

Abstract
Up to now, the field of liquid biopsies has focused on circulating tumour DNA and cells, though extracellular vesicles (EVs) have been of increasing interest in recent years. Thus, reported sources of tumour‐derived nucleic acids include leukocytes, platelets and apoptotic bodies (AB), as well as large (LEV) and small (SEV) EVs. Despite these competing claims, there has yet to be a standardized comparison of the tumour‐derived DNA associated with different components of blood. To address this issue, we collected twenty‐three blood samples from seventeen patients with pancreatic cancers of known mutant KRAS G12 genotype, and divided them into two groups based on the time of patient survival following sampling. After collecting red and white blood cells, we subjected 1 ml aliquots of platelet rich plasma to differential centrifugation in order to separate the platelets, ABs, LEVs, SEVs and soluble proteins (SP) present. We then confirmed the enrichment of specific blood components in each differential centrifugation fraction using electron microscopy, Western blotting, nanoparticle tracking analysis and bead‐based multiplex flow cytometry assays. By targeting wild type and tumour‐specific mutant KRAS alleles using digital PCR, we found that the levels of mutant KRAS DNA were highest in association with LEVs and SEVs early, and with SEVs and SP late in disease progression. Importantly, we established that SEVs were the most enriched in tumour‐derived DNA throughout disease progression, and verified this association using size exclusion chromatography. This work provides important direction for the rapidly expanding field of liquid biopsies by supporting an increased focus on EVs as a source of tumour‐derived DNA.
Keywords: apoptotic bodies, cancer diagnostics, ctDNA, digital PCR, exosomes, extracellular vesicles, liquid biopsy, microvesicles, pancreatic cancer, platelets
1. INTRODUCTION
The ability to access tumour‐derived DNA from the peripheral blood of cancer patients can provide real‐time information about an individual's disease without the need for invasive biopsy. This has raised the hope of future applications in early detection, disease monitoring and therapy targeting, though it is yet to have a profound effect on cancer patient care (Wan et al., 2017).
One of the greatest challenges facing the field of liquid biopsies is the minute amounts of tumour‐derived DNA present in blood, particularly at early stages of disease (Wan et al., 2017). To overcome this, PCR based methods, such as digital PCR, have been developed in order to reveal specific mutant alleles present at frequencies below 1 in 1000 molecules (Lee et al., 2019). Unfortunately, despite recent advances, untargeted and multiplexed detection methods are untenable at such low allele frequencies. (Zviran et al., 2020). To address the issue in this context requires a deeper understanding of the components of blood where tumour DNA is most concentrated (Foroni et al., 2020).
Blood is an incredibly complex mixture of different sized components, several of which have been suggested to be reservoirs of neoplastic material. For instance, circulating tumour cells can be of diverse sizes > 5 μm and are derived directly from tumours, but are rare and occur too late during disease progression to have broad diagnostic utility (Lin et al., 2018). In contrast, leukocytes, which are > 10 μm in diameter (Chennakrishnaiah et al., 2018), and platelets, which range from ∼1‐4 μm (Best et al., 2019), are derived from the haematopoietic system and have been suggested to take up tumour material from the extracellular environment and make it accessible in peripheral blood. However, the findings in leukocytes have yet to be confirmed in humans, while those regarding platelets have been questioned within the field (Brinkman et al., 2020; Chennakrishnaiah et al., 2018). Regardless, circulating tumour DNA (ctDNA) has been the most cited target of liquid biopsy research, and circulates in soluble protein (SP) complexes as single histone octamers, which are 11 nm across (Wan et al., 2017). Although it remains to be confirmed, ctDNA is thought to be released from dying cells, which may explain why its prevalence increases as cancer progresses towards terminal illness (Cescon et al., 2020; Lee et al., 2019).
When assessing sources of tumour‐derived material, one consideration is that most plasma used for diagnostics has undergone centrifugation (Cescon et al., 2020; Wan et al., 2017). This is important because centrifugation removes cells and different classes of extracellular vesicles (EV) dependent on the specific conditions used (Livshits et al., 2015). EVs are a diverse group of lipid membrane enclosed particles that have been shown to contain tumour‐derived DNA and are classified based on their biogenesis: Apoptotic bodies (AB) are formed during programmed cell death and range from ∼1‐4 μm (Bergsmedh et al., 2001). Microvesicles are released by membrane shedding at the cell surface and are ∼200 nm‐1 μm (Vagner et al., 2018). Lastly, exosomes are < 200 nm in size and derived by inward budding of the endolysosomal system. Although there remains debate as to the ability of exosomes to contain genomic DNA, there have been several studies demonstrating their utility and benefits in diagnostics (Allenson et al., 2017; Bernard et al., 2019; Jeppesen et al., 2019; Yokoi et al., 2019).
Despite the benefits of pre‐enriching tumour‐derived DNA before analysis, its abundance across the different components of blood has yet to be comprehensively compared. Pancreatic ductal adenocarcinoma (PDAC) is an ideal disease to study in this context due to its high levels of ctDNA and that over 70% of patients carry one of two specific mutations in KRAS codon 12 (Lee et al., 2019; Waters & Der, 2018). To study circulating DNA dynamics during disease progression, we separated the different components of blood from 23 samples, provided by 17 PDAC patients with known KRAS mutations, and divided them into two groups based on the time of patient survival after sampling. Using differential centrifugation and digital PCR, we have compared the amount of mutant and wild type KRAS DNA copies associated with red (RBC) and white blood cells (WBC), platelets, ABs, large EVs (LEV), small EVs (SEV), SP and a 10 kD‐filter flow through (FT) as a negative control. This analysis revealed that LEVs and SEVs were associated with the greatest amount of mutant KRAS DNA early in disease progression, while SEVs and SP contained the most in PDAC patients with advanced disease. Importantly, SEVs were associated with the greatest enrichment of mutant over wild type KRAS of all blood components, including platelet poor plasma (PPP). Thus, this comprehensive comparison suggests future liquid biopsy research should focus on EVs as the primary blood‐borne source of tumour‐derived DNA.
