

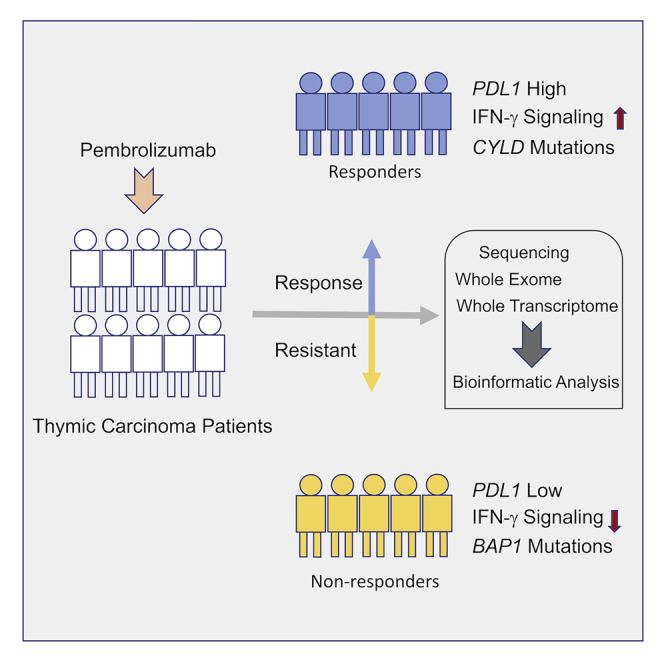
Summary
Thymic carcinoma is rare and has a poorer prognosis than thymomas. The treatment options are limited after failure of platinum-based chemotherapy. We previously performed a single-center phase II study of pembrolizumab in patients with advanced thymic carcinoma, showing a 22.5% response rate. Here, we characterize the genomic and transcriptomic profile of thymic carcinoma samples from 10 patients (5 non-responders versus 5 responders) in this cohort, with the main aim of identifying potential predictors of response to immunotherapy. We find that expression of PDL1 and alterations in genes or pathways that correlated with PD-L1 expression (CYLD and BAP1) could be potential predictors for response or resistance to immunotherapy in patients with advanced thymic carcinoma. Our study provides insights into potential predictive markers/pathways to select patients with thymic carcinoma for anti-PD-1 immunotherapy.
Keywords: thymic carcinoma, immune checkpoint inhibitors, whole exome sequencing, whole-transcriptome sequencing, predictors of response
Graphical abstract
Highlights
Expression of PDL1 is a predictor for response to immunotherapy in thymic carcinoma
Alterations of CYLD are exclusively found in tumor samples of the responders
Alterations of BAP1 are exclusively found in tumor samples of the non-responders
He et al. characterize the genomic and transcriptomic profile of thymic carcinoma samples from 10 patients treated with pembrolizumab. They find that expression of PDL1 and alterations in genes or pathways that correlated with PD-L1 expression could be potential predictors for response to immunotherapy in patients with advanced thymic carcinoma.
Introduction
Thymic carcinoma is a rare and highly aggressive malignancy derived from the thymic epithelial cells.1 They often metastasize to distant organs, and overall survival is much shorter than that of thymomas. Five-year survival rates for thymic carcinoma at stages I+II, III, and IV are 88.2%, 51.7%, and 37.6%, respectively, whereas they are 100%, 98.4%, 88.7%, 70.6%, and 52.8% for thymoma at stages I, II, III, IVA, and IVB, respectively.2 Often, thymic carcinomas are identified at an advanced stage resulting in poor prognosis.3 Platinum-based chemotherapy is the standard treatment for patients who are not operable, but responses are usually short lived in patients with advanced disease. There is a paucity of available treatments after failure of platinum-based chemotherapy. This is partly due to a poor understanding of the biology of these tumors.
The molecular drivers of thymic epithelial tumors (TETs) remain largely unknown. We previously identified a recurrent mutation in the GTF2I gene that is present in over 70% of type A and AB thymomas and rare in thymic carcinomas and identified recurrent mutations in TP53, CYLD, CDKN2A, BAP1, and PBRM1 genes in thymic carcinomas.4 Other reports described recurrent mutations in HRAS, NRAS, SETD2, FBXW7, and RB1 genes in thymic carcinoma.5,6 TETs have a very low average tumor mutation burden (TMB) compared to most adult tumor types, but thymic carcinomas have a higher TMB than thymomas.5 Recently, high PD-L1 expression has been reported in TETs,7, 8, 9 and immunotherapy, targeting PD-1/PD-L1, has shown activity in patients with TETs.10, 11, 12, 13, 14
We completed a single-center phase II study of the PD-1 antibody pembrolizumab in patients with advanced thymic carcinoma, for which 22.5% of 40 patients achieved a durable objective response.14 In that study, patients with a high expression of PD-L1 and a gamma-interferon signature in the tumor cells were more likely to respond to pembrolizumab.14 To better understand the molecular predictors of response to anti PD-1 therapy in patients with thymic carcinoma, we characterized the genomic profile of 10 patient samples (5 responders versus 5 non-responders) by using whole-exome sequencing and whole-transcriptome sequencing. We found that expression of PDL1 and alterations in genes or pathways that correlated with PD-L1 expression (CYLD and BAP1) could be potential predictors for response or resistance to immunotherapy for patients with advanced thymic carcinoma.
Results
A total of 10 patients with recurrent thymic carcinoma were included (Table 1; Figure S1). Four out of the five non-responders had progressive disease as best response and one had a stable disease of short duration (82 days), and all are deceased. Four of the five responders had a partial response and one a complete response, and all were alive at the time of manuscript publication. Three patients developed serious autoimmune disorders after pembrolizumab exposure, two were non-responders, and one was a responder.
Table 1.
Patient characteristics
| Patient ID | ECOG | Sex | Race | Histology | PD-L1 expression (%) | Response to pembrolizumab | Autoimmune disorder | Survival status |
|---|---|---|---|---|---|---|---|---|
| PD1-001 | 1 | Female | Caucasian | Poorly differentiated | 0 | PD | No | Deceased |
| PD1-003 | 0 | Female | Caucasian | Poorly differentiated | 10 | PD | Polymyositis, myocarditis, hepatitis | Deceased |
| PD1-019 | 0 | Female | Asian | Squamous | 5 | SD | Diabetes type I, pancreatitis, hepatitis | Deceased |
| PD1-020 | 0 | Male | Caucasian | Squamous | 0 | PD | No | Deceased |
| PD1-027 | 1 | Female | Caucasian | Poorly differentiated | 0 | PD | No | Deceased |
| PD1-004 | 1 | Female | Caucasian | Squamous | 80 | PR | Hepatitis | Alive |
| PD1-005 | 0 | Male | Black | Squamous | 80 | CR | No | Alive |
| PD1-011 | 0 | Male | Caucasian | Squamous | 80 | PR | No | Alive |
| PD1-017 | 0 | Male | Caucasian | Poorly differentiated | 10 | PR | No | Alive |
| PD1-032 | 0 | Male | Caucasian | Poorly differentiated | 15 | PR | No | Alive |
PD, progressive disease; SD, stable disease; PR, partial response; CR, complete response. ECOG, Eastern Cooperative Oncology Group.
