

Abstract
Protein glycosylation is one of the most common and diverse modifications. Aberrant protein glycosylation has been reported to associate with various diseases. High‐throughput and comprehensive characterization of glycoproteins is crucial for structural and functional studies of altered glycosylation in biological, physiological, and pathological processes. In this protocol, we detail a workflow for comprehensive analyses of intact glycopeptides (IGPs), glycosylation sites, and glycans from N‐linked glycoproteins. By utilizing liquid handling systems, our workflow could enrich IGPs in a high‐throughput manner while reducing sample processing time and human error involved in traditional proteomics sample processing techniques. Together, our workflow enables a high‐throughput enrichment of glycans, glycosites, and intact glycopeptides from complex biological or clinical samples. © 2021 The Authors. Current Protocols published by Wiley Periodicals LLC.
Basic Protocol 1: Enzymatic digestion of glycoproteins using C4‐tips
Basic Protocol 2: Intact glycopeptide analysis using C18/MAX‐tips
Basic Protocol 3: Glycan and glycosite analysis
Keywords: glycoproteomics, high‐throughput, mass spectrometry, sample processing
INTRODUCTION
Protein glycosylation is one of the most common and diverse post translational modifications, with as many as half of the proteins in the human body reported being glycosylated (Bennun et al., 2016; Khoury, Baliban, & Floudas, 2011). Aberrant glycosylation has also been reported to relate to different diseases, including various cancer types (An et al., 2006; Hu et al., 2020), heart disease (Nagai‐Okatani & Minamino, 2016), and rheumatoid arthritis (Ercan et al., 2010). Protein glycosylation has also been found to play pivotal roles in the pharmacokinetics of monoclonal antibodies and Fc fusion proteins (Higel, Seidl, Sörgel, & Friess, 2016). Recently, it was reported that afucosylated IgGs could potentially promote the exacerbation of coronavirus disease 2019 (COVID‐19) response (Larsen et al., 2020). Deciphering the mystery of protein glycosylation is crucial and necessary to improve our understanding for glycoprotein functions.
For mass spectrometry‐based glycoproteomic analysis, investigation of N‐linked glycosylation involves multiple steps, including proteolytic digestion, followed by enrichment and subsequent downstream identification of glycopeptides. Various methods have been utilized to characterize N‐linked glycosylation on different levels, which can be roughly separated into three categories: (1) glycosite analysis, (2) glycan profiling, or (3) site‐specific intact glycopeptide analysis. Glycosite analysis can be achieved using hydrazide chemistry and its automated method (Tian, Zhou, Elliott, Aebersold, & Zhang, 2007; Zhang, Li, Martin, & Aebersold, 2003), lectin enrichment (Kaji et al., 2003; Zielinska, Gnad, Wiśniewski, & Mann, 2010), or hydrophilic enrichment (Wada, Tajiri, & Yoshida, 2004). Glycan profiling can be analyzed on a native glycan level (Fujitani et al., 2013) via sialic acid derivatization (Shah et al., 2013) or permethylation (Kang, Mechref, Klouckova, & Novotny, 2005). Site‐specific intact glycopeptide analysis can be achieved using strategies such as solid‐phase extraction of N‐linked glycans and glycosite‐containing peptides (Sun et al., 2016), hydrophilic interaction liquid chromatography (HILIC) enrichment combined with spectral library search (Shu et al., 2020), and rapid analysis of glycopeptides by permethylation (Shajahan, Supekar, Heiss, Ishihara, & Azadi, 2017). For intact glycopeptide analysis, a mass spectrometry‐based method such as electron‐transfer/high‐energy collision dissociation (EThcD; Yu et al., 2017), stepped‐normalized collision energy (NCE) during higher‐energy collisional dissociation tandem mass spectrometry (HCD MS/MS) fragmentation (Yang, Yang, & Sun, 2018), intact O‐glycopeptide analysis using data‐independent acquisition (DIA) mode (Ye, Mao, Clausen, & Vakhrushev, 2019), or field asymmetric wave ion mobility spectrometry (FAIMS; Creese & Cooper, 2012) have also been reported to be efficient glycopeptide analysis strategies.
Site‐specific intact glycopeptide analysis is considered to be the most promising strategy to comprehensively characterize glycoproteins. However, it is challenging to readily analyze intact glycopeptides because enrichment methods, mass spectrometric analysis, and data analysis software still need to be further developed to ensure glycoproteome coverage and correct glycoform annotation. Furthermore, several challenges need to be addressed in order to increase sample processing throughput for large‐scale glycoproteomics characterization, including reducing the time span for sample processing, limiting sample loss during sample enrichment procedures (i.e, peptide desalting, sample dry down to reconstitute in appropriate liquid composition), and sample processing reproducibility.
In this protocol, we present a comprehensive intact glycopeptide, glycan, and glycosite analysis workflow (Chen et al., 2020) that can obtain the glycoproteomic information from standard protein, cell lysates, or even bodily fluids like human urine or serum. For urinary glycoproteomic analysis, a raw urine sample typically needs to undergo sample pre‐processing, for instance, ultracentrifugation, ultrafiltration, or buffer exchange, in order to concentrate proteins and remove contaminants from urine for subsequent enzymatic digestion. However, the presented C4‐tip and C18/MAX‐tip workflows do not need additional sample pre‐processing techniques; raw urine can be acidified and directly bound to the C4 resin digested into peptide mixtures. Utilizing a liquid handling system, intact glycopeptides can be enriched in a high‐throughput and highly reproducible manner, while allowing for orthogonal verification of glycosylation site identification via analysis of intact glycopeptides (IGPs) and deglycosylated N‐linked glycopeptides (Fig. 1). The presented enrichment workflow offers the flexibility of analyzing various levels of N‐linked glycoproteins (i.e., glycosite, glycans, and intact glycopeptides). Once intact glycopeptides are readily enriched, users can choose to further process the intact glycopeptides by PNGaseF into de‐N‐glycosylated peptides and glycans. Further separation using reverse‐phase C18 resin would allow separation of de‐N‐glycosylated peptides from glycans. By analysis of (1) intact glycopeptides, (2) de‐N‐glycosylated peptides, and (3) glycans, users would achieve (1) site‐specific intact glycopeptide analysis, (2) glycosite analysis, and (3) glycan profiling altogether by using this single workflow. Being a tip‐format, the workflow also offers a high degree of user flexibility, by allowing the users to process samples using a single pipet, a multichannel pipet, or liquid handling systems.
Figure 1.
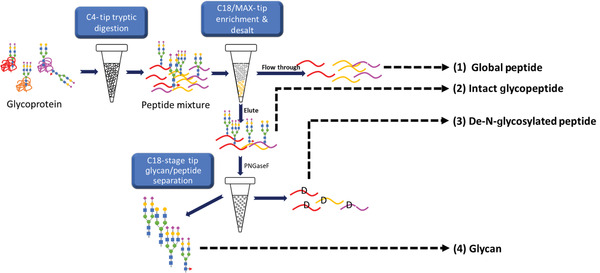
Workflow for C4‐tip and C18/MAX‐tip sample processing. Starting at the glycoprotein level, glycoproteins are first bound to C4‐tip, which undergo enzymatic digestion. Digested peptides are then loaded onto C18/MAX‐tip for intact glycopeptide (IGP) enrichment. (1) Non‐glycosylated peptides and (2) intact glycopeptides are sequentially eluted. Subsequently, an aliquot of the IGPs fraction can be subjected to PNGaseF digest. (3) Deglycosylated peptides, and (4) glycans are separated by C18‐stage tips. This figure is cited from our previous publication, Chen et al., 2020.
