

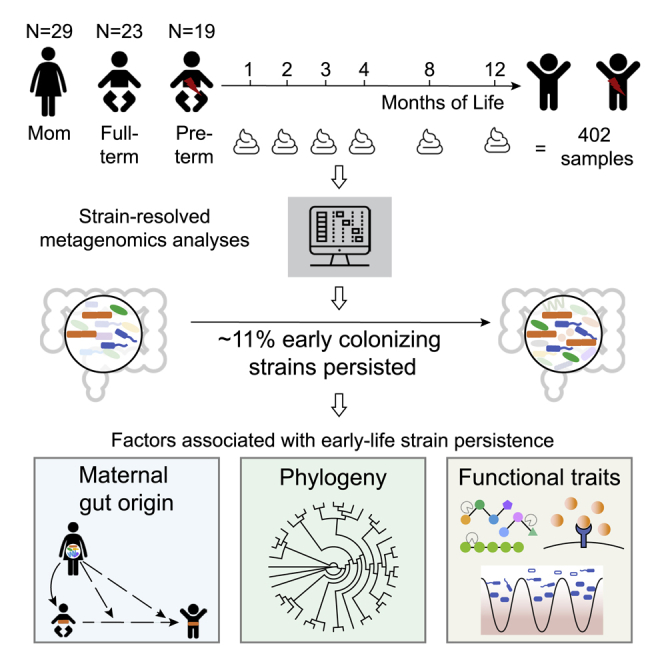
Summary
Gut microbiome succession affects infant development. However, it remains unclear what factors promote persistence of initial bacterial colonizers in the developing gut. Here, we perform strain-resolved analyses to compare gut colonization of preterm and full-term infants throughout the first year of life and evaluate associations between strain persistence and strain origin as well as genetic potential. Analysis of fecal metagenomes collected from 13 full-term and 9 preterm infants reveals that infants’ initially distinct microbiomes converge by age 1 year. Approximately 11% of early colonizers, primarily Bacteroides and Bifidobacterium, persist during the first year of life, and those are more prevalent in full-term, compared with preterm infants. Examination of 17 mother-infant pairs reveals maternal gut strains are significantly more likely to persist in the infant gut than other strains. Enrichment in genes for surface adhesion, iron acquisition, and carbohydrate degradation may explain persistence of some strains through the first year of life.
Keywords: strain-resolved metagenomics, Infant gut microbiome, community ecology, early-life gut colonization
Graphical abstract
Highlights
Strain-resolved analysis shows 11% of bacteria persist during the first year of life
Maternally acquired bacterial strains are more likely to persist in the infant gut
Certain persisting strains are enriched with functions such as surface attachment
Lou et al. use strain-resolved metagenomics to characterize preterm and full-term infant gut-microbiome succession during the first year of life. Approximately 11% of colonizing bacterial strains establish long-term residency, and many of these come from their mothers. Functions such as surface attachment may have facilitated retention in the gut.
Introduction
Microorganisms rapidly colonize the near-sterile infant gut during and shortly after birth.1 These early gut colonizers have important roles in the maturation of infants’ metabolic pathways, especially related to the immune system.2 Early life events, such as cesarean delivery and antibiotics administrations, which could disrupt microbial acquisition and assembly,3, 4, 5 have been associated with increased risks of developing diseases later in life, including asthma and metabolic syndrome.6, 7, 8
Certain bacterial commensals can persist within the adult gut for years.9, 10, 11 Infant gut microbiomes are less stable than adult microbiomes at the whole-community level,12,13 and fundamental questions remain regarding the persistence of their early colonizers. There is potential for long-term effect if the first colonizing strains, which are often hospital-associated pathogens in premature infants,14, 15, 16 persist as infants develop. Thus, it is important to analyze strain persistence and the sources and characteristics of persisting strains, as well as the time required for convergence of premature and full-term microbiomes.
Most studies on the infant microbiome have relied on 16S rRNA sequencing, which cannot resolve genomic differences beyond the species level. These studies have advanced our understanding of the early life gut microbiota assembly process.12,13,17 However, to answer questions regarding organism transmissions from various sources, organism persistence through early life, or sharing of organisms among individuals, whole-genome resolution is necessary. Robust detection of subtle genomic differences allows one to determine whether strains are identical or merely closely related and to distinguish commensal from pathogenic strains.18,19 Sequencing cultured isolates is one way to recover microbial genomes, but it is low throughput, targeted to particular taxa, and is unlikely to capture the full strain diversity present.20 Genome-resolved metagenomics circumvents the shortcomings of 16S rRNA and culture-dependent sequencing by generating genomes for essentially all microorganisms present in the gastrointestinal tracts of infants early in life without relying on culturing or any public reference genomes.21, 22, 23, 24 Recently, there have been several metagenomics studies examining strain sharing among family members, among unrelated infants, and within individuals over time.25, 26, 27, 28, 29, 30 However, these studies used public reference genomes and relied on read mapping to sets of species-specific marker genes for taxonomic characterization. This way of identifying organisms can ignore species that lack sufficient representative genomes in the public database and, therefore, one can only examine a subset of species and corresponding strains that are present in the database. These studies also used relatively non-stringent definitions of “identical” strains that ignore whole-genome information (i.e., considering single-nucleotide polymorphisms [SNPs] in marker genes only and/or measuring coding regions only), which may confuse closely related, but epidemiologically unconnected, strains.19
Here, we investigated early life gut microbiome assembly dynamics using genome-resolved metagenomics and relied on stringent, whole-genome comparisons to define two organisms as being the same. Our study targeted preterm and full-term infants born at the same hospital over a 3-year period and tracked their gut microbiome compositions to age 1 year. We also collected fecal samples from mothers at birth to identify transmission of strains between the infant and maternal gut microbiomes. In contrast to prior work in this area, we examined the early life gut microbiomes using de novo constructed microbial genomes, which allowed us to examine species without closely related representative genomes in public reference databases. Further, we applied a rigorous strain-level resolution when examining the succession of the gut microbiome, which allowed us to accurately track the persistence and gene content of strains colonizing the infant gut. Taken together, we determined that maternal origin, phylogeny, and functional potential of bacterial initial colonizers all contributed to strain persistence in infants. Insights regarding traits that enable gut microbiome residency during early life have implications for development of rational microbiome manipulations.
Results
Study design and sampling
In this study, we followed 23 full-term and 19 preterm infants from birth to age 1 year. A total of 402 fecal samples from these infants and their mothers were selected and subjected to deep metagenomic sequencing (∼3.5 tera base pairs [Tbp] of total sequence data in the form of 150 bp paired-end reads) (Figure S1). Reads were de novo assembled to recover 7,521 draft genome bins, which were further dereplicated at 98% whole-genome average nucleotide identity (gANI) to yield 1,005 genomes that represent unique microbial “subspecies.” We use the term “subspecies” as a taxonomic rank in between strain and species (Figure 1) (STAR Methods).
Figure 1.

Genome-resolved metagenomics pipeline
Fecal samples collected from infants and their mothers (1) were subjected to shotgun sequencing (2). Reads were subsequently processed and assembled into draft genomes (3), which were dereplicated at 98% gANI to result in 1,005 genomes that represented unique microbial subspecies (4). Reads from each individual sample were then mapped to 1,005 subspecies (5) for sample-specific genome detection and sample-specific relative abundance calculation (6). inStrain was run to identify same strains of the subspecies between samples of the same or different infants (7). See also Figure S1 and Tables S1 and S2.
Detection of identical strains was achieved using inStrain19 and was based on comparisons of read mapping to the same subspecies. A bacterial bin was considered identical in two samples if the compared region of the genome from both samples shared more than 99.999% population-level ANI (popANI) based on previously suggested thresholds19 (Figure 1). Our stringent definition of “strain” allowed us to discriminate between recent strain-transmission events and pairs of organisms that shared a recent evolutionary history but originated from distinct sources.
In addition to infant and maternal samples, we sequenced five negative reagent controls (one per extraction plate). The detection of common gut species in two negative control wells prompted us to thoroughly assess artifactual sequence sharing among the wells of all extraction plates. We concluded that the contamination observed in two negative control wells was a result of well-to-well contamination on those two extraction plates (STAR Methods). Given the importance of strain-level analyses, we rejected all samples on those plates (samples from 10 full-term and 10 preterm infants and 12 samples from mothers). No contamination was found in the other three extraction plates. Therefore, the 206 samples on these three plates (9 preterm and 13 full-term infants and 17 from their mothers) were used for downstream analyses (Figure S1). Metadata (Table S1) and sequencing data (Table S2) of those 22 infants and their mothers are provided.
Approximately 11% of bacterial early colonizers persisted throughout the first year of life
Infant fecal samples were grouped into seven windows of time (months 0, 1, 2, 3, 4, 8, and 12) based on infants’ chronological ages at the time of sample collection. Bacterial strains that arrived during the first 2 months of life were classified as early colonizers, and they were further subdivided into “persisters” or “non-persisters,” depending on whether they stayed within the infant gut beyond month 8 (persisters) or not (non-persisters) using the 99.999% popANI strain identity cutoff (STAR Methods) (Figure 2A).
Figure 2.

Certain bacterial strains persist in the infant gut from birth until near age 1 year
(A) (Top) Definition of persister (present both before month 2 and after month 8) and non-persister (present before month 2 and absent after month 4) bacterial strains. (Bottom) The decrease in the number of early colonizers (strains detected in the first 2 months of life) present at sequential time points.
(B) Percentage of early colonizers that persisted in the gut microbiomes of preterm and full-term infants. Light gray circles indicate values for individual infants. Salmon-red and sky-blue circles represent the mean values for preterm and full-term infants, respectively. The lines stretching out from the circles represent the upper and lower bounds of bootstrapped 95% CI of the mean values (∗p < 0.05).
(C) Percentage of early colonizers, persisters, and non-persisters by genus. Genera other than Bacteroides and Bifidobacterium are grouped into “Others.”
(D) Species composition of persisters in full-term (left) and preterm (right) infants. Bars are colored by phylum. The x axis is the percentage of the specific species in full-term (left) and preterm (right) persisters.
We found that 274 (47.7%) of the 575 bacterial subspecies detected across infants during the first year of life were early colonizers. Those 274 subspecies comprise 560 distinct strains; of which, 59 were persisters, and 501 were non-persisters (Figure 2A). The median residence time for persisters was 9.6 months (95% confidence interval [CI], 9.0–10.1 months), and the median residence time for non-persisters was 0.4 months (95% CI, 0.3–0.5 months). Of the non-persisters, ∼76% were not detected after month 2. Notably, the relative abundance of non-persisters was significantly less than that of persisters (p = 1.6e−19; Wilcoxon rank-sum test).