Mutational landscape
We performed whole-exome sequencing on 10 paired tumor/normal (blood) samples from thymic carcinoma patients. The average sequencing depth observed in the tumor samples and normal samples was 150× and 50×, respectively. PD1-027 was removed from the analyses because the quality of the data was poor. The pipeline detected both somatic and germline variants, and there was a total of 13,383 non-coding and 2,187 coding mutations (Figure 1A). These variants were further investigated using criteria described previously.15
Figure 1.

Summary of mutations and SNVs in the thymic carcinoma samples
(A) Summary of mutations, gene function, and sequence ontologies in samples of patients with thymic carcinoma (n = 9) (4 non-responders versus 5 responders). Distribution of indicated mutations by gene functions and sequence ontologies are displayed as pie charts.
(B) Number of SNVs with allelic frequency greater than 10% in samples of patients with thymic carcinoma (n = 9) are shown in the bar graph (4 non-responders versus 5 responders). See also Figures S1 and S2.
The TMB was calculated as the non-synonymous mutations/megabase (mut/Mb) of the whole-exome sequencing data. The TMB data ranged from 0.44 to 3.95 mut/Mb (Table 2). There was no significant difference in TMB between the responders and the non-responders (p = 0.4241). However, the two responders PD1-017 and PD1-032 showed significantly higher TMB than the rest of the responders (p < 0.0001) or the non-responders (p = 0.007). Next, the SNVs with allelic frequency greater than 10% were analyzed in the two groups. The non-responder group had an average of 44 SNVs with a total of 176, whereas the responder group showed a total of 397 variants (79.4 on average). However, consistent with the TMB data, most SNVs were detected in the two responder samples PD1-017 and PD1-032 with 165 and 154 SNVs, respectively (Figure 1B). The distribution of variants in the chromosomes was analyzed using a circos plot (Figure S2). The number of mutations in chromosomes 7 and 13 was more pronounced in the responders than in the non-responders, whereas that of chromosome 3 was greater in the non-responders.
Table 2.
Summary of tumor mutation burden
| Responder category | Sample | Tumor mutation burden (Mut/Mb) |
|---|---|---|
| Non-responder | PD1-001 | 1.05 |
| Non-responder | PD1-003 | 1.03 |
| Non-responder | PD1-019 | 0.95 |
| Non-responder | PD1-020 | 1.77 |
| Responder | PD1-004 | 0.74 |
| Responder | PD1-005 | 0.64 |
| Responder | PD1-011 | 0.44 |
| Responder | PD1-017 | 3.95 |
| Responder | PD1-032 | 3.92 |
The somatic variants observed in genes with known clinical significance or detected in the COSMIC database were further analyzed. Genes that are recurrently mutated in the non-responders include BAP1 and TP53. In the responders, CDKN2A, CYLD, and TET2 were mutated in more than one sample (Figure 2A; Table 3). All of these genes were reported as recurrently mutated genes in thymic carcinoma.16 TP53, the most frequently mutated gene in thymic carcinoma, was mutated in two non-responders (PD1-001 and PD1-003) and one responder (PD1-017). From our previous study, TP53 mutations were found in 13 samples, namely, in 11 non-responders (84.6%) and 2 responders (15.4%).14 Although TP53 mutations was not exclusively found in non-responders, it is interesting that TP53 mutations were found in more non-responders than responders in both studies. Moreover, mutations in BAP1 were found in three out of four samples in the non-responder group but none in the samples from the responders. We previously reported that BAP1 mutations were correlated with low PD-L1 expression in thymic carcinoma.14 Whether mutations of BAP1 may affect PD-L1 expression and therefore the responsiveness of thymic carcinoma to immunotherapy remains to be investigated.
Figure 2.

Mutational landscape of thymic carcinoma patients treated with pembrolizumab
(A) Clinically relevant somatic mutations as revealed by whole-exome sequencing (n = 9) (4 non-responders versus 5 responders). ∗, represents recurrently mutated genes in the responders. #, represents recurrently mutated genes in the non-responders.
(B) Copy number variations in the indicated samples (n = 9) (4 non-responders versus 5 responders). ∗, represents the genes with copy loss in the same samples.
(C) Number of genes that show germline mutations in each patient sample (n = 9) (4 non-responders versus 5 responders). Germline mutations were determined by whole-exome sequencing with blood-derived DNA samples. See also Figure S3 and Table S2.
Table 3.
Summary of somatic variants
| Gene | Variant for: |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Non-responder |
Responder |
||||||||
| PD1-001 | PD1-003 | PD1-019 | PD1-020 | PD1-004 | PD1-005 | PD1-011 | PD1-017 | PD1-032 | |
| ABL1 | p.E274K | ||||||||
| ACVR1 | p.V135A | ||||||||
| ACVR2A | p.E214K | ||||||||
| ARID1A | p.R2236fs | ||||||||
| BAP1 | p.L6fs | p.Q36∗ | p.E200∗ | ||||||
| BRAC1 | p.T276R | ||||||||
| CDKN2A | p.R80∗ | p.V51fs | |||||||
| CUX1 | p.D1400N | ||||||||
| CYLD | p.S331∗ | p.R850∗ | |||||||
| EP300 | p.M169I | ||||||||
| ERBB3 | p.V104L | ||||||||
| EZH2 | p.Y646F | ||||||||
| FANCG | p.E436Q | ||||||||
| FBXW7 | p.D607fs | ||||||||
| FGFR2 | p.S432L | ||||||||
| FGFR4 | p.R78H | ||||||||
| FLCN | p.R401H | ||||||||
| KAT6A | p.R1877H | ||||||||
| KMT2C | p.I4448fs | ||||||||
| KMT2D | p.P4241R | ||||||||
| NFKBIE | p.H240L | ||||||||
| PTPN13 | p.S1678L | ||||||||
| ROS1 | p.E395Q | ||||||||
| SETD2 | p.E1036∗ | ||||||||
| SOCS1 | p.A44V | ||||||||
| TET1 | p.V1312M | ||||||||
| TET2 | p.T1063fs | p.E1166∗/p.Q1083∗ | |||||||
| TP53 | p.C135Y | p.M237I | p.D148_153del | ||||||
| WAS | p.R94W | ||||||||
We also found unique alterations in the samples from the responders. CYLD mutations p.S331∗ and p.R850∗ occurred in the two responders PD1-004 and PD1-011, respectively, but not in the non-responders (Figure 2A). We previously showed that mutations of CYLD are associated with high PD-L1 expression in thymic carcinoma,14 and recently, we have demonstrated that downregulation of CYLD is associated with PD-L1 expression mediated by interferon-gamma in TET cells.17 Consistent with our previous findings, PD1-004 and PD1-011 showed higher mRNA expression of PD-L1 than the non-responders (p = 0.0031) (Table S1). Our finding suggests that CYLD mutations are positively correlated with PD-L1 expression and could be a potential predictor for response to immunotherapy.