Basic Protocol 1 provides detailed instruction on C4‐tip production and on‐tip enzymatic digestion of proteins using the C4‐tip. Basic Protocol 2 outlines instruction on C18/MAX‐tip production and intact glycopeptide enrichment using the C18/MAX‐tip. Basic Protocol 3 describes steps for de‐N‐glycopeptide sample processing by PNGaseF, glycan clean up by a porous graphitic carbon (PGC) NuTip, and also glycan profiling by matrix‐assisted laser desorption/ionization (MALDI).
CAUTION: Basic Protocols 1 and 2 describe steps to produce resin‐based tips; users are advised to operate in a fume hood or wear a mask to prevent inhalation of resins.
NOTE: Basic Protocols 1 and 2 are designed for users with liquid handling systems; users need to compile a procedure that matches the processing format of their individual liquid handling system (see Commentary, Time Considerations to construct instrument specific procedures). If users prefer to follow this protocol using a pipet or multichannel pipet, still follow Basic Protocols 1 and 2 but maintain the number of aspiration and dispersion amounts, as well as the overall reaction time.
Basic Protocol 1. ENZYMATIC DIGESTION OF GLYCOPROTEINS USING C4‐TIPS
While reverse‐phase separation resins such as C18 have been extensively used for peptide binding and subsequent desalting, the long alkyl chains of C18 would impede downstream protein elution steps. In our previous publication (Clark et al., 2019), we evaluated the use of reverse‐phase resin with shorter alkyl chains (i.e., C4) for use in urinary protein isolation paired with “on‐tip” protease digestion. Herein, we describe detailed steps for C4‐tip production and “on‐tip” protease digestion. Similar to traditional proteomics sample processing, Basic Protocol 1 should be used for enzymatic digestion of protein mixtures into peptides, with the benefits that on‐tip digestion can provide: Direct binding of urinary proteins, without doing sample preparation methods such as protein precipitation, ultrafiltration, or analytical ultracentrifugation. Users should skip Basic Protocol 1 and proceed with Basic Protocol 2 if focused on glycopeptide enrichment from peptide mixtures.
NOTE: For C4‐tips, we previously evaluated the binding capacity of the C4 resin and it is ∼400 µg of protein material per 30 mg of C4 resin (Clark et al., 2019). Users are advised to adjust the total amount of C4 resin according to the protein material being processed.
Materials
C4 Resin Beads (Separation Methods Technologies, cat. no. Ultra‐JH‐C4‐Resin)
Methanol (MeOH), Optima® LC/MS (Thermo Fisher Scientific, cat. no. A456‐4)
Acetonitrile (ACN), Optima® LC/MS (Thermo Fisher Scientific, cat. no. A955‐4)
Trifluoracetic acid (TFA), LC‐MS Grade (Thermo Fisher Scientific, cat. no. 85183)
Formic Acid, Optima® LC/MS (Thermo Fisher Scientific, cat. no. A117‐50)
50 mM triethylammonium bicarbonate (TEAB; see recipe)
Bond BreakerTM Tris 2‐carboxyethyl phosphine (TCEP) Solution (Thermo Fisher Scientific, cat. no. 77720)
Iodoacetamide (IAA; MilliporeSigma, cat. no. A3321‐10VL)
Lys C (Lysyl Endopeptidase®; FUJIFILM Wako Pure Chemical Corporation, cat. no. 129‐02541)
Trypsin (Promega, cat. no. V511X)
Digestion Buffer for C4 (see recipe)
Versette Automated Liquid Handling System (Thermo Fisher Scientific, cat. no. 650‐01‐BS)
Porex® 1/16 in. Fine Sheet (Interstate Specialty Products, cat. no. POR‐4900)
Harris Uni‐core disposable punch (I.D. 2 and 5 mm, MilliporeSigma)
300 µl D.A.R.T.S tips (Thermo Fisher Scientific, cat. no. 5516‐11)
Deepwell Plate, 96/500 µl, yellow border (Eppendorf, cat. no. 951031828)
SavantTM SpeedVacTM SC210A Concentrator (Thermo Fisher Scientific)
SavantTM SpeedVacTM Refrigerated Vapor Trap (Thermo Fisher Scientific RVT‐5105)
Production of C4‐tip
-
1
Weigh out 30 mg C4 resin beads/tip into an Eppendorf tube.
-
2
Use a Harris Uni‐core disposable punch (I.D. 2 mm) to punch 2‐mm holes in the 1/16 in. fine sheet and shoot in the D.A.R.T.S tips; make sure the flat side of the filter stays horizontal with the tip. Use a sterilized needle or iron wire to pack the filter firmly into the D.A.R.T.S tips; do not pack filter too tightly because it will affect downstream liquid aspiration and dispersion (Fig. 2).
We recommend using gravity to drop the iron wire ∼2 in. above the filter for ∼30 times to achieve reproducible results across all C4‐tips.
Figure 2.
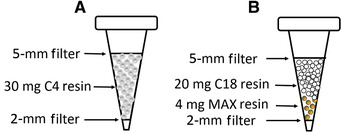
Schematic representation of C4‐tip and C18/MAX‐tip. By using a Harris Uni‐core disposable punch, punch 2‐mm holes in the 1/16 in. fine sheet and shoot in the D.A.R.T.S tips. Making sure the flat side of the filter stays horizontal with the tip, load (A) 30 mg C4 or (B) 4 mg MAX, followed by 20 mg C18 in the D.A.R.T.S tip, pack the tip by punching another 5‐mm hole in the 1/16 in. fine sheet and pack firmly on top. The resin should stay in the middle during the entire sample process procedure. If either the 5‐ or 2‐mm sheet is not placed horizontally, the resin may leak during the sample processing procedure.
-
3
Add 300 µl MeOH into Eppendorf tubes that contain C4 resin beads.
-
4
Vortex thoroughly and inject MeOH/C4 resin mixture into the D.A.R.T.S tips that contain 2‐mm filters. Rinse the Eppendorf tube again using 100 µl MeOH and inject into D.A.R.T.S tips to ensure C4 resin is all transferred to the D.A.R.T.S tip.
If using other liquid handling systems, the D.A.R.T.S tip might not be compatible with certain liquid handlers but the same packing method can still be implemented.
-
5
Punch 5‐mm holes using the Harris Uni‐core disposable punch (I.D. 5 mm) in the 1/16 in. fine sheet; the resin should be in between the 2‐ and 5‐mm filters.
-
6
Use tweezers or a flat shaped rod to push the 5‐mm filter firmly against the upper level of the resin.
We recommend marking a specific position on the flat shaped rod to push down the 5‐mm filter to the same depth across production of all tips.
-
7
Allow MeOH to flow through the bottom of the D.A.R.T.S tips naturally.
