

Abstract
Heat shock proteins (Hsps) are molecular chaperones that also play important roles in activation of the heat shock response (HSR). The HSR is an evolutionary conserved and protective mechanism that is used to counter abnormal physiological conditions, stressors, and disease states, such as those exemplified in cancer and/or neurodegeneration. In normal cells, heat shock factor-1 (HSF-1), the transcription factor that regulates the HSR, remains in a dormant multi-protein complex that is formed upon association with chaperones (Hsp90, Hsp70 etc.), co-chaperones, and client proteins. However, under cellular stress, HSF-1 dissociates from Hsp90 and induces the transcriptional upregulation of Hsp70 to afford protection against the encountered cellular stress. As a consequence of both peripheral and central neuropathies, cellular stress occurs and results in the accumulation of unfolded and/or misfolded proteins, which can be counterbalanced by activation of the HSR. Since Hsp90 is the primary regulator of the HSR, modulation of Hsp90 by small molecules represents an attractive therapeutic approach against both peripheral and central neuropathies.
Keywords: Neuropathy, Hsp90, heat shock response (HSR), HSF-1, chaperones, neurodegeneration, diabetic peripheral neuropathy (DPN)
Introduction
The Heat Shock Response (HSR) is a major cellular stress relief pathway that has been evolutionarily conserved across various species to refold denatured proteins1–11. Under stressful conditions (such as exposure to heat, toxic chemicals, radiation, etc.) the transcription factor, Heat Shock Factor-1 (HSF-1), activates the transcription of genes that encode for various chaperones (Hsps) that coordinate with one another to reverse cellular stress and refold denatured proteins12,13. Under normal conditions, chaperones maintain cellular homeostasis by folding nascent polypeptides into their correct three-dimensional conformations, as well as misfolded proteins into functional proteins14. However, Hsps also facilitate the clearance of misfolded proteins by chaperoning those substrates to the ubiquitin-proteasome pathway for subsequent degradation15,16. Along with these functions, Hsps also play an important role in autophagy and lysosomal degradation, which will be expanded upon later. Many pathological conditions, such as diabetes, cancer, dyslipidemia, neurodegenerative diseases, and aging can lead to a dysfunctional HSR and consequently, a loss of HSF-1 activation17,18. Researchers have also shown that diminished levels of Hsps (a direct result of deactivated HSF-1) can give rise to additional consequences of diabetes18,19, such as neuropathy19–22, retinopathy23,24, nephropathy25,26, cardiovascular diseases27,28, etc. In contrast, it has been shown that the induction of specific Hsps by small molecules can elicit neuroprotection21,22. This review will focus on the modulation of Hsps as a therapeutic option to treat both central and peripheral neuropathies.
Although some molecular chaperones are Hsps, not all chaperones are induced by the heat shock response or other cellular stresses29. The Heat shock protein family is categorized by molecular weight (in kDa); large heat shock proteins (L-Hsps) such as Hsp100, Hsp90, Hsp70, Hsp4030 and small heat shock proteins (S-Hsps) such as those with molecular weights between 12 – 43 kDa31. These molecular chaperones are also subcategorized into individual isoforms/paralogs32–34. Patients with neuropathy or related neurological disorders are known to express lower levels of both L-Hsps and S-Hsps, whereas the activation and/or overexpression of molecular chaperones has proven beneficial in several of these disease states31,35. For example, the upregulation of Hsp70 has been shown to reverse Diabetic Peripheral Neuropathy (DPN) by refolding aggregated and damaged proteins, which ultimately leads to the recovery of mitochondrial bioenergetics, dampening of pro-inflammatory cascades, and the reinnervation of nerve fibers35,36. Furthermore, patients with high Hsp27 levels manifest better nerve function as compared to those with lower levels37,38.
In stress-free normal cells, Hsp90 suppresses the transcriptional activity of HSF-1 by existing as an Hsp90-HSF-1 complex (Figure 1)39–42. However, under stressful conditions, such as glycemic insult, HSF-1 dissociates from Hsp90, undergoes trimerization, and enters the nucleus to induce the expression of antioxidant proteins and chaperones, such as Hsp7039,43. This response leads to the refolding of misfolded proteins and/or their clearance to alleviate cell stress44–49. In recent years, several small molecule Hsp90 modulators have been developed that can disrupt the Hsp90-HSF-1 complex.
Figure 1.
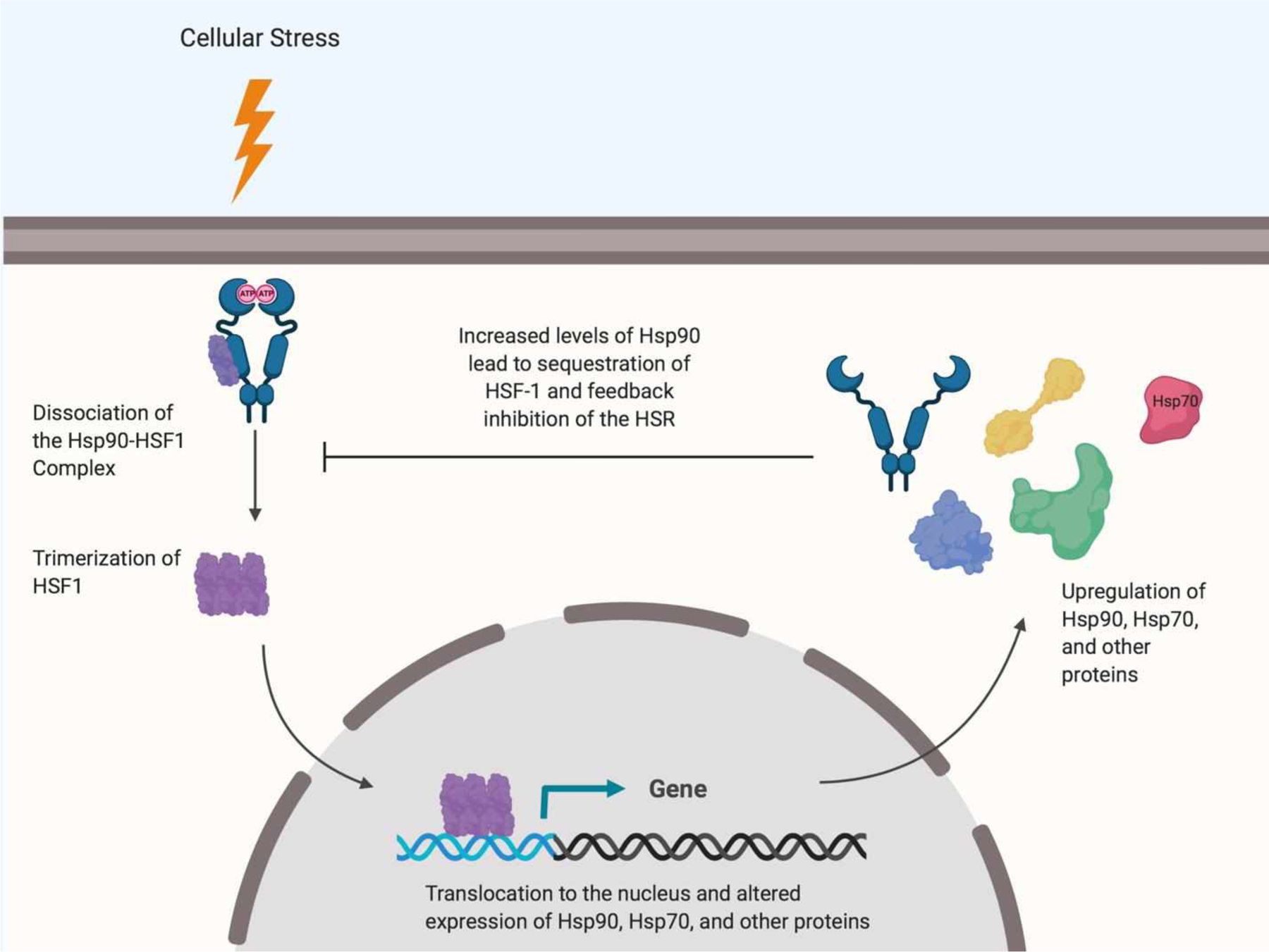
Mechanism of HSF-1-mediated transcriptional activation
Modulation of Hsp90 to Induce the HSR
The Hsp90 dimer is comprised of three domains: 1) An N-terminal ATP-binding domain (25 kDa), 2) a co-chaperone and client protein binding middle domain (33 kDa), and 3) a C-terminal dimerization domain (12 kDa) that is essential for maintaining the active homodimer (Figure 2)50,51. Hsp90 forms a series of complexes with co-chaperones, Hsp70, and client proteins in order to fulfill its chaperone activity. An interesting phenomenon that makes Hsp90 an intriguing and druggable target is that Hsp90 inhibitors have been shown to act as either cytotoxic or cytoprotective agents, depending on cell type and the cellular stressor18,21,22.
Figure 2.
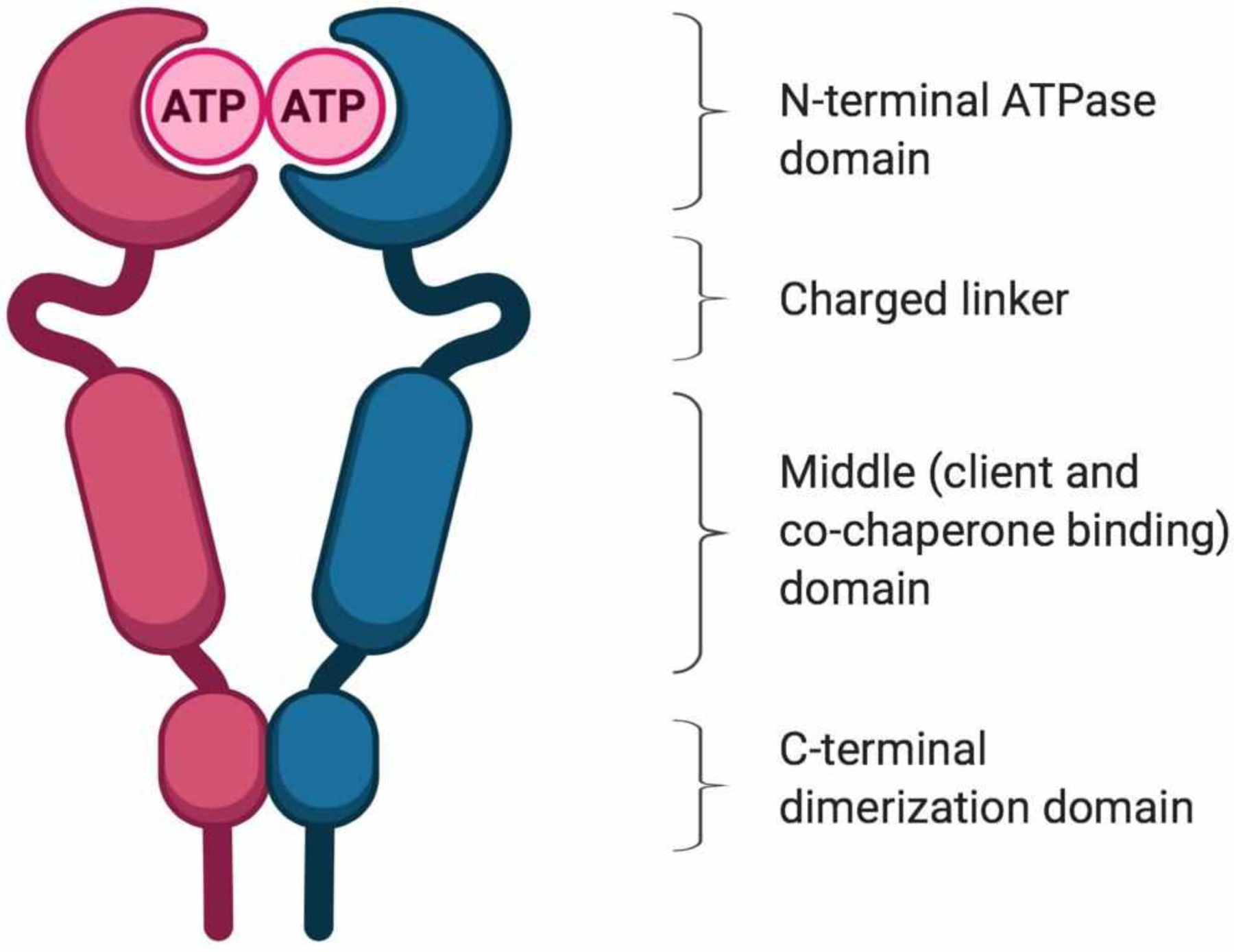
Structure of the Hsp90 homodimer
Hsp90 N-terminal inhibitors:
As mentioned earlier, the Hsp90 N-terminal domain contains an ATP-binding site that is responsible for the hydrolysis of ATP, which provides the energy necessary for the folding and release of Hsp90-dependent client proteins (Figure 3). In 2003, Kamal and co-workers demonstrated that Hsp90 remains in an uncomplexed and homodimeric state in normal cells; however in tumor cells, Hsp90 resides in a heteroprotein complex that manifests ~200-fold higher affinity for ATP than the homodimer alone52. A similar phenomenon was later described for Hsp90 in Alzheimer’s disease, wherein high-affinity Hsp90-CHIP (carboxy terminus of Hsp70–interacting protein) complexes were found to exist, which is in contrast to normal tissue53. In recent years, extensive work to characterize these high-affinity Hsp90 complexes that are responsible for cancer cell survival and disease progression has been pursued. While the normal function of the cellular proteome is maintained by an array of chaperones and enzymes, the chronic disease state manifests an epigenetically different chaperome in stressed cells54. In fact, the term “epichaperome” has been proposed to describe the unique heteroprotein complex present in stressed cells55. As a result, Hsp90 N-terminal inhibitors represent an attractive therapeutic opportunity that may impart selectivity for stressed cells as a consequence of these heteroprotein complexes and their differential binding affinities.
