

Abstract
Laryngopharyngeal reflux, a variant of gastroesophageal reflux disease, has been considered a risk factor in the development of hypopharyngeal cancer. Bile acids are frequently present in the gastroesophageal refluxate and their effect has been associated with inflammatory and neoplastic changes in the upper aerodigestive tract. Recent in vitro and in vivo studies have provided direct evidence of the role of acidic bile refluxate in hypopharyngeal carcinogenesis and documented the crucial role of NF-κB as a key mediator of early oncogenic molecular events in this process and also suggested a contribution of STAT3. Acidic bile can cause premalignant changes and invasive squamous cell cancer in the affected hypopharynx accompanied by DNA damage, elevated p53 expression and oncogenic mRNA and microRNA alterations, previously linked to head and neck cancer. Weakly acidic bile can also increase the risk for hypopharyngeal carcinogenesis by inducing DNA damage, exerting anti-apoptotic effects and causing precancerous lesions. The most important findings that strongly support bile reflux as an independent risk factor for hypopharyngeal cancer are presented in the current review and the underlying mechanisms are provided.
Keywords: laryngopharyngeal reflux, bile reflux, conjugated bile acids, deoxycholic acid, head and neck cancer, hypopharyngeal cancer; hypopharyngeal squamous cell carcinoma, in vivo, NF-κB, DNA damage
1. Introduction
Tobacco smoke, chronic alcohol use and infection with human papillomavirus type 16 (HPV-16) are known risk factors for laryngopharyngeal cancer (1–4); however, there is a growing interest in identifying other risk factors.
Laryngopharyngeal reflux (LPR), a variant of gastro-esophageal reflux disease (GERD), has also been considered a potential risk factor in the last decade that may exert a carcinogenic effect on the upper aerodigestive tract (5–11). Recent findings have clarified the role of bile reflux as an independent risk factor in hypopharyngeal carcinogenesis through preclinical and clinical models (12–18), and a model of the molecular mechanism of the bile-induced tumorigenic effect has been proposed (19–25). Bile is produced in the liver and stored in the gallbladder (26) and its primary role is to assist in lipid digestion and absorption. Regurgitation of duodenal bile contents into the stomach and esophagus is known as bile reflux. The present review provides the latest knowledge regarding the association of bile reflux with hypopharyngeal cancer.
2. Bile as a potential carcinogen
The caustic nature of bile has long been recognized (27). In ancient times, the medical theory that excess, deficiency or ectopic bile in the body may affect human health was first stated by the father of medicine, Hippocrates (27). This medical theory remained popular for centuries through the writings of Galen (129–201 AD) (28) but was decisively displaced newly published theories of cellular pathology by Virchow and Rather in 1858 (29). In the past century, in 1938-40, Cook (30) proposed the possible role of bile acids in cancer. Years later (1974–1993) several studies supported the role of bile acids as carcinogens causing gastrointestinal cancer (31–34). In parallel, clinical studies provided the first evidence of mixed gastric and bile (duodenal) fluids in refluxate of patients with GERD (35–37). During this period, Gotley et al (38) determined an increased amount of conjugated bile acids in 87% of aspirates using high-performance liquid chromatography, suggesting an association between bile and acid concentration and esophageal mucosal injury. Nehra et al (35), Kauer et al (39,40) and Domellof et al (41) also characterized the concentration and composition of bile fluid in aspirates of patients with GERD. Of note, Fein et al (42) were the first who provided solid evidence of bile fluid as an independent carcinogen in the gastrointestinal tract by using a rat model and demonstrating that bile acids are able to induce esophageal adenocarcinoma.
Until the present day, the clinical prevalence and magnitude of bile reflux have remained to be fully determined; however, there is growing evidence of bile contents in GERD refluxate (31,43–45). According to Covington et al (46), bile-containing enterogastric reflux is much more common than previously assumed. The increased information linking LPR and inflammatory/neoplastic disease of the upper aerodigestive tract as well as the lack of evidence for the carcinogenic effect of bile-containing refluxate into laryngopharynx led to further investigation.
Considerable research efforts to explore the carcinogenic effect of bile in the upper aerodigestive tract were made, including clinical and experimental studies. Galli et al (5) and Sasaki et al (11) suggested that during LPR, bile fluid reaches the epithelium of the upper aerodigestive tract, which may contribute to the development of inflammatory and neoplastic events. Furthermore, Lewin et al (47) reported a close association between LPR and patients with premalignant lesions or early carcinomas of the larynx. It is worth mentioning that although there have been efforts to link the effect of other gastroesophageal refluxate contents, such as pepsin, with pre-neoplastic events in the larynx and pharynx, the conclusions have been divergent (48–54). A recent study indicated a possible contributory effect of slightly acidic or neutral pepsin to the inflammatory and neoplastic effects of LPR (54), but there is still no direct evidence of carcinogenesis induced by pepsin.
At the beginning of 2016, a series of in vitro and in vivo experiments questioned the role of bile reflux in hypopharyngeal carcinogenesis (12–14). A study by our group from 2016 (13) established a murine model of wild-type mice, Mus musculus (C57BL/6J) and provided the first evidence that bile acids may cause preneoplastic lesions in the hypopharyngeal mucosa (HM). Using this model and long-term exposure to bile acids, the progressive mutagenic effect of biliary refluxate causing invasive cancer was subsequently observed (15,16). These and other studies provided direct evidence that bile fluid is a carcinogen, capable of inducing hypopharyngeal squamous cell carcinoma (HSCC) (15,16). The significance of these findings was in line with clinical findings derived from bile reflux-related HSCC (18). Specifically, a clinical pilot study demonstrated a characteristic bile-related molecular phenotype that was similarly identified in bile-exposed murine HM, which clearly differed from adjacent non-pathologic tissue (18).
3. Bile refluxate composition and acidity as critical factors of neoplastic events
Bile refluxate composition
According to Nehra et al (35) and Kauer et al (39,40), the majority of bile acids in esophageal aspirates of patients with GERD are conjugated with taurine or glycine and may be sulfated. Specifically, glycine conjugates of cholic, deoxycholic and chenodeoxycholic acids are the predominant bile acids aspirated from the esophagus of patients with GERD (the ratio of glycine to taurine conjugates in normal human bile is 3:1). It has also been indicated that unconjugated secondary bile acids, such as deoxycholic acid (DCA), may be present in the esophageal refluxate, particularly in patients with erosive esophagitis and Barrett's esophagus (35,41,55). It is known that pH affects the solubility of each bile component in various manners. At acidic pH (≤4), the conjugated bile acids are more un-ionized and therefore capable of penetrating or interacting with the cell membrane (taurine conjugates: pKa=1.8–1.9; glycine conjugates: pKa=4.3–5.2) (56). At pH <3.0, bile salts tend to precipitate, whereas between pH 5.5 and 7.0, most conjugated primary bile acids are found to be ionized and therefore relatively inactive. However, unconjugated secondary bile acid DCA remains unionized (pKa 5.5–6.2) and may therefore exert its harmful effects, causing mucosal injury even at a less acidic pH (12,16,17,55). According to Stamp (57), duodenal fluid, at a less acidic pH, may also contribute to gastrointestinal tract tumorigenesis. Specifically, glycine conjugates may remain un-charged and therefore be harmful to the epithelial cells at a less acidic pH. Ireland et al (58) indicated that duodenal fluid significantly contributes to the carcinogenic potential of methyl-N-amyl nitrosamine, particularly at less acidic pH. In addition, as bile acids are natural detergents when in high concentration, they may interact with the cell membrane even at a neutral pH.
Bile refluxate concentration
Although the bile composition is a crucial factor of bile reflux-related tumorigenesis, the concentration of bile may also potentiate its oncogenic effect. This view may be supported by previous in vivo studies indicating that the application of bile reflux components, chenodeoxycholic acid or DCA, at pharmacologic concentrations, both at a neutral pH of 7.0, to murine laryngopharyngeal mucosa was able to cause early premalignant changes, such as hyperplasia and dysplasia, as well as marked activation of NF-κB and its related oncogenic molecular phenotype (13,14).
Bile refluxate acidity
It appears that acidity of LPR refluxate is a critical factor for bile to induce a harmful effect on laryngopharyngeal mucosa (39,59,60). In the clinic, intraesophageal pH monitoring has been used extensively to identify reflux episodes. Although pH varies during gastroesophageal reflux episodes, according to Ulualp et al (61), 24-h ambulatory pH monitoring in the pharynx of patients with reflux laryngitis confirmed that a drop below pH 4.0 is common and is considered diagnostic of a reflux event, suggesting that acid may contribute to duodenogastric-induced inflammatory and neoplastic events. Lillemoe et al (62) demonstrated the injurious effect of the various duodenal components on rabbit esophageal mucosa at strongly acidic pH, supporting a synergism between bile and HCl. A study by our group from 2019 (17) documented that the tumorigenic effect of bile on hypopharyngeal cells is pH-dependent. Other studies from our group (13–15) also demonstrated that a strongly acidic pH (≤4.0) serves a critical role in the bile-induced tumorigenic effect in murine laryngopharyngeal mucosa. It was indicated that chronic intermittent exposure of murine HM to a mixture of bile salts at a strongly acidic pH of 3.0 was able to progressively induce premalignant changes, microinvasion and invasive squamous cell carcinoma, causing increased DNA damage and oncogenic molecular alterations (15). Specifically, it was demonstrated that histopathologic changes caused by acidic bile were accompanied by underlying molecular alterations, such as increased levels of i) oxidative DNA/RNA damage and double-strand break (DSB) markers, ii) p53 and cell proliferation markers (Ki67, cytokeratin 14, and p63), as well as iii) alterations of the expression of cell adhesion molecules, like E-Cadherin and β-catenin, and iv) activation of NF-κB and other cancer-related transcription factors, such as signal transducer and activator of transcription 3 (STAT3) (13–15). However, hypopharyngeal cells or mucosa exposed to the same mixture of conjugated primary bile acids at neutral pH (7.0) produced hyperplastic or mild dysplastic changes and significantly less intense underlying molecular changes compared to acidic bile salts (12,13,17). In parallel, it was indicated that chronic exposure to acid alone or concentrated glucose was not able to produce any histological changes (13,15). The negative or reduced effect of acid alone and/or bile salts at neutral pH, compared to acidic bile salts, indicates that the latter is particularly injurious.
Primary and secondary bile acids in refluxate
It is clear that the presence of conjugated primary bile acids in a highly acidic refluxate exerts a tumorigenic potency on the long-term exposed upper aerodigestive tract. This theory may explain findings from our group (13,15) indicating that chronic local exposure of murine HM to a mixture of conjugated primary bile acids, at concentrations previously measured in patients with GERD (35,40,43,35) at a strongly acidic pH (≤4.0), is able to progressively cause precancerous lesions and invasive cancer. Specifically, as taurine conjugates are active at low pH (≤4.0), it appears that taurine-conjugated bile acids may be responsible for the tumorigenic effect of bile at lower pH (15,17). There is also recent in vitro evidence supporting that acidity (pH ≤4.0) and bile composition may have a role in the progression of HSCC (63).
The above observations strongly support that controlling the pH during reflux episodes may have a protective effect by reducing the risk of bile-induced hypopharyngeal cancer. However, there is epidemiologic evidence that numerous patients with refractory GERD may also experience symptoms at a weakly acidic pH of 5.5–6.0 (60,64). Since unconjugated DCA and glycine-conjugated bile acids may be partially active at a weakly acidic pH, it appears that as the pH grows less acidic, approaching pH 5.5, the partially activated primary bile acids and the activated DCA may exert their influence (17). A recent study by our group (16) supported that DCA and glycine-conjugated bile acids are potent activators of DNA damage and oncogenic pathways in HM in a weakly acidic environment. Previous findings have demonstrated a similar association between DCA and its tumorigenic activity in the esophagus and colon (31,65,66). Regarding the hypopharynx, it has been documented that bile at a weakly acidic pH (5.0–5.5) with or without DCA, similarly to a strongly acidic pH 3.0, is able to increase the risk of bile-related hypopharyngeal neoplasia by promoting premalignant lesions, DNA damage and oncogenic molecular alterations, compared to controls (16). Of note, it was indicated that long-term exposure to a weakly acidic control (pH 5.5) was not able to induce any histological changes (16). This observation strongly supports that the oncogenic properties of biliary esophageal reflux on laryngopharyngeal mucosa may not be fully modified when antacid therapy is applied.
