

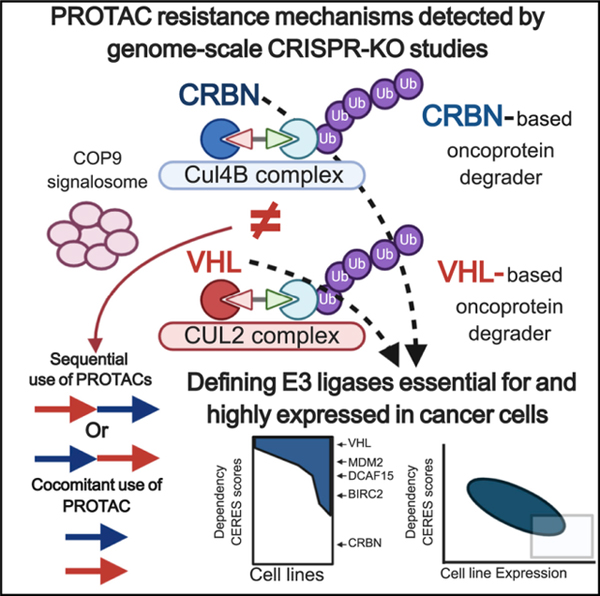
SUMMARY
Heterobifunctional proteolysis-targeting chimeric compounds leverage the activity of E3 ligases to induce degradation of target oncoproteins and exhibit potent preclinical antitumor activity. To dissect the mechanisms regulating tumor cell sensitivity to different classes of pharmacological “degraders” of oncoproteins, we performed genome-scale CRISPR-Cas9-based gene editing studies. We observed that myeloma cell resistance to degraders of different targets (BET bromodomain proteins, CDK9) and operating through CRBN (degronimids) or VHL is primarily mediated by prevention of, rather than adaptation to, breakdown of the target oncoprotein; and this involves loss of function of the cognate E3 ligase or interactors/regulators of the respective cullin-RING ligase (CRL) complex. The substantial gene-level differences for resistance mechanisms to CRBN- versus VHL-based degraders explains mechanistically the lack of cross-resistance with sequential administration of these two degrader classes. Development of degraders leveraging more diverse E3 ligases/CRLs may facilitate sequential/alternating versus combined uses of these agents toward potentially delaying or preventing resistance.
Graphical Abstract
In Brief
Genome-scale CRISPR gene editing studies by Shirasaki et al. reveal which genes confer myeloma cell resistance to PROTACs that leverage different E3 ligases to degrade various oncoproteins. The study provides a framework for sequential/alternating versus combined use of PROTACs, depending on which E3 ligase and oncoprotein they engage.
INTRODUCTION
Thalidomide and its immunomodulatory derivatives (IMIDs) recruit the E3 ubiquitin ligase Cereblon (CRBN) to ubiquitinate neo-morphic protein substrates (Krönke et al., 2014; Lu et al., 2014). This observation led to the development of “degronimids,” heterobifunctional compounds in which a thalidomide-like moiety is paired with one of many different small molecules to cause proteolysis of proteins binding to these latter moieties (Winter et al., 2015). The development of degronimids has renewed interest in the broader concept of heterobifunctional proteolysis-targeting chimeras (PROTACs). Targeting oncogenic proteins for intracellular degradation overcomes several potential limitations related to compounds that merely inhibit their function, including incomplete and transient target engagement by non-covalent inhibitors, the compensatory increase in levels of the target protein, or potential oncogenic functions by other uninhibited domains of the protein. Moreover, degronimids and other PROTACs exhibit sub-stoichiometric catalytic activity (Bondeson et al., 2015). Consequently, “degraders” can include molecules with limited inhibitory potency or even agonistic activity as long as they bind to their target selectively.
Given these advantages, degronimids and other PROTACs are being studied extensively in a broad spectrum of malignancies (Lu et al., 2015; Raina et al., 2016; Saenz et al., 2017; Winter et al., 2017). In this study, we performed genome-scale loss-of-function (LOF) screens with the clustered regularly interspaced short palindromic repeats (CRISPR) technology to identify mechanisms of resistance of multiple myeloma (MM) cells to degronimids targeting the bromodomain and extraterminal (BET) bromodomain proteins BRD4, BRD3, and BRD2 (dBET6) (Winter et al., 2017) or CDK9 (Thal-SNS-032) (Olson et al., 2018) and degraders of BRD4, BRD3, and BRD2, which operate through a different E3 ligase (von Hippel-Lindau disease tumor suppressor [VHL]), namely ARV-771 (Raina et al., 2016; Saenz et al., 2017; Sun et al., 2018) and MZ-1 (Gadd et al., 2017; Zengerle et al., 2015). This study sought to determine, in an unbiased manner, which mechanisms of resistance are common versus distinct for CRBN-versus VHL-mediated pharmacological degraders.
RESULTS
Preclinical Anti-myeloma Activity and Molecular Sequelae of Pharmacological BET Bromodomain Degradation
BRD4 regulates the expression and function of the c-Myc oncoprotein (Delmore et al., 2011), and the BET bromodomain inhibitor (BBI) JQ1 has preclinical in vitro and in vivo activity against MM(Delmore et al., 2011) and other neoplasms (e.g., Lovén et al., 2013; Zuber et al., 2011). However, BBIs cause compensatory upregulation of BRD4 protein levels (Lu et al., 2015; Winter et al., 2015), which could, in principle, attenuate the anti-tumor activity of BBIs and explain why JQ1 does not induce apoptosis in most cell types tested (Delmore et al., 2011; Zuber et al., 2011). Consistent with prior reports on the original BET bromodomain-targeting degronimid dBET1 (Winter et al., 2015, 2017) and its lead-optimized successor, dBET6 (Winter et al., 2017), we observed more potent in vitro anti-MM activity of dBET6 compared with dBET1 or JQ1 (Figures S1A and S1B) and thus focused on dBET6 (Figures S1C–S1K) as a representative degronimid against BET bromodomain proteins for the rest of our study.
We confirmed that dBET6 causes rapid depletion of BRD2, BRD3, BRD4, and c-Myc, whereas JQ1 causes less pronounced c-Myc depletion and a compensatory increase in BRD4 protein levels (Figures S1C–S1F). This pattern of effects of dBET6 is distinct from those induced by CRBN-based degraders of other targets; e.g., CDK9 degradation by Thal-SNS-032 (Figures S1G and S1H). In contrast to prior observations with JQ1 (Delmore et al., 2011), dBET6 induces a dose- and time-dependent proapoptotic response of MM.1S cells (Figure S1I,J). Exposure to dBET6 for as little as 4 h prior to drug washout is sufficient to commit MM.1S cells to death (Figures S1I and S1J). MM cells were responsive to dBET6 when co-cultured with bone marrow stromal cells (Figure S1K), which are known to induce resistance to diverse established or investigational agents in MM and other bone-homing neoplasms (McMillin et al., 2010, 2012a). Comparison of transcriptional (RNA sequencing [RNA-seq]) and proteomics (reverse-phase protein arrays [RPPAs]) profiles of dBET6 versus JQ1-treated MM.1S cells identified that dBET6 induced downregulation of a larger group of transcripts (Figure S2A) and more pronounced suppression of c-MYC transcript and protein levels (Figures S1F, S2B, and S2C). Importantly, dBET6 treatment at 4 h, but not JQ1, causes downregulation of Mcl-1 (Figure S2C), Cyclin B1 (data not shown), and BRD4 (Figures S1E and S2C), followed at later time points by suppression of Ser240/Ser244 S6 phosphorylation, induction of p21, and an increase in cleaved caspase-3 (Figure S2C). These differential effects on anti-apoptotic (e.g., Mcl-1) and pro-apoptotic (e.g., caspase cleavage) effectors could explain the induction of cell death/apoptosis by dBET6 but not JQ1. We observed a significantly lower tumor burden in dBET6- versus vehicle-treated mice in models of subcutaneous (s.c.) and disseminated, respectively, growth of human MM.1S cells in immunocompromised mice (Figures S2D and S2F). Prolongation of survival was observed in dBET6-treated mice with s.c. MM xenografts (Figure S2E) and only was minimally observed in the model of disseminated MM lesions (Figure S2G). Mice tolerated dBET6 treatment at the doses and schedules reported, but in other pilot experiments (data not shown), alternative regimens with lower or higher dose intensity had less efficacy or tolerability, respectively, suggesting that development of different preparations of dBET compounds with optimized delivery to the tumor sites is warranted.
Anti-tumor Activity of dBET6 in the Context of Decreased Responsiveness to Other Therapeutic Agents
We examined the activity of dBET6 against groups of MM cells with decreased responsiveness to other established or investigational therapeutic agents: (1) pools of MM.1S cells that exhibited decreased responsiveness to bortezomib or JQ1 (Figures S3A–S3C) after being transduced with a whole-genome library of single guide RNAs (sgRNAs) for CRISPR-based gene editing and then having received bortezomib or JQ1 treatment (Figures S3A and S3B) and (2) samples of patient-derived MM cells with variable patterns of exposure and resistance/refractoriness to currently available clinical treatments (Figure S3D). For both sets of studies, we observed that prior resistance to other therapies does not preclude responsiveness to dBET6 (Figures S3C and S3D). For instance, individual patient-derived samples treated with dBET6 demonstrated a broad range of sensitivities (Figure S3D) without any obvious clustering of the dose-response patterns depending on the disease status, e.g., relapsed/refractory, maintenance therapy, newly diagnosed, or monoclonal gammopathy of undetermined significance (MGUS).
CRISPR-Based Functional Genomic Characterization of Mechanisms of MM Cell Resistance to CRBN-Mediated Degraders
We performed genome-scale CRISPR-based gene editing screens (Figure S4) to identify candidate genes associated with resistance to dBET6. MM.1S-Cas9+ cells were transduced with pooled lentiviral particles of the GeCKOv2 sgRNA library and were treated with dBET6 (0.25 µM) versus control. After three rounds of dBET6 treatment, we observed outgrowth of dBET6-resistant cells (Figure 1A). PCR amplification and next-generation sequencing (NGS) quantified sgRNA barcodes in dBET6-resistant versus treatment-naive cells. We observed significant enrichment of sgRNAs targeting CRBN itself as well as other components or regulators of its cullin-RING ligase (CRL) complex, including members of the human COP9 signalosome (COPS7A, COPS7B, COPS2, COPS3, COPS8, GPS1, etc.), DDB1, or the E2 ubiquitin-conjugating enzyme UBE2G1 (Figure 1B). Transduction with several individual sgRNAs for COPS7B, COPS8, or CRBN (Figures 1C–1F) decreases the response of MM.1S cells to dBET6.
Figure 1. Resistance to dBET6 by Dysregulation of CRBN and Its Interactors/Regulators.
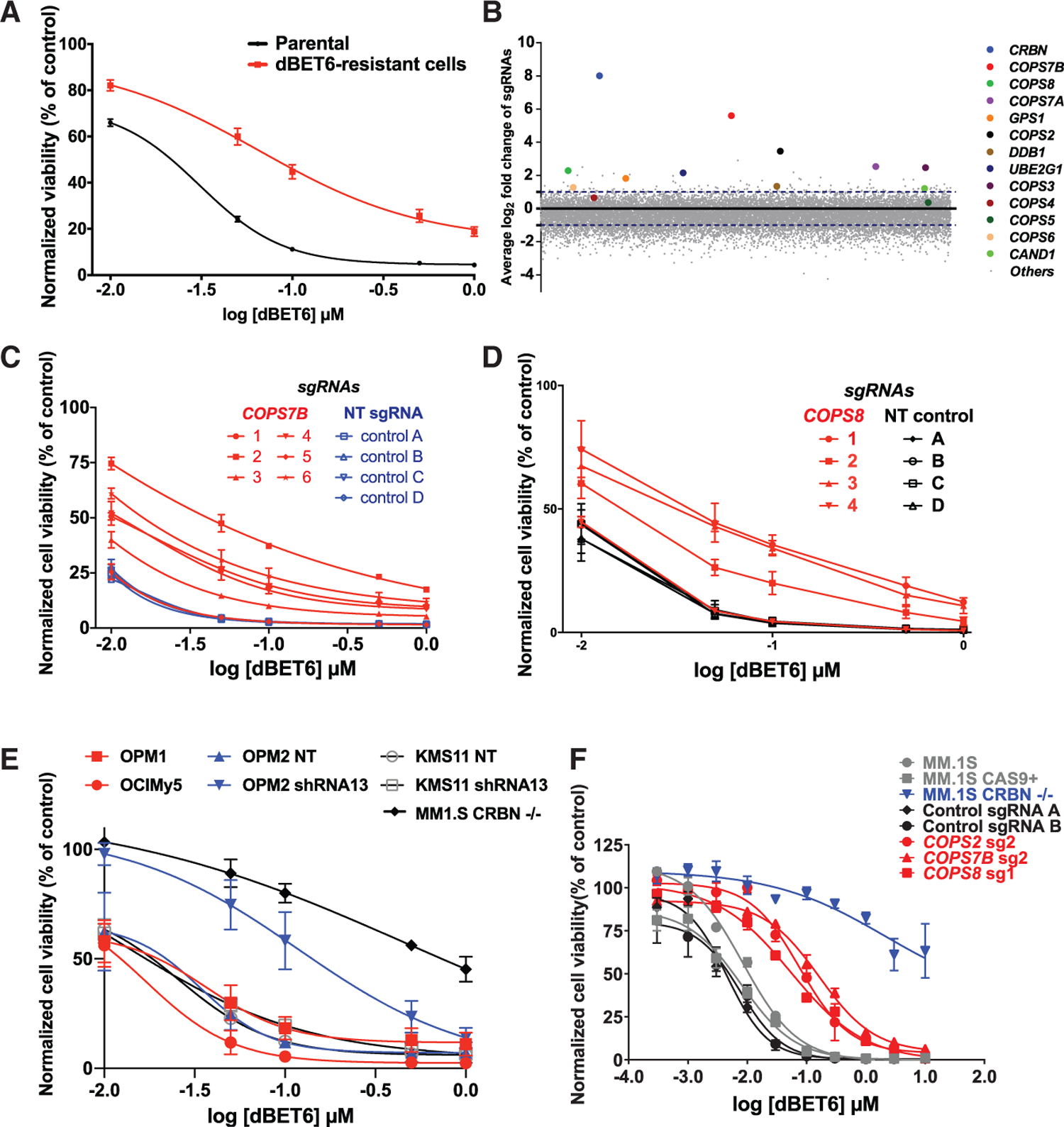
(A) dBET6 dose-response curves (CellTiter-Glo [CTG] assays, 48 h) for pools of MM.1S cells that survived repeated dBET6 treatments during the long-term genome-scale CRISPR gene editing screen versus dBET6-naive cells (2-way ANOVA; p < 0.05 in terms of drug dose and p < 0.05 in terms of status of prior treatment with dBET6 or no treatment).