Figure 3.
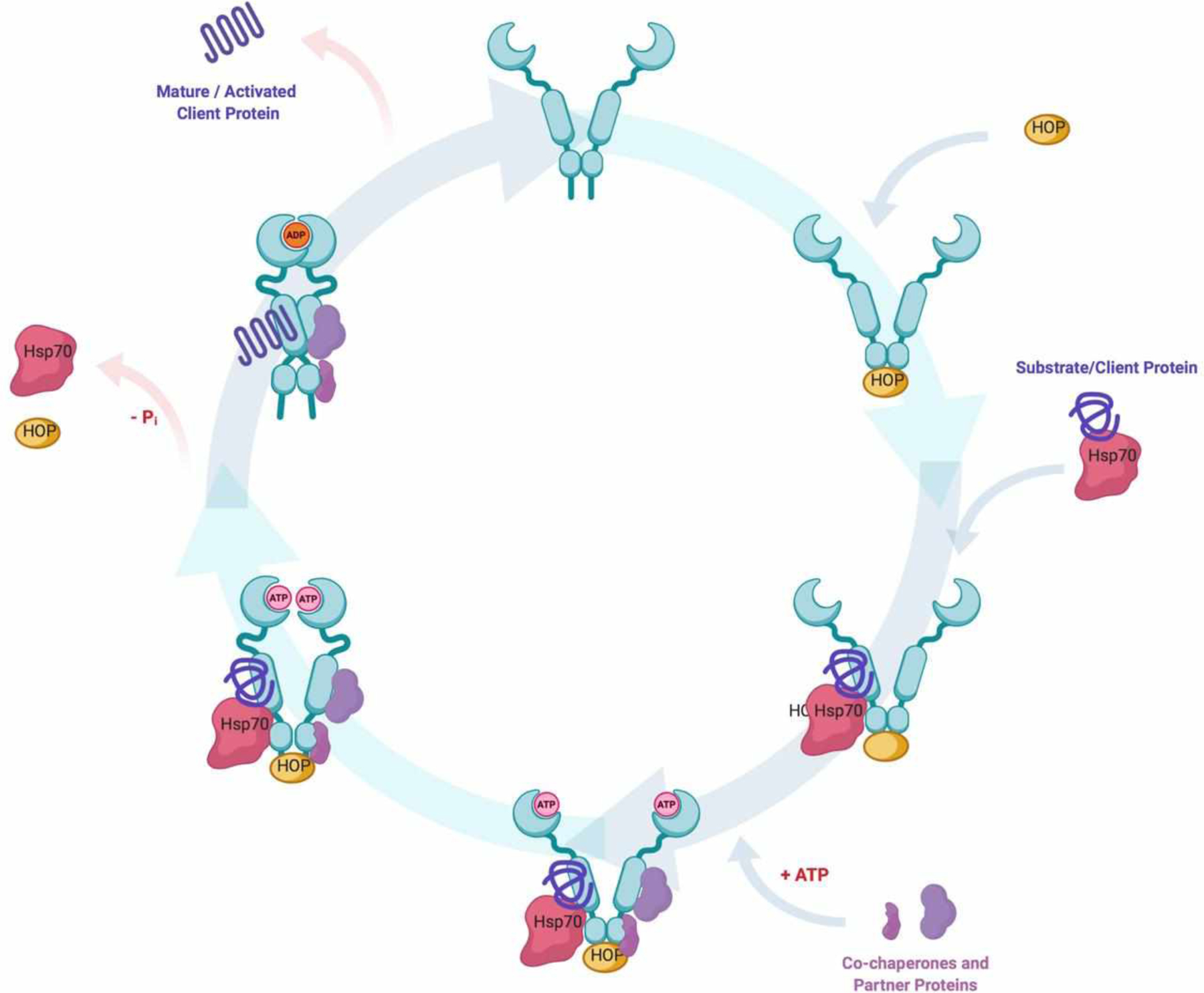
The Hsp90 ATPase/protein folding cycle
N-terminal inhibitors have demonstrated the ability to afford neuroprotection against diseases wherein neurodegeneration results from a protein folding disorder. For example, the Hsp90 N-terminal inhibitors 17-AAG56 (17-(Allylamino)-17-allylamino geldanamycin) (Figure 4) and several synthetic57,58, semi-synthetic59 and bio-engineered60 analogs of GDA (geldanamycin) (Figure 4) reverse the formation of β-amyloid and tau aggregation, while inhibiting the binding of tau to microtubules61. The increased levels of Hsp70 induced by these compounds prevent neuronal apoptosis, a common phenomenon found in patients with Alzheimer’s, Parkinson’s, and Huntington’s diseases62.
Figure 4.
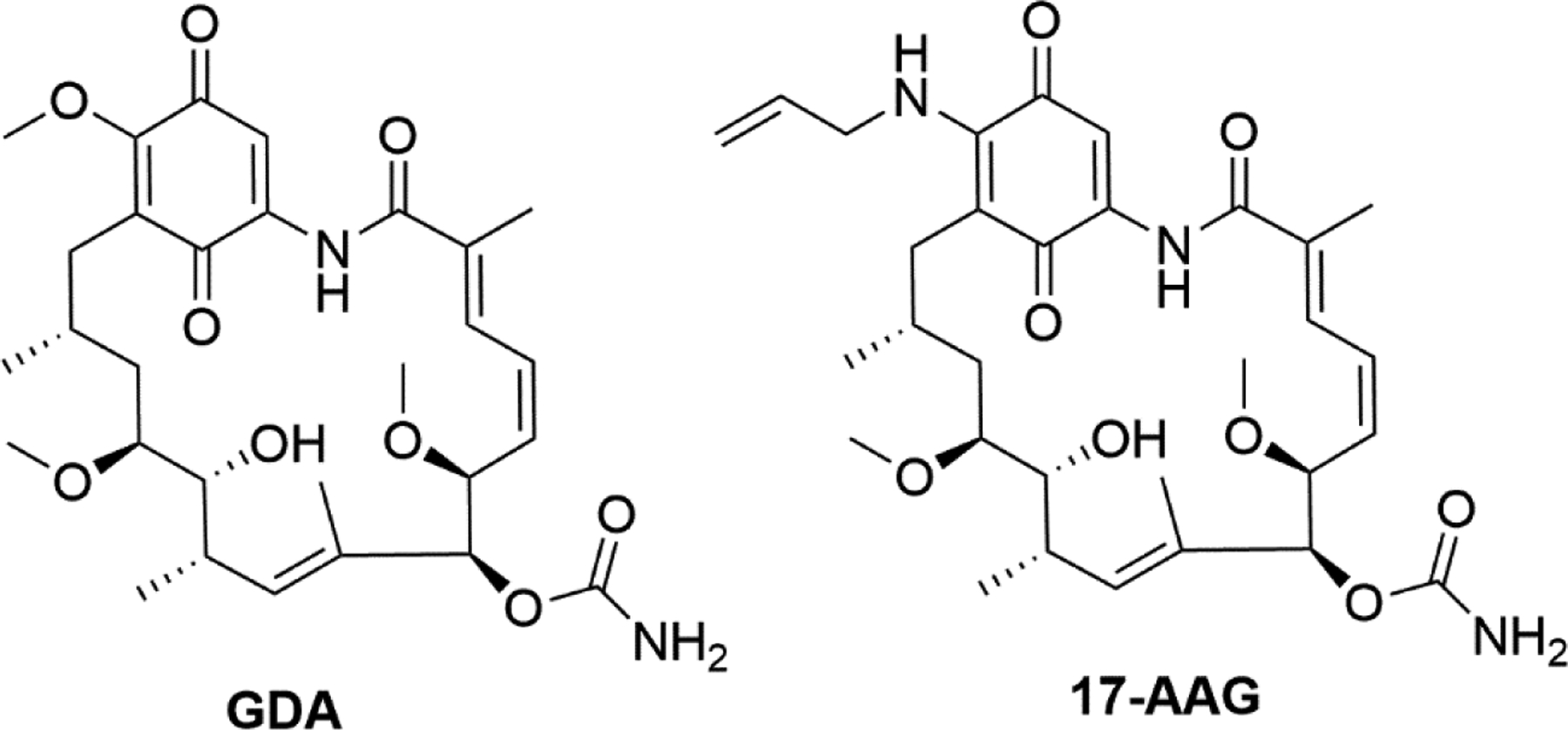
Structures of select N-terminal Hsp90 inhibitors
There are two isoforms of Hsp90 that reside in the cytosol and these include Hsp90α, which is the inducible isoform, and Hsp90β, which is constitutively expressed63. Recent studies have demonstrated that Hsp90α and Hsp90β exhibit differential binding affinities toward both client proteins and N-terminal inhibitors. For example, one resorcinol-based inhibitor, STA-9090, manifests higher affinity for Hsp90β than for Hsp90α, and it is well known that some client proteins are dependent upon a particular isoform for their conformational maturation as well64,65. In fact, it has been shown that Hsp90α binds HSF-1 with higher affinity, suggesting that Hsp90α inhibitors may be more effective at induction of the HSR, which could then manifest cytoprotective and/or neuroprotective activity65.
Hsp90 C-terminal inhibitors:
The size of the C-terminal domain of Hsp90 is approximately one half of the N-terminal domain and allosterically facilitates nucleotide exchange at the N-terminus, but does not exhibit ATPase activity66. This domain is primarily responsible for maintaining Hsp90’s dimeric form and coordinating interactions with Hsp90 partner proteins that contain a tetratricopeptide repeat (TPR)67,68. Despite the interest in Hsp90 function, no co-crystal structure of an inhibitor bound to the C-terminal domain has been solved. However, medicinal chemistry studies have shown that this region can be modulated by small molecules to promote cytotoxic or cytoprotective activities. In fact, two classes of novobiocin-based inhibitors have emerged for this domain. One class of compounds binds the Hsp90 C-terminal domain to disrupt interactions with Aha1, which then inhibits Aha1-stimulated ATPase activity69. For example, KU-174 (Figure 5) exhibits cytotoxicity against prostate cancer cell lines without induction of the HSR70. In contrast, the second class of C-terminal modulators induce the HSR without client protein degradation. This latter class of C-terminal modulators, such as A4, KU-32 and KU-596 (Figure 5), exhibit cytoprotective activities that are useful for the treatment of neurodegenerative disorders71,72. These distinct activities result from the existence of a large benzamide side chain that leads to cytotoxic activities, as opposed to the smaller acetamide side chain that manifests neuroprotective activity22. The C-terminal inhibitor, AEG3482 (Figure 5), an imidazothiadiazole sulfonamide, induces Hsp70 levels by activation of HSF-1. The increased levels of Hsp70 induced by AEG3482 prevent the induction of the c-jun N-terminal kinase (JNK) signaling cascade73, which is responsible for neuronal apoptosis62. KU-32, KU-596, and AEG3482 have undergone in vivo evaluation and have exhibited efficacy. The effect of KU-32 and KU-596 on neurons will be discussed.
Figure 5.
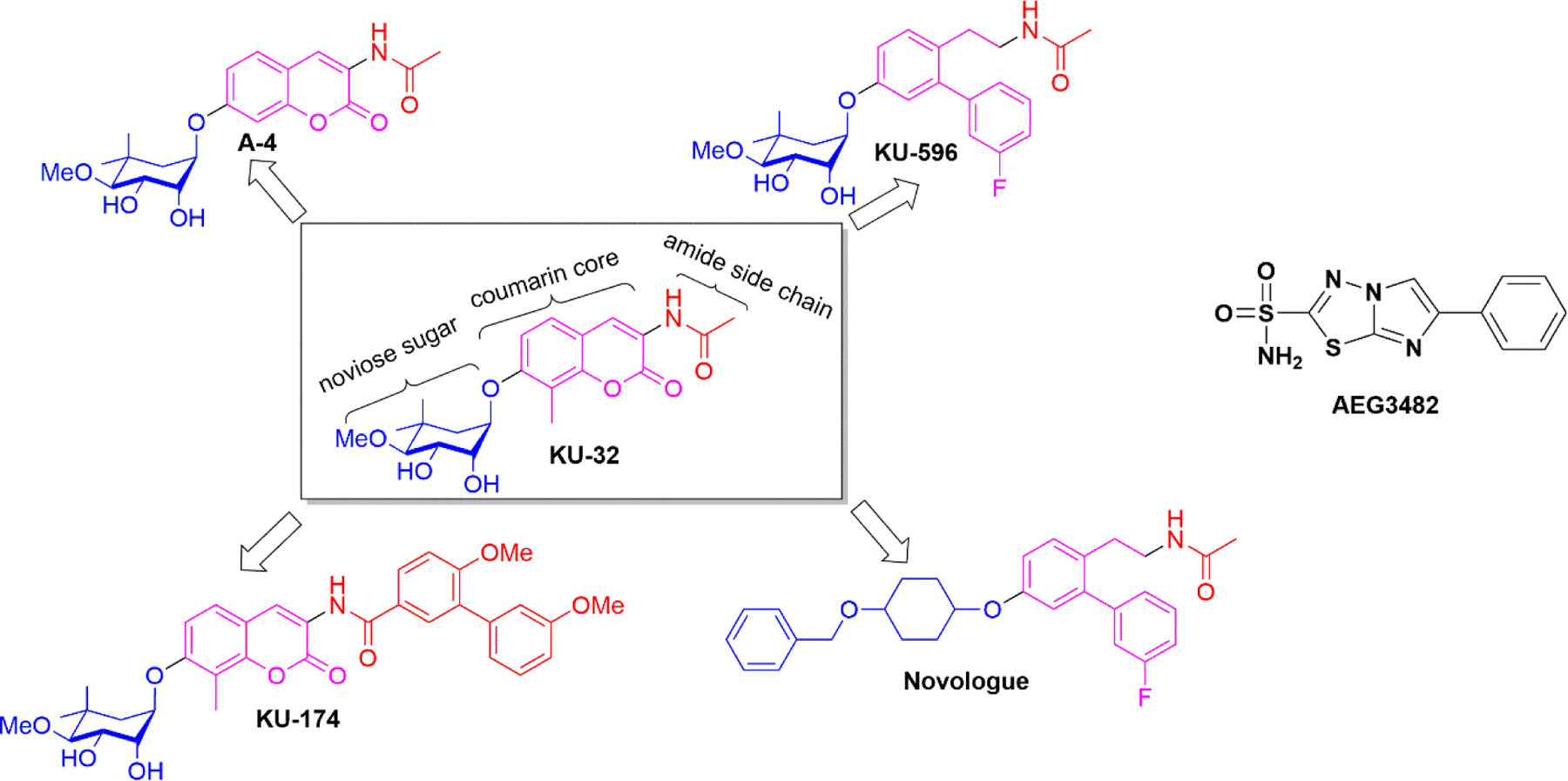
Structures of select C-terminal Hsp90 inhibitors
The middle domain:
While the Hsp90 C-terminal domain is responsible for binding proteins with TPR domains, the amphipathic middle domain is responsible for recognizing non-TPR co-chaperones and client protein substrates. Sato and co-workers were the first to demonstrate that the serine/threonine-specific protein kinase (Akt) binds Hsp90 at a location distinct from the N- or C-terminus74. Subsequent confirmation was achieved by solution of the co-crystal structure of the middle domain of Hsp90 in complex with Akt75. To our knowledge, no molecule that binds the Hsp90 middle domain and manifests neuroprotective activity has been reported to date.