Although further exploration with clinical evidence is expected to strengthen these previous preclinical observations, investigation of the mechanism by which bile refluxate exerts its oncogenic properties is expected to contribute not only to a better understanding of the pathophysiology of hypopharyngeal cancer but also to alternative therapeutic strategies for patients with refractory GERD, using specific inhibitors of relevant molecular pathways or bile receptors.
4. Key role of NF-κB in bile reflux-related hypopharyngeal carcinogenesis
Several epidemiologic studies have supported the role of LPR in the neoplasia of the upper aerodigestive tract (5–11,67,68). However, the exact mechanism of LPR-related laryngopharyngeal carcinogenesis has remained elusive and unexplored until the last decade (69–71). Studies including that by Huo et al (66) indicated that exposure of esophageal cells to DCA produced elevated levels of NF-κB in vitro, suggesting the role of NF-κB as a key molecule in esophageal cancer (72–75). Thus, these observations supported the hypothesis of a possible mechanistic role of NF-κB in cancer of extraesophageal sites, such as hypopharynx.
NF-κB is a transcriptional factor complex consisting of homo- and heterodimers of five members of the Rel family [RelA (p65), RelB, c-Rel, NF-κB1 (p50/p105) and NF-κB2 (p52/p100)] (76). The canonical pathway of NF-κB activation includes phosphorylation of IκB-α, which leads to nuclear translocation of the heterodimers p50/Rela or p50/cRel and consequent binding to the promoters of target genes and regulation of their expression. Constitutive activation of NF-κB has been observed in various cancer types, linking inflammation to the neoplastic transformation of the epithelium (77–79). In the initiation and progression of head and neck squamous cell carcinoma (HNSCC), several oncogenic pathways have been identified. These commonly include epidermal growth factor receptor (EGFR)/Ras/RAF/MAPK, PI3K/Akt1/mTOR, IΚΚ/NF-κB, STAT3 and Wnt/β-catenin (80–89). It has been indicated that HNSCC exhibits abundant NF-κB activation and several studies indicate that NF-κB is upregulated in premalignant lesions and invasive cancer (80,90–94).
The role of NF-κB in acidic bile reflux-related laryngopharyngeal carcinogenesis was first demonstrated in the last decade through in vitro and in vivo experimental models (12–25). A study by our group from 2017 (21) documented the key role of NF-κB in mediating acidic bile-induced oncogenic molecular events in human hypopharyngeal cells (HHCs). Subsequently in 2018, a study by our group (22) also demonstrated that NF-κB is a crucial factor in controlling the levels of small regulatory molecules, such as microRNA (miRNA/miR) markers, in HHCs. A series of in vitro and in vivo studies also suggested that NF-κB inhibition may prevent inflammatory and neoplastic events in HHCs and HM, including STAT3 activation and significant deregulations of several cancer-related genes and miRNA markers (19,23–25). Other non-specific stress factors, such as highly concentrated glucose or acidic pepsin, were not capable of inducing activation of genes with oncogenic function previously linked to HNSCC in vitro (12,13,53).
Bile reflux-induced NF-κB-related mRNA oncogenic phenotype
There is evidence that numerous types of cancer arise from sites of chronic inflammation (95). Specifically, a wide array of chronic inflammatory conditions predisposes susceptible cells of epithelial origin to neoplastic transformation (carcinomas) as a multistep process (focal proliferation of dysplastic cells with potential progression to malignant carcinoma). An example includes reflux esophagitis that may lead to DNA damage, development of Barrett's esophagus and esophageal carcinoma (96). In certain cases, the progenitors of the inflammation are known, such as bacterial infections or gastric acids that have been associated with increased risk of adenocarcinoma of the stomach and esophagus, respectively (97,98). It may be assumed that bile acids interact directly with HM, as described in a paragraph below, but may also be the progenitors of an LPR-induced chronic inflammatory microenvironment associated with an increased risk of hypopharyngeal cancer. It has been indicated that a chronic inflammatory microenvironment or harmful stimuli are able to induce a constitutive activation of NF-κB, which may lead to a cascade of molecular alterations. Specifically, constitutive activation of NF-κB may lead to subsequent transcriptional activation of genes that are implicated in a variety of signaling pathways via aberrant overexpression of cytokines, transcription factors and growth factor receptors, such as TNF-α, TLR and EGFR (76,77,79–89,93,94,99–103).
Preclinical studies from our group (12–25) documented that acidic bile is able to induce activation of NF-κB and significant overexpression of several cancer-related genes. Specifically, acidic bile was reported to induce significant transcriptional activation of anti-apoptotic BCL2 and other genes previously linked to HNSCC, such as STAT3, EGFR, WNT5A, TNF-α, ΔNp63, cREL, IL6, IL1β, AKT1 and PTGS2 (82–94,102–111) (Fig. 1). Furthermore, a clinical pilot study revealed that bile-related HSCC had significantly higher levels of NF-κB and differential expression of the above genes compared to the adjacent non-pathologic tissue or bile-negative HSCC, providing further evidence of the central role of NF-κB (18).
Figure 1.

The mechanistic role of NF-κB in the mRNA oncogenic phenotype induced by bile reflux in hypopharyngeal carcinogenesis. Acidic bile induces constitutive activation of NF-κB via TNF-α, EGFR or TLR, which promotes the transcriptional activation of genes with inflammatory, anti-apoptotic or oncogenic function, such as IL6, IL1β, TNF-α, BCL2L1, EGFR, cREL, STAT3, ΔNp63 and WNT5A. Activation of NF-κB under acidic bile exposure also induces overexpression of AKT1, suggesting acidic bile may contribute to the PI3K/AKT1 downstream pathway, which is frequently activated in HNSCC. In addition, NF-κB is able to upregulate the expression of PTGS2 (COX-2), supporting its regulatory role in inflammatory and cancer-related downstream signaling pathways. Finally, acidic bile-induced NF-κB activation may prevent the upregulation of wild-type TP53 expression.
Targeting NF-κB negatively affects the NF-κB signaling pathway and has been indicated to be an encouraging strategy to improve anticancer therapies (112). Several pharmacologic and dietary inhibitors of NF-κB are considered promising thera-peutic options, demonstrating chemo-preventive or chemo-sensitizing properties in head and neck cancer (99,112). BAY 11-7082 [(E)-3-(4-methylphenylsulphonyl)-1-propenenitrile] is a reliable inhibitor of the NF-κB pathway that has been widely used in numerous studies exploring the effect of NF-κB (112,113). It has been suggested that BAY 11-7082 offers the most rapid and potent anti-tumor effect among other NF-κB inhibitors (112) and it may be used as a sensitizer for anti-cancer treatment (114,115). Furthermore, curcumin is a turmeric natural supplement with known antioxidant, anti-inflammatory and anti-cancer properties, previously demonstrated to have potential chemo-preventive effects in head and neck malignancies (116), by blocking NF-κB activation and halting the proliferation of cancer cells (117) due to its pleiotropic properties (118).
Both BAY 11-7082 and dietary curcumin have been used in in vitro and in vivo experimental studies to investigate the underlying mechanism of bile reflux-induced carcinogenic effect into the hypopharynx. Studies by our group (19,25) suggested that application of BAY 11-7082 effectively suppressed cell proliferation rates, the activation of NF-κB and related oncogenic mRNA profiles induced by acidic bile exposure. This oncogenic phenotype included the significant overexpression of anti-apoptotic BCL2 and other genes implicated in the initiation and progression of HNSCC, including TNF-α, EGFR, STAT3, ΔNp63, cREL, IL6, IL1β, AKT1, PTGS2 and WNT5A (82–85,87,89–94,102–111) (Fig. 1). Parallel evidence that acidic bile stimulus is able to activate NF-κB and its related pathways in HHCs arose from the in vitro treatment with curcumin, which successfully blocked the transcriptional activity of NF-κB (20), similar to BAY 11-7082 (21,23). A study by our group from 2020 (24) documented the preventive and therapeutic properties of curcumin on murine HM against the acidic bile effect, thus shaping the future translational development of effective targeted therapies using topical non-pharmacologic inhibitors of NF-κB.
Strong evidence that NF-κB activation is able to influence the acidic bile-induced oncogenic mRNA profile inspired a further study on whether synchronizing its inhibition with acidic bile exposure is significant. Thus, a study by our group from 2019 (23) reported the temporal characteristics of NF-κB inhibition in blocking the acidic bile-induced oncogenic molecular events in HHCs. A series of studies also documented that topical application of BAY 11-7082 or curcumin to HM, either before, after or simultaneous to acidic bile exposure, successfully prevented or suppressed cell proliferation and NF-κB-related molecular events (19,24,25).
These results revealed that the upregulation of RELA, BCL2, STAT3, EGFR, WNT5A, TNF-α, IL6 and PTGS2 is directly promoted by acidic bile through NF-κB, shortly after its exposure (19,24,25) (Fig. 1), and strongly suggested that it may be clinically feasible to topically apply NF-κB inhibitors, without any precise synchronization with acidic bile exposure, to prevent acidic bile-induced oncogenic molecular changes.
Interactions between NF-κB activation and other factors
The application of NF-κB inhibitors also revealed important information about possible interactions between acidic bile-induced NF-κB activation and other central molecules in head and neck cancer (Fig. 1).
The NF-κB/STAT3 crosstalk has been indicated to be fundamental in inflammation-associated carcinogenesis in head and neck cancer (108,119,120). Application of NF-κB inhibitors successfully prevented the acidic bile-induced activation and nuclear translocation of STAT3, which is an important regulator of cell proliferation, and reduced the transcriptional levels of IL6 and STAT3, in treated hypopharyngeal cells (19–25) (Fig. 1). These data strongly support the theory that the acidic bile-induced activation of IL-6/STAT3 is NF-κB-dependent (108) (in a paragraph below, the role of STAT3 in bile-induced carcinogenesis is discussed). In addition, prior findings implied strong interactions between NF-κB and STAT3 in acidic bile-exposed premalignant HM (13,14), further supporting the theory that the inflammatory response induced by acidic bile may increase the risk of laryngopharyngeal cancer.
EGFR is frequently overexpressed in HNSCC (121,122). Crosstalk between NF-κB and downstream pathways of EGFR has been observed (123,124). Although the exact role of EGFR in bile-related hypopharyngeal carcinogenesis has remained to be elucidated, the application of NF-κB inhibitors resulted in the successful suppression of acidic bile-induced overexpression of EGFR, supporting the interactions between NF-κB and EGFR pathways during this process (Fig. 1). In addition, as both STAT3 and EGFR are important contributors to HNSCC pathogenesis (80,121), the above observations further emphasized the requirement to develop a therapeutic strategy for targeting NF-κB in head and neck malignancies and particularly in bile reflux-related HSCC.
Furthermore, NF-κB inhibition had a strong effect in suppressing the acidic bile-induced overexpression of WNT5A (Fig. 1), a factor related to cancer-associated inflammation and epithelial-to-mesenchymal transition (111), indicating that NF-κB is able to mediate acidic bile-induced changes in hypopharyngeal cell-cell interactions.
In addition, COX-2 is regularly highly overexpressed during inflammatory and neoplastic processes (125) and is significantly overexpressed in acidic bile-exposed HM (24,25). In vivo application of NF-κB inhibitor significantly abrogated the acidic bile-induced overexpression of PTGS2 (24,25) (Fig. 1), further supporting the regulatory role of NF-κB in early inflammatory and cancer-related pathways, such as COX-2 (126).
In addition to the above, the PI3K/Akt pathway (127) is one of the most frequently activated pathways in head and neck cancer (128). The successful suppression of acidic bile-induced AKT1 overexpression using topical application of NF-κB inhibitor on murine HM suggested that NF-κB may mediate acidic bile-induced deregulations of PI3K/Akt downstream pathways (127) (Fig. 1).