In addition, alterations in the CDKN2A gene occurred in four out of five samples. PD1-004 and PD1-011 had CDKN2A mutations (stop gain and frameshift), whereas PD1-005 and PD1-017 had copy number loss of CDKN2A (Figures 2A and 2B). Interestingly, the two responders (PD1-005 and PD1-017) who had copy loss of CDKN2A also showed MTAP copy loss (Figure 2B). Deficiency of the MTAP gene occurs in multiple tumor types, and this gene is frequently co-deleted with CDKN2A or CDKN2B genes.18 Our sequencing data from the two responders are consistent with those of previous reports.19,20 The MTAP gene encodes methylthioadenosine phosphorylase (MTAP), which is an important enzyme in the salvage of both adenine and methionine. MTAP cleaves methylthioadenosine (MTA) into 5-methylthioribose-1-phosphate.21 Loss of the MTAP gene results in production of MTA, which attenuates antitumor immunity.22 The role of MTAP loss in thymic carcinoma is unclear. In addition, two responders, namely, PD1-004 and PD1-005, harbored TET2 mutations and one responder, PD1-032, had a TET1 mutation. Deletion of TET2 in myeloid cells increased tumor-infiltrating T cells.23 Mutations in TET1 were shown as a potential predictor for response to immune checkpoint inhibitors (ICIs) in multiple tumor types, including non-small-cell lung cancer (NSCLC).24 Although the number of samples is small, the observation that mutations of TET2 and TET1 were only found in the responders is interesting, and the precise role of these genes in thymic carcinoma remains to be explored in a larger cohort of patients.
Germline variants were identified from the blood control samples (Figure 2C). PD1-005 had the highest number of germline variants (Figure 2C; Table S2). All nine samples, including both responders and non-responders, showed germline variants in APOB with unknown significance, and seven samples out of nine showed variants in CACNA1S (Table S2). These two genes are not frequently mutated in thymic carcinoma.
Moreover, we also found recurrently mutated genes with unknown clinical significance, including CKAP2 and YWHAE in the non-responders and NPTX2, NAA35, and TGFB2 in the responders (Figure S3). Interestingly, the non-responders who had a CKAP2 alteration also harbored TP53 mutations. CKAP2 was identified as a novel TP53 target gene.25 Overexpression of CKAP2 resulted in aneuploidy with aberrant centrosome numbers in the absence of TP53.25 Tumor aneuploidy was reported to correlate with a reduced response to immunotherapy.26 The role of these genes with unknown significance in response to immunotherapy for patients with advanced thymic carcinoma remains to be determined.
Differential gene expression and pathway enrichment analysis using RNA sequencing
The full transcriptional landscape of all samples was investigated by RNA sequencing. However, samples PD1-032 and PD1-019 were removed from the final analysis due to poor quality, leaving eight samples, including four responders and four non-responders (Figure S1). We identified a total of 2,801 differentially expressed genes (DEGs), including 1,341 upregulated and 1,460 downregulated DEGs, in the non-responders when compared to the responders, by using strict filtering parameters with a significance level of false discovery rate (FDR) of ≤0.1% and log fold change of 2 (Figure 3A; Table S3). To assess the pathways involved, we performed pathway analysis with gProfiler (https://biit.cs.ut.ee/gprofiler/gost),27 by using both the upregulated and the downregulated DEG lists of non-responders versus responders. Only three pathways were significantly enriched in the upregulated DEGs (Figure S4A; Table S4). However, none of them was related to immune response or tumorigenesis. Interestingly, 37 pathways were significantly enriched in the downregulated DEGs (Table S5), among which 10 pathways were related to immune response or tumorigenesis, including cytokine-cytokine receptor interaction, hematopoietic cell lineage, cell adhesion molecules, chemokine signaling pathway, intestinal immune network pathway, natural killer cell mediated cytotoxicity, tumor necrosis factor (TNF) signaling pathway, antigen processing and presentation, nuclear factor κB (NF-κB) signaling pathway, and Peroxisome proliferator-activated receptors (PPAR) signaling pathway (Figure 3B; Figure S4B; Table S5). We also performed the gene set enrichment analysis (GSEA) with Kyoto Encyclopedia of Genes and Genomes (KEGG) gene sets by using a pre-ranked DEG list and found 21 signaling pathways that were negatively enriched in the DEGs of the non-responders versus the responders (Table S6).28 A total of 13 of 21 pathways overlapped with those identified by gProfiler analysis (Tables S5 and S6). Particularly, cytokine-cytokine receptor interaction, natural-killer-cell-mediated cytotoxicity, and antigen processing and presentation pathways were revealed from both analyses, and they have been reported to be involved in resistance to immunotherapy (Figure S5A).29, 30, 31 Next, we assessed the expression of genes from each pathway above in samples from both the non-responders and the responders (Figure 3C; Figure S6). The expression of most genes in the indicated pathways was much lower in the samples from the non-responders than that in the samples from the responders, which is consistent with the result of the pathway analysis. In addition, we performed GSEA analyses with hallmark gene sets by using the pre-ranked DEGs of the non-responder versus the responders (Table S7). A total of 11 hallmark pathways were negatively enriched in the DEGs, among which 6 pathways were related to immune response and/or tumorigenesis. These pathways include interferon gamma response, inflammatory response, interferon alpha response, Tumor necrosis factor alpha (TNFA) signaling by NF-κB, interleukin-6 (IL-6)-JAK-STAT3 signaling, and IL2-STAT5-signaling (Figure 3D and 3E; Figure S5B). Moreover, we also assessed the expression of some immune checkpoint regulators in the samples from both the non-responders and the responders (Figure 3F; Table S1).32 In comparison to the responder samples, expression of most regulator genes was lower in the non-responder samples, except for CD276. Specifically, expression of PDL1 and CTLA4 was significantly lower in all non-responder samples than that in the responders. Expression of IDO1 was significantly higher in the responder samples, especially in PD1-004 and PD1-005. In addition, expression of TIGIT, CD96, and ICOS was slightly lower, albeit statistically significant, in the non-responder samples than that in the responder samples. Interestingly, the average expression of CD276 was slightly higher in the non-responder group than that in the responder group. PD1-001 and PD1-003 expressed a higher level of CD276 than all other samples (p = 0.0054).
Figure 3.

Determining the signaling pathways and molecular predictors in the non-responders and the responders using RNA sequencing (RNA-seq)
(A) Pie diagram showing the number of differentially expressed genes (DEGs) in the non-responders versus the responders, including 1,341 upregulated DEGs and 1,460 downregulated DEGs.
(B) Ten significantly enriched pathways in downregulated DEGs. Pathway analysis was performed with downregulated DEG list using gProfiler. Ten pathways related to immune response or tumorigenesis were selected and presented in a bar graph. The x axis represents −log2 (p value). ∗, represents the pathways being validated in (C).
(C) Heatmap of 10 representative DEGs involved in the indicated pathways (n = 8; 4 non-responders versus 4 responders). Heatmap with additional genes in the indicated pathway is shown in Figure S6.
(D) GSEA analyses with hallmark gene sets reveal negative enrichment of pathways in DEGs of the non-responders versus the responders. Six pathways related to immune response or tumorigenesis were selected and presented in a bar graph. The x axis represents Normalized enrichment score (NES) scores.
(E) GSEA plot of interferon gamma response pathway.
(F) Heatmap showing the expression pattern of representative immune checkpoint regulators in the indicated samples (n = 8; 4 non-responders versus 4 responders). ∗, stands for the genes from the DEG list that statistically significant. See also Figures S4–S6 and Tables S1, S3, S4, S5, S6, and S7.