Steps 1 to 7 describe the production of a single C4‐tip. If users are manufacturing multiple C4‐tips at the same time, we recommend weighing the appropriate amount of C4 material (30 mg/tip) in a small glass vial (preferably 20 ml) and adding MeOH in the following proportion: 30 mg C4 resin/200 µl MeOH (for example, for production of 20 tips, weigh out 600 mg of C4 resin, add 4,000 µl of MeOH). Add a magnetic stir bar and use a magnetic hot plate/stirrer to stir the resin/MeOH mixture. Make sure the mixture is homogenized and start to aspirate 200 µl/tip of mixture into the D.A.R.T.S tips, then follow steps 5 to 7.
Conditioning C4‐tips
-
8
Take out two racks, add 30 ml 50% ACN + 0.1% TFA, and 30 ml 0.1% TFA, separately.
The volume specification in step 8 is just to ensure C4‐tips can aspirate enough liquid, as long as the C4‐tip can be submerged to aspirate liquids; the specific volume should not be critical.
-
9
Use the liquid handling system to aspirate and disperse the C4‐tips in 50% ACN + 0.1% TFA for ten cycles.
-
10
Aspirate and disperse C4‐tips in 0.1% TFA for another ten cycles.
For the liquid handling system (i.e., Versette), one cycle (aspiration and dispersion) will take ∼2 min. If using other liquid handling systems or a multichannel pipet, procedures need to be changed accordingly to follow the overall processing time of specific steps.
Sample loading on C4‐tips
-
11
If the protein samples are dried, reconstitute sample into 300 µl with 1% formic acid.
For C4‐tips, we previously evaluated the binding capacity of the C4 resin as ∼400 µg of protein material per 30 mg of C4 resin (Clark et al., 2019). Users are advised to adjust the total amount of C4 resin according to the protein material being processed.
-
12
Vortex samples thoroughly.
-
13
Centrifuge samples at 900 × g for 10 min to remove insoluble proteins and precipitates.
-
14
Aliquot 300 µl supernatant into the 96‐well Deepwell plate; be cautious and do not aliquot precipitates.
-
15
Acidify samples with 20% formic acid until final concentration of formic acid is 1% to keep pH <3.
If samples are already at pH <3, this step can be skipped.
-
16
Using the liquid handling system, aspirate and disperse acidified protein solution using C4‐tips for 30 cycles.
For the sample loading procedure, we added an extra delay time of 1 min between aspiration and dispersion; therefore, one cycle would be ∼3 min. If using other liquid handling systems, procedures need to be changed accordingly to follow the overall processing time of specific steps.
Sample washing to remove inorganic salts, urea, and other contaminants
-
17
Remove contaminants and inorganic salts using 0.1% TFA; run for ten cycles.
-
18
Adjust the pH in each of the tips using 50 mM TEAB and run for ten cycles.
For downstream on‐tip digestion using a C4‐tip, the user must adjust the pH in each tip in order to maintain enzymatic activity, i.e., pH ∼7.6‐8.0. If TEAB is not compatible for the user's analysis, we recommend other pH buffers like ammonium bicarbonate.
“On‐tip” protein digestion using C4‐tip
-
19
Reduce protein disulfide bonds on bound proteins using 10 mM TCEP and run for twenty cycles.
-
20
Alkylate reduced cysteine residues on bound proteins using 15 mM IAA in the dark and run for twenty cycles.
IAA must be added in the dark because IAA is light sensitive.
-
21
Make a digestion buffer consisting of 50 mM TEAB in 30% ACN.
The composition of digestion buffer has been evaluated in our previous publication (Clark et al., 2019). We have found inclusion of 30% ACN in our digestion buffer resulted in a higher peptide recovery and also a higher trypsin enzyme activity.
-
22
Perform protease digestion on bound proteins by adding Lys C in digestion buffer (1:40 enzyme/protein) for 30 cycles.
-
23
Perform subsequent protease digestion on bound proteins by adding trypsin in digestion buffer (1:40 enzyme/protein) for another 120 cycles.
-
24
Elute digested peptide on a C4‐tip using 150 µl 50% ACN + 0.1% TFA and run for ten cycles.
-
25
Repeat step 24 to ensure higher recovery of digested peptides.
-
26
Pool solutions from steps 22, 23, 24, and 25 from the same C4‐tip.
-
27
Dry down digested peptides using a SpeedVac concentrator.
For SpeedVac processing to dry down the samples, we use the SpeedVac SC210A concentrator with settings of concentrator mode: “ON” and drying rate mode: “Low.”
-
28
Store samples at ‐20°C until ready to start Basic Protocol 2.
Steps 22, 23, 24, and 25 all contain digested peptides. After the last peptide elution step, solutions from steps 22 to 25 should be pooled together.
Basic Protocol 1 can also be directly applied for protease digestion of unmodified peptides. If users are focused on global proteomics, a further C18 stage tip clean up would be sufficient for global proteomic analysis on liquid chromatography‐tandem mass spectrometry (LC‐MS/MS).
Basic Protocol 2. INTACT GLYCOPEPTIDE ANALYSIS USING C18/MAX‐TIPS
With the advancement of mass spectrometry, high‐throughput sample preparation has been the focus of proteomics (Clark et al., 2019; Fu et al., 2018), while high‐throughput glycoproteomics has been somewhat limited to glycosite analysis using hydrazide chemistry (Berven, Ahmad, Clauser, & Carr, 2010; Chen, Shah, & Zhang, 2013). Traditionally, intact glycopeptides are enriched by hydrophilic chromatography (Sun et al., 2016; W. Yang et al., 2017) or lectin (Guo et al., 2015; Zhou et al., 2017), while sialylated glycopeptides are typically enriched by TiO2 (Kawahara et al., 2018; Palmisano et al., 2010), strong cationic exchange (SCX; Lewandrowski, Zahedi, Moebius, Walter, & Sickmann, 2007), immobilized metal affinity chromatography (IMAC) based on charge selection (Hu, Shah, Clark, Ao, & Zhang, 2018), or periodate‐oxidation of sialylated glycopeptides (Halim, Nilsson, Rüetschi, Hesse, & Larson, 2012; Nilsson et al., 2009). For periodate‐oxidation, sialic acid information is lost after de‐sialylation and release from hydrazide beads. With the limits described above, we developed a high‐throughput intact glycopeptide enrichment platform that would combine two distinct proteomics enrichment strategies (i.e., hydrophilic and hydrophobic) for simultaneous desalting and enrichment of glycopeptides. We've previously compared the enrichment efficiency and specificity of multiple enrichment methods (W. Yang et al., 2017), and reported mixed‐mode anionic exchange (MAX) showed higher yield of enrichment and great enrichment specificity. Hence, we further developed our glycopeptide enrichment platform using MAX resin (Chen et al., 2020). Basic Protocol 2 provides a step‐by‐step introduction of glycopeptide enrichment using a combination of hydrophilic and hydrophobic peptide enrichment strategies, i.e., C18 resin plus MAX resin. Utilizing liquid handling systems, users should expect enrichment of intact glycopeptide after ∼6 hr of processing time.