In summary, the NF-κB pathway is a core central pathway that interacts with multiple upstream and downstream signaling pathways linked to the carcinogenic process. Using both a specific NF-κB inhibitor, such as BAY 11-7082, and a more pleiotropic NF-κB inhibitor, such as curcumin, it was documented that the acidic bile-induced deregulations of cancer-related genes or inflammatory factors are mediated by the NF-κB (Fig. 1). Furthermore, as curcumin is able to prevent the bile-related anti-apoptotic effect independently of the pH status, it may have an advantage over other NF-κB inhibitors. Of note, curcumin specifically reduced a lower percentage of analyzed NF-κB signaling genes compared to BAY 11-7082 (25 vs. 85%) (20,21). Thus, curcumin may confer a clinical advantage by preventing generalized suppression of NF-κB signaling, which is essential to the basic metabolic function of healthy mucosa and thereby reducing global toxicity (24,129).
Bile reflux-induced NF-κB-related miRNA oncogenic phenotype
miRNA molecules have also been important in both inflammation and cancer (130), modulating the expression of genes by causing target mRNA degradation or inhibiting their translation (131). The expression levels of certain miRNAs, such as ‘oncomiRs’ and ‘tumor suppressor’ miRNAs, have been indicated to be altered in tumor cells compared to normal cells (upregulated or downregulated), and capable of contributing to carcinogenesis, thereby demonstrating a significant regulatory role in the multistep process of cancer initiation and progression (132).
There is further evidence that miRNA markers, such as ‘oncomiR’ miR-21 and ‘tumor suppressor’ miR-375, have a crucial role in the initiation and progression of HNSCC (14,22,133,134). Arantes et al (135) reported the fundamental role of miR-21 as a biomarker in head and neck carcinogenesis, while miR-375 has been proposed as a predictive biomarker for early diagnosis in laryngeal cancer (136). In addition, interactions between NF-κB and miRNA markers, such as miR-21, miR-34a and miR-451a, have been importantly described by others in human cancer cells, including HNSCC (104,137,138). Specifically, a cluster of miRNA markers was reported to be associated with NF-κB that may be associated with the aggressive biological behavior of HNSCC (104).
Explorations by our group (14,15,19,14) revealed that acidic bile caused deregulations of the expression of oncogenic miRNA markers, previously associated with laryngopharyngeal cancer (133–141). Specifically, the ‘oncomiRs’ miR-21, miR-192 and miR-155, and the ‘tumor suppressors’ miR-34a, miR-375, miR-451a, miR-99a and miR-504 were indicated to be significantly altered in exposed HHCs and murine laryngopharyngeal mucosa (14,15,19,14) (Fig. 2). Of note, results from our group (14,15) highlighted the role of miR-21 and miR-375 deregulations in acidic bile-related neoplasia.
Figure 2.

The mechanistic role of NF-κB in the miRNA oncogenic phenotype induced by bile reflux in hypopharyngeal carcinogenesis. Chronic stimulation of laryngopharyngeal mucosa by acidic bile induces constitutive activation of NF-κB, producing upregulation of ‘oncomiRs’ miR-21, miR-155 and miR-192, previously associated with oncogenic signaling pathways in head and neck cancer and GERD. The acidic bile-induced activation of NF-κB in treated hypopharyngeal mucosa is capable of downregulating ‘tumor suppressor’ miR-34a, miR-451a, miR-375, miR-99a and miR-504, are known to control the cell cycle and are frequently affected in head and neck cancer. Acidic bile-induced expression levels of ‘oncomiRs’ exhibited an inverse correlation with ‘tumor suppressor’ miRNAs that appears to be regulated by NF-κB. miRNA/miR, microRNA; GERD, gastroesophageal reflux disease; HNSCC, head and neck squamous cell carcinoma.
In addition, findings from HSCC tumor specimens from patients with documented bile reflux supported its strong association with upregulation of ‘oncomiR’ miR-21 and downregulation of ‘tumor suppressors’ miR-34a and particularly of miR-375, along with strong positivity for NF-κB (18) (Fig. 2). Bile reflux-associated HSCC was also associated with a marked reduction of ‘tumor suppressors’ miR-489, miR-504 and miR-99a compared with their adjacent non-pathologic tissue (18), suggesting their involvement in the onset and progression of HSCC (142,143). Finally, bile exposure-associated HSCC exhibited differential expression of miR-489 and miR-504, and particularly of miR-375, compared to bile-negative HSCC, which had significantly lower NF-κB levels. A previous view by our group (18) suggested that these miRNA markers may have a distinct role in biliary reflux-associated hypopharyngeal cancer.
Application of BAY 11-7082 in HHCs or murine HM was proven to prevent miRNA deregulations caused by acidic bile, providing insight into the interactions of transcriptionally active NF-κB with cancer-related miRNA markers (19,22,23,25). Specifically, it was demonstrated that BAY 11-7082 is able to effectively reverse the acidic bile-induced downregulation of ‘tumor suppressor’ miR-451a and miR-99a, and upregulation of ‘oncomiRs’ miR-21, miR-155 and miR-192 (19,22,23,25) (Fig. 2), which are considered important markers for poor prognosis in head and neck cancer or linked to gastroesophageal reflux (22,135,144–150). Other studies have also indicated that NF-κB has a direct regulatory effect on the expression of miR-21 and miR-155 through their binding promoters (151,152). Through in vitro and in vivo applications of BAY 11-7082 on hypopharyngeal cells and mucosa, respectively, a direct effect of acidic bile on the above miRNAs was demonstrated shortly after exposure (22,23,25). This observation proposed the use of these miRNAs as biomarkers of early neoplastic events in acidic bile-exposed HM, strongly supporting the role of NF-κB as a mediator in this process. In addition, topical in vivo application of BAY 11-7082 either before, after or simultaneous to acidic bile significantly inhibited the acidic bile-induced upregulation of ‘oncomiR’ miR-192 (19,25), previously associated with GERD (148), and downregulation of ‘tumor suppressor’ miR-504, a promising target for HSCC (142) (Fig. 2). This observation also suggested the utility of these miRNAs as biomarkers of early neoplastic events in acidic bile-exposed HM.
In summary, the above preclinical data provided evidence of the role of NF-κB as a regulator of miR-192, miR-21, miR-155, miR-451a, miR-375, miR-99a and miR-504 (Fig. 2), and proposed these miRNAs as potential therapeutic targets of bile-related mutagenic evolution in the HM.
Interactions between NF-κB-related mRNA and miRNA phenotypes
Previous studies suggested that interactions between miRNAs and mRNA molecules may be NF-κB-dependent during carcinogenesis (141,153,154). Rokavec et al (153) proposed that miRNA molecules, such as miR-34a, interact with STAT3 in an NF-κB-dependent manner. According to Tili et al (154), permanent upregulation of miR-155 may mediate a prolonged inflammatory reaction leading to cancer.
It has been proposed that acidic bile-induced NF-κB activation, BCL2 overexpression and significant alterations of oncogenic EGFR, STAT3, TNF-α, IL6, IL1β and WNT5A may directly or indirectly interact with cancer-related miRNA markers, such as ‘oncomiRs’ miR-21 and miR-155, as well as ‘tumor suppressors’ miR-34a, miR-375 and miR-451a (14–16,141) (Fig. 3). These observations suggest that inflammatory episodes caused by acidic bile may be associated with downstream oncogenic pathways and may be effectively prevented by NF-κB inhibition.
Figure 3.

Schematic representation of proposed interactions between acidic bile-induced NF-κB activation and alterations of cancer-related mRNA and miRNA phenotypes in hypopharyngeal cancer. NF-κB inhibition provided evidence of strong interactions between acidic bile-induced and cancer-related oncogenic mRNA and miRNA phenotypes in treated hypopharyngeal cells. miRNA/miR, microRNA.
NF-κB as a challenging target for cancer therapy
Recent findings documenting the crucial role of NF-κB in bile reflux-related hypopharyngeal carcinogenesis, pose a challenge for researchers and clinicians on how to identify patients who are more likely to benefit from NF-κB inhibition treatment. An NF-κB-related gene expression signature associated with bile reflux in patients with HSCC (Figs. 1 and 2) may provide a better prediction for inhibition selection and also allow the development of diagnostic and prognostic biomarkers of NF-κB inhibition response.
Although NF-κB targeted therapy has been already applied in clinical practice with promising results in anti-cancer therapy (129,155,156), including HNSCC treatment (157,158), there is an increasing effort in the pharmaceutical industry to develop advanced NF-κB inhibitors (129,159,160). In particular, research focuses on the identification of IKK/NF-κB inhibitors for targeted therapy that would prevent NF-κB activation without affecting other signaling pathways and selectively affect malignant cells rather than normal cells. As one of the major adverse effects of using NF-κB inhibitors as anticancer drugs is their ability to impair innate immunity when applied in excessive and prolonged periods (129,159–161), research should also focus on both minimizing systemic toxicity and prevention of long-term immunosuppression. Thus, an ideal inhibition of NF-κB should be transient and reversible, as well as effective when combined with other anti-cancer treatments. Prior in vivo explorations strongly supported the effectiveness of models using intermittent and topical treatment as opposed to prolonged and systemic treatment. Specifically, the marked efficacy of short-term topical treatment with NF-κB inhibitors, such as curcumin (2 mg/kg/day) and BAY 11-7082 (6.25 mg/kg/day), in suppressing bile reflux-induced early preneoplastic changes in the hypopharynx (19,24,25), strongly supports the view that non-systemic and transient NF-κB inhibition may be clinically feasible in preventing bile-reflux-related oncogenic effects. This area of research is progressing rapidly; however, another adverse effect is of high importance prior to the targeting of IKK or NF-κB in the clinic. This refers to the enhanced production of IL-1β and related cytokines by inhibitors of NF-κB activation during bacterial infections (161), suggesting the short-term use of NF-κB inhibitors in combination with antibiotics.
5. Role of STAT3 in bile reflux-related hypopharyngeal carcinogenesis
The STAT3 oncogene (162) is a transcription factor central to head and neck cancer (119,120,122). In addition to the significant role of NF-κB, recent preliminary data from our team indicate the important role of STAT3 in bile-related hypopharyngeal carcinogenesis by promoting early oncogenic molecular events, including cancer-related inflammatory molecules IL6, TNF-α and RELA (p65).
Specifically, using three different inhibitors with each blocking a different step of STAT3 upstream signaling, such as nifuroxazide, SI3-201 and STA-21 (163–165), preliminary data from our group were obtained regarding the mechanism of the effects of bile. In detail, acidic bile is able to induce constitutive activation of STAT3 that may not be exclusively dependent on JAK/STAT3 upstream signaling (165), but it may also be stimulated by alternative signaling, such as EGFR. Although targeting STAT3, either by its knockdown or its pharmacological inhibition, had a minimal effect on nuclear or total phosphorylated NF-κB (p65 S536) protein levels, it was observed to contribute, among others, to the transcriptional activation of NF-κB. As mentioned above, previous findings of NF-κB inhibition, using BAY 11-7082, had determined a role of NF-κB in acidic bile-induced activation of STAT3 (19–25).
All of these observations suggest possible molecular crosstalk between the NF-κB and STAT3 transcription factor associated signaling pathways in bile reflux-related inflammation and tumorigenesis in the hypopharynx, as similarly proposed in HNSCC (108,119,120).
6. Bile-induced DNA damage
One of the principal questions regarding the effects of bile on cellular physiology was how bile refluxate induces DNA damage. Dvorak et al (59), suggested that bile at acidic pH may potentially induce DNA damage in esophageal cells, speculating that chronic exposure to bile acids at low pH may result in increased genomic instability, abnormal cell signaling and resistance to apoptosis. According to Goldman et al (166), bile in combination with acid, but not acid alone, immediately activates all three isoforms of nitric oxide (NO) synthase, a family of enzymes catalyzing the production of NO, which links chronic inflammatory diseases and reactive oxygen/nitrogen species (ROS/RNS) with cancer. They also indicated that bile in combination with acid increased intracellular acidification and DNA damage in esophageal cells, which may lead to mutations and cancer progression (166). Bernstein et al (31,167) proposed that de-conjugated secondary bile acids, such as DCA, are capable of inducing DNA damage, giving rise to cancer due to the accumulation of mutations. Specifically, DCA was determined to induce increased intracellular production of ROS/RNS, resulting in increased oxidative stress and DNA damage (64,168,169).