(B) Average log2 fold change (log2FC) of normalized read counts for sgRNAs against CRBN and several of its interactors after repeated dBET6 treatments during the long-term genome-scale CRISPR gene editing screen.
(C and D) MM.1S-Cas9+ cells transduced with individual sgRNAs for COPS7B and COPS8 (versus control sgRNAs) were tested for in vitro response to dBET6 (48 h; 2-way ANOVA; p < 0.05 in terms of drug dose and p < 0.05 in terms of status of transduction with five of the sgRNAs for COPS7B or three sgRNAs for COPS8 versus non-targeting (NT) control sgRNAs).
(E) MM cell lines known to express low but detectable CRBN levels constitutively (OPM1 and OCI-My5) or after transduction with shRNAs against CRBN (KMS11, OPM2) were tested for their in vitro response to dBET6 (48 h).
(F) dBET6 dose-response curves (72 h) for MM.1S-Cas9+ cells transduced with sgRNAs against CRBN (MM.1S CRBN−/−), COPS7B, COPS2, COPS8, or non-targeting control sgRNAs.
(A) and (C)–(E) depict averages ± SE of 3 independent experiments (CTG) for each panel, with triplicates per condition. (F) depicts averages ± SE from a single experiment with triplicates per condition.
We performed a similar CRISPR-based genome-scale screen (Figures S4 and 2A) for resistance to the CRBN-based CDK9 degrader Thal-SNS-032 (Figures S1H and S1I). We again observed that Thal-SNS-032-resistant cells were enriched for sgRNAs against CRBN itself, COP9 signalosome complex genes (COPS7A, COPS7B, COPS2, COPS3, COPS8, GPS1, etc.), DDB1, or UBE2G1 (Figure 2B). These results were concordant with our dBET6 studies and a distinct CRISPR gene editing study (Sievers et al., 2018) on mechanisms of lenalidomide resistance of MM.1S cells. We again validated that several individual sgRNAs for these candidate genes (e.g., COPS7B, COPS2, DDB1, or COPS8; Figures 2C–2F, respectively) can decrease the Thal-SNS-032 responsiveness of MM.1S cells.
Figure 2. Resistance to a Degronimid against CDK9 (Thal-SNS-032) by Dysregulation of CRBN and Its Interactors/Regulators.
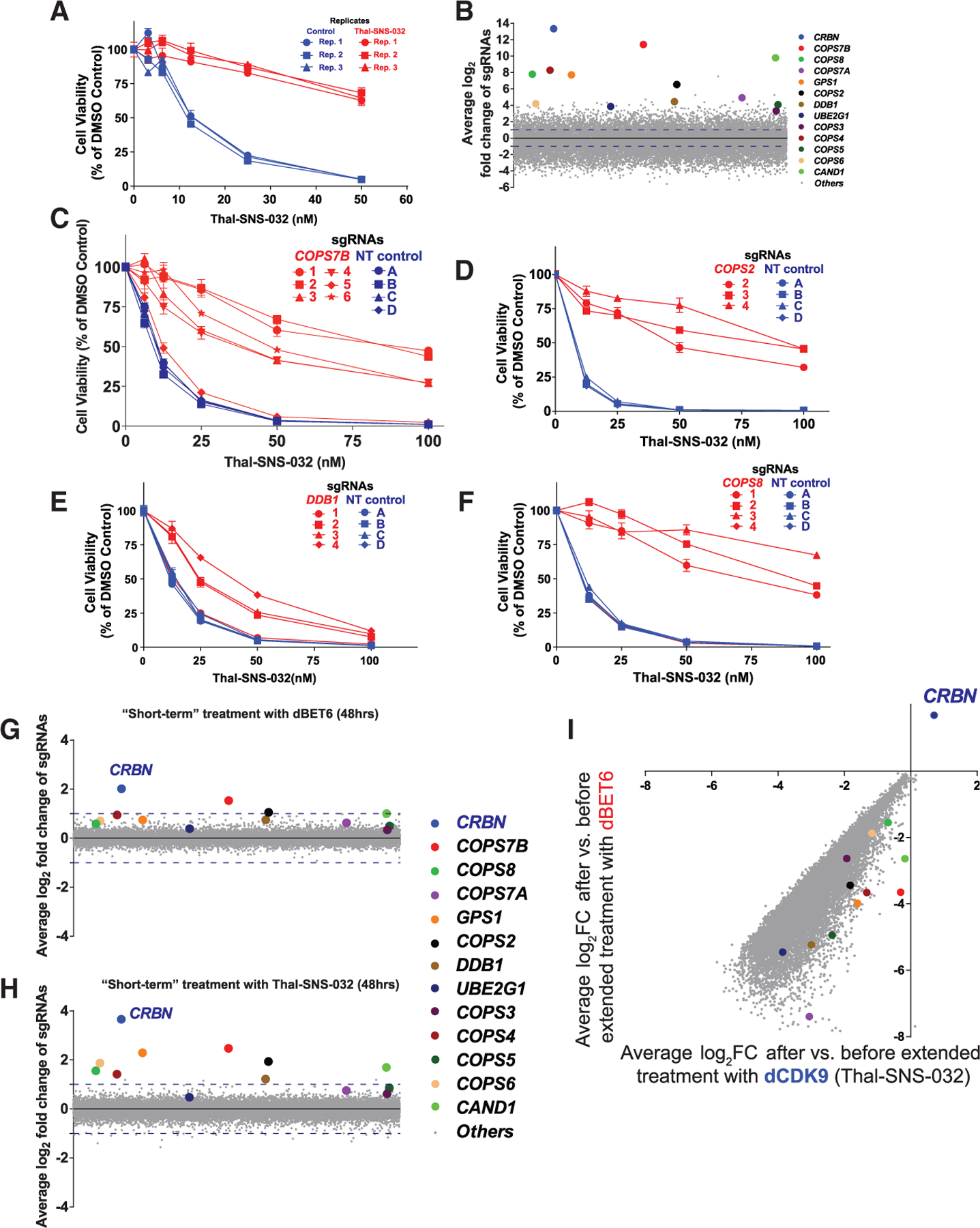
(A) Thal-SNS-032 dose-response curves (CTG assays, 72 h) for pools of MM1S cells that survived long-term Thal-SNS-032 treatments (versus Thal-SNS-032-naive cells) in the genome-scale CRISPR gene editing screen (3 replicate [Rep] pools for each respective population of cells; triplicates per pool, average ± SE).
(B) Average log2 fold change of normalized read counts for sgRNAs against CRBN and several of its interactors after long-term Thal-SNS-032 treatments in the genome-scale CRISPR gene editing screen.
(C–F) Thal-SNS-032 dose-response curves (72 h) for MM.1S-Cas9+ cells transduced with sgRNAs targeting individual genes (e.g., COPS7B, COPS2, DDB1, and COPS8; C–F, respectively) versus those transduced with control sgRNAs (CTG assays, averages ± SE of triplicates per experimental condition).
(G and H) Genome-scale CRISPR gene editing screen for genetic determinants of short-term MM.1S cell response to a single treatment with degronimids, (G) dBET6 (25 nM) or (H) Thal-SNS-032 (25 nM), for 48 h. Average log2 fold changes of normalized read counts for sgRNAs against CRBN and several of its interactors are highlighted.
(I) Pools of MM.1S-Cas9+ cells that had survived long-term treatment with Thal-SNS-032 in the context of the CRISPR gene editing screen in (B) received extended in vitro treatment with dBET6 (i.e., switch from one degronimid to another) or Thal-SNS-032 for an additional 2 weeks (Figure S4). Average log2 fold change of normalized read counts for sgRNAs after versus before extended treatment with each degronimid is depicted. Among genes associated with degronimid resistance after long-term treatment CRISPR screens, CRBN is the only one with significant sgRNA enrichment after extended degronimid treatment.
Decreased CRBN transcript levels or alternative splicing (Gandhi et al., 2014; Heintel et al., 2013), but typically not biallelic LOF (biallelic deletion or LOF mutations), are detected in tumor cells of individuals with MM with clinical resistance to thalidomide and its derivatives (Heintel et al., 2013; Zhu et al., 2011); i.e., the prototypical degronimids. We studied MM cell lines reported previously (Zhu et al., 2011) to have a decreased response to lenalidomide because of low but detectable levels of CRBN constitutively (OPM1 and OCI-MY5) or after transduction with short hairpin RNAs (shRNAs) against CRBN (KMS11 and OPM2). We observed that these lines (Figure 1E) remained, at least to some extent, responsive to dBET6. Therefore, low CRBN levels, which may not be sufficient for the usual therapeutic effects of IMIDs via degradation of IZKF1/3, may still allow CRBN-mediated degraders to maintain, at least in some cases, substantial levels of activity.
Kinetic Analyses of CRISPR Screens for Resistance to Degronimids Reveal Dynamics of LOF for CRBN versus Non-CRBN Resistance Genes
To examine the dynamics over time for MM cell populations with LOF for candidate degronimid resistance genes, we performed two additional types of genome-scale CRISPR studies (Figure S4): (1) “short-term” screens in which MM.1S-Cas9+ cells transduced with the genome-scale sgRNA library Brunello were exposed to short-term (48 h) treatments with dBET6 or Thal-SNS-032 (Figures 2G and 2H) and (2) “extended treatment” screens in which Thal-SNS-032-resistant MM.1S cell populations isolated from the initial “long-term” CRISPR/Cas9-based gene editing screen received for 2 weeks treatment with either Thal-SNS-032 (i.e., continuation of the treatment that led to isolation of these Thal-SNS-032-resistant cells) or dBET6 (Figure 2I). In both short-term degronimid screens, CRBN is the top most sgRNA-enriched gene, but with quantitatively less pronounced enrichment compared with screens involving long-term degronimid treatment (Figure 1B and Figure 2B). Thal-SNS-032-resistant MM.1S cells established from our long-term CRISPR screen and re-challenged with Thal-SNS-032 or dBET6 for an additional 2 weeks of in vitro culture led to further enrichment of MM cells with sgRNAs against CRBN (Figure 2I) but not against other “hits” identified from our earlier long-term screens with either degronimid.
CRISPR Studies to Identify Resistance Mechanisms to VHL-Based Degraders Targeting BRD2/3/4
Because in vitro resistance to CRBN-based degraders primarily involved LOF of CRBN and its regulators, we hypothesized that resistance to degraders that engage a different E3 ligase and CRL complex would involve a different set of candidate genes reflecting the composition of the latter complex. We thus performed additional CRISPR-based genome-scale gene editing studies for resistance against PROTACs causing BET protein degradation via the E3 ubiquitin ligase activity of VHL (ARV-771 [Raina et al., 2016; Saenz et al., 2017; Sun et al., 2018] and MZ-1 [Gadd et al., 2017; Zengerle et al., 2015]). In these ARV-771- or MZ-1-resistant cells, we observed (Figure 3A) enrichment of sgRNAs for CUL2, VHL itself, other members (e.g., RBX1, elongin B and C [TCEB2, TCEB1]; Kibel et al., 1995; Lonergan et al., 1998) of the CUL2 complex with VHL, as well as COP9 signalosome genes (COPS7B, COPS8) or UBE2R2. MM.1S cells with sgRNAs against COPS7B or COPS8 exhibited a decreased response to MZ-1 or ARV-771 (compared with parental MM.1S cells; Figures 3B and 3C), although this decrease was less pronounced than the one observed with CRBN-based degraders (Figures 1C, 1D, 2C, and 2F). Validation studies for additional genes documented decreased activity of VHL- but not CRBN-based degraders against cells with sgRNAs against VHL (MM.1S or KMS-11 cells; Figures S5A and S5B, respectively), TCEB1, TCEB2, CUL2, FBXW2 (MM.1S cells; Figure S5C), and UBE2R2 (MM.1S cells; Figures S5C and S5D). In contrast, no significant changes in ARV-771 activity was observed in cells with sgRNAs against CRBN (Figure S5A) or several olfactory receptor genes (Figures S5A–S5C) that are not expressed in MM cells and serve as additional negative controls.
Figure 3. Resistance to VHL-Based PROTACs by Dysregulation of the CUL2-VHL RING Ligase Complex and Its Interactors/Regulators.
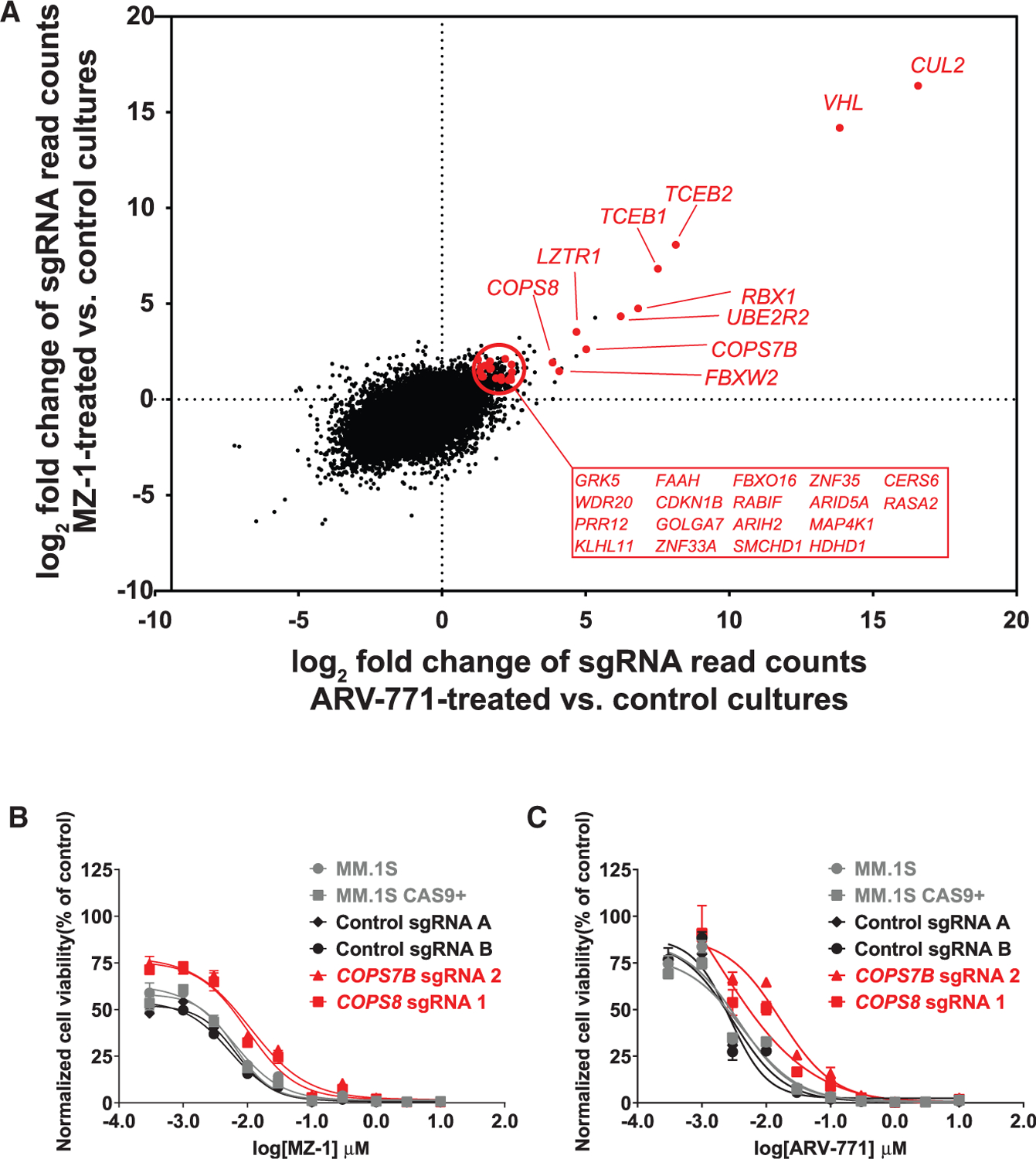
Genome-scale CRISPR gene editing screens were performed in MM.1S-Cas9+ cells receiving long-term treatment with VHL-based PROTACs of BRD2/3/4 (ARV-771 or MZ-1) until outgrowth of resistant cells could be confirmed.
