

Abstract
With the elucidation of a myriad anabolic and catabolic enzyme-catalyzed cellular pathways crisscrossing each other, an obvious question arose – how could these networks operate with maximal catalytic efficiency and minimal interference? A logical answer was the postulate of metabolic channeling, which in its simplest embodiment assumes that the product generated by one enzyme passes directly to a second without diffusion into the surrounding medium. This tight coupling of activities might increase a pathway’s metabolic flux and/ or serve to sequester unstable/ toxic/ reactive intermediates as well as prevent their access to other networks. Here we present evidence for this concept, commencing with enzymes that feature a physical molecular tunnel, to multi-enzyme complexes that retain pathway substrates through electrostatics or enclosures, and finally to metabolons that feature collections of enzymes assembled into clusters with variable stoichiometric composition. Lastly, we discuss the advantages of reversibly assembled metabolons in the context of the purinosome, the purine biosynthesis metabolon.
Keywords: Metabolic channeling, molecular tunnel, purinosome, metabolon, membrane-less compartmentalization
50 words Abstract:
The channeling of metabolic intermediates can increase pathway fluxes and protect metabolites. Mechanisms of channeling include direct physical tunnels, electrostatic trapping and enclosures, and membrane-less metabolic compartments or “metabolons”. We highlight examples of the above classes, finally focusing on the purinosome to discuss the advantages of dynamically assembled metabolons.
Classical understanding of how enzymatic reactions proceed in cells was based on a simple diffusive-mixing model. However, contemporary experimental evidence does not support this view (Luby-Phelps, 2013; Saks et al., 2008). Instead of being a dilute homogeneous mix of metabolites and the enzymes producing/ utilizing them, the cytosol appears to be a crowded micro-compartmentalized volume with anomalous diffusive properties. Indeed, completely diffusive mixing of metabolites and enzymes can potentially be detrimental to cell proliferation and survival if the flux of a critical metabolic pathway is limited by intermediate instability, toxicity, and/ or potential interference by competing metabolic pathways (Zhang and Fernie, 2021). Under selective pressure to overcome such limitations on biochemical pathways, metabolic channeling (Figure 1) has evolved in multiple manifestations.
Figure 1. Metabolic channeling in cascade reactions.
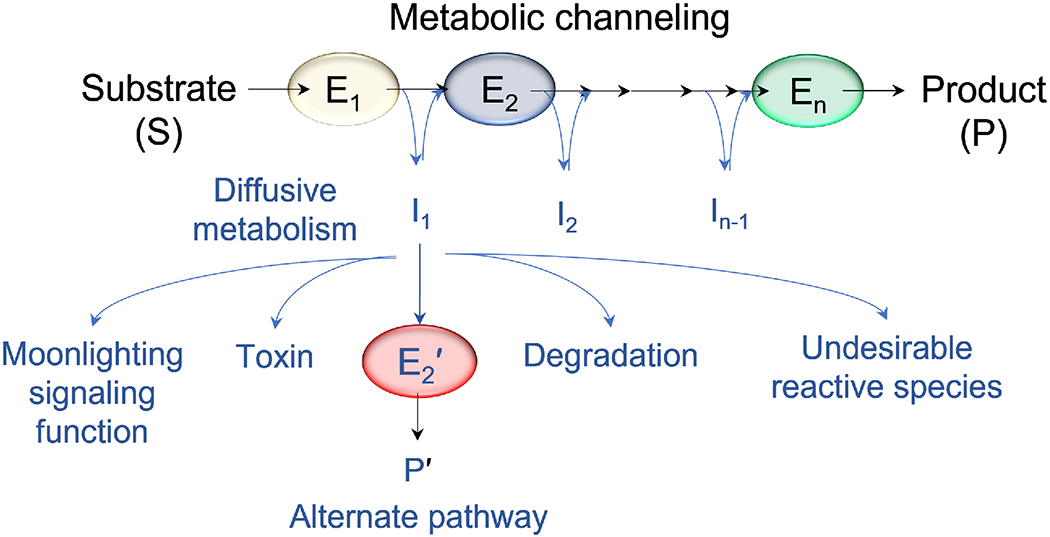
Unlike diffusive metabolism, metabolic channeling leads to intermediate sequestration, thus preventing accumulation of toxic, unstable, reactive intermediates, and /or depletion of intermediates by utilization in alternative pathways (P’). For intermediates with a signaling function, metabolic channeling could modulate such functions by regulating the abundance of free intermediates. Channeling leads to effective utilization of intermediates and higher pathway fluxes in response to cellular metabolic requirements. Abbreviation: E, enzymes, with successive enzymes in a pathway cascade numbered from E1 to En.
Metabolic channeling
Figure 2 shows the three different versions of metabolic channeling that circumvent free diffusion of intermediates: direct, proximity, and via multi-enzyme clustering (Castellana et al., 2014). These mechanisms increase retention of intermediates in the proximity of the involved enzyme active sites by their 1) sequestration and queuing in molecular tunnels and caged structures (Tsitkov et al., 2018), 2) electrostatic binding on the surface of the enzyme (Earl and Calabrese Barton, 2017), 3) or by the increase in escape times in phase-separated metabolons. It should be emphasized though that simple fusion of the enzymes of a pathway to form a multi-functional enzyme may not impart kinetic benefits, demonstrating that proximity of cascade enzymes does not automatically imply metabolic channeling (Castellana et al., 2014; Eun et al., 2014; Veraszto et al., 2020).
Figure 2. Different modes of metabolic channeling.
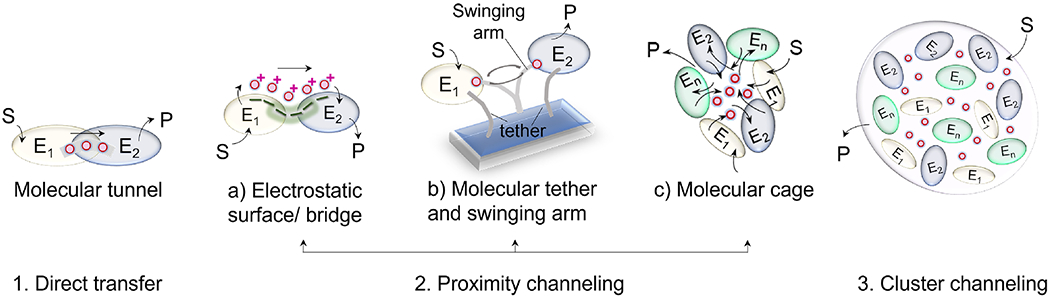
(1) Channeling via direct transfer by formation of contiguous molecular tunnels connecting two active sites. (2) Proximity channeling aided by (a) electrostatic interactions of the intermediate with the protein surfaces; (b) tethering the enzymes and a shared cofactor, such that the co-factor can swing back and forth and be effectively shared between the two active sites; and (c) formation of homo/ hetero oligomeric structures with a cavity that acts to increase the intermediate retention and accessibility for the cascade reactions. (3) Cluster channeling by formation of a metabolon. Abbreviations: S, substrate, P, product, E, enzymes, with successive enzymes in a pathway cascade numbered from E1 to En.
Direct transfer
Simulations of the direct transfer of intermediate between enzyme active sites predict that direct metabolic channeling can affect both the transient time to reach the pathway steady state (Heinrich and Schuster, 1991; Welch and Easterby, 1994; Wheeldon et al., 2016) as well as the steady-state intermediate pool sizes, although the latter effect is generally small (Easterby, 1989; Mendes et al., 1996; Poshyvailo et al., 2017) and a gain in flux at steady state is only observed when the reactions are diffusion limited (Kuzmak et al., 2019). Accordingly, the value of channeling contribution is generally not the overall pathway rate enhancement, which is ultimately limited by the turnover number of the slowest enzyme, but rather an increase in the metabolic yield by preventing the diffusive escape of the intermediates produced between successive enzymes. However, such simulations do not take into account the conformational plasticity of enzymes and the examples discussed below highlight the benefits of protein-protein interactions beyond what’s anticipated from simulations (Huang et al., 2001). Notably, direct channeling by engineering an intra/ inter-molecular tunnel between two cascade active sites has not yet been reported.
a). Tryptophan synthase: indole tunnel
Tryptophan synthase (TS) has two subunits (α and β) and catalyzes the last two reactions of tryptophan biosynthesis (Yanofsky and Crawford, 1972). The α subunit catalyzes the formation of indole and glyceraldehyde-3-phosphate (G3P) from indole-3-glycerol phosphate (IGP) and the β subunit uses the indole intermediate and serine to form tryptophan (Miles, 2001). The X-ray crystal structure of Salmonella typhimurium TS was first determined in 1988 (Hyde et al., 1988), and its structure-function relationship has been elucidated in the following years (Barends et al., 2008; Miles et al., 1986; Raboni et al., 2007; Rhee et al., 1996; Weyand et al., 2002). The tetrameric complex has a linear α/β/β/α arrangement, where two β subunits sit in the center of the complex and two α subunits locate at the opposite sides of the β2 dimer. The active site of the α subunit is located near the αβ interface, whereas that of the β subunit is buried in the center of the β2 dimer, with a closest distance between a pair of α and β subunit active sites around 25-30 Å (Hyde et al., 1988). Structural analysis revealed allosteric communication between α and β subunits resulting in the formation of a hydrophobic ‘indole tunnel’ that can accommodate up to four indole molecules and delivers indole from its site of production (subunit α) to its site of utilization (subunit β) preventing indole solvation and leakage (Figure 3a) (Hyde et al., 1988; Rhee et al., 1996). Furthermore, loop closure over the active site cavity and conformational changes in the tunnel domain residues are coordinated in the catalytic cycle to promote the entry of indole into the tunnel without loss to the bulk cytosol (Ahmed et al., 1991; Miles et al., 1986; Rhee et al., 1997).
