

Abstract
Most genetic variants for colorectal cancer (CRC) identified in genome-wide association studies (GWAS) are located in intergenic regions, implying pathogenic dysregulations of gene expression. However, comprehensive assessments of target genes in CRC remain to be explored. We conducted a multi-omics analysis using transcriptome and/or DNA methylation data from the Genotype-Tissue Expression, The Cancer Genome Atlas and the Colonomics projects. We identified 116 putative target genes for 45 GWAS-identified variants. Using summary-data-based Mendelian randomization approach (SMR), we demonstrated that the CRC susceptibility for 29 out of the 45 CRC variants may be mediated by cis-effects on gene regulation. At a cutoff of the Bonferroni-corrected PSMR < 0.05, we determined 66 putative susceptibility genes, including 39 genes that have not been previously reported. We further performed in vitro assays for two selected genes, DIP2B and SFMBT1, and provide functional evidence that they play a vital role in colorectal carcinogenesis via disrupting cell behavior, including migration, invasion and epithelial–mesenchymal transition. Our study reveals a large number of putative novel susceptibility genes and provides additional insight into the underlying mechanisms for CRC genetic risk loci.
Introduction
Genetic factors play an important role in the etiology of both sporadic and familial colorectal cancer (CRC). Multiple CRC moderate- or high-penetrance genes, including APC, MUTYH, MLH1, PMS2, MSH2, MSH6, PTEN, SMAD4, BMPR1A, POLE and POLD, have been identified as being responsible for CRC family syndromes, such as Lynch syndrome (1–3). In addition to these known moderate- or high-penetrance genes, ~150 genetic variants have been identified for CRC risk through genome-wide association studies (GWAS) (4–20). However, the underlying biological mechanisms and potential target genes for most of these GWAS-identified risk loci remain unclear.
Expression quantitative trait loci (eQTL) analyses have been widely used to identify putative target genes for a genetic variant based on the association between its genotype and gene expression levels. However, target genes that are reported by previous eQTL analyses that only use single transcriptome datasets may be subject to substantial type I or type II errors due to limited sample sizes. Our recent study showed that a meta-analysis that combined the regression results of eQTL analyses from different studies can improve target gene discovery. Using this approach, we reported 31 putative target genes from 83 lead single-nucleotide polymorphisms (SNPs) with the strongest association with CRC risk in a locus identified in European populations for CRC risk (21). However, a systematic analysis for previously reported GWAS-identified SNPs identified from all populations, especially those newly identified for CRC (20), is lacking. On the other hand, it has been well recognized that genetic effects on gene expression could be mediated through DNA methylation at CpG sites (22,23). Methylation quantitative trait loci (mQTL) analyses (24–26) have been used to evaluate the association of a SNP with DNA methylation levels at CpG sites of the nearest gene. This approach has been used widely to explore the target discovery of GWAS loci (22,24,27–29). However, such an mQTL analysis has not been conducted in previous CRC studies due to a lack of methylation data in colorectal normal tissues. Recently, colocalization analyses of GWAS association signals and eQTL data were conducted to illustrate the effects of a lead SNP on the risk of a phenotype that may be mediated by the expression of the identified target gene (26,30). Specifically, summary-data-based Mendelian randomization (SMR) has recently been developed to integrate summary-level multi-omics data from independent GWAS and eQTL (or mQTL) studies. Therefore, the exploration of target genes for lead SNPs through the integration of multi-omics data, including GWAS, gene expression and methylation data analysis, may provide further understanding of the biological mechanisms that contribute to cancer development.
In this study, we evaluated all previously reported risk genetic variants and characterized 131 lead SNPs for CRC (Supplementary Material, Table S1). We conducted an integrative multi-omics data analysis using the transcriptome data from the Genotype-Tissue Expression (GTEx) project and The Cancer Genome Atlas (TCGA), and both transcriptome and methylation data from the Colonomics project to identify putative susceptibility genes for CRC. Furthermore, we performed colocalization analyses of these identified genes and in vitro functional assays of two selected genes, DIP2B and SFMBT1, in multiple CRC cell lines. These findings provide additional insight into the underlying mechanisms for genetic susceptibility of CRC.
Results
Identification of putative target genes for lead SNPs using multi-omics data
We characterized 131 lead SNPs associated with CRC risk at P < 5.0 × 10−8 from the data in previous literature (Supplementary Material, Table S1). To identify potential target genes for lead SNPs, we conducted an eQTL analysis for lead SNPs and their nearby genes (±1 Mb region) using data from the GTEx, TCGA and Colonomics projects (see Materials and Methods). A further meta-analysis of the eQTL results revealed a total of 98 putative target genes for 39 lead SNPs at a Benjamini–Hochberg (BH)-corrected P < 0.05 (Fig. 1 and Supplementary Material, Table S2; see Materials and Methods). Of these putative target genes, 34 have been reported in previous eQTL studies. The remaining 64 have not been previously linked to CRC risk (Supplementary Material, Table S2).
Figure 1 .
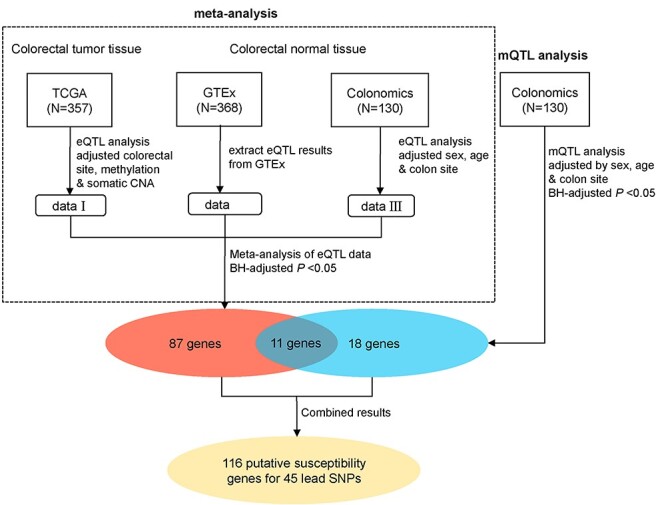
A workflow of the identification of putative target genes for CRC lead SNPs combining the results from the meta-analysis of eQTL and the mQTL analysis. The meta-eQTL analysis integrated the cis-eQTL results from TCGA, GTEx and Colonomics, whereas the mQTL analysis used data from Colonomics. The red oval indicates the putative target genes identified from meta-eQTL results. The blue oval indicates the target genes identified from the mQTL analysis and the yellow oval refers to unique putative target genes combining both results from the meta-eQTL analysis and mQTL analysis.