A greater percentage of early colonizers persisted throughout the first year of life in full-term infants than did so in preterm infants (p = 0.032, two-sided permutation test) (Figure 2B). This outcome was not confounded by the size or diversity of the initial populations that colonized preterm and full-term infants because no statistical difference was observed in either the total number of early colonizers or the alpha diversity of early colonizers between preterm and full-term infants (p = 0.22 and 0.76, respectively; Wilcoxon rank-sum test). To identify clinical variables that might contribute to strain persistence, we applied a generalized linear model (GLM) to evaluate the effect of prematurity (STAR Methods). We noted that a subset of clinical factors (i.e., Prolacta, a caloric fortifier received by most preterm infants and no full-term infants) were highly correlated and, thus, was confounded with term/preterm status (Figure S2). Hence, their contributions to strain persistence could not be quantified individually. Despite that, when controlling for term/preterm status, race, gender, feeding practices, breastfeeding cessation time, first solid-food introduction time, delivery mode, and antibiotic usage after month 2, we found that full-term status had a significant effect on the percentage of initial strains that persist in an infant (p = 0.00024; Poisson distribution GLM).
When considering all infants, Bacteroides and Bifidobacterium strains were more likely to persist than were strains of other bacterial genera (q = 7.8e−15 and 8.6e−05, respectively; Fisher’s exact test) (Figure 2C). At the species level, Bacteroides vulgatus and Bacteroides uniformis strains were more likely to persist than were strains of other bacterial species (q = 6.0e−6 and 1.6e−03, respectively; Fisher’s exact test). Meanwhile, strains of Veillonella and Clostridium were significantly less likely to persist than were strains of other genera (q = 0.023 and 0.023, respectively; Fisher’s exact test). These observations raised the question of whether the persisting and non-persisting strains differed between preterm and full-term infants. We found that persisting Bacteroides vulgatus and Bifidobacterium breve (q = 6.0e−06 and 0.0011, respectively; Fisher’s exact test) strains were significantly enriched in full-term infants, whereas Bacteroides uniformis and Escherichia coli persisting strains were enriched in preterm infants (q = 0.016 and 0.022, respectively; Fisher’s exact test) (Figure 2D).
Maternally derived strains are more likely to be persisters in the infant gut microbiome
To elucidate the influence of maternally derived intestinal strains on the development of the infant gut microbiome, we measured strain sharing between infants and their mothers. In this study, “vertical transmission” refers to bacterial strains being transmitted from the gut microbiomes of mothers to infants because no samples from other body sites were collected. Of the 22 infants in this study, we collected maternal fecal samples from 17 of them. Of those 17 infants, 9 of the 12 full-term and three of the five preterm infants inherited strains from their mothers. In total, there were 50 maternally sourced bacterial strains that were detected across 12 of 17 mother-infant pairs examined (4.4% of all identified maternal strains; Figure 3A).
Figure 3.

Maternally derived intestinal bacterial strains are more likely to be persisters in the infant gut
(A) Schematic of all 50 strains that were maternally transmitted to infants. Each row represents an infant-specific, maternally transmitted strain, and marks represent months in which the strain was detected. Shapes represent distinct strain identities (i.e., persisters, non-persisters, and late colonizers). Solid marks indicate ≥99.999% popANI between the infant and mother strains, and hollow marks indicate windows in which the identity of the infant and mother strains fell below the strain cutoff. Non-persisters of the same subspecies are connected via dashed lines, whereas persisters or late colonizers of the same subspecies are connected via solid lines. Double hashing indicates a non-persister early colonizer that was not maternally derived being replaced by a closely related maternal intestinal strain.
(B) Relative abundances of maternal subspecies that were and were not vertically transmitted to the infant gut microbiomes (∗∗∗p < 0.001). Each dot represents a subspecies detected from a maternal fecal sample.
(C) Phylum-level taxonomy of strains that were maternally transmitted to infants as well as all maternal gut strains.
(D) Percentage of maternally transmitted strains by genus and colored by phylum.
(E) Percentage of persisters (solid black) and non-persisters (dashed) derived from the maternal gut microbiomes and other sources.
Strains that were vertically transmitted were significantly more abundant in the maternal gut microbiomes than were strains that were not passed on to infants (p = 2.4e−16, Wilcoxon rank-sum test) (Figure 3B). Correspondingly, maternally acquired strains were also more abundant than were non-inherited strains in the infant gut microbiomes across all time windows (q < 0.001, Wilcoxon rank-sum test). Regardless of gestational age or delivery mode, Bacteroidetes were significantly enriched and Firmicutes were significantly depleted among maternally transmitted strains (q = 2.4e−09 and 2.4e−13, respectively; Fisher’s exact test) (Figures 3A and 3C). At the genus level, Bacteroides and Parasutterella were more likely to be acquired by infants from their mothers than were other bacterial genera (q = 2.0e−08 and 0.028, respectively; Fisher’s exact test) (Figures 3A and 3D). B. uniformis and B. vulgatus were the two most commonly observed species to be maternally transmitted in this cohort (q = 3.7e−05 and 0.0015, respectively; Fisher’s exact test).
Maternally transmitted strains were found to be significantly more likely to be persisters in the infant gut microbiomes than strains derived from other sources, suggesting strains acquired from the maternal gut microbiomes are likely to be well adapted to the infant gut (p = 4.0e−11, Fisher’s exact test) (Figures 3A and 3E). These maternally transmitted persisters were primarily Bacteroidetes, whereas persisters not detected in maternal fecal samples were mostly Firmicutes and Actinobacteria. Importantly, we detected new strains being transmitted from mothers to the infant gut microbiomes throughout the first year of life, suggesting vertical transmission is not limited to the intrapartum or postpartum periods (Figure 3A).
Non-related infants rarely shared bacterial strains
In addition to examining strain persistence and maternal strain transmission, we also searched for strain sharing between different infants in the study. When considering all possible pairs of individual infants, 18 of 231 infant pairs shared at least one bacterial strain (Figure 4A). Although most infant pairs shared no more than two bacterial strains, full-term infants 7 and 133 shared 11 strains (Figure 4). Our decontamination analysis ensured that this was not a result of cross-sample contamination. We, therefore, hypothesized, and later confirmed by searching medical record data, that these two infants were siblings, with infant 7 being born 2 years earlier. We examined the gut microbiomes of the siblings and their mother in greater detail in the next section. No other infants in our study were biologically related.
Figure 4.

A pair of siblings shared significantly more bacterial strains than non-related infants
(A) (Top) The number of strains that were shared between infant pairs. (Bottom) The percentage of the infant gut microbiome that shared near-identical (≥99.999% popANI) strains with other infants. “S” in both panels refers to the infants 7 and 133 sibling pair.
(B) Overview of year-1 gut microbiome compositions of the siblings. Bar height represents normalized subspecies relative abundance, and bars are colored by phylum. Sections of the same color with horizontal black lines correspond to individual subspecies of the same phylum. All maternal fecal samples were grouped into “Mom” on the x axis.
(C) Normalized relative abundance of the 11 strains shared between the siblings throughout the first year of life. Each shared strain is assigned to a unique color, and the rest of the strains were all colored in dark gray.
(D) Longitudinal detection of the 11 strains shared between the siblings. Colored squares on the left of the species names correspond to the barplot color scheme in (C). Each row represents a sibling shared strain and is colored based by phylum. Shapes represent distinct strain identities. Older and younger siblings are represented by darker and light blue rectangles, respectively (∗maternally transmitted strains). See also Figure S3.
Excluding comparisons between siblings, preterm infants were far more likely to share strains with other preterm infants than full-term infants were to share strains with other full-term infants (p = 4.6e−04, Fisher’s exact test) (Figure 4A). Most sharing among preterm infants occurred before the infants were discharged from the hospital, pointing to the hospital environment as a potential strain source. Clostridium butyricum was the most widely shared species among preterm infants, and one C. butyricum strain was shared by ≥5 non-related preterm infants based on pairwise comparisons.
A pair of siblings shared a significant number of strains throughout their first year of life
To further investigate strain sharing in the sibling gut microbiomes, we closely examined the gut microbial communities of full-term infants 7 and 133 and the two fecal samples provided two years apart by their mother (Figures 4B–4D). The two siblings were both born via cesarean section and were breastfed exclusively before weaning. During the first year of life, when compared with the older sibling, the younger one had fewer Proteobacteria and Verrucomicrobia subspecies and more Bacteroidetes. We speculated that changes in the gut microbiome of the mother around the time of birth of the second child, compared with the first, might explain the observed compositional differences between the siblings. Indeed, the mother’s gut microbiome was nearly twice as enriched in Bacteroidetes and contained about six times less abundance of Proteobacteria and no Verrucomicrobia around the time of second delivery compared with the first (Figure 4B).
The 11 bacterial strains that were shared by the siblings accounted for ∼20% of the overall gut microbiome of the older sibling and ∼50% of the gut microbial community of the younger sibling (Figure 4C). Interestingly, only one of the 11 shared strains (B. breve), not maternally derived, persisted in both siblings throughout most of their first year of life, and five of the 11 shared strains were classified as persisters only in the younger sibling (Figure 4D). No other infant pairs shared any bacterial persisters. Because most shared strains were late colonizers in the older sibling but were early colonizers in the younger sibling and they were mostly not detected in the gut microbiome of their mother, we hypothesize that strains may have been transmitted from the older to the younger sibling (Figure 4D).
Having collected two fecal samples from the same mother also allowed us to search for bacterial strains present in both samples. Of the 99 and 94 subspecies detected from the first and second maternal fecal samples, respectively, 12 (mostly Bacteroidetes) shared ≥99.999% popANI. Notably, these 12 strains constituted 20% of the maternal gut microbiome at the time of the birth of her first child and ∼50% of her gut microbiome 2 years later (Figure S3A). Of those 12 strains, a B. uniformis and a Megasphaera massiliensis strain were acquired by both siblings. Both of those strains were persisters in the younger sibling. Two of the other 10 maternal strains were detected when the younger sibling was 1 year old; another one of the 10 strains was detected in the older sibling at age 1 year (Figure S3B).
Diverse carbohydrate active enzymes are implicated in Bifidobacterium and Escherichia persistence
We next investigated whether specific capacities of early colonizers are associated with strain persistence. Specifically, we compared the gene content of persisters and non-persisters during the first 2 months of life to identify functional traits that could confer early colonizers with a persistence advantage (STAR Methods; Tables 1, S3, S4, and S5).
Table 1.
