B., Bloom S.R., D’Alessio D., Drucker D.J., Flatt P.R., Fritsche A., Gribble F., Grill H.J., Habener J.F. Glucagon-like peptide 1 (GLP-1) Mol. Metab. 2019;30:72–130. doi: 10.1016/j.molmet.2019.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drucker D.J., Buse J.B., Taylor K., Kendall D.M., Trautmann M., Zhuang D., Porter L., DURATION-1 Study Group Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: a randomised, open-label, non-inferiority study. Lancet. 2008;372:1240–1250. doi: 10.1016/S0140-6736(08)61206-4. [DOI] [PubMed] [Google Scholar]
- 6.Knudsen L.B., Lau J. The Discovery and Development of Liraglutide and Semaglutide. Front. Endocrinol. (Lausanne) 2019;10:155. doi: 10.3389/fendo.2019.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buckley S.T., Bækdal T.A., Vegge A., Maarbjerg S.J., Pyke C., Ahnfelt-Rønne J., Madsen K.G., Schéele S.G., Alanentalo T., Kirk R.K. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci. Transl. Med. 2018;10:eaar7047. doi: 10.1126/scitranslmed.aar7047. [DOI] [PubMed] [Google Scholar]
- 8.Drucker D.J. Advances in oral peptide therapeutics. Nat. Rev. Drug Discov. 2020;19:277–289. doi: 10.1038/s41573-019-0053-0. [DOI] [PubMed] [Google Scholar]
- 9.Aroda V.R., Rosenstock J., Terauchi Y., Altuntas Y., Lalic N.M., Morales Villegas E.C., Jeppesen O.K., Christiansen E., Hertz C.L., Haluzík M., PIONEER 1 Investigators PIONEER 1: Randomized Clinical Trial of the Efficacy and Safety of Oral Semaglutide Monotherapy in Comparison With Placebo in Patients With Type 2 Diabetes. Diabetes Care. 2019;42:1724–1732. doi: 10.2337/dc19-0749. [DOI] [PubMed] [Google Scholar]
- 10.Rodbard H.W., Rosenstock J., Canani L.H., Deerochanawong C., Gumprecht J., Lindberg S.O., Lingvay I., Søndergaard A.L., Treppendahl M.B., Montanya E., PIONEER 2 Investigators Oral Semaglutide Versus Empagliflozin in Patients With Type 2 Diabetes Uncontrolled on Metformin: The PIONEER 2 Trial. Diabetes Care. 2019;42:2272–2281. doi: 10.2337/dc19-0883. [DOI] [PubMed] [Google Scholar]
- 11.Rosenstock J., Allison D., Birkenfeld A.L., Blicher T.M., Deenadayalan S., Jacobsen J.B., Serusclat P., Violante R., Watada H., Davies M., PIONEER 3 Investigators Effect of Additional Oral Semaglutide vs Sitagliptin on Glycated Hemoglobin in Adults With Type 2 Diabetes Uncontrolled With Metformin Alone or With Sulfonylurea: The PIONEER 3 Randomized Clinical Trial. JAMA. 2019;321:1466–1480. doi: 10.1001/jama.2019.2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mosenzon O., Blicher T.M., Rosenlund S., Eriksson J.W., Heller S., Hels O.H., Pratley R., Sathyapalan T., Desouza C., PIONEER 5 Investigators Efficacy and safety of oral semaglutide in patients with type 2 diabetes and moderate renal impairment (PIONEER 5): a placebo-controlled, randomised, phase 3a trial. Lancet Diabetes Endocrinol. 2019;7:515–527. doi: 10.1016/S2213-8587(19)30192-5. [DOI] [PubMed] [Google Scholar]