Next, we conducted an mQTL analysis for lead SNPs using the methylation data from 130 colorectal normal tissues from the Colonomics project (see Materials and Methods). We showed that DNA methylation levels at CpG sites for a total of 29 genes were associated with 18 lead SNPs (Supplementary Material, Table S2). Of these genes, 11 were identified by our meta-analysis of eQTL results from gene expression data (Fig. 1 and Supplementary Material, Table S2). All of the remaining 18 genes have not been previously linked to CRC risk (Supplementary Material, Table S2).
Colocalization analysis
We next conducted a colocalization analysis for our identified putative target genes for 45 lead SNPs using the SMR approach (see Materials and Methods). Integrating the meta-analysis results from eQTL analyses and GWAS association signals, we showed that the effects of 23 lead SNPs on cancer risk may be mediated by regulating the expression of their putative target genes at a cutoff of Bonferroni-corrected PSMR < 0.05 (Supplementary Material, Table S3). Specifically, a total of 47 genes were identified for these lead SNPs. Although the other putative target genes did not reach the threshold of the Bonferroni correction in the SMR analysis, they showed nominal significance at P < 0.05 (Supplementary Material, Table S3). In addition, integrating the results from the mQTL analysis and GWAS association signals, we showed that the effects of 16 lead SNPs on cancer risk may be mediated by DNA methylations of the putative target genes, at a cutoff of the Bonferroni-corrected PSMR < 0.05 (Supplementary Material, Table S3). Specifically, a total of 27 genes were identified for these lead SNPs. In particular, a total of six lead SNPs and eight genes, ATF1, C11orf53, COLCA1, COLCA2, FADS2, FHL3, FUT2 and HLA-DRB6, were commonly detected in colocalization analyses from the meta-analysis of eQTL studies (Supplementary Material, Table S3). The findings suggest that these six lead SNPs of cancer risk may be mediated by the regulation of methylations and, consequently, altered gene expressions. Taken together, we demonstrated that the CRC susceptibility for 29 out of the 45 CRC variants (a total of 66 putative target genes) may be mediated by their cis-effects on the gene regulation via either DNA methylation or gene expression. Of these 66 putative target genes, including 39 have not been previously reported (Supplementary Material, Table S4). Functional enrichment analyses using an Ingenuity Pathway Analysis (IPA) revealed that these genes were significantly enriched in endocrine system disorders, and gastrointestinal, immunological and metabolic diseases (P < 0.01 for all).
In vitro functional assays for the putative oncogenes, DIP2B and SFMBT1
Our results showed that a lower predicted expression of the two genes, DIP2B and SFMBT1, may mediate the effect of the lead SNPs on a decreased risk of CRC (Supplementary Material, Tables S2 and S3). We further performed in vitro functional assays in different CRC cell lines to investigate putative oncogenic cellular functions of both selected genes (see Materials and Methods). We conducted quantitative polymerase chain reaction (qPCR) experiments to evaluate their relative expression levels in four CRC cell lines, RKO, SW480, SW620 and HT29. By evaluating their relative messenger RNA (mRNA) expression levels in these cell lines, we selected RKO and SW620 cells for functional assays (Fig. 2A). We designed two shRNA lentivirus transfection experiments for each of the genes, DIP2B and SFMBT1, in RKO and SW620 cells by constructing a stable cell line (see Materials and Methods).
Figure 2 .
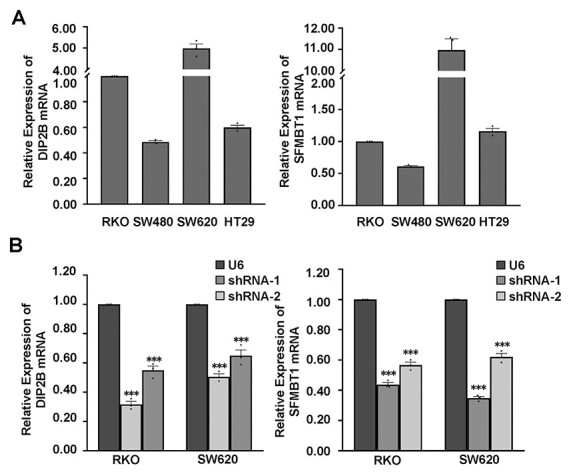
Knockdown of endogenous DIP2B and SFMBT1 in RKO and SW620 cell lines. (A) Quantitative RT-PCR analysis for DIP2B and SFMBT1 mRNA levels in various colorectal cancer cell lines; Beta-actin was used as a loading control. (B) The relative mRNA expression for target genes in RKO and SW620 cell lines transfected with U6 (control) and shRNA lentiviruses; Beta-actin was used as a loading control. The data are the mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
Using qPCR and western blot in both RKO and SW620 cells, we verified the high knockdown efficiency of both genes in the RKO and SW620 cells. Specifically, for both genes in both shRNA lentivirus transfections, we observed reductions of ~35–70% of the endogenous transcript and 38–64% of protein levels, respectively, in both cell lines (Figs 2B and 4C and D). Upon knockdown of both genes, both cell migration and invasion were significantly decreased compared with the control cell lines in transwell assays (Fig. 3A and B). Specifically, the gene DIP2B showed the most significant effects on cell migration in SW620 (Fig. 3A and B). In addition to cell migration, the gene SFMBT1 showed the most significant effects on cell invasion in SW620 (Fig. 3A and B). Furthermore, after the knockdown, the wound healing efficiency was decreased compared with the control cells (Fig. 4A and B), confirming that both genes indeed affect cell migration. A western blot analysis revealed decreased levels of β-catenin, vimentin and snail in target gene-depleted cell lines, suggesting that they crucially affect the epithelial–mesenchymal transition (EMT) (Fig. 4C and D and Supplementary Material, Fig. S2). This indicates that they may affect the metastasis of CRC by regulating EMT. Notably, the results from two independent short hairpin RNA (shRNA) assays for both genes are consistent in the RKO and SW620 cells. In addition to the effects of these cell behaviors, the results of the Cell Counting Kit-8 (CCK8) and colony formation assays showed that knockdown of both genes did not affect cell proliferation in two CRC cell lines (Supplementary Material, Fig. S1). In summary, our results suggest that the two putative susceptibility genes likely function as oncogenes, consistent with our observations from the eQTL and GWAS. These findings suggest that the disruption of this cell behavior may play a vital role in carcinogenesis and cancer susceptibility.