Immune signature analysis
To profile immune infiltration in the non-responder and the responder samples, we performed immune cell gene signature analyses with CIBERSORT, which allowed us to identify 22 immune subpopulations based on the expression signatures of 547 genes. We found that the non-responders had an increased fraction of M2 macrophages (p = 0.02), whereas the responders showed a higher fraction of CD4+ memory resting T cells (p = 0.01) and activated dendritic cells (p = 0.04) (Figures 4A and B). To validate the results from the CIBERSORT, we performed multiplex immunofluorescence staining with CD3, CD4, CD25, CD45RO, CD45RA, and CD163 antibodies. We found that there are more CD163+ cells in the non-responder tissue than that in the responder tissue (Figures 4C, 4E, and 4F), which is consistent with the increased fraction of M2 macrophages in the non-responders. Interestingly, most of CD3+ cells are CD4− in both the non-responder and the responder tissues (Figure S7). To investigate whether the CD3+CD4− cells are CD8+ cells, we next performed immunohistochemistry (IHC) staining with both CD8 and CD4 antibodies (Figure 4D). We found that there are more CD8+ cells than CD4+ cells in both the non-responder and the responder tissues (Figure 4I). Interestingly, there are more CD4+ cells in the responder tissues than in the non-responder tissues (Figures 4G and H).
Figure 4.

CIBERSORT analysis of immune gene signatures in the non-responders and the responders
(A and B) Different proportion of 22 types of immune cells that are associated with the samples were identified with CIBERSORT in both non-responders (A) and responders (B). (A) Result of CIBERSORT analysis in the non-responders. ∗, stands for the cell population that is significantly different in the non-responders, in comparison to the responders. (B) Result of CIBERSORT analysis in the responders. ∗, stands for the cell population that is significantly different in the responders, in comparison to the non-responders. The non-responders had higher fraction of M2 macrophages (p = 0.02), whereas the responders showed a higher fraction of CD4+ memory resting T cells (p = 0.01) and activated dendritic cells (p = 0.04).
(C) Representative images of IF staining with thymic carcinoma tissues from the non-responder and the responder using CD163 antibody (magenta). DAPI was used as a nuclear marker (dark blue). The scale bar represents 100 μm.
(D) Representative images of double IHC staining with thymic carcinoma tissues from the non-responder and the responder by using both CD8 (red) and CD4 (brown) antibodies. The scale bar represents 40 μm.
(E) Bar graph shows the percentage of CD163+ cells in both the non-responder and the responder groups, based on the IF staining. Six areas from each group were selected, and CD163+ and CD163− cells were counted using ImageJ. ∗∗∗∗p < 0.0001.
(F) Bar graph shows the percentage of CD163+ cells among CD163+ and CD3+ cells in both the non-responder and the responder groups, based on the IF staining. Six areas from each group were selected, and CD163+ and CD3+ cells were counted using ImageJ. ∗∗∗∗p < 0.0001.
(G) Bar graph showing the percentage of CD4+ cells in both the non-responder and the responder groups, based on IHC staining. Six areas from each group were selected, and CD4+ cells and CD4− cells were counted using ImageJ. ∗∗∗p < 0.001.
(H) Bar graph showing the percentage of CD4+ cells among CD4+ and CD8+ cells in both the non-responder and the responder groups, based on IHC staining. Six areas from each group were selected, and CD4+ cells and CD8+ cells were counted using ImageJ. ∗∗p < 0.01. The above data are presented as the mean ± SEM (n = 6).
(I) Bar graph showing the percentage of CD8+ and CD4+ cells in both the non-responder and the responder groups, based on the counting results from (H); the average percentage in each group was used to generate the graph. Also see Figure S7.
Discussion
ICIs have been approved to treat many malignancies.10, 11, 12, 13,33 However, most patients do not respond to the treatment, and there has been growing interest in identifying predictive markers for response to immunotherapy.34 The characterized predictive markers of response are PD-L1 expression and TMB, which have been used to guide treatment decisions for multiple tumor types.35, 36, 37 Many other potential predictors of response have also been identified, such as mutations in SERPINB3 and SERPINB4 genes, and high expression of CTLA4 along with interferon gamma target genes.38,39 In addition, predictors of resistance to immunotherapy have also been identified, such as a decrease in major histocompatibility complex class I (MHC class I) expression, impaired interferon gamma pathway, deregulation of wnt/beta-catenin pathway, loss of PTEN, and mutations in LKB1.30,40, 41, 42, 43 These predictors of response appear to be somewhat dependent on the tumor type. The predictors of response or resistance to immunotherapy remain largely unexplored for rare tumor types such as thymic carcinoma. Through whole-exome sequencing and whole-transcriptome sequencing, we characterized five responders and five non-responders with the aim of discovering potential predictors. Although the sample size is small, this exploratory analysis identified biomarkers that should be validated in larger cohorts of patients in the future.
Through whole-exome sequencing, we found that two responders, namely, PD1-017 and PD1-032, showed significantly higher TMB than either the rest of the responders (p < 0.0001) or the non-responders (p = 0.007). A TMB of 10 mut/Mb or more was reported as a predictor for better response to ICIs in NSCLC patients.44 The TMB of TETs is lower than that of most adult tumors and is similar to that of pediatric tumors for which only a limited number of genetic abnormalities are identified.5 The mean TMB was 0.48 mut/Mb for patients with TETs, whereas it was 9.1 mut/Mb for patients with lung adenocarcinoma.5,45 Although the TMBs of the two responders (3.92 and 3.95 mut/Mb) were much higher than the average TMB in TETs, the other two responders had low TMB. Therefore, whether the TMB can be a predictive marker for response needs to be further validated in a larger cohort of patients.
Whole-exome sequencing also allowed us to identify recurrent mutations that may predict response. Particularly, we found that alterations in genes that correlated with PD-L1 expression (CYLD and BAP1) are promising predictors for response or resistance to immunotherapy, although further validation in a larger cohort of patients is needed. CYLD mutations occurred in the two responders PD1-004 and PD1-011 but not in the non-responders (Figure 2A). We have reported that CYLD is recurrently mutated in these tumors (five responders), and it was positively correlated with PD-L1 expression in thymic carcinoma.14 More recently, we also reported that downregulation of CYLD was associated with PD-L1 expression mediated by interferon gamma in TET cells.17 In the current study, we demonstrated that, in comparison to the non-responders, the response of the interferon gamma pathway was enhanced in the responder and that mRNA expression of PD-L1 was also higher in the responders, especially in the samples with CYLD mutations (PD-1-004 and PD1-011) (Figure 3F). Our findings are consistent with the previous data and suggest that CYLD mutations could be a potential predictor of response to ICIs. Intriguingly, we found three out of four samples in the non-responder group that had mutations in the BAP1 gene (Figure 2A). The BAP1 gene encodes a deubiquitinating enzyme, which functions as a tumor suppressor and regulates multiple cellular pathways including cell cycle, cell differentiation, cell death, and DNA damage response.46, 47, 48 Loss of function of BAP1 induced chemoresistance of mesothelioma cells and was associated with an immunosuppressive microenvironment in uveal melanomas.32,49 Mutation of BAP1 in TETs has been reported previously4 and was correlated with low PD-L1 expression.14 It is worth noting that the status of the BAP1 gene in PD1-001 was inconsistent between whole-exome sequencing in the current study and targeted exome sequencing from previous study.14 Although mutations in EZH2, TP53, and WAS were found in both whole-exome and targeted exome sequencing, mutation of BAP1 was only revealed from whole-exome sequencing but not in the targeted exome sequencing. In addition, a GTF2I mutation was found in PD1-003 by targeted sequencing in our previous study but was not revealed by whole-exome sequencing in the current study. The inconsistency may be caused by the heterogeneity of the tumor.