Materials
Peptide samples (dried)
Preparative C18 125Å 55‐105 µm bulk packing material (Waters, cat. no. WAT020594)
Oasis® MAX Material (Waters, cat. no. 186007553)
Methanol, Optima® LC/MS (Thermo Fisher Scientific, cat. no. A456‐4)
Acetonitrile (ACN), Optima® LC/MS (Thermo Fisher Scientific, cat. no. A955‐4)
Water, Optima® LC/MS (Thermo Fisher Scientific, cat. no. W6‐4)
Trifluoracetic acid (TFA), LC‐MS Grade (Thermo Fisher Scientific, cat. no. 85183)
Formic Acid, Optima® LC/MS (Thermo Fisher Scientific, cat. no. A117‐50)
100 mM triethylammonium acetate buffer (TAAB; see recipe)
Versette Automated Liquid Handling System (Thermo Fisher Scientific, cat. no. 650‐01‐BS)
Porex® 1/16 in. Fine Sheet (Interstate Specialty Products, cat. no. POR‐4900)
Harris Uni‐core disposable punch (I.D. 2 and 5 mm, MilliporeSigma)
300 µl D.A.R.T.S tips (Thermo Fisher Scientific, cat. no. 5516‐11)
Sterilized needle or iron wire (to pack the filter)
Deepwell Plate 96/500 µl, yellow border (Eppendorf, cat. no. 951031828)
SavantTM SpeedVacTM SC210A Concentrator (Thermo Fisher Scientific)
SavantTM SpeedVacTM Refrigerated Vapor Trap (Thermo Fisher Scientific RVT‐5105)
Production of C18/MAX‐tip
-
1
Weigh out 4 mg MAX resin beads/tip in a small glass vial (preferably 20 ml).
-
2
Use a Harris Uni‐core disposable punch (I.D. 2 mm) to punch 2‐mm holes in the 1/16 in. fine sheet and shoot in the D.A.R.T.S tips; make sure the flat side of the filter stays horizontal with the tip. Use a sterilized needle or iron wire to pack the filter firmly into the D.A.R.T.S tips; do not pack the filter too tightly or it will affect downstream liquid aspiration and dispersion.
We recommend using gravity to drop the iron wire ∼2 in. above the filter for ∼30 times to achieve reproducible results across all C18/MAX‐tips.
-
3
Add methanol in the following proportion: 4 mg MAX resin/100 µl MeOH. (For example, for production of twenty tips, weigh out 80 mg of MAX resin and add 2,000 µl MeOH.)
-
4
Put a magnetic stir bar in the small glass vial containing the MAX resin.
-
5
Use a magnetic hot plate/stirrer to stir the resin/MeOH mixture. Make sure mixture is homogenized and start to aspirate 100 µl/tip of mixture into the D.A.R.T.S tips.
-
6
Weigh out 20 mg C18 resin beads/tip in a separate small glass vial (preferably 20 ml).
-
7
Add methanol in the following proportion: 20 mg C18 resin/200 µl MeOH. (For example, for production of twenty tips, weigh out 400 mg of MAX resin and add 4,000 µl MeOH.)
-
8
Put a magnetic stir bar in the small glass vial containing the C18 resin.
-
9
Use a magnetic hot plate/stirrer to stir the resin/MeOH mixture. Make sure mixture is homogenized and start to aspirate 200 µl/tip of mixture into the D.A.R.T.S tips that already contain MAX resin.
When adding the C18 resin inside D.A.R.T.S tips, make sure MAX resin is already settled. Add the MeOH/C18 resin directly on top of the MAX resin only after the MAX resin is settled in the tip.
-
10
Punch 5‐mm holes using the Harris Uni‐core disposable punch (I.D. 5 mm) in the 1/16 in. fine sheet.
The resin should be in between the 2‐ and 5‐mm filters.
-
11
Use tweezers or a flat shaped rod to push the 5‐mm filter firmly against the upper level of the resin.
-
12
Allow MeOH to flow through the bottom of the D.A.R.T.S tips naturally.
Conditioning C18/MAX‐tips
-
13
Take out four racks, add 30 ml 100% ACN, 100 mM TAAB, 95% ACN + 1% TFA, and 0.1% TFA, respectively.
The volume specification is just to ensure C18/MAX‐tips can aspirate enough liquid; as long as C18/MAX‐tips can be submerged to aspirate liquids, the specific volume should not be critical.
-
14
Use the Versette liquid handling system to aspirate and disperse the C18/MAX‐tips sequentially in 100% ACN, 100 mM TAAB, 95% ACN + 1% TFA, and 0.1% TFA for ten cycles.
We have reported that for a column‐based elution, the related position of C18 and MAX resin are optimized if they are stacked with C18 on top (G. Yang et al., 2020). However, because liquid handling systems would aspirate and disperse in an up‐down movement (unlike column‐based flow, which generally uses gravity to allow the elution wash to flow naturally). The relative position of C18 and MAX should not be critical.
Sample loading using C18/MAX‐tips
-
15
If the peptide samples are dried, reconstitute sample into 300 µl with 0.1% TFA.
-
16
Vortex samples thoroughly.
-
17
Centrifuge samples at 900 × g for 10 min to remove insolubilized proteins and precipitates.
-
18
Aliquot supernatant into a 96‐well Deepwell plate; be cautious and do not aliquot precipitates.
-
19
Acidify samples with 20% TFA until the final pH is <3.
If samples are already at pH <3, this step can be skipped.
-
20
Using the liquid handling system, aspirate and disperse the acidified peptide solution using C18/MAX‐tips for 25 cycles.
Unlike the C4‐tip sample loading steps, for this sample loading procedure, we did not add an extra delay time of 1 min between aspiration and dispersion; therefore, one cycle would still be ∼2 min. If using other liquid handling systems, procedures need to be changed accordingly to follow the overall processing time of specific steps.
The sample loading using 0.1% TFA is the sample loading condition for a typical C18 resin sample loading. Unmodified peptides and glycopeptides are able to bind to C18 material via hydrophobic interactions.
Sample washing/desalting using C18/MAX‐tips
-
21
Directly after the sample loading step, aspirate and disperse 0.1% TFA using the C18/MAX‐tip that contains peptide mixtures for ten cycles.
This step is to eliminate any unspecific binding of C18 material. Because the washing buffer is 0.1% TFA (i.e., does not contain organic solvents), peptide will not be eluted off the C18/MAX‐tip during this step.
Sample loading onto MAX material and elution of non‐glycosylated peptides
-
22
Aliquot 200 µl 95% ACN in 1% TFA into a new 96‐well Deepwell plate.
-
23
Using the liquid handling system, aspirate and disperse the C18/MAX‐tips for five cycles.
-
24
Repeat steps 22 and 23 two more times.
-
25
Collect eluant from steps 22 to 24; the eluant should contain the non‐glycosylated peptides.
95% ACN + 1% TFA is the intact glycopeptide sample loading condition which was evaluated in our previous publication (W. Yang et al., 2017). Simultaneously, 95% ACN will also allow for non‐glycosylated peptide to elute from the C18 material, while glycosylated peptide will still be retained on the MAX material via hydrophilic interactions.
Repeating steps 22 and 23 with fresh 95% ACN + 0.1% TFA is to ensure non‐glycosylated peptides are completely eluted off the MAX material. In our previous work (W. Yang et al., 2017), we observed ∼66% of enrichment specificity for MAX material when we compared percentage of peptide spectrum matches (PSMs) of all glycopeptides to the total PSMs of peptides and glycopeptides.