Figure 4 .
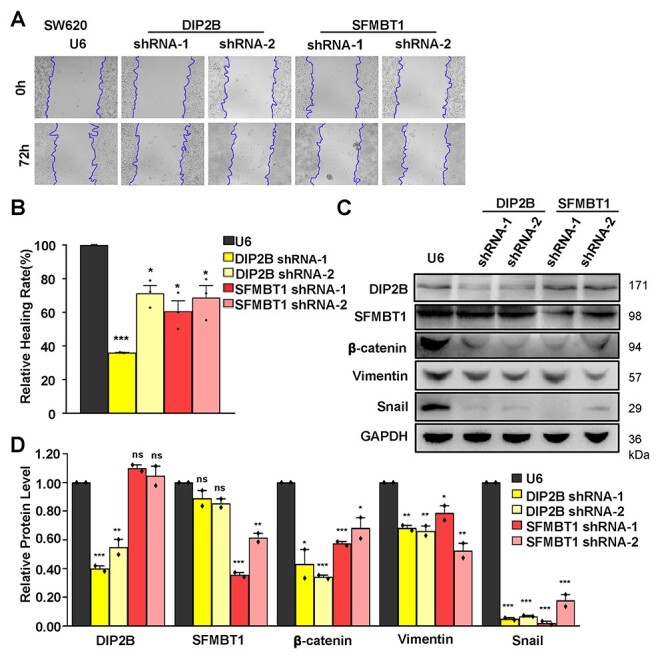
Wound healing assays and western blot analysis of the ability of cell migration and invasion in DIP2B and SFMBT1 knockdown cells. (A) Wound healing assays were performed in SW620 cells transfected with U6 and shRNA lentiviruses. Representative images are shown after 0 and 72 h (magnification ×10). The unhealed areas were measured and quantified by ImageJ software. Three independent experiments were performed. (B) Quantification of scratch areas in SW620 cells transfected with U6 and shRNA lentiviruses. Three images were taken for every membrane. (C) Total SW620 cells lysate of transfected cells with U6 (control) and shRNA lentiviruses were analyzed by western blot for the indicated proteins of EMT, DIP2B and SFMBT1. GAPDH was used as a loading control. (D) Quantification of the bands’ intensity in SW620 cells transfected with U6 and shRNA lentiviruses. Two independent experiments were performed. The data are the mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 3 .
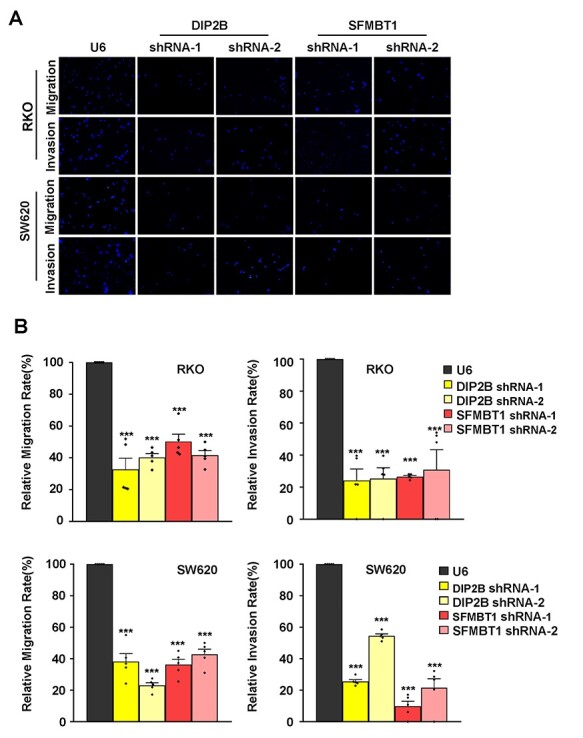
DIP2B and SFMBT1 knockdown affects cell migration and invasion in vitro by transwell assays. (A) Migration and invasion assays were performed in RKO and SW620 cell lines transfected with U6 (control) and shRNA lentiviruses. After 24 h, migratory or invasive cells were stained with DAPI and imaged with the excitation wavelengths of 360–400 nm by using a Leica DM4000 microscope (magnification ×20). The independent experiments were performed in triplicate. (B) Quantification of DAPI-positive cells on the membrane. Images were taken for every membrane. The data are the mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Here, we identified a total of 66 putative susceptibility genes, including 39 genes that have not been previously reported. We combined the regression results of eQTL analyses from three studies, the GTEx, TCGA and Colonomics projects, with the meta-analysis, which can significantly improve target gene discovery. In addition to the meta-analysis, for the first time, we conducted an mQTL analysis for all reported lead SNPs and identified an additional 18 putative CRC susceptibility genes. Furthermore, using SMR approach, we illustrated CRC susceptibility associated with a majority of lead SNPs (29 out of 45 lead SNPs) that are likely mediated by cis-effects on the regulation of their putative target genes. Our results provide strong statistical evidence to establish the underlying mechanism to link the risk loci, genes and CRC risk.
Our results showed that a reduced predicted expression of two genes, DIP2B and SFMBT1, confer a decreased risk of CRC. We verified their potential oncogenic function using in vitro functional assays in multiple CRC cells. Consistently, the gene SFMBT1 has been previously reported to regulate the EMT process as a histone reader subunit of the lysergic acid diethylamide 1 (LSD1) demethylase complex and is highly expressed in human cancer, including breast and lung cancer (31). SFMBT1 is essential for Snai1-dependent recruitment of LSD1 to chromatin, and subsequent H3K4me2 demethylation. The higher expression level of this gene is associated with a poorer prognosis (31). It should be noted that SFMBT1 was also identified as a putative susceptibility gene by our recent transcriptome-wide association analysis in CRC (32). Another gene, DIP2B, encodes a protein with a DNA methyltransferase 1 associated protein 1 binding domain, which suggests a role in DNA methylation machinery (33). DIP2B may also regulate the EMT process through changing DNA methylation, as shown by DIP2C, a homologous gene from a previous study (34).