The association between the transcriptome profile and response to immunotherapy has been studied in various tumor types, such as NSCLC, head and neck squamous cell carcinoma, melanoma, and renal cell carcinoma.50, 51, 52, 53, 54, 55 However, there are only a few studies in thymic carcinoma.12,14 Through RNA sequencing, we found that all the responders had significantly higher PDL1 expression than the non-responders. High expression of PD-L1 has been reported to predict response to immune checkpoint inhibitors in NSCLC and melanoma patients44,55, 56, 57 and is approved in some settings for selection of patients. Our finding in the current study is consistent with previous results. Through GSEA, we found that the interferon gamma response gene set was significantly reduced in the non-responders, in comparison to the responders. Impaired interferon gamma signaling pathways have been reported to be involved in resistance to immunotherapy,58 and an interferon-gamma-related mRNA profile has been used to predict clinical response to PD-1 blockade in melanoma and NSCLC patients.59, 60, 61 We previously performed NanoString gene expression profiling and found that the interferon gamma signature was higher in the responders than that in the non-responders, suggesting a predictive role of the interferon gamma pathway in response to anti-PD-1 in thymic carcinoma patients.14 In the current study, by using RNA sequencing and GSEA, we observed that the interferon gamma response gene set was downregulated in the non-responders, compared to the responders, which is consistent with our previous findings. However, Cho et al.12 did not find a correlation between interferon-gamma-related genes and clinical outcomes in their patient cohort. These conflicting observations may be caused by the different methods used. Further investigation with a larger cohort of patients is needed in the future. In addition, CIBERSORT allowed us to identify differences in immune infiltrates between responders and non-responders, and we validated the results by multiplex immunofluorescence (IF) and double-staining IHC. However, we were only able to validate the increased fraction of M2 macrophages in the non-responders in comparison to that in the responders. Immunohistochemistry has limitations in identifying many immune populations and is not very effective in capturing functional phenotypes.62 Other techniques, such as CYTOF, might be used for validation of CIBERSORT results.
Taken together, our study provides insights into potential predictive markers of response to anti-PD-1 immunotherapy in advanced thymic carcinoma. In the future, prospective validation of these findings will need to be performed. Targeted sequencing using a customized panel of genes, which are not usually included in most platforms, will likely be necessary.
Limitations of study
The sample size of the current study is small. A validation will be performed in larger cohorts of patients in future studies. Multiplex IF and IHC were performed to validate the CIBESORT results. However, these methods are unable to reliably identify many subtypes of immune cells. CYTOF might be a better method, which will be used in future studies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CD3 | Sigma | Cat# Sab5500058; RRID: AB_:2813777 |
| Anti-CD4 | Leica | Cat# PA0427 |
| Anti-CD25 | Leica | Cat# PA0305 |
| Anti-CD45RA | Abcam | Cat# Ab755; RRID: AB_305970 |
| Anti-CD45RO | Abcam | Cat# Ab23; RRID: AB_449887 |
| Anti-CD163 | Leica | Cat# PA0090 |
| Anti-CD8 | Leica | Cat# PA0183; RRID: AB_10555292 |
| Biological samples | ||
| Tumor and normal (blood) samples from the responders and the non-responders | Georgetown University | NCT02364076 |
| Critical commercial assays | ||
| Tempus xE Next-Generation sequencing | The Tempus | N/A |
| RNA sequencing | The Tempus | N/A |
| Deposited data | ||
| Tempus xE Next-Generation sequencing | This paper | GEO: GSE181815 |
| RNA sequencing | This paper | GEO: GSE181815 |
| Software and algorithms | ||
| Graphpad Prism 7.0 | Graphpad software | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Giuseppe Giaccone (gig4001@med.cornell.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Patient cohort
The 10 patient samples included in the study are summarized in Table 1 and Figure S1. The patients were selected from a single-arm single center phase 2 study of pembrolizumab in patients with recurrent thymic carcinoma who had progressed after at least one line of chemotherapy (NCT02364076).14 Patients with a history of autoimmune disease were excluded. Pembrolizumab 200 mg was given IV every 3 weeks for up to 2 years. In this phase II study 40 patients were enrolled, 9 patients experienced a major response and 6 developed severe autoimmune disorders. The selection of samples was based on availability of sufficient sample material to perform whole exome sequencing and whole transcriptome (RNA) sequencing. The selection of non-responders was also based on patients who either had progressive disease or short stable disease as best response.
Method details
Isolation of genomic DNA and RNA
Tumor DNA and RNA samples were extracted from formalin-fixed-paraffin-embedded (FFPE) tissue blocks of patients with thymic carcinoma using the DNA/RNA AllPrep Kit (QIAGEN, Valencia, CA), as described previously.4 Briefly, FFPE blocks were cut and subjected to hematoxylin and eosin (H&E) staining, and the tumor component was macrodissected from unstained slides that matched to the H&E staining slides. Control DNA for whole exome sequencing was extracted from paired patients’ peripheral blood samples using Genfind v2 kit (Beckman Coulter, Brea, CA).
Whole exome sequencing and data analysis
The whole exome library construction was performed using The Tempus xE hybrid capture Next-Generation sequencing panel (∼20,000 gene) consisting of IDT xGen LockDown probes. The panel was sequenced to a depth of at least 150x for tumor and 50x for germline using 126 bp paired end reads on the Illumina HiSeq 4000. The raw sequencing reads were processed to filter out low quality bases prior to alignment to the human genome and variant calling on the Seven Bridges Cloud Platform and additional variant analysis was performed using CRAVAT that maps variants to genes, identifies base and amino acid alterations, annotates pathogenic and variants of unknown significance, and reports on the depth of coverage and genomic tumor cellularity.
RNA sequencing
The library construction for RNA-seq was performed by Tempus using KAPA HiFi Library Amplification kit.15 The raw FASTQ files were filtered for high quality reads and aligned to Human Reference Genome Version GRCh38.84. The differential gene expression analysis was performed to compare the non-responder versus responder cohort, using EdgeR.63 Pathway enrichment analysis was performed with gProfiler and GSEA using the differentially expressed genes identified from the above analyses.27,28
Immune signature analysis
The transcriptome data was further analyzed using CIBERSORT62 that identifies the immune signature of the samples based on the immune cell fraction of the gene expression profile. The tumor infiltrating immune cell profile was analyzed using both responder versus non-responder cohorts. A set of 547 previously validated immune related genes were filtered out from the differentially expressed genes from the comparisons and input into the CIBERSORT analysis pipeline. The analysis identifies the different proportion of 22 immune cells that are associated with the samples.
