

Abstract
Striated preferentially expressed gene (SPEG), a member of the myosin light chain kinase family, is localized at the level of triad surrounding myofibrils in skeletal muscles. In humans, SPEG mutations are associated with centronuclear myopathy and cardiomyopathy. Using a striated muscle-specific Speg-knockout (KO) mouse model, we have previously shown that SPEG is critical for triad maintenance and calcium handling. Here, we further examined the molecular function of SPEG and characterized the effects of SPEG deficiency on triad and focal adhesion proteins. We used yeast two-hybrid assay, and identified desmin, an intermediate filament protein, to interact with SPEG and confirmed this interaction by co-immunoprecipitation. Using domain-mapping assay, we defined that Ig-like and fibronectin III domains of SPEG interact with rod domain of desmin. In skeletal muscles, SPEG depletion leads to desmin aggregates in vivo and a shift in desmin equilibrium from soluble to insoluble fraction. We also profiled the expression and localization of triadic proteins in Speg-KO mice using western blot and immunofluorescence. The amount of RyR1 and triadin were markedly reduced, whereas DHPRα1, SERCA1 and triadin were abnormally accumulated in discrete areas of Speg-KO myofibers. In addition, Speg-KO muscles exhibited internalized vinculin and β1 integrin, both of which are critical components of the focal adhesion complex. Further, β1 integrin was abnormally accumulated in early endosomes of Speg-KO myofibers. These results demonstrate that SPEG-deficient skeletal muscles exhibit several pathological features similar to those seen in MTM1 deficiency. Defects of shared cellular pathways may underlie these structural and functional abnormalities in both types of diseases.
Introduction
Centronuclear myopathies (CNMs) are a group of congenital myopathies characterized by clinical features of muscle weakness, increased central nuclei and genetic heterogeneity (1,2). The most common forms of CNMs have been attributed to X-linked recessive mutations in the MTM1 gene encoding myotubularin, autosomal dominant mutations in the DNM2 gene encoding dynamin 2 and the BIN1 gene encoding bridging integrator 1 and autosomal recessive mutations in BIN1, the RYR1 gene encoding ryanodine receptor 1, and the TTN gene encoding titin (3–8). Models to study the disease mechanisms that underlie these various forms of CNMs have been established in yeast (9,10), Caenorhabditis elegans (11–13), drosophila (14–16), zebrafish (17–19), mice (20–24) and dogs (25,26). Based on findings from these models, several pathogenic mechanisms have been suggested, including abnormalities of triads and excitation–contraction (E–C) coupling (17,27), as well as defects in autophagy (21,23), and of the neuromuscular junction (28,29), satellite cells (30), integrin trafficking (15), mitochondria and desmin cytoskeleton dynamics (31).
Our group has previously identified SPEG as the sixth gene associated with CNM (32) and found that SPEG deficiency leads to an abnormal triad structure and defective calcium handling in skeletal muscles (33), indicating an essential role of SPEG in triad maintenance and function. The SPEG gene encodes four distinct tissue-specific isoforms, including Spegα, Spegβ, Apeg1 and Bpeg (34). Spegα (250 kDa) and Spegβ (350 kDa) are the largest isoforms that are specific to striated muscles and share homology with the myosin light chain kinase (MLCK) family members, with characteristic immunoglobulin (Ig) and fibronectin domains. The Speg isoforms, along with obscurin-MLCK, are unique members of the MLCK family, containing two tandemly arranged serine/threonine kinase domains (34,35). SPEG exhibits a reticular distribution surrounding the myofibrils and colocalizes with proteins of the sarcoplasmic reticulum (SR) in skeletal muscles (32). Specifically, SPEG colocalizes with triadin at the junctional SR flanking desmin, which appears as a series of dots between the Z lines on longitudinal sections (32). SPEG shows a significant structural homology to obscurin (OBSCN), which is known to tether the longitudinal SR to maintain its structure (36), and we hypothesize that SPEG plays a similar role to maintain the triad integrity.
To decipher the functional significances of SPEG in skeletal muscles, we performed a yeast two-hybrid (Y2H) screening using the SPEG sequence as bait against a human fetal and adult skeletal muscle library and identified desmin as a SPEG-interacting partner. We further confirmed this interaction by a co-immunoprecipitation (co-IP) assay and mapped their interacting domains. In skeletal muscles, SPEG deficiency leads to an increased level of desmin aggregates and a shift in desmin equilibrium from the soluble to the insoluble fraction. In addition, the amount of RyR1 and triadin are markedly reduced in Speg-knockout (KO) muscles, whereas mislocalization of dihydropyridine receptor (DHPRα1), sarcoendoplasmic reticulum Ca2+ ATPase 1 (SERCA1) and triadin are noted. Further, Speg-KO muscles exhibited internalized vinculin and β1 integrin, the latter of which abnormally accumulates in early endosomes, indicating defective focal adhesion and membrane trafficking. These results demonstrate that SPEG-deficient skeletal muscles present similar pathological features to that found in MTM1-related myopathies, indicating shared pathways underlying these diseases.
Results
SPEG binds to desmin in skeletal muscles
To decipher the muscle-specific role of SPEG and the physiopathological mechanisms of SPEG-related CNM, we performed an Y2H screen using partial SPEG sequence [amino acids (aa) 2200–3267] as bait against a human fetal and adult skeletal muscle library. We identified desmin as a SPEG-interacting partner by isolating three different clones encoding portions of the desmin (aa 111–402) that include the central α-helical coiled-coil (‘rod’) domain (Fig. 1A). Desmin is the classical type III intermediate filament (IF) protein with a tripartite structure comprising a rod domain flanked by non-α-helical head and tail domains (Fig. 1A). The rod domain, formed by four α-helical segments (1A, 1B, 2A and 2B), is involved in protein–protein interactions and plays a critical role in desmin filament assembly and the formation of the extrasarcomeric cytoskeleton (37). The region of desmin (aa 179–228) that was contained in the three clones was part of the helical segment 1B domain.
Figure 1 .