2. MATERIAL AND METHODS
2.1. Research subjects
Patients with histologically confirmed pancreatic adenocarcinoma were recruited at the outpatient clinic of the Department for Gastrointestinal Cancer at Tema Cancer, Karolinska University Hospital Comprehensive Cancer Center. Patients > 18 years of age with locally advanced or metastatic disease who had no other concurrent malignancy were eligible for inclusion. Blood samples (∼16 ml) from all patients and healthy volunteers were obtained at inclusion in the study. If possible, repeat samples from the same patient were obtained in conjunction with planned follow‐up visits at the hospital. Patient and tumour characteristics were collected and managed using REDCap electronic data capture tools hosted at Karolinska Institutet (Harris et al., 2019). The clinical stage of the disease was classified according to the AJCC Cancer Staging Manual, 8th Edition (Amin et al., 2017). The performance status at inclusion was evaluated using the ECOG Scale (Oken et al., 1982). All samples and data in this study were pseudonymized. All individuals provided written informed consent before participation and research was approved by the Swedish Ethical Review Authority (Diary number: 2017/912‐31; 2018/952‐32).
2.2. Separating samples taken early and late in disease progression
In order to separate late‐stage PDAC patient samples from those taken earlier in disease progression, we calculated the number of days of patient survival after the date of each sampling (Supplementary Table S1) and divided them into two balanced groups (early and late, cut‐off date 250 days). Overall survival was measured from the date the sample was obtained to the date the patient deceased. For statistics, three patients who were alive were censored at last follow‐up and analysed in the early‐stage PDAC group.
2.3. Differential centrifugation and PPP
Whole blood was drawn into 4.5 ml 3.2% sodium citrate containing glass tubes (BD 369714). Within 2 h, samples were centrifuged at 120 × g for 20 min, 1 ml aliquots of plasma were made, 50 μl of RBCs were sampled from the cell pack and 25 μl of WBCs were removed from the buffy coat. Subsequently, plasma was serially centrifuged at 360 × g for 20 min (platelets), 3000 × g for 10 min (ABs) and 20000 × g for 10 min (LEVs), with the supernatant removed after each centrifugation and transferred to a clean microcentrifuge tube. The remaining plasma was then pooled and diluted in 0.2 μm filtered PBS prior to ultracentrifugation at 100000 × g for 90 min. After removing the SP supernatant, the SEV ultracentrifuge pellets were resuspended in 0.2 μm filtered PBS to prepare the same number of aliquots as for the other plasma derived fractions. The SP supernatant was then centrifuged at 4000 × g on 10 kD centrifugation filters (Amicon UFC901096) to concentrate the same number of 100 μl aliquots as the other plasma derived fractions. Finally, 100 μl aliquots of filter FT were also taken as negative controls. To analyse PPP, which is often used in clinics, we combined the AB, LEV, SEV and SP fractions from each early patient sample except P9, which did not have sufficient material remaining.
2.4. Electron microscopy
EV pellets from a healthy individual were diluted 1:10 in PBS and 8 μl were applied to Formvar/carbon‐coated 200‐mesh nickel grids (Agar Scientific, UK) for 1 min before droplets were dried using filter paper and washed with 8 μl distilled water for 1 min. The grids were then stained twice with 8 μl of 1% uranyl acetate for 1 min, followed by 1 min washes with distilled water and finally 10 min drying. Transmission electron microscope images of representative EVs were acquired using a Tecnai 12 Spirit BioTwin microscope (FEI Company) at 100 kV and digital images were captured using a 2kx2k Veleta CCD camera (Olympus Soft Imagining Solutions).
2.5. Western blot
One aliquot of each blood fraction from a healthy individual was run on GE Healthcare SpinTrap columns according to the manufacturer's instructions (GE Healthcare 28‐9480‐20). Samples were boiled at 95°C for 5 min in 0.5 M DTT, 0.4 M Sodium Carbonate, 8% SDS, 10% glycerol and sample dye before loading into 10‐well 4%–12% NuPAGE Bis‐Tris protein gels (Invitrogen NP0321BOX). Gels were run for 90 min at 120 mV and transferred to a nitrocellulose membrane using the iBlot system (Invitrogen LC2009). Membranes were incubated in Blocking Reagent (Li‐Cor 927–70001) for 1 h at room temperature, then antibodies in Blocking Reagent/PBST at 4°C overnight on a rocking tray. The antibodies used for this assay were Histone H3 (Santa Cruz FL‐136, 1:250), CD42A (Miltenyi 130‐100‐960, 1:150), cleaved‐CASP9 (Cell Signalling 9505S, 1:1000), BAX (Cell Signalling 2772S, 1:1000), CD9 (Abcam ab92726, 1:2000) and CD81 (Santa Cruz sc‐9158, 1:200). Membranes were washed 3 × 15 min in PBST before incubation with secondary antibodies in PBST (Li‐Cor 926–68020, 926–68071, 926–68079, 1:15 000). Signals were imaged on a Li‐Cor Odyssey Imager and exported from Li‐Cor Image Studio 5.2 software. The images displayed are representative of results obtained from at least two different individuals.