Multiplex immunofluorescence and immunohistochemistry staining
Multiplexed immunofluorescence (mIF) was performed by staining 4-μm-thick FFPE thymic carcinoma sections from both the non-responders and the responders in a BondRX automated stainer, as previously described.64 Tonsil tissues were used as positive control. One panel of primary antibody/fluorophore pairs was applied to all cases: (1) anti-CD3 (SP7, 1:100), (2) anti-CD4 (4B12, RTU), (3) anti-CD25 (4C9, RTU), (4) anti-CD45RO (UCH-L1, 1:2000), (5) anti-CD45RA (4KB5, 1: 1000), and (6) anti-CD163 antibody (10D6, RTU). Antibody/Opal fluor combinations were utilized as follows: CD3/480, CD4/620, CD25/570, CD45RO/520, CD45RA/690, and CD163/780. All slides were also stained with 4′,6-diamidino-2-phenylindole (DAPI) for nuclear identification. The images were scanned using the Vectra Polaris quantitative platform (Akoya Biosciences). In brief, whole slides scans were first performed at 20X resolution. Regions of interest were selected, and spectral unmixed in InForm VS 2.4. Unmixed images were analyzed using a combination of QuPath and ImageJ to generate quantitative outputs.
IHC double staining was performed with Leica biosystems BOND III system. Sequential staining with CD4 (PA0427, RTU, Leica) and CD8 (PA0183, RTU, Leica) antibodies was done based on the manufactory’s instruction. BOND polymer refine detection (DS9800) and BOND polymer refine Red detection (DS9390) kits were used to detect CD4 and CD8 respectively. Hematoxylin was used for counter staining. The images were scanned with Aperio system at 20X and regions of interest were selected and the images were analyzed using ImageJ to generate quantitative outputs.
Quantification and statistical analysis
Statistical significance between the two groups was calculated with a two-tailed Student’s t test, and a value of p < 0.05 was considered statistically significant. Statistical calculations were conducted using GraphPad Prism 7 software (GraphPad Software, San Diego, CA).
Additional Resources
The clinical identifier for this study is (NCT02364076).
Acknowledgments
This study was supported by The Eileen Fund (https://theeileenfund.org). We also thank the Tempus company for performing RNA sequencing and whole-exome sequencing for us and for providing reports for whole-exome sequencing.
Author contributions
Y.H. analyzed the RNA sequencing data, whole-exome sequencing data, and wrote the manuscript. A.R. analyzed whole-exome sequencing data and wrote the manuscript. Y.G. and K.B. analyzed the RNA sequencing and whole-exome sequencing data and provided bioinformatic support. G.G. designed the study and wrote and edited the manuscript.
Declaration of interests
The authors have declared that no competing interests exist.
Published: September 3, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100392.
Supplemental information
Data and code availability
Whole exome and transcriptome sequencing data have been deposited in GEO. Accession numbers are listed in the Key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Greene M.A., Malias M.A. Aggressive multimodality treatment of invasive thymic carcinoma. J. Thorac. Cardiovasc. Surg. 2003;125:434–436. doi: 10.1067/mtc.2003.133. [DOI] [PubMed] [Google Scholar]
- 2.Kondo K., Monden Y. Therapy for thymic epithelial tumors: a clinical study of 1,320 patients from Japan. Ann. Thorac. Surg. 2003;76:878–884, discussion 884–885. doi: 10.1016/s0003-4975(03)00555-1. [DOI] [PubMed] [Google Scholar]
- 3.Venuta F., Anile M., Diso D., Vitolo D., Rendina E.A., De Giacomo T., Francioni F., Coloni G.F. Thymoma and thymic carcinoma. Eur. J. Cardiothorac. Surg. 2010;37:13–25. doi: 10.1016/j.ejcts.2009.05.038. [DOI] [PubMed] [Google Scholar]
- 4.Petrini I., Meltzer P.S., Kim I.K., Lucchi M., Park K.S., Fontanini G., Gao J., Zucali P.A., Calabrese F., Favaretto A. A specific missense mutation in GTF2I occurs at high frequency in thymic epithelial tumors. Nat. Genet. 2014;46:844–849. doi: 10.1038/ng.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Radovich M., Pickering C.R., Felau I., Ha G., Zhang H., Jo H., Hoadley K.A., Anur P., Zhang J., McLellan M., Cancer Genome Atlas Network The Integrated Genomic Landscape of Thymic Epithelial Tumors. Cancer Cell. 2018;33:244–258.e10. doi: 10.1016/j.ccell.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saito M., Fujiwara Y., Asao T., Honda T., Shimada Y., Kanai Y., Tsuta K., Kono K., Watanabe S., Ohe Y., Kohno T. The genomic and epigenomic landscape in thymic carcinoma. Carcinogenesis. 2017;38:1084–1091. doi: 10.1093/carcin/bgx094. [DOI] [PubMed] [Google Scholar]
- 7.Katsuya Y., Fujita Y., Horinouchi H., Ohe Y., Watanabe S., Tsuta K. Immunohistochemical status of PD-L1 in thymoma and thymic carcinoma. Lung Cancer. 2015;88:154–159. doi: 10.1016/j.lungcan.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Padda S.K., Riess J.W., Schwartz E.J., Tian L., Kohrt H.E., Neal J.W., West R.B., Wakelee H.A. Diffuse high intensity PD-L1 staining in thymic epithelial tumors. J. Thorac. Oncol. 2015;10:500–508. doi: 10.1097/JTO.0000000000000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weissferdt A., Fujimoto J., Kalhor N., Rodriguez J., Bassett R., Wistuba I.I., Moran C.A. Expression of PD-1 and PD-L1 in thymic epithelial neoplasms. Mod. Pathol. 2017;30:826–833. doi: 10.1038/modpathol.2017.6. [DOI] [PubMed] [Google Scholar]
- 10.Yang Y., Ding L., Wang P. Dramatic response to anti-PD-1 therapy in a patient of squamous cell carcinoma of thymus with multiple lung metastases. J. Thorac. Dis. 2016;8:E535–E537. doi: 10.21037/jtd.2016.06.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zander T., Aebi S., Rast A.C., Zander A., Winterhalder R., Brand C., Diebold J., Gautschi O. Response to Pembrolizumab in a Patient with Relapsing Thymoma. J. Thorac. Oncol. 2016;11:e147–e149. doi: 10.1016/j.jtho.2016.07.018. [DOI] [PubMed] [Google Scholar]
- 12.Cho J., Kim H.S., Ku B.M., Choi Y.L., Cristescu R., Han J., Sun J.M., Lee S.H., Ahn J.S., Park K., Ahn M.J. Pembrolizumab for Patients With Refractory or Relapsed Thymic Epithelial Tumor: An Open-Label Phase II Trial. J. Clin. Oncol. 2019;37:2162–2170. doi: 10.1200/JCO.2017.77.3184. [DOI] [PubMed] [Google Scholar]