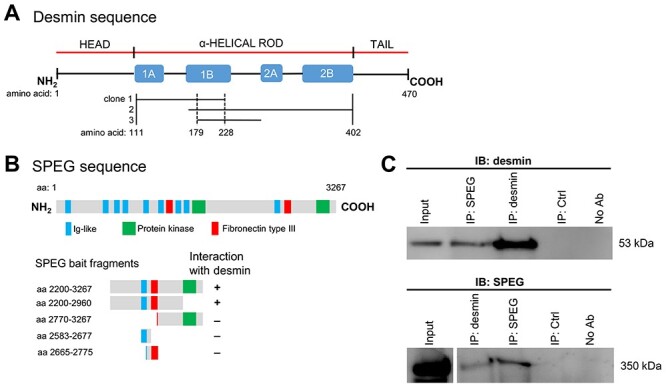
SPEG interacts with desmin in skeletal muscles. (A) Schematic of the three different desmin Y2H prey clones that interact with SPEG fragment bait. These clones encode portions of the desmin (aa 111–402, as denoted between vertical solid lines) that occupy the central α-helical rod domain. The region of desmin (aa 179−228, as denoted between vertical dotted lines) that was overlapped in these clones was part of the helical segment 1B domain. (B) A schematic of SPEGβ isoform illustrates the location of three types of domains (Ig-like, protein kinase and fibronectin type III domains) above the map of fragments used for deletion mapping of the region responsible for interactions with desmin. Deletion analysis of SPEG showed that the Ig-like-9 and fibronectin III-2 domains together are necessary to mediate the interaction with desmin. (C) SPEG and desmin co-immunoprecipitated from C2C12 myotube lysates with the use of rabbit anti-SPEG generated against a FLAG-tagged APEG-1 fusion protein (34) and anti-desmin antibodies.
To identify the SPEG domains that interact with desmin, we tested a peptide encompassing residues 168–402 of human desmin (part of the rod domain) for interaction with various SPEG fragments by using a direct 1-by-1 interaction assay. SPEG fragments containing amino acid residues 2200–2960, which include the Ig-like-9 and fibronectin III-2 domains, were confirmed to interact with desmin fragment (Fig. 1B). However, neither Ig-like nor fibronectin III domain alone in isolation showed interaction with desmin, suggesting that the critical binding region either overlaps the junction or includes binding sites from both SPEG domains. In support of the Y2H data, full-length SPEG and desmin co-immunoprecipitated with each other from differentiated C2C12 myotube lysates with the use of either anti-SPEG or anti-desmin antibodies (Fig. 1C).
Abnormal desmin aggregates in SPEG-deficient skeletal muscles
The expression levels of SPEG and desmin concomitantly increase during differentiation of myoblasts into myotubes (31,34). To address whether SPEG depletion affects desmin IF dynamics, we evaluated its expression and localization in Speg-KO skeletal muscles. The protein level of desmin was not significantly different in Speg-KO muscles as compared to litter-matched controls (Fig. 2A). However, desmin aggregates were observed in isolated muscles from 1-month-old Speg-KO mice (Fig. 2B and Supplementary Material, Fig. S1). Further, a shift in desmin equilibrium from the soluble to insoluble fraction was observed in Speg-KO muscles on immunoblot analysis (Fig. 2C), indicating a defect in desmin dynamics.
Figure 2 .
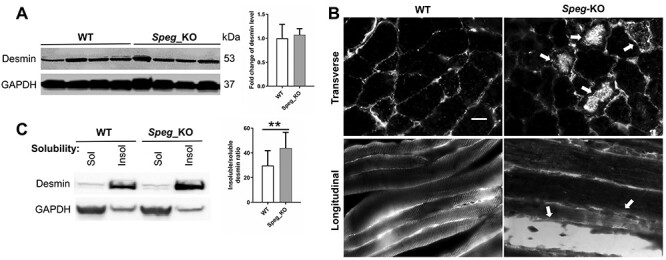
Effect of SPEG depletion on desmin expression and localization. (A) The protein expression level of desmin was not significantly changed in Speg-KO muscles compared with littermate controls. (B) Desmin aggregates in Speg-KO muscle biopsies. Arrows indicate the desmin aggregates. Scale bar, 20 μm. (C) Effect of SPEG deficiency on desmin expression and solubility (**P ˂ 0.01, n = 3).
SPEG colocalizes with DHPRα1, RyR1 and triadin and its deficiency causes defective expression and localization of triadic proteins
We have previously shown that SPEG colocalizes with triadin, a protein found in the terminal cisternae of the SR in human quadricep muscles (32). To further evaluate the localization of SPEG at triad, SPEG was co-stained in mouse tibialis anterior (TA) muscles with antibodies against DHPRα1 (T-tubule marker), RyR1 and triadin (markers for SR terminal cisternae). Our immunofluorescence results (Fig. 3) demonstrate that SPEG colocalizes with DHPRα1, RyR1 and triadin, confirming that SPEG is localized in the triad.
Figure 3 .

SPEG colocalizes with DHPRα1, RyR1 and triadin in WT skeletal muscles. Longitudinal sections from frozen WT mouse TA muscles were co-immunostained with mouse anti-DHPRα1, mouse anti-RyR1, mouse anti-triadin and rabbit anti-SPEG, respectively. As seen in merged images, SPEG staining colocalizes with DHPRα1, RyR1 and triadin. Scale bar, 20 μm.
SPEG deficiency leads to abnormal triad and defective calcium handling in skeletal muscles (33). Here, we further assessed the expression levels and distribution of multiple triadic proteins that are involved in these processes. We found that the protein expression levels of RyR1 and triadin were significantly reduced in Speg-KO muscles, whereas expression of DHPRα1, SERCA1 and junctophilin 1 (JP1) remained unchanged (Fig. 4A). We observed an abnormal accumulation of DHPRα1, SERCA1 and triadin in discrete areas of Speg-KO myofibers (Fig. 4B and Supplementary Material, Fig. S2), indicating disorganized T-tubules and triads after SPEG depletion.
Figure 4.

Effect of SPEG deficiency on the expression and localization of triadic proteins. (A) The protein expression levels of RyR1 and triadin were markedly reduced in the Speg-KO muscles compared to littermate controls (*P ˂ 0.05, n ≥ 3), whereas levels of DHPRα1, SERCA1 and JP1 were unchanged. (B) Transverse TA muscle sections stained for DHPRα1, RyR1, SERCA1, triadin and JP1. Abnormally accumulated DHPRα1 (denoted by arrowheads), SERCA1 (denoted by arrows) and triadin (denoted by asterisks) were observed in discrete areas of Speg-KO myofibers. Scale bar, 20 μm.