Elution of glycopeptides from C18/MAX‐tip
-
26
Aliquot 200 µl 50% ACN + 0.1% TFA into a new 96‐well Deepwell plate.
-
27
Using the liquid handling system, aspirate and disperse the C18/MAX‐tips for five cycles.
-
28
Repeat steps 26 and 27 two more times.
-
29
Collect eluant from steps 26 to 28; this eluant should contain the intact glycopeptides.
-
30
Dry down samples using a SpeedVac concentrator.
Basic Protocol 3. GLYCAN AND GLYCOSITE ANALYSIS
Basic Protocols 1 and 2 describe details for enzymatic digestion of protein mixtures and glycopeptide enrichment from global peptides. Basic Protocol 3 provides step‐by‐step instruction for N‐linked glycan cleavage using PNGaseF and downstream separation of glycans from de‐N‐glycosylated peptides as well as glycan clean up by a (PGC) NuTip. Following Basic Protocol 3, users can expect to receive (1) glycan information and (2) glycosite information by analyzing glycans and de‐N‐glycosylated peptides, separately. When comparing the intact glycopeptide results with glycosite results, a user can achieve an orthogonal verification of glycosylation sites by observing the 0.98 Da mass shift after deamidation of asparagine (N) into aspartic acid (D).
Additional Materials (see also Basic Protocol 1)
Acetonitrile (ACN), Optima® LC/MS (Thermo Fisher Scientific, cat. no. A955‐4)
Formic Acid (FA), Optima® LC/MS (Thermo Fisher Scientific, cat. no. A117‐50)
Triethylammonium bicarbonate (TEAB; MilliporeSigma, cat. no. T7408‐500ML)
Peptide‐N‐glycosidase F (PNGase F; New England BioLabs, cat. no. P0704L)
NuTip Carbon (Hypercarb), large 10‐200 µl (Glygen, cat. no. NT3CAR.96)
Solid Phase Extraction Disk (Empore, cat. no. 2215‐C18, Octadecyl)
Deepwell Plate 96/500 µl, yellow border (Eppendorf, cat. no. 951031828)
SavantTM SpeedVacTM SC210A Concentrator (Thermo Fisher Scientific)
SavantTM SpeedVacTM Refrigerated Vapor Trap (Thermo Fisher Scientific RVT‐5105)
Microcentrifuge (Eppendorf, cat. no. 5424)
MicroMix 5 Plate Shaker (DPC)
Cleaving N‐linked glycans using PNGaseF
-
1
If your glycopeptide samples are completely dried, resuspend them in 40 µl 0.1% TFA.
-
2
Vortex thoroughly for 30 min.
For vortexing of the IGPs, we are using a DPC MicroMix Plate Shaker with amplitude settings at 4, form settings at 3. If using a mini vortexer, we recommend vortexing at a low to moderate speed. For example, for a VWR standard mini vortexer, we do not recommend a vortex speed >6.
-
3
Aliquot 20 µl out into a new 96‐well Deepwell plate.
The purpose of this step is to preserve half of the samples as intact glycopeptides for intact‐glycopeptide analysis. If users are not interested in intact‐glycopeptide analysis, this step can be omitted. However, for the following steps, reaction volumes will have to be doubled.
-
4
Make the PNGaseF digestion buffer by adding 2 µl PNGaseF in 98 µl 100 mM TEAB.
According to protocols provided by New England BioLabs, 1 µl of PNGaseF contains 500 units of PNGaseF enzyme.
-
5
Add the PNGaseF/TEAB mixture in the original Deepwell plate that contains 20 µl of intact‐glycopeptide sample.
-
6
Let react at room temperature overnight using a shaker.
We recommend this reaction be performed in the original 96‐well Deepwell plate to save time and minimize sample loss during multiple sample transfer procedures.
Separating glycans from de‐N‐glycopeptides using C18 stage tips
There are multiple C18 stage tip protocols and manufacturers available to users. We pack our own C18 stage tips using a solid phase extraction disk (CDS EmporeTM).
-
7
Condition C18 stage tips with 100 µl MeOH, twice. Centrifuge at 1,500 rpm (211 × g) for 2 min.
-
8
Centrifuge 2 min at 1,800 rpm (304 × g) with 100 µl 50% ACN + 0.1% FA, twice.
-
9
Centrifuge 2 min under 1,800 rpm (304 × g) with 100 µl 0.1% FA, twice.
-
10
Load glycan/de‐N‐glycopeptide samples and centrifuge for 5 min at 1,000 rpm (94 × g), twice.
-
11
Wash for 2 min by centrifuging at 1,800 rpm (304 × g) with 100 µl 0.1% FA, twice.
-
12
Collect flow through from steps 10 and 11; this will be the glycan sample.
-
13
Elute de‐N‐glycopeptides using 75 µl 50% ACN + 0.1% FA, twice.
The specific parameters (time and spin rate) during stage tip procedures will vary according to the centrifuge and the tightness of the C18 packing material being packed. We highly recommend that users test out specific parameters prior to operating. We recommend not increasing the spin rate but increase the time of centrifuging, if parameters needed to be optimized.
Glycan purification using PGC NuTip carbon
-
14
Attach a PGC NuTip onto a 200‐µl pipet.
-
15
Aliquot 1 ml 80% ACN + 0.1% FA into an Eppendorf tube; denote as “conditioning buffer.”
-
16
Aliquot 1 ml 0.1% FA into an Eppendorf tube; denote as “binding buffer.”
-
17
Aspirate and disperse 50 µl conditioning buffer into an Eppendorf tube twenty times to condition the NuTip.
-
18
Aspirate and disperse 50 µl binding buffer into an Eppendorf tube twenty times to condition the NuTip.
-
19
Aspirate and disperse 50 µl of glycan samples twenty times to bind the glycan samples on the NuTip.
-
20
Aspirate and disperse 50 µl binding buffer into an Eppendorf tube twenty times to wash the glycan samples.
-
21
Aliquot 200 µl 80% ACN + 0.1% FA into an Eppendorf tube; denote as “releasing buffer.”
-
22
Aspirate and disperse 200 µl releasing buffer into an Eppendorf tube fifty times to elute glycan samples.
-
23
Dry down using a SpeedVac concentrator.
REAGENTS AND SOLUTIONS
Use HPLC‐grade water or double‐distilled water for all solutions.
Digestion buffer for C4‐tip
30 ml acetonitrile, Optima® LC/MS (Thermo Fisher Scientific, cat. no. A955‐4)
5 ml 1 M triethylammonium bicarbonate (MilliporeSigma, cat. no. T7408‐500ML)
65 ml water, Optima® LC/MS (Thermo Fisher Scientific, cat. no. W6‐4)
Prepare fresh prior to each experiment.
Triethylammonium acetate buffer (TAAB), 100 mM
20 ml 1 M triethylammonium acetate buffer (MilliporeSigma, cat. no. 90368‐500ML)
180 ml water, OptimaR LC/MS (Thermo Fisher Scientific, cat. no. W6‐4)
Reagent should be made fresh prior to each experiment.