Selected annotations that were significantly enriched in E. coli persisters
| Annotation definition | KO(s) | Pfam(s) | VF(s) |
|---|---|---|---|
| Surface adhesion | |||
| Antigen 43 | K12687 | agn43 | |
| CdiA (Putative filamentous hemagglutinin) |
K15125 |
PF05860, PF13332, |
cdiA |
| PF04829, PF15530, | |||
| PF03865, PF08479, | |||
| PF17287 | |||
| Iron acquisition | |||
| Yersiniabactin biosynthesis | K04781, K04783, | PF08242, PF08659, | ybtS, ybtP, ybtA, irp2, |
| K04784, K04785, | PF16197 | irp1, ybtU, ybtE, fyuA | |
| K04786, K05372, | |||
| K05373, K05374, | |||
| K15721 | |||
| Manganese/iron transport system, SitABCD |
K11604, K11605, |
sitA, sitB, sitC, sitD |
|
| K11606, K11607 | |||
| Bacterial toxins | |||
| Uropathogenic | PF01320, PF05638, PF06958 | usp | |
| Escherichia coli Colicin-Like (Usp) | |||
| Colibactin biosynthesis | K01071, K01426 | PF08659, PF13602, | clbA, clbB, clbC, clbE, |
| PF14765, PF16197, | clbF, clbG, clbH, clbI, | ||
| PF01425, PF08020, | clbL, clbM, clbN, clbO, clbQ, clbR | ||
| PF16197 | |||
Because the ability to metabolize a variety of carbohydrates is considered to be important for surviving in the gut,31,32 we hypothesized that persister genomes would be enriched with carbohydrate-active enzymes (CAZymes) when compared with non-persisters. To test our hypothesis, we annotated genes encoding CAZymes and measured the diversity of them in the genomes of persisters and non-persisters (STAR Methods).
Overall, persisters (Np = 59) had a significantly higher CAZyme Shannon diversity than non-persisters (Nnp = 501) (p = 4.1e−07; Wilcoxon rank-sum test). However, persisters in our study were primarily Bacteroides and Bifidobacterium, whose genomes are known to densely encode glycan-metabolizing genes.33, 34, 35 To address that potential taxonomy bias, we restricted our comparisons of CAZyme diversity to those between persisters and non-persisters from the same genus or species. Further, we required at least three persister and three non-persister strains for comparisons to retain statistical power. Of the five genera (Escherichia, Bifidobacterium, Klebsiella, Streptococcus, and Bacteroides) meeting those criteria, Escherichia (Np = 3, Nnp = 25) and Bifidobacterium (Np = 12, Nnp = 18) persisters encoded a greater CAZyme Shannon diversity when compared with their corresponding non-persisters (p = 0.030 and 0.014, respectively; two-sided permutation test) (Figure 5A). E. coli (Np = 3, Nnp = 25) was the only species that passed the filtering criteria, and its persisters encoded a significantly greater Shannon diversity of CAZymes than did non-persisters (p = 0.030; two-sided permutation test).
Figure 5.

Diverse carbohydrate active enzymes were detected in persisters of certain bacterial genera
(A and B) Comparison of CAZyme diversity (A) and coding density (B) per genome between persisters (solid color) and non-persisters (dashed) of the five bacterial genera that passed the filtering criteria (∗p < 0.05). Boxplots are colored by phylum.
(C) Comparison of CAZyme coding density per CAZyme family per genome between Bifidobacterium persisters (solid black) and non-persisters (dashed) (∗p < 0.05).
(D) Correlation between the number of genes coding for CAZymes (x axis) and unique CAZyme counts (y axis). Each dot represents a persister (left) or non-persister (right) genome and is colored by phylum.
We also examined CAZyme coding density in the genomes of persisters and non-persisters (STAR Methods) and found Bifidobacterium persisters dedicated a significantly higher percentage of their genomes to CAZymes than did Bifidobacterium non-persisters (p = 0.047; two-sided permutation test) (Figure 5B). This effect is largely due to the inclusion of glycosyltransferases (GTs) and carbohydrate esterase (CEs) (p = 0.012 and 0.012, respectively; two-sided permutation test) (Figure 5C). In addition, we measured the relationship between the number of genes encoding CAZymes and the number of unique CAZyme types detected in a genome. In general, both persisters and non-persisters showed a positive correlation between the number of genes encoding CAZymes and the number of unique CAZymes (Spearman correlation coefficient r = 0.96 and 0.97, respectively; p = 1.86e−33 and 8.20e−200, respectively) (Figure 5D). Notably, Bifidobacterium persisters had a higher ratio of CAZyme-coding genes to unique CAZymes than the related non-persisters had (p = 0.00076; Wilcoxon rank-sum test), suggesting that these strains generally encoded more duplicate copies of specific CAZyme families than their non-persisting counterparts did.
We next searched for specific CAZymes that were enriched in persisters or non-persisters of Bifidobacterium and Escherichia (STAR Methods) because these are the two genera that showed significant CAZyme diversity differences between their persisters and non-persisters. None of the CAZymes were enriched in non-persisters of either genus. Of the 109 CAZymes examined in Bifidobacterium early colonizers, six were significantly enriched in persisters (q < 0.05, Fisher’s exact test), and they were all predicted to participate in digesting dietary polysaccharides (Table S3). In Escherichia, 8 of 63 CAZymes examined were significantly enriched in persisters (q < 0.05, Fisher’s exact test). Most CAZymes such as GH33 and PL22 that were enriched in Escherichia persisters were involved in activities such as metabolizing small molecules, including sugar byproducts of mucin, and dietary polysaccharides degradation carried out by other community members (Table S4). Interestingly, GH153, a CAZyme predicted to be involved in biofilm formation,36 and GT107, a glycosyltransferase predicted to be involved in capsular polysaccharide biosynthesis,37 are also enriched in Escherichia persisters, suggesting Escherichia persisters might carry other traits that enable their stable gut colonization.
Surface adhesion and iron acquisition contribute to E. coli persistence
E. coli was the only species of Escherichia to be classified as an early colonizer. To identify other functions besides carbohydrate metabolism that could contribute to E. coli persistence in the infant gut, we compared the gene content of E. coli persisters and non-persisters present during the first 2 months of life using the Kyoto Encyclopedia of Genes and Genomes (KEGG), Pfam, transporter classification (TC), and E. coli virulence-associated gene (EcVG) databases (STAR Methods).
All three E. coli persisters in our study were detected from preterm infants. Specifically, one E. coli persister was detected in a preterm infant who survived two necrotizing enterocolitis (NEC) events, and that E. coli strain first appeared around the time when NEC recurred (Figure S4). Another two E. coli persisters were detected in two preterm infants before the onset of late-onset sepsis (LOS) (Figure S4). Blood cultures were drawn on the day of diagnosis from those two infants and they were both positive for E. coli (Table S1). Although a previous study has reported the translocation of E. coli from the gut to the bloodstream to be the cause of LOS in some infants,38 we could not confirm such a finding by comparing the gut and the bloodstream E. coli strains because no blood cultures were banked for sequencing.
Of the KEGG orthologies (KOs), Pfams, TC identifiers (TCIDs), and virulence factors (VFs) examined, 119 KOs, 140 Pfams, 37 TCIDs, and 72 VFs were significantly enriched in E. coli persisters (q < 0.05, Fisher’s exact test) (Tables 1 and S5). Notably, 4 KOs, 19 Pfams, 4 TCIDs, and 18 VFs were present in all three E. coli persisters and absent in all 25 E. coli non-persisters. These were primarily linked to genes involved in surface adhesion. For instance, CdiA and antigen 43 have been shown to enhance cell-cell aggregation and/or biofilm formation.39,40 Another function that was found in E. coli persisters only was biosynthesis of the toxin colibactin. Genes involved in colibactin synthesis are located on a 54-kilobase genomic island.41 We found 14 genes of that 19-gene cluster to be significantly enriched in E. coli persisters only, which prompted us to search for the presence of the complete colibactin biosynthesis gene cluster in E. coli persisters. Cluster detection via read mapping to de novo constructed E. coli representative genomes confirmed that a complete colibactin biosynthesis gene cluster was present in all E. coli persisters and absent in all E. coli non-persisters (STAR Methods).
We also identified genes for functions that were significantly enriched but not exclusively present in persisters. Many of these are involved in surface adhesion (e.g., type VI secretion system and biofilm biosynthesis). Also enriched was the uropathogenic Escherichia coli colicin-like protein (Usp), which has been postulated to be a bacteriocin against other E. coli strains and has also been shown to damage mammalian cells.42,43 Other enriched traits included sugar and amino acid metabolism (e.g., pectin-associated metabolism and d-serine detoxification and metabolism) and iron acquisition (e.g., manganese/iron transporters and siderophore production) (Tables 1 and S5).
We found adjacent biosynthesis gene clusters involved in the production of the siderophore yersiniabactin and the genotoxin colibactin exclusively in all three E. coli persisters (STAR Method) (Figure S5). The co-location of colibactin and yersiniabactin biosynthesis gene clusters has been found in both extraintestinal pathogenic strains and gut commensal isolates.41,44,45 These two gene clusters have been shown to be functionally interconnected via clbA, a gene from the colibactin gene cluster that also contributes to siderophore biosynthesis.46 How this genomic structure might influence the persistence of E. coli in the preterm infant gut and the onset of early life diseases remain to be determined.
We used comparative genomic analyses to verify that genes that are apparently absent or relatively uncommon in E. coli non-persisters were not simply missed because of missing genome fragments (STAR Methods). As expected, we detected insertions/deletions involving enriched/absent genes in otherwise syntenous regions. For instance, we found that genes involved in synthesis of the colicin-like protein (Usp) and its associated immunity protein, as well as a large region that encodes a type VI secretion system, were absent in otherwise syntenous regions of the persister and non-persister E. coli genomes (Figure S6).
Overall, we found E. coli persisters encoded a significantly higher percentage of virulence genes than did non-persisters (p = 0.0032; two-sided permutation test). Because many of these genes were involved in surface adhesion and iron acquisition, we assessed the importance of these two functions by measuring their density on the genomes of E. coli early colonizers (STAR Methods; Tables S6). We found E. coli persisters dedicated significantly higher percentages of their genomes to surface adhesion and iron acquisition than the non-persisters (p = 0.013 and 0.00030, respectively; two-sided permutation test), suggesting these two functions were particularly important for the persistence of E. coli in the infant gut.
Initially divergent gut microbiomes of full-term and preterm infants largely converged by age 1 year
To understand how early life gut microbiome assembly might differ between full-term and preterm infants at the community level, we measured the β-diversity of the two infant groups using the UniFrac distance47 (STAR Methods).
Weighted UniFrac, which considers the relative abundances of individual taxa, indicated that the gut microbiomes of preterm infants diverged from those of full-term infants between months 1 and 3. During that period, preterm infants’ gut microbiomes were disproportionately dominated by bacteria that are common in the hospital environment, including members of ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) pathogens48 (Figure 6A). However, convergence between the gut microbiomes of preterm and full-term infants began at month 3 and accelerated between months 4 and 8 (p = 2.0e−04, Wilcoxon rank-sum test). Overall, weighted UniFrac indicated that the microbiomes of full-term and preterm infants converged by age 1 year (p = 0.0024, Wilcoxon rank-sum test) (Figures 6B and S7A). Unweighted UniFrac, which excludes relative abundance, indicated that the gut microbiomes of preterm and full-term infants became similar between months 1 and 8 (p = 0.0059, Wilcoxon rank-sum test) but diverged rapidly after month 8. Altogether, unweighted UniFrac suggested that the preterm and full-term infant microbiomes became more distinct by age 1 year (p = 0.014, Wilcoxon rank-sum test) (Figures 6B and S7A). To evaluate the contrasting outcomes of the two UniFrac metrics, we also tested for convergence of gut microbiomes of full-term and preterm infants using Bray-Curtis distance (STAR Methods; Figure S7B). Consistent with weighted UniFrac, Bray-Curtis distance indicated that the gut microbial compositions of full-term and preterm infants largely converged by age 1 year (p = 1.7e−31, Wilcoxon rank-sum test).