Recent studies by our group (15,16,19) documented that conjugated bile acids led to the upregulation of γ-H2AX (pS139). γ-H2AX is a consistent marker of DNA DSBs (170,171), which was profoundly increased in bile-treated hypopharyngeal cells or HM at acidic pH, compared to neutral pH, acid alone or neutral control conditions (15,19). Subsequently, it was documented that bile-treated HM at weakly acidic pH, with or without DCA, also induced DNA damage in exposed HM (16). Specifically, premalignant and malignant lesions caused by acidic bile demonstrated increased levels of nuclear γ-H2AX, as well as DNA/RNA oxidative damage (Fig. 4) (15,16).
Figure 4.
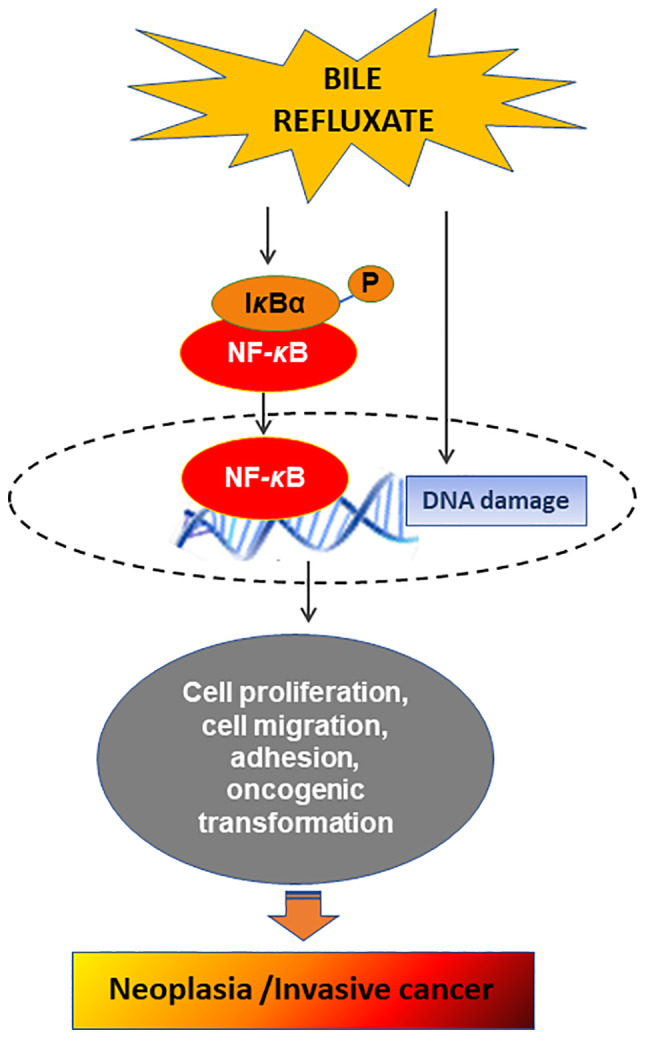
Schematic representation of the proposed mechanism of bile reflux-induced hypopharyngeal cancer. Bile refluxate is capable of inducing DNA damage, such as double-strand DNA breaks and oxidative damage and significant activation of NF-κB and its related anti-apoptotic pathways, leading to malignant transformation of laryngopharyngeal mucosa and invasive cancer.
According to previous findings, increased oxidative damage may result in high levels of ROS or DSBs incurring direct DNA damage (168,172,173), which may potentially lead to tumor-initiating mutations in head and neck cancer (174). All of these results advocate the theory that acidic bile-induced DNA damage may contribute to its mutated phenotype (Fig. 4). In addition, ROS is able to activate several cancer-associated signaling pathways, including NF-κB (164), concluding that acidic bile may contribute to evasion of apoptosis and/or proliferation of mutated hypopharyngeal epithelial cells, resulting in malignant lesions of HM.
In parallel, chronic exposure of murine hypopharyngeal epithelium to acidic bile was observed to induce a systematic release of inflammatory molecules, such as IL-6 and TNF-α, which are considered central to head and neck carcinogenesis (91). This systematic release of inflammatory molecules is able to maintain the constitutive release of other cancer-related cytokines in the microenvironment of the exposed epithelium. It is known that the chronic inflammatory microenvironment may lead to the production of activated inflammatory cells that may also serve as sources of ROS and reactive nitrogen intermediates (RNI), which are capable of inducing DNA damage and genomic instability and so promote mutations in neighboring epithelial cells (175,176). In addition, inflammatory cytokines contribute to increased intracellular ROS and RNI production in pre-malignant cells. In conclusion, the chronic inflammatory microenvironment caused by bile reflux may be one of the main factors in hypopharyngeal carcinogenesis.
7. Possible interactions of bile refluxate with hypopharyngeal mucosa
How acidic bile interacts with HM to exert its harmful effect, causing DNA damage and promoting NF-κB-related anti-apoptotic processes, leading to its malignant transformation, has remained to be fully elucidated. According to Li and Cao (177), bile acids may interact with membrane receptors, such as Takeda G-protein coupled receptor (TGR5). Their study also indicated that TGR5 is able to mediate bile reflux-induced DNA damage in esophageal cells (177). Another cell membrane receptor that is able to interact with bile acids is the sphingosine-1-phosphate receptor 1, known as S1PR1 or S1P1 (178,179). Both above-mentioned receptors were previously associated with lower esophageal cancer related to bile reflux (180,181). In particular, TGR5 was reported to be expressed in both adenocarcinoma and squamous cell carcinoma of the lower esophagus (181), while S1PR1 has been associated with squamous cell carcinoma of the head and neck (179).
Other studies provided evidence that bile acids are able to activate nuclear farnesoid X receptors (FXRs) (182,183), suggesting their contribution to pre-neoplastic changes (183) of the lower esophagus. Although several nuclear receptors (NRs) have been identified in the head and neck (184), an association between NRs and bile acids has not yet been described in HNSCC. Prior studies suggested a mutually antagonistic relationship between FXR and NF-κB activation (185,186) and proposed FXR receptors as useful targets for esophageal adenocarcinoma (182). However, the exact mechanism by which FXR affects the expression of proinflammatory molecules, such as NF-κB in either the lower or upper esophagus remains elusive and the role of FXR in bile reflux-related carcinogenesis deserves further exploration. Further investigation in the hypopharynx may identify specific receptors activated by acidic bile and clarify the role of NRs, such as FXR, and cell membrane receptors, such as TGR5 and S1PR1, in this process.
8. Conclusion
Recent in vitro and in vivo data provide evidence on bile reflux-associated hypopharyngeal carcinogenesis. The composition of biliary refluxate, such as conjugated bile acids, and acidity are pivotal factors in promoting DNA damage, as well as histologic and molecular changes in the HM, most likely through the constitutive activation of NF-κB. Chronic acidic bile exposure can cause increased oxidative damage, DSBs, overproduction of cytokines and cell-cell interaction changes, which are critical elements of tumor initiation and progression (187), possibly through derangements in both pre-neoplastic/neoplastic cells and their microenvironment. In parallel, acidic bile-induced constitutive activation of NF-κB can promote oncogenic mRNA and miRNA phenotypes, contributing to the proliferation of mutated cells and thus giving rise to the malignant transformation of the exposed HM. Primary data also support the contributing role of STAT3 in this process. Further investigation of the proposed mechanisms mediating bile-induced DNA damage, the tumor microenvironment and downstream oncogenic signaling pathways in HM, as well as the identification of specific receptors that may interact with bile, will contribute to innovative approaches to the diagnosis and prevention of laryngopharyngeal malignancies, as well as to the improvement of current therapeutic approaches to LPR-related carcinogenesis.
Acknowledgements
Not applicable.
Glossary
Abbreviations
- HM
hypopharyngeal mucosa
- HHCs
human normal hypopharyngeal cells
Funding
No funding was received.
Availability of data and materials
Data sharing is not applicable.
Authors' contributions
DPV and SGD were involved in the conceptualization of this review article. DPV, SGD, PGD and BLJ were involved in searching the literature. DPV, PGD and SGD were involved in the writing of the original draft. DPV, SGD, PGD and BLJ reviewed and edited the article. All authors have read and approved the final manuscript. Data authentication is not applicable.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Hashibe M, Boffetta P, Zaridze D, Shangina O, Szeszeni-Dabrowska N, Mates D, Fabiánová E, Rudnai P, Brennan P. Contribution of tobacco and alcohol to the high rates of squamous cell carcinoma of the supraglottis and glottis in Central Europe. Am J Epidemiol. 2007;165:814–820. doi: 10.1093/aje/kwk066. [DOI] [PubMed] [Google Scholar]
- 2.Curado MP, Hashibe M. Recent changes in the epidemiology of head and neck cancer. Curr Opin Oncol. 2009;21:194–200. doi: 10.1097/CCO.0b013e32832a68ca. [DOI] [PubMed] [Google Scholar]
- 3.Talamini R, Bosetti C, La Vecchia C, Dal Maso L, Levi F, Bidoli E, Negri E, Pasche C, Vaccarella S, Barzan L, Franceschi S. Combined effect of tobacco and alcohol on laryngeal cancer risk: A case-control study. Cancer Causes Control. 2002;13:957–964. doi: 10.1023/A:1021944123914. [DOI] [PubMed] [Google Scholar]
- 4.Pöschl G, Seitz HK. Alcohol and cancer. Alcohol Alcohol. 2004;39:155–165. doi: 10.1093/alcalc/agh057. [DOI] [PubMed] [Google Scholar]
- 5.Galli J, Cammarota G, De Corso E, Agostino S, Cianci R, Almadori G, Paludetti G. Biliary laryngopharyngeal reflux: A new pathological entity. Curr Opin Otolaryngol Head Neck Surg. 2006;14:128–132. doi: 10.1097/01.moo.0000193198.40096.be. [DOI] [PubMed] [Google Scholar]
- 6.Tutar H, Erdamar H, Köybaşioğlu A, Dinç AE, Ceylan A, Uslu S. Can bile acids be an etiological factor for laryngeal carcinoma? ORL J Otorhinolaryngol Relat Spec. 2011;73:156–161. doi: 10.1159/000327521. [DOI] [PubMed] [Google Scholar]
- 7.Geterud A, Bove M, Ruth M. Hypopharyngeal acid exposure: An independent risk factor for laryngeal cancer? Laryngoscope. 2003;113:2201–2205. doi: 10.1097/00005537-200312000-00030. [DOI] [PubMed] [Google Scholar]
- 8.Sereg-Bahar M, Jerin A, Hocevar-Boltezar I. Higher levels of total pepsin and bile acids in the saliva as a possible risk factor for early laryngeal cancer. Radiol Oncol. 2015;49:59–64. doi: 10.2478/raon-2014-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altman KW, Prufer N, Vaezi MF. A review of clinical practice guidelines for reflux disease: toward creating a clinical protocol for the otolaryngologist. Laryngoscope. 2011;121:717–723. doi: 10.1002/lary.22048. [DOI] [PubMed] [Google Scholar]
- 10.Assimakopoulos D, Patrikakos G. The role of gastroesophageal reflux in the pathogenesis of laryngeal carcinoma. Am J Otolaryngol. 2002;23:351–357. doi: 10.1053/ajot.2002.128038. [DOI] [PubMed] [Google Scholar]
- 11.Sasaki CT, Marotta J, Hundal J, Chow J, Eisen RN. Bile-induced laryngitis: Is there a basis in evidence? Ann Otol Rhinol Laryngol. 2005;114:192–197. doi: 10.1177/000348940511400304. [DOI] [PubMed] [Google Scholar]
- 12.Sasaki CT, Issaeva N, Vageli DP. In vitro model for gastroduodenal reflux-induced nuclear factor-kappaB activation and its role in hypopharyngeal carcinogenesis. Head Neck. 2016;38(Suppl 1):E1381–E1391. doi: 10.1002/hed.24231. [DOI] [PubMed] [Google Scholar]
- 13.Vageli DP, Prasad ML, Sasaki CT. Gastro-duodenal fluid induced nuclear Factor-κappaB activation and early pre-malignant alterations in murine hypopharyngeal mucosa. Oncotarget. 2016;7:5892–5908. doi: 10.18632/oncotarget.6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sasaki CT, Vageli DP. MiR-21, miR-155, miR-192, and miR-375 deregulations related to NF-kappaB activation in gastroduodenal Fluid-Induced early Preneoplastic lesions of laryngeal mucosa in vivo. Neoplasia. 2016;18:329–338. doi: 10.1016/j.neo.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sasaki CT, Doukas SG, Costa J, Vageli DP. The progressive mutagenic effects of acidic bile refluxate in hypopharyngeal squamous cell carcinogenesis: New insights. Cancers (Basel) 2020;12:1064. doi: 10.3390/cancers12051064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sasaki CT, Doukas SG, Doukas PG, Vageli DP. Weakly acidic bile is a risk factor for hypopharyngeal carcinogenesis evidenced by DNA damage, antiapoptotic function, and premalignant dysplastic lesions in vivo. Cancers (Basel) 2021;13:852. doi: 10.3390/cancers13040852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doukas SG, Cardoso B, Tower JI, Vageli DP, Sasaki CT. Biliary tumorigenic effect on hypopharyngeal cells is significantly enhanced by pH reduction. Cancer Med. 2019;8:4417–4427. doi: 10.1002/cam4.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sasaki CT, Doukas SG, Costa J, Vageli DP. Biliary reflux as a causal factor in hypopharyngeal carcinoma: New clinical evidence and implications. Cancer. 2019;125:3554–3565. doi: 10.1002/cncr.32369. [DOI] [PubMed] [Google Scholar]