We additionally conducted bioinformatics analyses of various epigenetic data to search the additional evidence of the identified putative target genes (See Materials and Methods). Of the 66 genes identified from the SMR analysis, we observed that a total of 41 genes (62.12%, 41 out of 66) showed potential evidence of cis-regulation by putative functional SNPs via promoter or enhancer–promoter interactions, which were supported by the data from proximal functional SNPs (n = 28), chromatin–chromatin interactions (n = 29) and FATOM5 (n = 9) (Supplementary Material, Table S5). Specifically, we observed that five genes, FUT2, HCG20, MAMSTR, PSORS1C1 and SMAD9, can be supported by the evidence of chromatin–chromatin interactions in CRC relevant cells and tissues using data from previous literature (35,36) (Supplementary Material, Tables S4 and S5). These provide an additional layer of evidence to support that the CRC susceptibility for the lead SNPs may be mediated by their cis-effects on gene regulation (Supplementary Material, Tables S4 and S5). Furthermore, we searched additional in vitro/vivo evidence for the identified putative target genes in previous studies in CRC and other cancer types. In particular, 18 of these 66 genes (27%), such as ATF1, CERS5, FADS2 and TOX2, have been verified to be involved in cancer-related functions in in vitro experiment studies of CRC or other cancer types (Supplementary Material, Table S4). It should be also noted that we observed that 23 of 66 genes exhibited at P < 0.05 in at least two studies, which provided strong statistical evidence of the associations (Supplementary Material, Table S4).
Our results show that a substantial proportion of genes (eight genes, 30% of genes from SMR in mQTL analysis) were identified in both mQTL and eQTL analyses with the classical inverse relationship, providing stronger evidence of these findings. In addition, of the remaining 19 genes, we have also observed that three genes, RASIP1, PNKD and TMBIM1, were supported by eQTL analysis at a nominal P value < 0.05 (Supplementary Material, Table S2). Although some of the mQTL discovery could be a complicated relationship with eQTL results due to many factors (i.e. distinct sample sizes and different genetic effects on DNA methylation and gene expression), in addition to the eQTL approach, we highlighted the importance of mQTL analysis in the discovery of target genes in genetic studies. It should be noted that we identified seven genes for GWAS-identified variants in Asian populations, whereas the multi-omics data were primarily generated in European populations (Supplementary Material, Table S3). Although there is a possibility for causative variants (or in strong linkage disequilibrium (LD)) shared in both Asian and European populations, the findings need to be further verified using data in Asian populations.
In conclusion, we identified a large number of putative susceptibility genes for lead SNPs supported by strong statistical evidence. We demonstrated the susceptibility of two selected genes, DIP2B and SFMBT1, using in vitro functional assays. Our study discovered underlying mechanisms for the association of genetic susceptibility risk loci with CRC and provided additional insight into the genetic and biological basis for the pathogenesis of this common cancer.
Materials and Methods
Data resources
For the SNPs reported from previous GWAS, each lead SNP with the best association in a locus with CRC risk has been systematically evaluated from the latest meta-analysis combing data from the Genetics and Epidemiology of Colorectal Cancer Consortium, the Colorectal Cancer Transdisciplinary Study and the Colon Cancer Family Registry (N = 125 K) (19). We included a total of 82 lead SNPs reported from the literature (Supplementary Material, Table S1). In addition, we included a total of 31 lead SNPs identified by 34 627 CRC cases and 71 379 controls of European ancestry from the COGENT (COlorectal cancer GENeTics) consortium (20) and an additional 18 GWAS-identified variants from Asian populations (Supplementary Material, Table S1). We characterized a total of 131 lead SNPs in our analysis (Supplementary Material, Table S1).
We downloaded TCGA colorectal adenocarcinoma data, including gene expression data (measured by median z score), DNA methylation data (Infinium Human Methylation 450) and somatic copy number alterations (SCNAs) data from cBioPortal. We also downloaded SNP data that were genotyped using the Affymetrix SNP 6.0 array in blood samples from the colon adenocarcinoma and rectum adenocarcinoma patients from TCGA’s data portal. We imputed SNP data using the reference genome from the 1000 Genomes project with the Minimac tool (37), implemented in the Michigan Imputation Server. Only common SNPs (minor allele frequency > 0.05) with high imputation quality (R2 > 0.3) were included. Genotype data of the nearby 1 Mb region for the GWAS-identified loci were extracted for our cis-eQTL analysis. We used a surrogate SNP in strong LD (R2 > 0.9) instead of the lead SNP if the lead SNP failed to meet the above criteria of either common or high imputation quality. In the end, a total of 357 tumor samples with gene expression, DNA methylation, SCNA and SNP data from TCGA were used for a downstream eQTL analysis. For GTEx data, we downloaded cis-eQTL results from normal transverse tissues (N = 368) in the GTEx database (version 8). For Colonomics data, the genotype, normalized gene expression and methylation data from the Colonomics project (N = 130, N = 37 for healthy colonic mucosa and N = 93 for normal mucosa adjacent to colorectal tumor tissues), have been described in previous literature (38,39).