Figure 6.

Community assembly dynamics of full-term and preterm infant gut microbiome during the first year of life
(A) Overview of year-1 gut microbiome compositions of the full-term and preterm infants. Bar height represents normalized subspecies relative abundance, and bars are colored by phylum. Sections of the same color with horizontal black lines correspond to individual subspecies of the same phylum. x axis represents months of life, and all maternal fecal samples were grouped into “Mom” on the x axis.
(B and C) Weighted (orange) and unweighted (blue) UniFrac comparing the compositional changes of the gut microbiomes of full-term and preterm infants (B) as well as of infants and mothers (C). x axis represents months of life. See also Figures S7A and S7B.
(D) Gut microbiome assembly trajectories of a representative full-term (left) and a representative preterm (right) infant. Shapes and colors represent distinct sample types. Samples belonging to the representative infant are colored green to distinguish them from samples from other infants, which are colored based on sample types and are in less-saturated colors. Samples are numbered according to infant age at the time of collection. PCA is based on weighted UniFrac distance. See also Figure S7C.
To further examine the maturation of the infant gut microbiome, we measured the β-diversity between the gut microbiomes of infants and mothers. Both weighted and unweighted UniFrac distances showed a gradual convergence between infants and mothers (Figures 6C and S7A). Development of gut microbiomes of preterm and full-term infants in the context of maternal microbiome composition was further examined via principal component analysis (PCA) (STAR Methods). Each infant’s assembly trajectory was visualized in a PCA by tracking the changes in composition between consecutive sampling time points (Figure 6D). The gut microbiomes of preterm infants, full-term infants, and their mothers formed distinct clusters in PCA space (permutational multivariate analysis of variance [PERMANOVA], p < 0.001) (Figure S7C). However, over time, the infant gut microbiomes all moved toward the PCA region in which the maternal samples were placed. Indeed, chronological age had a significant role in driving the gut microbiome changes for both full-term and preterm infants (PERMANOVA, p = 0.040 and p < 0.001, respectively). Notably, the trajectories of preterm infant microbiomes were different from those for full-term infants, possibly because their initial gut microbiomes were more distinct from the maternal gut microbiomes than those of full-term infants. Indeed, Jaccard dissimilarity comparing consecutive fecal metagenomes of each infant (STAR Methods) indicated that the changes between the early and late gut microbiomes of preterm infants were significantly larger than those of full-term infants (p = 0.0092; Wilcoxon rank-sum test).
Discussion
We conducted strain-resolved analyses to investigate ecological succession in the gut microbiomes of preterm and full-term infants, finding that ∼11% of bacterial early colonizers persisted through the first year of life. Our study used genome-resolved metagenomics to stringently identify persisting bacterial strains across all phyla in the early life gut microbiome and to investigate factors that are associated with strain persistence. Prior studies have identified the existence of persisting bacterial strains; however, many of those studies relied on isolation-based strategies,9,10,15,49 which can be biased toward cultivable lineages and strains. Several metagenomics-based studies have reported the detection of strains persisting in the infant gut over time,26,27,29 but they primarily focused on the influence of strain origin (i.e., maternally transmitted) on the fate of strains, relied on public reference genomes, and did not consider whole-genome information when defining strains, which may fail to discriminate between closely related, but epidemiologically unconnected, strains with genetic differences only resolvable with whole-genome comparisons.
We showed that most of the initial gut microbiome is transient, and only a small percentage of the early colonizers persist until age 1 year. This is in contrast with what has been reported in the adult gut microbiome.9,11 High strain turnover in the infant gut microbiome is not surprising. The initial microbial seeding of the near-sterile infant gut largely depends on the environment the infant is exposed to.50, 51, 52 Observation that non-persisters were significantly less abundant than persisters suggests that the transient presence of some early colonizers was, in part, due to neutral processes, such as ecological drift,53 because, by random chance, low-abundant organisms can be more easily driven to extinction by drift than high-abundant organisms can.51 The transient nature of some early colonizers could also be a reflection of them being poorly adapted to the gut environment, which is shaped by the host immune system, and some early colonizers including those that persist. Although constituting a small percentage of the early colonizers, persisters have the potential to shape the trajectory of the developing microbiome. Through priority effects, persisters can pose inhibitory and/or facilitative effects on late-arriving strains by niche preemption and/or modification.51,54 Although priority effects can also be exerted by non-persisting early colonizers,51 given their transient presence, the influence they have on the infant gut microbiome assembly is likely to be less significant when compared with those of persisters. In addition, the stable colonization of persisters implies their intimate interactions with the immune system. Early life microbial colonization is critical for the development of the immune system.52 It is plausible that persisters directly influence the maturation of the immune system, which can then further shape the infant gut microbiome assembly. The importance of persisting early colonizers in the early life gut microbiome, thus, motivated us to identify those strains and to investigate factors that contribute to their persistence.
We identified one important factor that seems to dictate whether an early colonizer is in the small subset of strains that persist beyond the first year of life. By analyzing maternal fecal samples collected around the time of birth, we determined that strains derived from the maternal gut (i.e., Bacteroides) are significantly more likely to persist than non-inherited strains. Our work extends previous maternal-transmission work conducted over a much shorter period and used a combination of consensus-SNP calling and gene-based approaches to identify mother-infant shared strains,26 a study that relied on analysis of rare SNPs of abundant species only,29 and a study that defined identical strains using SNP differences on species-specific marker genes only.27 Persistence of maternally transmitted strains could be a result of continuous seeding from the mother because we showed maternal transmission occurred through the first year of life. Persistence of maternal strains may also reflect their adaptation to the gut, which may include the metabolism of gut-associated nutrients and interaction with the infant immune system.2
Some bacterial taxa were far more likely than others to persist in the developing infant gut. Enrichment of Bacteroides persisters could be partially explained by their maternal gut origin. Bifidobacterium spp. were less-commonly detected in the maternal gut. Their high likelihood of persisting in the infant gut may be attributed to their high diversity and density of CAZymes; some of which degrade dietary polysaccharides.13,55 Our findings suggest that metabolic flexibility is crucial for Bifidobacterium persistence in the infant gut because it enables rapid adaptation when the infant’s diet shifts away from breast milk and/or formula. This is in line with prior work that proposed a link between plant polysaccharide metabolic capacity and the ability of a strain to adapt by shifting metabolism after introduction of solid food.32 In addition to metabolic flexibility, enrichment of glycosyltransferase-encoding genes, which can participate in capsular and/or exopolysaccharide biosynthesis, suggests that other functional traits, such as host adherence and resistance to bile acids,56 may also be important for Bifidobacterium persistence in the infant gut.
Flexible carbohydrate metabolism might be a common trait linked to persistence in the infant gut because Escherichia persisters also encoded a greater diversity of CAZymes than did respective non-persisters. However, as suggested with the persistence of Bifidobacterium, other factors likely influence persistence. We identified an enrichment of virulence factors, such as those coding for surface adhesion, iron acquisition, and colibactin biosynthesis in E. coli persisters, many of which are commonly carried by extraintestinal pathogenic Escherichia coli (ExPEC).57,58 It is plausible that these virulence factors enhance the competitiveness of E. coli in the gut without causing acute disease. Indeed, long-term intestinal colonization of commensal E. coli strains carrying virulence factors has been found in healthy individuals.49,57,59
Although we cannot state whether resident E. coli strains carrying virulence genes will have long-term negative effects on host health, their exclusive presence in preterm infants in this study implies that prematurity can affect the infant microbiome for a span of time. A recent comparison of the gut microbiomes of preterm and near-term infants noted that, even though infants’ gut microbiome compositions showed evidence of convergence, markers associated with prematurity remained by age 2 years.15 Using cultivation-independent genome-resolved strain analyses, we also found that, although initially distinct gut microbiomes of preterm and full-term infants largely converged by age 1 year, some differences remained. For instance, E. coli strains enriched in virulence genes were found in preterm infants only, which could result in differences in community assembly and immune system development. The persisting microbiome differences between full-term and preterm infants likely result from a combination of factors, including gestational age,17 early life antibiotic treatments,14,15 exposure to the hospital environment,15,21 and lack normal development of the immune system.60
By identifying and tracking individual strains in preterm and full-term infants through the first year of life and through careful analysis of strain-level functional potential, our study provides a fine-grained view of the early gut microbiome succession. By determining the types of strains that colonize in early life, where they come from, and what persistence-associated genetic traits they carry, we can better understand how the early life microbiome is assembled and gain insights into potential microbiome-based therapies when that assembly is disrupted.