2.6. Multiplex bead‐based EV surface protein profiling by flow cytometry
Prior to analysis, the 10 μl pellets of each non‐cell fraction and 10 μl of SP from a healthy individual were thawed and diluted in 0.2 μm filtered PBS (Gibco Invitrogen, pH 7.4) to a volume of 60 μl, while the platelet pellet was further diluted 1:10. All fractions were then subjected to bead‐based multiplex EV analysis (MACSPlex Exosome Kit, human, Miltenyi Biotec) using an optimized workflow as described previously (Wiklander et al., 2018). In brief, a final volume of 60 μl of each fraction was either loaded directly (AB, MV, EX and SP) or after appropriate dilution (platelets 1:10) onto wells of a pre‐wet and drained MACSPlex 96‐well 0.22 μm filter plate before 10 μl of MACSPlex Exosome Capture Beads were added to each well. Filter plates were incubated overnight at 450 rpm. Beads were washed by adding 200 μl of MACSPlex Buffer (MBP) to each well and the filter plate was put on a vacuum manifold with vacuum applied at ‐100 mBar (Sigma‐Aldrich, Germany) until all wells were drained. Next, 135 μl of MBP was added and EVs bound to capture beads were counterstained with a mix of APC‐conjugated anti‐CD9, anti CD63 and anti‐CD81 detection antibodies (5 μl each). Plates were incubated at 450 rpm for 1 h and then washed by adding 200 μl MPB to each well followed by draining on a vacuum manifold. After another washing step with 200 μl MPB, incubation at 450 rpm for 15 min and draining all wells again on a vacuum manifold, 150 μl of MPB was added to each well, beads were re‐suspended and transferred to a V‐bottom 96‐well microtiter plate (Thermo Scientific, USA) for flow cytometryic analysis on a MACSQuant Analyzer 10 flow cytometer (Miltenyi Biotec, Germany). FlowJo software (v10, FlowJo LLC) was used to analyse flow cytometric data according to the manufacturer's recommendations. Median fluorescence intensities (MFI) for all 39 capture bead subsets (37 specific markers and two internal IgG control) were background corrected by subtracting respective MFI values from matched non‐EV containing PBS controls that were otherwise treated exactly like EV‐containing samples. Data from CD56, CD105, CD326 and SSEA‐4 capture bead populations was excluded from analysis due to unusually high levels of background in samples and controls for one assay lot used.
2.7. DNA purification and quantification
DNA was extracted from aliquots of each blood fraction or reconstituted PPP by resuspending to a total volume of 500 μl in SDS lysis buffer (1% SDS, 1 M Tris‐HCl, 10 mM EDTA + 0.2 mg/ml Proteinase K) and incubating at 55°C for 30 min. 500 μl of 1:1:1 Phenol:Chloroform:Isoamyl alcohol was then added to each tube, shaken vigorously for 1 min and centrifuged at 12,000 × g for 5 min. A 400 μl of the aqueous phase was transferred to a tube containing 400 μl isopropanol, 40 μl 3 M sodium acetate pH 5.5 and 2 μl of Pellet Paint (Merck 69049). This was mixed thoroughly and incubated at ‐20°C overnight. After centrifugation for 30 min at 20000 × g at 4°C, the supernatant was removed and the pellet was washed twice with 500 μl 70% ethanol, followed by centrifugation at 20000 × g for 10 min. Pellets were dried at room temperature for 30 min and resuspended in 15 μl of Qiagen Elution Buffer. The DNA present in each fraction was measured using a Qubit High Sensitivity DNA Assay (Thermo Fisher Q32854).
2.8. Genotyping
To determine the KRAS genotype of each patient, histology slides with high tumour content were selected. The corresponding formalin‐fixed paraffin‐embedded tumour tissue samples were retrieved from the pathology archive, sectioned and DNA was extracted with the QIAamp DNA FFPE Tissue Kit (Qiagen 56404). Isolated genomic DNA was quantified with the Qubit High Sensitivity DNA Assay (Thermo Fisher Q32854) and sequenced at the Clinical Genomics facility at SciLifeLab (Solna, Sweden), using their GMCK Solid Cancer Panel. Each patient's KRAS status was determined from this. For those patients where sequencing was not successful due to insufficient material (P9, P12, P13 and P19), we used digital PCR on numerous blood components to target the KRAS G12D and G12V variants and determine their presence. Robust signals for G12D in P19 and for G12V in P9 allowed us to proceed with these.
2.9. Size exclusion chromatography (SEC)
Two aliquots of patient 1, 6, or 17–19 SEV or patient 1, 6 or 19 SP fractions were run on qEVoriginal 70 nm SEC columns (Izon SP1) according to the manufacturer's instructions with 0.2 μm filtered PBS. Briefly, after priming the column with PBS, samples were loaded, followed by 10 ml PBS. 3 ml of PBS was discarded before five 1.5 ml fractions were collected sequentially. Since nanoparticle tracking analysis (NTA) consumes significant amounts of sample, data for Figure 2e was produced using healthy control SEVs. Protein was measured for all patient samples to ensure it fit the pattern displayed, and DNA was purified as detailed above.
FIGURE 2.