- 13.Rajan A., Heery C.R., Thomas A., Mammen A.L., Perry S., O’Sullivan Coyne G., Guha U., Berman A., Szabo E., Madan R.A. Efficacy and tolerability of anti-programmed death-ligand 1 (PD-L1) antibody (Avelumab) treatment in advanced thymoma. J. Immunother. Cancer. 2019;7:269. doi: 10.1186/s40425-019-0723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giaccone G., Kim C., Thompson J., McGuire C., Kallakury B., Chahine J.J., Manning M., Mogg R., Blumenschein W.M., Tan M.T. Pembrolizumab in patients with thymic carcinoma: a single-arm, single-centre, phase 2 study. Lancet Oncol. 2018;19:347–355. doi: 10.1016/S1470-2045(18)30062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beaubier N., Tell R., Lau D., Parsons J.R., Bush S., Perera J., Sorrells S., Baker T., Chang A., Michuda J. Clinical validation of the tempus xT next-generation targeted oncology sequencing assay. Oncotarget. 2019;10:2384–2396. doi: 10.18632/oncotarget.26797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y., Thomas A., Lau C., Rajan A., Zhu Y., Killian J.K., Petrini I., Pham T., Morrow B., Zhong X. Mutations of epigenetic regulatory genes are common in thymic carcinomas. Sci. Rep. 2014;4:7336. doi: 10.1038/srep07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Umemura S., Zhu J., Chahine J.J., Kallakury B., Chen V., Kim I.K., Zhang Y.W., Goto K., He Y., Giaccone G. Downregulation of CYLD promotes IFN-γ mediated PD-L1 expression in thymic epithelial tumors. Lung Cancer. 2020;147:221–228. doi: 10.1016/j.lungcan.2020.07.018. [DOI] [PubMed] [Google Scholar]
- 18.Bertino J.R., Waud W.R., Parker W.B., Lubin M. Targeting tumors that lack methylthioadenosine phosphorylase (MTAP) activity: current strategies. Cancer Biol. Ther. 2011;11:627–632. doi: 10.4161/cbt.11.7.14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Illei P.B., Rusch V.W., Zakowski M.F., Ladanyi M. Homozygous deletion of CDKN2A and codeletion of the methylthioadenosine phosphorylase gene in the majority of pleural mesotheliomas. Clin. Cancer Res. 2003;9:2108–2113. [PubMed] [Google Scholar]
- 20.Chen Z.H., Zhang H., Savarese T.M. Gene deletion chemoselectivity: codeletion of the genes for p16(INK4), methylthioadenosine phosphorylase, and the alpha- and beta-interferons in human pancreatic cell carcinoma lines and its implications for chemotherapy. Cancer Res. 1996;56:1083–1090. [PubMed] [Google Scholar]
- 21.Harasawa H., Yamada Y., Kudoh M., Sugahara K., Soda H., Hirakata Y., Sasaki H., Ikeda S., Matsuo T., Tomonaga M. Chemotherapy targeting methylthioadenosine phosphorylase (MTAP) deficiency in adult T cell leukemia (ATL) Leukemia. 2002;16:1799–1807. doi: 10.1038/sj.leu.2402570. [DOI] [PubMed] [Google Scholar]
- 22.Gjuka D., Georgiou G., Stone E.M. The frequent tumor deletion of MTAP is a newly recognized potent immune checkpoint that is effectively reversed by a human methylthioadenosine degrading drug candidate. Mol. Cancer Ther. 2019;18:LB-A18. [Google Scholar]
- 23.Pan W., Zhu S., Qu K., Meeth K., Cheng J., He K., Ma H., Liao Y., Wen X., Roden C. The DNA Methylcytosine Dioxygenase Tet2 Sustains Immunosuppressive Function of Tumor-Infiltrating Myeloid Cells to Promote Melanoma Progression. Immunity. 2017;47:284–297.e5. doi: 10.1016/j.immuni.2017.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu H.X., Chen Y.X., Wang Z.X., Zhao Q., He M.M., Wang Y.N., Wang F., Xu R.H. Alteration in TET1 as potential biomarker for immune checkpoint blockade in multiple cancers. J. Immunother. Cancer. 2019;7:264. doi: 10.1186/s40425-019-0737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsuchihara K., Lapin V., Bakal C., Okada H., Brown L., Hirota-Tsuchihara M., Zaugg K., Ho A., Itie-Youten A., Harris-Brandts M. Ckap2 regulates aneuploidy, cell cycling, and cell death in a p53-dependent manner. Cancer Res. 2005;65:6685–6691. doi: 10.1158/0008-5472.CAN-04-4223. [DOI] [PubMed] [Google Scholar]
- 26.Davoli T., Uno H., Wooten E.C., Elledge S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. 2017;355:eaaf8399. doi: 10.1126/science.aaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sade-Feldman M., Jiao Y.J., Chen J.H., Rooney M.S., Barzily-Rokni M., Eliane J.P., Bjorgaard S.L., Hammond M.R., Vitzthum H., Blackmon S.M. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017;8:1136. doi: 10.1038/s41467-017-01062-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rieth J., Subramanian S. Mechanisms of Intrinsic Tumor Resistance to Immunotherapy. Int. J. Mol. Sci. 2018;19:E1340. doi: 10.3390/ijms19051340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsu J., Hodgins J.J., Marathe M., Nicolai C.J., Bourgeois-Daigneault M.C., Trevino T.N., Azimi C.S., Scheer A.K., Randolph H.E., Thompson T.W. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Invest. 2018;128:4654–4668. doi: 10.1172/JCI99317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Figueiredo C.R., Kalirai H., Sacco J.J., Azevedo R.A., Duckworth A., Slupsky J.R., Coulson J.M., Coupland S.E. Loss of BAP1 expression is associated with an immunosuppressive microenvironment in uveal melanoma, with implications for immunotherapy development. J. Pathol. 2020;250:420–439. doi: 10.1002/path.5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gong J., Chehrazi-Raffle A., Reddi S., Salgia R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J. Immunother. Cancer. 2018;6:8. doi: 10.1186/s40425-018-0316-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Havel J.J., Chowell D., Chan T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer. 2019;19:133–150. doi: 10.1038/s41568-019-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goodman A.M., Kato S., Bazhenova L., Patel S.P., Frampton G.M., Miller V., Stephens P.J., Daniels G.A., Kurzrock R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017;16:2598–2608. doi: 10.1158/1535-7163.MCT-17-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samstein R.M., Lee C.H., Shoushtari A.N., Hellmann M.D., Shen R., Janjigian Y.Y., Barron D.A., Zehir A., Jordan E.J., Omuro A. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019;51:202–206. doi: 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khunger M., Hernandez A.V., Pasupuleti V., Rakshit S., Pennell N.A., Stevenson J., Mukhopadhyay S., Schalper K., Velcheti V. Programmed Cell Death 1 (PD-1) Ligand (PD-L1) Expression in Solid Tumors As a Predictive Biomarker of Benefit From PD-1/PD-L1 Axis Inhibitors: A Systematic Review and Meta-Analysis. JCO Precis. Oncol. 2017;1:1–15. doi: 10.1200/PO.16.00030. [DOI] [PubMed] [Google Scholar]