The Nervous System
The nervous system plays an important role as a regulator of many bodily functions. It is divided into two major parts; the Central Nervous System (CNS) and the Peripheral Nervous System (PNS)76. The CNS consists of the brain and spinal cord, which receive information, coordinate function, and then influence other parts of our body. The PNS (consisting of nerves and ganglia) connects the limbs, organs and various parts of our bodies with the CNS. Signals from the brain and spinal cord are carried to the periphery by motor nerves, whereas sensory nerves relay the information from the periphery back to the brain. Together, the CNS acts as a “power house” and the PNS plays the role of “supply line and message carrier” as they work in concert with one another to maintain bodily functions77,78.
Based on a similar division of our nervous system, neuropathies are separated into two major classes as well; peripheral and central neuropathies (collectively called neurodegenerative diseases). The various pathological states and the roles played by Hsps in these diseases are described below as well as the small molecule modulators of Hsps that have demonstrated therapeutic potential.
Diabetic Peripheral Neuropathy (DPN)
According to the World Health Organization, 422 million people were living with diabetes around the globe in 2014. Secondary complications of diabetes mellitus include retinopathy, nephropathy, atherosclerosis and neuropathy18. Among these, neuropathy is a major complication that is present in ~50% of diabetic patients79. The manifestation of this neuropathy is diverse in nature. For example, one can have sensory numbness (reduced feeling or hypoalgesia in response to temperature change or pain), or in contrast, spontaneous sharp pain or hyperalgesia in the hands, feet and legs, as well as paresthesia and allodynia80. The latter is referred to as painful diabetic neuropathy (PDN), and is experienced by one third of diabetic neuropathy patients81. The most common phenotype manifested by ~75% of the diabetic neuropathy patients is a “change in sensation”81,82. This “change in sensation” occurs due to the neurodegeneration that begins at the distal ends of sensory neurons (axons) and progresses toward the proximal extremities when diabetes remains poorly managed for long periods of time.
Hyperglycemia, neuronal insulin deficiency or resistance, as well as dyslipidemia are the major contributors to DPN21. Other biochemical conditions can also cause nerve dysfunction and the degeneration of sensory fibers (unmyelinated C fibers or thinly myelinated Aδ sensory fibers) and aggravate DPN via oxidative/nitrosative stress, mitochondrial dysfunction, etc. Since the etiology of DPN is multifaceted and its presentation multi-symptomatic, a combinatorial therapeutic approach may be required, and at present, therapeutic options to treat DPN are limited to symptomatic relief22,83.
Managing blood glucose levels by controlling diet, exercise, medication, and insulin levels represent conventional approaches toward the management of DPN84. However, α-Lipoic acid (ALA) is an FDA approved therapeutic that can lessen some of the oxidative stress associated with DPN85. Other therapeutics under development include; 1) an aldose reductase inhibitor – ranirestat86,87, 2) a vascular endothelial growth factor gene transfer88, and 3) a protein kinase Cβ inhibitor – ruboxistaurin mesylate89–91. All of these therapeutic options have limitations, as they slow disease progression, but do not significantly reverse DPN pathology. Several reviews18,21,22,92–104 have been published that outline recent progress toward the elucidation of biochemical pathways that contribute to DPN pathogenesis, as well as some therapeutic strategies to modulate disease progression.
No direct correlation has been found between DPN and any specific misfolded protein; however, it is evident that hyperglycemia can induce protein misfolding by causing oxidative processes that lead to the modification of amino acid residues47,105,106. As such, experiments have confirmed that induction of the HSR can elicit a cytoprotective response in neurons against such glycemic/oxidative insults18,22. KU-32 (Figure 5) is a novel, novobiocin-based, C-terminal inhibitor that binds Hsp90 but does not disrupt Aha1-Hsp90 interactions. As a result, KU-32 induces the HSR and increases Hsp70 levels, which elicits a cytoprotective mechanism to overcome oxidative stress, mitochondrial degeneration and, as a result, affords neuroprotection against glycemic insult without altering blood glucose levels107–109. Another KU-32 inspired C-terminal inhibitor, KU-596 (Figure 5), has been developed to contain a meta-fluorinated biphenyl ring that replaces the coumarin core of KU-32. KU-596 works in a manner similar to KU-32 and also induces Hsp70 levels in hyperglycemic cells110–112. Both KU-32 and KU-596 were shown to restore sensory and motor neuron function in animal models of DPN. Notably, diabetic Hsp70 knockout (Hsp70 KO) mice were unresponsive to KU-596 treatment, highlighting the neuroprotective role played by Hsp70112 (Figure 6). KU-596 was licensed to Reata Pharmaceuticals and is currently awaiting Phase II clinical evaluation for the treatment of DPN. Mimics of the noviose sugar moiety present in KU-596 have also been investigated and several surrogates or “noviomimetics” have been synthesized and evaluated110,111. Recently, a benzyl containing novologue (Figure 5) was identified as the most potent Hsp70 inducer in a luciferase reporter assay111. Studies are now underway to further optimize this new class of compounds.
Figure 6.
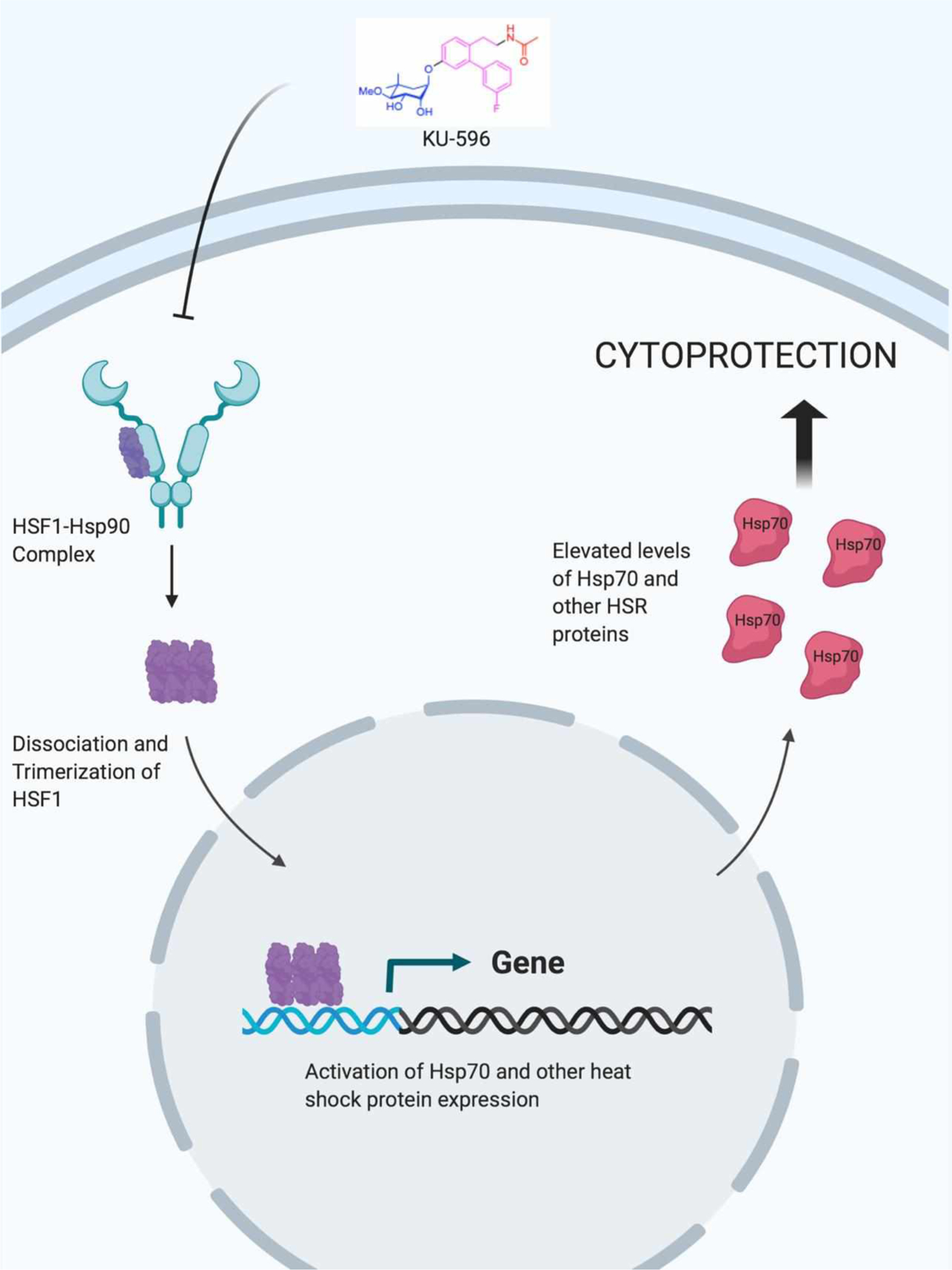
Mechanism of cytoprotection afforded by KU-596 elicitation of the HSR
KU-32 can also protect against 5-flurouracil (5-FU)-induced neuropathy. 5-FU is a commonly prescribed chemotherapeutic agent that is used against various cancers including breast, bowel, skin, stomach, oesophageal (gullet), and pancreatic113. Unfortunately, 5-FU causes chemobrain and cognitive impairment as a major adverse event114. Studies in rats revealed that 5-FU treatment along with KU-32 produced significant neuroprotection against 5-FU induced cognitive impairment115.
Central Nervous System Neuropathies / Neurodegeneration
The accumulation of misfolded proteins is a hallmark of several neurotoxic pathologies including Alzheimer’s disease (AD), Parkinson’s disease (PD), and other neurodegenerative diseases. Since Hsps regulate the folding, maturation, and clearance of more than 300 client proteins within the cell116,117, Hsps have emerged as promising therapeutic targets for the treatment of CNS disorders in which the aggregation of misfolded proteins leads to cellular stress, disruptions in signaling networks, and/or cell death. Microtubule-associated Protein Tau (MAPT, tau) is perhaps the most characterized and well known of the client proteins regulated by Hsps, as the aggregation of oligomeric tau drives the progression of a family of neurodegenerative syndromes collectively called tauopathies.
In 2002, Kakimura and coworkers discovered that Hsp90 levels are increased in both cytosolic and membranous fractions of AD brains and that Hsp90 colocalizes with amyloid plaques118; however, the significance of these findings and the contribution of Hsp90 to the pathology of AD and other tauopathies remained unresolved for several years. It has since been established that Hsp90 plays a role in the regulation of tau hyperphosphorylation as well as the seeding of tau fibrils through interactions with various co-chaperones, including FK506 Binding Protein 51 kDa (FKBP51, a peptidyl-prolyl cis-trans isomerase), and the Activator of Hsp90 ATPase Homolog 1 (Aha1, the only co-chaperone known to stimulate Hsp90’s ATPase activity). Levels of both Aha1 and FKBP51 correlate directly with Braak stage in human AD brains; however, the mechanisms by which these two co-chaperones cooperate with Hsp90 to facilitate pathological tau progression are quite different (Figure 7).
Figure 7.