- 13.Zinman B., Aroda V.R., Buse J.B., Cariou B., Harris S.B., Hoff S.T., Pedersen K.B., Tarp-Johansen M.J., Araki E., PIONEER 8 Investigators Efficacy, Safety, and Tolerability of Oral Semaglutide Versus Placebo Added to Insulin With or Without Metformin in Patients With Type 2 Diabetes: The PIONEER 8 Trial. Diabetes Care. 2019;42:2262–2271. doi: 10.2337/dc19-0898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grasset E., Puel A., Charpentier J., Collet X., Christensen J.E., Tercé F., Burcelin R. A Specific Gut Microbiota Dysbiosis of Type 2 Diabetic Mice Induces GLP-1 Resistance through an Enteric NO-Dependent and Gut-Brain Axis Mechanism. Cell Metab. 2017;25:1075–1090.e5. doi: 10.1016/j.cmet.2017.04.013. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki K., Akiyama M., Ishigaki K., Kanai M., Hosoe J., Shojima N., Hozawa A., Kadota A., Kuriki K., Naito M. Identification of 28 new susceptibility loci for type 2 diabetes in the Japanese population. Nat. Genet. 2019;51:379–386. doi: 10.1038/s41588-018-0332-4. [DOI] [PubMed] [Google Scholar]
- 16.Carlsson Petri K.C., Ingwersen S.H., Flint A., Zacho J., Overgaard R.V. Semaglutide s.c. Once-Weekly in Type 2 Diabetes: A Population Pharmacokinetic Analysis. Diabetes Ther. 2018;9:1533–1547. doi: 10.1007/s13300-018-0458-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geiser J.S., Heathman M.A., Cui X., Martin J., Loghin C., Chien J.Y., de la Peña A. Clinical Pharmacokinetics of Dulaglutide in Patients with Type 2 Diabetes: Analyses of Data from Clinical Trials. Clin. Pharmacokinet. 2016;55:625–634. doi: 10.1007/s40262-015-0338-3. [DOI] [PubMed] [Google Scholar]
- 18.Ingwersen S.H., Khurana M., Madabushi R., Watson E., Jonker D.M., Le Thi T.D., Jacobsen L.V., Tornøe C.W. Dosing rationale for liraglutide in type 2 diabetes mellitus: a pharmacometric assessment. J. Clin. Pharmacol. 2012;52:1815–1823. doi: 10.1177/0091270011430504. [DOI] [PubMed] [Google Scholar]
- 19.Drucker D.J., Habener J.F., Holst J.J. Discovery, characterization, and clinical development of the glucagon-like peptides. J. Clin. Invest. 2017;127:4217–4227. doi: 10.1172/JCI97233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drucker D.J. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 2018;27:740–756. doi: 10.1016/j.cmet.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 21.Baggio L.L., Drucker D.J. Glucagon-like peptide-1 receptors in the brain: controlling food intake and body weight. J. Clin. Invest. 2014;124:4223–4226. doi: 10.1172/JCI78371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malbert C.H., Chauvin A., Horowitz M., Jones K.L. Glucose Sensing Mediated by Portal Glucagon-Like Peptide 1 Receptor Is Markedly Impaired in Insulin-Resistant Obese Animals. Diabetes. 2021;70:99–110. doi: 10.2337/db20-0361. [DOI] [PubMed] [Google Scholar]
- 23.Burcelin R., Da Costa A., Drucker D., Thorens B. Glucose competence of the hepatoportal vein sensor requires the presence of an activated glucagon-like peptide-1 receptor. Diabetes. 2001;50:1720–1728. doi: 10.2337/diabetes.50.8.1720. [DOI] [PubMed] [Google Scholar]
- 24.Vahl T.P., Tauchi M., Durler T.S., Elfers E.E., Fernandes T.M., Bitner R.D., Ellis K.S., Woods S.C., Seeley R.J., Herman J.P., D’Alessio D.A. Glucagon-like peptide-1 (GLP-1) receptors expressed on nerve terminals in the portal vein mediate the effects of endogenous GLP-1 on glucose tolerance in rats. Endocrinology. 2007;148:4965–4973. doi: 10.1210/en.2006-0153. [DOI] [PubMed] [Google Scholar]