Cis-eQTL and mQTL analysis
The eQTL and mQTL analyses across studies were conducted based on a linear regression framework. We used the regression analysis with an adjustment for co-variants for each study. For the eQTL analysis using data in tumor tissues from TCGA, we used a linear regression analysis to evaluate the associations between lead SNPs and the normalized expression levels of nearby genes (within 1 Mb to the lead SNP), with an adjustment for colorectal location site, and methylation and SCNAs (40). For the analysis from GTEx, we extracted cis-eQTL results for lead SNPs and nearby genes (within 1 Mb to the lead SNP) from the GTEx database (V8) (41), based on normal tissues from the transverse colon. For both cis-eQTL and mQTL analyses using data from the Colonomics project, we used a linear regression analysis to evaluate the associations between lead SNPs and the normalized expression or methylation levels of nearby genes (within 1 Mb to the lead SNP), adjusted for sex, age and colon site (left/right). The P-values were further adjusted with the BH procedure, and the BH-adjusted P < 0.05 was applied to identify potential target genes for each lead SNP. In addition, we evaluated the association directions (beta value) of the results between eQTL and mQTL for the same lead SNPs and target genes (i.e. a gene annotated from a methylation at CpG). We filtered the associations for lead SNPs rs9271770 (HLA-DRB1), rs4360494 (SF3A3), rs3787089 (LIME1) and rs9831861 (ITIH4) in the analysis of mQTL due to the unexpected same association directions from the eQTL analysis.
Meta-analysis
Based on beta and P-values, we conducted a meta-analysis to integrate the summary cis-eQTL results from TCGA, GTEx and Colonomics projects, following previous studies (21,42). In brief, we calculated the z score from function qnorm(P/2)*sign(beta) and further converted the standard z score derived from sum(z*sqrt(N))/sqrt(sum(N)) with a normalized weighted sampled size. Here, beta and P-value were derived from eQTL results and N referred to the sample size for each study. The meta P-value was derived from the standard z score. The combined P-values were further adjusted with the BH and Bonferroni procedure, and the BH-adjusted P < 0.05 was applied to identify potential target genes for each lead SNP. All the analyses were implemented in R.
Colocalization analyses of GWAS association signals and eQTL/mQTL studies
A colocalization analysis for the identified putative target genes was conducted using the SMR approach (30) by integrating the GWAS summary statistics and the meta-analysis results of eQTL studies. Specifically, we have a statistic:
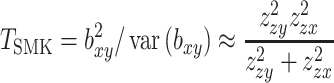 |
Here, 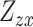 and
and 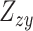 are the Z statistics for the GWAS summary statistics and meta-analysis of the cis-eQTL results, respectively.
are the Z statistics for the GWAS summary statistics and meta-analysis of the cis-eQTL results, respectively. 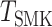 is the χ2 statistic, with one degree of freedom, which tests the significance of
is the χ2 statistic, with one degree of freedom, which tests the significance of 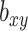 . Similarly, we performed a colocalization analysis by integrating the GWAS summary statistics data and the results from the mQTL analysis (defined as
. Similarly, we performed a colocalization analysis by integrating the GWAS summary statistics data and the results from the mQTL analysis (defined as 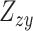 ). The significant colocalized signals were determined based on the threshold of the Bonferroni-corrected PSMR < 0.05.
). The significant colocalized signals were determined based on the threshold of the Bonferroni-corrected PSMR < 0.05.
Chromatin–chromatin interaction data analysis
We identified putative functional SNPs in strong LD (R2 > 0.8) for lead SNPs for target genes using 1000G Phase 1 European (or Asian if the lead SNP was identified from that population) population data from HaploReg v3 (43). The putative functional SNPs, with evidence of promoter or enhancer activities, annotated either from The Encyclopedia of DNA Elements (ENCODE) and/or the Roadmap Epigenomics Mapping Consortium (ROADMAP) projects, were selected for further analysis. Experimentally derived chromatin–chromatin interactions generated by High-through chromosome conformation capture (Hi-C); chromatin interaction analysis by paired-end tag sequencing (ChIA-PET); integrated method for predicting enhancer targets (IM-PET); were collected from 4DGenome (44). These data were generated from a total of 31 cell lines including human colorectal carcinoma cell line HCT116. We also analyzed data of chromatin–chromatin interactions generated in human colorectal relevant cells and tissues including HCT116, HT29, LoVo and Small Bowel, from the previous two studies (35,36). We used our in-house Perl script to examine the position of functional SNPs and ±2 kb flanking regions of the gene transcription start site to determine their chromatin–chromatin interactions. In addition, we also searched for evidence of target genes likely regulated by a lead SNP (i.e. located in an enhancer region) if there was a significant correlation of promoter–enhancer, using data from the FANTOM5 project.
Pathway enrichment analysis
For the identified high-confidence target genes, we examined their functional enrichment in the gene function category and biological pathways using the IPA tool. The top significant gene function categories and biological pathways were presented.
Cell culture
CRC cell lines, including RKO, SW480, SW620 and HT29, were obtained from the American Type Culture Collection. SW480, RKO cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 Medium, and SW620, HT-29 cells were cultured in Dulbecco's Modification of Eagle's Medium (DMEM) medium. All of the medium was supplemented with 10% fetal bovine serum (FBS) (Gibco, Cat No. 10099) and 1% penicillin/streptomycin (Gibco, Cat No. 15140). Cells were regularly maintained in a 37°C incubator with 5% CO2.
shRNAs lentivirus package and transfections
An shRNA fragment targeting the gene was generated using a pair of annealed primers and subcloned into the pLent-U6-RFP-Puro vector (Vigene Biosciences) via BamHI and MluI sites, and the resulting plasmid was confirmed by sequencing. The shRNA sequences were obtained from Sigma TRC shRNA libraries and the TRC library (http://www.broadinstitute.org/rnai/public/). Two putative susceptibility genes, DIP2B and SFMBT1, were selected for functional assays based on evaluating the top association results from (i) SMR results (Supplementary Material, Table S3), (ii) they were supported by the functional genomics data (Supplementary Material, Table S5) and (iii) no in vitro/in vivo experiments conducted in CRC (Supplementary Material, Table S4). Two shRNA sequences were designed to target DIP2B and SFMBT1 individually. The shRNA sequences are listed in Supplementary Material, Table S6. The viral production and infection is described in detail (http://www.addgene.org/tools/protocols/plko/). Packaged viruses were transfected to the cells with polybrene. After 72 h post-transfection, we performed a real-time qPCR analysis to verify the knockdown efficiency.
Reverse transcription PCR analysis
Cells were collected, washed twice with ice-cold phosphate-buffered saline (PBS) (Biosharp), and the total mRNA was extracted using an RNA plus reagent (TaKaRa, Cat No. 9109). Complementary DNA was synthesized using a PrimeScript RT reagent kit with genomic DNA eraser (TaKaRa Cat No. RR047A), according to the manufacturer’s instructions, and used as a template for the real-time qPCR. The primers used are listed in Supplementary Material, Table S7.