Limitations of study
Our study was underpowered to fully assess all confounding factors when conducting between group comparisons. For instance, we were unable to independently evaluate the effects of variables including Prolacta addition, birth weight, length of stay in hospital after birth, and early antibiotic administrations (before month 2) on strain persistence because many of these factors are tightly associated with prematurity. In addition, given the high percentage of preterm infants who survived NEC and LOS in our study, some of our preterm-related findings, including persisting E. coli strains, may not apply to healthy preterm infants. To expand on our observations and to address the relationship between persisting E. coli strains enriched with virulence factors and prematurity, future longitudinal studies recruiting larger and more-balanced cohorts of preterm infants are needed.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| DNeasy PowerSoil HTP 96 DNA isolation kit | QIAGEN | - |
| KAPA HyperPlus Kit | Roche | - |
| Deposited data | ||
| Metagenomic sequences of all infant and mother stool samples | This paper | NCBI BioProject: PRJNA698986 |
| Statistical script | This paper | GitHub: https://github.com/clarelou0128/R-statistical-scripts |
| Software and algorithms | ||
| bcl2fastq version 2.20 | - | https://support.illumina.com/downloads/bcl2fastq-conversion-software-v2-20.html |
| Sickle version 1.33 | - | https://github.com/najoshi/sickle |
| Bowtie2 version 2.3.5.1 | Langmead and Salzberg61 | https://github.com/BenLangmead/bowtie2 |
| IDBA-UD version 1.1.3 | Peng et al.62 | https://github.com/loneknightpy/idba |
| Prodigal version 2.6.3 | Hyatt et al.63 | https://github.com/hyattpd/Prodigal |
| MetaBAT version 2.12.1 | Kang et al.64 | https://bitbucket.org/berkeleylab/metabat/src/master/ |
| CONCOCT version 1.1.0 | Alneberg et al.65 | https://github.com/BinPro/CONCOCT |
| MaxBin version 2.2.7 | Wu et al.66 | https://sourceforge.net/projects/maxbin/ |
| DasTool version 1.1.1 | Sieber et al.67 | https://github.com/cmks/DAS_Tool |
| dRep version 2.6.2 | Olm et al.68 | https://github.com/MrOlm/drep |
| tRep version 0.5.3 | - | https://github.com/MrOlm/tRep |
| GTDB-Tk version 1.3.0 | Chaumeil et al.69 | https://github.com/Ecogenomics/GTDBTk |
| inStrain version 1.3.4 | Olm et al.19 | https://github.com/MrOlm/instrain |
| KofamKOALA | Aramaki et al.70 | https://www.genome.jp/tools/kofamkoala/ |
| run_dbcan version 2.0.11 | - | https://github.com/linnabrown/run_dbcan |
| HMMER version 3.3.2 | Eddy71 | http://hmmer.org/ |
| cath-resolve-hits version 0.16.5 | Lewis et al.72 | https://github.com/UCLOrengoGroup/cath-tools |
| BLASTP version 2.10.0 | - | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| SignalP version 5.0b | Armenteros et al.73 | http://www.cbs.dtu.dk/services/SignalP/ |
| TMHMM version 2.0 | Krogh et al.74 | https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 |
| antiSMASH version 5.1.2 | Blin et al.75 | https://github.com/antismash/antismash |
| ABRicate | - | https://github.com/tseemann/abricate |
| Geneious version 2020.2.4 | Kearse et al.76 | https://www.geneious.com/ |
| clinker version 0.0.20 | Gilchrist and Chooi77 | https://github.com/gamcil/clinker |
| FeGenie | Garber et al.78 | https://github.com/Arkadiy-Garber/FeGenie |
| RStudio | R | https://www.rstudio.com/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to the lead contact, Jillian F. Banfield (jbanfield@berkeley.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
This study was reviewed and approved by the University of Pittsburgh Human Research Protection Office (IRB STUDY19120040). This nested case-control observational study was originally designed to study the gut microbiomes of premature and full-term infants as well as the gut microbiomes of premature infants who developed NEC and/or LOS and age-matched premature infants over the first year of life. For these purposes, we enrolled a total of 183 infants (35 full-term infants and 148 preterm infants born before 34 weeks of gestation). The 148 preterm infants that were followed prospectively comprised 10 NEC infants including one that developed NEC twice, 5 LOS infants, 1 infant that developed both NEC and LOS, and 132 infants that did not develop NEC or LOS. For each infant with NEC or LOS, we identified a member of the cohort that was hospitalized concomitantly, had a similar age, and that had not been treated with antibiotics after the first week of life. However, some infants had to be excluded from our study due to patient withdrawal, missing samples at key time points, or low sample biomass. Ultimately, we acquired longitudinal samples from 23 full-term and 19 preterm (6 healthy controls, 8 NEC infants, 4 LOS infants and 1 infant that developed both NEC and LOS) from birth to age one (Figure S1).
Fecal samples from enrolled infants and their mothers were all collected at the UPMC Magee-Womens Hospital (Pittsburgh, PA) over the course of three years. While full-term infants were discharged from the hospital within 3 days after birth and received no perinatal antibiotics, all preterm infants received empiric antibiotics immediately following birth during an evaluation for early-onset sepsis and then spent their first 2-3 months in the hospital. In addition to infant fecal samples, we collected a single fecal sample from 28 mothers of 29 infants within the first two weeks after delivery. All samples were collected with parental consent and subjects were de-identified before the receipt of samples. Well-to-well contamination was identified on samples from 20 out of 42 infants (see section Identification of sources of contamination below), and genome-resolved metagenomics analyses were performed on the remaining 13 full-term and 9 preterm infants. De-identified metadata for the 22 infants whose samples were not contaminated is provided in Tables S1.
Method details
Sample collection and metagenomic sequencing
Throughout the first year of life, infant fecal samples were collected either at UPMC Magee-Womens Hospital by trained nurses or at home by parents provided with detailed collection instructions. Specifically, fresh infant stool samples were collected directly from infants while they were actively excreting or from diapers shortly after the stools were released. Maternal fecal samples were collected using a commode specimen collector, from which fecal samples were transferred into a collection tube. All stool samples collected at the hospital were immediately stored at −80°C following collections. Samples collected at home were stored in home freezers until they were picked up by research staff and transferred to the −80°C condition. DNA extraction of frozen fecal samples was performed via the QIAGEN DNeasy PowerSoil HTP 96 DNA isolation kit with modifications to the manufacturer’s protocol. For each 96-well extraction plate, a reagent-only negative control was included.
Metagenomic sequencing of collected infant and maternal fecal samples was performed in collaboration with the California Institute for Quantitative Biosciences at UC Berkeley (QB3-Berkeley). Library preparation on all samples was performed as previously described.79 Final sequence ready libraries were pooled into 2 subpools and visualized and quantified on the Advanced Analytical Fragment Analyzer. Four samples did not fit nicely into either subpool so their libraries were quantified separately. All libraries were then evenly pooled into a single pool and checked for pooling accuracy by sequencing on Illumina MiSeq Nano sequencing runs. The single pool was adjusted based on MiSeq sequencing run and sequenced on individual Illumina NovaSeq6000 150 paired-end sequencing lanes with 2% PhiX v3 spike-in controls. Post-sequencing bcl files were converted to demultiplexed fastq files per the original sample count with Illumina’s bcl2fastq v2.20 software.
Metagenomic assembly and gene prediction
Reads from all 402 samples were trimmed using Sickle (https://github.com/najoshi/sickle), and reads that mapped to the human genome with Bowtie261 under default settings were discarded. Reads from each sample were then assembled independently using IDBA-UD62 under default settings. Co-assemblies were also performed for each infant, in which reads from all samples of that infant were combined and assembled together. Scaffolds that are < 1 kb in length were discarded. On average, 93.2% of the sequencing reads (95% confidence interval, 92.4%–94.4%) were de novo assembled into scaffolds ≥ 1 kbp in length per sample. Remaining scaffolds were annotated using Prodigal63 to predict open reading frames using default metagenomic settings.
Metagenomic de novo binning
Pairwise cross-mapping was performed between all samples from each infant to generate differential abundance signals for binning. Each sample was binned independently using three automatic binning programs: metabat2,64 concoct65 and maxbin2.66 DasTool67 was then used to select the best bacterial bins from the combination of these three automatic binning programs. The resulting draft genome bins were dereplicated at 98% whole-genome average nucleotide identity (gANI) via dRep (v2.6.2),68 using a minimum completeness of 75%, maximum contamination of 10%, the ANImf algorithm, 98% secondary clustering threshold, and 25% minimum coverage overlap. Genomes with gANI ≥ 98% were classified as the same subspecies, and the genome with the highest score (as determined by dRep) was chosen as the representative genome from each subspecies. A total of 1005 genomes were selected to represent unique microbial “subspecies'' and they had an average of 96% completeness and 1.05% contamination.
Taxonomy assignment
The amino acid sequences of predicted genes of all assembled bins were searched against the UniProt100 database using the usearch ublast command with a maximum e-value of 0.0001. tRep (https://github.com/MrOlm/tRep/tree/master/bin) was used to convert identified taxIDs into taxonomic levels. Briefly, for each taxonomic level (species, genus, phylum, etc.), a taxonomic label was assigned to a bin if ≥ 50% of proteins had best hits to the same taxonomic label. GTDB-Tk (v1.3.0)69 was used to resolve taxonomic levels that could not be assigned by tRep.
Detection of subspecies and identification of strains using inStrain
Reads from each individual fecal sample were mapped to all 1005 representative subspecies (generated via dRep as described above) concatenated together using Bowtie2 under default settings. inStrain (v1.3.4) profile19 was run on all resulting mapping files using a minimum mapQ score of 0 and insert size of 160. Genomes with ≥ 0.5 breadth (meaning at least half of the nucleotides of the genome are covered by ≥ 1 read) in samples were considered to be present. inStrain compare was run to compare the genome similarity among all subspecies that were present in ≥ 2 samples. Specifically, inStrain compare was used under default settings to compare read mappings to the same genome in different pairs of samples. Samples were considered to share the same strain of the examined genome if the compared region of the genome from samples shared ≥ 99.999% population-level ANI (popANI). Only genomic areas with at least 5x coverage in samples were compared, and sample pairs with less than 50% of comparable regions of the genome were excluded (≥0.5 percent_genome_compared).
Genome metabolic annotation
Kyoto Encyclopedia of Genes and Genomes (KEGG) orthology groups (KOs) were assigned to predicted ORFs for all fecal metagenomes using KofamKOALA.70 Carbohydrate active enzymes (CAZymes) were assigned to all nucleotide sequences using run_dbcan.py (https://github.com/linnabrown/run_dbcan) against the dbCAN HMM (v9), DIAMOND (v0.9.31), and Hotpep (v2.0.8) databases with default settings. Final CAZyme domain annotations were the best hits based on the outputs of all three databases. Domains were also predicted using hmmsearch (v.3.3) (e-value cut-off 1 × 10−6) against the Pfam r32 database.80 The domain architecture of each protein sequence was resolved using cath-resolve-hits (v0.16.5) with default settings.72 The transporters were predicted both hmmsearch (same settings as the pfam prediction and domain architecture was resolved using cath-resolve-hits) and BLASTP (v2.10.0) (keeping the best hit, e-value cutoff 1e-20) against the Transporter Classification Database (TCDB) (downloaded in November 2020).81 SignalP (v.5.0b) (parameters, -f short gram+) was used to predict proteins’ putative cellular localization.73 Transmembrane helices in proteins were predicted via TMHMM (v.2.0) with default settings.74 Secondary metabolites were characterized using antiSMASH (v5.1.2) with default settings.75
To identify E. coli virulence factors, ABRicate (https://github.com/tseemann/abricate) was used under default settings to search all predicted protein sequences associated with E. coli persisters and non-persister genomes against the E. coli virulence-associated gene database (EcVGDB).82
Identification of sources of contamination
One negative reagent control (NC) was included in each 96-well DNA extraction plate, in which no material was added during the DNA extraction step. In total this study involved five extraction plates labeled P1 to P5. NCs were labeled by the plate number (i.e., NC1 refers to the negative control sample on the extraction plate 1). All five NC samples were subjected to the DNA extraction and sequencing the same as the fecal samples. Subspecies present in NC samples were detected via mapping reads from NC samples to all 1005 representative subspecies as described above. Subspecies detection limit was the same as described above. We found two NC samples (NC3 and NC4) had over 50% of their reads mapped to ∼60 out of 1005 representative subspecies. To search for subspecies that was unique to NC samples, we recovered draft genomes from all five NC samples and dRep (settings were the same as described above) was run on these genomes together with the 1005 dereplicated genomes recovered from fecal samples. Through this approach, we did not find any subspecies that were unique to NC samples.