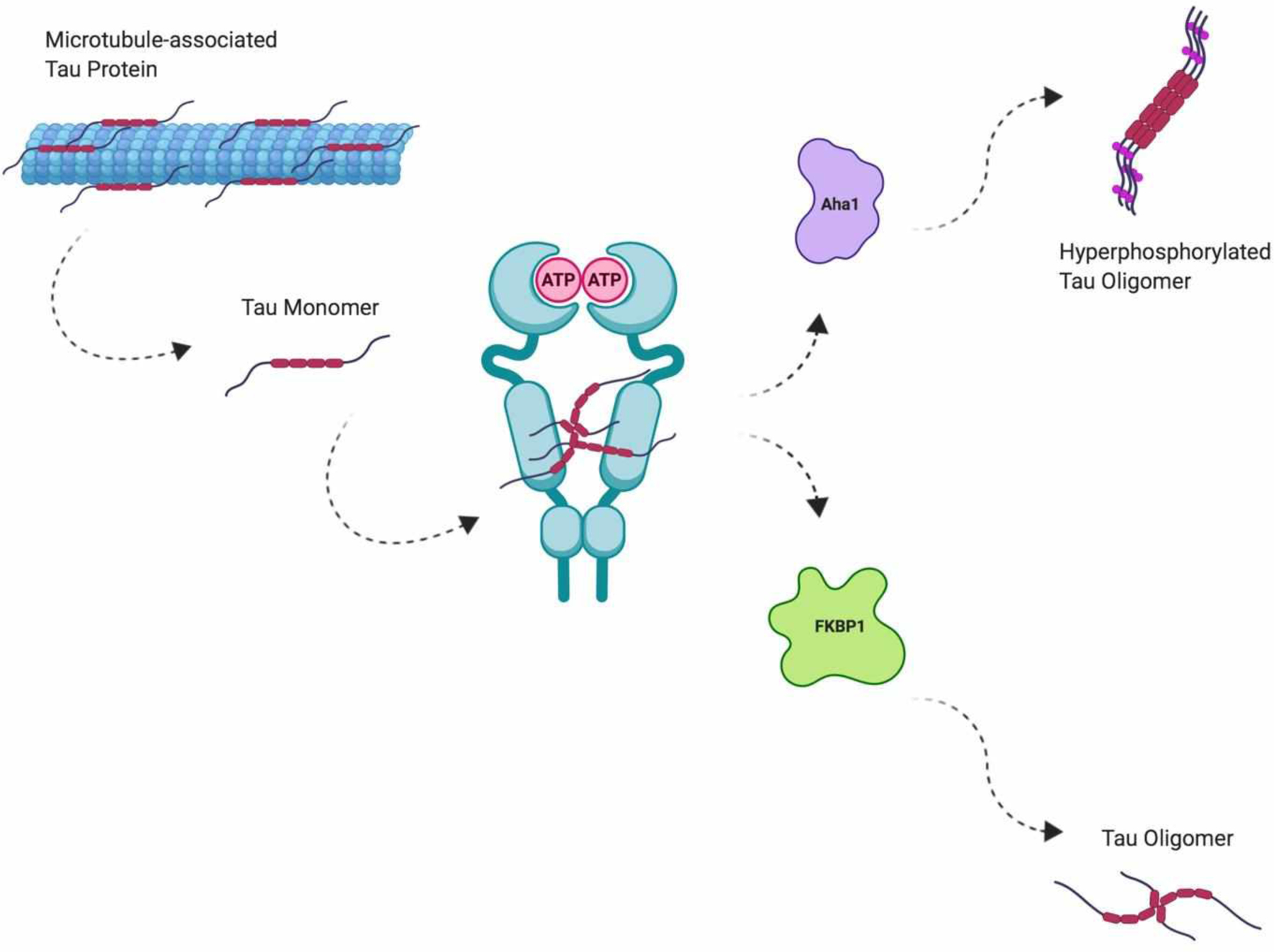
Hsp90-mediated tau oligomerization
Knockout of the FKBP5 gene (which encodes for FKBP51) reduced levels of tau in Fkbp5−/− mice, whereas the overexpression of FKBP51 by a viral vector in the rTg4510 tau mouse model disrupted the proteasomal clearance of tau, which led to the accumulation of oligomeric tau species, and eventually, neuronal cell death. The neurotoxic effects manifested by FKBP51 overexpression were attributed to increased interactions between FKBP51 and Hsp90, but precisely how the two proteins promote tau oligomerization was only recently elucidated. In a series of NMR-based structural investigations of the highly dynamic FKBP51/Hsp90/tau ternary complex, Hsp90 was shown to serve as a scaffold that orients the proline-rich region of tau into the FKBP51 PPIase catalytic site119. As tau hyperphosphorylation and aggregation have been shown to be dependent upon isomerization of its proline-rich region, it is believed that Hsp90 facilitates FKBP51-mediated proline isomerization of tau, which propagates tau fibril formation and leads to insoluble tau accumulation.
The mechanism by which Hsp90 interacts with Aha1 to modulate tau accumulation is less studied; however, combinations of both Aha1 and Hsp90 have been shown to be the most potent inducers of tau fibril formation identified to date. Shelton and coworkers demonstrated that the Aha1-E67K mutant, which is unable to bind Hsp90, is incapable of enhancing tau fibril formation; furthermore, these researchers determined that ATP is required for Aha1-stimulated tau aggregation120. These findings suggest that Aha1’s ability to control the Hsp90 ATPase cycle is critical for tau fibril formation – a proposition that was further supported by the fact that Aha1 overexpression in rTg4510 mice resulted in a significant increase in oligomeric and insoluble tau species, and subsequently led to neuronal loss and cognitive impairment. While the three-dimensional structure of the Aha1/Hsp90/tau complex has not yet been solved, there is precedence that disruption of the Aha1/Hsp90 complex can elicit a reduction in insoluble tau aggregation.
Hsp90, along with its co-chaperones and other smaller molecular chaperones, is also an important regulator of two major pathways that are associated with the clearance of misfolded and aggregated protein substrates – both of which are impaired in several central neuropathies. First, Hsp90 interacts with Heat Shock Cognate 71 kDa (Hsc70 / HSPA8) via the Hsc70-Hsp90 Organizer Protein (HOP) to regulate the Chaperone-Mediated Autophagy (CMA) pathway. Hsc70 is a chaperone that recognizes cytosolic proteins that contain a conserved KFERQ motif and transports them to the lysosomal membrane-bound, LAMP-2A receptor. There, LAMP-2A acts as a transporter to import protein substrates into the lysosome for their degradation and the recycling of amino acids121 (Figure 8). CMA is an important pathway for the clearance of oxidized cytosolic proteins, and several proteins associated with various neuropathic pathogeneses, such as tau and α-synuclein, are bonafide CMA substrates122,123. Interestingly, inhibition of Hsp90 with geldanamycin, a pan-Hsp90 inhibitor, was found to significantly activate the CMA response in IMR-90 cells124, suggesting that Hsp90 may attempt to refold these protein substrates prior to trafficking them to the lysosome for degradation.
Figure 8.
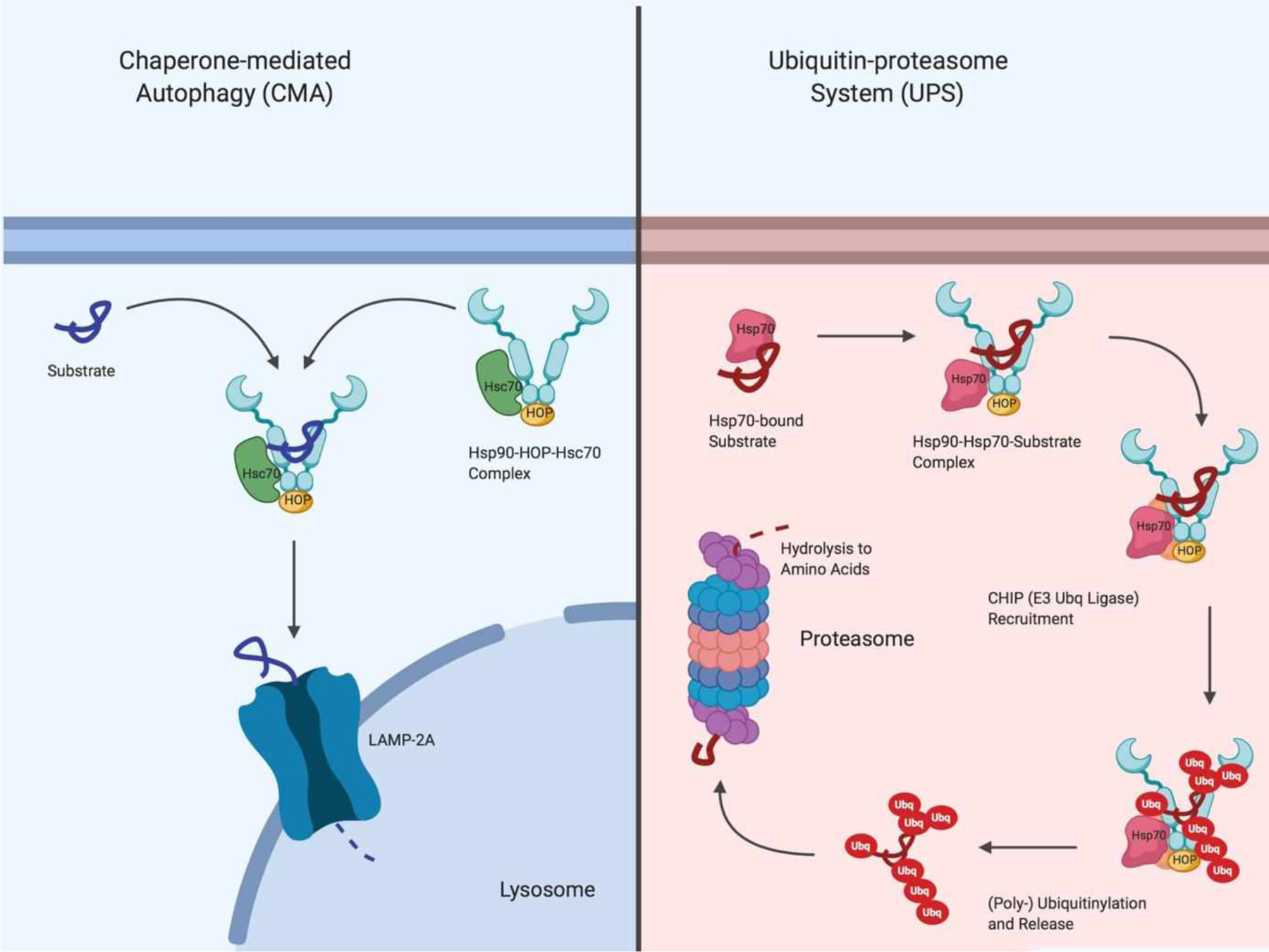
Chaperone-mediated autophagy versus the ubiquitin-proteasome system for degradation of protein substrates
Second, Hsp90 works in concert with Hsp70 and at least three ubiquitin ligases to mediate the proteolytic ubiquitin-proteasome system (UPS), which is the primary catabolism pathway responsible for the degradation of cellular proteins in mammals. Under normal conditions, Hsp90 binds client proteins that are delivered by Hsp70 and stabilizes them by preventing the exposure of hydrophobic residues that are present on client substrates, so that they can be properly folded into their active three-dimensional conformations. However, mutations or post-translational modifications to the protein substrate can destabilize its interaction with Hsp90. The destabilized complex recruits C-terminal Hsp70-interacting Protein (CHIP), an E3 ubiquitin ligase, to ubiquitinylate the mutated or modified client protein substrate. Consequently, the ubiquitinylated substrate is chaperoned to the proteasome for degradation and the recycling of amino acids (Figure 8). Ubiquitin, an 8 kDa cellular protein that serves as a marker for the proteasomal degradation of proteins, is elevated in the brains of patients with Alzheimer’s disease125 and other neurodegenerative disorders. The increased ubiquitinylation of cellular proteins is a consequence of the cell’s attempt to remove aberrant and aggregated proteins in these disease states. An example of an Hsp90 client protein whose clearance is mediated by the UPS is the Huntingtin (Htt) protein, the mutant form of which is known to aggregate and give rise to Huntington’s disease. Inhibition of Hsp90 disrupts the interaction between Hsp90 and Htt and ultimately promotes its clearance through the UPS126. This pathway has also been shown to be important for the degradation of post-translationally modified tau, α-synuclein, and other neurotoxic proteins whose clearance is disrupted in central neuropathies.
Smaller Hsps have also been studied in the context of central neuropathies. In particular, Hsp27 has been extensively investigated for its role in AD pathology. Hsp27 is significantly upregulated in astrocytes and degenerating neurons within AD brains127,128, interacts with amyloid-β in amyloid plaques129, and has been shown to interact physically with hyperphosphorylated tau and GSK3β, a known tau kinase130. Importantly, Hsp27 can be co-immunoprecipitated with tau antibodies from AD brains, but not from healthy brains131. The overexpression of Hsp27 in AD appears to play a role in the persistence of several cell cycle markers in AD130 and contributes to microtubule instability by mediating tau phosphorylation. However, it is unclear whether Hsp27 is a contributing factor to AD pathology or whether it plays a role in the cell’s effort to combat progression of the disease. In PD, Hsp27 appears to play a neuroprotective role, as the overexpression of Hsp27 manifests a potent anti-apoptotic effect against the damaging effects of wild-type and mutant forms of α-synuclein132.
Hsp27 and ɑB-crystallin, Two S-Hsps, in Neurodegeneration
Similarly, ɑB-crystallin is a polydisperse protein and a member of the S-Hsp family (HSPB5) that exhibits chaperone-like properties (including the ability to prevent denatured proteins from forming insoluble protein aggregates) and is also increased in AD brains and amyloid plaques129,133. Although ɑB-crystallin prevents the formation of Aβ1–40 fibrils, it has been shown that ɑB-crystallin can promote β-sheet formation of amyloid-β133 and increase Aβ1–40-associated toxicity134, presumably by promoting the nonfibrillar and highly toxic form of Aβ1–40. ɑB-crystallin has also been found to be the most abundant transcript in Multiple sclerosis lesions as compared to healthy brains168. Yet, others have found evidence that ɑB-crystallin confers a protective role against autoimmune encephalomyelitis135,136, stroke137, ischemic optic nerve neuropathy138, and myocardial infarction139. For these reasons, it has largely been accepted that ɑB-crystallin plays a role to combat neuropathic disease progression, but the specific role played by ɑB-crystallin plays appears to be disease state-specific.
Along with ɑB-crystallin, Hsp27 has been shown to be critically involved in the pathogenesis of diseases associated with amyloid deposition. Specifically, the N-terminal portion of Hsp27 and ɑB-crystallin have been shown to prevent amyloid fibril formation and confer cytoprotective activities140. Tau protein has also been shown to bind Hsp27 as well as to ɑB-crystallin, and these interactions are believed to be involved in the mechanism by which cells defend themselves from the type of neuropathic injury associated with pathological tau aggregation141. It appears that interactions between beta-strands on S-Hsps such as Hsp27 and ɑB-crystallin and beta-strands of aggregation-prone proteins can result in either stabilization of their structures, prevention of their aggregation, and/or facilitation of their proteolytic degradation142.