- 19.Sasaki CT, Doukas SG, Vageli DP. In Vivo Short-Term topical application of BAY 11-7082 prevents the acidic Bile-Induced mRNA and miRNA oncogenic phenotypes in exposed Murine Hypopharyngeal Mucosa. Neoplasia. 2018;20:374–386. doi: 10.1016/j.neo.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vageli DP, Doukas SG, Spock T, Sasaki CT. Curcumin prevents the bile reflux-induced NF-κB-related mRNA oncogenic phenotype, in human hypopharyngeal cells. J Cell Mol Med. 2018;22:4209–4220. doi: 10.1111/jcmm.13701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vageli DP, Doukas SG, Sasaki CT. Inhibition of NF-kappaB prevents the acidic bile-induced oncogenic mRNA phenotype, in human hypopharyngeal cells. Oncotarget. 2017;9:5876–5891. doi: 10.18632/oncotarget.23143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doukas SG, Vageli DP, Sasaki CT. NF-κB inhibition reverses acidic bile-induced miR-21, miR-155, miR-192, miR-34a, miR-375 and miR-451a deregulations in human hypopharyngeal cells. J Cell Mol Med. 2018;22:2922–2934. doi: 10.1111/jcmm.13591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doukas PG, Vageli DP, Doukas SG, Sasaki CT. Temporal characteristics of NF-κB inhibition in blocking bile-induced oncogenic molecular events in hypopharyngeal cells. Oncotarget. 2019;10:3339–3351. doi: 10.18632/oncotarget.26917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doukas SG, Doukas PG, Sasaki CT, Vageli D. The in vivo preventive and therapeutic properties of curcumin in bile reflux-related oncogenesis of the hypopharynx. J Cell Mol Med. 2020;24:10311–10321. doi: 10.1111/jcmm.15640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vageli DP, Kasle D, Doukas SG, Doukas PG, Sasaki CT. The temporal effects of topical NF-κB inhibition, in the in vivo prevention of bile-related oncogenic mRNA and miRNA phenotypes in murine hypopharyngeal mucosa: A preclinical model. Oncotarget. 2020;11:3303–3314. doi: 10.18632/oncotarget.27706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hofmann AF. Chemistry and enterohepatic circulation of bile acids. Hepatology. 1984;4(Suppl 5):4S–14S. doi: 10.1002/hep.1840040803. [DOI] [PubMed] [Google Scholar]
- 27.Greek Medicine, corp-author. http://www.greekmedicine.net/b_p/Four_Humors.html [Google Scholar]
- 28.Rather LJ. Disturbance of function (functio laesa): The legendary fifth cardinal sign of inflammation, added by Galen to the four cardinal signs of Celsus. Bull NY Acad Med. 1971;47:303–322. [PMC free article] [PubMed] [Google Scholar]
- 29.Virchow R, Rather LJ. Stanford University Press; Stanford, CA: 1958. Disease, Life, and Man: Selected Essays. [Google Scholar]
- 30.Cook JW. Carcinogenic chemical agents. Yale J Biol Med. 1938;11:1–13. [PMC free article] [PubMed] [Google Scholar]
- 31.Bernstein H, Bernstein C, Payne CM, Dvorakova K, Garewal H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res. 2005;589:47–65. doi: 10.1016/j.mrrev.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 32.Kuwahara A, Saito T, Kobayashi M. Bile acids promote carcinogenesis in the remnant stomach of rats. J Cancer Res Clin Oncol. 1989;115:423–428. doi: 10.1007/BF00393330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hill MJ. Bile flow and colon cancer. Mutat Res. 1990;238:313–320. doi: 10.1016/0165-1110(90)90023-5. [DOI] [PubMed] [Google Scholar]
- 34.Bayerdörffer E, Mannes GA, Ochsenkühn T, Dirschedl P, Wiebecke B, Paumgartner G. Unconjugated secondary bile acids in the serum of patients with colorectal adenomas. Gut. 1995;36:268–273. doi: 10.1136/gut.36.2.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nehra D, Howell P, Williams CP, Pye JK, Beynon J. Toxic bile acids in gastro-oesophageal reflux disease: Influence of gastric acidity. Gut. 1999;44:598–602. doi: 10.1136/gut.44.5.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vaezi MF, Richter JE. Double reflux: Double trouble. Gut. 1999;44:590–592. doi: 10.1136/gut.44.5.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vaezi MF, Singh S, Richter JE. Role of acid and duodenogastric reflux in esophageal mucosal injury: A review of animal and human studies. Gastroenterology. 1995;108:1897–1907. doi: 10.1016/0016-5085(95)90156-6. [DOI] [PubMed] [Google Scholar]
- 38.Gotley DC, Morgan AP, Cooper MJ. Bile acid concentrations in the refluxate of patients with reflux oesophagitis. Br J Surg. 1988;75:587–590. doi: 10.1002/bjs.1800750632. [DOI] [PubMed] [Google Scholar]
- 39.Kauer WK, Peters JH, DeMeester TR, Ireland AP, Bremner CG, Hagen JA. Mixed reflux of gastric and duodenal juices is more harmful to the esophagus than gastric juice alone. The need for surgical therapy re-emphasized. Ann Surg. 1995;222:525–531. doi: 10.1097/00000658-199522240-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kauer WK, Peters JH, DeMeester TR, Feussner H, Ireland AP, Stein HJ, Siewert RJ. Composition and concentration of bile acid reflux into the esophagus of patients with gastroesophageal reflux disease. Surgery. 1997;122:874–881. doi: 10.1016/S0039-6060(97)90327-5. [DOI] [PubMed] [Google Scholar]
- 41.Domellof L, Reddy BS, Weisburger JH. Microflora and deconjugation of bile acids in alkaline reflux after partial gastrectomy. Am J Surg. 1980;140:291–295. doi: 10.1016/0002-9610(80)90024-0. [DOI] [PubMed] [Google Scholar]
- 42.Fein M, Peters JH, Chandrasoma P, Ireland AP, Oberg S, Ritter MP, Bremner CG, Hagen JA, DeMeester TR. Duodenoesophageal reflux induces esophageal adenocarcinoma without exogenous carcinogen. J Gastrointest Surg. 1998;2:260–268. doi: 10.1016/S1091-255X(98)80021-8. [DOI] [PubMed] [Google Scholar]
- 43.McQuaid KR, Laine L, Fennerty MB, Souza R, Spechler SJ. Systematic review: The role of bile acids in the pathogenesis of gastro-oesophageal reflux disease and related neoplasia. Aliment Pharmacol Ther. 2011;34:146–165. doi: 10.1111/j.1365-2036.2011.04709.x. [DOI] [PubMed] [Google Scholar]
- 44.Oh DS, Hagen JA, Fein M, Bremner CG, Dunst CM, Demeester SR, Lipham J, Demeester TR. The impact of reflux composition on mucosal injury and esophageal function. J Gastrointest Surg. 2006;10:787–796. doi: 10.1016/j.gassur.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 45.Sweet MP, Patti MG, Hoopes C, Hays SR, Golden JA. Gastro-oesophageal reflux and aspiration in patients with advanced lung disease. Thorax. 2009;64:167–173. doi: 10.1136/thx.2007.082719. [DOI] [PubMed] [Google Scholar]
- 46.Covington MF, Krupinski E, Avery RJ, Kuo PH. Classification schema of symptomatic enterogastric reflux utilizing sincalide augmentation on hepatobiliary scintigraphy. J Nucl Med Technol. 2014;42:198–202. doi: 10.2967/jnmt.114.141168. [DOI] [PubMed] [Google Scholar]
- 47.Lewin JS, Gillenwater AM, Garrett JD, Bishop-Leone JK, Nguyen DD, Callender DL, Ayers GD, Myers JN. Characterization of laryngopharyngeal reflux in patients with premalignant or early carcinomas of the larynx. Cancer. 2003;97:1010–1014. doi: 10.1002/cncr.11158. [DOI] [PubMed] [Google Scholar]
- 48.Johnston N, Ondrey F, Rosen R, Hurley BP, Gould J, Allen J, DelGaudio J, Altman KW. Airway reflux. Ann N Y Acad Sci. 2016;1381:5–13. doi: 10.1111/nyas.13080. [DOI] [PubMed] [Google Scholar]
- 49.Adams J, Heintz P, Gross N, Andersen P, Everts E, Wax M, Cohen J. Acid/pepsin promotion of carcinogenesis in the hamster cheek pouch. Arch Otolaryngol Head Neck Surg. 2000;126:405–409. doi: 10.1001/archotol.126.3.405. [DOI] [PubMed] [Google Scholar]
- 50.Johnston N, Dettmar PW, Ondrey FG, Nanchal R, Lee SH, Bock JM. Pepsin: Biomarker, mediator, and therapeutic target for reflux and aspiration. Ann NY Acad Sci. 2018;1434:282–289. doi: 10.1111/nyas.13729. [DOI] [PubMed] [Google Scholar]
- 51.Johnston N, Wells CW, Samuels TL, Blumin JH. Pepsin in nonacidic refluxate can damage hypopharyngeal epithelial cells. Ann Otol Rhinol Laryngol. 2009;118:677–685. doi: 10.1177/000348940911800913. [DOI] [PubMed] [Google Scholar]
- 52.Del Negro A, Araújo MR, Tincani AJ, Meirelles L, Martins AS, Andreollo NA. Experimental carcinogenesis on the oropharyngeal mucosa of rats with hydrochloric acid, sodium nitrate and pepsin. Acta Cir Bras. 2008;23:337–342. doi: 10.1590/S0102-86502008000400007. [DOI] [PubMed] [Google Scholar]
- 53.Sasaki CT, Toman J, Vageli D. The in vitro effect of Acidic-Pepsin on nuclear factor KappaB activation and its related oncogenic effect on normal human hypopharyngeal cells. PLoS One. 2016;11:e0168269. doi: 10.1371/journal.pone.0168269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Doukas PG, Vageli DP, Sasaki CT, Judson BL. Pepsin promotes activation of epidermal growth factor receptor and downstream oncogenic pathways, at slightly acidic and neutral pH, in exposed hypopharyngeal cells. Int J Mol Sci. 2021;22:4275. doi: 10.3390/ijms22084275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goldstein JL, Schlesinger PK, Mozwecz HL, Layden TJ. Esophageal mucosal resistance. A factor in esophagitis. Gastroenterol Clin North Am. 1990;19:565–586. doi: 10.1016/S0889-8553(21)00657-9. [DOI] [PubMed] [Google Scholar]
- 56.Stamp D, Jenkins G. Jenkins GJ, Hardie L. Bile Acids: Toxicology and Bioactivity. Royal Society of Chemistry; Cambridge: 2008. An overview of bile-acid synthesis, chemistry and function. Print: Issues in Toxicology; 4. [DOI] [Google Scholar]
- 57.Stamp DH. Three hypotheses linking bile to carcinogenesis in the gastrointestinal tract: certain bile salts have properties that may be used to complement chemotherapy. Med Hypotheses. 2002;59:398–405. doi: 10.1016/S0306-9877(02)00125-1. [DOI] [PubMed] [Google Scholar]
- 58.Ireland AP, Peters JH, Smyrk TC, DeMeester TR, Clark GW, Mirvish SS, Adrian TE. Gastric juice protects against the development of esophageal adenocarcinoma in the rat. Ann Surg. 1996;224:358–370. doi: 10.1097/00000658-199609000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dvorak K, Payne CM, Chavarria M, Ramsey L, Dvorakova B, Bernstein H, Holubec H, Sampliner RE, Guy N, Condon A, et al. Bile acids in combination with low pH induce oxidative stress and oxidative DNA damage: Relevance to the pathogenesis of Barrett's oesophagus. Gut. 2007;56:763–771. doi: 10.1136/gut.2006.103697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kauer WK, Stein HJ. Role of acid and bile in the genesis of Barrett's esophagus. Chest Surg Clin N Am. 2002;12:39–45. doi: 10.1016/S1052-3359(03)00064-4. [DOI] [PubMed] [Google Scholar]