Defective focal adhesion proteins in Speg-KO skeletal muscles
In skeletal muscles, desmin IFs are linked to Z-discs and costameres, respectively, coupling sarcomeres with the muscle fiber plasma membrane. To determine the effects of SPEG deficiency on the organization of costameres, we evaluated the localization of several proteins forming the focal adhesion complex. We stained both transverse and longitudinal TA sections from wild-type (WT) and Speg-KO mice with antibodies against β1 integrin and vinculin. In WT muscles, vinculin and integrins localize at the sarcolemma, specifically at the costamere. Speg-KO muscles, however, exhibited internalized vinculin and β1 integrin (Fig. 5A and Supplementary Material, Fig. S3). The protein level of β1 integrin was slightly increased in Speg-KO mice compared to WT (Supplementary Material, Fig. S4), while the levels of vinculin were comparable in between them (Fig. 4).
Figure 5 .
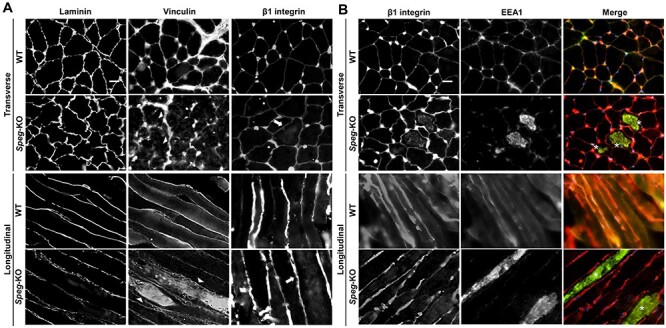
Abnormal localization of focal adhesion complex proteins associated with SPEG deficiency. (A) Transverse (upper panel) and longitudinal (lower panel) WT and Speg-KO TA muscles stained for the laminin, vinculin and β1 integrin. Arrowheads indicate internalized vinculin and arrows indicate internalized β1 integrin, respectively. Scale bar, 20 μm. (B) Transverse (upper panel) and longitudinal (lower panel) TA muscle sections stained for β1 integrin (red) and EEA1 (green). Asterisks indicate the colocalization between internalized β1 integrin and EEA1. Scale bar, 20 μm.
To investigate in which intracellular compartment β1 integrin accumulates, we labeled endosomes on muscle sections against early endosomal antigen 1 (EEA1), a marker of early endosomes, and found it to colocalize with β1 integrin (Fig. 5B and Supplementary Material, Fig. S3). EEA1-positive endosomes also aggregated in Speg-KO myofibers compared with WT (Fig. 5B). Overall, these results highlight that β1 integrin abnormally accumulates at early endosomes in Speg-KO muscles, indicating a defect in β1 integrin turnover.
We also examined the localization of dystrophin–glycoprotein complex proteins, including dystrophin and α-sarcoglycan (Supplementary Material, Fig. S5), and did not find any significant accumulations of such proteins in Speg-KO muscles. Further staining of α-actinin 2, a Z-line marker, and vimentin, one of the IF proteins that interacts with desmin and localizes at Z-line (38,39), revealed that the localization of these proteins appeared unaltered in the Speg-KO muscles (Supplementary Material, Fig. S6), suggesting an overall intact intracellular organization of myofibers.
Discussion
The physiological role of SPEG in skeletal muscle is yet to be deciphered. We have previously identified that SPEG is critical to maintaining triad structure and function, including calcium signaling, and that it interacts with MTM1, a phosphatase deficient in X-linked myotubular myopathy (XLMTM) (32,33). To further understand its relationship with other muscle proteins, we performed Y2H assay using SPEG fragment (aa 2200–3267) as a bait and identified desmin as its binding protein, which was further confirmed by co-IP. Molecular dissection of the interaction identified the Ig-like and fibronectin III domains of SPEG to interact with the rod domain of desmin. The desmin rod domain is essential for its assembly and binding with its molecular partners, such as IF and IF-associated proteins, and sarcomeric proteins (39).
SPEG-deficient skeletal muscles exhibited desmin aggregates, a major morphological hallmark of myofibrillar myopathies (MFMs) caused by mutations in genes encoding the myofibrillar proteins (myotilin, filamin C, Bcl-2-associated athanogene-3) or extramyofibrillar proteins (desmin, αB-crystallin, plectin) (40). The insights into the pathogenic mechanisms of MFMs indicate that the complex pathology is not solely due to direct effects of the altered protein but also secondary to interference in the interaction with binding partners, which influence the structural and functional organization of the extrasarcomeric cytoskeleton as well as intracellular signaling cascades (40). Interestingly, desmin IF aggregates have also been reported in another type of CNM, the myotubular myopathy (31). Prior studies have shown that MTM1 is bound to the 2B rod domain of desmin by its Rac1-induced recruitment domain and that MTM1 directly regulates desmin IF assembly in vitro (31). Furthermore, MTM1-deficient cells and skeletal muscles exhibited both increased desmin expression and its cytoplasmic aggregates (31). In this study, we identified that Ig-like-9 and fibronectin III-2 domains of SPEG bind with the rod domain of desmin, region of SPEG also found to interact with MTM1 (32). The importance of these domains in SPEG function is yet to be deciphered. SPEG-deficient skeletal muscles exhibit a significantly increased level of desmin aggregates, indicating that SPEG is critical for the maintenance of desmin IF network through its interaction with desmin and/or MTM1. Further studies are needed to decipher the specific role of SPEG in this process and to delineate their inter-relationship.
In addition to desmin IF disequilibrium, defects in focal adhesion have also been indicated as an important component of the pathological mechanisms in MTM1-related myopathy (15,41). In Drosophila, depletion of myotubularin (mtm) leads to increased integrin turnover at the sarcolemma and an accumulation of integrin with PI(3)P on endosomal-related membrane inclusions (15), indicating a role for mtm phosphatase activity in endocytic trafficking. Importantly, similar integrin localization defects were also found in human XLMTM myofibers (15) and Mtm1-KO muscles (41), which could be potentially explained by the fact that MTM1 phosphoinositide phosphatase activity is involved in the conversion of early to late or recycling endosomes (42). Additionally, accumulating data indicated that DNM2 may play a central role in focal adhesion disassembly (43), although whether DNM2 mutations related to CNMs cause focal adhesion defects is unknown. We show here that Speg-KO muscles exhibit internalized vinculin and β1 integrin and that β1 integrin abnormally accumulates at early endosomes. These results suggest that defective integrin trafficking and focal adhesion may account for one of the underlying mechanisms related to SPEG deficiency.