Detection of bacterial genomes in the NC3 and NC4 could be a result of index hopping, barcode bleeding, reagent contamination, and/or sample spillover (or “well-to-well contamination”). Since all samples were given Unique Dual Indexes, the observed contamination in NC3 and NC4 were unlikely to be a result of index hopping. We also eliminated the possibility of barcode bleeding by resequencing NC3 and NC4 alone. The possibility for reagent contamination to occur in our case was also unlikely since not only did we fail to detect any bacterial genomes in the rest of three NC samples, but we also did not find bacterial strains being shared over 50% of the samples either on the same extraction plates or across all five plates. We therefore hypothesized that the detection of intestinal bacterial genomes in NC3 and NC4 was a result of sample spillover within plates 3 and 4. Using the strain-resolved methods detailed above, we detected strain sharing across the extraction plates 3 and 4, but not with the rest of four plates. Given the reliance of our study on robust and accurate detection of strain sharing, we excluded from analysis all samples from plates 3 and 4. We were not able to resequence samples that were contaminated for this study. Beyond cost and lack of sufficient replacement samples, our laboratory was essentially closed for many months due to the pandemic and the sequencing facility diverted its capacity to COVID testing.
Detection of mother-to-infant vertical transmission
For each mother-infant pair, every fecal sample from the infant was compared to its maternal fecal sample to search for identical bacterial strains (≥99.999% popANI and ≥ 0.5 percent_genome_compared) via inStrain compare (described above). A strain was considered to be vertically transmitted if it was shared between the maternal fecal sample and at least one infant fecal sample.
Persister and non-persister detection
“Beginning-end” and “pairwise” approaches were used to identify persister and non-persister strains among early colonizers. The “beginning-end” approach searched for strains which shared ≥ 99.999% popANI between the first two months of life (≤month 2) and the last two sampling windows (around months 8 and 12). 54 persisters and 506 non-persisters were detected using this approach. The “pairwise approach” identified strains which shared ≥ 99.999% popANI across consecutive month windows (≤month 2 & month 3, month 3 & month 4, month 4 & ≥ month 8), yielding 36 persisters and 525 non-persisters. These two approaches combined resulted in the total identification of 59 persisters and 501 non-persisters across 22 infants (only 5 persistes were detected with the “pairwise approach” alone).
We chose to classify strains as persisters using the month 8 cutoff as we did not want to exclude persisters that would be missed due to lack of a month 12 sample (one infant) or poor genome recovery from month 12 samples (eight infants). We chose the cutoff of 99.999% popANI for persistence because we calculated that it is unlikely for a strain to acquire 40 SNPs in one year, given an average bacterial genome size of ∼4 Mbp and the expected rate of in situ bacterial evolution in the human gut (∼0.9 single-nucleotide polymorphisms (SNPs)/genome/year10).
Persister and non-persister functional enrichment analysis
Genes from persisters and non-persisters of each examined bacterial group (i.e., Bifidobacterium spp. and E. coli) were profiled via inStrain profile under default settings. Genes were considered to be present if they had ≥ 1x coverage across ≥ 70% of their length. Genes were annotated using the CAZy, KEGG, Pfam, Transporter Classification (TC) and E. coli virulence-associated gene (EcVG) databases as described above. Only annotations that were present in more than 65% of all persisters and less than 35% of all non-persisters as well as those that were present in less than 35% of all persisters and more than 65% of all non-persisters were kept for the enrichment analysis. Fisher’s exact test (as implemented using the Scipy module “scipy.stats.fisher_exact”) followed by false discovery rate (FDR) correction were run on genes annotated with each database (CAZy, KEGG, Pfam, TCDB and EcVFDB) independently to identify annotations from each database that were significantly enriched in persisters or non-persisters (q < 0.05).
To search for traits besides carbohydrate metabolism that were associated with E. coli persistence in the infant gut, genes with annotations that were significantly enriched in E. coli persisters or non-persisters from KEGG, Pfam, TC and EcVG databases were combined. Annotations were further verified using the UniProt100 and UniRef databases. In addition, we located genomic positions of differentially enriched annotations and used the functions of surrounding genes to improve the functional prediction for the gene of interest. The final datasheet listing annotations that were differentially enriched in E. coli persisters and non-persister is provided in Table S5. Annotations with p-values < 0.05 only (q-values > 0.05) are also provided in Table S5.
Detection of the complete colibactin biosynthesis gene cluster and its co-localization with the yersiniabactin biosynthesis gene clusters in E. coli persisters
Functional enrichment analysis (described above) revealed 14 out of 19 genes involved in colibactin biosynthesis were exclusively present in E. coli persisters. To confirm the presence of a complete colibactin synthesis cluster in all E. coli persisters, we located the gene cluster on the de novo constructed E. coli representative genome and manually inspected the read mapping on Geneious76 using reads from infant samples in which E. coli persisters were detected. No reads from non-persisters were mapped to the colibactin gene cluster.
On the same contig that we detected colibactin gene clusters, we also identified a complete yersiniabactin biosynthesis gene clusters, which were also found to be significantly enriched in E. coli persisters (Figure S5; Tables 1 and S5). The co-localization of colibactin and yersiniabactin biosynthesis gene clusters were verified to be present in all three E. coli persisters by inspecting the read mappings in Geneious.
Comparative genomic analysis on E. coli persisters and non-persisters
Infant-specific E. coli persister and non-persisters genomes that were from the same subspecies clusters as the dRep-chosen E. coli representative genomes were used to conduct comparative genomic analysis. Identification of matching scaffolds between E. coli persisters and non-persisters were achieved via BLAST. Specifically, scaffolds from E. coli persisters were compared to scaffolds from E. coli non-persisters using BLASTN (keeping the best hit, e-value cutoff 1e-10).
For each function that was found to be significantly enriched in E. coli persisters, we identified the scaffold from E. coli persisters in which the function was encoded on as well as the matching scaffold from E. coli non-persisters. Whole-scaffold alignments between persisters and non-persisters were performed in Geneious. Final alignments displayed in Figure S6 were created via clinker.77
Examination of coding density of surface adhesion and iron acquisition functions in E. coli persisters and non-persisters
To assess the coding density of surface adhesion and iron acquisition in E. coli, we first manually curated a list of KOs that were associated with either function based on extensive literature searches (Table S6). We then identified corresponding genes that were involved in either function. For iron acquisition, we further supplemented additional iron acquisition genes that were identified via FeGenie under default settings.78 For each E. coli persister and non-persister genome, coding density for either function was calculated by dividing the number of genes encoding surface adhesion or iron acquisition by the total number of genes.
Community diversity analysis
Since the earliest fecal sample was collected several days after birth for preterm infants and around the first month of life for full-term infants, all beta-diversity analysis between the two infant groups were conducted in the same chronological-age time frame (thus excluding any preterm samples taken before month 1). To measure convergence of the gut microbiomes, if not otherwise specified, a Wilcoxon rank-sum test was conducted to compare gut microbiomes at months 1 and 12. Modules from scikit-bio (http://scikit-bio.org/) were used to calculate weighted and unweighted UniFrac distances (“skbio.diversity.beta.weighted_unifrac” and “skbio.diversity.beta.unweighted_unifrac,” respectively), Bray-Curtis distance, and Jaccard dissimilarity (both were implemented via “skbio.diversity.beta_diversity”). To calculate UniFrac distances, a phylogenetic tree was constructed by comparing all 1005 dereplicated bacterial subspecies to each other using dRep cluster with a mash sketch size of 10,000.
Principal components analysis
Principal components analysis (PCA) (performed using scikit-learn [https://scikit-learn.org]) was conducted based on the relative abundance of bacterial subspecies in each fecal metagenome as assessed using weighted UniFrac distance. Significance of the clustering by variables (i.e., mode of delivery, prematurity, and feeding type) was determined by Permutational Multivariate Analysis of Variance (PERMANOVA) with 1000 permutations (as implemented using the scikit-bio module “skbio.stats.distance.permanova”).
Quantification and statistical analysis
Two-group univariate comparisons
Statistical significance for was calculated using Fisher’s exact test (as implemented using the Scipy module “scipy.stats.fisher_exact”), Wilcoxon rank-sum test (as implemented using the Scipy module “scipy.stats.ranksums”) and two-sided permutation test with 9999 permutations (in-house R script) as reported in the main text and in the STAR Methods. All multiple comparisons were false discovery rate (FDR) corrected with a threshold of q < 0.05.
Multivariate statistical analyses
Two-sided permutation test with 9999 permutations comparing the percentage of persisting early colonizers among infants indicated that full-term infants had more persisters than preterm infants (Figure 2B). To assess whether the outcome was confounded by other clinical variables, we developed a statistical model that takes into account and controls for all clinical data collected from infants enrolled in our study (in-house R script). We first evaluated the correlation between each pair of variables and found that some are confounded by the sampling design (e.g., all preterm babies received empiric antibiotics immediately following birth and had Prolacta added to their diet). Therefore, it is not possible to quantify the influence of those effects independently. In addition, variables including birth weight, extent of hospital stay, whether had NEC and/or LOS, weaning starting time and antibiotic administrations before month 2 are highly correlated with preterm delivery and therefore cannot be quantified individually in our study. We therefore performed our statistical analyses excluding these preterm-associated variables and controlling for other clinical factors (term/preterm status, gender, feeding practices (BRM only versus BRM plus formula), and antibiotics administrations after month 2). To show whether full-term status had a significant impact on the percentage of persisters in an infant, a generalized linear model (GLM) with a Poisson family was performed using R.
Acknowledgments
We thank Rohan Sachdeva, Ka Ki Lily Law, and Shufei Lei for the technical support; Raphaël Méheust and Jacob West-Roberts for assistance in bioinformatics tools; Alexandra Sheppeck for fecal sample collection; Yun Song for helpful suggestions; and Adair Borges for comments on the manuscript. We are also grateful for all the families that participated in this study. For funding support, we acknowledge NIH award RAI092531A to J.F.B and M.J.M and Chan Zuckerberg Biohub support to J.F.B.
Author contributions
Y.C.L., M.R.O., M.J.M., and J.F.B. designed the study; B.A.F. performed DNA extractions of fecal samples; R.B. supervised the enrollment of infants; Y.C.L. coordinated the acquisition of, and performed analysis on, the metagenomics data; Y.C.L. and S.D. conducted statistical modeling; M.R.O, S.D., and A.C.-C. assisted with functional enrichment analyses; Y.C.L and J.F.B. wrote the manuscript, and all authors contributed to the manuscript revisions.
Declaration of interests
J.F.B. is a cofounder of Metagenomi. The other authors declare no completing interests.
Published: September 7, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100393.