- 25.Aulinger B.A., Perabo M., Seeley R.J., Parhofer K.G., D’Alessio D.A. Rapid hepatic metabolism blunts the endocrine action of portally infused GLP-1 in male rats. Am. J. Physiol. Endocrinol. Metab. 2020;318:E189–E197. doi: 10.1152/ajpendo.00298.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song Y., Koehler J.A., Baggio L.L., Powers A.C., Sandoval D.A., Drucker D.J. Gut-Proglucagon-Derived Peptides Are Essential for Regulating Glucose Homeostasis in Mice. Cell Metab. 2019;30:976–986.e3. doi: 10.1016/j.cmet.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lagou V., Jiang L., Ulrich A., Zudina L., González K.S.G., Balkhiyarova Z., Faggian A., Chen S., Todorov P., Sharapov S. Random glucose GWAS in 493,036 individuals provides insights into diabetes pathophysiology, complications and treatment stratification. medRxiv. 2021 doi: 10.1101/2021.04.17.21255471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu M., Wang K., Liu H., Cao R. GLP1R variant is associated with response to exenatide in overweight Chinese Type 2 diabetes patients. Pharmacogenomics. 2019;20:273–277. doi: 10.2217/pgs-2018-0159. [DOI] [PubMed] [Google Scholar]
- 29.Jensterle M., Pirš B., Goričar K., Dolžan V., Janež A. Genetic variability in GLP-1 receptor is associated with inter-individual differences in weight lowering potential of liraglutide in obese women with PCOS: a pilot study. Eur. J. Clin. Pharmacol. 2015;71:817–824. doi: 10.1007/s00228-015-1868-1. [DOI] [PubMed] [Google Scholar]
- 30.Kristensen S.L., Rørth R., Jhund P.S., Docherty K.F., Sattar N., Preiss D., Køber L., Petrie M.C., McMurray J.J.V. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019;7:776–785. doi: 10.1016/S2213-8587(19)30249-9. [DOI] [PubMed] [Google Scholar]
- 31.Marso S.P., Bain S.C., Consoli A., Eliaschewitz F.G., Jódar E., Leiter L.A., Lingvay I., Rosenstock J., Seufert J., Warren M.L., SUSTAIN-6 Investigators Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016;375:1834–1844. doi: 10.1056/NEJMoa1607141. [DOI] [PubMed] [Google Scholar]
- 32.Husain M., Birkenfeld A.L., Donsmark M., Dungan K., Eliaschewitz F.G., Franco D.R., Jeppesen O.K., Lingvay I., Mosenzon O., Pedersen S.D., PIONEER 6 Investigators Oral Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2019;381:841–851. doi: 10.1056/NEJMoa1901118. [DOI] [PubMed] [Google Scholar]
- 33.Petri K.C.C., Ingwersen S.H., Flint A., Zacho J., Overgaard R.V. Exposure-response analysis for evaluation of semaglutide dose levels in type 2 diabetes. Diabetes Obes. Metab. 2018;20:2238–2245. doi: 10.1111/dom.13358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilding J.P., Overgaard R.V., Jacobsen L.V., Jensen C.B., le Roux C.W. Exposure-response analyses of liraglutide 3.0 mg for weight management. Diabetes Obes. Metab. 2016;18:491–499. doi: 10.1111/dom.12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Neil P.M., Birkenfeld A.L., McGowan B., Mosenzon O., Pedersen S.D., Wharton S., Carson C.G., Jepsen C.H., Kabisch M., Wilding J.P.H. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet. 2018;392:637–649. doi: 10.1016/S0140-6736(18)31773-2. [DOI] [PubMed] [Google Scholar]
- 36.Iepsen E.W., Zhang J., Thomsen H.S., Hansen E.L., Hollensted M., Madsbad S., Hansen T., Holst J.J., Holm J.C., Torekov S.S. Patients with Obesity Caused by Melanocortin-4 Receptor Mutations Can Be Treated with a Glucagon-like Peptide-1 Receptor Agonist. Cell Metab. 2018;28:23–32.e3. doi: 10.1016/j.cmet.2018.05.008. [DOI] [PubMed] [Google Scholar]