(A) Average log2 fold change (3 experimental Reps per condition) of normalized read counts of sgRNAs for each gene in ARV-771- or MZ-1-treated MM.1S cells (versus common vehicle control MM.1S cells). Genes highlighted in red exhibited sgRNA enrichment (3–4 of 4 sgRNAs per gene, p < 0.05 [rank aggregation algorithm], log2FC > 1.0) in both screens and are expressed at Reads Per Kilobase of transcript per Million reads mapped (RPKM) > 1.0 based on RNA-seq data from CCLE on MM.1S cells. (B and C) MM.1S-Cas9+ cells transduced with sgRNAs against COPS7B or COPS8 (or non-targeting control sgRNAs) were tested for their in vitro response to MZ-1 (B) and ARV-771 (C) (72 h). Shown are averages ± SE from a single experiment with triplicates for each condition.
Sequential versus Concomitant Exposure to Degraders Operating through Different E3 Ligases
In our functional genomics studies, different individual genes, which reflect common pathways, emerge as genomic determinants of the responses to PROTACs targeting the same oncoprotein through different E3 ligases. We thus reasoned that sequential administration of PROTACs operating through different E3 ligases, even when targeting the same oncoprotein, could delay or prevent development of resistance compared with sequential administration of PROTACs targeting different oncoproteins via the same E3 ligase. To address this question, we examined pools of MM cells that had survived CRISPR-based studies after (1) long-term treatment with Thal-SNS-032 or long-term treatment with Thal-SNS-032 followed by (2) extended treatment with Thal-SNS-032 or (3) extended treatment with dBET6 versus (4) populations of drug-naive cells that remained in culture during the long-term or extended treatments with these CRBN-based degraders and were collected at the end of the respective studies. Each of these cell populations (3 replicate pools for each population) were exposed to dBET6, Thal-SNS-032, ARV-771, or MZ-1. Pools of MM cells that had survived previous treatment with the CRBN-based degraders were, compared with treatment-naive cells, significantly less responsive to repeat treatment with either CRBN-based degrader but highly responsive to both VHL-based degraders (Figure 4A). Consistent with these observations, pools of MM cells from our genome-scale CRISPR screens that had survived long-term exposure to ARV-771 and then extended treatment with MZ-1 exhibited similar responses to dBET6 as treatment-naive cells (Figure 4B).
Figure 4. Sequential versus Concomitant Administration of CRBN- and VHL-Based PROTACs.
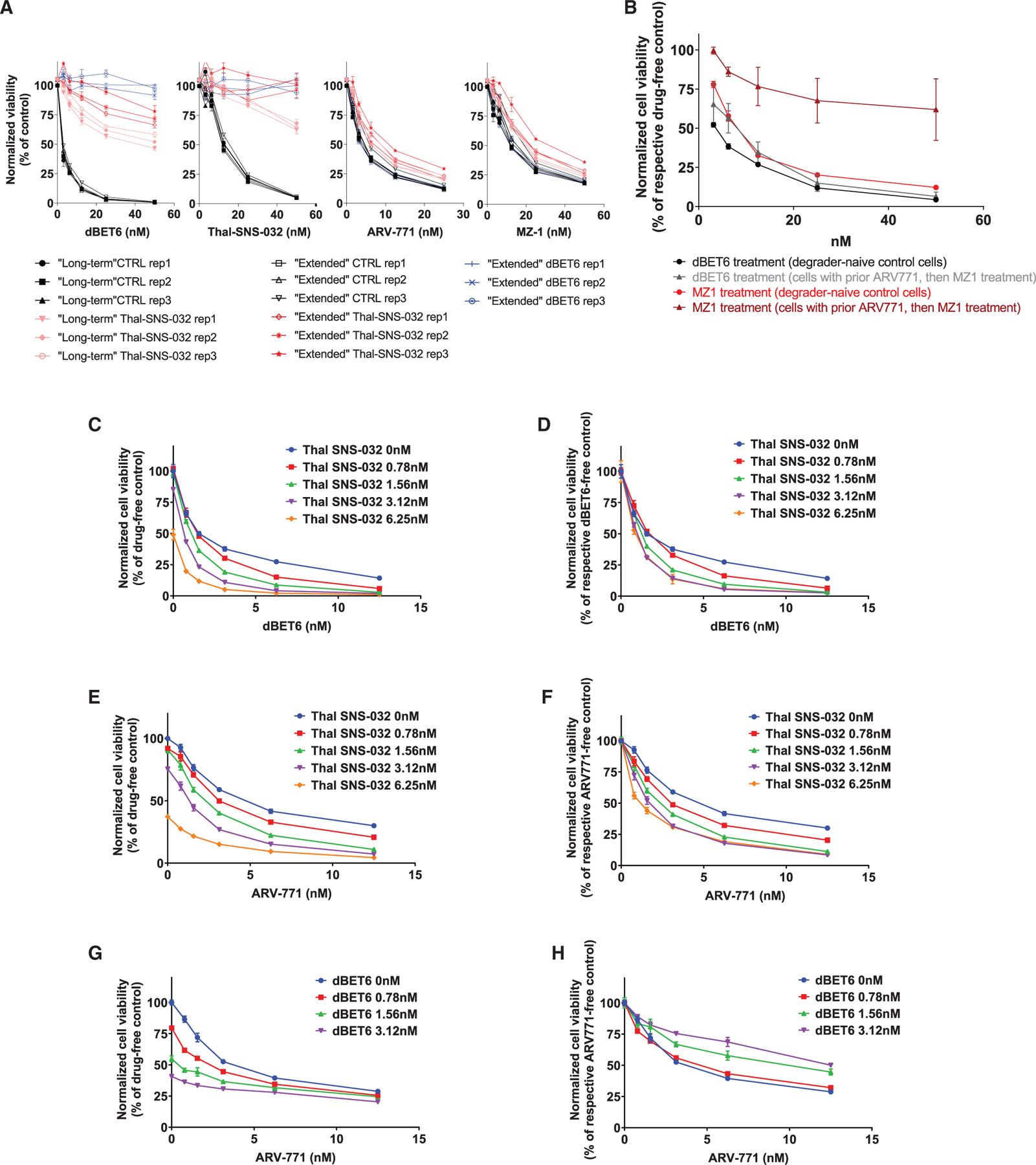
(A) Dose-response curves (CTG assays, 72 h) for dBET6, Thal-SNS-032, ARV-771, or MZ-1 treatment of pools (3 Rep pools for each population) of MM.1S-Cas9+ cells that survived in CRISPR-based studies after (1) long-term treatment with Thal-SNS-032 or long-term treatment with Thal-SNS-032 followed by extended treatment with (2) Thal-SNS-032 or (3) dBET6 versus (4) populations of drug-naive control (CTRL) cells that remained in culture during long-term or extended treatments and were collected at the end of the respective studies.
(B) Pools of MM.1S-Cas9+ cells that survived, in genome-scale CRISPR-based studies, long-term treatment with ARV-771 and then extended MZ-1 treatment were tested for their response to MZ-1 or dBET6 (CTG assay, 72 h. All experimental conditions received non-cytotoxic concentration of MDR1 inhibitor [see Methods] to prevent potential confounding by nonspecific MDR1 effects on response to all degraders).
(C–H) Combined administration of CRBN- or VHL-based PROTACs. MM.1S-Cas9+ cells were exposed simultaneously to the indicated concentrations of (C and D) Thal-SNS-032 plus dBET6, (E and F) Thal-SNS-032 plus ARV-771, and (G and H) dBET6 plus ARV-771. Cell viability (CTG assays, 72 h) results are depicted, for each combination, as percent viable cells compared with drug-free controls (C, E, and G) or the respective dBET6- or ARV-771-free cultures for each Thal-SNS-032 or dBET6 dose level (D, F, and H).
Shown are averages ± SE of triplicates per condition in each experiment.
The results from sequential administration of PROTACs leveraging different E3 ligases raised the question whether concomitant administration of such PROTACs would also lead to enhanced antitumor activity. We thus examined the simultaneous exposure of MM.1S cells to Thal-SNS-032 plus dBET6 (Figures 4C and 4D), Thal-SNS-032 plus ARV-771 (Figures 4E and 4F), and dBET6 plus ARV-771 (Figures 4G and 4H). For the first two of these combinations, increasing concentrations of Thal-SNS-032 were associated with a decrease in the percent viability of dBET6- or ARV-771-treated cells compared with drug-free controls (Figures 4C and 4E) or with the respective dBET6- or ARV-771-free cultures for each Thal-SNS-032 dose level (Figures 4D and 4F). In contrast, for the third combination, increasing dBET6 concentrations were associated with decreased percent viability of ARV-771-treated cells compared with drug-free control cells (Figure 4G) but lower relative effect compared with the respective ARV-771-free cultures for each dBET6 dose level (Figure 4H). These results indicate that concomitant exposure to two PROTACs targeting the same oncoprotein through different E3 ligases may lead to antagonistic effects. These observations may represent a version of the so-called “hook effect” (Bondeson et al., 2015; Buckley et al., 2015; Burslem et al., 2018; Lu et al., 2015; Ohoka et al., 2018; Olson et al., 2018; Schiedel et al., 2017; Winter et al., 2015); high concentrations of an individual degrader may lead to increased levels of the individual binary complexes between degrader-target protein and degrader-E3 ligase, preventing formation of the ternary complexes between target protein-PROTAC-E3 ligase that are required for target ubiquitination and eventual degradation. Simultaneous treatment with two PROTACs targeting the same protein via different E3 ligases presumably creates a similar, perhaps exacerbated, hook effect with two different ternary complexes (one for each degrader-E3 ligase pair) and four possible binary complexes that compete against each other to prevent target protein ubiquitination.
Functional Genomics Landscape for Degrader Resistance Genes in Human Tumor Cell Lines
To complement our CRISPR-based gene editing studies, we also performed CRISPR-based gene activation screens to define genes associated with PROTAC resistance. Pools of MM.1S cells expressing a dCas9-VP64 fusion were transduced with the Calabrese genome-scale library of sgRNAs for P65-HSF (heat-shock factor 1)-mediated activation of the respective genes. Cells were then exposed (similar to the long-term CRISPR-based gene-editing LOF studies) to successive rounds of dBET6 or ARV-771 treatment, allowing re-growth between treatments and until in vitro drug sensitivity tests confirmed selection of pools of MM.1S cells with a significant right shift of their dose-response curve (compared with degradernaive controls) for the respective treatment. ABCB1, the gene for the MDR1 transporter, was identified as the only gene with sgRNA enrichment in CRISPR activation studies for both dBET6 and ARV-771 (Figures S6A and S6B), reflecting the role of these large hydrophobic molecules as MDR1 substrates. In the absence of other plausible degrader resistance hits identified from these gain-of-function studies, we focused our attention on the functional genomics implications of results from our CRISPR LOF studies. Notably, LOF events recurrently detected in MM patients and/or typically associated with high-risk MM (e.g., for TP53, PTEN, negative regulators of the cell cycle, etc.; Walker et al., 2015) are not enriched in our CRISPR screens among cells surviving PROTAC treatment (Figures S6C and S6D; data not shown), suggesting that this class of compounds may exhibit activity against tumor cells with prognostically adverse genetic features.
LOF CRISPR screens of our groups identified CRBN and several other highly concordant hits associated with resistance to degronimids against multiple targets (BET bromodomains and CDK9 in the current study; and IKZF1/3 [Sievers et al., 2018]). We thus examined whether these genes are associated with potential mechanisms of resistance to thalidomide derivatives in clinically annotated molecular profiling data of individuals with MM (Multiple Myeloma Research Foundation [MMRF] CoMMpass study; Foulk et al., 2018; Kowalski et al., 2016; Miller et al., 2017). Unlike the association of CRBN expression with clinical outcome, other hits from our CRISPR studies were neither downregulated nor mutated in baseline (pre-treatment) samples from individuals with MM with an inferior clinical outcome (e.g., shorter progression-free survival or overall survival) after receiving IMID-containing regimens (regardless of whether they contained proteasome inhibitors) or in samples collected from individuals with MM who relapsed after the initial response to such IMID-containing regimens (data not shown).
We reasoned that, compared with other hits from our CRISPR studies, LOF for CRBN may have a distinct role in clinical resistance to IMIDs because it does not have a major adverse effect on proliferation and survival of MM cells. Indeed, most non-CRBN hits from our degronimid resistance screens, but not CRBN itself, have significant depletion of their sgRNAs in cell lines from MM and other neoplasms cultured in the absence of drug treatment (Figure S7). These results suggest that LOF of CRBN may provide MM cells with an advantage in the context of degronimid/IMID treatment because CRBN plays a central role in the mechanism of action of these agents and its LOF, unlike most other candidate degronimid resistance genes, does not confer to tumor cells a “fitness cost” in the form of attenuated proliferation or survival in the absence of treatment.
Functional Genomics Landscape for E3 Ligases in Human Tumor Cell Lines
So far only a few (e.g., CRBN, VHL, MDM2 [Schneekloth et al., 2008] DCAF15 [Han et al., 2017; Uehara et al., 2017], and BIRC2 [Ohoka et al., 2018]) of the ~600 known or presumed E3 ligases (Medvar et al., 2016; Nguyen et al., 2016) have been leveraged for generation of PROTAC molecules, and most E3 ligases have not yet been formally examined for such a role. Having observed that the redundant nature of CRBN in tumor cell lines in vitro may influence the patterns and dynamics of response versus resistance to CRBN-mediated degraders, we examined the dependency landscape of known E3 ligases across a broad range of neoplasms based on results of genome-scale CRISPR essentiality screens in vitro. Human tumor cell lines include a spectrum of E3 ligases whose CRISPR knockout (KO) suppresses in vitro growth for the large majority (e.g., VHL) or sizeable subsets (e.g., MDM2, BIRC2, and DCAF15) of human cancer cell lines as well as other E3 ligases that, similar to CRBN, are universally redundant for in vitro cell viability and proliferation (Figure 5A).