Supplemental information
Data and code availability
-
•
Metagenomics sequencing reads reported in this paper are available under NCBI BioProject: PRJNA698986; SRA: SRR13622550–SRR13622957. Metagenome assembled genomes have been deposited at GenBank: JAGYZD000000000–JAHALT000000000.
-
•
R script used for two-sided permutation test and generalized linear model is available on GitHub: https://github.com/clarelou0128/R-statistical-scripts.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
Reference
- 1.Robertson R.C., Manges A.R., Finlay B.B., Prendergast A.J. The human microbiome and child growth—first 1000 days and beyond. Trends Microbiol. 2019;27:131–147. doi: 10.1016/j.tim.2018.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Wang S., Ryan C.A., Boyaval P., Dempsey E.M., Ross R.P., Stanton C. Maternal vertical transmission affecting early-life microbiota development. Trends Microbiol. 2020;28:28–45. doi: 10.1016/j.tim.2019.07.010. [DOI] [PubMed] [Google Scholar]
- 3.Baumann-Dudenhoeffer A.M., D’Souza A.W., Tarr P.I., Warner B.B., Dantas G. Infant diet and maternal gestational weight gain predict early metabolic maturation of gut microbiomes. Nat. Med. 2018;24:1822–1829. doi: 10.1038/s41591-018-0216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shao Y., Forster S.C., Tsaliki E., Vervier K., Strang A., Simpson N., Kumar N., Stares M.D., Rodger A., Brocklehurst P. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature. 2019;574:117–121. doi: 10.1038/s41586-019-1560-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yassour M., Vatanen T., Siljander H., Hämäläinen A.-M., Härkönen T., Ryhänen S.J., Franzosa E.A., Vlamakis H., Huttenhower C., Gevers D., DIABIMMUNE Study Group Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci. Transl. Med. 2016;8:343ra81. doi: 10.1126/scitranslmed.aad0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bisgaard H., Li N., Bonnelykke K., Chawes B.L.K., Skov T., Paludan-Müller G., Stokholm J., Smith B., Krogfelt K.A. Reduced diversity of the intestinal microbiota during infancy is associated with increased risk of allergic disease at school age. J. Allergy Clin. Immunol. 2011;128:646–652.e1–5. doi: 10.1016/j.jaci.2011.04.060. [DOI] [PubMed] [Google Scholar]
- 7.Arrieta M.-C., Stiemsma L.T., Dimitriu P.A., Thorson L., Russell S., Yurist-Doutsch S., Kuzeljevic B., Gold M.J., Britton H.M., Lefebvre D.L., CHILD Study Investigators Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci. Transl. Med. 2015;7:307ra152. doi: 10.1126/scitranslmed.aab2271. [DOI] [PubMed] [Google Scholar]
- 8.Tamburini S., Shen N., Wu H.C., Clemente J.C. The microbiome in early life: implications for health outcomes. Nat. Med. 2016;22:713–722. doi: 10.1038/nm.4142. [DOI] [PubMed] [Google Scholar]
- 9.Faith J.J., Guruge J.L., Charbonneau M., Subramanian S., Seedorf H., Goodman A.L., Clemente J.C., Knight R., Heath A.C., Leibel R.L. The long-term stability of the human gut microbiota. Science. 2013;341:1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao S., Lieberman T.D., Poyet M., Kauffman K.M., Gibbons S.M., Groussin M., Xavier R.J., Alm E.J. Adaptive evolution within gut microbiomes of healthy people. Cell Host Microbe. 2019;25:656–667.e8. doi: 10.1016/j.chom.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schloissnig S., Arumugam M., Sunagawa S., Mitreva M., Tap J., Zhu A., Waller A., Mende D.R., Kultima J.R., Martin J. Genomic variation landscape of the human gut microbiome. Nature. 2013;493:45–50. doi: 10.1038/nature11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yatsunenko T., Rey F.E., Manary M.J., Trehan I., Dominguez-Bello M.G., Contreras M., Magris M., Hidalgo G., Baldassano R.N., Anokhin A.P. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koenig J.E., Spor A., Scalfone N., Fricker A.D., Stombaugh J., Knight R., Angenent L.T., Ley R.E. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. USA. 2011;108(Suppl 1):4578–4585. doi: 10.1073/pnas.1000081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gibson M.K., Wang B., Ahmadi S., Burnham C.-A.D., Tarr P.I., Warner B.B., Dantas G. Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nat. Microbiol. 2016;1:16024. doi: 10.1038/nmicrobiol.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gasparrini A.J., Wang B., Sun X., Kennedy E.A., Hernandez-Leyva A., Ndao I.M., Tarr P.I., Warner B.B., Dantas G. Persistent metagenomic signatures of early-life hospitalization and antibiotic treatment in the infant gut microbiota and resistome. Nat. Microbiol. 2019;4:2285–2297. doi: 10.1038/s41564-019-0550-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raveh-Sadka T., Firek B., Sharon I., Baker R., Brown C.T., Thomas B.C., Morowitz M.J., Banfield J.F. Evidence for persistent and shared bacterial strains against a background of largely unique gut colonization in hospitalized premature infants. ISME J. 2016;10:2817–2830. doi: 10.1038/ismej.2016.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.La Rosa P.S., Warner B.B., Zhou Y., Weinstock G.M., Sodergren E., Hall-Moore C.M., Stevens H.J., Bennett W.E., Jr., Shaikh N., Linneman L.A. Patterned progression of bacterial populations in the premature infant gut. Proc. Natl. Acad. Sci. USA. 2014;111:12522–12527. doi: 10.1073/pnas.1409497111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brito I.L., Alm E.J. Tracking strains in the microbiome: insights from metagenomics and models. Front. Microbiol. 2016;7:712. doi: 10.3389/fmicb.2016.00712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olm M.R., Crits-Christoph A., Bouma-Gregson K., Firek B.A., Morowitz M.J., Banfield J.F. inStrain profiles population microdiversity from metagenomic data and sensitively detects shared microbial strains. Nat. Biotechnol. 2021;39:727–736. doi: 10.1038/s41587-020-00797-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Rossum T., Ferretti P., Maistrenko O.M., Bork P. Diversity within species: interpreting strains in microbiomes. Nat. Rev. Microbiol. 2020;18:491–506. doi: 10.1038/s41579-020-0368-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brooks B., Olm M.R., Firek B.A., Baker R., Thomas B.C., Morowitz M.J., Banfield J.F. Strain-resolved analysis of hospital rooms and infants reveals overlap between the human and room microbiome. Nat. Commun. 2017;8:1814. doi: 10.1038/s41467-017-02018-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olm M.R., Bhattacharya N., Crits-Christoph A., Firek B.A., Baker R., Song Y.S., Morowitz M.J., Banfield J.F. Necrotizing enterocolitis is preceded by increased gut bacterial replication, Klebsiella, and fimbriae-encoding bacteria. Sci. Adv. 2019;5:eaax5727. doi: 10.1126/sciadv.aax5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olm M.R., Brown C.T., Brooks B., Firek B., Baker R., Burstein D., Soenjoyo K., Thomas B.C., Morowitz M., Banfield J.F. Identical bacterial populations colonize premature infant gut, skin, and oral microbiomes and exhibit different in situ growth rates. Genome Res. 2017;27:601–612. doi: 10.1101/gr.213256.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brooks B., Firek B.A., Miller C.S., Sharon I., Thomas B.C., Baker R., Morowitz M.J., Banfield J.F. Microbes in the neonatal intensive care unit resemble those found in the gut of premature infants. Microbiome. 2014;2:1. doi: 10.1186/2049-2618-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vatanen T., Plichta D.R., Somani J., Münch P.C., Arthur T.D., Hall A.B., Rudolf S., Oakeley E.J., Ke X., Young R.A. Genomic variation and strain-specific functional adaptation in the human gut microbiome during early life. Nat. Microbiol. 2019;4:470–479. doi: 10.1038/s41564-018-0321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferretti P., Pasolli E., Tett A., Asnicar F., Gorfer V., Fedi S., Armanini F., Truong D.T., Manara S., Zolfo M. Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microbe. 2018;24:133–145.e5. doi: 10.1016/j.chom.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Podlesny D., Fricke W.F. Strain inheritance and neonatal gut microbiota development: a meta-analysis. Int. J. Med. Microbiol. 2021;311:151483. doi: 10.1016/j.ijmm.2021.151483. [DOI] [PubMed] [Google Scholar]
- 28.Asnicar F., Manara S., Zolfo M., Truong D.T., Scholz M., Armanini F., Ferretti P., Gorfer V., Pedrotti A., Tett A., Segata N. Studying vertical microbiome transmission from mothers to infants by strain-level metagenomic profiling. mSystems. 2017;2:e00164. doi: 10.1128/mSystems.00164-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korpela K., Costea P., Coelho L.P., Kandels-Lewis S., Willemsen G., Boomsma D.I., Segata N., Bork P. Selective maternal seeding and environment shape the human gut microbiome. Genome Res. 2018;28:561–568. doi: 10.1101/gr.233940.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yassour M., Jason E., Hogstrom L.J., Arthur T.D., Tripathi S., Siljander H., Selvenius J., Oikarinen S., Hyöty H., Virtanen S.M. Strain-level analysis of mother-to-child bacterial transmission during the first few months of life. Cell Host Microbe. 2018;24:146–154.e4. doi: 10.1016/j.chom.2018.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El Kaoutari A., Armougom F., Gordon J.I., Raoult D., Henrissat B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 2013;11:497–504. doi: 10.1038/nrmicro3050. [DOI] [PubMed] [Google Scholar]
- 32.Fischbach M.A., Sonnenburg J.L. Eating for two: how metabolism establishes interspecies interactions in the gut. Cell Host Microbe. 2011;10:336–347. doi: 10.1016/j.chom.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sela D.A., Mills D.A. Nursing our microbiota: molecular linkages between bifidobacteria and milk oligosaccharides. Trends Microbiol. 2010;18:298–307. doi: 10.1016/j.tim.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcobal A., Barboza M., Sonnenburg E.D., Pudlo N., Martens E.C., Desai P., Lebrilla C.B., Weimer B.C., Mills D.A., German J.B., Sonnenburg J.L. Bacteroides in the infant gut consume milk oligosaccharides via mucus-utilization pathways. Cell Host Microbe. 2011;10:507–514. doi: 10.1016/j.chom.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wexler A.G., Goodman A.L. An insider’s perspective: bacteroides as a window into the microbiome. Nat. Microbiol. 2017;2:17026. doi: 10.1038/nmicrobiol.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Little D.J., Pfoh R., Le Mauff F., Bamford N.C., Notte C., Baker P., Guragain M., Robinson H., Pier G.B., Nitz M. PgaB orthologues contain a glycoside hydrolase domain that cleaves deacetylated poly-β(1,6)-N-acetylglucosamine and can disrupt bacterial biofilms. PLoS Pathog. 2018;14:e1006998. doi: 10.1371/journal.ppat.1006998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doyle L., Ovchinnikova O.G., Myler K., Mallette E., Huang B.-S., Lowary T.L., Kimber M.S., Whitfield C. Biosynthesis of a conserved glycolipid anchor for Gram-negative bacterial capsules. Nat. Chem. Biol. 2019;15:632–640. doi: 10.1038/s41589-019-0276-8. [DOI] [PubMed] [Google Scholar]