- 61.Ulualp SO, Roland PS, Toohill RJ, Shaker R. Prevalence of gastroesophagopharyngeal acid reflux events: An evidence-based systematic review. Am J Otolaryngol. 2005;26:239–244. doi: 10.1016/j.amjoto.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 62.Lillemoe KD, Gadacz TR, Harmon JW. Bile absorption occurs during disruption of the esophageal mucosal barrier. J Surg Res. 1983;35:57–62. doi: 10.1016/0022-4804(83)90126-9. [DOI] [PubMed] [Google Scholar]
- 63.Sasaki CT, Hajek M, Doukas SG, Vageli DP. The role of bile reflux and its related NF-κB activated pathway in progression of hypopharyngeal squamous cell cancer. Oral Oncol. 2020;105:104668. doi: 10.1016/j.oraloncology.2020.104668. [DOI] [PubMed] [Google Scholar]
- 64.Hemmink GJ, Bredenoord AJ, Weusten BL, Monkelbaan JF, Timmer R, Smout AJ. Esophageal pH-impedance monitoring in patients with therapy-resistant reflux symptoms: ‘On’ or ‘off’ proton pump inhibitor? Am J Gastroenterol. 2008;103:2446–2453. doi: 10.1111/j.1572-0241.2008.02033.x. [DOI] [PubMed] [Google Scholar]
- 65.Bernstein H, Payne CM, Bernstein C, Schneider J, Beard SE, Crowley CL. Activation of the promoters of genes associated with DNA damage, oxidative stress, ER stress and protein malfolding by the bile salt, deoxycholate. Toxicol Lett. 1999;108:37–46. doi: 10.1016/S0378-4274(99)00113-7. [DOI] [PubMed] [Google Scholar]
- 66.Huo X, Juergens S, Zhang X, Rezaei D, Yu C, Strauch ED, Wang JY, Cheng E, Meyer F, Wang DH, et al. Deoxycholic acid causes DNA damage while inducing apoptotic resistance through NF-κB activation in benign Barrett's epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2011;301:G278–G286. doi: 10.1152/ajpgi.00092.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Langevin SM, Michaud DS, Marsit CJ, Nelson HH, Birnbaum AE, Eliot M, Christensen BC, McClean MD, Kelsey KT. Gastric reflux is an independent risk factor for laryngopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prev. 2013;22:1061–1068. doi: 10.1158/1055-9965.EPI-13-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Coca-Pelaz A, Rodrigo JP, Takes RP, Silver CE, Paccagnella D, Rinaldo A, Hinni ML, Ferlito A. Relationship between reflux and laryngeal cancer. Head Neck. 2013;35:1814–1818. doi: 10.1002/hed.23208. [DOI] [PubMed] [Google Scholar]
- 69.Attwood SE, Smyrk TC, DeMeester TR, Mirvish SS, Stein HJ, Hinder RA. Duodenoesophageal reflux and the development of esophageal adenocarcinoma in rats. Surgery. 1992;111:503–510. [PubMed] [Google Scholar]
- 70.Fein M, Fuchs KH, Stopper H, Diem S, Herderich M. Duodenogastric reflux and foregut carcinogenesis: Analysis of duodenal juice in a rodent model of cancer. Carcinogenesis. 2000;21:2079–2084. doi: 10.1093/carcin/21.11.2079. [DOI] [PubMed] [Google Scholar]
- 71.Miwa K, Hattori T, Miyazaki I. Duodenogastric reflux and foregut carcinogenesis. Cancer. 1995;75(Suppl 6):S1426–S1432. doi: 10.1002/1097-0142(19950315)75:6+<1426::AID-CNCR2820751506>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 72.Fang Y, Chen H, Hu Y, Djukic Z, Tevebaugh W, Shaheen NJ, Orlando RC, Hu J, Chen X. Gastroesophageal reflux activates the NF-κB pathway and impairs esophageal barrier function in mice. Am J Physiol Gastrointest Liver Physiol. 2013;305:G58–G65. doi: 10.1152/ajpgi.00438.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McAdam E, Haboubi HN, Griffiths AP, Baxter JN, Spencer-Harty S, Davies C, Jenkins GJ. Reflux composition influences the level of NF-κB activation and upstream kinase preference in oesophageal adenocarcinoma cells. Int J Cancer. 2015;136:527–535. doi: 10.1002/ijc.29029. [DOI] [PubMed] [Google Scholar]
- 74.Bus P, Siersema PD, van Baal JW. Cell culture models for studying the development of Barrett's esophagus: A systematic review. Cell Oncol (Dordr) 2012;35:149–161. doi: 10.1007/s13402-012-0076-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hormi-Carver K, Zhang X, Zhang HY, Whitehead RH, Terada LS, Spechler SJ, Souza RF. Unlike esophageal squamous cells, Barrett's epithelial cells resist apoptosis by activating the nuclear factor-κB pathway. Cancer Res. 2009;69:672–677. doi: 10.1158/0008-5472.CAN-08-3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 77.Wang H, Cho CH. Effect of NF-kappaB signaling on apoptosis in chronic inflammation-associated carcinogenesis. Curr Cancer Drug Targets. 2010;10:593–599. doi: 10.2174/156800910791859425. [DOI] [PubMed] [Google Scholar]
- 78.DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012;246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- 79.Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. 2013;12:86. doi: 10.1186/1476-4598-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nottingham LK, Yan CH, Yang X, Si H, Coupar J, Bian Y, Cheng TF, Allen C, Arun P, Gius D, et al. Aberrant IKKα and IKKβ cooperatively activate NF-κB and induce EGFR/AP1 signaling to promote survival and migration of head and neck cancer. Oncogene. 2014;33:1135–1147. doi: 10.1038/onc.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stadler ME, Patel MR, Couch ME, Hayes DN. Molecular biology of head and neck cancer: Risks and pathways. Hematol Oncol Clin North Am. 2008;22:1099–1124. doi: 10.1016/j.hoc.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009;45:324–334. doi: 10.1016/j.oraloncology.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.King KE, Ponnamperuma RM, Allen C, Lu H, Duggal P, Chen Z, Van Waes C, Weinberg WC. The p53 homologue DeltaNp63alpha interacts with the nuclear factor-kappaB pathway to modulate epithelial cell growth. Cancer Res. 2008;68:5122–5131. doi: 10.1158/0008-5472.CAN-07-6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jackson-Bernitsas DG, Ichikawa H, Takada Y, Myers JN, Lin XL, Darnay BG, Chaturvedi MM, Aggarwal BB. Evidence that TNF-TNFR1-TRADD-TRAF2-RIP-TAK1-IKK pathway mediates constitutive NF-kappaB activation and proliferation in human head and neck squamous cell carcinoma. Oncogene. 2007;26:1385–1397. doi: 10.1038/sj.onc.1209945. [DOI] [PubMed] [Google Scholar]
- 85.Dong J, Jimi E, Zeiss C, Hayden MS, Ghosh S. Constitutively active NF-kappaB triggers systemic TNFalpha-dependent inflammation and localized TNFalpha-independent inflammatory disease. Genes Dev. 2010;24:1709–1717. doi: 10.1101/gad.1958410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Foxwell BM, Bondeson J, Brennan F, Feldmann M. Adenoviral transgene delivery provides an approach to identifying important molecular processes in inflammation: Evidence for heterogenecity in the requirement for NF-kappaB in tumour necrosis factor production. Ann Rheum Dis. 2000;59(Suppl 1):i54–i59. doi: 10.1136/ard.59.suppl_1.i54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Guyer RA, Macara IG. Loss of the polarity protein PAR3 activates STAT3 signaling via an atypical protein kinase C (aPKC)/NF-κB/interleukin-6 (IL-6) axis in mouse mammary cells. J Biol Chem. 2015;290:8457–8468. doi: 10.1074/jbc.M114.621011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhao Y, Zhang C, Huang Y, Yu Y, Li R, Li M, Liu N, Liu P, Qiao J. Up-regulated expression of WNT5a increases inflammation and oxidative stress via PI3K/AKT/NF-κB signaling in the granulosa cells of PCOS patients. J Clin Endocrinol Metab. 2015;100:201–211. doi: 10.1210/jc.2014-2419. [DOI] [PubMed] [Google Scholar]
- 89.Bo H, Zhang S, Gao L, Chen Y, Zhang J, Chang X, Zhu M. Upregulation of Wnt5a promotes epithelial-to-mesenchymal transition and metastasis of pancreatic cancer cells. BMC Cancer. 2013;13:496. doi: 10.1186/1471-2407-13-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Klein JD, Grandis JR. The molecular pathogenesis of head and neck cancer. Cancer Biol Ther. 2010;9:1–7. doi: 10.4161/cbt.9.1.10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Allen CT, Ricker JL, Chen Z, Van Waes C. Role of activated nuclear factor-kappaB in the pathogenesis and therapy of squamous cell carcinoma of the head and neck. Head Neck. 2007;29:959–971. doi: 10.1002/hed.20615. [DOI] [PubMed] [Google Scholar]
- 92.Loercher A, Lee TL, Ricker JL, Howard A, Geoghegen J, Chen Z, Sunwoo JB, Sitcheran R, Chuang EY, Mitchell JB, et al. Nuclear factor-kappaB is an important modulator of the altered gene expression profile and malignant phenotype in squamous cell carcinoma. Cancer Res. 2004;64:6511–6523. doi: 10.1158/0008-5472.CAN-04-0852. [DOI] [PubMed] [Google Scholar]
- 93.Chung CH, Parker JS, Ely K, Carter J, Yi Y, Murphy BA, Ang KK, El-Naggar AK, Zanation AM, Cmelak AJ, et al. Gene expression profiles identify epithelial-to-mesenchymal transition and activation of nuclear factor-kappaB signaling as characteristics of a high-risk head and neck squamous cell carcinoma. Cancer Res. 2006;66:8210–8218. doi: 10.1158/0008-5472.CAN-06-1213. [DOI] [PubMed] [Google Scholar]
- 94.Lee TL, Yang XP, Yan B, Friedman J, Duggal P, Bagain L, Dong G, Yeh NT, Wang J, Zhou J, et al. A novel nuclear factor-kappaB gene signature is differentially expressed in head and neck squamous cell carcinomas in association with TP53 status. Clin Cancer Res. 2007;13:5680–5691. doi: 10.1158/1078-0432.CCR-07-0670. [DOI] [PubMed] [Google Scholar]
- 95.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Souza RF. From reflux esophagitis to esophageal adenocarcinoma. Dig Dis. 2016;34:483–490. doi: 10.1159/000445225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wroblewski LE, Peek RM, Jr, Wilson KT. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin Microbiol Rev. 2010;23:713–739. doi: 10.1128/CMR.00011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shaheen N, Ransohoff DF. Gastroesophageal reflux, Barrett esophagus, and esophageal cancer: Scientific review. JAMA. 2002;287:1972–1981. doi: 10.1001/jama.287.15.1972. [DOI] [PubMed] [Google Scholar]
- 99.Vander Broek R, Snow GE, Chen Z, Van Waes C. Chemoprevention of head and neck squamous cell carcinoma through inhibition of NF-kappaB signaling. Oral Oncol. 2014;50:930–941. doi: 10.1016/j.oraloncology.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 101.Lee TL, Yeh J, Friedman J, Yan B, Yang X, Yeh NT, Van Waes C, Chen Z. A signal network involving coactivated NF-kappaB and STAT3 and altered p53 modulates BAX/BCL-XL expression and promotes cell survival of head and neck squamous cell carcinomas. Int J Cancer. 2008;122:1987–1998. doi: 10.1002/ijc.23324. [DOI] [PubMed] [Google Scholar]