We have previously demonstrated that SPEG deficiency is associated with an abnormal triad structure, defective E–C coupling and calcium mishandling (33), features that are also revealed in mouse models of human diseases caused by MTM1, DNM2 and BIN1 mutations (18,19,27). In this study, we observed aberrant localization of triadic proteins, including DHPRα1, SERCA1 and triadin, from immunofluorescence analyses of Speg-KO muscles, similar to what has been seen in other CNM-related diseases (44). Remarkably, we found significantly reduced levels of RyR1 and triadin in SPEG-deficient skeletal muscles as compared to controls. Recessive mutations in RYR1 are a common cause of congenital myopathies with central nuclei (4), which are associated with hypomorphic expression of RyR1 and other triadic proteins including triadin (45–47,48). Reduction of RyR1 protein has been reported in the absence of calpain-3 (49,50). Calpain-3 is the muscle-specific member of a family of proteolytic enzymes that bind to triad components, such as RyR1, thereby play a stabilizing role for RyR1 at the triad (49). Reduction in RyR1 has also been seen in MTM1 deficiency (27), and the postulated underlying mechanisms include epigenetic changes, such as altered levels of muscle-specific microRNAs and upregulation of histone deacetylase-4 (51). Recent studies in cardiac muscles have identified that SPEG interacts with and phosphorylates RyR2, a cardiac isoform of ryanodine receptor, at the dyads, thereby reducing RyR2-mediated SR Ca2+ release (52,53). While we suspect that SPEG interacts with and stabilizes RyR1 at the triad, the mechanisms of RyR1 reduction associated with SEPG deficiency in our study are yet to be deciphered.
In summary, we have shown that SPEG interacts with desmin and that its deficiency leads to similar pathological changes to those found in XLMTM, including triadic protein abnormalities, desmin aggregates, focal adhesion defects and reduced RyR1 level (Table 1). These findings suggest that mechanistic pathway(s), including endosome trafficking and triad maintenance, may underlie the pathogenesis of SPEG deficiency and MTM1-related myopathies, and therefore, similar therapeutic approaches may be applied to both disorders.
Table 1.
Histopathological and molecular findings in Speg- and Mtm1-mutant mouse models
| Findings | Speg-KO (Spegfl/fl: MCK-cre+) | Mtm1-KO (Mtm1−/y) |
|---|---|---|
| Triad structure and function | Poor integrity, low number of triads and calcium mishandling (33) | Fewer triads and abnormal longitudinally oriented T-tubules, impaired SR calcium release (27) |
| Triadic protein abnormalities | Reduced RyR1 and triadin protein, mislocalization of DHPRα1, SERCA1 and triadin (this study) | A 3-fold reduction of RyR1 protein, ~30% decrease of DHPRα1 and 6-fold increase of DHPRβ1 in microsomal preparations (27); disorganization of DHPR and RyR1 labeling (44) |
| Desmin aggregation | Cytoplasmic desmin aggregates and increased insolubility (this study) | Increased protein level and insolubility of desmin, desmin aggregation (31) |
| Focal adhesion defects | Cytoplasmic accumulation of vinculin and β1 integrin, accumulation of β1 integrin at early endosomes (this study) | Increased protein levels of laminin, vinculin and β1 integrin; internalized vinculin and β1 integrin; and accumulation of β1 integrin at early endosomes (41) |
| Others | Fewer satellite cells and delayed muscle regeneration in response to injury (58) | Abnormalities in the number and behavior of myogenic cells with decreased proliferation and increased apoptosis (30) |
Materials and Methods
Yeast two-hybrid
Y2H screening was performed by Hybrigenics, S.A. (Paris, France) (http://www.hybrigenics-services.com) as previously described (32). The coding sequence for aa 2200–3267 of human SPEG (NM_005876.4) was PCR-amplified and cloned into pB66 as a C-terminal fusion to the Gal4 DNA-binding domain (Gal4-SPEG). The construct was checked by sequencing and used as a bait to screen a random-primed Human Adult/Fetal Skeletal Muscle cDNA library constructed into pP6. Fifty million clones (5-fold the complexity of the library) were screened using a mating approach with YHGX13 (Y187 ade2-101::loxP-kanMX-loxP, matα) and CG1945 (mata) yeast strains as previously described (54). 324 His+ colonies were selected on a medium lacking tryptophan, leucine and histidine. The prey fragments of the positive clones were amplified by PCR and sequenced at their 5′ and 3′ junctions. The resulting sequences were used to identify the corresponding interacting proteins in the GenBank database (NCBI) using a fully automated procedure. A confidence score (PBS, for Predicted Biological Score) was attributed to each interaction as previously described (55). Three clones encoding the Human class III IF desmin were identified in the initial screen.
Direct 1-by-1 interaction assay
To identify the SPEG domains that interact with desmin, the coding sequence of the human SPEG fragments (aa 2200–2960, aa 2770–3267, aa 2583–2677 and aa 2665–2775) was PCR-amplified and cloned into pB66 as a C-terminal fusion to the Gal4 DNA-binding domain (Gal4-SPEG). The fragment corresponding to the protein desmin (aa 168–402, NM_005876.4) was extracted from the ULTImate Y2H™ screening of SPEG (aa 2200–3267) with the human Adult/Fetal Skeletal Muscle library. The prey fragment is cloned in frame with the Gal4 activation domain into plasmid pP6. All constructs were checked by sequencing. Bait and prey constructs were transformed in the yeast haploid cells CG1945 (mata) and YHGX13 (Y187 ade2-101::loxP-kanMX-loxP, matα), respectively. The diploid yeast cells were obtained using a mating protocol with both yeast strains (54), based on the HIS3 reporter gene (growth assay without histidine). As negative controls, the bait plasmid was tested in the presence of empty prey vector (pP7) and all prey plasmids were tested with the empty bait vector (pB66). The interaction between SMAD and SMURF is used as positive control (56).
Co-immunoprecipitation
Whole-cell extracts from C2C12 myotubes were obtained by homogenization in Co-IP buffer (10% NP-40, 20% 20 mm NaF, 1% Triton X-100) supplemented with complete protease inhibitor tablet (Roche Applied Science, Indianapolis, IN) and 1 mm leupeptin and 1 mm pepstatin A (Sigma-Aldrich, St. Louis, MO). Cells were collected and lysed at 4°C for 30 min with rock. After centrifugation (16 000×g, 20 min), the soluble fractions were collected and concentration measured using a colorimetric BCA assay (23 225; ThermoFisher Scientific, Waltham, MA). Soluble homogenates were pre-cleared with Dynabead Protein G beads (ThermoFisher Scientific) for 1 h and supernatants were incubated with the specific antibodies directed against the protein of interest at 4°C for 12–24 h. Dynabead Protein G beads were then added for 2 h to capture the immune complex. Beads were washed three times with Co-IP buffer supplemented with 0.1% CHAPS. For all experiments, two negative controls consisted of a sample lacking the primary antibody and a sample incubated with another primary antibody from the same serotype as the antibody of interest. Resulting beads were eluted with Laemmli buffer and submitted to SDS-PAGE followed by western blot.
Animals
All studies were approved by the Institutional Animal Care and Use Committee at Children’s Hospital Boston (Boston, MA). Speg-KO mice have been generated as previously described (33). In brief, the homozygous Speg-conditional KO mice (Spegfl/fl) were bred with male transgenic mice (MCK-Cre+) who have the Cre recombinase driven by muscle creatine kinase promoter, with Cre activity observed in skeletal and cardiac muscles. Spegfl/fl: MCK-cre+ mice were the Speg-KO mice that were utilized for further experiments. Littermates with Spegfl/fl: MCK-cre−, Spegfl/wt: MCK-cre+ and Spegfl/wt: MCK-cre− genotypes were used as WT controls.
Immunoblot analysis
Skeletal muscles from Speg-KO and WT littermate mice were dissected, snap frozen in isopentane and stored at −80°C until analysis. Protein isolation and western blot procedures were performed as described previously (57). Proteins were probed with antibody against rabbit anti-SPEG (1:500 dilution, from Dr Mark Perrella, USA) (32), rabbit anti-desmin (Ab8592, 1:1000 dilution, Abcam, Cambridge, MA), rabbit anti-RyR1 (1:500 dilution, from Dr Isabelle Marty, France), rabbit anti-triadin (1:500 dilution, from Dr Isabelle Marty), rabbit anti-DHPRα1 (1:500 dilution, from Dr Isabelle Marty), mouse anti-SERCA1 (Ab2819, 1:1000 dilution, Abcam), rabbit anti-junctophilin-1 (JP1, 40-5100, 1:500 dilution, ThermoFisher Scientific), rat anti-integrin β1 (MAB1997, 1:1000 dilution, Sigma-Aldrich), rabbit anti-laminin (L9393, 1:100 dilution, Sigma-Aldrich), mouse anti-vinculin (66305-1-IG, 1:1000 dilution, Proteintech, Chicago, IL) and mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; MA5-15738, 1:1000 dilution, ThermoFisher Scientific). Secondary horseradish peroxidase-conjugated antibodies against mouse (7076S, 1:2000 dilution, Cell Signaling Technology, Danvers, MA, USA) and against rabbit (7074S, 1:2000 dilution, Cell Signaling Technology) were detected using enhanced chemiluminescence. Quantification of protein levels normalized to GAPDH/vinculin was performed using the program Quantity One, version 4.2.1 (Bio-Rad Laboratories, Inc., Hercules, CA) on an Image Station 440 (Kodak DS; Eastman Kodak Co., Rochester, NY).
Immunofluorescence
Transversal and longitudinal sections (8-μm thick) of isopentane-frozen TA muscle were used for Immunofluorescence. M.O.M. (Mouse on Mouse) Blocking Reagent (MKB-2213-1, Vector Laboratories, Burlingame, CA) was used to block the endogenous mouse Ig staining. Immunofluorescence was performed by standard protocol using mouse anti-desmin (MA5-13259, 1:50 dilution, ThermoFisher Scientific), mouse anti-DHPRα1 antibody (CACNA1S, Ab2862, 1:50 dilution, Abcam), mouse anti-RyR1 (R129, 1:500 dilution, Sigma-Aldrich), rabbit anti-RyR1 (1:100 dilution, from Dr Isabelle Marty), mouse anti-SERCA1 (Ab2819, 1:500 dilution, Abcam), mouse anti-triadin (IIG12, 1:50 dilution, Developmental Studies Hybridoma Bank, Iowa City, IA), rabbit anti-junctophilin-1 (JP1, 40-5100, 1:50 dilution, ThermoFisher Scientific), rabbit anti-laminin (L9393, 1:30 dilution, Sigma-Aldrich), mouse anti-vinculin (66305-1-IG, 1:100 dilution, Proteintech), rat anti-integrin β1 (MAB1997, 1:100 dilution, Sigma-Aldrich), mouse anti-EEA1 (sc-137 130, 1:100 dilution, Santa Cruz Biotechnology, Dallas, TX), rabbit anti-vimentin (5741S, 1:100 dilution, Cell Signaling Technology), rabbit anti-α-actinin-2 (4B3, 1:100 dilution, from Dr Alan Beggs, USA), mouse anti-dystrophin (NCL-DYS2, 1:20 dilution, Leica Biosystems, Buffalo Grove, IL), mouse anti-α-sarcoglycan (NCL-L-a-SARC, 1:100 dilution, Leica Biosystems) and rabbit anti-SPEG (12472-T16, 1:100 dilution, SinoBiological, Beijing, China) for primary antibodies. Goat anti-mouse IgG (H + L) secondary antibody-Alexa Fluor 594 (A-11005, 1:1000 dilution, ThermoFisher Scientific), goat anti-rabbit IgG (H + L) secondary antibody-Alexa Fluor 488 (A-11008, 1:1000 dilution, ThermoFisher Scientific), goat anti-mouse IgG (H + L) secondary antibody-Alexa Fluor 488 (A-11017, 1:1000 dilution, ThermoFisher Scientific) and goat anti-rat IgG (H + L) secondary antibody-Alexa Fluor 594 (A-11007, 1:1000 dilution, ThermoFisher Scientific) were used for secondary antibodies. Slides were coverslipped using Vectashield Mounting Medium with DAPI (H-1200, Vector Laboratories). Images were captured using a Nikon Eclipse 90i microscope in conjunction with NIS-Elements AR software (Nikon Instruments Inc., NY).
Desmin solubility assay
Equal weight of triceps muscles was homogenized with a Bullet blender at full speed for 5 min in ice-cold T-PER Tissue Protein Extraction Reagent (ThermoFisher Scientific) supplemented with Halt Protease Inhibitor Cocktail (ThermoFisher Scientific). Muscle extracts were incubated on ice for 30 min and centrifuged for 20 min at 16 000×g at 4°C. Pellets were collected as the insoluble material and solubilized in extraction buffer (50 mm Tris-Cl pH 7.5, 50 mm NaCl, 5 mm EDTA, 5 mm EGTA, 1 mm DTT, 0.5% Triton X-100) supplemented with 8 M urea and protease inhibitor.
Data analysis and statistics
Results were analyzed with GraphPad Prism (v.8.0; GraphPad Software, San Diego, CA) and expressed as mean ± standard deviation. Student’s t-test was used to determine statistically significant differences between WT and Speg-KO groups.
Conflict of Interest statement. None declared.
Supplementary Material
{kind=link}
Funding
National Institute of Arthritis and Musculoskeletal and Skin Diseases of National Institute of Health (R01 AR068429 to P.B.A.).
References
- 1.Tasfaout, H., Cowling, B.S. and Laporte, J. (2018) Centronuclear myopathies under attack: a plethora of therapeutic targets. J. Neuromuscul. Dis., 5, 387–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jungbluth, H., Wallgren-Pettersson, C. and Laporte, J. (2008) Centronuclear (myotubular) myopathy. Orphanet J. Rare Dis., 3, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ceyhan-Birsoy, O., Agrawal, P.B., Hidalgo, C., Schmitz-Abe, K., DeChene, E.T., Swanson, L.C., Soemedi, R., Vasli, N., Iannaccone, S.T., Shieh, P.B. et al. (2013) Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology, 81, 1205–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilmshurst, J.M., Lillis, S., Zhou, H., Pillay, K., Henderson, H., Kress, W., Muller, C.R., Ndondo, A., Cloke, V., Cullup, T. et al. (2010) RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann. Neurol., 68, 717–726. [DOI] [PubMed] [Google Scholar]
- 5.Nicot, A.S., Toussaint, A., Tosch, V., Kretz, C., Wallgren-Pettersson, C., Iwarsson, E., Kingston, H., Garnier, J.M., Biancalana, V., Oldfors, A. et al. (2007) Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat. Genet., 39, 1134–1139. [DOI] [PubMed] [Google Scholar]
- 6.Bohm, J., Biancalana, V., Malfatti, E., Dondaine, N., Koch, C., Vasli, N., Kress, W., Strittmatter, M., Taratuto, A.L., Gonorazky, H. et al. (2014) Adult-onset autosomal dominant centronuclear myopathy due to BIN1 mutations. Brain, 137, 3160–3170. [DOI] [PubMed] [Google Scholar]
- 7.Bitoun, M., Maugenre, S., Jeannet, P.Y., Lacene, E., Ferrer, X., Laforet, P., Martin, J.J., Laporte, J., Lochmuller, H., Beggs, A.H. et al. (2005) Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat. Genet., 37, 1207–1209. [DOI] [PubMed] [Google Scholar]
- 8.Laporte, J., Hu, L.J., Kretz, C., Mandel, J.L., Kioschis, P., Coy, J.F., Klauck, S.M., Poustka, A. and Dahl, N. (1996) A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat. Genet., 13, 175–182. [DOI] [PubMed] [Google Scholar]
- 9.Cebollero, E., van der Vaart, A., Zhao, M., Rieter, E., Klionsky, D.J., Helms, J.B. and Reggiori, F. (2012) Phosphatidylinositol-3-phosphate clearance plays a key role in autophagosome completion. Curr. Biol., 22, 1545–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parrish, W.R., Stefan, C.J. and Emr, S.D. (2004) Essential role for the myotubularin-related phosphatase Ymr1p and the synaptojanin-like phosphatases Sjl2p and Sjl3p in regulation of phosphatidylinositol 3-phosphate in yeast. Mol. Biol. Cell, 15, 3567–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neukomm, L.J., Nicot, A.S., Kinchen, J.M., Almendinger, J., Pinto, S.M., Zeng, S., Doukoumetzidis, K., Tronchere, H., Payrastre, B., Laporte, J.F. et al. (2011) The phosphoinositide phosphatase MTM-1 regulates apoptotic cell corpse clearance through CED-5-CED-12 in C. elegans. Development, 138, 2003–2014. [DOI] [PubMed] [Google Scholar]
- 12.Zou, W., Lu, Q., Zhao, D., Li, W., Mapes, J., Xie, Y. and Wang, X. (2009) Caenorhabditis elegans myotubularin MTM-1 negatively regulates the engulfment of apoptotic cells. PLoS Genet., 5, e1000679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dang, H., Li, Z., Skolnik, E.Y. and Fares, H. (2004) Disease-related myotubularins function in endocytic traffic in Caenorhabditis elegans. Mol. Biol. Cell, 15, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Velichkova, M., Juan, J., Kadandale, P., Jean, S., Ribeiro, I., Raman, V., Stefan, C. and Kiger, A.A. (2010) Drosophila Mtm and class II PI3K coregulate a PI(3)P pool with cortical and endolysosomal functions. J. Cell Biol., 190, 407–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ribeiro, I., Yuan, L., Tanentzapf, G., Dowling, J.J. and Kiger, A. (2011) Phosphoinositide regulation of integrin trafficking required for muscle attachment and maintenance. PLoS Genet., 7, e1001295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chin, Y.H., Lee, A., Kan, H.W., Laiman, J., Chuang, M.C., Hsieh, S.T. and Liu, Y.W. (2015) Dynamin-2 mutations associated with centronuclear myopathy are hypermorphic and lead to T-tubule fragmentation. Hum. Mol. Genet., 24, 5542–5554. [DOI] [PubMed] [Google Scholar]