Hsp90 as a Therapeutic Target for Central Neuropathies
Small molecules that modulate Hsp90’s inherent ATPase activity have shown great promise in preclinical models of neurodegeneration, but this success has not yet translated into the clinic. Dickey and coworkers143 were the first to demonstrate that Hsp90 inhibitors able to cross the blood-brain barrier could decrease tau protein levels in vitro. Later, it was discovered that Hsp90 inhibitors enhance the degradation of phosphorylated tau through a mechanism involving the Carboxy Terminus of Hsp70-interacting Protein (CHIP), a tau ubiquitin ligase. In fact, disruption of the Hsp90/CHIP-mediated refolding complex led to decreased levels of phosphorylated tau in a murine model of tauopathy. Furthermore, the Hsp90 inhibitor, 17-AAG, promoted the degradation of Akt/PKB, an upstream regulator of tau kinase Microtubule Affinity-regulating Kinase 2 (PAR1/MARK2). Hence, Hsp90 inhibitors demonstrated the ability to modulate specific interactions between Hsp90 and CHIP, which serve to regulate pathways implicated in neurodegenerative tauopathies.
Geldanamycin, an N-terminal pan-Hsp90 inhibitor, manifested neuroprotective activity in cells that express mutant forms of tau via the increased expression of Hsp90 and Hsp70, which occurs upon N-terminal inhibition61. Similarly, A4, a novobiocin-based C-terminal Hsp90 inhibitor, protected primary neurons against amyloid-beta-induced neurotoxicity by inducing the expression of Hsp70 without concomitant inhibition of the chaperone machinery72. Clearly, the modulation of Hsp90 and Hsp70 expression represents a viable and promising approach for the treatment of central neuropathies in which the pathologies result from the accumulation of misfolded/aggregated proteins.
By 2008, Hsp90 had emerged as one of the most promising targets for the treatment of Alzheimer’s and Parksinson’s diseases. Consequently, a class of novobiocin analogs was developed and screened for their ability to protect differentiated SH-SY5Y cells from amyloid-beta-induced cell death via Hsp90 C-terminal inhibition. Data from these experiments showed several of these compounds to manifest neuroprotective activity at low nanomolar concentrations144. Not surprisingly, novobiocin-based small molecules continue to be actively pursued for neuroprotection against pathological species of amyloid and tau in neurodegenerative diseases.
Studies performed on mouse models of tauopathy have suggested that hyperphosphorylated tau species are the primary drivers of cognitive deficits in central neuropathies and that Hsp90 plays a key role in regulating the phosphorylation of tau via several mechanisms145. First, it has been shown that Hsp90 inhibition can directly reduce the tau kinase activities of Cdk5146 and Akt147 and subsequently lower levels of tau aggregates in both cellular and mouse models of tauopathy. Consequently, direct inhibition of Hsp90 represents a therapeutic opportunity to decrease hyperphosphorylated tau species through disruption of tau kinase activities148. Second, Hsp90 influences the stability of hyperphosphorylated tau through interactions with Cdc37, a co-chaperone that co-localizes and interacts directly with tau and tau kinases in neuronal cells. It has been shown that Cdc37 knockdown in HeLa cells significantly alters the phosphorylation state of tau due to the reduced stability of tau kinases149. Lastly, Hsp90 appears to regulate the phosphorylation status of tau through interactions with a number of cellular phosphatases. In particular, two Hsp90 co-chaperones have been shown to dephosphorylate tau, PP5 and CacyBP/SIP150–152. A recent study found the levels of CacyBP/SIP to be significantly increased in regions of the brain that are implicated in several neurodegenerative diseases153. Another study suggested that impaired PP5 activity also contributes to tau hyperphosphorylation in AD brains150. Therefore, small molecules that modulate interactions between Hsp90 and these co-chaperones to regulate tau phosphorylation may also represent a therapeutic option to treat patients with AD or other tauopathies.
Small molecules that target Hsp90 and exploit its modulation of the UPS may also be therapeutically useful. The administration of geldanamycin has been shown to inhibit huntingtin protein aggregation in cellular models of Huntington’s disease154 and rescue dopaminergic neurons from degeneration in Drosophila models of Parkinson’s disease155, presumably through mechanisms involving the UPS. Hsp90 inhibition has also been shown to promote the proteasomal clearance of Htt126, suggesting that Hsp90 inhibition may be a therapeutic strategy to reduce the mutant Htt accumulation in Huntington’s disease.
Small molecule modulators of Hsp90 have demonstrated promising activities in in vivo models of central neuropathies as well. For instance, one CNS-permeable Hsp90 inhibitor, OS47720, was found to promote dendritic spine formation and rescue spatial learning and memory in a Tg2576 mouse model of Alzheimer’s disease via an HSF1-dependent mechanism156. More recently, a vaccine consisting of Grp94 and α-synuclein was shown to suppress PD-associated microgliosis in the substantia nigra and striatum in a chronic MPTP murine model of PD157, suggesting that formulations consisting of disease-related misfolded proteins and Hsps – specifically, isoforms of Hsp90 – may be beneficial for the treatment of central neuropathies that involve misfolded proteins. Lastly, the compound PU-AD, an orally administered brain-penetrable inhibitor of Hsp90 epichaperomes discovered by the Chiosis laboratory, is being advanced by Samus Therapeutics to Phase 1 clinical evaluation in Alzheimer’s disease. In preclinical studies, PU-AD promoted neuronal survival by preventing the aggregation and hyperphosphorylation of tau, while stimulating its degradation158.
Aha1 as a Therapeutic Target for CNS Neuropathies
Disruption of the Aha1/Hsp90 complex has emerged as an alternative therapeutic strategy and may provide an additional opportunity to overcome the poor blood-brain barrier permeability and toxicities associated with direct inhibitors of Hsp90 and Hsp70. Shelton et al. were the first to demonstrate that disruption of the Aha1/Hsp90 complex with small molecules can dramatically reduce tau fibril seeding and formation of insoluble tau species both in vitro and in cultured cells120. Importantly, the molecules were shown to manifest neuroprotective activity without inhibition of the Hsp90-mediated refolding process. Such data provides evidence that strategic disruption of the interaction between Hsp90 and Aha1 can reduce tau accumulation without affecting Hsp90’s ability to fold other client proteins. Therefore, inhibition of the Aha1/Hsp90 complex represents a promising therapeutic strategy for the development of small molecules to treat AD and other tauopathies.
A select number of small molecule Aha1/Hsp90 disruptors have been discovered to date (Figure 9). In the aforementioned study, KU-177 demonstrated the ability to disrupt interactions between Hsp90 and Aha1 in co-immunoprecipitation experiments and ablated Aha1-driven enhancement of Hsp90-dependent tau aggregation120. Using a FRET-based screen, Stiegler and coworkers identified another small molecule, HAM-1, that disrupts the Aha1/Hsp90 complex and selectively inhibits Aha1-stimulated ATPase activity of Hsp90, but not Hsp90’s basal ATPase activity in the absence of Aha1159. In contrast to the well-characterized interaction site between the N-terminal domain of Aha1 and middle domain of Hsp9075, NMR studies revealed that HAM-1 binds the Hsp90 N-terminus at a site that serves as a transient Aha1 C-terminal domain interaction site. This second interaction site is in close proximity to Hsp90’s ATP-binding site and the interaction between the two proteins at this site is transient only enough to allow for stabilization of a rate-limiting, closed state of the Hsp90 ATPase cycle160,161.
Figure 9.
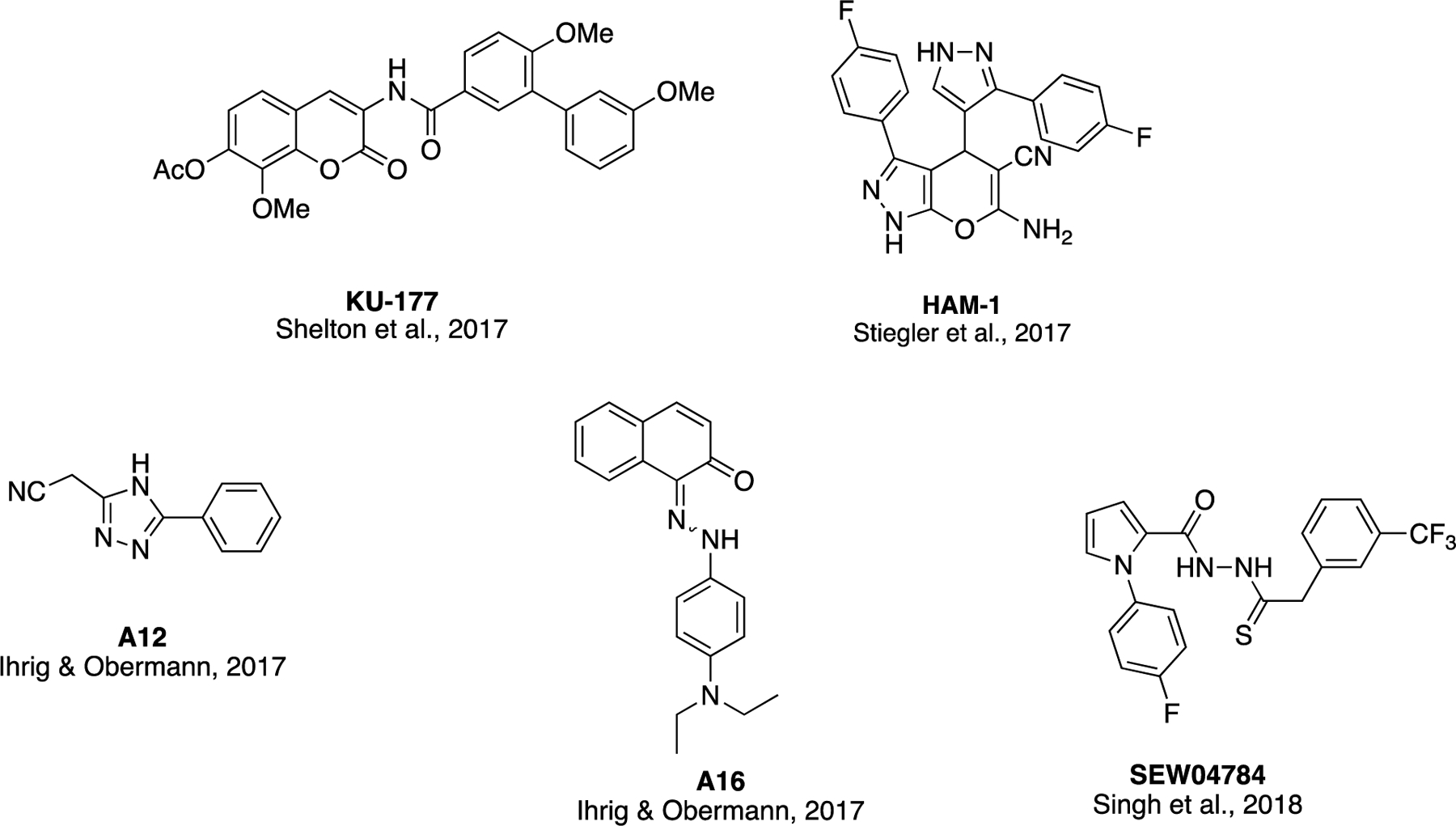
Structures of select Aha1/Hsp90 disruptors
Two other Aha1/Hsp90 complex disruptors, A12 and A16, were identified by amplified luminescence proximity homogeneous assays (Alpha)162. Both of these compounds restored chloride channel activity in cells expressing mutant cystic fibrosis transmembrane conductance regulator (CFTR) protein and may be further developed to treat cystic fibrosis, as Aha1 appears to play a disruptive role in CFTRΔ508 degradation during cystic fibrosis pathology163,164. A quinaldine red ATPase assay was used to identify one final compound, SEW04784 that binds to the C-terminal domain of Aha1 and disrupts its interaction with Hsp90165. This compound inhibits the Aha1-stimulated ATPase activity of Hsp90, but not Hsp90’s basal ATPase activity.
HSR Induction and Hsp70 as Therapeutic Targets for CNS Neuropathies
Markers of HSR induction, including increased expression of Hsp70, have been found to accumulate in plaques and neurofibrillary tangles, and have been detected in the brains of patients with AD166,167. Interestingly, Hsp70 appears to combat the early stages of AD pathogenesis, as preparations of recombinant Hsp70, its co-chaperone Hsp40, and Hsp90 can block the assembly of amyloid-β oligomers, although these preparations exhibited little effect during amyloid-β fibrillar assembly168. In the same study, researchers discovered that the anti-aggregation activity of Hsp70 could be enhanced by pharmacological stimulation of Hsp70 or conversely, inhibited by ATPᵧS, a non-hydrolyzable ATP analog.
In PD, Hsp70, Hsp90, Hsp60, Hsp40, and Hsp27 have been detected in Lewy bodies extracted from patients with cortical Lewy body disease169. It is believed that the sequestration of these molecular chaperones into Lewy bodies results in their cellular depletion, and the subsequent loss of chaperone activity may lead to degeneration155. In agreement with this hypothesis, HSR induction and subsequent elevations in Hsp70 levels protected against α-synuclein-induced cell death in a yeast model of PD170 and prevented cell death in the MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) model of PD171,172. The exact role played by Hsp70 in PD – in particular, its contributions to α-synuclein-induced toxicity – remains unclear. However, it has been shown that Hsp70 binds α-synuclein filaments to mediate their inhibitory effects on the proteasome173. Hsp70 has also been shown to bind prefibrillar α-synuclein species and prevent key steps of α-synuclein aggregation174. It is clear that stimulation of Hsp70 manifests neuroprotective activity against α-synuclein-associated toxicity, which is supported by the fact that the expression of Hsp70 reduces aggregate formation and toxicity that is induced by C-terminally truncated forms of α-synuclein in cells. Furthermore, Hsp70 overexpression that results upon crossing human α-synuclein transgenic mice with transgenic mice overexpressing rat Hsp70 resulted in a significant reduction of insoluble α-synuclein aggregates175. In summary, agents whose activities result in HSR induction, enhancement (or restore) Hsp70 expression, and/or stimulation of chaperone activity are being actively pursued for their ability to protect neurons against toxicities associated with misfolded proteins in both AD and PD.