- 38.Carl M.A., Ndao I.M., Springman A.C., Manning S.D., Johnson J.R., Johnston B.D., Burnham C.-A.D., Weinstock E.S., Weinstock G.M., Wylie T.N. Sepsis from the gut: the enteric habitat of bacteria that cause late-onset neonatal bloodstream infections. Clin. Infect. Dis. 2014;58:1211–1218. doi: 10.1093/cid/ciu084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruhe Z.C., Townsley L., Wallace A.B., King A., Van der Woude M.W., Low D.A., Yildiz F.H., Hayes C.S. CdiA promotes receptor-independent intercellular adhesion. Mol. Microbiol. 2015;98:175–192. doi: 10.1111/mmi.13114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trunk T., Khalil H.S., Leo J.C. Bacterial autoaggregation. AIMS Microbiol. 2018;4:140–164. doi: 10.3934/microbiol.2018.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nougayrède J.-P., Homburg S., Taieb F., Boury M., Brzuszkiewicz E., Gottschalk G., Buchrieser C., Hacker J., Dobrindt U., Oswald E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313:848–851. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 42.Nipič D., Podlesek Z., Budič M., Črnigoj M., Žgur-Bertok D. Escherichia coli uropathogenic-specific protein, Usp, is a bacteriocin-like genotoxin. J. Infect. Dis. 2013;208:1545–1552. doi: 10.1093/infdis/jit480. [DOI] [PubMed] [Google Scholar]
- 43.Parret A.H.A., De Mot R. Escherichia coli’s uropathogenic-specific protein: a bacteriocin promoting infectivity? Microbiology (Reading) 2002;148:1604–1606. doi: 10.1099/00221287-148-6-1604. [DOI] [PubMed] [Google Scholar]
- 44.Putze J., Hennequin C., Nougayrède J.-P., Zhang W., Homburg S., Karch H., Bringer M.-A., Fayolle C., Carniel E., Rabsch W. Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect. Immun. 2009;77:4696–4703. doi: 10.1128/IAI.00522-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wami H., Wallenstein A., Sauer D., Stoll M., von Bünau R., Oswald E., Müller R., Dobrindt U. Diversity and prevalence of colibactin- and yersiniabactin encoding mobile genetic elements in enterobacterial populations: insights into evolution and co-existence of two bacterial secondary metabolite determinants. bioRxiv. 2021 doi: 10.1101/2021.01.22.427840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin P., Marcq I., Magistro G., Penary M., Garcie C., Payros D., Boury M., Olier M., Nougayrède J.-P., Audebert M. Interplay between siderophores and colibactin genotoxin biosynthetic pathways in Escherichia coli. PLoS Pathog. 2013;9:e1003437. doi: 10.1371/journal.ppat.1003437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lozupone C., Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rice L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect. Dis. 2008;197:1079–1981. doi: 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 49.Nowrouzian F.L., Oswald E. Escherichia coli strains with the capacity for long-term persistence in the bowel microbiota carry the potentially genotoxic pks island. Microb. Pathog. 2012;53:180–182. doi: 10.1016/j.micpath.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 50.Palmer C., Bik E.M., DiGiulio D.B., Relman D.A., Brown P.O. Development of the human infant intestinal microbiota. PLoS Biol. 2007;5:e177. doi: 10.1371/journal.pbio.0050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sprockett D., Fukami T., Relman D.A. Role of priority effects in the early-life assembly of the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2018;15:197–205. doi: 10.1038/nrgastro.2017.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gensollen T., Iyer S.S., Kasper D.L., Blumberg R.S. How colonization by microbiota in early life shapes the immune system. Science. 2016;352:539–544. doi: 10.1126/science.aad9378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hubbell S.P. Princeton University Press; 2001. The Unified Neutral Theory of Biodiversity and Biogeography (MPB-32) [DOI] [PubMed] [Google Scholar]
- 54.Martínez I., Maldonado-Gomez M.X., Gomes-Neto J.C., Kittana H., Ding H., Schmaltz R., Joglekar P., Cardona R.J., Marsteller N.L., Kembel S.W. Experimental evaluation of the importance of colonization history in early-life gut microbiota assembly. eLife. 2018;7:e36521. doi: 10.7554/eLife.36521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bäckhed F., Roswall J., Peng Y., Feng Q., Jia H., Kovatcheva-Datchary P., Li Y., Xia Y., Xie H., Zhong H. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17:852. doi: 10.1016/j.chom.2015.05.012. [DOI] [PubMed] [Google Scholar]
- 56.Fanning S., Hall L.J., Cronin M., Zomer A., MacSharry J., Goulding D., Motherway M.O., Shanahan F., Nally K., Dougan G., van Sinderen D. Bifidobacterial surface-exopolysaccharide facilitates commensal-host interaction through immune modulation and pathogen protection. Proc. Natl. Acad. Sci. USA. 2012;109:2108–2113. doi: 10.1073/pnas.1115621109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nowrouzian F.L., Wold A.E., Adlerberth I. Escherichia coli strains belonging to phylogenetic group B2 have superior capacity to persist in the intestinal microflora of infants. J. Infect. Dis. 2005;191:1078–1083. doi: 10.1086/427996. [DOI] [PubMed] [Google Scholar]
- 58.Wold A.E., Caugant D.A., Lidin-Janson G., de Man P., Svanborg C. Resident colonic Escherichia coli strains frequently display uropathogenic characteristics. J. Infect. Dis. 1992;165:46–52. doi: 10.1093/infdis/165.1.46. [DOI] [PubMed] [Google Scholar]
- 59.Nowrouzian F.L., Adlerberth I., Wold A.E. Enhanced persistence in the colonic microbiota of Escherichia coli strains belonging to phylogenetic group B2: role of virulence factors and adherence to colonic cells. Microbes Infect. 2006;8:834–840. doi: 10.1016/j.micinf.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 60.Melville J.M., Moss T.J.M. The immune consequences of preterm birth. Front. Neurosci. 2013;7:79. doi: 10.3389/fnins.2013.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peng Y., Leung H.C.M., Yiu S.M., Chin F.Y.L. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 63.Hyatt D., Chen G.-L., Locascio P.F., Land M.L., Larimer F.W., Hauser L.J. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kang D.D., Li F., Kirton E., Thomas A., Egan R., An H., Wang Z. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 2019;7:e7359. doi: 10.7717/peerj.7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alneberg J., Bjarnason B.S., de Bruijn I., Schirmer M., Quick J., Ijaz U.Z., Lahti L., Loman N.J., Andersson A.F., Quince C. Binning metagenomic contigs by coverage and composition. Nat. Methods. 2014;11:1144–1146. doi: 10.1038/nmeth.3103. [DOI] [PubMed] [Google Scholar]
- 66.Wu Y.-W., Simmons B.A., Singer S.W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2016;32:605–607. doi: 10.1093/bioinformatics/btv638. [DOI] [PubMed] [Google Scholar]
- 67.Sieber C.M.K., Probst A.J., Sharrar A., Thomas B.C., Hess M., Tringe S.G., Banfield J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018;3:836–843. doi: 10.1038/s41564-018-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Olm M.R., Brown C.T., Brooks B., Banfield J.F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017;11:2864–2868. doi: 10.1038/ismej.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chaumeil P.-A., Mussig A.J., Hugenholtz P., Parks D.H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2019:btz848. doi: 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aramaki T., Blanc-Mathieu R., Endo H., Ohkubo K., Kanehisa M., Goto S., Ogata H. KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics. 2020;36:2251–2252. doi: 10.1093/bioinformatics/btz859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eddy S.R. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 72.Lewis T.E., Sillitoe I., Lees J.G. cath-resolve-hits: a new tool that resolves domain matches suspiciously quickly. Bioinformatics. 2019;35:1766–1767. doi: 10.1093/bioinformatics/bty863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Almagro Armenteros J.J., Tsirigos K.D., Sønderby C.K., Petersen T.N., Winther O., Brunak S., von Heijne G., Nielsen H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019;37:420–423. doi: 10.1038/s41587-019-0036-z. [DOI] [PubMed] [Google Scholar]
- 74.Krogh A., Larsson B., von Heijne G., Sonnhammer E.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 75.Blin K., Shaw S., Steinke K., Villebro R., Ziemert N., Lee S.Y., Medema M.H., Weber T. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019;47(W1):W81–W87. doi: 10.1093/nar/gkz310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gilchrist C.L.M., Chooi Y.-H. Clinker & clustermap.js: automatic generation of gene cluster comparison figures. Bioinformatics. 2021 doi: 10.1093/bioinformatics/btab007. Published online January 18, 2021. [DOI] [PubMed] [Google Scholar]
- 78.Garber A.I., Nealson K.H., Okamoto A., McAllister S.M., Chan C.S., Barco R.A., Merino N. FeGenie: A Comprehensive Tool for the Identification of Iron Genes and Iron Gene Neighborhoods in Genome and Metagenome Assemblies. Front. Microbiol. 2020;11:37. doi: 10.3389/fmicb.2020.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Olm M.R., West P.T., Brooks B., Firek B.A., Baker R., Morowitz M.J., Banfield J.F. Genome-resolved metagenomics of eukaryotic populations during early colonization of premature infants and in hospital rooms. Microbiome. 2019;7:26. doi: 10.1186/s40168-019-0638-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.El-Gebali S., Mistry J., Bateman A., Eddy S.R., Luciani A., Potter S.C., Qureshi M., Richardson L.J., Salazar G.A., Smart A. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47(D1):D427–D432. doi: 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saier M.H., Jr., Tran C.V., Barabote R.D. TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 2006;34:D181–D186. doi: 10.1093/nar/gkj001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Biggel M., Xavier B.B., Johnson J.R., Nielsen K.L., Frimodt-Møller N., Matheeussen V., Goossens H., Moons P., Van Puyvelde S. Horizontally acquired papGII-containing pathogenicity islands underlie the emergence of invasive uropathogenic Escherichia coli lineages. Nat. Commun. 2020;11:5968. doi: 10.1038/s41467-020-19714-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Metagenomics sequencing reads reported in this paper are available under NCBI BioProject: PRJNA698986; SRA: SRR13622550–SRR13622957. Metagenome assembled genomes have been deposited at GenBank: JAGYZD000000000–JAHALT000000000.
-
•
R script used for two-sided permutation test and generalized linear model is available on GitHub: https://github.com/clarelou0128/R-statistical-scripts.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.