- 102.Wheeler SE, Suzuki S, Thomas SM, Sen M, Leeman-Neill RJ, Chiosea SI, Kuan CT, Bigner DD, Gooding WE, Lai SY, Grandis JR. Epidermal growth factor receptor variant III mediates head and neck cancer cell invasion via STAT3 activation. Oncogene. 2010;29:5135–5145. doi: 10.1038/onc.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yan B, Yang X, Lee TL, Friedman J, Tang J, Van Waes C, Chen Z. Genome-wide identification of novel expression signatures reveal distinct patterns and prevalence of binding motifs for p53, nuclear factor-kappaB and other signal transcription factors in head and neck squamous cell carcinoma. Genome Biol. 2007;8:R78. doi: 10.1186/gb-2007-8-5-r78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yan B, Li H, Yang X, Shao J, Jang M, Guan D, Zou S, Van Waes C, Chen Z, Zhan M. Unraveling regulatory programs for NF-kappaB, p53 and microRNAs in head and neck squamous cell carcinoma. PLoS One. 2013;8:e73656. doi: 10.1371/journal.pone.0073656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sriuranpong V, Park JI, Amornphimoltham P, Patel V, Nelkin BD, Gutkind JS. Epidermal growth factor receptor-independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin 6/gp130 cytokine system. Cancer Res. 2003;63:2948–2956. [PubMed] [Google Scholar]
- 106.Yang X, Lu H, Yan B, Romano RA, Bian Y, Friedman J, Duggal P, Allen C, Chuang R, Ehsanian R, et al. ΔNp63 versatility regulates a Broad NF-κB gene program and promotes squamous epithelial proliferation, migration, and inflammation. Cancer Res. 2011;71:3688–3700. doi: 10.1158/0008-5472.CAN-10-3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Du J, Romano RA, Si H, Mattox A, Bian Y, Yang X, Sinha S, Van Waes C, Chen Z. Epidermal overexpression of transgenic ΔNp63 promotes type 2 immune and myeloid inflammatory responses and hyperplasia via NF-κB activation. J Pathol. 2014;232:356–368. doi: 10.1002/path.4302. [DOI] [PubMed] [Google Scholar]
- 108.Squarize CH, Castilho RM, Sriuranpong V, Pinto DS, Jr, Gutkind JS. Molecular cross-talk between the NFkappaB and STAT3 signaling pathways in head and neck squamous cell carcinoma. Neoplasia. 2006;8:733–746. doi: 10.1593/neo.06274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yap LF, Ahmad M, Zabidi MM, Chu TL, Chai SJ, Lee HM, Lim PV, Wei W, Dawson C, Teo SH, et al. Oncogenic effects of WNT5A in Epstein-Barr virus associated nasopharyngeal carcinoma. Int J Oncol. 2014;44:1774–1780. doi: 10.3892/ijo.2014.2342. [DOI] [PubMed] [Google Scholar]
- 110.Sherwood V, Chaurasiya SK, Ekström EJ, Guilmain W, Liu Q, Koeck T, Brown K, Hansson K, Agnarsdóttir M, Bergqvist M, et al. WNT5A-mediated β-catenin-independent signalling is a novel regulator of cancer cell metabolism. Carcinogenesis. 2014;35:784–794. doi: 10.1093/carcin/bgt390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Asem MS, Buechler S, Wates RB, Miller DL, Stack MS. Wnt5a Signaling in Cancer. Cancers (Basel) 2016;8:79. doi: 10.3390/cancers8090079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nakanishi C, Toi M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5:297–309. doi: 10.1038/nrc1588. [DOI] [PubMed] [Google Scholar]
- 113.Meng Z, Lou S, Tan J, Xu K, Jia Q, Zheng W. Nuclear factor-kappa B inhibition can enhance apoptosis of differentiated thyroid cancer cells induced by 131I. PLoS One. 2012;7:e33597. doi: 10.1371/journal.pone.0033597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lin Y, Bai L, Chen W, Xu S. The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin Ther Targets. 2010;14:45–55. doi: 10.1517/14728220903431069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Van Waes C. Nuclear factor-kappaB in development, prevention, and therapy of cancer. Clin Cancer Res. 2007;13:1076–1082. doi: 10.1158/1078-0432.CCR-06-2221. [DOI] [PubMed] [Google Scholar]
- 116.Wilken R, Veena MS, Wang MB, Srivatsan ES. Curcumin: A review of anti-cancer properties and therapeutic activity in head and neck squamous cell carcinoma. Mol Cancer. 2011;10:12. doi: 10.1186/1476-4598-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.LoTempio MM, Veena MS, Steele HL, Ramamurthy B, Ramalingam TS, Cohen AN, Chakrabarti R, Srivatsan ES, Wang MB. Curcumin suppresses growth of head and neck squamous cell carcinoma. Clin Cancer Res. 2005;11:6994–7002. doi: 10.1158/1078-0432.CCR-05-0301. [DOI] [PubMed] [Google Scholar]
- 118.Naksuriya O, Okonogi S, Schiffelers RM, Hennink WE. Curcumin nanoformulations: A review of pharmaceutical properties and preclinical studies and clinical data related to cancer treatment. Biomaterials. 2014;35:3365–3383. doi: 10.1016/j.biomaterials.2013.12.090. [DOI] [PubMed] [Google Scholar]
- 119.Fan Y, Mao R, Yang J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell. 2013;4:176–185. doi: 10.1007/s13238-013-2084-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gaykalova DA, Manola JB, Ozawa H, Zizkova V, Morton K, Bishop JA, Sharma R, Zhang C, Michailidi C, Considine M, et al. NF-κB and stat3 transcription factor signatures differentiate HPV-positive and HPV-negative head and neck squamous cell carcinoma. Int J Cancer. 2015;137:1879–1889. doi: 10.1002/ijc.29558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Khaznadar SS, Khan M, Schmid E, Gebhart S, Becker ET, Krahn T, von Ahsen O. EGFR overexpression is not common in patients with head and neck cancer. Cell lines are not representative for the clinical situation in this indication. Oncotarget. 2018;9:28965–28975. doi: 10.18632/oncotarget.25656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Johnson DE, Burtness B, Leemans CR, Lui VW, Bauman JE, Grandis JR. Head and neck squamous cell carcinoma. Nat Rev Dis Primers. 2020;6:92. doi: 10.1038/s41572-020-00224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vander Broek R, Mohan S, Eytan DF, Chen Z, Van Waes C. The PI3K/Akt/mTOR axis in head and neck cancer: Functions, aberrations, cross-talk, and therapies. Oral Dis. 2015;21:815–825. doi: 10.1111/odi.12206. [DOI] [PubMed] [Google Scholar]
- 124.Li Z, Yang Z, Passaniti A, Lapidus RG, Liu X, Cullen KJ, Dan HC. A positive feedback loop involving EGFR/Akt/mTORC1 and IKK/NF-kB regulates head and neck squamous cell carcinoma proliferation. Oncotarget. 2016;7:31892–31906. doi: 10.18632/oncotarget.7441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Harris RE. Cyclooxygenase-2 (cox-2) and the inflammogenesis of cancer. Subcell Biochem. 2007;42:93–126. doi: 10.1007/1-4020-5688-5_4. [DOI] [PubMed] [Google Scholar]
- 126.Lim JW, Kim H, Kim KH. Nuclear factor-kappaB regulates cyclooxygenase-2 expression and cell proliferation in human gastric cancer cells. Lab Invest. 2001;81:349–360. doi: 10.1038/labinvest.3780243. [DOI] [PubMed] [Google Scholar]
- 127.Meng F, Liu L, Chin PC, D'Mello SR. Akt is a downstream target of NF-kappa B. J Biol Chem. 2002;277:29674–29680. doi: 10.1074/jbc.M112464200. [DOI] [PubMed] [Google Scholar]
- 128.Ahmad A, Biersack B, Li Y, Kong D, Bao B, Schobert R, Padhye SB, Sarkar FH. Targeted regulation of PI3K/Akt/mTOR/NF-κB signaling by indole compounds and their derivatives: Mechanistic details and biological implications for cancer therapy. Anticancer Agents Med Chem. 2013;13:1002–1013. doi: 10.2174/18715206113139990078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Baud V, Karin M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8:33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tili E, Michaille JJ, Croce CM. MicroRNAs play a central role in molecular dysfunctions linking inflammation with cancer. Immunol Rev. 2013;253:167–184. doi: 10.1111/imr.12050. [DOI] [PubMed] [Google Scholar]
- 131.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 132.Svoronos AA, Engelman DM, Slack FJ. OncomiR or tumor suppressor? The duplicity of microRNAs in cancer. Cancer Res. 2016;76:3666–3670. doi: 10.1158/0008-5472.CAN-16-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hu A, Huang JJ, Xu WH, Jin XJ, Li JP, Tang YJ, Huang XF, Cui HJ, Sun GB. MiR-21 and miR-375 microRNAs as candidate diagnostic biomarkers in squamous cell carcinoma of the larynx: Association with patient survival. Am J Transl Res. 2014;6:604–613. [PMC free article] [PubMed] [Google Scholar]
- 134.Hu A, Huang JJ, Xu WH, Jin XJ, Li JP, Tang YJ, Huang XF, Cui HJ, Sun GB, Li RL, Duan JL. MiR-21/miR-375 ratio is an independent prognostic factor in patients with laryngeal squamous cell carcinoma. Am J Cancer Res. 2015;5:1775–1785. [PMC free article] [PubMed] [Google Scholar]
- 135.Arantes LM, Laus AC, Melendez ME, de Carvalho AC, Sorroche BP, De Marchi PR, Evangelista AF, Scapulatempo-Neto C, de Souza Viana L, Carvalho AL. MiR-21 as prognostic biomarker in head and neck squamous cell carcinoma patients undergoing an organ preservation protocol. Oncotarget. 2017;8:9911–9921. doi: 10.18632/oncotarget.14253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wu Y, Yu J, Ma Y, Wang F, Liu H. MiR-148a and miR-375 may serve as predictive biomarkers for early diagnosis of laryngeal carcinoma. Oncol Lett. 2016;12:871–878. doi: 10.3892/ol.2016.4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li J, Wang K, Chen X, Meng H, Song M, Wang Y, Xu X, Bai Y. Transcriptional activation of microRNA-34a by NF-kappa B in human esophageal cancer cells. BMC Mol Biol. 2012;13:4. doi: 10.1186/1471-2199-13-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li HP, Zeng XC, Zhang B, Long JT, Zhou B, Tan GS, Zeng WX, Chen W, Yang JY. MiR-451 inhibits cell proliferation in human hepatocellular carcinoma through direct suppression of IKK-β. Carcinogenesis. 2013;34:2443–2451. doi: 10.1093/carcin/bgt206. [DOI] [PubMed] [Google Scholar]
- 139.Zhou P, Zeng F, Liu J, Lv D, Liu S. Correlation betweenmir-21 expression and laryngeal carcinoma risks: A meta-analysis. J Evid Based Med. 2016;9:32–37. doi: 10.1111/jebm.12184. [DOI] [PubMed] [Google Scholar]
- 140.Shen Z, Zhan G, Ye D, Ren Y, Cheng L, Wu Z, Guo J. MicroRNA-34a affects the occurrence of laryngeal squamous cell carcinoma by targeting the antiapoptotic gene survivin. Med Oncol. 2010;29:2473–2480. doi: 10.1007/s12032-011-0156-x. [DOI] [PubMed] [Google Scholar]
- 141.Chen Y, Xian PF, Yang L, Wang SX. MicroRNA-21 promotes proliferation of fibroblast-like synoviocytes through mediation of NF-κB nuclear translocation in a rat model of collagen-induced rheumatoid arthritis. Biomed Res Int. 2016;2016:9279078. doi: 10.1155/2016/9279078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kikkawa N, Kinoshita T, Nohata N, Hanazawa T, Yamamoto N, Fukumoto I, Chiyomaru T, Enokida H, Nakagawa M, Okamoto Y, Seki N. MicroRNA-504 inhibits cancer cell proliferation via targeting CDK6 in hypopharyngeal squamous cell carcinoma. Int J Oncol. 2014;44:2085–2092. doi: 10.3892/ijo.2014.2349. [DOI] [PubMed] [Google Scholar]
- 143.Kikkawa N, Hanazawa T, Fujimura L, Nohata N, Suzuki H, Chazono H, Sakurai D, Horiguchi S, Okamoto Y, Seki N. MiR-489 is a tumour-suppressive miRNA target PTPN11 in hypopharyngeal squamous cell carcinoma (HSCC) Br J Cancer. 2010;103:877–884. doi: 10.1038/sj.bjc.6605811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fukumoto I, Kinoshita T, Hanazawa T, Kikkawa N, Chiyomaru T, Enokida H, Yamamoto N, Goto Y, Nishikawa R, Nakagawa M, et al. Identification of tumour suppressive microRNA-451a in hypopharyngeal squamous cell carcinoma based on microRNA expression signature. Br J Cancer. 2014;111:386–394. doi: 10.1038/bjc.2014.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hui AB, Lenarduzzi M, Krushel T, Waldron L, Pintilie M, Shi W, Perez-Ordonez B, Jurisica I, O'Sullivan B, Waldron J, et al. Comprehensive microRNA profiling for head and neck squamous cell carcinomas. Clin Cancer Res. 2010;16:1129–1139. doi: 10.1158/1078-0432.CCR-09-2166. [DOI] [PubMed] [Google Scholar]