Figure 5. Functional Genomic Landscape of E3 Ligases as Dependencies in Human Tumor Cells.
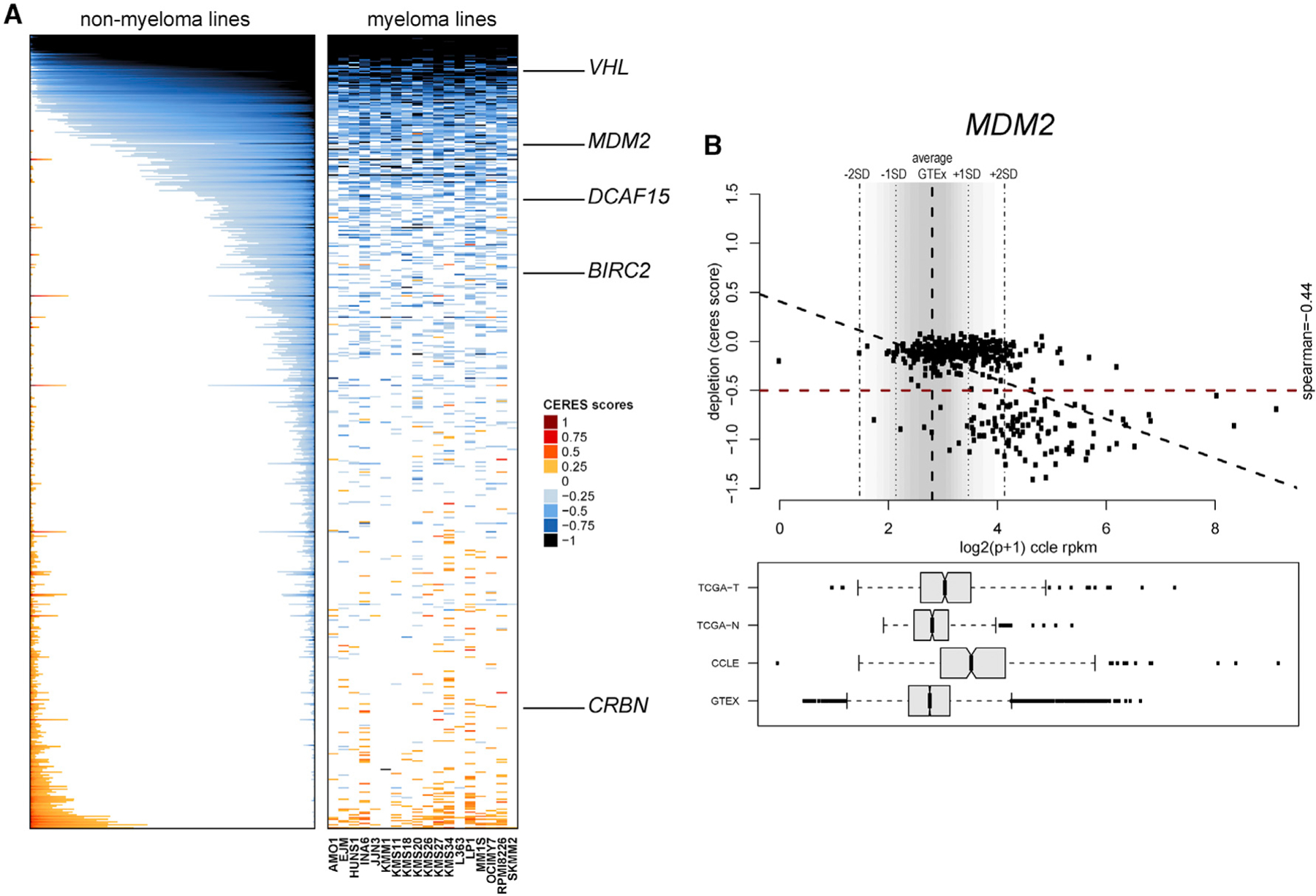
(A) Color-coded heatmaps depict CERES scores as quantitative metric of essentiality for E3 ligase genes in human tumor cell lines based on CRISPR gene editing screens (AVANA sgRNA library) in vitro in the absence of drug treatment. CERES scores for MM cell lines are depicted as a matrix (right side of graph) of cell lines (in columns) and genes (in rows), whereas data for non-MM lines are depicted for each gene (row) in stacked bars to visualize CERES scores in descending order (from left to right) for each gene. Black or dark blue indicates negative CERES scores compatible with pronounced sgRNA depletion in a given cell line. Genes are ranked from top to bottom in descending order of the average CERES scores in all tumor cell lines. Examples of key E3 ligases leveraged for development of PROTACs are highlighted.
(B) Example of evaluation of an E3 ligase (in this case MDM2) for the relationship between transcript levels in tumor versus normal cells and the gene’s status as a dependency. Data from the GTEx database were used to define the distribution (average ± 2 SD) of MDM2 transcript levels across a broad range of normal tissues. In the bottom panel, transcript levels from The Cancer Genome Atlas (TCGA; normal tissues [TCGA-N] and tumors [TCGA-T]) study and cell lines from the Cancer Cell Line Encyclopedia (CCLE) panel and GTEx are included for comparative purposes, and data are presented in boxplots (representing the average and interquartile range in each group; error bars represent the upper and lower limits of the 95% confidence interval, whereas individual dots represent samples with outlier expression). The upper part of (B) highlights the relationship between transcript levels (based on RNA-seq data from the CCLE) and CERES scores for MDM2 in tumor cell lines. The bottom right quadrant of the top panel highlights tumor cell lines with MDM2 transcript levels above the average + 2 SD of transcript levels in normal tissues of GTEx (high expressors) and CERES scores of less than ‒0.5. Linear correlation analysis examines the inverse association of high MDM2 transcript levels and low CERES scores (Spearman correlation coefficient = ‒0.44, p < 0.05).
When both the E3 ligase and target protein that interact with a PROTAC are broadly expressed in normal tissues, toxicities may conceivably ensue. We thus examined the landscape of E3 ligase expression versus dependency in different tumor types while also considering the distribution of E3 ligase transcript expression across a broad range of normal tissues. We identified E3 ligases for which more than 25% of lines across cancers express transcript levels higher than the average + 2 SD of expression in normal tissues (Genotype-Tissue Expression [GTEx] database) and examined whether these E3 ligases are essential for a large percentage of their “high expressor” cell lines. E3 ligases with such frequent co-occurrence of essentiality and high expressor status (compared with most normal tissues) (Figures S8A–S8E) include VHL, genes with known roles in tumor cell proliferation or survival (e.g., members of the anaphase promoting complex/cyclosome [APC/C]; Turnell et al., 2005), and also other E3 ligases (e.g., KCMF1 and RNF4), which, to our knowledge, have not been examined extensively as candidates for potential design of PROTACs. Notably, MDM2 also exhibits pronounced essentiality among its high expressor cell lines (Figures 5B and S8A), but this relationship applies to p53 wild-type cell lines (Figures S8B–S8D), consistent with the role of MDM2 as an E3 ligase for p53.
DISCUSSION
Degronimids and other heterobifunctional pharmacological degraders are designed to deplete, rather than simply inhibit, the action of a therapeutic target and exhibit preclinical antitumor activity against multiple tumor types (Bondeson et al., 2018; Gadd et al., 2017; Gechijian et al., 2018; Han et al., 2017; Lebraud and Heightman, 2017; Lebraud et al., 2016; Lu et al., 2015; Ohoka et al., 2018; Olson et al., 2018; Robb et al., 2017; Saenz et al., 2017; Schneekloth et al., 2008; Uehara et al., 2017; Winter et al., 2015, 2017; Wurz et al., 2018; Zengerle et al., 2015; Zhang et al., 2018; Zhou et al., 2018). Our study extends these observations and reports that CRBN-mediated degradation of BET bromodomain proteins exhibited in vitro activity even against MM cells resistant to other clinically applied agents or against pools of MM cells that had survived treatment with JQ1 or bortezomib in the context of genome-scale CRISPR-based gene editing studies.
Through genome-scale CRISPR-based studies, we determined that the top individual LOF events conferring resistance to PROTACs operating via CRBN or VHL did not represent a compensatory mechanism or “work-around” for the loss of the respective oncoprotein target(s) but, rather, dysregulation of the degradation machinery itself. Indeed, the top hits from CRISPR KO studies for CRBN-based degraders against different oncoproteins were CRBN itself and, to a quantitatively lesser extent, other members (e.g., DDB1, GPS1, and UBE2G1) or regulators (e.g., COP9 signalosome genes) of the Cullin 4A-RING-CRBN ligase complex (CRL4CRBN) that catalyzes ubiquitination of target(s) for degronimids. These observations are consistent with other CRISPR studies of our groups (Sievers et al., 2018) on candidate resistance genes to lenalidomide and pomalidomide, the prototypical degronimids that target IKZF1 and IKF3. Similarly, in CRISPR KO studies with two VHL-mediated PROTACs against BRD2/3/4, the top hits were components or regulators of the CRL2VHL complex, including CUL2 and VHL themselves and, to a lesser extent, TCEB1/TCEB2 (elongin C/B), RBX1, UBE2R2, and the COP9 signalosome genes COPS7B and COPS8. Notably, LOF events detected recurrently in individuals with MM and/or typically associated with high-risk MM (e.g., for TP53, PTEN, negative regulators of cell cycle, etc.; Walker et al., 2015) are not enriched among PROTAC-resistant MM cells in our CRISPR screens, indicating that the anti-MM activity of these agents may be capable of overriding the biological effect of key prognostically adverse genetic features in this disease.
Most individual genes associated with tumor cell resistance to CRBN-versus VHL-based PROTACs are different for these 2classes of PROTACs but have striking functional overlap. This conceivably reflects the distinct composition and, potentially, regulation of CRL complexes associated with the respective E3 ligases. For instance, DDB1 and elongin B/C (TCEB2/TCEB1) preferentially associate with CRL complexes of CUL4A/B and CUL2, respectively (Kibel et al., 1995; Lonergan et al., 1998; Shiyanov et al., 1999), explaining why their LOF causes resistance correspondingly to CRBN- or VHL-mediated degraders but not both. LOF of the COP9 signalosome genes COPS7B and COPS8 confers a decreased response to CRBN-and VHL-mediated degraders, but this effect is quantitatively more pronounced for CRBN-based degraders, suggesting possible differences in how the COP9 signalosome regulates CRL2 versus CRL4A complexes and potentially others.
The pathway-level similarities versus individual gene-level differences in resistance mechanisms for CRBN- and VHL-mediated PROTACs have major potential translational implications. First, our study indicates cross-resistance between sequential exposure to PROTACs targeting different oncoproteins through the same E3 ligase (e.g., CRBN-based PROTACs against BET bromodomain proteins or CDK9) but not between PROTACs operating through different E3 ligases and, possibly, through different CRL complexes. Interestingly, concurrent administration of PROTACs operating via the same E3 ligase against different oncoproteins (e.g., CRBN-based degraders against BET bromodomain proteins or CDK9) or through different E3 ligases against different oncoproteins (e.g., CRBN-based degrader against CDK9 and VHL-based degrader against BET bromodomain proteins) leads to enhanced anti-tumor activity. In contrast, concurrent treatment with PROTACs operating through different E3 ligases against the same oncoprotein (e.g., CRBN- and VHL-based degraders against BET bromodomain proteins) attenuates their anti-tumor effect, perhaps reflecting a variation of the so-called “hook effect” (Bondeson et al., 2015; Buckley et al., 2015; Burslem et al., 2018; Lu et al., 2015; Ohoka et al., 2018; Olson et al., 2018; Schiedel et al., 2017; Winter et al., 2015). These observations indicate that administration of multiple different PROTACs may have highly contextual outcomes, depending on the relative timing (sequential versus concurrent) of their administration and which specific E3 ligase(s) and oncoprotein target(s) are involved. Importantly, our finding of lack of cross-resistance between sequential use of PROTACs targeting the same protein through different E3 ligases/CRL complexes highlights the value of developing more PROTACs, leveraging as many different E3 ligases as possible from each type of ligase complexes.
Inactivation of the COP9 signalosome is identified in our studies as a common mechanism of decreased tumor cell response to CRBN- and VHL-based PROTACs. How COP9 signalosome function may alter E3 ligase activity and, hence, the antitumor activity of degraders remains an area of active investigation. It has been proposed that inhibition of the COP9 signalosome or CAND1 may inactivate CRL function by disrupting the dynamic cycle of assembly, disassembly, and remodeling of CRLs (Dubiel, 2009). This impaired CRL plasticity leads to constitutive E3 ligase activity (Mayor-Ruiz et al., 2019), which, in turn, leads to auto-ubiquitination of E3 ligases or, potentially, other components of the CRL complex (Fischer et al., 2011; Hotton and Callis, 2008). To our knowledge, selective pharmacological activators of the COP9 signalosome have not been reported. Therefore, to prevent PROTAC resistance because of COP9 signalosome inactivation, other approaches may be needed, including identification of selective molecular vulnerabilities of tumor cells with LOF of the COP9 signalosome (versus their wild-type counter-parts) toward designing “synthetic lethal” strategies against COP9 signalosome inactivation.
CRBN- and VHL-based PROTACs exhibited an intriguing quantitative difference; LOF for CRBN was consistently the top enriched hit in all configurations of degronimid resistance studies, whereas LOF for VHL exhibited 4- to 8-fold lower enrichment compared with CUL2, the top hit in our studies of long-term treatment with VHL-mediated PROTACs. These differences could reflect an aggregate effect of the fitness benefit versus cost associated with LOF for each gene. CRBN is universally dispensable for in vitro survival and proliferation of human cancer cell lines in the absence of a PROTAC; therefore, its LOF conceivably confers a minimal fitness cost because it protects tumor cells from degronimids while conferring no disadvantage in cell growth. In contrast, CRISPR LOF of VHL impairs in vitro growth for the large majority of VHL-proficient human cancer cell lines (Figure S7; Bindra et al., 2002; Gamper et al., 2012; Welford et al., 2010; Young et al., 2008), and this fitness cost conceivably attenuates the overall fitness benefit associated with the protection from VHL-based PROTAC treatment. These findings suggest that it may be therapeutically advantageous to design PROTACs leveraging ubiquitin ligase complexes whose function contributes to cell growth rather than those that are redundant or act as tumor suppressors. Accordingly, we identified several E3 ligases whose transcript expression in large subsets of human cancer cell lines was more than 2 SD above the average expression in normal tissues and were also required for growth of most of these cell lines. These included VHL, MDM2 (for p53 wild-type cell lines), and several other known or presumed E3 ligases not yet exploited for degrader design.