- 17.Dowling, J.J., Vreede, A.P., Low, S.E., Gibbs, E.M., Kuwada, J.Y., Bonnemann, C.G. and Feldman, E.L. (2009) Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet., 5, e1000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibbs, E.M., Davidson, A.E., Telfer, W.R., Feldman, E.L. and Dowling, J.J. (2014) The myopathy-causing mutation DNM2-S619L leads to defective tubulation in vitro and in developing zebrafish. Dis. Model. Mech., 7, 157–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith, L.L., Gupta, V.A. and Beggs, A.H. (2014) Bridging integrator 1 (Bin1) deficiency in zebrafish results in centronuclear myopathy. Hum. Mol. Genet., 23, 3566–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buj-Bello, A., Laugel, V., Messaddeq, N., Zahreddine, H., Laporte, J., Pellissier, J.F. and Mandel, J.L. (2002) The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc. Natl. Acad. Sci. U. S. A., 99, 15060–15065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Durieux, A.C., Vassilopoulos, S., Laine, J., Fraysse, B., Brinas, L., Prudhon, B., Castells, J., Freyssenet, D., Bonne, G., Guicheney, P. et al. (2012) A centronuclear myopathy--dynamin 2 mutation impairs autophagy in mice. Traffic, 13, 869–879. [DOI] [PubMed] [Google Scholar]
- 22.Durieux, A.C., Vignaud, A., Prudhon, B., Viou, M.T., Beuvin, M., Vassilopoulos, S., Fraysse, B., Ferry, A., Laine, J., Romero, N.B. et al. (2010) A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum. Mol. Genet., 19, 4820–4836. [DOI] [PubMed] [Google Scholar]
- 23.Fetalvero, K.M., Yu, Y., Goetschkes, M., Liang, G., Valdez, R.A., Gould, T., Triantafellow, E., Bergling, S., Loureiro, J., Eash, J. et al. (2013) Defective autophagy and mTORC1 signaling in myotubularin null mice. Mol. Cell. Biol., 33, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fraysse, B., Guicheney, P. and Bitoun, M. (2016) Calcium homeostasis alterations in a mouse model of the dynamin 2-related centronuclear myopathy. Biol. Open., 5, 1691–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bohm, J., Vasli, N., Maurer, M., Cowling, B.S., Shelton, G.D., Kress, W., Toussaint, A., Prokic, I., Schara, U., Anderson, T.J. et al. (2013) Altered splicing of the BIN1 muscle-specific exon in humans and dogs with highly progressive centronuclear myopathy. PLoS Genet., 9, e1003430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beggs, A.H., Bohm, J., Snead, E., Kozlowski, M., Maurer, M., Minor, K., Childers, M.K., Taylor, S.M., Hitte, C., Mickelson, J.R. et al. (2010) MTM1 mutation associated with X-linked myotubular myopathy in Labrador retrievers. Proc. Natl. Acad. Sci. U. S. A., 107, 14697–14702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Qusairi, L., Weiss, N., Toussaint, A., Berbey, C., Messaddeq, N., Kretz, C., Sanoudou, D., Beggs, A.H., Allard, B., Mandel, J.L. et al. (2009) T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proc. Natl. Acad. Sci. U. S. A., 106, 18763–18768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dowling, J.J., Joubert, R., Low, S.E., Durban, A.N., Messaddeq, N., Li, X., Dulin-Smith, A.N., Snyder, A.D., Marshall, M.L., Marshall, J.T. et al. (2012) Myotubular myopathy and the neuromuscular junction: a novel therapeutic approach from mouse models. Dis. Model. Mech., 5, 852–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robb, S.A., Sewry, C.A., Dowling, J.J., Feng, L., Cullup, T., Lillis, S., Abbs, S., Lees, M.M., Laporte, J., Manzur, A.Y. et al. (2011) Impaired neuromuscular transmission and response to acetylcholinesterase inhibitors in centronuclear myopathies. Neuromuscul. Disord., 21, 379–386. [DOI] [PubMed] [Google Scholar]
- 30.Lawlor, M.W., Alexander, M.S., Viola, M.G., Meng, H., Joubert, R., Gupta, V., Motohashi, N., Manfready, R.A., Hsu, C.P., Huang, P. et al. (2012) Myotubularin-deficient myoblasts display increased apoptosis, delayed proliferation, and poor cell engraftment. Am. J. Pathol., 181, 961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hnia, K., Tronchere, H., Tomczak, K.K., Amoasii, L., Schultz, P., Beggs, A.H., Payrastre, B., Mandel, J.L. and Laporte, J. (2011) Myotubularin controls desmin intermediate filament architecture and mitochondrial dynamics in human and mouse skeletal muscle. J. Clin. Invest., 121, 70–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agrawal, P.B., Pierson, C.R., Joshi, M., Liu, X., Ravenscroft, G., Moghadaszadeh, B., Talabere, T., Viola, M., Swanson, L.C., Haliloglu, G. et al. (2014) SPEG interacts with myotubularin, and its deficiency causes centronuclear myopathy with dilated cardiomyopathy. Am. J. Hum. Genet., 95, 218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huntoon, V., Widrick, J.J., Sanchez, C., Rosen, S.M., Kutchukian, C., Cao, S., Pierson, C.R., Liu, X., Perrella, M.A., Beggs, A.H. et al. (2018) SPEG-deficient skeletal muscles exhibit abnormal triad and defective calcium handling. Hum. Mol. Genet., 27, 1608–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsieh, C.M., Fukumoto, S., Layne, M.D., Maemura, K., Charles, H., Patel, A., Perrella, M.A. and Lee, M.E. (2000) Striated muscle preferentially expressed genes alpha and beta are two serine/threonine protein kinases derived from the same gene as the aortic preferentially expressed gene-1. J. Biol. Chem., 275, 36966–36973. [DOI] [PubMed] [Google Scholar]
- 35.Sutter, S.B., Raeker, M.O., Borisov, A.B. and Russell, M.W. (2004) Orthologous relationship of obscurin and Unc-89: phylogeny of a novel family of tandem myosin light chain kinases. Dev. Genes Evol., 214, 352–359. [DOI] [PubMed] [Google Scholar]
- 36.Lange, S., Ouyang, K., Meyer, G., Cui, L., Cheng, H., Lieber, R.L. and Chen, J. (2009) Obscurin determines the architecture of the longitudinal sarcoplasmic reticulum. J. Cell Sci., 122, 2640–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bar, H., Mucke, N., Kostareva, A., Sjoberg, G., Aebi, U. and Herrmann, H. (2005) Severe muscle disease-causing desmin mutations interfere with in vitro filament assembly at distinct stages. Proc. Natl. Acad. Sci. U. S. A., 102, 15099–15104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lazarides, E., Granger, B.L., Gard, D.L., O'Connor, C.M., Breckler, J., Price, M. and Danto, S.I. (1982) Desmin- and vimentin-containing filaments and their role in the assembly of the Z disk in muscle cells. Cold Spring Harb. Symp. Quant. Biol., 46, 351–378. [DOI] [PubMed] [Google Scholar]