Triethylammonium bicarbonate (TEAB), 50 mM
10 ml 1 M triethylammonium bicarbonate (MilliporeSigma, cat. no. T7408‐500ML)
190 ml water, OptimaR LC/MS (Thermo Fisher Scientific, cat. no. W6‐4)
Reagent should be made fresh prior to each experiment.
COMMENTARY
Background Information
Recently, the glycoproteomics field has drastically advanced, from initial discovery of several hundred intact glycopeptides to the detection of >20,000 unique intact glycopeptides (Shu et al., 2020). The mass spectrometry (MS) instrumentation, sample preparation techniques, and data analysis software for intact glycopeptide analysis are all coming of age, which have made characterization of large‐scale cohorts possible. With isobaric tags like isobaric tag for relative and absolute quantitation (iTRAQ), tandem mass tag (TMT), or downstream intact glycopeptide analysis software like GPQuest, Byonic, or MAGIC emerging, the rate‐limiting step seems to shift towards the sample preparation aspect. Therefore, implementation of high‐throughput sample processing for intact glycopeptide is a must. Over the years, high‐throughput glycoproteomics has been somewhat limited to glycosite analysis using hydrazide chemistry (Berven et al., 2010; Chen et al., 2013) or glycan profiling (Yang, Clark, Liu, Li, & Zhang, 2017). This protocol introduces a comprehensive workflow by combining two of our previous publications (Chen et al., 2020; Clark et al., 2019), such that users can process protein samples of intact glycopeptides in a high‐throughput manner. This protocol can offer rapid sample processing, while still retaining highly reproducible characteristics. Users can expect to obtain intact glycopeptide samples starting from protein within 3 days. The C4‐tip and C18/MAX‐tip can also be utilized for standard protein, bodily fluids, cell lysates, or tissue samples.
Critical Parameters and Troubleshooting
To successfully enrich glycopeptide samples, users must make sure of the following: During the sample binding procedure, users must be mindful that the pH of the binding condition is <3 to ensure optimum binding for both C4 and C18/MAX. When processing bodily fluids or tissue samples, it is critical to do a rigorous centrifuge step—3,000 rpm (900 × g) for 10 min‐to remove any sediments that would impede downstream sample processing. It is also crucial to consider the evaporation of organic solvents during the liquid handling system operation, especially during the intact‐glycopeptide enrichment steps (when 95% ACN + 1% TFA is used). Do not let high volumes of organic solvents remain excessively long in the liquid handling system; this can be avoided by using fresh eluant multiple times during Basic Protocol 2, steps 22 to 25. (For example, by using 3 × 5 cycles during these steps, organic solvents will evaporate more than if eluted in 1 × 15 cycles, thus users are encouraged to use 3 × 5 cycles instead of 1 × 15 cycles.) Furthermore, liquid will evaporate at a faster rate due to the temperature rising during the liquid handling system operation; this can be avoided by attaching a heater‐cooler thermoblock inside the liquid handling system.
Understanding Results
Glycan, glycosite, and intact‐glycopeptide analysis on the standard protein fetuin can be used for orthogonal verification of a glycosylation site using the C4‐tip and C18/MAX‐tip workflow.
To demonstrate the feasibility of the C4‐tip and C18/MAX‐tip workflow, we first used the standard protein fetuin, started from the protein level, to non‐glycosylated peptide, intact glycopeptide, glycan, and de‐N‐glycopeptide. After PNGaseF digest of N‐linked glycans, users can expect a deamidation change from asparagine (N) to aspartic acid (D). By comparison of de‐N‐glycopeptide with non‐glycosylated peptide, the glycosylation site can be further verified via comparison of an intact‐glycopeptide spectrum and a de‐glycopeptide spectrum (Fig. 3).
Figure 3.
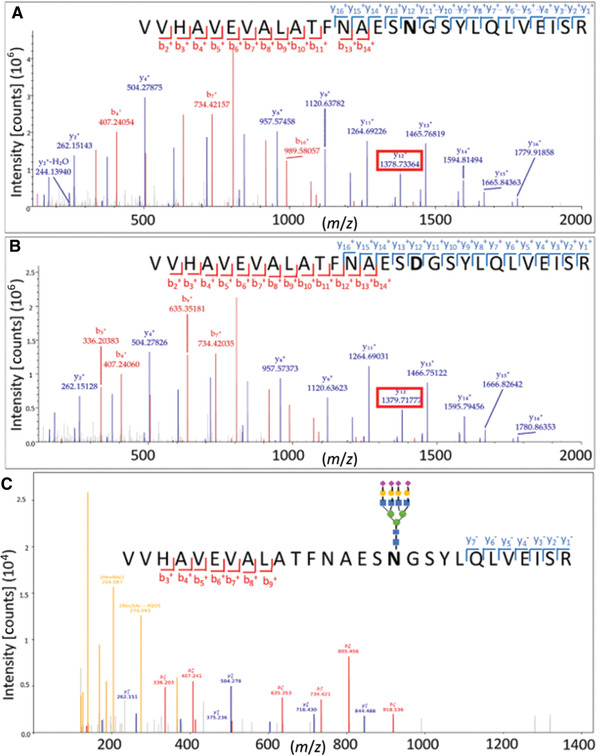
Comparison of (A) non‐glycosylated peptide, (B) de‐N‐glycopeptide, and (C) intact glycopeptide for standard protein fetuin, peptide: VVHAVEVALATFNAESN#GSYLQLVEISR. The site of glycosylation and Asn to Asp transition are indicated by #. Glycosylation site can be determined by observing +0.98 Da shift after PNGaseF digest, as seen in y12+ ion of these spectrums. Intact‐glycopeptide spectrum can provide orthogonal verification of a glycosylation site, while further providing glycan information. This figure is cited from our previous publication Chen et al., 2020.
Glycan, glycosite, and intact‐glycopeptide analysis on human urine
Urine is an attractive sample source for urological diseases because of its proximity, sample availability, and non‐invasiveness during sample collection. However, urine also contains several components that will confound proteomic analysis, such as a high concentration of urea, inorganic salts, or other biomolecules. Unlike traditional urine proteomics sample processing, use of C4 resin does not need additional sample pre‐processing, such as protein precipitation, buffer exchange, ultrafiltration, or ultracentrifugation. The above‐mentioned sample pre‐process methodologies cannot be readily adapted for high‐throughput urinary protein sample preparation; C4 resin, on the other hand, can be adapted into liquid handling systems with ease.
Besides standard protein, the C4‐tip and C18/MAX‐tip workflow can also be used for human bodily fluids, such as human serum, blood, or urine. Similar to fetuin, the user can expect to obtain non‐glycosylated peptides and intact glycopeptides by using the C4‐tip and C18/MAX‐tip workflow, while further PNGaseF digestion could provide orthogonal verification of glycosylation sites.
However, since normal urine protein concentration can vary from 0 to 14 mg/dL, one might expect the peptide or intact glycopeptide identification rate to vary according to the initial urine protein concentration.