Figure 3. Examples of direct and proximity channeling.
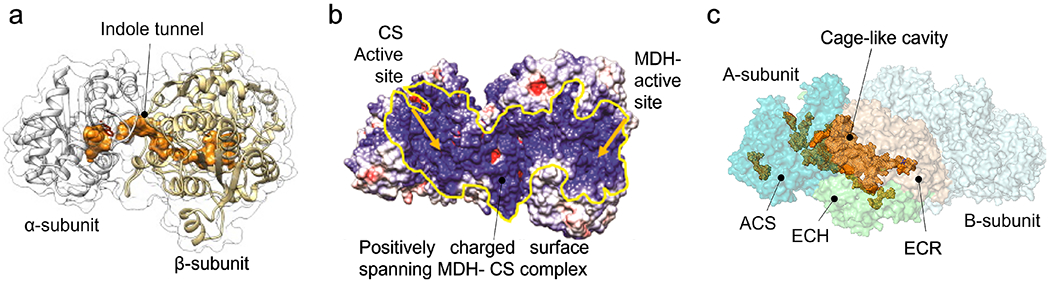
Illustrations showing (a) indole tunnel in tryptophan synthase (adapted from (Fleming, Schupfner et al. 2018)). Indole is produced in the α-subunits and is directly channeled to the β-subunits by sequestration and the indole tunnel; (b) the charged surface of the MDH-CS complex enables electrostatic binding of the intermediate, OAA, and its delivery to the CS active site (adapted from (Bulutoglu, Garcia et al. 2016)). Yellow arrows indicate the location of the two active sites; (c) molecular cage-like cavity in the propionyl-CoA synthase (PDB ID 6EQO) connecting the active sites of acyl-CoA synthetase (ACS, teal), enoyl-CoA hydratase (ECH, limegreen) enoyl-CoA reductase (ECR, wheat) domains of A-subunit are shown. B-subunit is shown in lightcyan. Figure created in PyMOL 1.7.4.5 and cavity dimensions were determined by PCASA 1.1 (http://g6altair.sci.hokudai.ac.jp/g6/service/pocasa/).
In vitro kinetic studies (Anderson et al., 1995; Anderson et al., 1991) failed to trap indole in a steady-state experiment, supporting a direct indole transfer mechanism. Rapid quench flow and stopped flow analysis of the α and β reactions in isolation versus the mixture of the two enzymes revealed that when mixed, the α and β reactions show a 150- and a 250-fold rate enhancement, respectively, compared to each isolated reaction. This rate difference was attributed to a reciprocal conformational communication between the two subunits as they bind the substrate and perform catalysis. Furthermore, based on structural analysis (Anderson et al., 1995; Anderson et al., 1991), residues critical for indole tunnel formation and inter-subunit communication were identified. Replacement of these residues by site-directed mutagenesis showed: a) impaired indole channeling resulting in equilibration of indole with the bulk solvent and detection in a single turnover reaction, b) disruption of inter-subunit conformational communication, and c) a significant reduction (up to 300-fold) in the rate of tryptophan synthesis compared to the wild type enzyme.
b). GMP synthetase: ammonia tunnel
Guanosine monophosphate synthetase (GMPS) catalyzes the reaction of xanthosine monophosphate (XMP) with ammonia (NH3) to form GMP, the last step in guanosine nucleotide synthesis (Buchanan, 1973). The enzyme’s glutamine amidotransferase (GAT) activity hydrolyses glutamine to produce ammonia, which is then utilized by the ATP pyrophosphatase (ATPPase) for GMP synthesis. The two activities are either located on two domains of a single polypeptide or on different subunits that interact for coordinated catalysis (Grimaldi et al., 2000; Maruoka et al., 2010; Tesmer et al., 1996). Interestingly, the binding of substrate XMP and its activation by adenylylation in the ATPPase domain triggers a series of loop movements and conformational changes leading to GAT activation and establishment of a molecular tunnel (30 Å) between the two active sites (Bhat et al., 2011; Patel et al., 1977; Raushel et al., 2003; Tesmer et al., 1996; Vetter and Wittinghofer, 1999; Zalkin and Smith, 1998; Zyk et al., 1969). At physiological pH ~7.5, hydration of ammonia to ammonium is highly favored, but the pH dependence of the ATPPase activity shows that, in this reaction, ammonia is the reactive species and not the ammonium ion (Bhat et al., 2011). This observation highlights the importance of NH3 sequestration for GMPS activity. Extensive structural (Amaro et al., 2007; Myers et al., 2005; Tesmer et al., 1996; von der Saal et al., 1985; Welin et al., 2013) and kinetic examination (Oliver et al., 2013) of the enzyme reveals that the interdomain/ subunit communication not only ensures that the GAT domain only produces ammonia upon interaction with the catalytically competent state of the ATPPase active site, but it also ensures sequestration of ammonia from the bulk solvent and undesirable reactions. A similar direct metabolite channeling has also been observed in other amidotransferases, including the two de novo purine biosynthesis (DNPB) enzymes, amidophosphoribosyl transferase (PPAT) and phosphoribosylamine glycinamide synthetase (PFAS), discussed later in the article (Buchanan, 1973).
Proximity channeling
While several examples of natural and artificial enzyme systems show proximity channeling (PC), i.e., high probability of intermediate processing by a proximal active site in the absence of a physical tunnel, proximity in space alone is insufficient to ensure metabolic channeling. This distinction is exemplified by the fungal aromatic complex (AROM complex) (Hawkins et al., 1993; Veraszto et al., 2020). This polyfunctional complex is formed by natural fusion of enzymes and catalyzes five consecutive reaction steps in the aromatic amino acid synthesis pathway. Despite proximity of the active sites, both in vivo and in vitro kinetics on Neurospora crassa and Chaetomium thermophilum AROM complex shows no evidence of metabolic channeling. In contrast, there are notable natural and engineered examples when additional biochemical and biophysical features of the participating enzymes facilitate proximity channeling and increase the pathway throughput (Bugada et al., 2018; Dubey and Tripathi, 2021). The first PC class is based on electrostatic interactions between the intermediate and protein surface (Figure 2). The second one features molecular tethers that enable transfer of intermediates between the participating proteins. Finally, a third class features entrapment of intermediates by a multi-enzyme assembly acting as a cage.
To clarify the requirements for proximity channeling, McCammon and coworkers performed a Brownian dynamics simulation on a hypothetical two enzyme model and assessed the effect of distance and orientation of the subsequent active sites on proximity channeling. The model incorporated electrostatic binding in an ‘active zone’ close to the active site of the recipient enzyme. Distance between the active zones was varied from 5-50 Å and their orientation angle from 0-180°. Active zone orientation had a marked effect on channeling and reaction probability. As the distance between active zones reached 25 Å, electrostatic binding of the intermediate failed to accrue any metabolic benefits (Bauler et al., 2010). Importantly, retention mechanisms for the intermediate become necessary as the distance between consecutive active site increases beyond ~10 Å (Wheeldon et al., 2016).
a). Intermediate sequestration by electrostatic surfaces
To explain the discrepancies between the in vivo abundance of oxaloacetic acid (OAA) and the observed TCA cycle flux, a mechanism involving direct interaction of the enzyme’s malate dehydrogenase (MDH, converts malate to OAA) and citrate synthase (CS, converts OAA to citrate) was proposed (Noor et al., 2014; Srere et al., 1973). Srere and coworkers observed that the immobilization of malate dehydrogenase (MDH) and citrate synthase (CS) promoted inter-protein interaction and, furthermore, their immobilization in close proximity led to a slight kinetic advantage (Srere et al., 1973). This MDH-CS inter-protein interaction and channeled uptake of OAA was promoted in the presence of polyethylene glycol (PEG) (Datta et al., 1985; Halper and Srere, 1977). If the two enzymes were genetically fused (Lindbladh et al., 1994), the lag time for the reaction was reduced and the competition by aspartate aminotransferase (AAT), an alternate OAA utilizing enzyme, was curtailed, thus providing further support for a channeling mechanism.