The development of degronimids and other heterobifunctional PROTACs (Gechijian et al., 2018; Nabet et al., 2018; Olson et al., 2018; Winter et al., 2015, 2017; Xu et al., 2018) offers new chemical probes targeting key oncoproteins through a relatively limited set of E3 ligase binders. Our study indicates that LOF events that induce resistance to degraders prevent actual degradation of the target oncoprotein rather than enable adaptation to its loss. To prevent resistance to oncoprotein degraders, development of more heterobifunctional compounds that leverage a variety of E3 ligases and CRL complexes must be pursued. Empirical testing in vitro and in vivo will help inform how different classes of PROTACs could be best used simultaneously or in sequential/alternating therapy.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Constantine Mitsiades (Constantine_Mitsiades@dfci.harvard.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Data and code used in this study are available via the Lead Contact upon reasonable request. RNA sequencing data supporting the current study have been deposited to Gene Expression Omnibus with accession number GEO: GSE162205.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
We examined the biological responses of human MM cells to the degronimids dBET6 (Winter et al., 2017) and Thal-SNS-032 (Olson et al., 2018), as well as the VHL-mediated degraders of BRD4/3/2, ARV-771 (Raina et al., 2016; Saenz et al., 2017; Sun et al., 2018) and MZ-1 (Gadd et al., 2017; Zengerle et al., 2015), using protocols similar to those described in prior studies on drug sensitivity testing assays, such as CTG (Delmore et al., 2011) or CS-BLI for tumor cell monoculture versus co-culture with BMSCs (McMillin et al., 2010, 2012a); Annexin V/PI staining for assessment of apoptotic cell death (Delmore et al., 2011); RNA-sequencing (Wan et al., 2017), immunoblotting (Matthews et al., 2015; Newbold et al., 2013), reverse phase protein array (RPPA) studies (Li et al., 2017); as well as in vivo efficacy studies in xenografts established in NSG mice after subcutaneous or intravenous (Delmore et al., 2011; McMillin et al., 2010, 2011, 2012b; Mitsiades et al., 2008) injection of human MM.1S cells. More detailed information on these experimental procedures, as well as the design of our genome-scale CRISPR-based gene editing screens for genes whose loss of function confers resistance to CRBN- or VHL-mediated degraders is outlined below.
Cell culture
The human cell lines (MM.1S, RPMI-8226, OPM-2, LP-1, OPM-1, JJN3, L363, AMO-1, OCI-My5, KMS-20, KMS-26, KMS-27) were obtained from ATCC, JCRB, or DSMZ. MM cell lines OPM-2 and KMS11 expressing non-targeting control shRNA or shRNA.CRBN#13 (Zhu et al., 2011, 2013) were a gift from Keith Stewart (Mayo Clinic, AZ, USA). MM.1S CRBN−/−cells, generated by CRISPR-knockout (Lu et al., 2014) were kindly provided by the laboratory of Dr William Kaelin (DFCI, Boston, MA, USA). The MM.1S-Cas9+ cells were generated by the laboratory of Dr Benjamin Ebert (DFCI). KMS11-Cas9+ cells (transduced with pLX 311-Cas9 [Addgene #96924] construct) were obtained from the Broad Institute. The human stromal cell line HS27A was obtained from ATCC. All human MM cell lines were cultured in RPMI 1640 medium supplemented with L-glutamine (Life Technologies, Carlsbad, CA, USA), FBS (10%) (Gemini Bioproducts, Woodland, CA), 20 I.U./mL penicillin and 20μg/mL streptomycin (Fisher Scientific, Springfield, NJ, USA) and cultured at 37°C, with 5% CO2.
Patient-derived samples
Bone marrow aspirates (1–4mL) from individuals with MM (newly diagnosed, smoldering, relapsed/refractory, maintenance treatment) or MGUS were collected after patients provided informed consent and based on tissue collection protocol approved by the Dana-Farber Cancer Institute Institutional Review Board. The age and sex of the respective patients was not available. Deidentified samples were processed for separation of CD138+ plasma cells, using CD138-positive selection beads, and immunomagnetic separation, as per kit instructions (EasySep, StemCell, Cambridge, MA). Patient-derived tumor cells were cultured in RPMI 1640 medium supplemented with L-glutamine (Life Technologies, Carlsbad, CA, USA), FBS (10%) (Gemini Bioproducts, Woodland, CA), 20 I.U./mL penicillin and 20µg/mL streptomycin (Fisher Scientific, Springfield, NJ, USA) and hIL-6 (Thermo Fisher Scientific, Waltham, MA, USA 1–5ng/mL).
Mice
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) female (6–8 week-old) mice were purchased from Jackson Laboratory. Mice were bred and maintained in individual ventilated cages and fed with autoclaved food and water at Dana-Farber Cancer Institute Animal Facility. Animal studies were performed according to a protocol approved by the Dana-Farber Cancer Institute Animal Care and Use Committee.
METHOD DETAILS
Immunoblotting
MM.1S cells were seeded in 6-well plates at 3×106 cells/well (~24h prior to addition of drug). After the addition of dBET6, JQ1 or Thal-SNS-032 cells were incubated, harvested after 4 h using trypsin, washed in ice-cold PBS and immediately frozen (‒80°C). Cell pellets were thawed on ice, lysed using RIPA buffer (ThermoFisher) with protease/phosphatase inhibitor cocktail (Cell Signaling Technology, Danvers, MA) by incubating on ice for 30 min. The lysates were clarified by spinning at 10,000 g for 10 min at 4°C and the concentration of the lysate was determined using BCA protocol (ThermoFisher).
Samples were prepared for western blot using LDS sample buffer (NuPage, Invitrogen, Carlsbad, CA, USA) with sample reducing agent (NuPage, Invitrogen) and heated to 95°C for 5 min. Samples were loaded (20 µg/sample) into Tris-acetate or Bis-Tris gels (NuPage, Invitrogen) and run as per kit instructions using appropriate running buffers. Gels were transferred as described (Matthews et al., 2015) onto PVDF membrane using SDS-based transfer buffer (NuPage, Invitrogen). Membranes were blocked (5% skim milk; or 5% bovine serum albumin in TBS-T) for at least 30 min then probed with primary antibodies overnight (4°C). Secondary antibodies were incubated for 1 h at RT (5% skim milk in TBS-T) prior to incubation in ECL (ThermoFisher). Each protein of interest (BRD2, BRD3, BRD4, c-myc and CDK9) was assessed in a separate gel and corresponding membrane (which was simultaneously incubated with antibodies for both the target protein of interest and GAPDH, to provide loading control within the same blot). Visualization of western blotting results was performed by C-DiGit®Blot Scanner (LI-COR Biotechnology, Lincoln, NE)
Assessment of cell viability
Cell viability and growth of human MM cell lines were assessed using CellTiter-Glo (CTG) assay (Promega, Madison, WI) per kit instructions for monocultures of MM cells; whereas Compartment-Specific Bioluminescence Imaging (CS-BLI) was performed as described (Delmore et al., 2011; McMillin et al., 2010, 2011, 2012a, 2012b) for MM cell cocultures with stromal cells versus respective cell monocultures. Briefly, MM cells were seeded (5×103 or 1×103 cells/well) into 96- or 384-well opaque plates, in supplemented media (40 or 50 µL) and incubated for 24 h prior to addition of drug. For co-culture experiments, HS27A immortalized bone marrow stromal cells were pre-seeded (5×103 cells/well) into each well in media prior (24 h) to the addition of MM cells. At each time point (24 h, 48 h, and/or 72 h) CTG reagent (10% volume) (or, for CS-BLI, luciferin) was added to each well, and plates were read using a microplate reader (BioTek Synergy 2, BioTek, Winooski, VA). CTG assays were performed to quantify the response to various treatments for pools of MM cells which had been transduced with whole-genome libraries of sgRNAs for CRISPR-based gene editing and then survived exposure to different therapeutic agents, including Thal-SNS-032 (Brunello sgRNA library); ARV-771 or MZ-1 (Brunello), dBET6 (GeCKOv2), bortezomib (GeCKOv2) or JQ1 (GeCKOv2). Combination treatments of dergader-naive cells (e.g., dBET6 plus Thal-SNS-032, Thal-SNS-032 plus ARV-771, or dBET6 plus ARV-771) were performed at the concentrations indicated in respective figures and assessed by CTG at 72 h.
Assessment of apoptosis
For the assessment of apoptosis, MM.1S cells were seeded into 6-well plates (2×106 cells/well) in medium supplemented with 10% FBS and penicillin/streptomycin 24 h prior to treatment with drug (e.g., dBET6) or respective vehicle. After the end of treatment, cells were harvested prior to staining with Annexin V-FITC and PI (BD, Bedford, MA). Apoptosis was assessed immediately using flow cytometry (LSR II, BD, Bedford, MA).
Collection and ex vivo treatment of patient-derived tumor cells
Samples were processed for separation of CD138+ plasma cells using CD138-positive selection beads, and immunomagnetic separation, as per kit instructions (EasySep, StemCell, Cambridge, MA). Sorted cells were immediately plated into 96-well plates (100 µL media) and cultured overnight prior to treatment or immediately treated with dBET6. Viability was assessed using CTG (48 h) and each treatment was repeated up to 3 times (n = 3 biological replicates).
RNA-sequencing
MM.1S cells were seeded in 6-well plates (2×106/well) and cultured overnight prior to treatment. After 12 h, dBET6 (100 nM or 500 nM) or JQ1 (500 nM) was added to the cultures, then cells were harvested after 4 h and immediately frozen (‒80°C). In order to assess transcriptional modulation by dBET6, RNA was extracted as previously described (Matthews et al., 2015) and analyzed using next-generation sequencing by the Molecular Biology Core Facilities (MBCF, DFCI). All samples and corresponding untreated controls were measured in triplicates and sequenced with technical replication for each sample.
Reverse phase protein array (RPPA)
MM.1S cells were seeded in 6-well plates (2×106/well) and cultured overnight prior to treatment. After 12 h, dBET6 (100 nM) or JQ1 (500 nM) was added to the cells, then cells were harvested after 4h or 18 h and immediately frozen (‒80°C). The experiment was performed in triplicates for each treatment condition and untreated controls. Protein lysates of these samples were processed by the Functional Proteomics RPPA Core Facility of the MD Anderson Cancer Center (Houston, TX), based on protocols and procedures outlined on the website of the facility (https://www.mdanderson.org/research/research-resources/core-facilities/functional-proteomics-rppa-core/rppa-process.html) and similarly to prior reports (Li et al., 2017).
In vivo dBET6 treatment
Subcutaneous MM xenograft model
Luciferase-positive MM.1S cells (10×106 Luc-GFP-MM.1S in matrigel) were transplanted into the right flank of NSG mice (100µL). Tumor size was measured using calipers. dBET6, prepared daily in Solutol (5%), or vehicle control was injected IP into mice when tumor size reached approximately 100 mm3. Treatments were carried out daily for 8 days and tumor size was assessed using calipers. Mice were culled when tumors reached 2 cm in any dimension or when moribund.
Xenograft model of diffuse MM lesions
Luciferase-positive MM.1S cells (1×106 Luc-GFP-MM.1S) were injected via the tail vein (IV) into NSG mice. Mice were imaged (BLI) after 1 week and mice with detectable tumor burden were randomly assigned to receive treatment with dBET6 or vehicle control daily for approximately 12 days. BLI was undertaken weekly and mice were euthanized when moribund or upon signs of hind-limb paralysis.
Animal studies were performed according to a protocol approved by the Dana-Farber Cancer Institute Animal Care and Use Committee.
CRISPR/Cas9-based gene editing or gene activation screens to identify candidate mechanisms of tumor cell resistance to degraders
We performed genome-scale CRISPR/Cas9 gene-editing screens, similarly to previous studies (Doench et al., 2016; Meyers et al., 2017; Shalem et al., 2014; Wang et al., 2017), and in 3 configurations, which involved: i) “short-term” (48 h) treatments with either dBET6 or Thal-SNS-032, followed by tumor cell collection at the end of the treatment; ii) “long-term” studies with successive rounds of treatment (e.g. with dBET6, Thal-SNS-032, ARV-771, or MZ-1) of pools of MM.1S-Cas9+ cells transduced with the respective sgRNA library, allowing regrowth between treatments, and until in vitro drug sensitivity testing confirmed the selection of pools of MM.1S cells with significant shift-to-the-right of their dose-response curve (compared to degrader-naive controls) for the respective treatment; and iii) extended dergader treatment screens, in which e.g., Thal-SNS-032-resistant MM.1S-Cas9+ cell populations isolated from the initial long-term CRISPR/Cas9-based gene editing screen continued receiving additional degronimid treatment for 2 weeks, with either Thal-SNS-032 (i.e., a continuation of the initial treatment that had led to the isolation of these resistant cells) or dBET6. Similar extended treatment studies were performed for VHL-based degraders (initial long-term screen with ARV-771 followed by extended treatment with MZ-1).
These genome-scale CRISPR gene editing screens were performed with MM.1S-Cas9+ cells (transduced with lentiviral construct for SpCas9 and kindly provided by Quinlan Sievers [Ebert Lab, BWH/DFCI]) which were transduced with pooled lentiviral particles for genome-scale sgRNA libraries, specifically GeCKOv2 for the long-term dBET6 screen; and Brunello, for other genome-wide CRIPSR gene editing studies of degrader-treated cells.
We also performed genome-scale CRISPR-based gene-activation screens, which followed the long-term configuration of the gene editing studies. Specifically, MM.1S-dCas9-VP64 cells (generated after transduction with lentiviral construct for dCas9-VP64 [Addgene]) were transduced with pooled lentiviral particles for the Calabrese genome-scale sgRNA library, and underwent treatment with dBET6 or ARV-771 until in vitro drug sensitivity testing confirmed the selection of pools of MM.1S cells with significant shift-to-the-right of their dose-response curve (compared to drug-naive controls) for the respective degrader.
Production of viral particles for genome-scale CRISPR screens
Lenti-X-293T cells (Clontech, Mountain View, CA) were plated in T175 culture flasks (0.6×106 cells/mL) in DMEM (Life Technologies) with FBS (10%) for 24 h. After aspiration of cell medium, OPTI-MEM (6 mL) and Lipofectamine 2000 (100 µL; Life Technologies) were added to each flask plus packaging plasmids psPAX2 (20 µg) and MD2.G (10 µg) and plasmid preps of each of the sub-libraries (V2.1, V2.2) of the GeCKOv2 genome-scale sgRNA library; the Brunello sgRNA library; or the Calabrese sgRNA library (20 µg per prep). The GeCKOv2 library was kindly provided to us by Offir Shalem and Feng Zhang (Zhang Lab, Broad Institute of MIT and Harvard). Plasmid preps for the Brunello and Calabrese sgRNA libraries were purchased from Addgene. The transfected Lenti-X-293T cells were incubated at 37°C (20 min), topped up with fresh media (25 mL), and then refreshed again after 16 h. Viral supernatants were collected after 24 h and stored at ‒80°C prior to use.