- 39.Clemen, C.S., Herrmann, H., Strelkov, S.V. and Schroder, R. (2013) Desminopathies: pathology and mechanisms. Acta Neuropathol., 125, 47–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schroder, R. and Schoser, B. (2009) Myofibrillar myopathies: a clinical and myopathological guide. Brain Pathol., 19, 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lionello, V.M., Nicot, A.S., Sartori, M., Kretz, C., Kessler, P., Buono, S., Djerroud, S., Messaddeq, N., Koebel, P., Prokic, I. et al. (2019) Amphiphysin 2 modulation rescues myotubular myopathy and prevents focal adhesion defects in mice. Sci. Transl. Med., 11, eaav1866. [DOI] [PubMed] [Google Scholar]
- 42.Ketel, K., Krauss, M., Nicot, A.S., Puchkov, D., Wieffer, M., Muller, R., Subramanian, D., Schultz, C., Laporte, J. and Haucke, V. (2016) A phosphoinositide conversion mechanism for exit from endosomes. Nature, 529, 408–412. [DOI] [PubMed] [Google Scholar]
- 43.Brinas, L., Vassilopoulos, S., Bonne, G., Guicheney, P. and Bitoun, M. (2013) Role of dynamin 2 in the disassembly of focal adhesions. J. Mol. Med. (Berl), 91, 803–809. [DOI] [PubMed] [Google Scholar]
- 44.Toussaint, A., Cowling, B.S., Hnia, K., Mohr, M., Oldfors, A., Schwab, Y., Yis, U., Maisonobe, T., Stojkovic, T., Wallgren-Pettersson, C. et al. (2011) Defects in amphiphysin 2 (BIN1) and triads in several forms of centronuclear myopathies. Acta Neuropathol., 121, 253–266. [DOI] [PubMed] [Google Scholar]
- 45.Lawal, T.A., Todd, J.J. and Meilleur, K.G. (2018) Ryanodine receptor 1-related myopathies: diagnostic and therapeutic approaches. Neurotherapeutics, 15, 885–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amburgey, K., Bailey, A., Hwang, J.H., Tarnopolsky, M.A., Bonnemann, C.G., Medne, L., Mathews, K.D., Collins, J., Daube, J.R., Wellman, G.P. et al. (2013) Genotype-phenotype correlations in recessive RYR1-related myopathies. Orphanet J. Rare Dis., 8, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee, C.S., Hanna, A.D., Wang, H., Dagnino-Acosta, A., Joshi, A.D., Knoblauch, M., Xia, Y., Georgiou, D.K., Xu, J., Long, C. et al. (2017) A chemical chaperone improves muscle function in mice with a RyR1 mutation. Nat. Commun., 8, 14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rokach, O., Sekulic-Jablanovic, M., Voermans, N., Wilmshurst, J., Pillay, K., Heytens, L., Zhou, H., Muntoni, F., Gautel, M., Nevo, Y. et al. (2015) Epigenetic changes as a common trigger of muscle weakness in congenital myopathies. Hum. Mol. Genet., 24, 4636–4647. [DOI] [PubMed] [Google Scholar]
- 49.DiFranco, M., Kramerova, I., Vergara, J.L. and Spencer, M.J. (2016) Attenuated Ca(2+) release in a mouse model of limb girdle muscular dystrophy 2A. Skelet. Muscle, 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kramerova, I., Kudryashova, E., Wu, B., Ottenheijm, C., Granzier, H. and Spencer, M.J. (2008) Novel role of calpain-3 in the triad-associated protein complex regulating calcium release in skeletal muscle. Hum. Mol. Genet., 17, 3271–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bachmann, C., Jungbluth, H., Muntoni, F., Manzur, A.Y., Zorzato, F. and Treves, S. (2017) Cellular, biochemical and molecular changes in muscles from patients with X-linked myotubular myopathy due to MTM1 mutations. Hum. Mol. Genet., 26, 320–332. [DOI] [PubMed] [Google Scholar]
- 52.Quick, A.P., Wang, Q., Philippen, L.E., Barreto-Torres, G., Chiang, D.Y., Beavers, D., Wang, G., Khalid, M., Reynolds, J.O., Campbell, H.M. et al. (2017) SPEG (striated muscle preferentially expressed protein kinase) is essential for cardiac function by regulating junctional membrane complex activity. Circ. Res., 120, 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Campbell, H.M., Quick, A.P., Abu-Taha, I., Chiang, D.Y., Kramm, C.F., Word, T.A., Brandenburg, S., Hulsurkar, M., Alsina, K.M., Liu, H.B. et al. (2020) Loss of SPEG inhibitory phosphorylation of ryanodine receptor Type-2 promotes atrial fibrillation. Circulation, 142, 1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fromont-Racine, M., Rain, J.C. and Legrain, P. (1997) Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nat. Genet., 16, 277–282. [DOI] [PubMed] [Google Scholar]
- 55.Formstecher, E., Aresta, S., Collura, V., Hamburger, A., Meil, A., Trehin, A., Reverdy, C., Betin, V., Maire, S., Brun, C. et al. (2005) Protein interaction mapping: a Drosophila case study. Genome Res., 15, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Colland, F., Jacq, X., Trouplin, V., Mougin, C., Groizeleau, C., Hamburger, A., Meil, A., Wojcik, J., Legrain, P. and Gauthier, J.M. (2004) Functional proteomics mapping of a human signaling pathway. Genome Res., 14, 1324–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Agrawal, P.B., Joshi, M., Savic, T., Chen, Z. and Beggs, A.H. (2012) Normal myofibrillar development followed by progressive sarcomeric disruption with actin accumulations in a mouse Cfl2 knockout demonstrates requirement of cofilin-2 for muscle maintenance. Hum. Mol. Genet., 21, 2341–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li, Q., Lin, J., Rosen, S.M., Zhang, T., Kazerounian, S., Luo, S. and Agrawal, P.B. (2020) Striated preferentially expressed protein kinase (SPEG)-deficient skeletal muscles display fewer satellite cells with reduced proliferation and delayed differentiation. Am. J. Pathol., 190, 2453–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.