- 38.Riaz N., Havel J.J., Kendall S.M., Makarov V., Walsh L.A., Desrichard A., Weinhold N., Chan T.A. Recurrent SERPINB3 and SERPINB4 mutations in patients who respond to anti-CTLA4 immunotherapy. Nat. Genet. 2016;48:1327–1329. doi: 10.1038/ng.3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mo X., Zhang H., Preston S., Martin K., Zhou B., Vadalia N., Gamero A.M., Soboloff J., Tempera I., Zaidi M.R. Interferon-γ Signaling in Melanocytes and Melanoma Cells Regulates Expression of CTLA-4. Cancer Res. 2018;78:436–450. doi: 10.1158/0008-5472.CAN-17-1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skoulidis F., Goldberg M.E., Greenawalt D.M., Hellmann M.D., Awad M.M., Gainor J.F., Schrock A.B., Hartmaier R.J., Trabucco S.E., Gay L. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018;8:822–835. doi: 10.1158/2159-8290.CD-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peng W., Chen J.Q., Liu C., Malu S., Creasy C., Tetzlaff M.T., Xu C., McKenzie J.A., Zhang C., Liang X. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016;6:202–216. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao J., Shi L.Z., Zhao H., Chen J., Xiong L., He Q., Chen T., Roszik J., Bernatchez C., Woodman S.E. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016;167:397–404.e9. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spranger S., Bao R., Gajewski T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 44.Ready N., Hellmann M.D., Awad M.M., Otterson G.A., Gutierrez M., Gainor J.F., Borghaei H., Jolivet J., Horn L., Mates M. First-Line Nivolumab Plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer (CheckMate 568): Outcomes by Programmed Death Ligand 1 and Tumor Mutational Burden as Biomarkers. J. Clin. Oncol. 2019;37:992–1000. doi: 10.1200/JCO.18.01042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berland L., Heeke S., Humbert O., Macocco A., Long-Mira E., Lassalle S., Lespinet-Fabre V., Lalvée S., Bordone O., Cohen C. Current views on tumor mutational burden in patients with non-small cell lung cancer treated by immune checkpoint inhibitors. J. Thorac. Dis. 2019;11:S71–S80. doi: 10.21037/jtd.2018.11.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pan H., Jia R., Zhang L., Xu S., Wu Q., Song X., Zhang H., Ge S., Xu X.L., Fan X. BAP1 regulates cell cycle progression through E2F1 target genes and mediates transcriptional silencing via H2A monoubiquitination in uveal melanoma cells. Int. J. Biochem. Cell Biol. 2015;60:176–184. doi: 10.1016/j.biocel.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 47.Sime W., Niu Q., Abassi Y., Masoumi K.C., Zarrizi R., Køhler J.B., Kjellström S., Lasorsa V.A., Capasso M., Fu H., Massoumi R. BAP1 induces cell death via interaction with 14-3-3 in neuroblastoma. Cell Death Dis. 2018;9:458. doi: 10.1038/s41419-018-0500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu H., Pak H., Hammond-Martel I., Ghram M., Rodrigue A., Daou S., Barbour H., Corbeil L., Hébert J., Drobetsky E. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc. Natl. Acad. Sci. USA. 2014;111:285–290. doi: 10.1073/pnas.1309085110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okonska A., Bühler S., Rao V., Ronner M., Blijlevens M., van der Meulen-Muileman I.H., de Menezes R.X., Wipplinger M., Oehl K., Smit E.F. Functional Genomic Screen in Mesothelioma Reveals that Loss of Function of BRCA1-Associated Protein 1 Induces Chemoresistance to Ribonucleotide Reductase Inhibition. Mol. Cancer Ther. 2020;19:552–563. doi: 10.1158/1535-7163.MCT-19-0356. [DOI] [PubMed] [Google Scholar]
- 50.Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ascierto M.L., McMiller T.L., Berger A.E., Danilova L., Anders R.A., Netto G.J., Xu H., Pritchard T.S., Fan J., Cheadle C. The Intratumoral Balance between Metabolic and Immunologic Gene Expression Is Associated with Anti-PD-1 Response in Patients with Renal Cell Carcinoma. Cancer Immunol. Res. 2016;4:726–733. doi: 10.1158/2326-6066.CIR-16-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daud A.I., Loo K., Pauli M.L., Sanchez-Rodriguez R., Sandoval P.M., Taravati K., Tsai K., Nosrati A., Nardo L., Alvarado M.D. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J. Clin. Invest. 2016;126:3447–3452. doi: 10.1172/JCI87324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang E., Bedognetti D., Marincola F.M. Prediction of response to anticancer immunotherapy using gene signatures. J. Clin. Oncol. 2013;31:2369–2371. doi: 10.1200/JCO.2013.49.2157. [DOI] [PubMed] [Google Scholar]
- 54.Prat A., Navarro A., Paré L., Reguart N., Galván P., Pascual T., Martínez A., Nuciforo P., Comerma L., Alos L. Immune-Related Gene Expression Profiling After PD-1 Blockade in Non-Small Cell Lung Carcinoma, Head and Neck Squamous Cell Carcinoma, and Melanoma. Cancer Res. 2017;77:3540–3550. doi: 10.1158/0008-5472.CAN-16-3556. [DOI] [PubMed] [Google Scholar]
- 55.Conroy J.M., Pabla S., Nesline M.K., Glenn S.T., Papanicolau-Sengos A., Burgher B., Andreas J., Giamo V., Wang Y., Lenzo F.L. Next generation sequencing of PD-L1 for predicting response to immune checkpoint inhibitors. J. Immunother. Cancer. 2019;7:18. doi: 10.1186/s40425-018-0489-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Velcheti V., Chandwani S., Chen X., Pietanza M.C., Piperdi B., Burke T. Outcomes of first-line pembrolizumab monotherapy for PD-L1-positive (TPS ≥50%) metastatic NSCLC at US oncology practices. Immunotherapy. 2019;11:1541–1554. doi: 10.2217/imt-2019-0177. [DOI] [PubMed] [Google Scholar]
- 57.Fumet J.D., Richard C., Ledys F., Klopfenstein Q., Joubert P., Routy B., Truntzer C., Gagné A., Hamel M.A., Guimaraes C.F. Prognostic and predictive role of CD8 and PD-L1 determination in lung tumor tissue of patients under anti-PD-1 therapy. Br. J. Cancer. 2018;119:950–960. doi: 10.1038/s41416-018-0220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jenkins R.W., Barbie D.A., Flaherty K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer. 2018;118:9–16. doi: 10.1038/bjc.2017.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaderbhaï C., Tharin Z., Ghiringhelli F. The Role of Molecular Profiling to Predict the Response to Immune Checkpoint Inhibitors in Lung Cancer. Cancers (Basel) 2019;11:E201. doi: 10.3390/cancers11020201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ayers M., Lunceford J., Nebozhyn M., Murphy E., Loboda A., Kaufman D.R., Albright A., Cheng J.D., Kang S.P., Shankaran V. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017;127:2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ribas A., Robert C., Hodi F.S., Wolchok J.D., Joshua A.M., Hwu W.J., Weber J.S., Zarour H.M., Kefford R., Loboda A. Association of response to programmed death receptor 1 (PD-1) blockade with pembrolizumab (MK-3475) with an interferon-inflammatory immune gene signature. J. Clin. Oncol. 2015;33(15_suppl) 3001-3001. [Google Scholar]
- 62.Chen B., Khodadoust M.S., Liu C.L., Newman A.M., Alizadeh A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018;1711:243–259. doi: 10.1007/978-1-4939-7493-1_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patel S.S., Lipschitz M., Pinkus G.S., Weirather J.L., Pozdnyakova O., Mason E.F., Inghirami G., Hasserjian R.P., Rodig S.J., Weinberg O.K. Multiparametric in situ imaging of NPM1-mutated acute myeloid leukemia reveals prognostically-relevant features of the marrow microenvironment. Mod. Pathol. 2020;33:1380–1388. doi: 10.1038/s41379-020-0498-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole exome and transcriptome sequencing data have been deposited in GEO. Accession numbers are listed in the Key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.