Time Considerations
The C4‐tip (Basic Protocol 1) can be completed in ∼15 hr, which includes 1 hr of C4‐tip conditioning, 1.5 hr of sample loading, 1 hr of sample washing, 1 hr of reduction using TCEP, 1 hr of alkylation using IAA: Take out the tips that contain the bound proteins from the liquid handling systems, aliquot ∼200 µl of 50 mM TEAB on top of each C4‐tip, seal the C4‐tips that contain the bound proteins using Parafilm, and store at 4°C. TEAB and IAA can be dispersed the next morning right before enzymatic digestion.
The C18/MAX (Basic Protocol 2) can be completed in ∼6 hr, which includes 2 hr of C18/MAX‐tip conditioning, 1 hr of sample loading, 1 hr of sample washing and desalting, 0.5 hr of non‐glycosylated peptide elution, and 0.5 hr of intact glycopeptide elution.
Basic Protocol 3 can be achieved in ∼15 hr, which includes 12 hr of overnight PNGaseF digestion, 2 hr of C18 stage tips procedure for glycan/de‐N‐glycopeptide separation, and 1 hr of glycan purification.
Basic Protocols 1, 2, and 3 all include multiple procedures to dry samples using a SpeedVac concentrator. These procedures are not included in the time consideration calculations because different reaction volume, instrumentation, and sample size can contribute to the overall processing time.
Author Contributions
Shao‐Yung Chen: Conceptualization, investigation, methodology, project administration, writing: original draft, David Clark: Conceptualization, methodology, Hui Zhang: Conceptualization, funding acquisition, resources, supervision
Conflict of Interest
The authors declare no competing financial interest.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health, National Cancer Institute, the Early Detection Research Network (EDRN, U01CA152813), and the Clinical Proteomic Tumor Analysis Consortium (CPTAC, U24CA210985).
Chen, S., Clark, D. J., & Zhang, H. (2021). High‐throughput analyses of glycans, glycosites, and intact glycopeptides using C4‐and C18/MAX‐tips and liquid handling system. Current Protocols, 1, e186. doi: 10.1002/cpz1.186
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Literature Cited
- An, H. J., Miyamoto, S., Lancaster, K. S., Kirmiz, C., Li, B., Lam, K. S., … Lebrilla, C. B. (2006). Profiling of glycans in serum for the discovery of potential biomarkers for ovarian cancer. Journal of Proteome Research, 5(7), 1626–1635. doi: 10.1021/pr060010k. [DOI] [PubMed] [Google Scholar]
- Bennun, S. V., Hizal, D. B., Heffner, K., Can, O., Zhang, H., & Betenbaugh, M. J. (2016). Systems glycobiology: Integrating glycogenomics, glycoproteomics, glycomics, and other ‘omics data sets to characterize cellular glycosylation processes. Journal of Molecular Biology, 428(16), 3337–3352. doi: 10.1016/j.jmb.2016.07.005. [DOI] [PubMed] [Google Scholar]
- Berven, F. S., Ahmad, R., Clauser, K. R., & Carr, S. A. (2010). Optimizing performance of glycopeptide capture for plasma proteomics. Journal of Proteome Research, 9(4), 1706–1715. doi: 10.1021/pr900845m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J., Shah, P., & Zhang, H. (2013). Solid phase extraction of N‐linked glycopeptides using hydrazide tip. Analytical Chemistry, 85(22), 10670–10674. doi: 10.1021/ac401812b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S.‐Y., Dong, M., Yang, G., Zhou, Y., Clark, D. J., Lih, T. M., … Zhang, H. (2020). Glycans, glycosite, and intact glycopeptide analysis of N‐linked glycoproteins using liquid handling systems. Analytical Chemistry, 92(2), 1680–1686. doi: 10.1021/acs.analchem.9b03761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, D. J., Hu, Y., Schnaubelt, M., Fu, Y., Ponce, S., Chen, S. Y., … Zhang, H. (2019). Simple tip‐based sample processing method for urinary proteomic analysis. Analytical Chemistry, 91(9), 5517–5522. doi: 10.1021/acs.analchem.8b05234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creese, A. J., & Cooper, H. J. (2012). Separation and identification of isomeric glycopeptides by high field asymmetric waveform ion mobility spectrometry. Analytical Chemistry, 84(5), 2597–2601. doi: 10.1021/ac203321y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan, A., Cui, J., Chatterton, D. E. W., Deane, K. D., Hazen, M. M., Brintnell, W., … Lee, D. M. (2010). Aberrant IgG galactosylation precedes disease onset, correlates with disease activity, and is prevalent in autoantibodies in rheumatoid arthritis. Arthritis and Rheumatism, 62(8), 2239–2248. doi: 10.1002/art.27533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Q., Kowalski, M. P., Mastali, M., Parker, S. J., Sobhani, K., Van Den Broek, I., … Van Eyk, J. E. (2018). Highly reproducible automated proteomics sample preparation workflow for quantitative mass spectrometry. Journal of Proteome Research, 17(1), 420–428. doi: 10.1021/acs.jproteome.7b00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujitani, N., Furukawa, J., Araki, K., Fujioka, T., Takegawa, Y., Piao, J., … Shinohara, Y. (2013). Total cellular glycomics allows characterizing cells and streamlining the discovery process for cellular biomarkers. Proceedings of the National Academy of Sciences of the United States of America, 110(6), 2105–2110. doi: 10.1073/pnas.1214233110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Z., Liu, X., Li, M., Shao, C., Tao, J., Sun, W., & Li, M. (2015). Differential urinary glycoproteome analysis of type 2 diabetic nephropathy using 2D‐LC‐MS/MS and iTRAQ quantification. Journal of Translational Medicine, 13(1), 1–17. doi: 10.1186/s12967-015-0712-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim, A., Nilsson, J., Rüetschi, U., Hesse, C., & Larson, G. (2012). Human urinary glycoproteomics; attachment site specific analysis of N‐ and O‐linked glycosylations by CID and ECD. Molecular and Cellular Proteomics, 11(4), 1–17. doi: 10.1074/mcp.M111.013649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higel, F., Seidl, A., Sörgel, F., & Friess, W. (2016). N‐glycosylation heterogeneity and the influence on structure, function and pharmacokinetics of monoclonal antibodies and Fc fusion proteins. European Journal of Pharmaceutics and Biopharmaceutics, 100, 94–100. doi: 10.1016/j.ejpb.2016.01.005. [DOI] [PubMed] [Google Scholar]
- Hu, Y., Pan, J., Shah, P., Ao, M., Thomas, S. N., Liu, Y., … Shi, Z. (2020). Integrated proteomic and glycoproteomic characterization of human high‐grade serous ovarian carcinoma. Cell Reports, 33(3)108276. doi: 10.1016/j.celrep.2020.108276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Y., Shah, P., Clark, D. J., Ao, M., & Zhang, H. (2018). Reanalysis of global proteomic and phosphoproteomic data identified a large number of glycopeptides. Analytical Chemistry, 90(13), 8065–8071. doi: 10.1021/acs.analchem.8b01137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji, H., Saito, H., Yamauchi, Y., Shinkawa, T., Taoka, M., Hirabayashi, J., … Isobe, T. (2003). Lectin affinity capture, isotope‐coded tagging and mass spectrometry to identify N‐linked glycoproteins. Nature Biotechnology, 21(6), 667–672. doi: 10.1038/nbt829. [DOI] [PubMed] [Google Scholar]