- 37.Bækdal T.A., Breitschaft A., Navarria A., Hansen C.W. A randomized study investigating the effect of omeprazole on the pharmacokinetics of oral semaglutide. Expert Opin. Drug Metab. Toxicol. 2018;14:869–877. doi: 10.1080/17425255.2018.1488965. [DOI] [PubMed] [Google Scholar]
- 38.Edgerton D.S., Moore M.C., Gregory J.M., Kraft G., Cherrington A.D. Importance of the route of insulin delivery to its control of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2021;320:E891–E897. doi: 10.1152/ajpendo.00628.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pieber T.R., Bode B., Mertens A., Cho Y.M., Christiansen E., Hertz C.L., Wallenstein S.O.R., Buse J.B., PIONEER 7 Investigators Efficacy and safety of oral semaglutide with flexible dose adjustment versus sitagliptin in type 2 diabetes (PIONEER 7): a multicentre, open-label, randomised, phase 3a trial. Lancet Diabetes Endocrinol. 2019;7:528–539. doi: 10.1016/S2213-8587(19)30194-9. [DOI] [PubMed] [Google Scholar]
- 40.Pratley R., Amod A., Hoff S.T., Kadowaki T., Lingvay I., Nauck M., Pedersen K.B., Saugstrup T., Meier J.J., PIONEER 4 Investigators Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): a randomised, double-blind, phase 3a trial. Lancet. 2019;394:39–50. doi: 10.1016/S0140-6736(19)31271-1. [DOI] [PubMed] [Google Scholar]
- 41.Kapitza C., Nosek L., Jensen L., Hartvig H., Jensen C.B., Flint A. Semaglutide, a once-weekly human GLP-1 analog, does not reduce the bioavailability of the combined oral contraceptive, ethinylestradiol/levonorgestrel. J. Clin. Pharmacol. 2015;55:497–504. doi: 10.1002/jcph.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Overgaard R.V., Delff P.H., Petri K.C.C., Anderson T.W., Flint A., Ingwersen S.H. Population Pharmacokinetics of Semaglutide for Type 2 Diabetes. Diabetes Ther. 2019;10:649–662. doi: 10.1007/s13300-019-0581-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu C., Zhang J., Zhou H. Confirmatory analysis for phase III population pharmacokinetics. Pharm. Stat. 2011;10:14–26. doi: 10.1002/pst.403. [DOI] [PubMed] [Google Scholar]
- 44.Overgaard R.V., Ingwersen S.H., Tornøe C.W. Establishing Good Practices for Exposure-Response Analysis of Clinical Endpoints in Drug Development. CPT Pharmacometrics Syst. Pharmacol. 2015;4:565–575. doi: 10.1002/psp4.12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Original data may be reviewed consistent with conditions within the informed consent signed by participants in the PIONEER and SUSTAIN clinical trial programs, upon contacting the authors, upon request to the lead contact, Daniel Drucker.
-
•
No new code was generated in this study. For the oral semaglutide pharmacokinetics model, parameterization and NONMEM computer program settings for parameter estimation are provided in the Methods S1 and S2.
-
•
The patient level data reported in this study cannot be deposited in a public repository because of limitations associated with the informed consent for the SUSTAIN and PIONER trials.
-
•
To request access, contact the Lead contact DanieljDrucker@gmail.com.
-
•
All of the data reported in this manuscript will be shared upon contact of the lead author Danieljdrucker@gmail.com.
-
•
Any additional information required to re-analyze the data reported in this work paper is available from the Lead Contact upon request.