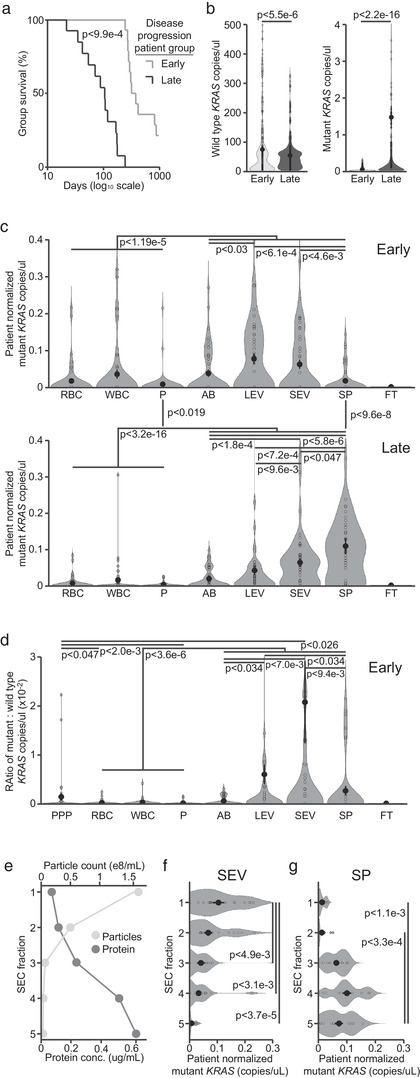
Blood components are associated with distinct amounts of wild type and mutant KRAS DNA in PDAC patients. (a) Kaplan‐Meier curves showing the days of survival following blood sampling for 14 early and 13 late group samples. (b) Copies/μl dPCR reaction mix of wild type (left) and mutant (right) KRAS DNA found in all blood components of early and late group patients. (c) Patient normalized copies/μl of mutant KRAS DNA found in each blood component in early (top) and late (bottom) group patients. (d) Ratio of mutant : wild type KRAS DNA copies/μl for each blood component and platelet poor plasma (PPP) in early patients. (e) Particle count and protein concentration for the five SEC fractions derived from two pooled SEV fractions. (f) Copies/μl of mutant KRAS DNA found in each SEC fraction of patient SEVs. (g) Copies/μl of mutant KRAS DNA found in each SEC fraction of patient SP. Statistics for a&b are derived from two‐tailed unequal variance t‐tests between indicated groups. Statistics for c–g are derived from two‐tailed unpaired Mann‐Whitney‐Wilcoxon tests between indicated groups
2.10. Digital PCR
The QuantStudio 3D Digital PCR 20k Chip system was used to run the Thermo Fisher Taqman Hs000000050_rm (KRAS G12V) or Hs000000051_rm (KRAS G12D) assay as appropriate for each patient according to the manufacturer's instructions. Each sample was run on two separate chips, which together consumed the DNA extracted from one whole fraction aliquot. Each chip was analysed in two orientations on a QuantStudio 3D Digital PCR System for a total of four readings per sample. Data was analysed using the QuantStudio 3D AnalysisSuite. Thresholds were set using universal standards for all fractions from each individual to call only single positive wells for FAM and VIC. The copies/μl presented in Supplementary Table S1 and Figure 2b represent the copies of DNA present per μl of dPCR reaction mix. This can be multiplied by 22 in order to approximate the copies of mutant or wild type KRAS present in each blood component from 1 ml of patient plasma, 50 μl RBCs or 25 μl WBCs. Copies/μl were patient normalized by dividing the values for each individual sample by the total amount of DNA for the patient they derived from. The ratio of mutant to wild type KRAS copies/μl was calculated within each individual chip reading.
2.11. Statistics
Statistical comparisons between group survival and wild type DNA levels were performed using two‐tailed unequal‐variance t‐tests. Comparisons between mutant DNA levels or ratios of mutant : wild type DNA were performed using two‐tailed unpaired Mann‐Whitney‐Wilcoxon tests, as these data sets did not exhibit a normal distribution pattern. All p‐values are displayed according to the figure legends and lines inset in figures.
3. RESULTS
To perform a comprehensive comparison of the potential DNA‐bearing components in circulation, we devised a differential centrifugation protocol based on a combination of previously published work (Figure 1a; Best et al., 2019; Livshits et al., 2015). To characterize the blood fractions isolated and confirm their hypothesized contents, we began by performing transmission electron microscopy on the EV fractions. This analysis confirmed an enrichment of huge vesicles (1‐4 um) in the AB, and large EVs (150 nm‐1 μm) in the LEV, fractions. These classes of EV were absent in SEV samples, which instead contained abundant small EVs (50‐150 nm) (Figure 1b). Next, we implemented Western blotting against known markers of cellular nuclei (H3; Senshu et al., 1985), platelets (CD42A; Berndt et al., 1985) and apoptosis (cleaved‐CASP9 and BAX; Kuida et al., 1998; Westphal et al., 2014), as well as one microvesicle (CD9) and one exosome (CD81) enriched tetraspanin (Figure 1c; Kowal et al., 2016). These analyses suggested that our differential centrifugation protocol efficiently enriched for the desired blood components within the hypothesized fractions.
FIGURE 1.
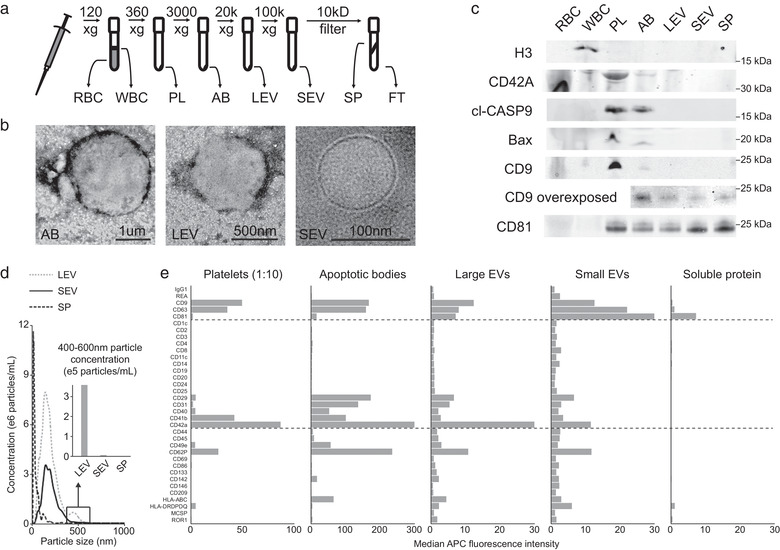
Differential centrifugation of whole blood efficiently separates different cell and vesicle populations. (a) Schematic of the differential centrifugation protocol used to separate red (RBC) and white blood cells (WBC), platelets (PL), apoptotic bodies (AB), large (LEV) and small extracellular vesicles (SEV), as well as soluble protein (SP) and flow through (FT). (b) Representative transmission electron microscopy images of vesicles enriched in AB, LEV and SEV fractions. Corresponding scale bars are inset. (c) Representative Western blots of blood components for histone H3 (H3), CD42A, cleaved Caspase 9 (CASP9), BAX, CD9 and CD81. A CD9 blotted membrane with vesicular and SP components was additionally overexposed. Molecular weight is indicated beside each blot. (d) Nanoparticle tracking analysis data showing the concentration of particles/ml at sizes ranging from 0–1 μm in LEV, SEV and SP fractions. Inset is a quantification of the concentration of particles from 400–600 nm found in each fraction. (d) Median CD9/63/81 fluorescence for 35 capture antibody coated beads from the MACSplex vesicle surface protein flow cytometry assay in the different blood components. The platelet fraction was diluted 1:10 before analysis
To confirm the distinct particle sizes within the LEV, SEV and SP fractions, we performed NTA on these components. This assay demonstrated that LEVs contained more particles between 400 and 600 nm in size than SEVs, while SP contained very few particles of any size (Figure 1d). To differentiate between proteins in soluble form and those present on an EV surface, we turned to a bead‐based multiplex flow cytometry assay (Wiklander et al., 2018). This assay confirmed that the relative enrichments of CD42A, CD9 and CD81 demonstrated by Western blotting were reflective of their abundances on EVs and platelets. In contrast, Western blotting implied the presence of higher levels of CD9 and CD81 in SP than the flow cytometry method requiring dual surface binding (Figure 1e).
To begin our clinical investigation into the associations of circulating tumour‐derived DNA at different stages of disease progression, we recruited a cohort of 21 PDAC patients. We then characterized their clinical presentation, TNM stage, ECOG performance status and CA19‐9 levels (Supplementary Table S1) (Amin et al., 2017; Oken et al., 1982). We obtained 27 blood samples from these patients and separated them into two groups, termed early‐ and late‐stage progression, based on the time of patient survival following sampling (Figure 2a, Supplementary Table S1). This division was supported by the clinical characteristics recorded for each patient (Supplementary Table S1). To assess the amounts of tumour and healthy cell‐derived DNA associated with each blood component, we utilized digital PCR systems that could distinguish the wild type and mutant KRAS sequences in the 11 early and 12 late progression samples from the 17 patients with confirmed G12D or G12V mutations. Overall, the early patient group had significantly more total copies of circulating wild type KRAS DNA than the late group, while the opposite was true for mutant KRAS DNA (Figure 2b). To compare the relative distribution of DNA in circulation between patients with varying absolute levels, we normalized the copies/μl of DNA in each blood component to the total amount in all fractions for each patient. In all patients, wild type KRAS DNA was most abundant in WBCs, followed by platelets and RBCs, and finally the vesicular and SP fractions, though these differences were less pronounced in late‐stage patients (Supplementary Figure S1A, Supplementary Table S1). In contrast, mutant KRAS DNA was least prevalent in platelets and blood cells, while vesicles and SP contained significantly more, in both patient groups (Figure 2c). Interestingly, LEVs and SEVs were linked with significantly more mutant KRAS DNA than other blood fractions in early patients, while SP contained the most in late‐stage patients (Figure 2c). However, this change was solely due to a significant increase in SP mutant KRAS in late patients, as SEVs remained associated with significantly more than any other blood fraction in this patient group (Figure 2c). Importantly, the total amount of DNA ( 25.5 vs. cutpoint 22.2 ng/ml) and the copies of mutant KRAS ( 46.2 vs. cutpoint 86.4 copies/ml) in SP were comparable to those from previous work on PDAC ctDNA (Supplementary Table S1; Liu et al., 2019).
To assess the enrichment of tumour‐derived DNA within each blood component, and compare this with PPP, we analysed the ratio of mutant to wild type KRAS DNA in each sample. In the early patient group, this was over 3‐fold increased and significantly higher in SEVs than any other fraction, including PPP (Figure 2d). In late‐stage patients, LEVs, SEVs and SP all showed similar enrichments of mutant DNA (Supplementary Figure S1B). To confirm the association of SEV DNA with vesicles, and free histone‐bound DNA with SP, we performed SEC to separate them components within these fractions. We found that both mutant and wild type DNA eluted predominantly with the SEC fractions containing the most particles for SEVs, and the most protein for SP (Figure 2e‐g; Supplementary Figure S1C‐D).
4. DISCUSSION
Liquid biopsy aims to provide information about solid tumours in a non‐invasive manner. However, clinical application has been lagging due to the challenge of detecting tumour‐derived material, particularly at early stages of disease. Thus, finding the component of blood associated with the highest concentration of tumour‐derived DNA is important for future progress in the field of liquid biopsies. In this work, we attempt to address this systematically by separating blood from PDAC patients into seven well described components and analysing the enrichment of mutant KRAS DNA in each (Figure 1a).
To characterize the different blood components obtained by differential centrifugation fractionation, we performed electron microscopy, Western blotting, nanoparticle tracking and multiplex bead‐based EV flow‐cytometry analysis (Figure 1b‐e). Although histone‐associated DNA is known to be present in several components of blood, histone H3 was only detectable within the WBC fraction (Cescon et al., 2020; Vagner et al., 2018; Wan et al., 2017). CD42A is a specific marker of megakaryocytes and platelets that was found to be most highly enriched in the platelet fraction by both Western blot and multiplex bead‐based EV analysis (Berndt et al., 1985). Cleaved CASP9 (Kuida et al., 1998) and BAX (Westphal et al., 2014) are markers of cellular apoptosis, and were found specifically in the platelet and AB fractions (Figure 1c). Although the relative levels of CD42A and cleaved CASP9 confirmed our desired enrichment for platelets and ABs, this data revealed abundant apoptotic contamination within the 360 x g centrifugation pellet commonly used for platelet isolation (Best et al., 2019; Brinkman et al., 2020).
Our analyses showed that ABs and LEVs contained large vesicles that were absent in the SEV fraction (Figure 1b&d). This was reflected by a higher ratio of CD9:CD81 in ABs and LEVs than SEVs (Figure 1c&e). This agrees with previous work (Kowal et al., 2016) and is reinforced by the presence of CD9 protein at the cell membrane, where microvesicles form, and the enrichment of CD81 protein within the endosomal system, which gives rise to exosomes (Ryu et al., 2000). These findings are also influenced by CD9 expression on platelet vesicles (Görgens et al., 2019), but support the conclusion that microvesicles are enriched in LEVs, while exosomes are the primary component of SEVs. Though SEVs are also present in the LEV fraction, it is important to recognize the relative volumes and surface areas of these vesicles when considering their molecular associations. Interestingly, we also observed greater amounts of CD9 and CD81 in SP by Western blot than was detected by bead‐based flow cytometry (Figure 1e). Due to the requirement for dual surface antibody binding in the flow cytometry assay, this suggests that the majority of CD9 and CD81 in SP is vesicle free, which presents an important consideration when using Western blot to characterize vesicle proteins (Wiklander et al., 2018). Although differential centrifugation is not a purification method, our results suggest that it enriches for the appropriate components in each fraction, including ctDNA in SP, enough to support our conclusions.
Liquid biopsy research has focused on the study of circulating tumour cells, ctDNA, and EVs, although comparisons between these have been rare (Nanou et al., 2020; Wan et al., 2017). However, ctDNA is primarily a feature of late‐stage disease and is rarely separated from EVs in clinical or research settings. This has likely contributed to the variable results reported in ctDNA publications, as inconsistencies in EV content amplifies differences due to centrifugation, DNA isolation and analysis techniques (Bergsmedh et al., 2001). ctDNA is thought to arise during the process of cell death, as DNA is sheared and apoptotic bodies lyse in circulation (Wan et al., 2017). Our data supports this interpretation, as the highest raw correlation between mutant DNA quantities in two components was observed between ABs and SP. Interestingly, SEV mutant DNA quantities were most correlated to those of LEVs, suggesting that EV DNA may arise by a distinct mechanism that precedes tumour‐cell death (Supplementary Table S1).
When analysing KRAS DNA abundance, we found that WBCs, RBCs and platelets contained the least mutant DNA throughout disease progression (Figure 2c). Although detectable in specific instances, and particularly in late‐stage patients (Supplementary Table S1), this did not agree with recent publications showing an enrichment of tumour‐derived nucleic acids in WBCs (Chennakrishnaiah et al., 2018) or platelets (Best et al., 2019). Instead, we found that LEVs and SEVs were associated with significantly more mutant KRAS DNA than the other fractions in early‐stage patients, while we observed the most mutant DNA in connection with SEVs and SP in patients later in disease progression. Interestingly, this change was driven by a drastic increase in the quantity of mutant DNA in late‐stage patient SP (Figure 2c). Thus, it was important that we could confirm these results using SEC, though even this is unable to fully separate EVs from the largest species of soluble lipoproteins (Figure 2e‐g). Since these patients had significantly shorter survival than early group patients, this agrees with previous findings that ctDNA is primarily a feature of late‐stage disease. Although this limits the diagnostic utility of ctDNA, our results suggest that EV‐associated tumour DNA is present early in disease progression and could thus play a role in the clinical monitoring of PDAC if properly isolated. Importantly, due to their relatively low levels of wild type DNA, we identify SEVs as the most tumour DNA enriched blood component – notably above that of PPP or SP in early‐stage patients. Since EVs can be further separated based on the transmembrane markers of their cells of origin, finding that SEVs are the most enriched component of blood in PDAC‐derived DNA provides an important lead toward making liquid biopsy monitoring standard practice throughout clinical oncology.
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest.
Supporting information
Supplementary Figure 1. (A) Patient normalized copies/μl of wild type KRAS DNA found in each blood component in early (top) and late (bottom) group patients. Only statistical comparison between the non‐significant (n.s.) groups indicated resulted in P‐values > 0.05. (B) Ratio of mutant : wild type KRAS DNA copies/μl for each blood component in late patients. (C) Copies/μl of wild type KRAS DNA found in each SEC fractions of patient SEVs. (D) Copies/μl of wild type KRAS DNA found in SEC fractions from patient SP. Statistics in A, C and D are derived from two‐tailed unequal variance t‐tests between indicated groups. Statistics for B are derived from two‐tailed unpaired Mann‐Whitney‐Wilcoxon tests between indicated groups.
Table S1
ACKNOWLEDGEMENTS
The authors thank all patients and their families for their participation. We would like to acknowledge Pia Raja Pakse and Lylie Mbuye for collection of blood samples and logistic support and Georgia Kokaraki for DNA preparation from archival tissue and logistic support. AG is an International Society for Advancement of Cytometry Marylou Ingram Scholar. This work was supported by funding from StratCan, the Swedish Gastro‐Oncological Society (to MK), the Olof and Signe Wallenius Foundation, Lennart Glans Stiftelsen (to JML), CIMED (to JML and SEA), the Swedish Research Council, the Swedish Foundation for Strategic Research (to SEA), the Tornspiran Foundation, Lars Hierta's Memory Foundation, and the Swedish Childhood Cancer Fund (to DWH).
Hagey, D. W., Kordes, M., Görgens, A., Mowoe, M. O., Nordin, J. Z., Moro, C. F., Löhr, J.‐M., & EL Andaloussi, S. (2021). Extracellular vesicles are the primary source of blood‐borne tumour‐derived mutant KRAS DNA early in pancreatic cancer. Journal of Extracellular Vesicles, 10, e12142. 10.1002/jev2.12142
Daniel W. Hagey and Maximilian Kordes contributed equally to this study as first authors.
J.‐Matthias Löhr and Samir EL Andaloussi contributed equally to this study as last authors.