Outlook
Heat shock proteins (Hsps) are evolutionarily conserved proteins that play a critical role in cells by “chaperoning” newly formed polypeptides as well as minimizing protein aggregation through the refolding of denatured proteins or regulating their degradation through the UPS or CMA. While Hsps maintain cellular homeostasis in normal cells, they form distinct “epichaperome” complexes in diseased cells. In fact, their abnormal function or expression level (elevation or downregulation) can be observed in many pathological conditions, including both peripheral and central neuropathies. In this review, the biological roles played by the heat shock proteins during the pathology of such diseases as well as the therapeutic potential that small molecule inhibitors and/or modulators of heat shock proteins exhibit for the treatment of these diseases is summarized. Hsp regulation continues to be an active and compelling area of research, and may offer therapeutic opportunities to treat additional neuropathies in the future.
Acknowledgements
BMK is a fellow of the Chemistry-Biochemistry-Biology Interface (CBBI) Program at the University of Notre Dame, supported by the NIH Training Grant T32GM075762 from the National Institute of General Medical Sciences. BSJB is supported by The National Institutes of Health [CA213586] [N5075311]. Images were created with BioRender.
Author Biosketches
Subhabrata Chaudhury, Ph.D. is a native Bengali from India. After receiving his bachelor’s degree from University of Calcutta and master’s degree from IIT – Kharagpur, he came to work with Prof. William A. Donaldson at Marquette University, where he earned his Ph.D. degree in organic chemistry. Upon earning his Ph.D., Dr. Chaudhury worked as Post-doctoral Research Scholar (USA), Research Associate (UK), and Team Leader (industries in India) in three different continents. Presently, he is a scientist in Dr. Brian Blagg’s laboratory at the University of Notre Dame.
Bradley M. Keegan earned his B.S. with honors in chemistry (concentrations in biochemistry and premedicine) and M.S. in biomedical sciences with a thesis in integrative physiology and pharmacology from Wake Forest University. Prior to joining the Blagg laboratory at the University of Notre Dame, he served as a co-founder and Chief Science Officer of EncepHeal Therapeutics. He is currently a CBBI Predoctoral Research Fellow at the University of Notre Dame, where his doctoral research focuses on synthesizing and evaluating small molecule modulators of the Hsp90 chaperome for the reduction of tau aggregation in Alzheimer’s and Parkinson’s diseases.
Brian S. J. Blagg, Ph.D. received a B.A. in chemistry and environmental studies from Sonoma State University in 1994. He then went on to earn a Ph.D. in organic chemistry from the University of Utah in the laboratory of Dale Poulter. Following the completion of his Ph.D., he served as a NIH post-doctoral fellow at the Scripps Research Institute, where he studied in Dale Boger’s laboratory until 2002. Dr. Blagg began his independent research career as a medicinal chemistry professor at the University of Kansas, where he remained until 2017. In 2017, Dr. Blagg became the Director of the Warren Family Research Center for Drug Discovery and Development at the University of Notre Dame and the Charles Huisking Professor of Chemistry and Biochemistry. His research focuses on the design, synthesis, and evaluation of Hsp90 inhibitors.
References
- 1.Goldschmidt R Gen und Außeneigenschaft. Zeitschrift Für Induktive Abstammungs- Und Vererbungslehre 1935;69(1):38–69. [Google Scholar]
- 2.Ritossa F A new puffing pattern induced by temperature shock and DNP in drosophila. Experientia 1962;18(12):571–573. [Google Scholar]
- 3.Morimoto R, Tissieres A, Georgopoulos C. Stress Proteins in Biology and Medicine. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1990. [DOI] [PubMed] [Google Scholar]
- 4.Tissières A, Mitchell HK, Tracy UM. Protein synthesis in salivary glands of Drosophila melanogaster: relation to chromosome puffs. J Mol Biol 1974;84(3):389–98. [DOI] [PubMed] [Google Scholar]
- 5.Kelley PM, Schlesinger MJ. The effect of amino acid analogues and heat shock on gene expression in chicken embryo fibroblasts. Cell 1978;15(4):1277–86. [DOI] [PubMed] [Google Scholar]
- 6.Lemaux PG, Herendeen SL, Bloch PL, Neidhardt FC. Transient rates of synthesis of individual polypeptides in E. coli following temperature shifts. Cell 1978;13(3):427–34. [DOI] [PubMed] [Google Scholar]
- 7.Ashburner M, Bonner JJ. The induction of gene activity in drosophilia by heat shock. Cell 1979;17(2):241–54. [DOI] [PubMed] [Google Scholar]
- 8.Peterson NS, Moller G, Mitchell HK. Genetic mapping of the coding regions for three heat-shock proteins in Drosophila melanogaster. Genetics 1979;92(3):891–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McAlister L, Finkelstein DB. Heat shock proteins and thermal resistance in yeast. Biochem Bioph Res Co 1980;93(3):819–24. [DOI] [PubMed] [Google Scholar]
- 10.Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet 1988;22(1):631–77. [DOI] [PubMed] [Google Scholar]
- 11.Richter K, Haslbeck M, Buchner J. The Heat Shock Response: Life on the Verge of Death. Mol Cell 2010;40(2):253–266. [DOI] [PubMed] [Google Scholar]
- 12.Pockley AG. Heat shock proteins as regulators of the immune response. Lancet Lond Engl 2003;362(9382):469–76. [DOI] [PubMed] [Google Scholar]
- 13.Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biology 2004;5(10):781–91. [DOI] [PubMed] [Google Scholar]
- 14.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biology 2010;11(7):515–28. [DOI] [PubMed] [Google Scholar]
- 15.Jolly C, Morimoto RI. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J Natl Cancer I 2000;92(19):1564–72. [DOI] [PubMed] [Google Scholar]
- 16.Caplan AJ, Mandal AK, Theodoraki MA. Molecular chaperones and protein kinase quality control. Trends Cell Biol 2007;17(2):87–92. [DOI] [PubMed] [Google Scholar]
- 17.Gomez-Pastor R, Burchfiel ET, Thiele DJ. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat Rev Mol Cell Biology 2017;19(1):4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Padmalayam I The heat shock response: its role in pathogenesis of type 2 diabetes and its complications, and implications for therapeutic intervention. Discov Med 2014;18(97):29–39. [PubMed] [Google Scholar]
- 19.Hooper PL, Hooper JJ. Loss of defense against stress: diabetes and heat shock proteins. Diabetes Technol The 2005;7(1):204–8. [DOI] [PubMed] [Google Scholar]
- 20.Hooper PL, Hooper PL. Inflammation, heat shock proteins, and type 2 diabetes. Cell Stress Chaperon 2008;14(2):113–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farmer KL, Li C, Dobrowsky RT. Diabetic peripheral neuropathy: should a chaperone accompany our therapeutic approach? Pharmacol Rev 2012;64(4):880–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dobrowsky RT. Targeting the Diabetic Chaperome to Improve Peripheral Neuropathy. Curr Diabetes Rep 2016;16(8):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Losiewicz MK, Fort PE. Diabetes impairs the neuroprotective properties of retinal alpha-crystallins. Invest Ophth Vis Sci 2011;52(9):5034–42. [DOI] [PubMed] [Google Scholar]
- 24.Reddy VS, Raghu G, Reddy SS, Pasupulati AK, Suryanarayana P, Reddy GB. Response of small heat shock proteins in diabetic rat retina. Invest Ophth Vis Sci 2013;54(12):7674–82. [DOI] [PubMed] [Google Scholar]
- 25.Lazaro I, Oguiza A, Recio C, Mallavia B, Madrigal-Matute J, Blanco J, Egido J, Martin-Ventura J-L, Gomez-Guerrero C. Targeting HSP90 Ameliorates Nephropathy and Atherosclerosis Through Suppression of NF-κB and STAT Signaling Pathways in Diabetic Mice. Diabetes 2015;64(10):3600–13. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H-M, Dang H, Kamat A, Yeh C-K, Zhang B-X. Geldanamycin derivative ameliorates high fat diet-induced renal failure in diabetes. Plos One 2012;7(3):e32746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madrigal-Matute J, Martin-Ventura JL, Blanco-Colio LM, Egido J, Michel J-B, Meilhac O. Heat-shock proteins in cardiovascular disease. Adv Clin Chem 2011;541–43. [DOI] [PubMed] [Google Scholar]
- 28.Karpe PA, Tikoo K. Heat shock prevents insulin resistance-induced vascular complications by augmenting angiotensin-(1–7) signaling. Diabetes 2013;63(3):1124–39. [DOI] [PubMed] [Google Scholar]
- 29.Finka A, Mattoo RUH, Goloubinoff P. Meta-analysis of heat- and chemically upregulated chaperone genes in plant and human cells. Cell Stress Chaperon 2010;16(1):15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zuo D, Subjeck J, Wang X-Y. Unfolding the Role of Large Heat Shock Proteins: New Insights and Therapeutic Implications. Front Immunol 2016;775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun Y, MacRae TH. The small heat shock proteins and their role in human disease. Febs J 2005;272(11):2613–2627. [DOI] [PubMed] [Google Scholar]
- 32.Brocchieri L, Macario EC de, Macario AJL. hsp70 genes in the human genome: Conservation and differentiation patterns predict a wide array of overlapping and specialized functions. Bmc Evol Biol 2008;8(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radons J The human HSP70 family of chaperones: where do we stand? Cell Stress Chaperon 2016;21(3):379–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiosis G Heat Shock Proteins in Disease - From Molecular Mechanisms to Therapeutics. Curr Top Med Chem 2016;16(25):2727–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Atalay M, Oksala N, Lappalainen J, Laaksonen DE, Sen CK, Roy S. Heat shock proteins in diabetes and wound healing. Curr Protein Pept Sc 2009;10(1):85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daugaard M, Rohde M, Jäättelä M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. Febs Lett 2007;581(19):3702–10. [DOI] [PubMed] [Google Scholar]
- 37.Akbar MT, Lundberg AMC, Liu K, Vidyadaran S, Wells KE, Dolatshad H, Wynn S, Wells DJ, Latchman DS, Belleroche J de. The neuroprotective effects of heat shock protein 27 overexpression in transgenic animals against kainate-induced seizures and hippocampal cell death. J Biological Chem 2003;278(22):19956–65. [DOI] [PubMed] [Google Scholar]
- 38.Stetler RA, Gao Y, Signore AP, Cao G, Chen J. HSP27: mechanisms of cellular protection against neuronal injury. Curr Mol Med 2009;9(7):863–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morimoto RI. Cells in stress: transcriptional activation of heat shock genes. Sci New York N Y 1993;259(5100):1409–10. [DOI] [PubMed] [Google Scholar]
- 40.Trinklein ND, Murray JI, Hartman SJ, Botstein D, Myers RM. The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Mol Biol Cell 2003;15(3):1254–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neef DW, Jaeger AM, Thiele DJ. Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat Rev Drug Discov 2011;10(12):930–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vihervaara A, Sistonen L. HSF1 at a glance. J Cell Sci 2014;127(Pt 2):261–6. [DOI] [PubMed] [Google Scholar]
- 43.Batista-Nascimento L, Neef DW, Liu PCC, Rodrigues-Pousada C, Thiele DJ. Deciphering human heat shock transcription factor 1 regulation via post-translational modification in yeast. Plos One 2011;6(1):e15976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rangaraju S, Madorsky I, Pileggi JG, Kamal A, Notterpek L. Pharmacological induction of the heat shock response improves myelination in a neuropathic model. Neurobiol Dis 2008;32(1):105–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koren J, Jinwal UK, Jin Y, O’Leary J, Jones JR, Johnson AG, Blair LJ, Abisambra JF, Chang L, Miyata Y, Cheng AM, Guo J, Cheng JQ, Gestwicki JE, Dickey CA. Facilitating Akt clearance via manipulation of Hsp70 activity and levels. J Biological Chem 2009;285(4):2498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li C, Ma J, Zhao H, Blagg BSJ, Dobrowsky RT. Induction of heat shock protein 70 (Hsp70) prevents neuregulin-induced demyelination by enhancing the proteasomal clearance of c-Jun. Asn Neuro 2012;4(7):e00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chittoor-Vinod VG, Lee S, Judge SM, Notterpek L. Inducible HSP70 is critical in preventing the aggregation and enhancing the processing of PMP22. Asn Neuro 2015;7(1):175909141556990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hentze N, Breton LL, Wiesner J, Kempf G, Mayer MP. Molecular mechanism of thermosensory function of human heat shock transcription factor Hsf1. Elife 2016;5e11576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kijima T, Prince TL, Tigue ML, Yim KH, Schwartz H, Beebe K, Lee S, Budzynski MA, Williams H, Trepel JB, Sistonen L, Calderwood S, Neckers L. HSP90 inhibitors disrupt a transient HSP90-HSF1 interaction and identify a noncanonical model of HSP90-mediated HSF1 regulation. Sci Rep-uk 2018;8(1):6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peterson LB, Blagg BSJ. To fold or not to fold: modulation and consequences of Hsp90 inhibition. Future Med Chem 2009;1(2):267–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li L, Wang L, You Q-D, Xu X-L. Heat Shock Protein 90 Inhibitors: An Update on Achievements, Challenges, and Future Directions. J Med Chem [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 52.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003;425(6956):407. [DOI] [PubMed] [Google Scholar]