In a Brownian dynamics simulation of the MDH-CS complex by Elcock et al. electrostatic binding acted to increase the transport efficiency of the intermediate OAA between the MDH dimer and CS dimer (Elcock and McCammon, 1996). The active sites of MDH and CS are about 60 Å apart, but without a continuous molecular tunnel connecting the two. Instead, positively charged residues from the two enzymes form a partly solvent-exposed electrostatic surface that aids preferential binding of the negatively charged intermediate OAA (Figure 3b). Recent studies have confirmed these findings and provided further proof for the cytosolic MDH-CS complex through in vivo and in vitro chemical cross-linking. Their work further suggests that the presence of OAA promotes MDH-CS complexation, upon which the two enzymes undergo significant conformational rearrangement and charge redistribution to promote OAA binding and transport (Bulutoglu et al., 2016; Wiegand and Remington, 1986; Wu et al., 2015).
b). Proximity channeling by engineered electrostatic bridge
Informed by the aforementioned publications, Liu et al. synthesized a putative channeling complex featuring the enzyme hexokinase (HK) that catalyzes the phosphorylation of glucose to glucose-6-phosphate (G6P) and the enzyme glucose-6-phosphate dehydrogenase (G6PDH) that catalyzes G6P oxidation (Liu et al., 2017). The two enzymes were linked by a cationic poly-lysine bridge to shuttle the negatively charged intermediate, G6P. From molecular dynamics simulations, lysine was chosen over arginine and histidine, and the peptide length and composition were optimized based on the adsorption time and surface mobility of G6P on the bridge surface. An optimal balance between adsorption and mobility enables the shuttling of G6P by minimizing its diffusion into bulk solvent but retaining its mobility on the linkage surface. HK and G6PDH were joined by two sets of peptide bridges, penta-lysine (K5) as an electrostatic pathway and penta-glycine (G5) – the neutral bridge as the control to differentiate surface channeling from a pure proximity effect. After injection of glucose, the lag time (τ) to attain the steady state for K5 (τ =70 ± 6 s) was shorter than the control G5 (τ =105 ± 1 s) and the two uncoupled enzymes (τ =103 ± 10 s). The effect on the reaction lag time attained by K5 bridging was lost in the presence of high ionic strength salt solution, confirming that metabolic channeling aided by weak electrostatic interaction could be disrupted.
c). Proximity channeling by artificial swing arm and molecular tethering
In another notable example, Fu et al. built a two-enzyme system, tethering G6PDH and MDH to a DNA scaffold with a poly(T)20 oligonucleotide linked NAD+ at its center point to mimic a mobile swing arm (Fu et al., 2014; Perham, 2000). G6PDH oxidizes G6P, simultaneously reducing NAD+ to NADH, which is then used as a cofactor by MDH. The distance from these two enzymes to the NAD+ arm was optimized at 7 nm. The complete swing arm assembly G6PDH-NAD+-MDH shows a 90 fold increase in specific activity for the two-step reaction relative to a system where tethered enzymes receive freely diffusive NAD+ at the same concentration. Since G6PDH has higher activity than MDH, expansion of the model to surround each G6PDH by two or four MDH enzymes (NAD+-MDH)2/ (NAD+-MDH)4 provided additional 2-3 fold kinetic advantage compared to the initial model with equimolar abundance of each enzyme. Additionally, the tethered cofactor was less available to lactate dehydrogenase (LDH), which competes with MDH for access to NADH, relative to the untethered cofactor, underscoring the beneficial characteristics of metabolic channeling.
d). Intermediate sequestration by structural cages
3-hydroxypropionate (3-OHP) is converted to propionyl-CoA (PC) by the enzyme propionyl-CoA synthase as a part of the 3-hydroxypropionate cycle, an autotrophic CO2 fixation pathway in some phototrophic eubacteria and chemotrophic archaebacteria (Alber and Fuchs, 2002). The multifunctional homodimeric enzyme consists of three domains- acyl-CoA synthase (ACS), enoyl-CoA hydratase (ECH), and enoyl- CoA reductase (ECR). It was proposed that the enzyme might carry out the reactions in a channeled manner given that the one of the pathway intermediates, acrylyl-CoA, is highly toxic. X-ray structure analysis revealed that at the center of each monomer is a ~33 nm3 cavity that connects the three active centers in each monomer (Figure 3c) (Bernhardsgrutter et al., 2018). The cavity is lined by positively charged residues, increasing the CoA-ester intermediate retention in the cavity and the negatively charged residues surrounding the openings minimize their loss to the bulk solvent. Reaction time course measurements found acrylyl-CoA to be undetectable or very low, indicating sequestration of acrylyl-CoA. Furthermore, isotope labeling competition assays showed that at steady state, the enzyme preferentially processed the internally- generated intermediates to form the product over the intermediates supplemented in the bulk solvent. The inaccessibility of the externally supplemented intermediates was attributed to inter-domain communication which triggers a ‘closed’ conformation of the enzyme during the catalytic cycle. This restricts loss of the internally generated intermediates and prevents the externally available intermediates from binding the enzyme.
Interestingly, in other examples of acyl-CoA ester processing enzymes, molecular cage formation is accompanied with covalent tethering of the intermediate to promote the intermediate sequestration. FAS is a large multifunctional enzyme that catalyzes the synthesis and elongation of fatty acids facilitated by a prosthetic group derived from CoA. Fungal and mammalian FAS are giant complexes containing eight functional domains. Fungal FAS (Jenni et al., 2007; Lomakin et al., 2007) is a 2.6 MDa “barrel shape” complex consisting of two different subunits: both α and β subunit contains four functional domains each. These eight functional domains carry out all of the fatty acid biosynthesis steps including activation, priming, multiple cycles of elongation, and termination, generating fatty acid chains with 16 or 18 carbons (Lynen et al., 1980). While channeling and its metabolic consequences have not been directly demonstrated, the FAS complex structure shows a molecular cage-like cavity, which has been proposed to restrict free diffusion of the intermediates and to aid channeled synthesis by creation of a reaction chamber. Based on the crystal structure of yeast FAS, six α-subunits form a ring/ wheel shape in the center with two β-trimers sitting on the top and bottom surfaces and creating six reaction chambers. A key feature in FAS is the covalent tethering of the activated thiol ester intermediates to the acyl carrier protein (ACP), arguably to avoid premature product release due to hydrolysis of the thioester linkage (Beld et al., 2015). Intermediate sequestration also limits side-reactions, and the size of the reaction chamber controls the fatty acid chain length. In each reaction chamber, the mobile ACP domain, part of the α-subunit, is able to reach the active sites of all the functional domains through a swinging movement and trigger inter-domain communication (Lomakin et al., 2007). Such inter-domain communication and tethering of the intermediate that results in metabolic channeling is also observed in polyketide synthase (Khosla et al., 2014; Robbins et al., 2016). Different from the fungal FAS, mammalian FAS only has one 270 kDa polypeptide with all the functional domains on it and a different structural arrangement of the domains (Smith and Tsai, 2007). Similarly, the structure of the pyruvate dehydrogenase complex, comprised of multiple copies of pyruvate dehydrogenase, dihydrolipoamide transacetylase, and dihydrolipoamide dehydrogenase also harbors a large molecular cage-like cavity that is proposed to trap the intermediates and disallow their equilibration with the bulk cytosol (Patel and Roche, 1990).
These examples highlight the different intermediate retention mechanisms for achieving proximity channeling.
Metabolons
A different mode of metabolic channeling called cluster channeling was suggested based on the early research on mitochondrial respiratory chain enzyme complexation and localization in the mitochondrial membrane fraction and similar observations on complexation of enzymes involved in other central metabolic pathways (Ernster and Schatz, 1981; L J Reed and Cox, 1966). The aggregates of enzymes catalyzing two or more steps of a pathway were termed ‘multienzyme complexes’ by Reed (Reed, 1974); Wilson coined the term ‘ambiquitous enzymes’ to describe kinetically distinct pools of enzymes that reversibly partition between soluble and membrane- bound forms (Wilson, 1978); McConkey called the transient macromolecular interactions in vivo ‘quinary complexs’ (McConkey, 1982); and Srere, defined ‘metabolon’ as the “supramolecular complex of sequential metabolic enzymes and cellular structural elements” (Srere, 1985; Srere, 1987). Building on these concepts, we define a metabolon as a dynamic enzyme complex carrying out the sequential steps of a metabolic pathway by cluster channeling, where an intermediate can be processed by any of the multiple copies of each enzyme instead of dependence on the nearest one. Metabolons involve multivalent interactions between protein surfaces, probably accompanied with liquid-liquid phase separation, leading to membrane-less microcompartments. The assembly may further be stabilized by non-enzymatic components- accessory proteins and chaperone machinery; interaction with cellular structural elements (membranes and cytoskeleton); and non-membranous compartmentalization by liquid-liquid phase separation (Kastritis and Gavin, 2018; Lynch et al., 2020; Lyon et al., 2021; Prouteau and Loewith, 2018; Schmitt and An, 2017; Strom and Brangwynne, 2019; Sweetlove and Fernie, 2018; Zhang et al., 2021).