Lentiviral transductions with sgRNA libraries
For screens with the GeCKOv2 sgRNA library (Sanjana et al., 2014; Shalem et al., 2014), tumor cell transductions were performed in batches of 8×107 cells per sub-library for each replicate. Cells were incubated for 16 h in cell medium containing polybrene (2µg/mL; Santa Cruz Biotechnology), 10 mM HEPES (pH 7.4) (Sigma-Aldrich) and viral prep (4 mL) diluted to achieve a MOI of 0.4. After the end of the incubation with the viral preps, cells were washed and incubated for an additional day. Transduced cells were cultured at an initial density of 1×106 cells/mL and were treated with puromycin (1 μg/mL) for up to 14 days immediately after transduction. After puromycin selection, MM.1S-Cas9+ cells transduced with each GeCKOv2 sub-library were plated at 60×106 cells per flask (T175, 100 mL) to enable estimated average coverage (number of cells per sgRNA) of ~1000X and were sub-cultured at four to five-day intervals to prevent confluence. At each passage, cells were harvested, washed with PBS (Corning, NY), pelleted (5000 rpm, 5 min, 4°C) and frozen as cell pellets (‒80°C) for next generation sequencing, and also replated at 60×106 cells.
For the genome-scale screens with the Brunello sgRNA library (Doench et al., 2016), tumor cell transductions were performed in batches of 5×107 cells per library per replicate. Cells were incubated (18 h) in cell medium containing polybrene (5 µg/mL; Santa Cruz Biotechnology), 10 mM HEPES (pH 7.4) (Sigma-Aldrich) and viral prep (30 mL) diluted 1:1. Transduced cells were cultured at an initial density of 1×106 cells/mL in puromycin-free media for 2 days and then were treated with puromycin (1 μg/mL) for up to 5 days. After stable transduction, pooled MM.1S cells were plated at 40×106 cells per flask (T175, 100 mL) to enable estimated average coverage of ~500X and were sub-cultured at three- to four-day intervals to prevent confluence. At each passage, cells were harvested, washed with PBS (Corning), pelleted (5000 rpm, 5 min, 4°C) and frozen as cell pellets (‒80°C) for next generation sequencing, or also replated at 40×106 cells.
For the genome-scale screen with the Calabrese sgRNA library, tumor cells were transduced in batches of 3×107 cells per sublibrary for triplicates. Cells were incubated (18 h) in cell medium containing polybrene (4 µg/mL; Santa Cruz Biotechnology), 10 mM HEPES (pH 7.4) (Sigma-Aldrich) and viral prep (30 mL) diluted 1:1. Transduced cells were cultured at an initial density of 1×106 cells/mL in puromycin-free media for 2 days and then were treated with puromycin (1 µg/mL) for up to 7 days. After stable transduction, pooled MM.1S cells were plated at 30×106 cells per flask (T175, 100 mL) to enable estimated average coverage of ~500X and were sub-cultured at three- to four-day intervals to prevent confluence. At each passage, cells were harvested, washed with PBS (Corning), pelleted (5000 rpm, 5 min, 4°C) and frozen as cell pellets (‒80°C) for next generation sequencing or replated at 30×106 cells per sub-library.
Generation of drug-resistant populations of tumor cells harboring CRISPR-based gene editing or activation
As outlined earlier, we generated through our pooled genome-scale CRISPR-based gene editing studies treatment-resistant/tolerant tumor cell populations after (i) “long-term” treatment with dBET6, Thal-SNS-032, ARV-771, MZ-1, JQ1 or Bortezomib; (ii) “short-term” (48h) treatments with either dBET6 or Thal-SNS-032; (iii) “extended treatment” with dBET6 or Thal-SNS-032, for Thal-SNS-032-resistant MM.1S-Cas9+ cell populations isolated from a “long-term” Thal-SNS-032 treatment screen; and (iv) extended treatment with MZ-1 in MM.1S-Cas9+ cell populations isolated from a long-term ARV-771 treatment. All treatments were performed in triplicates per experimental condition.
For “long-term” treatment screens with dBET6, JQ1 and Bortezomib, drug treatments were carried out on MM.1S-Cas9+ cells transduced with pools of the GeCKOv2 sgRNA sub-libraries (≤14 days post-puromycin, ~60×106/ flask, ~1000X coverage, 100 mL, n = 3 individual flasks per sgRNA sub-library) 24 h after seeding, as follows: a) for dBET6, cells were treated with drug (0.25 µM) for 24 h prior to complete drug washout. In total, MM.1S-Cas9+ cells were treated 3 times with dBET6; b) for Bortezomib, cells were treated with drug (0.025 µM) for 24 h prior to drug washout. In total, MM.1S-Cas9+ cells were treated 3–4 times with Bortezomib; c) for JQ1, cells were treated with drug for 72 h (0.5 µM) prior to drug washout. In total, MM.1S-Cas9+ cells were treated 3–4 times with JQ1; or d) appropriate vehicle controls treated per individual drug protocols. Following treatments, all cells were serially cultured without drug and allowed to regrow for as long as required to enable reseeding (~60×106 cells per flask, 100 mL) and retreatment with individual drugs. Concomitant with retreatment, remaining cells were frozen (‒80°C; 20% FBS/DMSO) for later analysis or assessed for drug resistance/sensitivity using CTG assays. Between treatments, and in order to maintain the health of cultures, dead cells were periodically removed using Ficoll centrifugation (1,500 rpm for 10–20 min).
For long-term treatment screens with Thal-SNS-032, ARV-771 and MZ-1, treatment of MM.1S-Cas9+ cells (40×106, ~500X coverage, in triplicates for drug treatments [25 nM] or for vehicle control) transduced with the Brunello sgRNA library started 4 weeks post-puromycin selection. Cells were serially passaged every 3–4 days to prevent confluence, each time replating at ~500X coverage (~100 mL/replicate) and at the same time replenishing each compound or vehicle. Cells were periodically assayed, e.g. typically at least once weekly, for drug sensitivity/resistance using CTG. Drug treatments were adjusted according to cell numbers in the flasks and CTG assessment of responsiveness. Drug treatments were paused when cell numbers fell below 10 × 106 per flask, to allow cells to recover and regrow, and treatments were resumed when cell numbers reached ~40×10 6 again. After 4 weeks of incubation with Thal-SNS-032 and 6 weeks of incubation with ARV-771 or MZ-1 treatment, cells were collected for next generation sequencing; whereas a portion of Thal-SNS-032-resistant cells were processed for extended treatment screens involving 2 additional weeks of continuous treatment (compound replenished every 3–4 days) with either: a) Thal-SNS-032 or b) switch to dBET6 treatment. Similarly, a portion of cells that survived the long-term ARV-771 treatment was processed for extended treatment with MZ-1. These latter cell pools contained cells overexpressing MDR1 (data not shown): in dose-response analyses of these pools with different degraders, subtoxic concentrations of MDR1 inhibitor (HM30181, 100nM) were added in all experimental conditions to prevent confounding by MDR1 status on assessment of the intrinsic degrader sensitivity of these cells. Pools of cells isolated after sequential long-term and extended treatments with degronimids did not exhibit evidence of MDR1 overexpression (data not shown), and thus MDR1 inhibitor use was not required in dose-response analyses of different degraders in these cell pools.
For short-term treatment screens with Thal-SNS-032 or dBET6, MM.1S-Cas9+ cells (80×106) transduced with the Brunello sgRNA library were treated with Thal-SNS-032 (25nM), dBET6 (25nM), or vehicle control for 48 h. Cells were immediately harvested (30×106) and sgRNA distribution assessed, by next generation sequencing.
For genome-scale CRISPR activation screens, we started treatment of MM.1S-dCas9-VP64 cells (30×106, ~500X coverage) transduced with the Calabrese sgRNA library 2 weeks post-puromycin selection with dBET6 (50nM), ARV-771 (50nM), or vehicle control, similar to the approaches for the gene editing screens with long-term treatments described above. After 5 weeks of culture and dBET6 or ARV-771 treatment, cells were collected for next generation sequencing to quantify the distribution of sgRNAs in dBET6- or ARV-771-treated cells versus their respective treatment-naive controls.
Next-generation sequencing
Preparation of DNA for next-generation sequencing was undertaken using a two-step PCR protocol as described (Shalem et al., 2014). Briefly, DNA was extracted from frozen cell pellets (~2 or 3×107 cells; Blood & Cell Culture DNA Midi Kit or Maxi Kit, QIAGEN) per manufacturer’s instructions. DNA concentration was quantified by UV-spectroscopy (NanoDrop 8000; ThermoFisher Scientific).
In the first PCR, sgRNA loci were selectively amplified from a total of 130 µg or 160 µg of genomic DNA (10 μg DNA per sample x 13 reactions, 100µL volume for GeCKOv2 and Calabrese library; or 16 reactions for Brunello library) using primers described in Table S1 and Phusion® High-Fidelity DNA Polymerase (New England Biolabs, Beverly, MA). A second PCR was performed using 5µL of the pooled Step 1 PCR product per reaction (typically 1 reaction per 10,000 sgRNAs of the respective library; 100µL reaction volume) to attach Illumina adapters and to barcode samples (Table S1). Primers for the second PCR included a staggered forward primer (to increase sequencing complexity) and an 8bp barcode on the reverse primer for multiplexing of disparate biological samples (Table S1). PCR replicates were combined, gel normalized (2% w/v) and pooled, then the entire sample run on a gel for size extraction. The bands containing the amplified and barcoded sgRNA sequences (approximately 350–370bp) were excised and DNA extracted (QIA-quick Gel Extraction Kit,QIAGEN). Multiplexed samples were then sequenced at the Molecular Biology Core Facility (Dana-Farber Cancer Institute) and/or the Genomics Platform (Broad Institute) using an Illumina NextSeq 500 (Illumina, San Diego, CA).
CRISPR/Cas9-based gene knockouts with individual sgRNAs to validate candidate resistance genes
Individual sgRNAs against several candidate degrader resistance genes (e.g., COPS2, COPS8, DDB1) were designed using the Broad Institute sgRNA design portal (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). Non-targeting control sgRNAs and COPS7B sgRNAs were from the GeCKOv2 sgRNA library. The sgRNAs generated for each gene are listed in Table S2). All sgRNAs were synthesized by CustomArray Inc (Bothell, WA). Cloning was performed similar to previous studies, e.g., (Shalem et al., 2014) and protocols published by (e.g., https://media.addgene.org/cms/filer_public/4f/ab/4fabc269–56e2–4ba5–92bd-09dc89c1e862/zhang_lenticrisprv2_and_lentiguide_oligo_cloning_protocol_1.pdf) using a pHKO9 vector contaning puromycin resistance gene. Briefly, to clone the sgRNA guide sequence, plasmids were digested with BsmBI (New England Biolabs, Ipswich, MA) at 55°C for 1 h. Oligonucleotides for each sgRNA sequence were phosphorylated using T4 Ligation Buffer and T4 polynucleotide kinase (New England Biolabs) at 37°C for 30 min and then annealed by heating to 95°C for 5 min and cooling to 25°C at 1.5°C/minute. Using T4 ligation buffer and T4 ligase (New England Biolabs), annealed oligos were ligated into gel purified vectors (QIAGEN) at 65°C for 10 min. 2.5 µg of the ligation product were transformed in 30 µL E.coli electrocompetent cells (Lucigen E.cloni 10 G ELITE PLUS, Invitrogen). Subsequently, 500 µL of transformed cells were plated on LB Agar-AmpR (ThermoFisher Scientific) and incubated overnight at 37°C. Three colonies per plate were then picked and inoculated in a mini-prep culture (QIAGEN). The product was then digested with BsmBI and XhoI (New England Biolabs) at 37°C for 1 h and compared to the uncut control on 1% agarose gel. One out of three colonies (10 µL) was then precultured in 3 mL LB Broth-AmpR and shaken at 37°C for 6 h. Afterward it was transferred to a 250 mL LB Broth-AmpR flask and incubated overnight at 37°C. Each culture was then inoculated into a maxi-prep culture (QIAGEN)
Also, sgRNAs for CRBN, VHL, TCEB1, TCEB2, UBE2R2, CUL2, FBXW2, and olfactory receptor genes were selected among the respective sgRNAs included in the Brunello library and were cloned using pLVX-hyg-sgRNA1 Vector system (Takara Bio USA, CA) following to manufacturer’s manual (https://www.takarabio.com/documents/User%20Manual/Lenti/Lenti-X_CRISPR-Cas9_System_User_Manual_121316.pdf)).
Production of viral particles for individual sgRNAs
Lenti-X-293T cells (Clontech, Mountain View, CA) were plated in 6-well plates (1.5×106 cells/well) in DMEM (Life Technologies) with FBS (10%) for 24 h. After aspiration of cell medium, OPTI-MEM (6 mL) and Lipofectamine 2000 (100 µL; Life Technologies) were added to each flask plus packaging plasmids psPAX2 (5 µg) and MD2.G (2.5 µg) and plasmid preps of each of the constructs (1300 ng per prep). The transfected Lenti-X-293T cells were incubated at 37°C (20 min), topped up with fresh media (2 mL per well), and then refreshed again after 16 h. Viral supernatants were collected after 24h and stored at ‒80°C prior to use.
Lentiviral transductions with constructs for individual sgRNAs
500×103 to 1×106 MM.1S-Cas9+ or KMS11-Cas9+ cells were plated in 50 µL of complete RPMI1640 medium per well in a 24-well plate, and additional 100 µL of complete RPMI1640 medium were added to cover the well completely. Cells were incubated in cell medium containing polybrene (5 to 8 µg/mL; Santa Cruz Biotechnology), and viral prep. 24 h after addition of viral preps, media were changed and, after another 48 h, puromycin or hygromycin B selection (1 to 2 µg/ml or 350 µg/ml per well, respectively, depending on the vector used) was started. After 7 or 14 days, respectively, of antibiotic selection, transduced MM cells were collected and expanded in T75 flasks.
Dose-response assays in cells with individual sgRNAs
CRISPR knockout cells and non-targeting controls were plated in 96-well or 384-well plates as described and treated with dBET6 at the concentrations stated for each experiment; and viability was assessed after 48 or 72 h using CTG. In the case of Thal-SNS-032, MZ-1, or ARV-771, cells were plated in 384-well plates and treated at concentrations described in the respective experiments and viability assays were performed at 72 h.