- 53.Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, Patterson C, Dickson DW, Nahman NS, Hutton M, Burrows F, Petrucelli L. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest 2007;117(3):648–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taldone T, Ochiana SO, Patel PD, Chiosis G. Selective targeting of the stress chaperome as a therapeutic strategy. Trends Pharmacol Sci 2014;35(11):592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodina A, Wang T, Yan P, Gomes ED, Dunphy MPS, Pillarsetty N, Koren J, Gerecitano JF, Taldone T, Zong H, Caldas-Lopes E, Alpaugh M, Corben A, Riolo M, Beattie B, Pressl C, Peter RI, Xu C, Trondl R, Patel HJ, Shimizu F, Bolaender A, Yang C, Panchal P, Farooq MF, Kishinevsky S, Modi S, Lin O, Chu F, Patil S, Erdjument-Bromage H, Zanzonico P, Hudis C, Studer L, Roboz GJ, Cesarman E, Cerchietti L, Levine R, Melnick A, Larson SM, Lewis JS, Guzman ML, Chiosis G. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 2016;538(7625):397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schulte TW, Neckers LM. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemoth Pharm 1998;42(4):273–9. [DOI] [PubMed] [Google Scholar]
- 57.Tadtong S, Meksuriyen D, Tanasupawat S, Isobe M, Suwanborirux K. Geldanamycin derivatives and neuroprotective effect on cultured P19-derived neurons. Bioorg Med Chem Lett 2006;17(10):2939–43. [DOI] [PubMed] [Google Scholar]
- 58.Maroney AC, Marugan JJ, Mezzasalma TM, Barnakov AN, Garrabrant TA, Weaner LE, Jones WJ, Barnakova LA, Koblish HK, Todd MJ, Masucci JA, Deckman IC, Galemmo RA, Johnson DL. Dihydroquinone ansamycins: toward resolving the conflict between low in vitro affinity and high cellular potency of geldanamycin derivatives. Biochemistry-us 2006;45(17):5678–85. [DOI] [PubMed] [Google Scholar]
- 59.Lee K, Ryu JS, Jin Y, Kim W, Kaur N, Chung SJ, Jeon Y-J, Park J-T, Bang JS, Lee HS, Kim TY, Lee JJ, Hong Y-S. Synthesis and anticancer activity of geldanamycin derivatives derived from biosynthetically generated metabolites. Org Biomol Chem 2007;6(2):340–8. [DOI] [PubMed] [Google Scholar]
- 60.Patel K, Piagentini M, Rascher A, Tian Z-Q, Buchanan GO, Regentin R, Hu Z, Hutchinson CR, McDaniel R. Engineered biosynthesis of geldanamycin analogs for Hsp90 inhibition. Chem Biol 2004;11(12):1625–33. [DOI] [PubMed] [Google Scholar]
- 61.Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Chaperones increase association of tau protein with microtubules. P Natl Acad Sci Usa 2003;100(2):721–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gallo KA. Targeting HSP90 to halt neurodegeneration. Chem Biol 2006;13(2):115–6. [DOI] [PubMed] [Google Scholar]
- 63.Sreedhar AS, Kalmár E, Csermely P, Shen Y-F. Hsp90 isoforms: functions, expression and clinical importance. Febs Lett 2004;562(1–3):11–5. [DOI] [PubMed] [Google Scholar]
- 64.Wang Y, Trepel JB, Neckers LM, Giaccone G. STA-9090, a small-molecule Hsp90 inhibitor for the potential treatment of cancer. Curr Opin Investigational Drugs Lond Engl 2000 2010;11(12):1466–76. [PubMed] [Google Scholar]
- 65.Prince TL, Kijima T, Tatokoro M, Lee S, Tsutsumi S, Yim K, Rivas C, Alarcon S, Schwartz H, Khamit-Kush K, Scroggins BT, Beebe K, Trepel JB, Neckers L. Client Proteins and Small Molecule Inhibitors Display Distinct Binding Preferences for Constitutive and Stress-Induced HSP90 Isoforms and Their Conformationally Restricted Mutants. Plos One 2015;10(10):e0141786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marcu MG, Schulte TW, Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J Natl Cancer I 2000;92(3):242–8. [DOI] [PubMed] [Google Scholar]
- 67.Carrello A, Ingley E, Minchin RF, Tsai S, Ratajczak T. The common tetratricopeptide repeat acceptor site for steroid receptor-associated immunophilins and hop is located in the dimerization domain of Hsp90. J Biological Chem 1999;274(5):2682–9. [DOI] [PubMed] [Google Scholar]
- 68.Das AK, Cohen PW, Barford D. The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein-protein interactions. Embo J 1998;17(5):1192–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hall JA, Forsberg LK, Blagg BSJ. Alternative approaches to Hsp90 modulation for the treatment of cancer. Future Med Chem 2014;6(14):1587–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eskew JD, Sadikot T, Morales P, Duren A, Dunwiddie I, Swink M, Zhang X, Hembruff S, Donnelly A, Rajewski RA, Blagg BS, Manjarrez JR, Matts RL, Holzbeierlein JM, Vielhauer GA. Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. Bmc Cancer 2011;11(1):468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Verkhratsky A, Sofroniew MV, Messing A, deLanerolle NC, Rempe D, Rodríguez JJ, Nedergaard M. Neurological diseases as primary gliopathies: a reassessment of neurocentrism. Asn Neuro 2012;4(3):AN20120010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ansar S, Burlison JA, Hadden MK, Yu XM, Desino KE, Bean J, Neckers L, Audus KL, Michaelis ML, Blagg BSJ. A non-toxic Hsp90 inhibitor protects neurons from Aβ-induced toxicity. Bioorg Med Chem Lett 2007;17(7):1984–1990. [DOI] [PubMed] [Google Scholar]
- 73.Salehi AH, Morris SJ, Ho W-C, Dickson KM, Doucet G, Milutinovic S, Durkin J, Gillard JW, Barker PA. AEG3482 is an antiapoptotic compound that inhibits Jun kinase activity and cell death through induced expression of heat shock protein 70. Chem Biol 2006;13(2):213–23. [DOI] [PubMed] [Google Scholar]
- 74.Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. P Natl Acad Sci Usa 2000;97(20):10832–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Meyer P, Prodromou C, Hu B, Vaughan C, Roe SM, Panaretou B, Piper PW, Pearl LH. Structural and functional analysis of the middle segment of hsp90: implications for ATP hydrolysis and client protein and cochaperone interactions. Mol Cell 2003;11(3):647–58. [DOI] [PubMed] [Google Scholar]
- 76.Tortora G, Derrickson B. Principles of Anatomy and Physiology, 15th Edition. J. Wiley; 2016. [Google Scholar]
- 77.Kandel E, Schwartz J, Jessell TM, Siegelbaum SA, Hudspeth AJ, Mack S. Principles of Neural Science, Fifth Edition. McGraw-Hill Professional; n.d. [Google Scholar]
- 78.Standring S Gray’s Anatomy, 39th Edition: The Anatomical Basis of Clinical Practice. Churchill Livingstone; 2005. [Google Scholar]
- 79.Harati Y Diabetic neuropathies: unanswered questions. Neurol Clin 2007;25(1):303–17. [DOI] [PubMed] [Google Scholar]
- 80.Ramos KM, Jiang Y, Svensson CI, Calcutt NA. Pathogenesis of spinally mediated hyperalgesia in diabetes. Diabetes 2007;56(6):1569–76. [DOI] [PubMed] [Google Scholar]
- 81.Callaghan BC, Cheng HT, Stables CL, Smith AL, Feldman EL. Diabetic neuropathy: clinical manifestations and current treatments. Lancet Neurology 2012;11(6):521–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Callaghan BC, Price RS, Feldman EL. Distal Symmetric Polyneuropathy: A Review. Jama 2015;314(20):2172–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Griebeler ML, Morey-Vargas OL, Brito JP, Tsapas A, Wang Z, Leon BGC, Phung OJ, Montori VM, Murad MH. Pharmacologic interventions for painful diabetic neuropathy: An umbrella systematic review and comparative effectiveness network meta-analysis. Ann Intern Med 2014;161(9):639–49. [DOI] [PubMed] [Google Scholar]
- 84.Tesfaye S, Vileikyte L, Rayman G, Sindrup SH, Perkins BA, Baconja M, Vinik AI, Boulton AJM, Neuropathy TEP on D. Painful diabetic peripheral neuropathy: consensus recommendations on diagnosis, assessment and management. Diabetes Metabolism Res Rev 2011;27(7):629–38. [DOI] [PubMed] [Google Scholar]
- 85.Ziegler D, Ametov A, Barinov A, Dyck PJ, Gurieva I, Low PA, Munzel U, Yakhno N, Raz I, Novosadova M, Maus J, Samigullin R. Oral treatment with alpha-lipoic acid improves symptomatic diabetic polyneuropathy: the SYDNEY 2 trial. Diabetes Care 2006;29(11):2365–70. [DOI] [PubMed] [Google Scholar]
- 86.Bril V, Buchanan RA. Aldose reductase inhibition by AS-3201 in sural nerve from patients with diabetic sensorimotor polyneuropathy. Diabetes Care 2004;27(10):2369–75. [DOI] [PubMed] [Google Scholar]
- 87.Bril V, Buchanan RA. Long-term effects of ranirestat (AS-3201) on peripheral nerve function in patients with diabetic sensorimotor polyneuropathy. Diabetes Care 2006;29(1):68–72. [DOI] [PubMed] [Google Scholar]
- 88.Leinninger GM, Vincent AM, Feldman EL. The role of growth factors in diabetic peripheral neuropathy. J Peripher Nerv Syst Jpns 2004;9(1):26–53. [DOI] [PubMed] [Google Scholar]
- 89.Beckman JA, Goldfine AB, Gordon MB, Garrett LA, Creager MA. Inhibition of protein kinase Cbeta prevents impaired endothelium-dependent vasodilation caused by hyperglycemia in humans. Circ Res 2002;90(1):107–11. [DOI] [PubMed] [Google Scholar]
- 90.Tuttle KR, Bakris GL, Toto RD, McGill JB, Hu K, Anderson PW. The effect of ruboxistaurin on nephropathy in type 2 diabetes. Diabetes Care 2005;28(11):2686–90. [DOI] [PubMed] [Google Scholar]
- 91.Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res 2010;106(8):1319–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 2005;6(1):11–22. [DOI] [PubMed] [Google Scholar]
- 93.Chittoor VG, Sooyeon L, Rangaraju S, Nicks JR, Schmidt JT, Madorsky I, Narvaez DC, Notterpek L. Biochemical characterization of protein quality control mechanisms during disease progression in the C22 mouse model of CMT1A. Asn Neuro 2013;5(5):e00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vincent AM, Russell JW, Low P, Feldman EL. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr Rev 2004;25(4):612–28. [DOI] [PubMed] [Google Scholar]
- 95.Vincent AM, Hinder LM, Pop-Busui R, Feldman EL. Hyperlipidemia: a new therapeutic target for diabetic neuropathy. J Peripher Nerv Syst Jpns 2009;14(4):257–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pop-Busui R, Sima A, Stevens M. Diabetic neuropathy and oxidative stress. Diabetes Metabolism Res Rev 2006;22(4):257–73. [DOI] [PubMed] [Google Scholar]
- 97.Calcutt NA, Backonja MM. Pathogenesis of pain in peripheral diabetic neuropathy. Curr Diabetes Rep 2007;7(6):429–34. [DOI] [PubMed] [Google Scholar]
- 98.Zochodne DW. Diabetes mellitus and the peripheral nervous system: manifestations and mechanisms. Muscle Nerve 2007;36(2):144–66. [DOI] [PubMed] [Google Scholar]
- 99.Zochodne DW. Diabetic polyneuropathy: an update. Curr Opin Neurol 2008;21(5):527–33. [DOI] [PubMed] [Google Scholar]
- 100.Edwards JL, Vincent AM, Cheng HT, Feldman EL. Diabetic neuropathy: mechanisms to management. Pharmacol Therapeut 2008;120(1):1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Calcutt NA, Cooper ME, Kern TS, Schmidt AM. Therapies for hyperglycaemia-induced diabetic complications: from animal models to clinical trials. Nat Rev Drug Discov 2009;8(5):417–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Finnerup NB, Sindrup SH, Jensen TS. The evidence for pharmacological treatment of neuropathic pain. Pain 2010;150(3):573–81. [DOI] [PubMed] [Google Scholar]
- 103.Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Sign 2010;12(4):537–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Malik RA, Veves A, Tesfaye S, Smith G, Cameron N, Zochodne D, Lauria G, Neuropathy TCP on D. Small fibre neuropathy: role in the diagnosis of diabetic sensorimotor polyneuropathy. Diabetes Metabolism Res Rev 2011;27(7):678–84. [DOI] [PubMed] [Google Scholar]