The ability of metabolons consisting of clusters of subsequent pathway enzymes (Figure 2) to protect intermediates and increase pathway flux has been modeled using reaction-diffusion equations (Buchner et al., 2013; Castellana et al., 2014; Hinzpeter et al., 2017; Hinzpeter et al., 2019) and by multiple scattering theory (Gopich, 2021). These models have become especially pertinent in view of the increased recognition of liquid condensates in cells. From such models, one can calculate the metabolic pathway efficiency as a function of enzyme numbers, kinetic rates, and condensate size, including the relative enzyme stoichiometries required for optimal efficiency (maximum pathway output flux). All models consider the trade-off between the effective channeling of reaction intermediates (favored by larger condensates) and access to substrates (favored by smaller condensates).
Purinosome: the de novo purine biosynthesis metabolon
The prime example to date of a cellular metabolon is the purinosome, which catalyzes de novo purine biosynthesis (DNPB) (Figure 4). Commencing with the substrate 5-ribosyl-1-pyrophosphate (PRPP), the pathway produces inosine monophosphate (IMP), which is partitioned to adenosine monophosphate (AMP) and guanosine monophosphate (GMP) (Pareek et al., 2021). The biosynthesis to IMP proceeds through ten intermediates and is catalyzed by six enzymes, some of which are multifunctional (Figure 4a). The pathway requires substrates such as glycine, aspartic acid, and formate (for production of the folate cofactor, formyl-THF), which are furnished by the mitochondria. Preliminary data also implicates the cytosolic methylene tetrahydrofolate dehydrogenase (MTHFD1) needed to produce the folate cofactor as a member of the purinosome (Pareek et al., 2020; Smith et al., 1980). DNPB also proceeds by a parallel diffusive pathway, albeit with low efficiency (Figure 4b) (Pareek et al., 2020).
Figure 4. Purine synthesis and its dependency on mitochondrial metabolism.
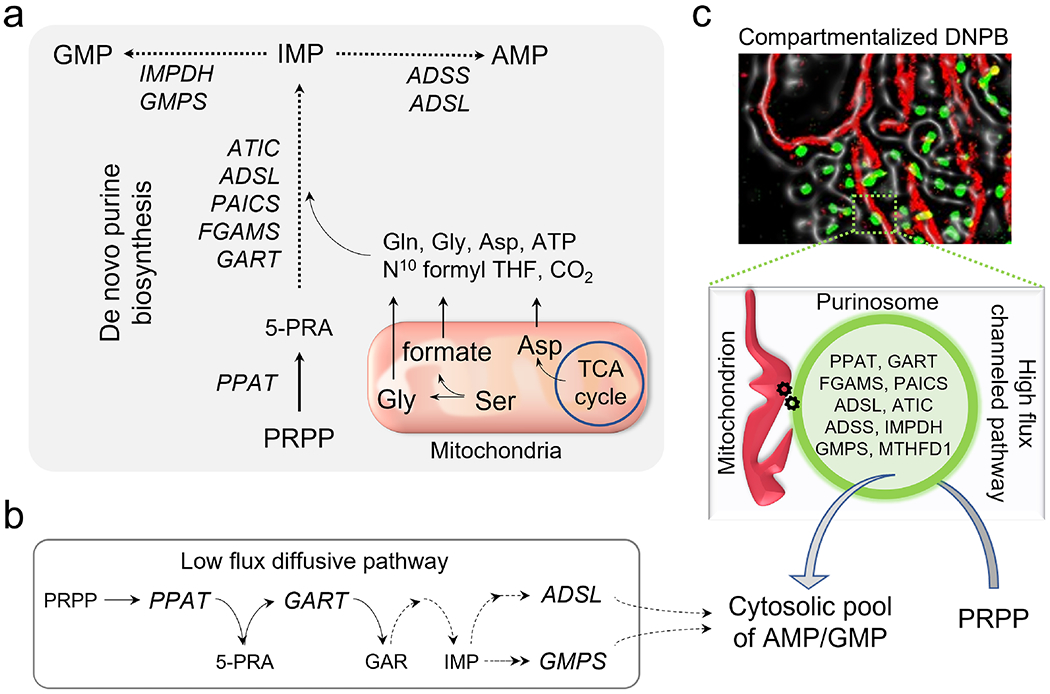
(a)De novo purine biosynthesis (DNPB) converts phosphoribosyl pyrophosphate (PRPP) to inosine monophosphate (IMP) which is bifurcated into the products adenosine monophosphate (AMP) and guanosine monophosphate (GMP) employing nine enzymes: amidophosphoribosyl transferase (PPAT), trifunctional phosphoribosylglycinamide formyltransferase (GART), phoshoribosylformylglycinimidine transferase (PFAS/ FGAMS), bifunctional phosphoribosylaminoimidazole carboxylase and phosphoribosyl aminoimidazole succinocarboxamide synthase (PAICS), bifunctional adenylosuccinate lyase (ADSL), bifunctional 5-aminoimidazole-4-carboxamide nucleotide formyltransferase/ IMP cyclohydroxylase (ATIC), adenylosuccinate synthetase (ADSS), IMP dehydrogenase (IMPDH), GMP synthetase (GMPS). Generation of the pathway substrates glycine (Gly), aspartic acid (Asp), and cofactor N 10formyl-tetrahydrofolate (N10 formyl THF) depends on mitochondrial metabolism. Solid arrows: single reaction step, dashed arrows: multiple steps in the cascade. (b) Under purine depletion, HeLa cells perform diffusive DNPB with only a low flux contribution. Abbreviations: 5-phosphoribosylamine (5-PRA) and glycinamide ribonucleotide (GAR). (c) Channeled synthesis utilizing mitochondria (labeled in red) associated purinosomes (labeled in green) visualized by fluorescence imaging in HeLa cells is the major contributor to the overall DNPB flux. Note the inclusion in the purinosome of methylenetetrahydrofolate dehydrogenase/cyclohydrolase (cytosolic isoform MTHFD1). The factors responsible for stabilizing purinosomes on mitochondrial membranes are not yet known.
Fluorescence imaging shows purinosomes localize at the microtubule/mitochondria interface, a result supported by a proximity-ligation assay, and an isotopically-labeled substrate-incorporation assay, both performed in unmanipulated HeLa cells expressing proteins at endogenous level (Pareek et al., 2021) (Figure 4c). High resolution-gas cluster ion beam secondary-ion mass spectrometry (GCIB SIMS) imaging following isotopically labeled metabolite labeling in HeLa cells showed that all the intermediates from the initial PRPP substrate to IMP are sequestered within the purinosome (Doigneaux et al., 2020; Pareek et al., 2021; Pareek et al., 2020; Pedley and Benkovic, 2017). The biosynthesis is accelerated at least seven-fold relative to a cytosolic diffusive mechanism. Moreover, the partitioning of IMP is directed towards AMP in the purinosome, whereas the competing enzymes are expected to favor GMP in the diffusive mechanism (Pareek et al., 2020). Thus, purinosomes possess all the properties anticipated for a channeled enzyme cluster. It is notable that so far, the attempts to purify the native complex or reconstitute it in vitro have not been successful, implying the weak and dynamic nature of interactions that hold these enzymes together. The inherent dynamics of the complex is proposed to be controlled by post-translation modifications that alter the surface properties and protein-protein interactions of these enzymes (Pareek et al., 2021).
In a transient transfection model, DNPB enzyme cluster size ranges from 100 nm- 1.5 μm in diameter, and the numbers per cell reach to 100 or more, although, given the complexities associated with such models, such estimates are only tentative (Chan et al., 2018; Chan et al., 2015; Doigneaux et al., 2020). The difficulty in visualizing these clusters at endogenous protein level implies that the size of functional purinosomes may be much smaller than these estimates; larger puncta may represent aggregates and non-functional storage bodies, an artifact of protein overexpression of the tagged proteins (Schmitt et al., 2016; Zhao et al., 2014); and PLA and mass spectrometry imaging of HeLa cells estimates 10-30 purinosomes/ cell at endogenous protein levels (Doigneaux et al., 2020; Pareek et al., 2020). To understand the distribution of DNPB enzymes at endogenous level in HeLa cells, we theoretically examined the partitioning of PPAT, the rate limiting enzyme in the pathway (Box 1). PPAT is the least abundant DNPB enzyme and catalyzes the first committed step of PRPP to 5-PRA conversion, albeit with a low kcat/KM, and produces an unstable reaction product (5-PRA half-life ~5 sec at 37 °C), a clear pathway bottleneck that purinosomes-mediated metabolic channeling must overcome (Itzhak et al., 2016; Mueller et al., 1994; Nagaraj et al., 2011). From the estimation in Box 1, the size of purinosomes may only reach ≤ 200-300 nm, and the number of enzyme molecules/ purinosome may range from ~103 to 105/purinosome, assuming equimolar distribution of all 10 enzymes constituting the purinosome. In line with the cluster channeling model, our estimation of the metabolite abundance (≥106/ purinosome) appears to be significantly higher than the estimated enzyme abundance/ purinosome (Pareek et al., 2020). The exact mechanisms underpinning the sizes and abundances of purinosomes in different cells remains unclear, although the data so far suggests heterogeneity in size and constitution of the complex. One can anticipate purinosome size control by variations in the stoichiometric abundance of different enzymes and/ or the number of copies of each enzyme/ purinosome while keeping stoichiometry constant. Additionally, interaction of two of the DNPB enzymes, PPAT and PFAS, with HSP90/70, post-translation modifications of DNPB enzymes, and different signaling pathways are thought to involved in controlling the spatial and temporal dynamics of the purinosome assembly, although the exact mechanism has not been elucidated (Pareek et al., 2021; Pedley and Benkovic, 2017).
Several important issues remain unresolved. How are purinosomes formed and maintained? What is the complete composition and stoichiometry of the enzymes within purinosomes? What is the precise role of the Hsp90/70 chaperone system – does it assist in protein folding and/or in assembly and maintenance of purinosomes? In the absence of a lipid or protein/RNA shell, is a diffusion barrier achieved to prevent loss of the metabolic intermediates, yet allow entrance of needed cofactors? The determinants of branch-point processing of IMP to favor AMP over GMP formation by purinosomes are not known: do they derive from the ratio of the partitioning enzymes or regulation of their respective activities in the purinosome? We note that all of the enzymes in the pathway are multimeric – what role does oligomerization play in the assembly of purinosomes? We speculate that inter-molecular signaling and conformational changes induced by protein-protein interactions between different DNPB enzymes may introduce another level of pathway flux control. Answers to such questions will greatly facilitate the understanding of metabolon-based metabolic regulation in cells and the design of artificial systems.
Perspective
A general consensus exists that channeling provides a means of sequestering metabolic intermediates, protecting them from degradation or undesirable processing. Sequestering may also prevent damage from the toxicity of some intermediates. Since multiple forms of channeling are possible – direct, proximity, and metabolon – it is reasonable to ask what distinct advantages might each form provide? These advantages could relate to more direct aspects of metabolism including speed, yield, and pool sizes, as well as considerations of regulation and evolvability. Here we briefly consider the distinct types of channeling from this point of view and highlight some open questions.
Direct channeling.
Intuitively, direct channels may provide the least “leaky” form of channeling. By confining intermediates in a physical tunnel, the mixing of intermediates with the rest of the cytoplasm may be reduced to negligible levels. Similarly, the close proximity of the two active sites can lead to extremely fast processing of intermediates. Hence, direct channeling may be particularly advantageous for unstable or highly reactive intermediates. Another interesting possible advantage is highlighted by the case of tryptophan synthase and several amidotransferases: the physical contact between enzymes required to form a tunnel allows allosteric enhancement of catalytic rates, preventing production of intermediates in the absence of the tunnel. One obvious disadvantage of direct channeling is the fixed stoichiometry of active sites, typically 1:1. This means that whichever process is slower will be rate limiting, with no option to balance rates by adjusting enzyme stoichiometry. A second limitation is that direct channeling does not readily generalize to longer pathways – multistep pathways would not only require multiple connected tunnels but also some means of avoiding “clogging” by a backward flux of intermediates. From an evolutionary perspective, direct channels may be “hard” to evolve – requiring extensive close-fitting protein-protein interfaces, a protected tunnel of the right radius and surface properties (including the need to exclude undesirable molecules), and dedicated routes for access to substrates and escape of products. For the same reasons, direct channels may also be hard for evolution to co-opt to different pathways or modify. For example, even in the case of direct channeling of the same intermediate molecule, ammonia, in each case, the molecular tunnel seems to have evolved independently (Raushel et al., 1999).
Proximity channeling.
As presented in the examples above, electrostatic or covalent “tethering” of intermediates enables high probability transfer of an intermediate between two active sites, even in the absence of a dedicated tunnel. Thus, proximity channeling offers many of the same advantages as direct channeling. Moreover, the lack of a tunnel may allow different stoichiometries than 1:1, particularly in the case of a flexible covalent tether, which can also facilitate multistep pathways (Fu et al., 2014; Jenni et al., 2007; Perham, 2000). That said, proximity channeling is likely to be more leaky than direct channeling, particularly in the case of electrostatic tethering where intermediates must compete with other charged metabolites/ions occupying the path between active sites (Huang et al., 2018; Liu et al., 2017). Covalent tethering overcomes these limitations but requires additional chemistry to attach metabolites to the tether and release them as final products. From an evolutionary perspective, proximity channeling also faces many of the same challenges as direct channeling.
Metabolons.
The formation of clusters of pathway enzymes offers a qualitatively distinct form of channeling and, correspondingly, a distinct set of advantages. The main distinction is that, in a metabolon, intermediates are no longer likely to be processed by the nearest downstream enzyme to their point of production, but rather may be processed by any such enzyme within the cluster. This immediately relieves the burden of fixed stoichiometry: differences in catalytic rates can be simply compensated by including different ratios of enzymes in the cluster. Moreover, clustering naturally extends to multistep pathways, as adding additional steps simply requires adding additional enzymes to the cluster. The ability to dynamically form and dissolve clusters, as observed for the purinosome, also offers a strong regulatory handle on processing, including shunting of fluxes at metabolic branch points (Castellana et al., 2014; Hinzpeter et al., 2019). Insofar as metabolons may form via LLPS, this regulation can be quite sensitive, i.e., a small change in the “stickiness” of components due to post-translational modification, oligomerization, temperature change, etc. can lead to rapid formation or dissolution of condensates, without change in overall protein levels (Kastritis and Gavin, 2018; Lyon et al., 2021; Prouteau and Loewith, 2018; Soding et al., 2020). Finally, the formation of clusters also facilitates localization of enzyme activity within the cell – the observed proximity of purinosomes to mitochondria, which are the sources of purine building blocks, can be orchestrated by relatively weak, but high-avidity interactions. The most notable disadvantage of metabolons may be the leakage of intermediates. Since these are free to diffuse within a cluster they can also diffuse out of the cluster, where they are subject to degradation or misprocessing. This diffusive loss of flux is exacerbated for long pathways, since each intermediate in the pathway has its own chance to escape the cluster. This loss of flux by diffusive escape may limit the use of metabolons to cases where enzyme density can be high, enzyme activity rapid, diffusion of intermediates slow, and/or clusters large – all of which can help prevent the diffusive escape of intermediates. From an evolutionary perspective, metabolons may be relatively easier to evolve than direct or proximity channels. Indeed, the ubiquity of condensates in cells highlights the multiplicity of possible evolutionary routes that lead to phase separation. That said, while low affinity multivalent interactions may be easier to evolve, a functional metabolon can only be achieved by enriching specific enzymes while excluding others.
A lot of central and secondary metabolic pathways are anticipated to be organized in the form of metabolons and untargeted in situ proximity detection techniques as well as computational approaches to predict molecular interactions can uncover these (Gingras et al., 2019; Kerbler et al., 2021; Li et al., 2010; Mateus et al., 2020; Xu et al., 2021). However, given the heterogeneity in the size, abundance, composition, subcellular localization, strength of the involved protein-protein interactions, the functional and structural characterization of such complexes will further require application of a slew of targeted in vitro, in vivo and in situ characterization techniques (Bassard and Halkier, 2018; Laursen et al., 2016; Pareek et al., 2020).
Open questions.
While interesting questions remain concerning the detailed operation of direct and proximity channels, including the optimal design of the latter, we focus here on some of the many questions concerning the prevalence, operation, and governing principles of metabolons. A central question regarding metabolons is what drives clustering? In many cases of LLPS, post-translational modifications such as phosphorylation/dephosphorylation, acetylation/ deacetylation regulate phase separation (Liu et al., 2019; Sang et al., 2021; Soding et al., 2020). This is a natural possibility for metabolons, but not the only possibility – conformational changes of the enzymes themselves upon substrate binding could also induce clustering. A related question is how the enzyme stoichiometry within metabolons is determined. Proper stoichiometry can optimize total flux, but are the correct ratios hard-coded into protein levels and modifications, or are relative enzyme levels tuned by feedback, e.g. based on the degree of saturation of each enzyme type? Insofar as diffusive escape of intermediates may be a limiting factor in metabolon operation, it is natural to ask whether diffusive barriers around metabolons can limit this escape. Even in the absence of lipid membranes, diffusive barriers to trap metabolites can be constructed from protein shells, as in the carboxysome (Long et al., 2018), as well as other materials such as starch granules (Toyokawa et al., 2020). However, in some cases, such diffusion barriers can be a “double-edged sword” because they may also slow access to substrates (Mangan et al., 2016). We noted above that direct and proximity channeling allow for allosteric catalytic activation, e.g. the mutual activation of the α and β subunits of tryptophan synthase. An open question is whether the activity of enzymes within metabolons can be similarly stimulated even in the absence of strong, stable interactions? Such stimulation could in principle occur even due to weak transient interactions, e.g. with intrinsically disordered domains as are commonly associated with LLPS. Finally, the formation and localization of metabolons may not be unrelated. Recent studies of LLPS in proximity to membranes and cytoskeletal elements invoke the idea of “prewetting” (Fries et al., 2020; Wiegand and Hyman, 2020), i.e. even when a bulk dense phase is unstable, a favorable surface can nucleate and stabilize finite clusters, perhaps simultaneously providing localization and size control of metabolons.