- 146.Tai J, Xiao X, Huang ZG, Yu ZK, Chen XH, Zhou WG, Chen XJ, Rao YS, Fang JG, Ni X. MicroRNAs regulate epithelial-mesenchymal transition of supraglottic laryngeal cancer. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2013;48:499–503. (In Chinese) [PubMed] [Google Scholar]
- 147.Zhao XD, Zhang W, Liang HJ, Ji WY. Overexpression of miR-155 promotes proliferation and invasion of human laryngeal squamous cell carcinoma via targeting SOCS1 and STAT3. PLoS One. 2013;8:e56395. doi: 10.1371/journal.pone.0056395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Luzna P, Gregar J, Uberall I, Radova L, Prochazka V, Ehrmann J., Jr Changes of microRNAs-192, 196a and 203 correlate with Barrett's esophagus diagnosis and its progression compared to normal healthy individuals. Diagn Pathol. 2011;6:114. doi: 10.1186/1746-1596-6-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bus P, Siersema PD, Verbeek RE, van Baal JW. Upregulation of miRNA-143, −145, −192, and −194 in esophageal epithelial cells upon acidic bile salt stimulation. Dis Esophagus. 2014;27:591–600. doi: 10.1111/dote.12112. [DOI] [PubMed] [Google Scholar]
- 150.Chen D, Cabay RJ, Jin Y, Wang A, Lu Y, Shah-Khan M, Zhou X. MicroRNA deregulations in head and neck squamous cell carcinomas. J Oral Maxillofac Res. 2013;4:e2. doi: 10.5037/jomr.2013.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yang Z, Fang S, Di Y, Ying W, Tan Y, Gu W. Modulation of NF-κB/miR-21/PTEN pathway sensitizes non-small cell lung cancer to cisplatin. PLoS One. 2015;10:e0121547. doi: 10.1371/journal.pone.0121547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Xiao T, Ling M, Xu H, Luo F, Xue J, Chen C, Bai J, Zhang Q, Wang Y, Bian Q, Liu Q. NF-κB-regulation of miR-155, via SOCS1/STAT3, is involved in the PM2.5-accelerated cell cycle and proliferation of human bronchial epithelial cells. Toxicol Appl Pharmacol. 2019;377:114616. doi: 10.1016/j.taap.2019.114616. [DOI] [PubMed] [Google Scholar]
- 153.Rokavec M, Öner MG, Li H, Jackstadt R, Jiang L, Lodygin D, Kaller M, Horst D, Ziegler PK, Schwitalla S, et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest. 2014;124:1853–1867. doi: 10.1172/JCI73531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tili E, Croce CM, Michaille JJ. MiR-155: On the crosstalk between inflammation and cancer. Int Rev Immunol. 2009;28:264–284. doi: 10.1080/08830180903093796. [DOI] [PubMed] [Google Scholar]
- 155.Bharti AC, Donato N, Singh S, Aggarwal BB. Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor-kappa B and IkappaBalpha kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood. 2003;101:1053–1062. doi: 10.1182/blood-2002-05-1320. [DOI] [PubMed] [Google Scholar]
- 156.Dai Y, Pei XY, Rahmani M, Conrad DH, Dent P, Grant S. Interruption of the NF-kappaB pathway by Bay 11-7082 promotes UCN-01-mediated mitochondrial dysfunction and apoptosis in human multiple myeloma cells. Blood. 2004;103:2761–2770. doi: 10.1182/blood-2003-09-3037. [DOI] [PubMed] [Google Scholar]
- 157.Monisha J, Roy NK, Bordoloi D, Kumar A, Golla R, Kotoky J, Padmavathi G, Kunnumakkara AB. Nuclear factor kappa B: A potential target to persecute head and neck cancer. Curr Drug Targets. 2017;18:232–253. doi: 10.2174/1389450117666160201112330. [DOI] [PubMed] [Google Scholar]
- 158.Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa B system: A treasure trove for drug development. Nat Rev Drug Discov. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- 159.Ramadass V, Vaiyapuri T, Tergaonkar V. Small molecule NF-κB pathway inhibitors in clinic. Int J Mol Sci. 2020;21:5164. doi: 10.3390/ijms21145164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, Göktuna SI, Neuenhahn M, Fierer J, Paxian S, et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007;130:918–931. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/S0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 162.Geiger JL, Grandis JR, Bauman JE. The STAT3 pathway as a therapeutic target in head and neck cancer: Barriers and innovations. Oral Oncol. 2016;56:84–92. doi: 10.1016/j.oraloncology.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nelson EA, Walker SR, Kepich A, Gashin LB, Hideshima T, Ikeda H, Chauhan D, Anderson KC, Frank DA. Nifuroxazide inhibits survival of multiple myeloma cells by directly inhibiting STAT3. Blood. 2008;112:5095–5102. doi: 10.1182/blood-2007-12-129718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Siddiquee KA, Gunning PT, Glenn M, Katt WP, Zhang S, Schrock C, Sebti SM, Jove R, Hamilton AD, Turkson J. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem Biol. 2007;2:787–798. doi: 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]
- 165.Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15:234–248. doi: 10.1038/nrclinonc.2018.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Goldman A, Shahidullah M, Goldman D, Khailova L, Watts G, Delamere N, Dvorak K. A novel mechanism of acid and bile acid-induced DNA damage involving Na+/H+ exchanger: Implication for Barrett's oesophagus. Gut. 2010;59:1606–1616. doi: 10.1136/gut.2010.213686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bernstein H, Bernstein C, Payne CM, Dvorak K. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J Gastroenterol. 2009;15:3329–3340. doi: 10.3748/wjg.15.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lechner S, Müller-Ladner U, Schlottmann K, Jung B, McClelland M, Rüschoff J, Welsh J, Schölmerich J, Kullmann F. Bile acids mimic oxidative stress induced upregulation of thioredoxin reductase in colon cancer cell lines. Carcinogenesis. 2002;23:1281–1288. doi: 10.1093/carcin/23.8.1281. [DOI] [PubMed] [Google Scholar]
- 169.Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–5694. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rothkamm K, Löbrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci USA. 2003;100:5057–5062. doi: 10.1073/pnas.0830918100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nikitaki Z, Hellweg CE, Georgakilas AG, Ravanat JL. Stress-induced DNA damage biomarkers: Applications and limitations. Front Chem. 2015;3:35. doi: 10.3389/fchem.2015.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Tsantoulis PK, Kotsinas A, Sfikakis PP, Evangelou K, Sideridou M, Levy B, Mo L, Kittas C, Wu XR, Papavassiliou AG, Gorgoulis VG. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene. 2008;27:3256–3264. doi: 10.1038/sj.onc.1210989. [DOI] [PubMed] [Google Scholar]
- 173.Choudhari SK, Chaudhary M, Gadbail AR, Sharma A, Tekade S. Oxidative and antioxidative mechanisms in oral cancer and precancer: A review. Oral Oncol. 2014;50:10–18. doi: 10.1016/j.oraloncology.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 174.Janssens S, Tschopp J. Signals from within: The DNA-damage-induced NF-kappaB response. Cell Death Differ. 2006;13:773–784. doi: 10.1038/sj.cdd.4401843. [DOI] [PubMed] [Google Scholar]
- 175.Kawanishi S, Ohnishi S, Ma N, Hiraku Y, Murata M. Crosstalk between DNA damage and inflammation in the multiple steps of carcinogenesis. Int J Mol Sci. 2017;18:1808. doi: 10.3390/ijms18081808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tafani M, Sansone L, Limana F, Arcangeli T, De Santis E, Polese M, Fini M, Russo MA. The interplay of reactive oxygen species, hypoxia, inflammation, and sirtuins in cancer initiation and progression. Oxid Med Cell Longev. 2016;2016:3907147. doi: 10.1155/2016/3907147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Li D, Cao W. Bile acid receptor TGR5, NADPH Oxidase NOX5-S and CREB mediate bile acid-induced DNA damage in Barrett's esophageal adenocarcinoma cells. Sci Rep. 2016;6:31538. doi: 10.1038/srep31538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Nema R, Vishwakarma S, Agarwal R, Panday RK, Kumar A. Emerging role of sphingosine-1-phosphate signaling in head and neck squamous cell carcinoma. Onco Targets Ther. 2016;9:3269–3280. doi: 10.2147/OTT.S99989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tamashiro PM, Furuya H, Shimizu Y, Iino K, Kawamori T. The impact of sphingosine kinase-1 in head and neck cancer. Biomolecules. 2013;3:481–513. doi: 10.3390/biom3030481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Liu R, Li X, Hylemon PB, Zhou H. Conjugated bile acids promote invasive growth of esophageal adenocarcinoma cells and cancer stem cell expansion via sphingosine 1-phosphate receptor 2-mediated yes-associated protein activation. Am J Pathol. 2018;188:2042–2058. doi: 10.1016/j.ajpath.2018.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Pang C, LaLonde A, Godfrey TE, Que J, Sun J, Wu TT, Zhou Z. Bile salt receptor TGR5 is highly expressed in esophageal adenocarcinoma and precancerous lesions with significantly worse overall survival and gender differences. Clin Exp Gastroenterol. 2017;10:29–37. doi: 10.2147/CEG.S117842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Guan B, Li H, Yang Z, Hoque A, Xu X. Inhibition of farnesoid X receptor controls esophageal cancer cell growth in vitro and in nude mouse xenografts. Cancer. 2013;119:1321–1329. doi: 10.1002/cncr.27910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yu JH, Zheng JB, Qi J, Yang K, Wu YH, Wang K, Wang CB, Sun XJ. Bile acids promote gastric intestinal metaplasia by upregulating CDX2 and MUC2 expression via the FXR/NF-κB signaling pathway. Int J Oncol. 2019;54:879–892. doi: 10.3892/ijo.2019.4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Schweitzer A, Knauer SK, Stauber RH. Nuclear receptors in head and neck cancer: Current knowledge and perspectives. Int J Cancer. 2010;126:801–809. doi: 10.1002/ijc.24968. [DOI] [PubMed] [Google Scholar]
- 185.Gadaleta RM, Oldenburg B, Willemsen EC, Spit M, Murzilli S, Salvatore L, Klomp LW, Siersema PD, van Erpecum KJ, van Mil SW. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-κB signaling in the intestine. Biochim Biophys Acta. 2011;1812:851–858. doi: 10.1016/j.bbadis.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 186.Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology. 2008;48:1632–1643. doi: 10.1002/hep.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wang Z, Li Y, Sarkar FH. Signaling mechanism(s) of reactive oxygen species in epithelial-mesenchymal transition reminiscent of cancer stem cells in tumor progression. Curr Stem Cell Res Ther. 2010;5:74–80. doi: 10.2174/157488810790442813. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable.