

Abstract
The unprecedented coverage offered by next-generation sequencing (NGS) technology has facilitated the assessment of the population complexity of intra-host RNA viral populations at an unprecedented level of detail. Consequently, analysis of NGS datasets could be used to extract and infer crucial epidemiological and biomedical information on the levels of both infected individuals and susceptible populations, thus enabling the development of more effective prevention strategies and antiviral therapeutics. Such information includes drug resistance, infection stage, transmission clusters and structures of transmission networks. However, NGS data require sophisticated analysis dealing with millions of error-prone short reads per patient. Prior to the NGS era, epidemiological and phylogenetic analyses were geared toward Sanger sequencing technology; now, they must be redesigned to handle the large-scale NGS datasets and properly model the evolution of heterogeneous rapidly mutating viral populations. Additionally, dedicated epidemiological surveillance systems require big data analytics to handle millions of reads obtained from thousands of patients for rapid outbreak investigation and management. We survey bioinformatics tools analyzing NGS data for (i) characterization of intra-host viral population complexity including single nucleotide variant and haplotype calling; (ii) downstream epidemiological analysis and inference of drug-resistant mutations, age of infection and linkage between patients; and (iii) data collection and analytics in surveillance systems for fast response and control of outbreaks.
Keywords: quasispecies, next-generation sequencing, variant calling, haplotype calling, outbreak investigation, surveillance systems
Introduction
Due to error-prone replication, RNA viruses mutate at rates estimated to be as high as 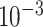 substitutions per nucleotide per replication cycle [1]. Since mutations are generally well tolerated, such viruses exist in infected hosts as ‘quasispecies’—a term used by virologists to describe populations of closely related genomic variants [2–5]. Genetic heterogeneity of viral quasispecies has major biological implications, contributing to the efficiency of virus transmission, tissue tropism, virulence, disease progression and the emergence of drug/vaccine-resistant variants [6–10].
substitutions per nucleotide per replication cycle [1]. Since mutations are generally well tolerated, such viruses exist in infected hosts as ‘quasispecies’—a term used by virologists to describe populations of closely related genomic variants [2–5]. Genetic heterogeneity of viral quasispecies has major biological implications, contributing to the efficiency of virus transmission, tissue tropism, virulence, disease progression and the emergence of drug/vaccine-resistant variants [6–10].
With the advent of next-generation sequencing (NGS) technologies, molecular epidemiology and virology are undergoing a fundamental transformation that promises to revolutionize our approach to epidemiological data analysis, disease prevention and treatment [11–14]. NGS has already shown its potential to advance epidemiological practices and it is steadily moving into clinical practices. There are numerous examples of successful applications of NGS for studying viruses such as coronavirus [15], influenza [16–21], HIV [22–27], hepatitis [28–32], Ebola [33, 34], Zika [35] and other viruses [36].
NGS allows sequencing with the unprecedented coverage, which is crucial for characterizing intra-host viral population complexity. However, inferring and analyzing the viral population from NGS data are computationally challenging and require specialized, highly sophisticated computational tools [37]. Even for NGS technologies offering very deep coverage, the presence of sequencing errors makes it difficult to distinguish between rare variants and sequencing errors. Additionally, low intra-host viral diversity complicates assembling whole-genome sequences that are necessary for the unique identification of viral haplotypes. Therefore, the analysis of heterogeneous virus populations was complemented by technological developments.
The viral population reconstructed from NGS data can be further used for the detection of drug resistance in the patients’ samples as well as the age of infection. The importance of this detection is constantly growing [38], especially for influenza [39], hepatitis C virus (HCV) [40] and HIV [41, 42], because of the high prevalence of these diseases in the population. As for HIV, there is an additional problem. Since HIV has no cure, its treatment can only slow down its progression, and the development of drug resistance creates the risk of losing a drug forever as a treatment option for the patient. This is further complicated by the increasing longevity of HIV patients and the prevalence of the disease among the general population. Since viruses exist as a swarm of haplotypes, it is crucial to detect minority drug-resistant populations.
The haplotypes inferred from NGS data can also be very effective for outbreak investigation. Millions of viral variants that are carried in the samples of thousands of infected individuals can be analyzed with the help of NGS. Molecular data collected from densely sampled outbreaks in large high-risk communities are of particular interest since it allows for the first time to study the evolution of heterogeneous intra-host viral populations within a single evolutionary space under frequent transmissions between hosts [43–45]. The growing knowledge about social network structures and progress in the development of methods for the collection of large volumes of socio-behavioral and geographic data gives us new information about the conditions of disease spread [46–48]. The availability of such large-scale datasets provides a new opportunity to implement massive molecular surveillance and forecasting of viral diseases [49–55]. Deployment of massive molecular surveillance programs intends to facilitate our understanding of virus evolution, which may enable the development of more effective public health intervention strategies. To be effective, molecular surveillance and forecasting should analyze unprecedented amounts of heterogeneous biomedical data. This requires extensive computational methods for processing, integrating and analyzing big data, i.e. both epidemiological and molecular. In addition, this requires new mathematical models that allow for describing, understanding and predicting complex multidimensional-linear disease dynamics.
The remainder of the review will discuss the pipeline of software tools for primary and secondary NGS data analysis constituting a sequencing-based molecular surveillance system (see Figure 1). The primary NGS data analysis consists of error correction, consensus assembly/selection, read alignment and inference of intra-host viral population including single nucleotide variant calling and haplotype reconstruction. The secondary NGS data analysis includes intra-host analysis such as detection of drug resistance and estimations of the age of infection as well as inter-host analysis such as outbreak detection and investigation. Finally, we review existing molecular surveillance systems that integrate all the above analyses.
Figure 1 .

A molecular surveillance pipeline for software tools for primary and secondary viral NGS data analysis.
Primary analysis of viral NGS data
Primary analysis can be partitioned into two major steps: (i) basic primary analysis which starts with error correction followed by identification of the consensus sequence and read mapping and (ii) characterization of the intra-host viral population complexity by calling SNVs and haplotype variants in the viral sample.
Basic primary analysis
The error correction of viral sequencing reads is a notoriously difficult task. The standard error correction tools tuned to correct reads from a human genome do not perform well for viral genomes since viral haplotypes differ only slightly between themselves [56]. There are several error-correction tools that have been proposed specifically to handle viral sequencing samples [57–59]. A Bayesian probabilistic clustering approach [57] integrates error correction with SNV and haplotype calling, while KEC [58] is a k-mer counting-based approach that identifies erroneous k-mers by analyzing the distributions of k-mer frequencies. A more sophisticated random forest classifier MultiRes [59] can be used to distinguish between erroneous and rare k-mers.
Identification of the consensus sequence can be either picked from existing reference genomes or de novo assembled to avoid reference biases. The reference-based identification of the consensus relies on the existence of closely related genomic sequences. NGS reads are aligned to the reference sequence with a significant number of mismatches. To avoid reference biases, the aligned reads are used for updating each position of the reference genome with the base most frequent in reads and re-aligning reads to the consensus [60, 61]. The drawback of this approach is that selecting the reference genome is not a well-formalized procedure.
De novo assemblers are based on de Bruijn graphs such as VICUNA and overlap graphs such as SAVAGE [26, 62–65]. SAVAGE constructs an overlap graph with vertices representing reads and/or contigs and edges connecting two reads/contigs belonging to the same haplotypic sequence. Statistically, well-calibrated groups of reads/contigs are then efficiently used for reconstruction of the individual haplotypes from this overlap graph. SAVAGE has an additional advantage over VICUNA since it builds multiple haplotype contigs rather than a single consensus. De novo assemblers require much higher memory and time resources than reference-based identification of the consensus.
A recent tool, SHIVER [66], combines the reference-based and de novo approaches by using both reads and contigs assembled from those reads for HIV sequencing. Contigs are compared with the existing references, wherein some are spliced and some are removed as contaminants. After the closest existing reference is identified, it is updated to the consensus by well-mapped reads that do not match contaminants.
Single nucleotide variant calling
The natural advantage of NGS versus Sanger sequencing is its ability to identify low-frequency mutations (i.e. <20%) that are particularly relevant in the context of drug resistance [67–69]. The main challenge for SNV calling is to distinguish between sequencing errors and low-frequency true SNVs. All existing methods apply a particular error model to estimate the probability that an observed mismatch with the consensus is an error and qualify it as an SNV if this probability is low enough.
Below, we briefly describe widely known tools [37] and recently developed tools. VarScan [70] reports SNVs that are deeply covered by the reads with high quality. A similar approach with improved codon-based filtration is introduced by VirVarSeq [71] of SNV. The method LoFreq [72] derives sequencing error probability from a Phred-scaled quality value and optimizes estimation of P-value. V-Phaser [73, 74] introduces a basic primary analysis and error model, which takes into account the simultaneous occurrence of pairs of SNV in the same reads. V-Phaser 2 [74] specifies this model for Illumina reads. Pairs of mutations are explored by CoVaMa [75] using a linkage disequilibrium model. An accurate analysis of linked SNV pairs independent of error rate is proposed by CliqueSNV [76], which also contains an efficient implementation of the SNV-pair analysis. ViVan [77] and ViVaMBC [78] are based on maximum likelihood models. MinVar [79] and SiNPle [80] utilize the Poisson–Binomial distribution and Bayesian model respectively. Validation of MinVar on Illumina Miseq samples and shows that SNVs with the frequency of at least 5% are reliably identified without introducing false positives. PASeq [81] and Hydra Web [22] are web-based publicly available tools that are thoroughly tested for identifying mutations with frequencies 20% and 5%. Interestingly, SNV calling for viral data is very similar to somatic mutation calling and the quality of algorithms for both problems can be compared [80].
Table 1 describes the list of tools analyzing viral NGS data for SNV calling. For each tool, we specify the SNV detection method and whether it requires a reference.
Table 1.
SNV calling software tools for viral NGS data
| SNV calling tools | Year | System | De novo/Ref based | Pair-end reads | SNV detection method | Tool availability |
|---|---|---|---|---|---|---|
| VarScan [70] | 2009 | Java | Ref | + | Read coverage | http://varscan.sourceforge.net/ |
| LoFreq [72] | 2012 | Linux | Ref | + | Poisson binomial distribution | https://csb5.github.io/lofreq/ |
| Vphaser [73] | 2012 | Linux | Ref | − | Bernoulli phasing model | https://www.broadinstitute.org/viral-genomics/v-phaser |
| Vphaser2 [74] | 2013 | Linux | Ref | + | Bernoulli phasing model | https://www.broadinstitute.org/viral-genomics/v-phaser-2 |
| ViVan [77] | 2015 | — | Ref | + | Maximum likelihood | http://www.vivanbioinfo.org |
| ViVaMBC [78] | 2015 | R | Ref | + | Maximum likelihood | https://sourceforge.net/projects/vivambc/ |
| VirVarSeq [71] | 2015 | Linux | Ref | + | Codon-level quality filtration | https://sourceforge.net/projects/virtools/?source=directory |
| CoVaMa [75] | 2015 | Python | Ref | + | Linkage disequilibrium | https://sourceforge.net/projects/covama/ |
| MinVar [79] | 2017 | Python | Ref | + | Poisson binomial distribution | http://git.io/minvar |
| MultiRes [59] | 2017 | Linux | De novo | + | Frame-based model | https://github.com/raunaq-m/MultiRes |
| CliqueSNV [76] | 2018 | Java | Ref | + | Linkage of SNV pairs | https://github.com/vtsyvina/CliqueSNV |
| SiNPle [80] | 2019 | Linux | Ref | + | Bayesian model | https://mallorn.pirbright.ac.uk:4443/gitlab/drcyber/SiNPle |
| PASeq | Web | https://paseq.org/ | ||||
| Hydra Web | Web | https://hydra.canada.ca/pages/home?lang=en-CA | ||||
| SmartGen | Web | https://www.smartgene.com/mod_hiv.html |
Viral haplotype variant calling
Rather than determining variation in a single position, the haplotype calling is required to find the haplotypes spanning the entire viral genome or amplicons of special interest. The haplotypes and their frequencies are more informative than SNVs for detecting drug resistance that can non-linearly depend on accumulated SNVs. Haplotypes are also used for significantly more accurate detection of transmission clusters and outbreak sources.
Note that haplotype frequency reconstruction is considered to be a simpler problem as soon as haplotypes are inferred. The expectation–maximization algorithm based on the estimation of the probability that a given read has been emitted by a given haplotype has been shown to be sufficiently reliable with accuracy growing with the sequencing depth [60, 82].
The first haplotype reconstruction tools were read-graph based with vertices corresponding to reference-mapped reads and edges connecting reads that agree on their overlap [83, 84]. Many tools followed this idea [60, 82, 85–92] significantly improving the quality of reconstruction [37, 93]. But all these tools usually are not fast enough to handle recently available multi-million read data sets.
Probabilistic modeling of the sequencing process and/or viral haplotype generation [94–98] was shown to be an attractive alternative to the read-graph approach. The most successful tool among probabilistic tools is PredictHaplo [96] that exhibits high specificity and can reconstruct haplotypes with frequency over 10%. Hierarchical-clustering of reads (especially long PacBio reads) has been suggested in [99], and recent methods, aBayesQR [100], combined probabilistic modeling with clustering making the Bayesian approach computationally tractable.
Novel scalable tools handling millions of reads and improving over existing tools are actively developed in multiple labs. CliqueSNV [76] efficiently recognizes groups of linked SNVs and constructs an SNV graph, where SNVs are nodes and edges connect linked SNVs. It can assemble close viral haplotypes with frequencies as low as 0.1% from Illumina and PacBio reads.
It is necessary to separately note de novo haplotype callers, i.e. tools that de novo assemble multiple distinct haplotypes rather than a consensus. Currently, there exist three de novo assemblers MLEHaplo [98], SAVAGE [65] and PEHaplo [92]. The advantage of these tools is that they do not introduce reference biases.
Recently, 12 NGS haplotype callers were tested using viral populations simulated under realistic evolutionary dynamics but without error simulation [101]. In contrast to other simulations, the number of haplotypes was very large (216-1,185) and each frequency was small (<7%). Under such stressful conditions, PreditHaplo and CliqueSNV showed certain advantages over other reference-based methods and PEHaplo among de novo assemblers. It is also very important to distinguish low-frequency haplotypes from similar high-frequency haplotypes coexisting in the same intra-host viral population. Therefore, it is critical to validate haplotype reconstruction tools on benchmarks containing such pairs of similar haplotypes.
Table 2 describes the list of tools analyzing viral NGS data for haplotype calling. For each tool, we specify (i) whether it is a de novo method or requires a reference, (ii) sequencing error handling, (iii) the method for haplotype assembly and (iv) the method for haplotype frequency estimation.
Table 2.
Haplotype calling software tools for viral NGS data
| Haplotyping tools | Year | System | De novo/Ref based | Pair-end reads | Sequencing error handling | Haplotype assembly method | Haplotype frequency estimation method | Output sequences | Tool availability |
|---|---|---|---|---|---|---|---|---|---|
| Shorah [82] | 2011 | Linux | Ref | + | Probabilistic clustering | Minimal path cover | EM | Full haplotypes | https://github.com/cbg-ethz/shorah |
| ViSpA [60] | 2011 | Linux | Ref | − | Binomial model | Max-bandwidth path | EM | Full haplotypes | http://alan.cs.gsu.edu/NGS/?q=content/vispa |
| QColors [86] | 2012 | — | De novo | − | — | Overlap graph + Conflict graph | — | Full haplotypes | — |
| QuRe [87] | 2012 | Java | Ref | + | Poison model | Multinomial distribution matching | Read coverage | Full haplotypes | https://sourceforge.net/projects/qure/ |
| bioa [85] | 2012 | Linux | Ref | − | k-mer-based error correction | Maximum Bandwidth Path | Fork balancing | Full haplotypes | http://alan.cs.gsu.edu/vira/index.html |
| Vicuna [63] | 2012 | Linux | De novo | + | Read count | — | — | Consensus + contigs | https://www.broadinstitute.org/viral-genomics/vicuna |
| QuasiRecomb [95] | 2013 | Linux | Ref | + | Hidden Markov model | Hidden Markov model | Hidden Markov model | Full haplotypes | https://github.com/cbg-ethz/QuasiRecomb |
| Vira (AmpMCF) [88] | 2013 | Linux | Ref | − | — | Multicommodity flows | Normalized flow size | Full haplotypes | http://alan.cs.gsu.edu/vira/index.html |
| ShotMCF [88] | 2013 | JAVA | Ref | − | Binomial model | Max-bandwidth path + Multicommodity flows | EM + normalized flow size | Full haplotypes | http://alan.cs.gsu.edu/NGS/?q=content/shotmcf |
| BAsE-Seq [61] | 2014 | − | Ref | + | Poisson binomial distribution model | Clustering of reads by SNVs | Read coverage | Full haplotypes | — |
| VGA [90] | 2014 | Linux | Ref | + | Requires high-fidelity sequencing protocol | Min-graph coloring | EM | Full haplotypes | http://genetics.cs.ucla.edu/vga/ |
| HaploClique [89] | 2014 | Linux | Ref | + | — | Max-clique enumeration | Normalized read count | Full haplotypes | https://github.com/cbg-ethz/haploclique |
| PredictHaplo [96] | 2014 | Linux | Ref | + | Dirichlet Process Mixture Model | Dirichlet Process Mixture Model | Dirichlet Process Mixture Model | Full haplotypes | https://bmda.dmi.unibas.ch/software.html |
| IVA [64] | 2015 | Linux | De novo | − | Read count | — | — | Contigs | https://sanger-pathogens.github.io/iva/ |
| MLEHaplo [98] | 2015 | Linux | De novo | + | — | Maximum likelihood | — | Full haplotypes | https://github.com/raunaq-m/MLEHaplo |
| ViQuaS [91] | 2015 | Linux | Ref | + | Chimeric error correction | Multinomial distribution matching | Read count | Full haplotypes | https://sourceforge.net/projects/viquas/ |
| SAVAGE [65] | 2017 | Linux | De novo | + | Overlap fuzzy matching error correction | Enumerating cliques in overlap graph | EM | Contigs | https://bitbucket.org/jbaaijens/savage/ |
| aBayesQR [100] | 2017 | Linux | Ref | + | Cluster coverage by reads | Bayesian inference | Bayesian inference | Full haplotypes | https://github.com/SoYeonA/aBayesQR |
| RegressHaplo [97] | 2017 | R | Ref | + | — | Penalized regression | Penalized regression | Full haplotypes | https://github.com/SLeviyang/RegressHaplo |
| 2SNV [99] | 2017 | Java | Ref | − | Linkage of SNV pairs | Hierarchical clustering of reads by SNVs | EM | Full haplotypes | http://alan.cs.gsu.edu/NGS/?q=content/2snv |
| PEHaplo [92] | 2018 | Linux | De novo | + | Overlap error correction | Path finding in overlap graph | — | Contigs | https://github.com/chjiao/PEHaplo |
| Shiver [66] | 2018 | Linux | De novo + ref | + | BLAST database match | — | — | Consensus | https://github.com/ChrisHIV/shiver |
| CliqueSNV [76] | 2018 | JAVA | Ref | + | Linkage of SNV pairs | Clique enumeration and merging | EM | Full haplotypes | https://github.com/vtsyvina/CliqueSNV |
Secondary analysis of viral NGS data
Secondary NGS analysis addresses three tasks: (i) predicting of drug resistance that takes SNV and haplotypes obtained during primary analysis and determine whether they are drug-resistant or not; (ii) determining the recency of the infection, i.e. predicting the moment in the past when patient was infected; (iii) outbreak investigation, i.e. determining the borders of outbreak, finding the source of infection and reconstruction of infection spread paths.
Predicting drug resistance
Certain haplotypes and mutations that are found during the primary NGS should be analyzed for drug resistance. This is especially important for viruses such as HIV [102], HCV [103], influenza [39] and others [104]. For HIV, the detection of drug resistance is especially relevant since HIV patients have to adhere to a treatment for the span of their lives. If a patient develops HIV drug resistance, they will be required to switch to a different line of treatment, and these treatments may be less studied and of a higher risk to the patient’s health. Additionally, the number of drug-resistant mutations in the patient is constantly growing as well as the number of drug-resistant patients in the outbreak [105]. This makes the task of tracking HIV drug resistance a more onerous one [106].
Detection of drug resistance is typically associated with matching genome mutations with the efficiency of drugs [104]. Usually, different mutations have different resistance power and often mutations work collectively [107], so the process of finding correlations between mutations and drug resistance is non-linear [108]. The comprehensive overview of computational approaches to drug-resistant HIV mutations can be found in [109]. Most of the tools are aimed at Sanger sequencing data since NGS data has only been accumulating for a short period of time. Sanger sequencing allows the detection of mutations with frequencies >25% which has low benefits for the clinical application [110, 111]. NGS increases the sensitivity and lowers the frequency threshold up to 1–5% [112].
There are two main challenges in the detection of drug resistance that depends on the results of primary NGS data analysis. They are connected with the accuracy of detecting minority mutations and haplotypes. The first problem is that if there is a minor drug-resistant mutation, the haplotypes with this mutation will have an advantage over other haplotypes dealing with drug pressure. As a result, these drug-resistant haplotypes will begin to dominate over time [102, 113]. The second problem is that drug resistance is connected with haplotypes rather than with the mutations themselves, but haplotypes are harder to detect and so the drug resistance analysis can be significantly improved with more sensitive haplotyping tools [114].
Currently, tools for detecting drug resistance are modeled to handle Sanger sequencing data accumulated in designated databases [109]. The limitation of Sanger data is that only the major haplotype and SNVs with frequency at least 20% can be reconstructed. This hurts the performance of the most efficient drug resistance prediction tools that are based on machine-learning [31, 114–118]. Such tools would rather take into account all patient’s haplotypes [114, 119] to overcome Sanger sequencing limitations by generating all possible haplotypes with given SNVs, e.g. 10 SNVs make 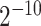 = 1024 different haplotypes.
= 1024 different haplotypes.
The number of HIV patients sequenced with NGS is beginning to grow very fast. Since NGS can detect rare SNPs and haplotypes, drug resistance can be predicted more accurately [107, 109]. We expect that the number of NGS samples to train these models will grow much faster after the Food and Drug Administration authorizes the first NGS test for detecting HIV-1 drug resistance mutations [120]. Recent clinical studies showed up to 2.7-fold improvement for detecting drug resistance with utilizing NGS data [69, 121–126] to antiretroviral therapy such as zidovudine (see Table 3). Zidovudine was designed to target the conserved domain of retroviral transcriptase. Mutations of amino acids localized at hydrophilic regions may result in conformation change of tertiary structure and block the targeted sites of zidovudine. Combining the evolutionary analytics with conformation dynamics of the retroviral transcriptase can potentially help to develop novel drugs. Therefore, it is critical to develop appropriate statistical models of the evolutionary dynamics of HIV retroviral transcriptase. One of the promising approaches to take into account the HIV protease 3D structure is based on Voronoi diagrams [114].
Table 3.
Detection of drug-resistant mutations in clinical studies: NGS versus Sanger sequencing
| Study | Patients group | Patients number | Collection date | Region | DRM detection: NGS/Sanger (fold) |
|---|---|---|---|---|---|
| Metzner et al. [121] | Acute patients | 49 | 1999–2003 | Germany | 2.0 |
| Fisher et al. [122] | Infants after PMTCT failure | 15 | 2006–2009 | South Africa | 2.5 |
| Alidjinou et al. [123] | ART-naive patients | 48 | 2013–2015 | France | 2.7 |
| Tzou et al. [69] | Undisclosed | 177 | 2001–2016 | Undisclosed | 1.2 |
| Fokam et al. [124] | Vertically infected children | 18 | 2015 | Cameroon | 1.7 |
| Derache et al. [126] | ART-naive patients | 1148 | 2012–2016 | South Africa | 1.4 |
| Derache et al. [125] | Patients failing first line ART | 1287 | 2012–2016 | South Africa | 2.0 |
Estimating infection recency
Over 80% of untreated cases of HCV infection becomes chronic. This impedes the timely diagnosis of the disease, due to the fact that the infection often does not manifest any clinical symptoms in its early stages. Currently, there are no diagnostic assays to determine the stage of HCV infection. Therefore, distinguishing recently infected patients from chronically infected patients using computational methods would be highly advantageous for both personalized therapeutic purposes and for epidemiological surveillance, e.g. for detection of incident HCV cases. Similarly, detection of the age of HIV infection is crucial for HIV-1 surveillance and the understanding of viral pathogenesis [127].
Measuring the time since infection using genomic data has recently been addressed in several studies [127–131]. The simpler version of this problem is infection staging, i.e. distinguishing between recent and chronic infections using viral sequences sampled by NGS. A number of methods establish an age or stage of HIV or HCV infection using various measures of the population structure [127–131]. An underlying assumption of such methods is that intra-host viral evolution is associated with continuous genetic diversification. This results in the existence of a correlation between genetic heterogeneity of quasispecies and the age of quasispecies, which allows for the use of properly calibrated diversity measures as age markers.
Recently, groups of comprehensive features accounting for population diversity, population genetics, topological, information-theoretical and physico-chemical properties of quasispecies populations were integrated using sophisticated machine-learning-based techniques [130, 132]. These methods take into account recent observations in the evolution of viruses, such as HCV, resulting in a gradual intra-host adaptation that is accompanied by a decrease in heterogeneity and an increase in negative selection [30, 133–135].
Outbreak investigation
Detection and investigation of viral outbreaks are the primary epidemiological tasks. Historically, epidemiological investigations have been based on in-field surveys of epidemiological settings and interviews with persons potentially involved in pathogen spread. However, such methods are time- and labor-consuming and the data obtained are prone to various socio-behavioral biases. Analysis of viral genomic data provides alternative unbiased machinery for outbreak investigations and quantification of major factors responsible for disease spread [136].
It should be noted that in the recent decade, the rich variety of tools for inferring epidemiological parameters has been developed within the field of viral phylodynamics [137, 138]. In addition, there are a plethora of methods for outbreak investigations that combine various types of genomic and epidemiological data [139–145]. Despite being highly effective in many settings, these tools are currently not intended for application to NGS data and usually do not support calculations with extremely large genomic datasets. Therefore in this article, we concentrate on tools specifically designed to handle heterogeneous intra-host viral populations using NGS.
The primary task in the outbreak investigation is the detection of transmission clusters. The main challenge, here, is the development and implementation of evolutionary distance measures between intra-host viral populations that reflect the epidemiological relations between the hosts. These distances can be efficiently calculated and combined with a broad variety of clustering techniques and phylogenetic and network-based methods [46, 146]. Distances between consensus sequences that are still often used for epidemiological investigations provide only very coarse estimates of evolutionary distances and lose significant signal encoded in quasispecies structure. In particular, outbreak distances between viral variants from certain hosts can be comparable or even higher than distances between variants from different hosts. For example, for HIV-1, the recommended inter-host threshold for detecting transmission clusters in pol region is in a range of 0.5–1.5% [136], while the nucleotide genetic variability inside hosts can be as high as 5% [147].
Analysis of quasispecies populations reconstructed from NGS data drastically improves the estimation of evolutionary distances. Pioneering NGS-based study for HCV outbreak investigations [148] proposed to measure the distance between samples as the distance between the closest pair of haplotypes from different samples. Even this simple method has been shown to significantly outperform the consensus-based approach [148]. Similar techniques have been applied to HIV [50]. Despite the simplicity of the metric, its calculation is challenging for extremely large NGS datasets, since its naive implementation requires a pairwise comparison of sequences from all pairs of patients. To address this challenge, several filtering techniques have been proposed [149, 150]. In consecutive studies [43, 44, 131, 151], more sophisticated distance measures for quasispecies populations have been proposed. In particular, Melnyk et al. [151] avoid reconstruction of haplotypes and/or phylogenetic trees by utilizing k-mer-based approach. Specifically, each viral sample is represented by a corresponding k-mer distribution, the distance between pairs of k-mers is computed over a single de Bruijn graph of all k-mers, and the distance between populations is identified with the earth mover’s distance (EMD) between two k-mer distributions.
The next step of the bioinformatics pipeline for epidemiological analysis is an investigation of viral transmissions inside each transmission cluster. It includes a prediction of possible transmission directions, detection of the source or ‘superspreader’ of an outbreak and inference of transmission networks indicating who infected whom. QUENTIN [43] and VOICE [44] estimate the distance between quasispecies populations as the analogue of a cover for a Markov-type model of viral evolution and choose the direction of transmission from a sample A to sample B based on minimum evolution principle, i.e. if it requires less evolution time than the time for evolving from sample A to B. In Romero-Severson et al. [151], it is proposed to identify the transmission directions by phylogenetic analysis and detection of paraphyletic, polyphyletic and monophyletic relations between sampled intra-host variants from different hosts. This idea has been further developed and implemented in Phyloscanner [152].
Both QUENTIN and Phyloscanner also allow reconstructing viral transmission networks. QUENTIN does it via Bayesian inference and Markov chain Monte Carlo sampling, with the likelihood of a transmission network being defined using general properties of social networks relevant to the infection dissemination. Phyloscanner relies on a maximum-parsimony approach and assigns ancestral hosts to internal nodes of a viral phylogeny containing quasispecies populations from different hosts by minimizing the number of transmission events while taking into account possible contaminations, multiple infections and presence of unsampled hosts.
Before determining the source of the outbreak, it is critical to decide whether the source of the outbreak is present among sequenced samples [151]. Finding the source of an outbreak is quite important for outbreak disruption. The papers [43, 44, 151] validated their approaches on Centers for Disease Control and Prevention (CDC) data for HCV outbreaks with the known sources and showed that the source prediction accuracy is ~90%. But before determining the source of the outbreak, it is critical to decide whether the source of the outbreak is present among sequenced samples [151]. This problem is quite difficult and has been addressed for the first time in [151].
Table 4 describes the list of tools analyzing viral NGS data for outbreak investigation including identification of (i) transmission clusters, (ii) transmission direction, (iii) source of infection, (iv) presence of source and (v) transmission network. For each tool, we indicate which of five tasks are addressed by which tool.
Table 4.
Outbreak investigation software tools for viral NGS data
| Tool | Year | System | Algorithm | Transmission clusters | Transmission direction | Transmission network | Source of infection | Presence of source | Tool availability |
|---|---|---|---|---|---|---|---|---|---|
| MinDist [148] | 2016 | — | Distance based | + | − | − | + | − | — |
| RED [44] | 2017 | Matlab | Clustering | + | + | − | + | − | https://bitbucket.org/osaofgsu/red |
| VOICE [44] | 2017 | Linux | Simulation based | + | + | − | + | − | https://bitbucket.org/osaofgsu/voicerep |
| PhyloScanner [152] | 2017 | Linux | Phylogeny | + | + | + | + | − | https://github.com/BDI-pathogens/phyloscanner |
| Quentin [43] | 2017 | Matlab | Simulation based | + | + | + | + | − | https://github.com/skumsp/QUENTIN |
| Signature-sj [150] | 2018 | Java | k-mers | + | − | − | − | − | https://github.com/vtsyvina/signature-sj |
| k-mer EMD [151] | 2019 | Linux | k-mer based distance | + | + | − | + | + | |
| https://github.com/amelnyk34/kemd |
Molecular surveillance systems and databases
The advent of NGS technologies makes possible, for the first time, the deployment of molecular epidemiological surveillance systems that are intended to analyze and infer the dynamics of epidemics and outbreaks in real or almost real time using computational analysis of viral genomic data [50, 51]. Such systems are characterized by a broad bioinformatics functionality including the processing of raw sequencing data, sequence alignment, phylogeny or network construction, transmission history inference and visualization. The number of computational molecular surveillance systems is currently being developed and deployed. One of the widely cited systems is Nextstrain [153] that allows for phylodynamics analysis and interactive visualization of the evolution of a variety of pathogens. The Nextstrain incorporates several computational tools for alignment, phylogenetic inference, reconstruction, dating and geographic localization of transmission events. However, currently, a toolkit of Nextstrain is not intended for the analysis of NGS data and intra-host viral populations, although its open-source architecture makes possible incorporation of such methods in the future. The library of tools for viral epidemiological data analysis developed and maintained by the R Epidemics Consortium [154] also should be mentioned. It includes R statistical packages for handling, visualizing and analyzing outbreak data, but has similar limitations.
Two surveillance systems that support NGS data are specifically tailored for HIV and viral hepatitis and are recommended and/or maintained by the CDC. These systems are HIV-Trace [50] and Global Hepatitis Outbreak Surveillance Technology (GHOST) [51], and they are based on high-throughput bioinformatics pipelines for genetic relatedness analysis. They allow estimates of genetic distances between intra-host populations sampled from HIV-infected individuals, use these distances to detect possible transmission linkages between the individuals, reconstruct and visualize transmission clusters and genetic relatedness networks. Both systems can work with haplotypes obtained from NGS data and are scalable for extremely large datasets produced by Illumina MiSeq and other sequencing platforms. In particular, GHOST employs several efficient k-mer-based filtering techniques for viral sequence similarity queries, which allow for the elimination of an exhaustive comparison of all pairs of viral haplotypes and allow processing of NGS data from a given HCV outbreak in minutes [150].
Another important issue is the creation of curated databases that contain both genomic and epidemiological data and can be used for the validation of new computational molecular epidemiology tools. Some previously published papers [43, 44] provide links to datasets that can be used for these purposes, but, to the best of our knowledge, large systematically curated collections of such datasets are yet to be created. In this context, Pangea HIV consortium efforts on curated analysis for HIV outbreaks in the African region [52] are very important. At this moment, they maintain a collection of >18 000 HIV NGS samples that can be used for outbreak investigations and data-driven design of prevention strategies.
Conclusions
The NGS extracts quantitatively and qualitatively more information from patients’ viral samples than the Sanger sequencing. But the extraction of this information requires sophisticated algorithms and software tools. In this article, we have reviewed bioinformatics methods and tools for NGS data analysis in viral epidemiology, which can be partitioned into the following three categories (see Figure 1):
Primary sequencing data analysis that consists of main strain reconstruction, read alignment and characterization of intra-host viral population structure including SNV and haplotype calling.
Secondary sequencing data analysis that employs reconstructed viral populations for predicting drug resistance, estimating recency of infection and outbreak investigation, including transmission cluster detection and identification of transmission direction and outbreak sources.
Molecular surveillance systems that provide a software environment for combined primary and secondary analysis of viral NGS data in real time.
In summary, NGS-based characterization of intra-host viral population structures is advanced enough and is getting ready to be used in epidemiological and clinical studies. This claim is supported by the number of recently published studies that use quasispecies analysis for outbreak investigation and transmission inference [49, 155, 156]. Inferred intra-host viral population structure can facilitate accurate answers to essential epidemiological questions about drug resistance, recency of infection, transmission clusters and outbreak sources. Future NGS-based surveillance systems should employ big data analytics to combine enormous amounts of sequencing and epidemiological data for the timely detection of outbreaks and the design of efficient public health intervention strategies.
Key Points
Analysis of intra-host viral populations sampled by NGS was shown to provide important epidemiological and clinical information.
Genetic characterization of intra-host viral populations offers a new framework for studies on drug resistance, identification of transmission clusters, sources of infection in outbreaks and time of infection inception.
Application of molecular data generated by NGS in combination with epidemiological information is a key to future improvement in public health surveillance.
Sergey Knyazev is a PhD student in computer science at Georgia State University, Atlanta, GA, USA. He received the MS degree in applied mathematics at Saint Petersburg Academic University, Saint Petersburg, Russia. He develops methods for analyzing viral genomic sequencing data.
Lauren Hughes received her BA degree in English from the University of Georgia, Athens, GA, USA. She is currently pursuing her BS degree in mathematics and computer science and an MS degree in geosciences at Georgia State University, Atlanta, GA, USA.
Pavel Skums received a PhD degree in computer science at Belarusian State University, Belarus in 2007. In 2010–16, he was a research fellow in the Centers for Disease Control and Prevention, and in 2016, he joined Georgia State University, Atlanta, GA, USA as an assistant professor.
Alexander Zelikovsky received a PhD degree in computer science at Belarusian State University, Belarus in 1989. He joined Georgia State University, Atlanta, GA, USA in 1999, where he is currently a distinguished university professor.
Funding
This work has been partially supported by National Institute of Health Grant R01.
References
- 1.Drake JW, Holland JJ. Mutation rates among RNA viruses. Proc Natl Acad Sci USA 1999;96:13910–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Domingo E, Holland JJ. RNA virus mutations and fitness for survival. Annu Rev Microbiol 1997;51:151–78. [DOI] [PubMed] [Google Scholar]
- 3.Domingo E, Martínez-Salas E, Sobrino F, et al. The quasispecies (extremely heterogeneous) nature of viral RNA genome populations: biological relevance—a review. Gene 1985;40:1–8. [DOI] [PubMed] [Google Scholar]
- 4.Eigen M, McCaskill J, Schuster P. Molecular quasi-species. J Phys Chem 1988;92:6881–91. [Google Scholar]
- 5.Martell M, Esteban JI, Quer J, et al. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: quasispecies nature of HCV genome distribution. J Virol 1992;66:3225–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beerenwinkel N, Sing T, Lengauer T, et al. Computational methods for the design of effective therapies against drug resistant HIV strains. Bioinformatics 2005;21:3943–50. [DOI] [PubMed] [Google Scholar]
- 7.Douek DC, Kwong PD, Nabel GJ. The rational design of an AIDS vaccine. Cell 2006;124:677–81. [DOI] [PubMed] [Google Scholar]
- 8.Gaschen B, Taylor J, Yusim K, et al. Diversity considerations in HIV-1 vaccine selection. Science 2002;296:2354–60. [DOI] [PubMed] [Google Scholar]
- 9.Holland JJ, De La Torre JC, Steinhauer DA. RNA virus populations as Quasispecies. Curr Top Microbiol Immunol 1992;176:1–20. [DOI] [PubMed] [Google Scholar]
- 10.Rhee S-Y, Liu TF, Holmes SP, et al. HIV-1 subtype B protease and reverse transcriptase amino acid covariation. PLoS Comput Biol 2007;3:e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Capobianchi MR, Giombini E, Rozera G. Next-generation sequencing technology in clinical virology. Clin Microbiol Infect 2013;19:15–22. [DOI] [PubMed] [Google Scholar]
- 12.Cruz-Rivera M, Forbi JC, Yamasaki LHT, et al. Molecular epidemiology of viral diseases in the era of next generation sequencing. J Clin Virol 2013;57:378–80. [DOI] [PubMed] [Google Scholar]
- 13.Gwinn M, MacCannell D, Armstrong GL. Next-generation sequencing of infectious pathogens. JAMA 2019;321:893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Polonsky JA, Baidjoe A, Kamvar ZN, et al. Outbreak analytics: a developing data science for informing the response to emerging pathogens. Philos Trans R Soc Lond B Biol Sci 2019;374:20180276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen Z, Xiao Y, Kang L, et al. Genomic diversity of SARS-CoV-2 in coronavirus disease 2019 patients. Clin Infect Dis 2020. doi: 10.1093/cid/ciaa203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sobel Leonard A, McClain MT, Smith GJD, et al. Deep sequencing of influenza a virus from a human challenge study reveals a selective bottleneck and only limited Intrahost genetic diversification. J Virol 2016;90:11247–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGinnis J, Laplante J, Shudt M, et al. Corrigendum to ‘next generation sequencing for whole genome analysis and surveillance of influenza a viruses’ [J. Clin. Virol. 79 (2016) 44–50]. J Clin Virol 2017;93:65. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Moore NE, Deng Y-M, et al. MinION nanopore sequencing of an influenza genome. Front Microbiol 2015;6:766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rutvisuttinunt W, Chinnawirotpisan P, Simasathien S, et al. Simultaneous and complete genome sequencing of influenza a and B with high coverage by Illumina MiSeq platform. J Virol Methods 2013;193:394–404. [DOI] [PubMed] [Google Scholar]
- 20.Vemula SV, Zhao J, Liu J, et al. Current approaches for diagnosis of influenza virus infections in humans. Viruses 2016;8:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischer N, Indenbirken D, Meyer T, et al. Evaluation of unbiased next-generation sequencing of RNA (RNA-seq) as a diagnostic method in influenza virus-positive respiratory samples. J Clin Microbiol 2015;53:2238–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jair K, McCann CD, Reed H, et al. Validation of publicly-available software used in analyzing NGS data for HIV-1 drug resistance mutations and transmission networks in a Washington, DC. Cohort PLoS One 2019;14:e0214820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cornelissen M, Gall A, Vink M, et al. From clinical sample to complete genome: comparing methods for the extraction of HIV-1 RNA for high-throughput deep sequencing. Virus Res 2017;239:10–6. [DOI] [PubMed] [Google Scholar]
- 24.Boltz VF, Rausch J, Shao W, et al. Ultrasensitive single-genome sequencing: accurate, targeted, next generation sequencing of HIV-1 RNA. Retrovirology 2016;13:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chabria SB, Gupta S, Kozal MJ. Deep sequencing of HIV: clinical and research applications. Annu Rev Genomics Hum Genet 2014;15:295–325. [DOI] [PubMed] [Google Scholar]
- 26.Henn MR, Boutwell CL, Charlebois P, et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog 2012;8:e1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fischer W, Ganusov VV, Giorgi EE, et al. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS One 2010;5:e12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomson E, Ip CLC, Badhan A, et al. Comparison of next-generation sequencing Technologies for Comprehensive Assessment of full-length hepatitis C viral genomes. J Clin Microbiol 2016;54:2470–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Welzel TM, Bhardwaj N, Hedskog C, et al. Global epidemiology of HCV subtypes and resistance-associated substitutions evaluated by sequencing-based subtype analyses. J Hepatol 2017;67:224–36. [DOI] [PubMed] [Google Scholar]
- 30.Campo DS, Dimitrova Z, Yamasaki L, et al. Next-generation sequencing reveals large connected networks of intra-host HCV variants. BMC Genomics 2014;15(Suppl 5):S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fourati S, Pawlotsky J-M. Virologic tools for HCV drug resistance testing. Viruses 2015;7:6346–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roll M, Norder H, Magnius LO, et al. Nosocomial spread of hepatitis B virus (HBV) in a haemodialysis unit confirmed by HBV DNA sequencing. J Hosp Infect 1995;30:57–63. [DOI] [PubMed] [Google Scholar]
- 33.Quick J, Loman NJ, Duraffour S, et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016;530:228–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoenen T, Groseth A, Rosenke K, et al. Nanopore sequencing as a rapidly deployable Ebola outbreak tool. Emerg Infect Dis 2016;22:331–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quick J, Grubaugh ND, Pullan ST, et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc 2017;12:1261–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woolhouse M, Scott F, Hudson Z, et al. Human viruses: discovery and emergence. Philos T R Soc B 2012;367:2864–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Posada-Cespedes S, Seifert D, Beerenwinkel N. Recent advances in inferring viral diversity from high-throughput sequencing data. Virus Res 2017;239:17–32. [DOI] [PubMed] [Google Scholar]
- 38.McKeegan KS, Borges-Walmsley MI, Walmsley AR. Microbial and viral drug resistance mechanisms. Trends Microbiol 2002;10:S8–14. [DOI] [PubMed] [Google Scholar]
- 39.Pizzorno A, Abed Y, Boivin G. Influenza drug resistance. Semin Respir Crit Care Med 2011;32:409–22. [DOI] [PubMed] [Google Scholar]
- 40.Lontok E, Harrington P, Howe A, et al. Hepatitis C virus drug resistance-associated substitutions: state of the art summary. Hepatology 2015;62:1623–32. [DOI] [PubMed] [Google Scholar]
- 41.Beyrer C, Pozniak A. HIV drug resistance—an emerging threat to epidemic control. N Engl J Med 2017;377:1605–7. [DOI] [PubMed] [Google Scholar]
- 42.Wensing AM, Calvez V, Ceccherini-Silberstein F, et al. 2019 update of the drug resistance mutations in HIV-1. Top Antivir Med 2019;27:111–21. [PMC free article] [PubMed] [Google Scholar]
- 43.Skums P, Zelikovsky A, Singh R, et al. QUENTIN: reconstruction of disease transmissions from viral quasispecies genomic data. Bioinformatics 2018;34:163–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glebova O, Knyazev S, Melnyk A, et al. Inference of genetic relatedness between viral quasispecies from sequencing data. BMC Genomics 2017;18:918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Melnyk A, Knyazev S, Vannberg F, et al. Using earth Mover’s distance for viral outbreak investigations. 2019. doi: 10.1101/628859. [DOI] [PMC free article] [PubMed]
- 46.Campbell EM, Jia H, Shankar A, et al. Detailed transmission network analysis of a large opiate-driven outbreak of HIV infection in the United States. J Infect Dis 2017;216:1053–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peters PJ, Pontones P, Hoover KW, et al. HIV infection linked to injection use of Oxymorphone in Indiana, 2014-2015. N Engl J Med 2016;375:229–39. [DOI] [PubMed] [Google Scholar]
- 48.Latkin C, Yang C, Srikrishnan AK, et al. The relationship between social network factors, HIV, and hepatitis C among injection drug users in Chennai, India. Drug Alcohol Depen 2011;117:50–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ratmann O, Grabowski MK, Hall M, et al. Inferring HIV-1 transmission networks and sources of epidemic spread in Africa with deep-sequence phylogenetic analysis. Nat Commun 2019;10:1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kosakovsky Pond SL, Weaver S, Leigh Brown AJ, et al. HIV-TRACE (TRAnsmission cluster engine): a tool for large scale molecular epidemiology of HIV-1 and other rapidly evolving pathogens. Mol Biol Evol 2018;35:1812–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Longmire AG, Sims S, Rytsareva I, et al. GHOST: global hepatitis outbreak and surveillance technology. BMC Genomics 2017;18:916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abeler-Dörner L, Grabowski MK, Rambaut A, et al. PANGEA-HIV 2: Phylogenetics and networks for generalised epidemics in Africa. Curr Opin HIV AIDS 2019;14:173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuiken C, Korber B, Shafer RW. HIV sequence databases. AIDS Rev 2003;5:52–61. [PMC free article] [PubMed] [Google Scholar]
- 54.Organization and financing of public health services in Europe . In: Rechel B, Jakubowski E, McKee M, et al. (eds). European Observatory on Health Systems and Policies (Health Policy Series, No. 50.), Copenhagen, Denmark, 2018. https://www.ncbi.nlm.nih.gov/books/NBK535724/. [PubMed]
- 55.Bourgeois AC, Edmunds M, Awan A, et al. HIV in Canada-surveillance report, 2016. Can Commun Dis Rep 2017;43:248–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mitchell K, Mandric I, Brito J, et al. Benchmarking of computational error-correction methods for next-generation sequencing data. Genome Biol 2020;21. doi: 10.1186/s13059-020-01988-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zagordi O, Geyrhofer L, Roth V, et al. Deep sequencing of a genetically heterogeneous sample: local haplotype reconstruction and read error correction. J Comput Biol 2010;17:417–28. [DOI] [PubMed] [Google Scholar]
- 58.Skums P, Dimitrova Z, Campo DS, et al. Efficient error correction for next-generation sequencing of viral amplicons. BMC Bioinformatics 2012;13(Suppl 10):S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Malhotra R, Jha M, Poss M, et al. A random forest classifier for detecting rare variants in NGS data from viral populations. Comput Struct Biotechnol J 2017;15:388–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Astrovskaya I, Tork B, Mangul S, et al. Inferring viral quasispecies spectra from 454 pyrosequencing reads. BMC Bioinformatics 2011;12(Suppl 6):S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hong LZ, Hong S, Wong HT, et al. BAsE-Seq: a method for obtaining long viral haplotypes from short sequence reads. Genome Biol 2014;15. doi: 10.1186/s13059-014-0517-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Warren RL, Sutton GG, Jones SJM, et al. Assembling millions of short DNA sequences using SSAKE. Bioinformatics 2007;23:500–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang X, Charlebois P, Gnerre S, et al. De novo assembly of highly diverse viral populations. BMC Genomics 2012;13:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hunt M, Gall A, Ong SH, et al. IVA: accurate de novo assembly of RNA virus genomes. Bioinformatics 2015;31:2374–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baaijens JA, El Aabidine AZ, Rivals E, et al. De novo assembly of viral quasispecies using overlap graphs. Genome Res 27:835–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wymant C, Blanquart F, Golubchik T, et al. Easy and accurate reconstruction of whole HIV genomes from short-read sequence data with shiver. Virus Evol 2018;4:vey007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bellecave P, Recordon-Pinson P, Papuchon J, et al. Detection of low-frequency HIV type 1 reverse transcriptase drug resistance mutations by ultradeep sequencing in naive HIV type 1-infected individuals. AIDS Res Hum Retroviruses 2014;30:170–3. [DOI] [PubMed] [Google Scholar]
- 68.Arias A, López P, Sánchez R, et al. Sanger and next generation sequencing approaches to evaluate HIV-1 virus in blood compartments. Int J Environ Res Public Health 2018;15:pii: E1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tzou PL, Ariyaratne P, Varghese V, et al. Comparison of an in vitro diagnostic next-generation sequencing assay with sanger sequencing for HIV-1 genotypic resistance testing. J Clin Microbiol 2018;56:pii: e00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koboldt DC, Chen K, Wylie T, et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009;25:2283–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Verbist BMP, Thys K, Reumers J, et al. VirVarSeq: a low-frequency virus variant detection pipeline for Illumina sequencing using adaptive base-calling accuracy filtering. Bioinformatics 2015;31:94–101. [DOI] [PubMed] [Google Scholar]
- 72.Wilm A, Aw PPK, Bertrand D, et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 2012;40:11189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Macalalad AR, Zody MC, Charlebois P, et al. Highly sensitive and specific detection of rare variants in mixed viral populations from massively parallel sequence data. PLoS Comput Biol 2012;8:e1002417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang X, Charlebois P, Macalalad A, et al. V-Phaser 2: variant inference for viral populations. BMC Genomics 2013;14:674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Routh A, Chang MW, Okulicz JF, et al. CoVaMa: co-variation mapper for disequilibrium analysis of mutant loci in viral populations using next-generation sequence data. Methods 2015;91:40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Knyazev S, Tsyvina V, Melnyk A, et al. CliqueSNV: scalable reconstruction of intra-host viral populations from ngs reads. bioRxiv 2018. doi: 10.1101/264242. [DOI] [Google Scholar]
- 77.Isakov O, Bordería AV, Golan D, et al. Deep sequencing analysis of viral infection and evolution allows rapid and detailed characterization of viral mutant spectrum. Bioinformatics 2015;31:2141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Verbist B, Clement L, Reumers J, et al. ViVaMBC: estimating viral sequence variation in complex populations from illumina deep-sequencing data using model-based clustering. BMC Bioinformatics 2015;16:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huber M, Metzner KJ, Geissberger FD, et al. MinVar: a rapid and versatile tool for HIV-1 drug resistance genotyping by deep sequencing. J Virol Methods 2017;240:7–13. [DOI] [PubMed] [Google Scholar]
- 80.Ferretti L, Tennakoon C, Silesian A, et al. SiNPle: fast and sensitive variant calling for deep sequencing data. Genes 2019;10:pii: E561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Noguera-Julian M. HIV drug resistance testing—the quest for point-of-care. EBioMedicine 2019;50:11–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zagordi O, Bhattacharya A, Eriksson N, et al. ShoRAH: estimating the genetic diversity of a mixed sample from next-generation sequencing data. BMC Bioinformatics 2011;12:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Eriksson N, Pachter L, Mitsuya Y, et al. Viral population estimation using pyrosequencing. PLoS Comput Biol 2008;4:e1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Westbrooks K, Astrovskaya I, Campo D, et al. HCV Quasispecies assembly using network flows. Bioinformatics Res Appl 4983:159–70. [Google Scholar]
- 85.Mancuso N, Tork B, Skums P, et al. Reconstructing viral quasispecies from NGS amplicon reads. In Silico Biol 2011;11:237–49. [DOI] [PubMed] [Google Scholar]
- 86.Huang A, Kantor R, DeLong A, et al. QColors: an algorithm for conservative viral quasispecies reconstruction from short and non-contiguous next generation sequencing reads. In Silico Biol. 2011;11:193–201. doi: 10.3233/ISB-2012-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Prosperi MCF, Salemi M. QuRe: software for viral quasispecies reconstruction from next-generation sequencing data. Bioinformatics 2012;28:132–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Skums P, Mancuso N, Artyomenko A, et al. Reconstruction of viral population structure from next-generation sequencing data using multicommodity flows. BMC Bioinformatics 2013;14:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Töpfer A, Marschall T, Bull RA, et al. Viral quasispecies assembly via maximal clique enumeration. PLoS Comput Biol 2014;10:e1003515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mangul S, Wu NC, Mancuso N, et al. Accurate viral population assembly from ultra-deep sequencing data. Bioinformatics 2014;30:i329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jayasundara D, Saeed I, Maheswararajah S, et al. ViQuaS: an improved reconstruction pipeline for viral quasispecies spectra generated by next-generation sequencing. Bioinformatics 2015;31:886–96. [DOI] [PubMed] [Google Scholar]
- 92.Chen J, Zhao Y, Sun Y. De novo haplotype reconstruction in viral quasispecies using paired-end read guided path finding. Bioinformatics 2018;34:2927–35. [DOI] [PubMed] [Google Scholar]
- 93.Mandoiu I, Zelikovsky A. Computational Methods for Next Generation Sequencing Data Analysis, Hoboken, NJ: John Wiley & Sons, 2016, ISBN: 978-1-118-16948-3. [Google Scholar]
- 94.Jojic V, Hertz T, Jojic N. Population sequencing using short reads: HIV as a case study. Pac Symp Biocomput 2008;114–25. [PubMed] [Google Scholar]
- 95.Töpfer A, Zagordi O, Prabhakaran S, et al. Probabilistic inference of viral quasispecies subject to recombination. J Comput Biol 2013;20:113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Prabhakaran S, Rey M, Zagordi O, et al. HIV haplotype inference using a propagating Dirichlet process mixture model. IEEE/ACM Trans Comput Biol Bioinform 2014;11:182–91. [DOI] [PubMed] [Google Scholar]
- 97.Leviyang S, Griva I, Ita S, et al. A penalized regression approach to haplotype reconstruction of viral populations arising in early HIV/SIV infection. Bioinformatics 2017;33:2455–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Malhotra R, Wu MMS, Rodrigo A, et al. Maximum likelihood de novo reconstruction of viral populations using paired end sequencing data. arXiv 2015. doi: https://arxiv.org/abs/1502.04239. [DOI] [PubMed] [Google Scholar]
- 99.Artyomenko A, Wu NC, Mangul S, et al. Long single-molecule reads can resolve the complexity of the influenza virus composed of rare, closely related mutant variants. J Comput Biol 2017;24:558–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ahn S, Vikalo H. aBayesQR: a Bayesian method for reconstruction of viral populations characterized by low diversity. J Comput Biol 2018;25:637–48. [DOI] [PubMed] [Google Scholar]
- 101.Eliseev A, Gibson KM, Avdeyev P, et al. Evaluation of haplotype callers for next-generation sequencing of viruses. Infect Genet Evol 2020;82:104277. doi: 10.1016/j.meegid.2020.104277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu TF, Shafer RW. Web resources for HIV type 1 genotypic-resistance test interpretation. Clin Infect Dis 2006;42:1608–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rosenthal P. Faculty of 1000 evaluation for hepatitis C virus drug resistance-associated substitutions: state of the art summary. Hepatology 2015;62((5)):1623–32. [DOI] [PubMed] [Google Scholar]
- 104.Irwin KK, Renzette N, Kowalik TF, et al. Antiviral drug resistance as an adaptive process. Virus Evol 2016;2:vew014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gibson KM, Steiner MC, Kassaye S, et al. Corrigendum: a 28-year history of HIV-1 drug resistance and transmission in Washington, DC. Front Microbiol 2019;10. doi: 10.3389/fmicb.2019.02590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Assefa Y, Gilks CF. Second-line antiretroviral therapy: so much to be done. Lancet HIV 2017;4:e424–5. [DOI] [PubMed] [Google Scholar]
- 107.Flynn WF, Chang MW, Tan Z, et al. Deep sequencing of protease inhibitor resistant HIV patient isolates reveals patterns of correlated mutations in gag and protease. PLoS Comput Biol 2015;11:e1004249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Feder AF, Rhee S-Y, Holmes SP, et al. More effective drugs lead to harder selective sweeps in the evolution of drug resistance in HIV-1. Elife 2016;5. doi: 10.7554/eLife.10670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Riemenschneider M, Heider D. Current approaches in computational drug resistance prediction in HIV. Curr HIV Res 2016;14:307–15. [DOI] [PubMed] [Google Scholar]
- 110.Larder BA, Kohli A, Kellam P, et al. Quantitative detection of HIV-1 drug resistance mutations by automated DNA sequencing. Nature 1993;365:671–3. [DOI] [PubMed] [Google Scholar]
- 111.Döring M, Büch J, Friedrich G, et al. geno2pheno[ngs-freq]: a genotypic interpretation system for identifying viral drug resistance using next-generation sequencing data. Nucleic Acids Res 2018;46:W271–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hamers RL, Paredes R. Next-generation sequencing and HIV drug resistance surveillance. Lancet HIV 2016;3:e553–4. [DOI] [PubMed] [Google Scholar]
- 113.Johnson JA, Li J-F, Wei X, et al. Minority HIV-1 drug resistance mutations are present in antiretroviral treatment–Naïve populations and associate with reduced treatment efficacy. PLoS Med 2008;5:e158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pawar SD, Freas C, Weber IT, et al. Analysis of drug resistance in HIV protease. BMC Bioinformatics 2018;19:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Obermeier M, Pironti A, Berg T, et al. HIV-GRADE: a publicly available, rules-based drug resistance interpretation algorithm integrating bioinformatic knowledge. Intervirology 2012;55:102–7. [DOI] [PubMed] [Google Scholar]
- 116.Woods CK, Brumme CJ, Liu TF, et al. Automating HIV drug resistance genotyping with RECall, a freely accessible sequence analysis tool. J Clin Microbiol 2012;50:1936–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Beerenwinkel N, Däumer M, Oette M, et al. Geno2pheno: estimating phenotypic drug resistance from HIV-1 genotypes. Nucleic Acids Res 2003;31:3850–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shafer RW. Rationale and uses of a public HIV drug-resistance database. J Infect Dis 2006;194:S51–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cashin K, Gray LR, Harvey KL, et al. Reliable genotypic tropism tests for the major HIV-1 subtypes. Sci Rep 2015;5. doi: 10.1038/srep08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Case Medical Research . FDA authorizes marketing of first next-generation sequencing test for detecting HIV-1 drug resistance mutations. Case Med Res 2019. https://www.fda.gov/news-events/press-announcements/fda-authorizes-marketing-first-next-generation-sequencing-test-detecting-hiv-1-drug-resistance. [Google Scholar]
- 121.Metzner KJ, Rauch P, Walter H, et al. Detection of minor populations of drug-resistant HIV-1 in acute seroconverters. AIDS 2005;19:1819–25. [DOI] [PubMed] [Google Scholar]
- 122.Fisher RG, Smith DM, Murrell B, et al. Next generation sequencing improves detection of drug resistance mutations in infants after PMTCT failure. J Clin Virol 2015;62:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Alidjinou EK, Deldalle J, Hallaert C, et al. RNA and DNA sanger sequencing versus next-generation sequencing for HIV-1 drug resistance testing in treatment-naive patients. J Antimicrob Chemother 2017;72:2823–30. [DOI] [PubMed] [Google Scholar]
- 124.Fokam J, Bellocchi MC, Armenia D, et al. Next-generation sequencing provides an added value in determining drug resistance and viral tropism in Cameroonian HIV-1 vertically infected children. Medicine 2018;97:e0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Derache A, Iwuji CC, Danaviah S, et al. Predicted antiviral activity of tenofovir versus abacavir in combination with a cytosine analogue and the integrase inhibitor dolutegravir in HIV-1-infected south African patients initiating or failing first-line ART. J Antimicrob Chemother 2019;74:473–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Derache A, Iwuji CC, Baisley K, et al. Impact of next-generation sequencing defined human immunodeficiency virus pretreatment drug resistance on virological outcomes in the ANRS 12249 treatment-as-prevention trial. Clin Infect Dis 2019;69:207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Carlisle LA, Turk T, Kusejko K, et al. Viral diversity based on next-generation sequencing of HIV-1 provides precise estimates of infection Recency and time since infection. J Infect Dis 2019;220:254–65. [DOI] [PubMed] [Google Scholar]
- 128.Montoya V, Olmstead AD, Janjua NZ, et al. Differentiation of acute from chronic hepatitis C virus infection by nonstructural 5B deep sequencing: a population-level tool for incidence estimation. Hepatology 2015;61:1842–50. [DOI] [PubMed] [Google Scholar]
- 129.Astrakhantseva IV, Campo DS, Araujo A, et al. Differences in variability of hypervariable region 1 of hepatitis C virus (HCV) between acute and chronic stages of HCV infection. In Silico Biol 2011;11:163–73. [DOI] [PubMed] [Google Scholar]
- 130.Baykal PI, Artyomenko A, Ramachandran S, et al. Assessment of HCV infection stage as recent or chronic using multi-parameter analysis and machine learning. 2017 IEEE 7th International Conference on Computational Advances in Bio and Medical Sciences (ICCABS) 2017; 1–1. doi: 10.1109/ICCABS.2017.8114316. [DOI]
- 131.Basodi S, Baykal PI, Zelikovsky A, et al. Analysis of heterogeneous genomic samples using image normalization and machine learning. Submitted 2019. doi: 10.1101/642108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Basodi S, Icer PB, Skums P, et al. Classification of HCV infections through sequence image normalization. 2017 IEEE 7th International Conference on Computational Advances in Bio and Medical Sciences (ICCABS), 2017. doi: 10.1109/ICCABS.2017.8114313. [DOI]
- 133.Ramachandran S, Campo DS, Dimitrova ZE, et al. Temporal variations in the hepatitis C virus intrahost population during chronic infection. J Virol 2011;85:6369–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gismondi MI, Díaz Carrasco JM, Valva P, et al. Dynamic changes in viral population structure and compartmentalization during chronic hepatitis C virus infection in children. Virology 2013;447:187–96. [DOI] [PubMed] [Google Scholar]
- 135.Domingo-Calap P, Segredo-Otero E, Durán-Moreno M, et al. Social evolution of innate immunity evasion in a virus. Nat Microbiol 2019;4:1006–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Oster AM, France AM, Panneer N, et al. Identifying clusters of recent and rapid HIV transmission through analysis of molecular surveillance data. J Acquir Immune Defic Syndr 2018;79:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rasmussen DA, Volz EM, Koelle K. Phylodynamic inference for structured epidemiological models. PLoS Comput Biol 2014;10:e1003570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Volz EM, Koelle K, Bedford T. Viral phylodynamics. PLoS Comput Biol 2013;9:e1002947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Klinkenberg D, Backer JA, Didelot X, et al. Simultaneous inference of phylogenetic and transmission trees in infectious disease outbreaks. PLoS Comput Biol 2017;13:e1005495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jombart T, Eggo RM, Dodd PJ, et al. Reconstructing disease outbreaks from genetic data: a graph approach. Heredity 2011;106:383–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.De Maio N, Wu C-H, Wilson DJ. SCOTTI: efficient reconstruction of transmission within outbreaks with the structured coalescent. PLoS Comput Biol 2016;12:e1005130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jombart T, Cori A, Didelot X, et al. Bayesian reconstruction of disease outbreaks by combining epidemiologic and genomic data. PLoS Comput Biol 2014;10:e1003457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mollentze N, Nel LH, Townsend S, et al. A Bayesian approach for inferring the dynamics of partially observed endemic infectious diseases from space-time-genetic data. Proc R Soc B 2014;281:20133251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Morelli MJ, Thébaud G, Chadœuf J, et al. A Bayesian inference framework to reconstruct transmission trees using epidemiological and genetic data. PLoS Comput Biol 2012;8:e1002768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ypma RJF, van Ballegooijen WM, Wallinga J. Relating phylogenetic trees to transmission trees of infectious disease outbreaks. Genetics 2013;195:1055–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Alroy-Preis S, Daly ER, Adamski C, et al. Large outbreak of hepatitis C virus associated with drug diversion by a healthcare technician. Clin Infect Dis 2018;67:845–53. [DOI] [PubMed] [Google Scholar]
- 147.Salemi M. The intra-host evolutionary and population dynamics of human immunodeficiency virus type 1: a phylogenetic perspective. Infect Dis Rep 2013;5:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Campo DS, Xia G-L, Dimitrova Z, et al. Accurate genetic detection of hepatitis C virus transmissions in outbreak settings. J Infect Dis 2016;213:957–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rytsareva I, Campo DS, Zheng Y, et al. Efficient detection of viral transmissions with next-generation sequencing data. BMC Genomics 2017;18:372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tsyvina V, Campo DS, Sims S, et al. Fast estimation of genetic relatedness between members of heterogeneous populations of closely related genomic variants. BMC Bioinformatics 2018;19:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Romero-Severson EO, Bulla I, Leitner T. Phylogenetically resolving epidemiologic linkage. Proc Natl Acad Sci USA 2016;113:2690–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wymant C, Hall M, Ratmann O, et al. PHYLOSCANNER: inferring transmission from within- and between-host pathogen genetic diversity. Mol Biol Evol 2018;35:719–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hadfield J, Megill C, Bell SM, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 2018;34:4121–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.RECON-R Epidemics Consortium . R epidemics consortium. https://www.repidemicsconsortium.org/.
- 156.Akiyama MJ, Lipsey D, Ganova-Raeva L, et al. A phylogenetic analysis of HCV transmission, relapse, and reinfection among people who inject drugs receiving opioid agonist therapy. J Infect Dis 2020. doi: 10.1093/infdis/jiaa100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ramachandran S, Thai H, Forbi JC, et al. A large HCV transmission network enabled a fast-growing HIV outbreak in rural Indiana, 2015. EBioMedicine 2018;37:374–81. [DOI] [PMC free article] [PubMed] [Google Scholar]