Acknowledgements:
SJB acknowledges support by NIH grant GM024129-40. VP thanks the Huck Institutes of Life Sciences, Penn State for financial support. NSW acknowledges support in part by the National Science Foundation, through the Center for the Physics of Biological Function (PHY-1734030) and NIH grant R01GM140032.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Ahmed SA, Ruvinov SB, Kayastha AM, and Miles EW (1991). Mechanism of mutual activation of the tryptophan synthase alpha and beta subunits. Analysis of the reaction specificity and substrate-induced inactivation of active site and tunnel mutants of the beta subunit. J Biol Chem 266, 21548–21557. [PubMed] [Google Scholar]
- Alber BE, and Fuchs G (2002). Propionyl-coenzyme A synthase from Chloroflexus aurantiacus, a key enzyme of the 3-hydroxypropionate cycle for autotrophic CO2 fixation. J Biol Chem 277, 12137–12143. [DOI] [PubMed] [Google Scholar]
- Amaro RE, Sethi A, Myers RS, Davisson VJ, and Luthey-Schulten ZA (2007). A Network of Conserved Interactions Regulates the Allosteric Signal in a Glutamine Amidotransferase. Biochemistry 46, 2156–2173. [DOI] [PubMed] [Google Scholar]
- Anderson KS, Kim AY, Quillen JM, Sayers E, Yang X-J, and Miles EW (1995). Kinetic Characterization of Channel Impaired Mutants of Tryptophan Synthase (*). Journal of Biological Chemistry 270, 29936–29944. [DOI] [PubMed] [Google Scholar]
- Anderson KS, Miles EW, and Johnson KA (1991). Serine modulates substrate channeling in tryptophan synthase. A novel intersubunit triggering mechanism. J Biol Chem 266, 8020–8033. [PubMed] [Google Scholar]
- Barends TR, Domratcheva T, Kulik V, Blumenstein L, Niks D, Dunn MF, and Schlichting I (2008). Structure and mechanistic implications of a tryptophan synthase quinonoid intermediate. Chembiochem 9, 1024–1028. [DOI] [PubMed] [Google Scholar]
- Bassard JE, and Halkier BA (2018). How to prove the existence of metabolons? Phytochem Rev 17, 211–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauler P, Huber G, Leyh T, and McCammon JA (2010). Channeling by Proximity: The Catalytic Advantages of Active Site Colocalization Using Brownian Dynamics. J Phys Chem Lett 1, 1332–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beld J, Lee DJ, and Burkart MD (2015). Fatty acid biosynthesis revisited: structure elucidation and metabolic engineering. Mol Biosyst 11, 38–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardsgrutter I, Vogeli B, Wagner T, Peter DM, Cortina NS, Kahnt J, Bange G, Engilberge S, Girard E, Riobe F, et al. (2018). The multicatalytic compartment of propionyl-CoA synthase sequesters a toxic metabolite. Nat Chem Biol 14, 1127–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat JY, Venkatachala R, Singh K, Gupta K, Sarma SP, and Balaram H (2011). Ammonia Channeling in Plasmodium falciparum GMP Synthetase: Investigation by NMR Spectroscopy and Biochemical Assays. Biochemistry 50, 3346–3356. [DOI] [PubMed] [Google Scholar]
- Buchanan JM (1973). The amidotransferases. Adv Enzymol Relat Areas Mol Biol 39, 91–183. [DOI] [PubMed] [Google Scholar]
- Buchner A, Tostevin F, and Gerland U (2013). Clustering and optimal arrangement of enzymes in reaction-diffusion systems. Phys Rev Lett 110, 208104. [DOI] [PubMed] [Google Scholar]
- Bugada LF, Smith MR, and Wen F (2018). Engineering Spatially Organized Multienzyme Assemblies for Complex Chemical Transformation. Acs Catal 8, 7898–7906. [Google Scholar]
- Bulutoglu B, Garcia KE, Wu F, Minteer SD, and Banta S (2016). Direct Evidence for Metabolon Formation and Substrate Channeling in Recombinant TCA Cycle Enzymes. ACS Chem Biol 11, 2847–2853. [DOI] [PubMed] [Google Scholar]
- Castellana M, Wilson MZ, Xu YF, Joshi P, Cristea IM, Rabinowitz JD, Gitai Z, and Wingreen NS (2014). Enzyme clustering accelerates processing of intermediates through metabolic channeling. Nat Biotechnol 32, 1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CY, Pedley AM, Kim D, Xia C, Zhuang X, and Benkovic SJ (2018). Microtubule-directed transport of purine metabolons drives their cytosolic transit to mitochondria. Proc Natl Acad Sci U S A 115, 13009–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CY, Zhao H, Pugh RJ, Pedley AM, French J, Jones SA, Zhuang X, Jinnah H, Huang TJ, and Benkovic SJ (2015). Purinosome formation as a function of the cell cycle. Proc Natl Acad Sci U S A 112, 1368–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta A, Merz JM, and Spivey HO (1985). Substrate channeling of oxalacetate in solid-state complexes of malate dehydrogenase and citrate synthase. J Biol Chem 260, 15008–15012. [PubMed] [Google Scholar]
- Doigneaux C, Pedley AM, Mistry IN, Papayova M, Benkovic SJ, and Tavassoli A (2020). Hypoxia drives the assembly of the multienzyme purinosome complex. J Biol Chem 295, 9551–9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey NC, and Tripathi BP (2021). Nature Inspired Multienzyme Immobilization: Strategies and Concepts. ACS Applied Bio Materials 4, 1077–1114. [DOI] [PubMed] [Google Scholar]
- Earl E, and Calabrese Barton S (2017). Simulation of intermediate transport in nanoscale scaffolds for multistep catalytic reactions. Phys Chem Chem Phys 19, 15463–15470. [DOI] [PubMed] [Google Scholar]
- Easterby JS (1989). The analysis of metabolite channelling in multienzyme complexes and multifunctional proteins. Biochem J 264, 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elcock AH, and McCammon JA (1996). Evidence for Electrostatic Channeling in a Fusion Protein of Malate Dehydrogenase and Citrate Synthase. Biochemistry 35, 12652–12658. [DOI] [PubMed] [Google Scholar]
- Ernster L, and Schatz G (1981). Mitochondria: a historical review. J Cell Biol 91, 227s–255s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eun C, Kekenes-Huskey PM, Metzger VT, and McCammon JA (2014). A model study of sequential enzyme reactions and electrostatic channeling. J Chem Phys 140, 105101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming JR, Schupfner M, Busch F, Basel A, Ehrmann A, Sterner R, and Mayans O (2018). Evolutionary Morphing of Tryptophan Synthase: Functional Mechanisms for the Enzymatic Channeling of Indole. J Mol Biol 430, 5066–5079. [DOI] [PubMed] [Google Scholar]
- Fries MR, Stopper D, Skoda MWA, Blum M, Kertzscher C, Hinderhofer A, Zhang F, Jacobs RMJ, Roth R, and Schreiber F (2020). Enhanced protein adsorption upon bulk phase separation. Sci Rep 10, 10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Yang YR, Johnson-Buck A, Liu M, Liu Y, Walter NG, Woodbury NW, and Yan H (2014). Multi-enzyme complexes on DNA scaffolds capable of substrate channelling with an artificial swinging arm. Nat Nanotechnol 9, 531–536. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Abe KT, and Raught B (2019). Getting to know the neighborhood: using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr Opin Chem Biol 48, 44–54. [DOI] [PubMed] [Google Scholar]
- Gopich IV (2021). Cluster Channeling in Cascade Reactions. J Phys Chem B 125, 2061–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi C, Dutertre M, and Simonet J (2000). Genetic organization and polymorphism of the guaA gene encoding the GMP synthetase in Lactobacillus rhamnosus. Current Microbiology 40, 245–249. [DOI] [PubMed] [Google Scholar]
- Halper LA, and Srere PA (1977). Interaction between citrate synthase and mitochondrial malate dehydrogenase in the presence of polyethylene glycol. Arch Biochem Biophys 184, 529–534. [DOI] [PubMed] [Google Scholar]
- Hawkins AR, Moore JD, and Lamb HK (1993). The molecular biology of the pentafunctional AROM protein. Biochem Soc Trans 21, 181–186. [DOI] [PubMed] [Google Scholar]
- Heinrich R, and Schuster S (1991). Is metabolic channelling the complicated solution to the easy problem of reducing transient times? J Theor Biol 152, 57–61. [DOI] [PubMed] [Google Scholar]
- Hinzpeter F, Gerland U, and Tostevin F (2017). Optimal Compartmentalization Strategies for Metabolic Microcompartments. Biophys J 112, 767–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinzpeter F, Tostevin F, and Gerland U (2019). Regulation of reaction fluxes via enzyme sequestration and co-clustering. J R Soc Interface 16, 20190444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Holden HM, and Raushel FM (2001). Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu Rev Biochem 70, 149–180. [DOI] [PubMed] [Google Scholar]