Migration and invasion assay
Cell migration and invasion assays were conducted using the 24-well chambers with 8 mm pore polycarbonate membranes (Costar). A total of 1 × 105 cells were plated into the upper chamber. The bottom medium contained RPMI-1640 medium and supplemented with 10% FBS. After 24 h, the invading membranes were washed twice with PBS, fixed with 4% paraformaldehyde (PFA) for 20 mins and stained with 1% 4,6-diamino-2-phenyl indole (DAPI). The membranes stained with DAPI were imaged with the excitation wavelengths of 360–400 nm. A paired t-test was used to derive P-value from the comparison between knockdown and control in migration and invasion assays (*P < 0.05, **P < 0.01, ***P < 0.001).
Wound healing assay
Cell migration was assessed by wound healing assays. Cells were plated on six-well plates and wounded by manual scratching with a 10 μl pipette tip after cells attached. Seventy-two hours after incubation, cells were washed twice with PBS and images of scratch areas were taken using an AXIO Observer A1.
Cell proliferation and colony formation assay
To determine whether knockdown affects cell proliferation ability, the shRNA expressing cells or control cells were plated into 96-well plates, respectively. We used CCK8 kit to determine the proliferation of these cells according to manufacturer’s instructions during the day. The indicated cells were trypsinized into a single-cell suspension and seeded into six-well plates at densities of 1000 cells per well. After 10 days, these colonies were washed and fixed with 4% PFA for 30 mins at room temperature, and further stained with Coomassie Blue Staining Solution (Beyotime Biotechnology, Cat No. P0017B). Clones containing at least 50 cells were considered one formation. All experiments were performed three times.
Western blot analysis
Western blot analyses were conducted regularly. Cells were lysed in a cold lysis buffer with protease inhibitor cocktail (Roche). Cell lysate was centrifuged at 12 000 RPM for 15 min, boiled in a 5× sample loading buffer at 95–100°C for 5 min, separated on a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and blotted onto 0.22 μm polyvinylidene fluoride membranes. The membranes were incubated with DIP2B antibody (Bethyl Laboratories, Cat. A304-638A), SFMBT1 antibody (Santa Cruz, Cat. sc-135 559), beta-catenin antibody (Proteintech, Cat. 51 067-2-AP), Vimentin antibody (Abcam, Cat. D21H3), Snail antibody (Abcam, Cat. C15D3) and GAPDH antibody (Proteintech, Cat. 60 004-1-Ig), respectively. The membranes were developed using electrochemiluminescent solution (Thermo). The images were taken by ChemiDoc MP (Biorad) and quantified by Image Lab 5.2.0 software.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
The authors thank TCGA, GTEx, ENCODE and ROADMAP for providing valuable data resources for the research. The authors thank Marshal Younger for assistance with editing and manuscript preparation. The data analyses were conducted using the Advanced Computing Center for Research and Education at Vanderbilt University.
Conflict of Interest statement. None declared.
Contributor Information
Yuan Yuan, The Fourth Affiliated Hospital, Zhejiang University School of Medicine, Jinhua, Zhejiang 322000, China.
Jiandong Bao, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN 37203, USA; College of Life Sciences, Fujian Agriculture and Forestry University, Fuzhou, Fujian 350002, China.
Zhishan Chen, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN 37203, USA.
Anna Díez Villanueva, Unit of Biomarkers and Susceptibility, Oncology Data Analytics Program, Catalan Institute of Oncology (ICO); Colorectal Cancer Group, ONCOBELL Program, Bellvitge Biomedical Research Institute (IDIBELL); Consortium for Biomedical Research in Epidemiology and Public Health (CIBERESP); Faculty of Medicine, Department of Clinical Sciences, University of Barcelona, Barcelona 08908, Spain.
Wanqing Wen, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN 37203, USA.
Fangqin Wang, The Fourth Affiliated Hospital, Zhejiang University School of Medicine, Jinhua, Zhejiang 322000, China.
Dejian Zhao, Departments of Genetics, Neurology and Neuroscience, Albert Einstein College of Medicine, Bronx, NY 10461, USA.
Xianghui Fu, Division of Endocrinology and Metabolism, State Key Laboratory of Biotherapy, West China Hospital, Chengdu, Sichuan 610041, China.
Qiuyin Cai, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN 37203, USA.
Jirong Long, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN 37203, USA.
Xiao-ou Shu, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN 37203, USA.
Deyou Zheng, Departments of Genetics, Neurology and Neuroscience, Albert Einstein College of Medicine, Bronx, NY 10461, USA.
Victor Moreno, Unit of Biomarkers and Susceptibility, Oncology Data Analytics Program, Catalan Institute of Oncology (ICO); Colorectal Cancer Group, ONCOBELL Program, Bellvitge Biomedical Research Institute (IDIBELL); Consortium for Biomedical Research in Epidemiology and Public Health (CIBERESP); Faculty of Medicine, Department of Clinical Sciences, University of Barcelona, Barcelona 08908, Spain.
Wei Zheng, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN 37203, USA.
Weiqiang Lin, The Fourth Affiliated Hospital, Zhejiang University School of Medicine, Jinhua, Zhejiang 322000, China.
Xingyi Guo, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN 37203, USA; Department of Biomedical Informatics, Vanderbilt University School of Medicine, Nashville, TN 37203, USA.
Funding
US National Institutes of Health (R37 CA227130); research development fund from Vanderbilt University Medical Center to X.G.; US National Institutes of Health (R01 CA18821 to W.Z.); National Natural Science Foundation of China (31470776, 81670651 to W.L.); National Key R&D Program of China (2018YFC2000400 to W.L.); Science Fund for Distinguished Young Scholars of Fujian Agriculture and Forestry University (XJQ201511 to J.B.).