Competition assay using amplicon sequencing of CRISPR-induced insertions-deletions (indel analysis)
MM.1S-Cas9+ cells transduced with sgRNAs for either OR2S2 or UBE2R2 were mixed at 9:1 ratios. Mixtures of cells were then continuously treated with ARV-771 (15 nM) or DMSO for 14 days. ARV-771 or DMSO were replenished every three or four days. Samples were collected at baseline and at day 14. DNA was extracted from collected cells by Qiamp DNA Mini Kit. For each sample, 500 ng of DNA were extracted and used for PCR amplification with primers flanking the DNA cut-site of the UBE2R2 sgRNA. PCR products underwent gel electrophoresis and bands of the expected amplicon size were extracted using the QIAquick Gel Extraction Kit. Amplicon sequencing was performed by the MGH CCIB DNA Core facility (https://dnacore.mgh.harvard.edu/new-cgi-bin/site/pages/crispr_sequencing_main.jsp).
The indel and frameshift analysis was performed with the command-line version of CRISPResso2 using the input of paired-end fastq files, amplicon sequence, targeting sgRNA sequence and a string of comma-delimited exon subsequences of the amplicon (Clement et al., 2019).
To generate the input sequence data for CRISPResso2, the primers of the amplicon and the corresponding targeting sgRNA were mapped to the human genome using bowtie2 (Langmead and Salzberg, 2012). We retrieved the genome coordinates of all Ensemble exons contained within the respective amplicon coordinates using biomaRt and trimmed overlapping exon coordinates to the respective amplicon coordinates. The amplicon sequence and exon subsequences were retrieved using the Ensemble REST API (http://grch37.rest.ensembl.org/).
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-sequencing differential gene expression analysis
Mapping of single-end RNA-sequencing reads to the human genome HG38.84 and readcount estimation for each sample was performed using STAR (https://github.com/alexdobin/STAR/releases) on the Orchestra research computing cluster (Harvard Medical School). We used the GTF annotation file Homo_sapiens.GRCh38.84.gtf from the Ensembl database (https://www.ensembl.org/). Ensembl identifiers were mapped to gene symbols retrieved from the Biomart database (version mar2016). Differential gene expression analysis for dBET6- or JQ1-treated samples versus controls was performed using deseq2 (Love et al., 2014). For heatmap visualization, we estimated log2fold changes on the replicate level of the samples with drug treatment to the respective average readcount of the untreated controls.
Reverse phase protein array (RPPA) analysis
We mapped protein antibody labels to gene symbols available from Antibody Information and Protocols at MDACC (https://www.mdanderson.org/research/research-resources/core-facilities/functional-proteomics-rppa-core/antibody-information-and-protocols.html). The RPPA linear normalized data matrix comprised 303 proteins. For data preprocessing, results from technical replicates were averaged for each protein. The differential protein expression analysis for dBET6- or JQ1-treated samples versus controls were performed using the limma moderated t-statistic (Ritchie et al., 2015).
Analysis of genome-scale CRISPR screens
Sequencing data on PCR-amplified sgRNAs sequences were processed using cutadapt (v.1.9.1, (Martin, 2011)) to remove the 5′ staggered primer adapter (added to increase the read complexity for the Illumina sequencing procedure) and 3′ adapter from the raw reads. We generated a count matrix of uniquely matching reads, allowing at most one mismatch to the respective sgRNA library based on bowtie2 read mappings (Langmead and Salzberg, 2012). The trimmed reads (20mers) were aligned to the respective sgRNA library using bowtie2 using the parameter settings–norc–local -D 20 -R 3 -N 0 -L 10 -i S,1,0.5 -p 6 for a highly sensitive alignment search. For each sample we filtered the bowtie2 alignments for unique matching reads with at most 1 base mismatch and estimated the respective count frequencies for each sgRNA. The feature count matrix and sgRNA matrix formatting was performed in the script language bash and in R. For the purpose of comparison, we also generated the feature count matrix based on a perfect match criteria of the reads to the sgRNA library using the count function from the MAGeCK software (Li et al., 2014). Technical replicates from the sequencing runs were merged by summation of the read counts. For CRISPR libraries with two sub-libraries (GeCKOV2 and Calabrese), the data of paired samples of the sub-libraries A and B (each containing three sgRNAs per gene) were joined after total readcount normalization for the 3 replicates of the respective experimental conditions. Statistical analysis for the enrichment and depletion of the sgRNAs was performed using MAGeCK (Li et al., 2014). For the gene-level rank aggregation of the sgRNAs in the MAGeCK algorithm, the non-targeting sgRNAs of the respective libraries were used as control distribution. Results of CRISPR screens in Figures 1B, 2B, 2G, and 2H are depicted with the same random order of genes on the x axes.
Gene essentiality and molecular profiling data
CERES scores (Meyers et al., 2017), as metrics of essentiality, were examined for genes of interest (e.g., “hits” from our CRISPR screens for PROTAC resistance regulators; and known or presumed E3 ligases) based on results from genome-scale in vitro CRISPR/Cas9 gene knockout screens of the Dependency Map program (http://www.depmap.org/) performed with the AVANA sgRNA library in myeloma and non-myeloma lines. Results presented in this study reflect the 18Q4 release. The cell line HuNS1 is listed within the MM lines, based on publicly available information from cell line banks, such as ATCC (https://www.atcc.org), but is also reported to exhibit lymphoblastoid features and its relevance as a potential model of MM is not conclusive. The observations of this study on the patterns of essentiality for different genes of interest are not affected by inclusion or not of HuNS1 among MM lines and are also consistent with data derived from other subsequent releases of data from the Dependency Map screen (data not shown). Known or presumed E3 ligases were identified based on information from 2 publicly available databases (http://140.138.144.145/~ubinet/browseE3.php [Nguyen et al., 2016] and https://hpcwebapps.cit.nih.gov/ESBL/Database/E3-ligases/ [Medvar et al., 2016]). Molecular profiling data on tumor cell lines were downloaded from the Cancer Cell Line Encyclopedia (CCLE) portal (https://portals.broadinstitute.org/ccle/data). Gene expression data on patient-derived tumors and normal tissues were downloaded, respectively, from the TCGA (https://gdac.broadinstitute.org/) and the GTEx database (http://gtexportal.org/home/datasets).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-c-Myc | Santa Cruz Biotechnology | Cat# sc-40; RRID: AB_2857941 |
| Rabbit polyclonal anti-BRD2 | Bethyl Laboratories | Cat# A302–583A; RRID: AB_2034829 |
| Rabbit polyclonal anti-BRD3 | Bethyl Laboratories | Cat# A302–368A; RRID: AB_1907251 |
| Rabbit polyclonal anti-BRD4 | Bethyl Laboratories | Cat# A301–985A-M; RRID: AB_2631450 |
| Rabbit monoclonal anti-CDK9 (C12F7) | Cell Signaling Technology | Cat# 2316; RRID: AB_2291505 |
| Rabbit monoclonal anti-GAPDH (14C10) | Cell Signaling Technology | Cat# 3683; RRID: AB_1642205 |
| HRP conjugated | ||
| Horse anti-mouse HRP conjugated | Cell Signaling Technology | Cat# 7076S; RRID: AB_330924 |
| Goat anti-rabbit HRP conjugated | Cell Signaling Technology | Cat# 7074P2; RRID: AB_2099233 |
| Annexin V-FITC | BD Biosciences | Cat# 556547; RRID: AB_2869082 |
|
| ||
| Biological Samples | ||
|
| ||
| Patient-derived bone marrow samples | Jerome Lipper Multiple Myeloma Center – Dana Farber Cancer Institute | N/A |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Propidium Iodide staining solution | BD Biosciences | Cat# 556463 |
| dBET6 | J Bradner’s lab | N/A |
| MZ-1 | Tocris Bioscience | Cat# 61545 |
| ARV-771 | MedChemexpress | Cat# HY-100972 |
| JQ1 | J. Bradner’s lab | N/A |
| Bortezomib | Thermo Fisher Scientific | Cat# 507419 |
| Thal-SNS-032 | N Gray’s lab | N/A |
| HM30181 | EMD Millipore | Cat# 533794 |
| Human recombinant interleukin 6 (IL6) | Thermo Fisher Scientific | Cat# 10395HNAE25 |
| Beetle Luciferin, Potassium Salt | Promega | Cat# E1605 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat# G7572 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| Blood & Cell Culture DNA Midi Kit | QIAGEN | Cat# 13343 |
| Blood & Cell Culture DNA Maxi Kit | QIAGEN | Cat# 13362 |
| QIAquick Gel Extraction Kit | QIAGEN | Cat# 28706 |
| QIamp DNA Mini Kit | QIAGEN | Cat# 51304 |
|
| ||
| Deposited Data | ||
|
| ||
| Gene Expression Omnibus | GSE162205 | N/A |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| MM.1S | ATCC | Cat# CRL-2974 |
| RPMI-8226 | DSMZ | Cat# ACC-538 |
| OPM-1 | Teru Hideshima, Anderson Lab, DFCI | N/A |
| OPM-2 | DSMZ | Cat# ACC-50 |
| JJN3 | DSMZ | Cat# ACC-541 |
| L363 | DSMZ | Cat# ACC-49 |
| AMO-1 | DSMZ | Cat# ACC-538 |
| OCI-My5 | Ontario Cancer Institute (OCI); Toronto; Canada | N/A |
| KMS-20 | JCRB | JCRB1196 |
| KMS-26 | JCRB | JCRB1187 |
| KMS-27 | JCRB | JCRB1188 |
| LP-1 | DSMZ | Cat# ACC-41 |
| HS27A | ATCC | Cat# 50188912FP |
| OPM-2.shRNA.CRBN and non-targeting control | K Stewart’s lab | N/A |
| KMS11.shRNA.CRBN and non-targeting control | K Stewart’s lab | N/A |
| MM.1S CRBN−/−(CRISPR-knockout) | W Kaelin’s lab | N/A |
| MM.1S-Cas9+ | B Ebert’s lab | N/A |
| KMS11-Cas9+ | Broad Institute, MIT | N/A |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | Cat# 005557 |
|
| ||
| Oligonucleotides | ||
|
| ||
| List of primers – see Table S1 | IDT | N/A |
| List of sgRNAs for single-gene knock-out – see Table S2 | IDT | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| lentiCas9-Blast | Addgene | RRID: Addgene_52962 |
| lentiGuide-Puro | Addgene | RRID: Addgene_52963 |
| psPAX2 | Addgene | RRID: Addgene_12260 |
| pMD2.G | Addgene | RRID: Addgene_12259 |
| GeCKO v2 human sgRNA library | Addgene | Zhang Lab, Broad Institute of MIT and Harvard |
| Lenti dCAS9-VP64_Blast | Addgene | RRID: Addgene_61425 |
| pLVX-hyg-sgRNA1 | Takara | Cat# 632630 |
| Brunellohuman sgRNA library | Addgene | RRID: Addgene_73179 |
| Human CRISPR Activation Pooled sgRNA Library (Calabrese library) | Addgene | RRID: Addgene_61425 |
|
| ||
| Software and Algorithms | ||
|
| ||
| MAGeCK | Li et al., 2014 | https://sourceforge.net/projects/mageck/ |
| PRISM 6 | GraphPad | https://www.graphpad.com |
| FlowJo V9.7.6 | Tree Star | https://www.flowjo.com/ |
| Biorender | https://www.biorender.com/ | |
Highlights.
PROTAC resistance via disruption of rather than adaptation to oncoprotein degradation
PROTACs using different E3s/CRLs: resistance via similar pathways but different genes
Result of using two PROTACs depends on E3, target, and sequential versus concurrent use
E3s essential for and highly expressed in tumor cells are useful for future PROTACs
ACKNOWLEDGMENTS
The authors would like to thank William G. Kaelin (DFCI) and Cory Johannessen (Broad Institute) for insightful scientific comments on this work and Jeffrey D. Sorrell (DFCI) for his contribution to the organizational planning for this study. This work was supported by grants NIH R01 CA050947 (to C.S.M.), CA179483 (to C.S.M. and N.S.G.), CA196664 (to C.S.M. and R.W.J.G.), CA180475 (to J.D.L. and C.S.M.), and U01 CA225730 (to C.S.M.), and U01CA176058 (to W.C.H.) and by the de Gunzburg Myeloma Research Fund (to C.S.M. and P.G.R.), the Ludwig Center at Harvard (to C.S.M.), the International Myeloma Foundation (to R.S. and G.M.M.), the Lauri Strauss Leukemia Foundation (to R.S.), a Leukemia and Lymphoma Society (LLS) Scholar Award (to C.S.M.), the LLS Translational Research Program (to C.S.M. and R.W.J.G.), the LLS Quest for Cure Program (to C.S.M. and R.W.J.G.), the Multiple Myeloma Research Foundation (MMRF) Answer Fund (to C.S.M., J.D.L., and L.H.B.), the MMRF Translational Network of Excellence (to C.S.M.), an MMRF Epigenetics Program Project grant (to J.D.L. and C.S.M.), the Shawna Ashlee Corman Investigatorship in Multiple Myeloma Research (to C.S.M.), the Cobb Family Myeloma Research Fund (to C.S.M.), the Chambers Family Advanced Myeloma Research Fund (to C.S.M. and P.G.R.), Department of Defense grants W81XWH-15–1-0012 (to C.S.M.) and W81XWH-15–1-0013 (to A.C.C.), the American-Australian Association (to G.M.M), Associazione Italiana per la Ricerca sul Cancro (to S.G.), and the Claudia Adams Barr Program in Innovative Basic Cancer Research at Dana-Farber Cancer Institute (to M.S., E.D., and C.S.M.). D.L.B. was a Merck Fellow of the Damon Runyon Cancer Research Foundation (DRG219614). Q.L.S. was supported by award T32GM007753 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. RPPA analyses were conducted by the Functional Proteomics RPPA Core facility, which is supported by MD Anderson Cancer Center support grant 5 P30 CA016672–40.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108532.
DECLARATION OF INTERESTS
F.V. receives research funding from Novo Ventures. P.G.R. reports research support from Oncopeptides, Celgene/BMS, and Takeda and is an advisory committee member for Karyopharm, Oncopeptides, Celgene/BMS, Takeda, Janssen, Sanofi, Secura Bio, and GSK. E.S.F. is a founder, science advisory board member, and equity holder in Civetta, Jengu (board member), and Neo-morph; an equity holder in C4; and a consultant to Astellas, Novartis, Deerfield, and EcoR1. The Fischer lab receives or has received research funding from Novartis, Astellas, Ajax, and Deerfield. N.P.K. is a consultant to Epiphanes, Inc. W.C.H. is a consultant for Thermo Fisher, Solvasta, MPM Capital, Tyra Biosciences, Jubilant Therapeutics, Frontier Medicines, RAPPTA Therapeutics, and Parexel and is a founder of and advisor to KSQ Therapeutics. L.H.B. receives research funding from AstraZeneca and is a consultant for AstraZeneca and Genentech. B.L.E. has received research funding from Celgene and Deerfield. He has received consulting fees from GRAIL, and he serves on the scientific advisory boards for and holds equity in Skyhawk Therapeutics and Exo Therapeutics. J.E.B. is an author on United States patent applications licensed from the Dana-Farber Cancer Institute to C4 Therapeutics (TPD) and Tensha Therapeutics (now Roche; BRD4 inhibition). He is now an executive of and shareholder in Novartis AG. N.S.G. is a scientific founder, member of the scientific advisory board and equity holder in C4 Therapeutics, Syros Pharmaceuticals, Petra Pharmaceuticals, Ravenna, Inception, Allorion, Jengu, and Soltego (board member) and is the inventor on intellectual property licensed to these entities. R.S., G.M.M., S.G., R.d.M.S., and C.S.M. are authors on a patent application on the use of degraders. C.S.M. also discloses consultant/honoraria from Fate Therapeutics, Ionis Pharmaceuticals and FI-MECS; employment of a relative with Takeda; and research funding from Janssen/Johnson & Johnson, TEVA, EMD Serono, Abbvie, Arch Oncology, Karyopharm, Sanofi, and Nurix.