- Kang, P., Mechref, Y., Klouckova, I., & Novotny, M. V. (2005). Solid‐phase permethylation of glycans for mass spectrometric analysis. Rapid Communications in Mass Spectrometry, 19(23), 3421–3428. doi: 10.1002/rcm.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara, R., Ortega, F., Rosa‐Fernandes, L., Guimarães, V., Quina, D., Nahas, W., … Palmisano, G. (2018). Distinct urinary glycoprotein signatures in prostate cancer patients. Oncotarget, 9(69), 33077–33097. doi: 10.18632/oncotarget.26005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury, G. A., Baliban, R. C., & Floudas, C. A. (2011). Proteome‐wide post‐translational modification statistics: Frequency analysis and curation of the swiss‐prot database. Scientific Reports, 1(1), 90. doi: 10.1038/srep00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, M. D., de Graaf, E. L., Sonneveld, M. E., Plomp, H. R., Nouta, J., Hoepel, W., … Vidarsson, G. (2020). Afucosylated IgG characterizes enveloped viral responses and correlates with COVID‐19 severity. Science, 371(6532), eabc8378. doi: 10.1126/science.abc8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandrowski, U., Zahedi, R. P., Moebius, J., Walter, U., & Sickmann, A. (2007). Enhanced N‐glycosylation site analysis of sialoglycopeptides by strong cation exchange prefractionation applied to platelet plasma membranes. Molecular and Cellular Proteomics, 6(11), 1933–1941. doi: 10.1074/mcp.M600390-MCP200. [DOI] [PubMed] [Google Scholar]
- Nagai‐Okatani, C., & Minamino, N. (2016). Aberrant glycosylation in the left ventricle and plasma of rats with cardiac hypertrophy and heart failure. PLoS ONE, 11(6), e0150210. doi: 10.1371/journal.pone.0150210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson, J., Rüetschi, U., Halim, A., Hesse, C., Carlsohn, E., Brinkmalm, G., & Larson, G. (2009). Enrichment of glycopeptides for glycan structure and attachment site identification. Nature Methods, 6(11), 809–811. doi: 10.1038/nmeth.1392. [DOI] [PubMed] [Google Scholar]
- Palmisano, G., Lendal, S. E., Engholm‐Keller, K., Leth‐Larsen, R., Parker, B. L., & Larsen, M. R. (2010). Selective enrichment of sialic acid‐containing glycopeptides using titanium dioxide chromatography with analysis by HILIC and mass spectrometry. Nature Protocols, 5(12), 1974–1982. doi: 10.1038/nprot.2010.167. [DOI] [PubMed] [Google Scholar]
- Shah, P., Yang, S., Sun, S., Aiyetan, P., Yarema, K. J., & Zhang, H. (2013). Mass spectrometric analysis of sialylated glycans with use of solid‐phase labeling of sialic acids. Analytical Chemistry, 85(7), 3606–3613. doi: 10.1021/ac3033867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajahan, A., Supekar, N. T., Heiss, C., Ishihara, M., & Azadi, P. (2017). Tool for rapid analysis of glycopeptide by permethylation via one‐pot site mapping and glycan analysis. Analytical Chemistry, 89, 56. doi: 10.1021/acs.analchem.7b01730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu, Q., Li, M., Shu, L., An, Z., Wang, J., Lv, H., … Yang, F. (2020). Large‐scale identification of n‐linked intact glycopeptides in human serum using HILIC enrichment and spectral library search. Molecular and Cellular Proteomics, 19(4), 672–689. doi: 10.1074/mcp.RA119.001791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, S., Shah, P., Eshghi, S. T., Yang, W., Trikannad, N., Yang, S., … Zhang, H. (2016). Comprehensive analysis of protein glycosylation by solid‐phase extraction of N‐linked glycans and glycosite‐containing peptides. Nature Biotechnology, 34(1), 84–88. doi: 10.1038/nbt.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, Y., Zhou, Y., Elliott, S., Aebersold, R., & Zhang, H. (2007). Solid‐phase extraction of N‐linked glycopeptides. Nature Protocols, 2(2), 334–339. doi: 10.1038/nprot.2007.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada, Y., Tajiri, M., & Yoshida, S. (2004). Hydrophilic affinity isolation and MALDI multiple‐stage tandem mass spectrometry of glycopeptides for glycoproteomics. Analytical Chemistry, 76(22), 6560–6565. doi: 10.1021/ac049062o. [DOI] [PubMed] [Google Scholar]
- Yang, G., Höti, N., Chen, S.‐Y., Zhou, Y., Wang, Q., Betenbaugh, M., & Zhang, H. (2020). One‐step enrichment of intact glycopeptides from glycoengineered chinese hamster ovary cells. Frontiers in Chemistry, 8, 240. doi: 10.3389/fchem.2020.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H., Yang, C., & Sun, T. (2018). Characterization of glycopeptides using a stepped higher‐energy C‐trap dissociation approach on a hybrid quadrupole orbitrap. Rapid Communications in Mass Spectrometry, 32(16), 1353–1362. doi: 10.1002/rcm.8191. [DOI] [PubMed] [Google Scholar]
- Yang, S., Clark, D., Liu, Y., Li, S., & Zhang, H. (2017). High‐throughput analysis of N‐glycans using AutoTip via glycoprotein immobilization. Scientific Reports, 7(1), 10216. doi: 10.1038/s41598-017-10487-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, W., Shah, P., Hu, Y., Toghi Eshghi, S., Sun, S., Liu, Y., & Zhang, H. (2017). Comparison of enrichment methods for intact N‐ and O‐linked glycopeptides using strong anion exchange and hydrophilic interaction liquid chromatography. Analytical Chemistry, 89(21), 11193–11197, doi: 10.1021/acs.analchem.7b03641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, Z., Mao, Y., Clausen, H., & Vakhrushev, S. Y. (2019). Glyco‐DIA: A method for quantitative O‐glycoproteomics with in silico‐boosted glycopeptide libraries. Nature Methods, 16(9), 902–910. doi: 10.1038/s41592-019-0504-x. [DOI] [PubMed] [Google Scholar]
- Yu, Q., Wang, B., Chen, Z., Urabe, G., Glover, M. S., Shi, X., … Li, L. (2017). Electron‐transfer/higher‐energy collision dissociation (EThcD)‐enabled intact glycopeptide/glycoproteome characterization. Journal of the American Society for Mass Spectrometry, 28(9), 1751–1764. doi: 10.1007/s13361-017-1701-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H., Li, X.‐J., Martin, D. B., & Aebersold, R. (2003). Identification and quantification of N‐linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nature Biotechnology, 21(6), 660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- Zhou, J., Yang, W., Hu, Y., Hö, N., Liu, Y., Shah, P., … Zhang, H. (2017). Site‐specific fucosylation analysis identifying glycoproteins associated with aggressive prostate cancer cell lines using tandem affinity enrichments of intact glycopeptides followed by mass spectrometry, Analytical Chemistry, 89(14), 7623–7630. doi: 10.1021/acs.analchem.7b01493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinska, D. F., Gnad, F., Wiśniewski, J. R., & Mann, M. (2010). Precision mapping of an in vivo N‐glycoproteome reveals rigid topological and sequence constraints. Cell, 141(5), 897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.