- Huang YM, Huber GA, Wang N, Minteer SD, and McCammon JA (2018). Brownian dynamic study of an enzyme metabolon in the TCA cycle: Substrate kinetics and channeling. Protein Sci 27, 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde CC, Ahmed SA, Padlan EA, Miles EW, and Davies DR (1988). Three-dimensional structure of the tryptophan synthase alpha 2 beta 2 multienzyme complex from Salmonella typhimurium. J Biol Chem 263, 17857–17871. [PubMed] [Google Scholar]
- Itzhak DN, Tyanova S, Cox J, and Borner GH (2016). Global, quantitative and dynamic mapping of protein subcellular localization. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenni S, Leibundgut M, Boehringer D, Frick C, Mikolásek B, and Ban N (2007). Structure of Fungal Fatty Acid Synthase and Implications for Iterative Substrate Shuttling. Science 316, 254–261. [DOI] [PubMed] [Google Scholar]
- Kastritis PL, and Gavin AC (2018). Enzymatic complexes across scales. Essays Biochem 62, 501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbler SM, Natale R, Fernie AR, and Zhang Y (2021). From Affinity to Proximity Techniques to Investigate Protein Complexes in Plants. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosla C, Herschlag D, Cane DE, and Walsh CT (2014). Assembly line polyketide synthases: mechanistic insights and unsolved problems. Biochemistry 53, 2875–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmak A, Carmali S, von Lieres E, Russell AJ, and Kondrat S (2019). Can enzyme proximity accelerate cascade reactions? Sci Rep 9, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed, a. LJ, and Cox DJ (1966). Macromolecular Organization of Enzyme Systems. Annual Review of Biochemistry 35, 57–84. [Google Scholar]
- Laursen T, Borch J, Knudsen C, Bavishi K, Torta F, Martens HJ, Silvestro D, Hatzakis NS, Wenk MR, Dafforn TR, et al. (2016). Characterization of a dynamic metabolon producing the defense compound dhurrin in sorghum. Science 354, 890–893. [DOI] [PubMed] [Google Scholar]
- Li X, Wu M, Kwoh CK, and Ng SK (2010). Computational approaches for detecting protein complexes from protein interaction networks: a survey. BMC Genomics 11 Suppl 1, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindbladh C, Rault M, Hagglund C, Small WC, Mosbach K, Buelow L, Evans C, and Srere PA (1994). Preparation and kinetic characterization of a fusion protein of yeast mitochondrial citrate synthase and malate dehydrogenase. Biochemistry 33, 11692–11698. [DOI] [PubMed] [Google Scholar]
- Liu C, Knudsen GM, Pedley AM, He J, Johnson JL, Yaron TM, Cantley LC, and Benkovic SJ (2019). Mapping Post-Translational Modifications of de Novo Purine Biosynthetic Enzymes: Implications for Pathway Regulation. J Proteome Res 18, 2078–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Hickey DP, Guo J-Y, Earl E, Abdellaoui S, Milton RD, Sigman MS, Minteer SD, and Calabrese Barton S (2017). Substrate Channeling in an Artificial Metabolon: A Molecular Dynamics Blueprint for an Experimental Peptide Bridge. Acs Catal 7, 2486–2493. [Google Scholar]
- Lomakin IB, Xiong Y, and Steitz TA (2007). The Crystal Structure of Yeast Fatty Acid Synthase, a Cellular Machine with Eight Active Sites Working Together. Cell 129, 319–332. [DOI] [PubMed] [Google Scholar]
- Long BM, Hee WY, Sharwood RE, Rae BD, Kaines S, Lim YL, Nguyen ND, Massey B, Bala S, von Caemmerer S, et al. (2018). Carboxysome encapsulation of the CO2-fixing enzyme Rubisco in tobacco chloroplasts. Nat Commun 9, 3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luby-Phelps K (2013). The physical chemistry of cytoplasm and its influence on cell function: an update. Mol Biol Cell 24, 2593–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch EM, Kollman JM, and Webb BA (2020). Filament formation by metabolic enzymes-A new twist on regulation. Curr Opin Cell Biol 66, 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynen F, Engeser H, Foerster E-C, Fox JL, Hess S, Kresze G-B, Schmitt T, Schreckenbach T, Siess E, Wieland F, et al. (1980). On the Structure of Fatty Acid Synthetase of Yeast. European Journal of Biochemistry 112, 431–442. [DOI] [PubMed] [Google Scholar]
- Lyon AS, Peeples WB, and Rosen MK (2021). A framework for understanding the functions of biomolecular condensates across scales. Nat Rev Mol Cell Biol 22, 215–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan NM, Flamholz A, Hood RD, Milo R, and Savage DF (2016). pH determines the energetic efficiency of the cyanobacterial CO2 concentrating mechanism. Proc Natl Acad Sci U S A 113, E5354–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruoka S, Horita S, Lee WC, Nagata K, and Tanokura M (2010). Crystal Structure of the ATPPase Subunit and Its Substrate-Dependent Association with the GATase Subunit: A Novel Regulatory Mechanism for a Two-Subunit-Type GMP Synthetase from Pyrococcus horikoshii OT3. Journal of Molecular Biology 395, 417–429. [DOI] [PubMed] [Google Scholar]
- Mateus A, Kurzawa N, Becher I, Sridharan S, Helm D, Stein F, Typas A, and Savitski MM (2020). Thermal proteome profiling for interrogating protein interactions. Mol Syst Biol 16, e9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConkey EH (1982). Molecular evolution, intracellular organization, and the quinary structure of proteins. Proc Natl Acad Sci U S A 79, 3236–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes P, Kell DB, and Westerhoff HV (1996). Why and when channelling can decrease pool size at constant net flux in a simple dynamic channel. Biochim Biophys Acta 1289, 175–186. [DOI] [PubMed] [Google Scholar]
- Miles EW (2001). Tryptophan synthase: A multienzyme complex with an intramolecular tunnel. The Chemical Record 1, 140–151. [DOI] [PubMed] [Google Scholar]
- Miles EW, Phillips RS, Yeh HJ, and Cohen LA (1986). Isomerization of (3S)-2,3-dihydro-5-fluoro-L-tryptophan and of 5-fluoro-L-tryptophan catalyzed by tryptophan synthase: studies using fluorine-19 nuclear magnetic resonance and difference spectroscopy. Biochemistry 25, 4240–4249. [DOI] [PubMed] [Google Scholar]
- Mueller EJ, Meyer E, Rudolph J, Davisson VJ, and Stubbe J (1994). N5-carboxyaminoimidazole ribonucleotide: evidence for a new intermediate and two new enzymatic activities in the de novo purine biosynthetic pathway of Escherichia coli. Biochemistry 33, 2269–2278. [DOI] [PubMed] [Google Scholar]
- Myers RS, Amaro RE, Luthey-Schulten ZA, and Davisson VJ (2005). Reaction coupling through interdomain contacts in imidazole glycerol phosphate synthase. Biochemistry 44, 11974–11985. [DOI] [PubMed] [Google Scholar]
- Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Paabo S, and Mann M (2011). Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor E, Bar-Even A, Flamholz A, Reznik E, Liebermeister W, and Milo R (2014). Pathway Thermodynamics Highlights Kinetic Obstacles in Central Metabolism. Plos Comput Biol 10, el003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver JC, Linger RS, Chittur SV, and Davisson VJ (2013). Substrate activation and conformational dynamics of guanosine 5’-monophosphate synthetase. Biochemistry 52, 5225–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareek V, Pedley AM, and Benkovic SJ (2021). Human de novo purine biosynthesis. Crit Rev Biochem Mol Biol 56, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareek V, Tian H, Winograd N, and Benkovic SJ (2020). Metabolomics and mass spectrometry imaging reveal channeled de novo purine synthesis in cells. Science 368, 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MS, and Roche TE (1990). Molecular biology and biochemistry of pyruvate dehydrogenase complexes. Faseb j 4, 3224–3233. [DOI] [PubMed] [Google Scholar]
- Patel N, Moyed HS, and Kane JF (1977). Properties of xanthosine 5’-monophosphate-amidotransferase from Escherichia coli. Archives of Biochemistry and Biophysics 178, 652–661. [DOI] [PubMed] [Google Scholar]
- Pedley AM, and Benkovic SJ (2017). A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem Sci 42, 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perham RN (2000). Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annu Rev Biochem 69, 961–1004. [DOI] [PubMed] [Google Scholar]