REFERENCES
- Bindra RS, Vasselli JR, Stearman R, Linehan WM, and Klausner RD (2002). VHL-mediated hypoxia regulation of cyclin D1 in renal carcinoma cells. Cancer Res 62, 3014–3019. [PubMed] [Google Scholar]
- Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, et al. (2015). Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol 11, 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S, Wang J, Hamman BD, Ishchenko A, and Crews CM (2018). Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol 25, 78–87.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DL, Raina K, Darricarrere N, Hines J, Gustafson JL, Smith IE, Miah AH, Harling JD, and Crews CM (2015). HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol 10, 1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP, Toure M, Dong H, Qian Y, Wang J, et al. (2018). The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol 25, 67–77.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement K, Rees H, Canver MC, Gehrke JM, Farouni R, Hsu JY, Cole MA, Liu DR, Joung JK, Bauer DE, and Pinello L (2019). CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol 37, 224–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubiel W (2009). Resolving the CSN and CAND1 paradoxes. Mol. Cell 35, 547–549. [DOI] [PubMed] [Google Scholar]
- Fischer ES, Scrima A, Böhm K, Matsumoto S, Lingaraju GM, Faty M, Yasuda T, Cavadini S, Wakasugi M, Hanaoka F, et al. (2011). The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 147, 1024–1039. [DOI] [PubMed] [Google Scholar]
- Foulk B, Schaffer M, Gross S, Rao C, Smirnov D, Connelly MC, Chaturvedi S, Reddy M, Brittingham G, Mata M, et al. ; MMRF CoMMpass Network (2018). Enumeration and characterization of circulating multiple myeloma cells in patients with plasma cell disorders. Br. J. Haematol 180, 71–81. [DOI] [PubMed] [Google Scholar]
- Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ, Zengerle M, and Ciulli A (2017). Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol 13, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper AM, Qiao X, Kim J, Zhang L, DeSimone MC, Rathmell WK, and Wan Y (2012). Regulation of KLF4 turnover reveals an unexpected tissue-specific role of pVHL in tumorigenesis. Mol. Cell 45, 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi AK, Mendy D, Waldman M, Chen G, Rychak E, Miller K, Gaidarova S, Ren Y, Wang M, Breider M, et al. (2014). Measuring cereblon as a biomarker of response or resistance to lenalidomide and pomalidomide requires use of standardized reagents and understanding of gene complexity. Br. J. Haematol 164, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gechijian LN, Buckley DL, Lawlor MA, Reyes JM, Paulk J, Ott CJ, Winter GE, Erb MA, Scott TG, Xu M, et al. (2018). Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat. Chem. Biol 14, 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC, Xie Y, Williams NS, and Nijhawan D (2017). Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 356, eaal3755. [DOI] [PubMed] [Google Scholar]
- Heintel D, Rocci A, Ludwig H, Bolomsky A, Caltagirone S, Schreder M, Pfeifer S, Gisslinger H, Zojer N, Jäger U, and Palumbo A (2013). High expression of cereblon (CRBN) is associated with improved clinical response in patients with multiple myeloma treated with lenalidomide and dexamethasone. Br. J. Haematol 161, 695–700. [DOI] [PubMed] [Google Scholar]
- Hotton SK, and Callis J (2008). Regulation of cullin RING ligases. Annu. Rev. Plant Biol 59, 467–489. [DOI] [PubMed] [Google Scholar]
- Kibel A, Iliopoulos O, DeCaprio JA, and Kaelin WG Jr. (1995). Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science 269, 1444–1446. [DOI] [PubMed] [Google Scholar]
- Kowalski J, Dwivedi B, Newman S, Switchenko JM, Pauly R, Gutman DA, Arora J, Gandhi K, Ainslie K, Doho G, et al. (2016). Gene integrated set profile analysis: a context-based approach for inferring biological endpoints. Nucleic Acids Res 44, e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, et al. (2014). Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebraud H, and Heightman TD (2017). Protein degradation: a validated therapeutic strategy with exciting prospects. Essays Biochem 61, 517–527. [DOI] [PubMed] [Google Scholar]
- Lebraud H, Wright DJ, Johnson CN, and Heightman TD (2016). Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci 2, 927–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao W, Akbani R, Liu W, Ju Z, Ling S, Vellano CP, Roebuck P, Yu Q, Eterovic AK, et al. (2017). Characterization of Human Cancer Cell Lines by Reverse-phase Protein Arrays. Cancer Cell 31, 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonergan KM, Iliopoulos O, Ohh M, Kamura T, Conaway RC, Conaway JW, and Kaelin WG Jr. (1998). Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol. Cell. Biol 18, 732–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong KK, Bradner JE, and Kaelin WG Jr. (2014). The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, and Crews CM (2015). Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol 22, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- Matthews GM, Mehdipour P, Cluse LA, Falkenberg KJ, Wang E, Roth M, Santoro F, Vidacs E, Stanley K, House CM, et al. (2015). Functional-genetic dissection of HDAC dependencies in mouse lymphoid and myeloid malignancies. Blood 126, 2392–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor-Ruiz C, Jaeger MG, Bauer S, Brand M, Sin C, Hanzl A, Mueller AC, Menche J, and Winter GE (2019). Plasticity of the Cullin-RING Ligase Repertoire Shapes Sensitivity to Ligand-Induced Protein Degradation. Mol. Cell 75, 849–858.e8. [DOI] [PubMed] [Google Scholar]
- McMillin DW, Delmore J, Weisberg E, Negri JM, Geer DC, Klippel S, Mitsiades N, Schlossman RL, Munshi NC, Kung AL, et al. (2010). Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nat. Med 16, 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillin DW, Delmore J, Negri J, Ooi M, Klippel S, Miduturu CV, Gray NS, Richardson PG, Anderson KC, Kung AL, and Mitsiades CS (2011). Microenvironmental influence on pre-clinical activity of polo-like kinase inhibition in multiple myeloma: implications for clinical translation. PLoS ONE 6, e20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillin DW, Delmore J, Negri JM, Vanneman M, Koyama S, Schlossman RL, Munshi NC, Laubach J, Richardson PG, Dranoff G, et al. (2012a). Compartment-Specific Bioluminescence Imaging platform for the high-throughput evaluation of antitumor immune function. Blood 119, e131–e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillin DW, Jacobs HM, Delmore JE, Buon L, Hunter ZR, Monrose V, Yu J, Smith PG, Richardson PG, Anderson KC, et al. (2012b). Molecular and cellular effects of NEDD8-activating enzyme inhibition in myeloma. Mol. Cancer Ther 11, 942–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvar B, Raghuram V, Pisitkun T, Sarkar A, and Knepper MA (2016). Comprehensive database of human E3 ubiquitin ligases: application to aqua-porin-2 regulation. Physiol. Genomics 48, 502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller A, Asmann Y, Cattaneo L, Braggio E, Keats J, Auclair D, Lonial S, Russell SJ, and Stewart AK; MMRF CoMMpass Network (2017). High somatic mutation and neoantigen burden are correlated with decreased progression-free survival in multiple myeloma. Blood Cancer J 7, e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiades CS, Ocio EM, Pandiella A, Maiso P, Gajate C, Garayoa M, Vilanova D, Montero JC, Mitsiades N, McMullan CJ, et al. (2008). Aplidin, a marine organism-derived compound with potent antimyeloma activity in vitro and in vivo. Cancer Res 68, 5216–5225. [DOI] [PubMed] [Google Scholar]
- Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, Leggett AL, Erb MA, Lawlor MA, Souza A, et al. (2018). The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol 14, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbold A, Matthews GM, Bots M, Cluse LA, Clarke CJ, Banks KM, Cullinane C, Bolden JE, Christiansen AJ, Dickins RA, et al. (2013). Molecular and biologic analysis of histone deacetylase inhibitors with diverse specificities. Mol. Cancer Ther 12, 2709–2721. [DOI] [PubMed] [Google Scholar]
- Nguyen VN, Huang KY, Weng JT, Lai KR, and Lee TY (2016). UbiNet: an online resource for exploring the functional associations and regulatory networks of protein ubiquitylation. Database (Oxford) 2016, baw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka N, Morita Y, Nagai K, Shimokawa K, Ujikawa O, Fujimori I, Ito M, Hayase Y, Okuhira K, Shibata N, et al. (2018). Derivatization of inhibitor of apoptosis protein (IAP) ligands yields improved inducers of estrogen receptor a degradation. J. Biol. Chem 293, 6776–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson CM, Jiang B, Erb MA, Liang Y, Doctor ZM, Zhang Z, Zhang T, Kwiatkowski N, Boukhali M, Green JL, et al. (2018). Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol 14, 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, Wang J, Chen X, Dong H, Siu K, et al. (2016). PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 113, 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb CM, Contreras JI, Kour S, Taylor MA, Abid M, Sonawane YA, Zahid M, Murry DJ, Natarajan A, and Rana S (2017). Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem. Commun. (Camb.) 53, 7577–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenz DT, Fiskus W, Qian Y, Manshouri T, Rajapakshe K, Raina K, Coleman KG, Crew AP, Shen A, Mill CP, et al. (2017). Novel BET protein proteolysis-targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia 31, 1951–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiedel M, Herp D, Hammelmann S, Swyter S, Lehotzky A, Robaa D, Olah J, Ovadi J, Sippl W, and Jung M (2017). Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem 61, 482–491. [DOI] [PubMed] [Google Scholar]
- Schneekloth AR, Pucheault M, Tae HS, and Crews CM (2008). Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett 18, 5904–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiyanov P, Nag A, and Raychaudhuri P (1999). Cullin 4A associates with the UV-damaged DNA-binding protein DDB. J. Biol. Chem 274, 35309–35312. [DOI] [PubMed] [Google Scholar]
- Sievers QL, Gasser JA, Cowley GS, Fischer ES, and Ebert BL (2018). Genome-wide screen identifies cullin-RING ligase machinery required for lenalidomide-dependent CRL4CRBN activity. Blood 132, 1293–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Fiskus W, Qian Y, Rajapakshe K, Raina K, Coleman KG, Crew AP, Shen A, Saenz DT, Mill CP, et al. (2018). BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia 32, 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnell AS, Stewart GS, Grand RJ, Rookes SM, Martin A, Yamano H, Elledge SJ, and Gallimore PH (2005). The APC/C and CBP/p300 cooperate to regulate transcription and cell-cycle progression. Nature 438, 690–695. [DOI] [PubMed] [Google Scholar]
- Uehara T, Minoshima Y, Sagane K, Sugi NH, Mitsuhashi KO, Yamamoto N, Kamiyama H, Takahashi K, Kotake Y, Uesugi M, et al. (2017). Selective degradation of splicing factor CAPERa by anticancer sulfonamides. Nat. Chem. Biol 13, 675–680. [DOI] [PubMed] [Google Scholar]
- Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM, Proszek PZ, Johnson DC, Kaiser MF, Melchor L, et al. (2015). Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J. Clin. Oncol 33, 3911–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Wen H, Li Y, Lyu J, Xi Y, Hoshii T, Joseph JK, Wang X, Loh YE, Erb MA, et al. (2017). ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 543, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Krall EB, Aguirre AJ, Kim M, Widlund HR, Doshi MB, Sicinska E, Sulahian R, Goodale A, Cowley GS, et al. (2017). ATXN1L, CIC, and ETS Transcription Factors Modulate Sensitivity to MAPK Pathway Inhibition. Cell Rep 18, 1543–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welford SM, Dorie MJ, Li X, Haase VH, and Giaccia AJ (2010). Renal oxygenation suppresses VHL loss-induced senescence that is caused by increased sensitivity to oxidative stress. Mol. Cell. Biol 30, 4595–4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, and Bradner JE (2015). DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, Reyes JM, di Iulio J, Souza A, Ott CJ, et al. (2017). BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol. Cell 67, 5–18.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurz RP, Dellamaggiore K, Dou H, Javier N, Lo MC, McCarter JD, Mohl D, Sastri C, Lipford JR, and Cee VJ (2018). A “Click Chemistry Platform” for the Rapid Synthesis of Bispecific Molecules for Inducing Protein Degradation. J. Med. Chem 61, 453–461. [DOI] [PubMed] [Google Scholar]
- Xu L, Chen Y, Mayakonda A, Koh L, Chong YK, Buckley DL, Sandanaraj E, Lim SW, Lin RY, Ke XY, et al. (2018). Targetable BET proteins-and E2F1-dependent transcriptional program maintains the malignancy of glioblastoma. Proc. Natl. Acad. Sci. USA 115, E5086–E5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AP, Schlisio S, Minamishima YA, Zhang Q, Li L, Grisanzio C, Signoretti S, and Kaelin WG Jr. (2008). VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat. Cell Biol 10, 361–369. [DOI] [PubMed] [Google Scholar]
- Zengerle M, Chan KH, and Ciulli A (2015). Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol 10, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Lee HC, Shirazi F, Baladandayuthapani V, Lin H, Kuiatse I, Wang H, Jones RJ, Berkova Z, Singh RK, et al. (2018). Protein targeting chimeric molecules specific for bromodomain and extra-terminal motif family proteins are active against pre-clinical models of multiple myeloma. Leukemia 32, 2224–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Hu J, Xu F, Chen Z, Bai L, Fernandez-Salas E, Lin M, Liu L, Yang CY, Zhao Y, et al. (2018). Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J. Med. Chem 61, 462–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YX, Braggio E, Shi CX, Bruins LA, Schmidt JE, Van Wier S, Chang XB, Bjorklund CC, Fonseca R, Bergsagel PL, et al. (2011). Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 118, 4771–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YX, Kortuem KM, and Stewart AK (2013). Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leuk. Lymphoma 54, 683–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. (2011). RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data and code used in this study are available via the Lead Contact upon reasonable request. RNA sequencing data supporting the current study have been deposited to Gene Expression Omnibus with accession number GEO: GSE162205.