REFERENCES
- Allenson, K., Castillo, J., San Lucas, F. A., Scelo, G., Kim, D. U., Bernard, V., Davis, G., Kumar, T., Katz, M., Overman, M. J., Foretova, L., Fabianova, E., Holcatova, I., Janout, V., Meric‐Bernstam, F., Gascoyne, P., Wistuba, I., Varadhachary, G., Brennan, P.…Alvarez, H. (2017). High prevalence of mutant KRAS in circulating exosome‐derived DNA from early‐stage pancreatic cancer patients. Annals of Oncology, 28(4), 741–747. 10.1093/annonc/mdx004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin, M. B., Edge, S., Greene, F., Byrd, D. R., Brookland, R. K., Washington, M. K., Gershenwald, J. E., Compton, C. C., & Hess, K. R. et al. (Eds.) (2017). AJCC Cancer Staging Manual (8th edition). Springer International Publishing: American Joint Commission on Cancer. [Google Scholar]
- Bergsmedh, A., Szeles, A., Henriksson, M., Bratt, A., Folkman, M. J., Spetz, A. ‐. L., & Holmgren, L. (2001). Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proceedings of the National Academy of Sciences of the United States of America, 98(11), 6407–6411. 10.1073/pnas.101129998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard, V., Kim, D. U., San Lucas, F. A., Castillo, J., Allenson, K., Mulu, F. C., Stephens, B. M., Huang, J., Semaan, A., Guerrero, P. A., Kamyabi, N., Zhao, J., Hurd, M. W., Koay, E. J., Taniguchi, C. M., Herman, J. M., Javle, M., Wolff, R., Katz, M.…Alvarez, H. A. (2019). Circulating Nucleic Acids Are Associated With Outcomes of Patients With Pancreatic Cancer. Gastroenterology, 156(1), 108–118.e4. 10.1053/j.gastro.2018.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt, M. C., Gregory, C., Kabral, A., Zola, H., Fournier, D., & Castaldi, P. A. (1985). Purification and preliminary characterization of the glycoprotein Ib complex in the human platelet membrane. European Journal of Biochemistry FEBS, 151(3), 637–649. 10.1111/j.1432-1033.1985.tb09152.x [DOI] [PubMed] [Google Scholar]
- Best, M. G., In ’T Veld, S. G. J. G., Sol, N., & Wurdinger, T. (2019). RNA sequencing and swarm intelligence‐enhanced classification algorithm development for blood‐based disease diagnostics using spliced blood platelet RNA. Nature Protocols, 14(4), 1206–1234. 10.1038/s41596-019-0139-5 [DOI] [PubMed] [Google Scholar]
- Brinkman, K., Meyer, L., Bickel, A., Enderle, D., Berking, C., Skog, J., & Noerholm, M. (2020). Extracellular vesicles from plasma have higher tumour RNA fraction than platelets. Journal of Extracellular Vesicles, 9(1), 1741176. 10.1080/20013078.2020.1741176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cescon, D. W., Bratman, S. V., Chan, S. M., & Siu, L. L. (2020). Circulating tumor DNA and liquid biopsy in oncology. Nat Cancer 1, 276–290 10.1038/s43018-020-0043-5 [DOI] [PubMed] [Google Scholar]
- Chennakrishnaiah, S., Meehan, B., D'asti, E., Montermini, L., Lee, T. ‐. H., Karatzas, N., Buchanan, M., Tawil, N., Choi, D., Divangahi, M., Basik, M., & Rak, J. (2018). Leukocytes as a reservoir of circulating oncogenic DNA and regulatory targets of tumor‐derived extracellular vesicles. Journal of Thrombosis and Haemostasis, 16(9), 1800–1813. 10.1111/jth.14222 [DOI] [PubMed] [Google Scholar]
- Foroni, C., Zarovni, N., Bianciardi, L., Bernardi, S., Triggiani, L., Zocco, D., Venturella, M., Chiesi, A., Valcamonico, F., & Berruti, A. (2020). When less is more: Specific capture and analysis of tumor exosomes in plasma increases the sensitivity of liquid biopsy for comprehensive detection of multiple androgen receptor phenotypes in advanced prostate cancer patients. Biomedicines, 8(5), 131. 10.3390/biomedicines8050131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görgens, A., Bremer, M., Ferrer‐Tur, R., Murke, F., Tertel, T., Horn, P. A., Thalmann, S., Welsh, J. A., Probst, C., Guerin, C., Boulanger, C. M., Jones, J. C., Hanenberg, H., Erdbrügger, U., Lannigan, J., Ricklefs, F. L., El‐Andaloussi, S., & Giebel, B. (2019). Optimisation of imaging flow cytometry for the analysis of single extracellular vesicles by using fluorescence‐tagged vesicles as biological reference material. Journal of Extracellular Vesicles, 8(1), 1587567. 10.1080/20013078.2019.1587567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, P. A., Taylor, R., Minor, B. L., Elliott, V., Fernandez, M., O'neal, L., Mcleod, L., Delacqua, G., Delacqua, F., Kirby, J., & Duda, S. N. (2019). The REDCap consortium: Building an international community of software partners. Journal of Biomedical Informatics. 95, 103208, 10.1016/j.jbi.2019.103208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen, D. K., Fenix, A. M., Franklin, J. L., Higginbotham, J. N., Zhang, Q., Zimmerman, L. J., Liebler, D. C., Ping, J., Liu, Q., Evans, R., Fissell, W. H., Patton, J. G., Rome, L. H., Burnette, D. T., & Coffey, R. J. (2019). Reassessment of exosome composition. Cell, 177(2), 428–445.e18. 10.1016/j.cell.2019.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal, J., Arras, G., Colombo, M., Jouve, M., Morath, J. P., Primdal‐Bengtson, B., Dingli, F., Loew, D., Tkach, M., & Théry, C. (2016). Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proceedings of the National Academy of Sciences of the United States of America, 113(8), E968–E977. 10.1073/pnas.1521230113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida, K., Haydar, T. F., Kuan, C.‐Y., Gu, Y., Taya, C., Karasuyama, H., Su, M. S.‐S., Rakic, P., & Flavell, R. A. (1998). Reduced apoptosis and cytochrome c‐mediated caspase activation in mice lacking caspase 9. Cell, 94(3), 325–337. 10.1016/s0092-8674(00)81476-2 [DOI] [PubMed] [Google Scholar]