- Poshyvailo L, von Lieres E, and Kondrat S (2017). Does metabolite channeling accelerate enzyme-catalyzed cascade reactions? PLoS One 12, e0172673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prouteau M, and Loewith R (2018). Regulation of Cellular Metabolism through Phase Separation of Enzymes. Biomolecules 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raboni S, Mozzarelli A, and Cook PF (2007). Control of ionizable residues in the catalytic mechanism of tryptophan synthase from Salmonella typhimurium. Biochemistry 46, 13223–13234. [DOI] [PubMed] [Google Scholar]
- Raushel FM, Thoden JB, and Holden HM (1999). The amidotransferase family of enzymes: molecular machines for the production and delivery of ammonia. Biochemistry 38, 7891–7899. [DOI] [PubMed] [Google Scholar]
- Raushel FM, Thoden JB, and Holden HM (2003). Enzymes with molecular tunnels. Acc Chem Res 36, 539–548. [DOI] [PubMed] [Google Scholar]
- Reed LJ (1974). Multienzyme complexes. Accounts of Chemical Research 7, 40–46. [Google Scholar]
- Rhee S, Parris KD, Ahmed SA, Miles EW, and Davies DR (1996). Exchange of K+ or Cs+ for Na+ induces local and long-range changes in the three-dimensional structure of the tryptophan synthase alpha2beta2 complex. Biochemistry 35, 4211–4221. [DOI] [PubMed] [Google Scholar]
- Rhee S, Parris KD, Hyde CC, Ahmed SA, Miles EW, and Davies DR (1997). Crystal structures of a mutant (betaK87T) tryptophan synthase alpha2beta2 complex with ligands bound to the active sites of the alpha- and beta-subunits reveal ligand-induced conformational changes. Biochemistry 36, 7664–7680. [DOI] [PubMed] [Google Scholar]
- Robbins T, Liu YC, Cane DE, and Khosla C (2016). Structure and mechanism of assembly line polyketide synthases. Curr Opin Struct Biol 41, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saks V, Beraud N, and Wallimann T (2008). Metabolic compartmentation - a system level property of muscle cells: real problems of diffusion in living cells. Int J Mol Sci 9, 751–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang L, Ju HQ, Yang Z, Ge Q, Zhang Z, Liu F, Yang L, Gong H, Shi C, Qu L v et al. (2021). Mitochondrial long non-coding RNA GAS5 tunes TCA metabolism in response to nutrient stress. Nat Metab 3, 90–106. [DOI] [PubMed] [Google Scholar]
- Schmitt DL, and An S (2017). Spatial Organization of Metabolic Enzyme Complexes in Cells. Biochemistry 56, 3184–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt DL, Cheng YJ, Park J, and An S (2016). Sequestration-Mediated Downregulation of de Novo Purine Biosynthesis by AMPK. ACS Chem Biol 11, 1917–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GK, Mueller WT, Wasserman GF, Taylor WD, and Benkovic SJ (1980). Characterization of the enzyme complex involving the folate-requiring enzymes of de novo purine biosynthesis. Biochemistry 19, 4313–4321. [DOI] [PubMed] [Google Scholar]
- Smith S, and Tsai SC (2007). The type I fatty acid and polyketide synthases: a tale of two megasynthases. Nat Prod Rep 24, 1041–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soding J, Zwicker D, Sohrabi-Jahromi S, Boehning M, and Kirschbaum J (2020). Mechanisms for Active Regulation of Biomolecular Condensates. Trends Cell Biol 30, 4–14. [DOI] [PubMed] [Google Scholar]
- Srere PA (1985). The metabolon. Trends in Biochemical Sciences 10, 109–110. [Google Scholar]
- Srere PA (1987). Complexes of sequential metabolic enzymes. Annu Rev Biochem 56, 89–124. [DOI] [PubMed] [Google Scholar]
- Srere PA, Mattiasson B, and Mosbach K (1973). An Immobilized Three-Enzyme System: A Model for Microenvironmental Compartmentation in Mitochondria. Proceedings of the National Academy of Sciences 70, 2534–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, and Brangwynne CP (2019). The liquid nucleome - phase transitions in the nucleus at a glance. J Cell Sci 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweetlove LJ, and Fernie AR (2018). The role of dynamic enzyme assemblies and substrate channelling in metabolic regulation. Nat Commun 9, 2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesmer JJG, Klem TJ, Deras ML, Davisson VJ, and Smith JL (1996). The crystal structure of GMP synthetase reveals a novel catalytic triad and is a structural paradigm for two enzyme families. Nature Structural Biology 3, 74–86. [DOI] [PubMed] [Google Scholar]
- Toyokawa C, Yamano T, and Fukuzawa H (2020). Pyrenoid Starch Sheath Is Required for LCIB Localization and the CO2-Concentrating Mechanism in Green Algae. Plant Physiol 182, 1883–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsitkov S, Pesenti T, Palacci H, Blanchet J, and Hess H (2018). Queueing Theory-Based Perspective of the Kinetics of “Channeled” Enzyme Cascade Reactions. Acs Catal 8, 10721–10731. [Google Scholar]
- Veraszto HA, Logotheti M, Albrecht R, Leitner A, Zhu HB, and Hartmann MD (2020). Architecture and functional dynamics of the pentafunctional AROM complex. Nat Chem Biol 16, 973–978. [DOI] [PubMed] [Google Scholar]
- Vetter IR, and Wittinghofer A (1999). Nucleoside triphosphate-binding proteins: different scaffolds to achieve phosphoryl transfer. Quarterly Reviews of Biophysics 32, 1–56. [DOI] [PubMed] [Google Scholar]
- von der Saal W, Crysler CS, and Villafranca JJ (1985). Positional isotope exchange and kinetic experiments with Escherichia coli guanosine-5’-monophosphate synthetase. Biochemistry 24, 5343–5350. [DOI] [PubMed] [Google Scholar]
- Welch GR, and Easterby JS (1994). Metabolic channeling versus free diffusion: transition-time analysis. Trends Biochem Sci 19, 193–197. [DOI] [PubMed] [Google Scholar]
- Welin M, LehtiÖ L, Johansson A, Flodin S, Nyman T, Trésaugues L, Hammarström M, Gräslund S, and Nordlund P (2013). Substrate Specificity and Oligomerization of Human GMP Synthetase. Journal of Molecular Biology 425, 4323–4333. [DOI] [PubMed] [Google Scholar]
- Weyand M, Schlichting I, Herde P, Marabotti A, and Mozzarelli A (2002). Crystal structure of the beta Ser178--> Pro mutant of tryptophan synthase. A “knock-out” allosteric enzyme. J Biol Chem 277, 10653–10660. [DOI] [PubMed] [Google Scholar]
- Wheeldon I, Minteer SD, Banta S, Barton SC, Atanassov P, and Sigman M (2016). Substrate channelling as an approach to cascade reactions. Nat Chem 8, 299–309. [DOI] [PubMed] [Google Scholar]
- Wiegand G, and Remington SJ (1986). Citrate synthase: structure, control, and mechanism. Annu Rev Biophys Biophys Chem 15, 97–117. [DOI] [PubMed] [Google Scholar]
- Wiegand T, and Hyman AA (2020). Drops and fibers - how biomolecular condensates and cytoskeletal filaments influence each other. Emerg Top Life Sci 4, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JE (1978). Ambiquitous enzymes: Variation in intracellular distribution as a regulatory mechanism. Trends in Biochemical Sciences 3, 124–125. [Google Scholar]
- Wu F, Pelster LN, and Minteer SD (2015). Krebs cycle metabolon formation: metabolite concentration gradient enhanced compartmentation of sequential enzymes. Chemical Communications 51, 1244–1247. [DOI] [PubMed] [Google Scholar]
- Xu Y, Fan X, and Hu Y (2021). In vivo interactome profiling by enzyme-catalyzed proximity labeling. Cell Biosci 11, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanofsky C, and Crawford IP (1972). Tryptophan Synthetase. In The Enzymes, Boyer PD, ed. (Academic Press; ), pp. 1–31. [Google Scholar]
- Zalkin H, and Smith JL (1998). Enzymes utilizing glutamine as an amide donor. Adv Enzymol Relat Areas Mol Biol 72, 87–144. [DOI] [PubMed] [Google Scholar]
- Zhang JZ, Mehta S, and Zhang J (2021). Liquid-liquid phase separation: a principal organizer of the cell’s biochemical activity architecture. Trends Pharmacol Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, and Fernie AR (2021). Metabolons, enzyme-enzyme assemblies that mediate substrate channeling, and their roles in plant metabolism. Plant Commun 2, 100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao A, Tsechansky M, Ellington AD, and Marcotte EM (2014). Revisiting and revising the purinosome. Mol Biosyst 10, 369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zyk N, Citri N, and Moyed HS (1969). Conformative response of xanthosine 5’-phosphate aminase. Biochemistry 8, 2787–2794. [DOI] [PubMed] [Google Scholar]