References
- 1.Palles, C., Cazier, J.B., Howarth, K.M., Domingo, E., Jones, A.M., Broderick, P., Kemp, Z., Spain, S.L., Guarino, E., Salguero, I. et al. (2013) Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet., 45, 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Network (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature, 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fearon, E.R. (2011) Molecular genetics of colorectal cancer. Annu. Rev. Pathol., 6, 479–507. [DOI] [PubMed] [Google Scholar]
- 4.Zeng, C., Matsuda, K., Jia, W.H., Chang, J., Kweon, S.S., Xiang, Y.B., Shin, A., Jee, S.H., Kim, D.H., Zhang, B. et al. (2016) Identification of susceptibility loci and genes for colorectal cancer risk. Gastroenterology, 150, 1633–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michailidou, K., Beesley, J., Lindstrom, S., Canisius, S., Dennis, J., Lush, M.J., Maranian, M.J., Bolla, M.K., Wang, Q., Shah, M. et al. (2015) Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet., 47, 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Tassan, N.A., Whiffin, N., Hosking, F.J., Palles, C., Farrington, S.M., Dobbins, S.E., Harris, R., Gorman, M., Tenesa, A., Meyer, B.F. et al. (2015) A new GWAS and meta-analysis with 1000Genomes imputation identifies novel risk variants for colorectal cancer. Sci. Rep., 5, 10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang, H., Burnett, T., Kono, S., Haiman, C.A., Iwasaki, M., Wilkens, L.R., Loo, L.W., Van Den Berg, D., Kolonel, L.N., Henderson, B.E. et al. (2014) Trans-ethnic genome-wide association study of colorectal cancer identifies a new susceptibility locus in VTI1A. Nat. Commun., 5, 4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmit, S.L., Schumacher, F.R., Edlund, C.K., Conti, D.V., Raskin, L., Lejbkowicz, F., Pinchev, M., Rennert, H.S., Jenkins, M.A., Hopper, J.L. et al. (2014) A novel colorectal cancer risk locus at 4q32.2 identified from an international genome-wide association study. Carcinogenesis, 35, 2512–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang, B., Jia, W.H., Matsuda, K., Kweon, S.S., Matsuo, K., Xiang, Y.B., Shin, A., Jee, S.H., Kim, D.H., Cai, Q. et al. (2014) Large-scale genetic study in East Asians identifies six new loci associated with colorectal cancer risk. Nat. Genet., 46, 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Figueiredo, J.C., Hsu, L., Hutter, C.M., Lin, Y., Campbell, P.T., Baron, J.A., Berndt, S.I., Jiao, S., Casey, G., Fortini, B. et al. (2014) Genome-wide diet-gene interaction analyses for risk of colorectal cancer. PLoS Genet., 10, e1004228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whiffin, N., Hosking, F.J., Farrington, S.M., Palles, C., Dobbins, S.E., Zgaga, L., Lloyd, A., Kinnersley, B., Gorman, M., Tenesa, A. et al. (2014) Identification of susceptibility loci for colorectal cancer in a genome-wide meta-analysis. Hum. Mol. Genet., 23, 4729–4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang, B., Jia, W.H., Matsuo, K., Shin, A., Xiang, Y.B., Matsuda, K., Jee, S.H., Kim, D.H., Cheah, P.Y., Ren, Z. et al. (2014) Genome-wide association study identifies a new SMAD7 risk variant associated with colorectal cancer risk in East Asians. Int. J. Cancer, 135, 948–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters, U., Jiao, S., Schumacher, F.R., Hutter, C.M., Aragaki, A.K., Baron, J.A., Berndt, S.I., Bezieau, S., Brenner, H., Butterbach, K. et al. (2013) Identification of genetic susceptibility loci for colorectal tumors in a genome-wide meta-analysis. Gastroenterology, 144, 799–807.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunlop, M.G., Dobbins, S.E., Farrington, S.M., Jones, A.M., Palles, C., Whiffin, N., Tenesa, A., Spain, S., Broderick, P., Ooi, L.Y. et al. (2012) Common variation near CDKN1A, POLD3 and SHROOM2 influences colorectal cancer risk. Nat. Genet., 44, 770–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peters, U., Hutter, C.M., Hsu, L., Schumacher, F.R., Conti, D.V., Carlson, C.S., Edlund, C.K., Haile, R.W., Gallinger, S., Zanke, B.W. et al. (2012) Meta-analysis of new genome-wide association studies of colorectal cancer risk. Hum. Genet., 131, 217–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tomlinson, I.P., Carvajal-Carmona, L.G., Dobbins, S.E., Tenesa, A., Jones, A.M., Howarth, K., Palles, C., Broderick, P., Jaeger, E.E., Farrington, S. et al. (2011) Multiple common susceptibility variants near BMP pathway loci GREM1, BMP4, and BMP2 explain part of the missing heritability of colorectal cancer. PLoS Genet., 7, e1002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui, R., Okada, Y., Jang, S.G., Ku, J.L., Park, J.G., Kamatani, Y., Hosono, N., Tsunoda, T., Kumar, V., Tanikawa, C. et al. (2011) Common variant in 6q26-q27 is associated with distal colon cancer in an Asian population. Gut, 60, 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoffman, J.D., Graff, R.E., Emami, N.C., Tai, C.G., Passarelli, M.N., Hu, D., Huntsman, S., Hadley, D., Leong, L., Majumdar, A. et al. (2017) Cis-eQTL-based trans-ethnic meta-analysis reveals novel genes associated with breast cancer risk. PLoS Genet., 13, e1006690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huyghe, J.R., Bien, S.A., Harrison, T.A., Kang, H.M., Chen, S., Schmit, S.L., Conti, D.V., Qu, C., Jeon, J., Edlund, C.K. et al. (2019) Discovery of common and rare genetic risk variants for colorectal cancer. Nat. Genet., 51, 76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Law, P.J., Timofeeva, M., Fernandez-Rozadilla, C., Broderick, P., Studd, J., Fernandez-Tajes, J., Farrington, S., Svinti, V., Palles, C., Orlando, G. et al. (2019) Association analyses identify 31 new risk loci for colorectal cancer susceptibility. Nat. Commun., 10, 2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen, Z., Wen, W., Beeghly-Fadiel, A., Shu, X.O., Diez-Obrero, V., Long, J., Bao, J., Wang, J., Liu, Q., Cai, Q. et al. (2019) Identifying putative susceptibility genes and evaluating their associations with somatic mutations in human cancers. Am. J. Hum. Genet., 105, 477–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bell, J.T., Pai, A.A., Pickrell, J.K., Gaffney, D.J., Pique-Regi, R., Degner, J.F., Gilad, Y. and Pritchard, J.K. (2011) DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol., 12, R10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baylin, S.B. and Jones, P.A. (2016) Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol., 8, a019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hannon, E., Gorrie-Stone, T.J., Smart, M.C., Burrage, J., Hughes, A., Bao, Y., Kumari, M., Schalkwyk, L.C. and Mill, J. (2018) Leveraging DNA-methylation quantitative-trait loci to characterize the relationship between methylomic variation, gene expression, and complex traits. Am. J. Hum. Genet., 103, 654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schubeler, D. (2015) Function and information content of DNA methylation. Nature, 517, 321–326. [DOI] [PubMed] [Google Scholar]