- Lee, J. ‐. S., Park, S. S., Lee, Y. K., Norton, J. A., & Jeffrey, S. S. (2019). Liquid biopsy in pancreatic ductal adenocarcinoma: Current status of circulating tumor cells and circulating tumor DNA. Molecular Oncology, 13(8), 1623–1650. 10.1002/1878-0261.12537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, E., Cao, T., Nagrath, S., & King, M. R. (2018). Circulating tumor cells: Diagnostic and therapeutic applications. Annual Review of Biomedical Engineering, 20, 329–352. 10.1146/annurev-bioeng-062117-120947 [DOI] [PubMed] [Google Scholar]
- Liu, X., Liu, L., Ji, Y., Li, C., Wei, T., Yang, X., Zhang, Y., Cai, X., Gao, Y., Xu, W., Rao, S., Jin, D., Lou, W., Qiu, Z., & Wang, X. (2019). Enrichment of short mutant cell‐free DNA fragments enhanced detection of pancreatic cancer. EBioMedicine, 41, 345–356. 10.1016/j.ebiom.2019.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livshits, M. A., Khomyakova, E., Evtushenko, E. G., Lazarev, V. N., Kulemin, N. A., Semina, S. E., Generozov, E. V., & Govorun, V. M. (2015). Isolation of exosomes by differential centrifugation: Theoretical analysis of a commonly used protocol. Scientific Reports, 5, 17319. 10.1038/srep17319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanou, A., Miller, M. C., Zeune, L. L., De Wit, S., Punt, C. J. A., Groen, H. J. M., Hayes, D. F., De Bono, J. S., & Terstappen, L. W. M. M. (2020). Tumour‐derived extracellular vesicles in blood of metastatic cancer patients associate with overall survival. British Journal of Cancer, 122(6), 801–811. 10.1038/s41416-019-0726-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oken, M. M., Creech, R. H., Tormey, D. C., Horton, J., Davis, T. E., Mcfadden, E. T., & Carbone, P. P. (1982). Toxicity and response criteria of the Eastern Cooperative Oncology Group. American Journal of Clinical Oncology, 5(6), 649–656. [PubMed] [Google Scholar]
- Ryu, F., Takahashi, T., Nakamura, K., Takahashi, Y., Kobayashi, T., Shida, S., Kameyama, T., & Mekada, E. (2000). Domain analysis of the tetraspanins: Studies of CD9/CD63 chimeric molecules on subcellular localization and upregulation activity for diphtheria toxin binding. Cell Structure and Function, 25(5), 317–327. 10.1247/csf.25.317 [DOI] [PubMed] [Google Scholar]
- Senshu, T., Akiyama, K., Ohsawa, T., & Takahashi, K. (1985). Immunochemical measurement of histone H3 in non‐nucleosomal compartments of cultured mammalian cells. European Journal of Biochemistry FEBS, 146(2), 261–266. 10.1111/j.1432-1033.1985.tb08648.x [DOI] [PubMed] [Google Scholar]
- Vagner, T., Spinelli, C., Minciacchi, V. R., Balaj, L., Zandian, M., Conley, A., Zijlstra, A., Freeman, M. R., Demichelis, F., De, S., Posadas, E. M., Tanaka, H., & Di Vizio, D. (2018). Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. Journal of Extracellular Vesicles, 7(1), 1505403. 10.1080/20013078.2018.1505403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan, J. C. M., Massie, C., Garcia‐Corbacho, J., Mouliere, F., Brenton, J. D., Caldas, C., Pacey, S., Baird, R., & Rosenfeld, N. (2017). Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nature Reviews Cancer, 17(4), 223–238. 10.1038/nrc.2017.7 [DOI] [PubMed] [Google Scholar]
- Waters, A. M., & Der, C. J. (2018). KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harbor perspectives in medicine, 8(9), a031435. 10.1101/cshperspect.a031435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal, D., Kluck, R. M., & Dewson, G. (2014). Building blocks of the apoptotic pore: How Bax and Bak are activated and oligomerize during apoptosis. Cell Death and Differentiation, 21(2), 196–205. 10.1038/cdd.2013.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiklander, O. P. B., Bostancioglu, R. B., Welsh, J. A., Zickler, A. M., Murke, F., Corso, G., Felldin, U., Hagey, D. W., Evertsson, B., Liang, X. ‐. M., Gustafsson, M. O., Mohammad, D. K., Wiek, C., Hanenberg, H., Bremer, M., Gupta, D., Björnstedt, M., Giebel, B., Nordin, J. Z.…Görgens, A. (2018). Systematic methodological evaluation of a multiplex bead‐based flow cytometry assay for detection of extracellular vesicle surface signatures. Frontiers in Immunology, 9, 1326. 10.3389/fimmu.2018.01326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi, A., Villar‐Prados, A., Oliphint, P. A., Zhang, J., Song, X., De Hoff, P., Morey, R., Liu, J., Roszik, J., Clise‐Dwyer, K., Burks, J. K., O'halloran, T. J., Laurent, L. C., & Sood, A. K. (2019). Mechanisms of nuclear content loading to exosomes. Science Advances, 5(11), eaax8849. 10.1126/sciadv.aax8849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zviran, A., Schulman, R. C., Shah, M., Hill, S. T. K., Deochand, S., Khamnei, C. C., Maloney, D., Patel, K., Liao, W., Widman, A. J., Wong, P., Callahan, M. K., Ha, G., Reed, S., Rotem, D., Frederick, D., Sharova, T., Miao, B., Kim, T.…Landau, D. A. (2020). Genome‐wide cell‐free DNA mutational integration enables ultra‐sensitive cancer monitoring. Nature Medicine, 26(7), 1‐11. 10.1038/s41591-020-0915-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. (A) Patient normalized copies/μl of wild type KRAS DNA found in each blood component in early (top) and late (bottom) group patients. Only statistical comparison between the non‐significant (n.s.) groups indicated resulted in P‐values > 0.05. (B) Ratio of mutant : wild type KRAS DNA copies/μl for each blood component in late patients. (C) Copies/μl of wild type KRAS DNA found in each SEC fractions of patient SEVs. (D) Copies/μl of wild type KRAS DNA found in SEC fractions from patient SP. Statistics in A, C and D are derived from two‐tailed unequal variance t‐tests between indicated groups. Statistics for B are derived from two‐tailed unpaired Mann‐Whitney‐Wilcoxon tests between indicated groups.
Table S1