- 26.Wu, Y., Zeng, J., Zhang, F., Zhu, Z., Qi, T., Zheng, Z., Lloyd-Jones, L.R., Marioni, R.E., Martin, N.G., Montgomery, G.W. et al. (2018) Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun., 9, 918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olsson, A.H., Volkov, P., Bacos, K., Dayeh, T., Hall, E., Nilsson, E.A., Ladenvall, C., Ronn, T. and Ling, C. (2014) Genome-wide associations between genetic and epigenetic variation influence mRNA expression and insulin secretion in human pancreatic islets. PLoS Genet., 10, e1004735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hannon, E., Weedon, M., Bray, N., O'Donovan, M. and Mill, J. (2017) Pleiotropic effects of trait-associated genetic variation on DNA methylation: utility for refining GWAS loci. Am. J. Hum. Genet., 100, 954–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qi, T., Wu, Y., Zeng, J., Zhang, F., Xue, A., Jiang, L., Zhu, Z., Kemper, K., Yengo, L., Zheng, Z. et al. (2018) Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat. Commun., 9, 2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu, Z., Zhang, F., Hu, H., Bakshi, A., Robinson, M.R., Powell, J.E., Montgomery, G.W., Goddard, M.E., Wray, N.R., Visscher, P.M. et al. (2016) Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet., 48, 481–487. [DOI] [PubMed] [Google Scholar]
- 31.Tang, M., Shen, H., Jin, Y., Lin, T., Cai, Q., Pinard, M.A., Biswas, S., Tran, Q., Li, G., Shenoy, A.K. et al. (2013) The malignant brain tumor (MBT) domain protein SFMBT1 is an integral histone reader subunit of the LSD1 demethylase complex for chromatin association and epithelial-to-mesenchymal transition. J. Biol. Chem., 288, 27680–27691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo, X., Lin, W., Wen, W., Huyghe, J., Bien, S., Cai, Q., Harrison, T., Chen, Z., Qu, C., Bao, J. et al. (2020) Identifying novel susceptibility genes for colorectal cancer risk from a transcriptome-wide association study of 125,478 subjects. Gastroenterology [online. doi: 10.1053/j.gastro.2020.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winnepenninckx, B., Debacker, K., Ramsay, J., Smeets, D., Smits, A., FitzPatrick, D.R. and Kooy, R.F. (2007) CGG-repeat expansion in the DIP2B gene is associated with the fragile site FRA12A on chromosome 12q13.1. Am. J. Hum. Genet., 80, 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larsson, C., Ali, M.A., Pandzic, T., Lindroth, A.M., He, L. and Sjoblom, T. (2017) Loss of DIP2C in RKO cells stimulates changes in DNA methylation and epithelial-mesenchymal transition. BMC Cancer, 17, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jung, I., Schmitt, A., Diao, Y., Lee, A.J., Liu, T., Yang, D., Tan, C., Eom, J., Chan, M., Chee, S. et al. (2019) A compendium of promoter-centered long-range chromatin interactions in the human genome. Nat. Genet., 51, 1442–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orlando, G., Law, P.J., Cornish, A.J., Dobbins, S.E., Chubb, D., Broderick, P., Litchfield, K., Hariri, F., Pastinen, T., Osborne, C.S. et al. (2018) Promoter capture Hi-C-based identification of recurrent noncoding mutations in colorectal cancer. Nat. Genet., 50, 1375–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Howie, B., Fuchsberger, C., Stephens, M., Marchini, J. and Abecasis, G.R. (2012) Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet., 44, 955–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Closa, A., Cordero, D., Sanz-Pamplona, R., Sole, X., Crous-Bou, M., Pare-Brunet, L., Berenguer, A., Guino, E., Lopez-Doriga, A., Guardiola, J. et al. (2014) Identification of candidate susceptibility genes for colorectal cancer through eQTL analysis. Carcinogenesis, 35, 2039–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moreno, V., Alonso, M.H., Closa, A., Valles, X., Diez-Villanueva, A., Valle, L., Castellvi-Bel, S., Sanz-Pamplona, R., Lopez-Doriga, A., Cordero, D. et al. (2018) Colon-specific eQTL analysis to inform on functional SNPs. Br. J. Cancer, 119, 971–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo, X., Lin, W., Bao, J., Cai, Q., Pan, X., Bai, M., Yuan, Y., Shi, J., Sun, Y., Han, M.R. et al. (2018) A comprehensive cis-eQTL analysis revealed target genes in breast cancer susceptibility loci identified in genome-wide association studies. Am. J. Hum. Genet., 102, 890–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.GTEx Consortium, Laboratory, Date Analysis & Coordinating Center—Analysis Working Group, Statistical Methods groups—Analysis Working Group, Enhancing GTEx groups, NIH Common Fund, NIH/NCI, NIH/NHGRI, NIH/NIMH, NIH/NIDA et al. (2017) Genetic effects on gene expression across human tissues. Nature, 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borenstein, M., Hedges, L.V., Higgins, J.P.T. and Rothstein, H.R. (2009) Introduction to Meta-Analysis. Wiley, Chichester, UK. [Google Scholar]
- 43.Ward, L.D. and Kellis, M. (2016) HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res., 44, D877–D881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teng, L., He, B., Wang, J. and Tan, K. (2015) 4DGenome: a comprehensive database of chromatin interactions. Bioinformatics, 31, 2560–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.