- 105.Urban MJ, Li C, Yu C, Lu Y, Krise JM, McIntosh MP, Rajewski RA, Blagg BSJ, Dobrowsky RT. Inhibiting heat-shock protein 90 reverses sensory hypoalgesia in diabetic mice. Asn Neuro 2010;2(4):e00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang L, Zhao H, Blagg BSJ, Dobrowsky RT. C-terminal heat shock protein 90 inhibitor decreases hyperglycemia-induced oxidative stress and improves mitochondrial bioenergetics in sensory neurons. J Proteome Res 2012;11(4):2581–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ma J, Farmer KL, Pan P, Urban MJ, Zhao H, Blagg BSJ, Dobrowsky RT. Heat shock protein 70 is necessary to improve mitochondrial bioenergetics and reverse diabetic sensory neuropathy following KU-32 therapy. J Pharmacol Exp Ther 2013;348(2):281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kusuma BR, Zhang L, Sundstrom T, Peterson LB, Dobrowsky RT, Blagg BSJ. Synthesis and evaluation of novologues as C-terminal Hsp90 inhibitors with cytoprotective activity against sensory neuron glucotoxicity. J Med Chem 2012;55(12):5797–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ma J, Pan P, Anyika M, Blagg BSJ, Dobrowsky RT. Modulating Molecular Chaperones Improves Mitochondrial Bioenergetics and Decreases the Inflammatory Transcriptome in Diabetic Sensory Neurons. Acs Chem Neurosci 2015;6(9):1637–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Anyika M, McMullen M, Forsberg LK, Dobrowsky RT, Blagg BSJ. Development of Noviomimetics as C-Terminal Hsp90 Inhibitors. Acs Med Chem Lett 2015;7(1):67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Forsberg LK, Anyika M, You Z, Emery S, McMullen M, Dobrowsky RT, Blagg BSJ. Development of noviomimetics that modulate molecular chaperones and manifest neuroprotective effects. Eur J Med Chem 2018;143(Med. Res. Rev. 36 1 2016):1428–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang X, Li C, Fowler SC, Zhang Z, Blagg BSJ, Dobrowsky RT. Targeting Heat Shock Protein 70 to Ameliorate c-Jun Expression and Improve Demyelinating Neuropathy. Acs Chem Neurosci 2017;9(2):381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 2003;3(5):330–8. [DOI] [PubMed] [Google Scholar]
- 114.Johnson MR, Diasio RB. Importance of dihydropyrimidine dehydrogenase (DPD) deficiency in patients exhibiting toxicity following treatment with 5-fluorouracil. Adv Enzyme Regul 2001;41(1):151–7. [DOI] [PubMed] [Google Scholar]
- 115.Sofis MJ, Jarmolowicz DP, Kaplan SV, Gehringer RC, Lemley SM, Garg G, Blagg BS, Johnson MA. KU32 prevents 5-fluorouracil induced cognitive impairment. Behav Brain Res 2017;329186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Röhl A, Rohrberg J, Buchner J. The chaperone Hsp90: changing partners for demanding clients. Trends Biochem Sci 2013;38(5):253–62. [DOI] [PubMed] [Google Scholar]
- 117.Echeverría PC, Bernthaler A, Dupuis P, Mayer B, Picard D. An interaction network predicted from public data as a discovery tool: application to the Hsp90 molecular chaperone machine. Plos One 2011;6(10):e26044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kakimura JI, Kitamura Y, Takata K, Umeki A, Suzuki S, Shigaki K, Taniguchi T, Nomura Y, Gebicke-Haerter PJ, Ith MA, Peryy G, Shimohama S. Microglial activation and amyloid-β clearance induced by exogenous heat-shock proteins1. Faseb J 2002;16(6):601–603. [DOI] [PubMed] [Google Scholar]
- 119.Oroz J, Blair LJ, Zweckstetter M. Dynamic Aha1 co-chaperone binding to human Hsp90. Protein Sci Publ Protein Soc 2019;28(9):1545–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shelton LB, Baker JD, Zheng D, Sullivan LE, Solanki PK, Webster JM, Sun Z, Sabbagh JJ, Nordhues BA, Koren J, Ghosh S, Blagg BSJ, Blair LJ, Dickey CA. Hsp90 activator Aha1 drives production of pathological tau aggregates. Proc National Acad Sci 2017;114(36):9707–9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dice JF. Chaperone-mediated autophagy. Autophagy 2007;3(4):295–9. [DOI] [PubMed] [Google Scholar]
- 122.Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow E-M, Cuervo AM, Mandelkow E. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 2009;18(21):4153–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Sci New York N Y 2004;305(5688):1292–5. [DOI] [PubMed] [Google Scholar]
- 124.Finn PF, Mesires NT, Vine M, Dice JF. Effects of small molecules on chaperone-mediated autophagy. Autophagy 2005;1(3):141–5. [DOI] [PubMed] [Google Scholar]
- 125.Wang GP, Khatoon S, Iqbal K, Grundke-Iqbal I. Brain ubiquitin is markedly elevated in Alzheimer disease. Brain Res 1991;566(1–2):146–51. [DOI] [PubMed] [Google Scholar]
- 126.Baldo B, Weiss A, Parker CN, Bibel M, Paganetti P, Kaupmann K. A screen for enhancers of clearance identifies huntingtin as a heat shock protein 90 (Hsp90) client protein. J Biological Chem 2011;287(2):1406–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yoo BC, Kim SH, Cairns N, Fountoulakis M, Lubec G. Deranged expression of molecular chaperones in brains of patients with Alzheimer’s disease. Biochem Bioph Res Co 2001;280(1):249–58. [DOI] [PubMed] [Google Scholar]
- 128.Dabir DV, Trojanowski JQ, Richter-Landsberg C, Lee VM-Y, Forman MS. Expression of the small heat-shock protein alphaB-crystallin in tauopathies with glial pathology. Am J Pathology 2004;164(1):155–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shinohara H, Inaguma Y, Goto S, Inagaki T, Kato K. Alpha B crystallin and HSP28 are enhanced in the cerebral cortex of patients with Alzheimer’s disease. J Neurol Sci 1993;119(2):203–8. [DOI] [PubMed] [Google Scholar]
- 130.Björkdahl C, Sjögren MJ, Zhou X, Concha H, Avila J, Winblad B, Pei J. Small heat shock proteins Hsp27 or αB‐crystallin and the protein components of neurofibrillary tangles: Tau and neurofilaments. J Neurosci Res 2008;86(6):1343–1352. [DOI] [PubMed] [Google Scholar]
- 131.Gorantla NV, Chinnathambi S. Tau Protein Squired by Molecular Chaperones During Alzheimer’s Disease. J Mol Neurosci 2018;66(3):356–368. [DOI] [PubMed] [Google Scholar]
- 132.Zourlidou A, Smith MDP, Latchman DS. HSP27 but not HSP70 has a potent protective effect against alpha-synuclein-induced cell death in mammalian neuronal cells. J Neurochem 2004;88(6):1439–48. [DOI] [PubMed] [Google Scholar]
- 133.Liang JJ. Interaction between beta-amyloid and lens alphaB-crystallin. Febs Lett 2000;484(2):98–101. [DOI] [PubMed] [Google Scholar]
- 134.Stege GJ, Renkawek K, Overkamp PS, Verschuure P, Rijk AF van, Reijnen-Aalbers A, Boelens WC, Bosman GJ, Jong WW de. The molecular chaperone alphaB-crystallin enhances amyloid beta neurotoxicity. Biochem Bioph Res Co 1999;262(1):152–6. [DOI] [PubMed] [Google Scholar]
- 135.Ousman SS, Tomooka BH, Noort JM van, Wawrousek EF, O’Connor KC, Hafler DA, Sobel RA, Robinson WH, Steinman L. Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature 2007;448(7152):474–9. [DOI] [PubMed] [Google Scholar]
- 136.Han MH, Hwang S-I, Roy DB, Lundgren DH, Price JV, Ousman SS, Fernald GH, Gerlitz B, Robinson WH, Baranzini SE, Grinnell BW, Raine CS, Sobel RA, Han DK, Steinman L. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 2008;451(7182):1076–81. [DOI] [PubMed] [Google Scholar]
- 137.Arac A, Brownell SE, Rothbard JB, Chen C, Ko RM, Pereira MP, Albers GW, Steinman L, Steinberg GK. Systemic augmentation of alphaB-crystallin provides therapeutic benefit twelve hours post-stroke onset via immune modulation. P Natl Acad Sci Usa 2011;108(32):13287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pangratz-Fuehrer S, Kaur K, Ousman SS, Steinman L, Liao YJ. Functional rescue of experimental ischemic optic neuropathy with αB-crystallin. Eye Lond Engl 2011;25(6):809–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Velotta JB, Kimura N, Chang SH, Chung J, Itoh S, Rothbard J, Yang PC, Steinman L, Robbins RC, Fischbein MP. αB-crystallin improves murine cardiac function and attenuates apoptosis in human endothelial cells exposed to ischemia-reperfusion. Ann Thorac Surg 2011;91(6):1907–13. [DOI] [PubMed] [Google Scholar]
- 140.Selig EE, Zlatic CO, Cox D, Mok Y-F, Gooley PR, Ecroyd H, Griffin MDW. N- and C-terminal regions of αB-crystallin and Hsp27 mediate inhibition of amyloid nucleation, fibril binding, and fibril disaggregation. J Biological Chem 2020;295(29):9838–9854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Baughman HER, Pham T-HT, Adams CS, Nath A, Klevit RE. Release of a disordered domain enhances HspB1 chaperone activity toward tau. P Natl Acad Sci Usa 2020;117(6):2923–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Muranova LK, Ryzhavskaya AS, Sudnitsyna MV, Shatov VM, Gusev NB. Small Heat Shock Proteins and Human Neurodegenerative Diseases. Biochem Biokhimiia 2019;84(11):1256–1267. [DOI] [PubMed] [Google Scholar]
- 143.Dickey C, Eriksen J, Kamal A, Burrows F, Kasibhatla S, Eckman C, Hutton M, Petrucelli L. Development of a High Throughput Drug Screening Assay for the Detection of Changes in Tau Levels - Proof of Concept with HSP90 inhibitors. Curr Alzheimer Res 2005;2(2):231–238. [DOI] [PubMed] [Google Scholar]
- 144.Lu Y, Ansar S, Michaelis ML, Blagg BSJ. Neuroprotective activity and evaluation of Hsp90 inhibitors in an immortalized neuronal cell line. Bioorgan Med Chem 2009;17(4):1709–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Salminen A, Ojala J, Kaarniranta K, Hiltunen M, Soininen H. Hsp90 regulates tau pathology through co-chaperone complexes in Alzheimer’s disease. Prog Neurobiol 2010;93(1):99–110. [DOI] [PubMed] [Google Scholar]
- 146.Luo W, Dou F, Rodina A, Chip S, Kim J, Zhao Q, Moulick K, Aguirre J, Wu N, Greengard P, Chiosis G. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc National Acad Sci 2007;104(22):9511–9516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biological Chem 2002;277(42):39858–66. [DOI] [PubMed] [Google Scholar]
- 148.Li L, Wang L, You Q-D, Xu X-L. Heat shock protein 90 (Hsp90) Inhibitors: An Update on Achievements, Challenges, and Future Directions. J Med Chem [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 149.Jinwal UK, Trotter JH, Abisambra JF, Koren J, Lawson LY, Vestal GD, O’Leary JC, Johnson AG, Jin Y, Jones JR, Li Q, Weeber EJ, Dickey CA. The Hsp90 kinase co-chaperone Cdc37 regulates tau stability and phosphorylation dynamics. J Biological Chem 2011;286(19):16976–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Liu F, Grundke-Iqbal I, Iqbal K, Gong C-X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. European J Neurosci 2005;22(8):1942–50. [DOI] [PubMed] [Google Scholar]
- 151.Wasik U, Schneider G, Mietelska-Porowska A, Mazurkiewicz M, Fabczak H, Weis S, Zabke C, Harrington CR, Filipek A, Niewiadomska G. Calcyclin binding protein and Siah-1 interacting protein in Alzheimer’s disease pathology: neuronal localization and possible function. Neurobiol Aging 2012;34(5):1380–8. [DOI] [PubMed] [Google Scholar]
- 152.Góral A, Bieganowski P, Prus W, Krzemień-Ojak Ł, Kądziołka B, Fabczak H, Filipek A. Calcyclin Binding Protein/Siah-1 Interacting Protein Is a Hsp90 Binding Chaperone. Plos One 2016;11(6):e0156507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Góral A, Bartkowska K, Djavadian RL, Filipek A. CacyBP/SIP, a Hsp90 binding chaperone, in cellular stress response. Int J Biochem Cell Biology;(Methods 352005): [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 154.Sittler A, Lurz R, Lueder G, Priller J, Lehrach H, Hayer-Hartl MK, Hartl FU, Wanker EE. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum Mol Genet 2001;10(12):1307–15. [DOI] [PubMed] [Google Scholar]
- 155.Auluck PK, Chan HYE, Trojanowski JQ, Lee VMY, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Sci New York N Y 2001;295(5556):865–8. [DOI] [PubMed] [Google Scholar]
- 156.Wang B, Liu Y, Huang L, Chen J, Li JJ, Wang R, Kim E, Chen Y, Justicia C, Sakata K, Chen H, Planas A, Ostrom RS, Li W, Yang G, McDonald MP, Chen R, Heck DH, Liao F-F. A CNS-permeable Hsp90 inhibitor rescues synaptic dysfunction and memory loss in APP-overexpressing Alzheimer’s mouse model via an HSF1-mediated mechanism. Mol Psychiatr 2016;22(7):990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Villadiego J, Labrador‐Garrido A, Franco JM, Leal‐Lasarte M, Genst EJD, Dobson CM, Pozo D, Toledo‐Aral JJ, Roodveldt C. Immunization with α‐synuclein/Grp94 reshapes peripheral immunity and suppresses microgliosis in a chronic Parkinsonism model. Glia 2018;66(1):191–205. [DOI] [PubMed] [Google Scholar]
- 158.Samus Therapeutics. (website: www.samustherapeutics.com)
- 159.Stiegler SC, Rübbelke M, Korotkov VS, Weiwad M, John C, Fischer G, Sieber SA, Sattler M, Buchner J. A chemical compound inhibiting the Aha1–Hsp90 chaperone complex. J Biol Chem 2017;292(41):17073–17083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hessling M, Richter K, Buchner J. Dissection of the ATP-induced conformational cycle of the molecular chaperone Hsp90. Nat Struct Mol Biol 2009;16(3):287–293. [DOI] [PubMed] [Google Scholar]
- 161.Retzlaff M, Hagn F, Mitschke L, Hessling M, Gugel F, Kessler H, Richter K, Buchner J. Asymmetric Activation of the Hsp90 Dimer by Its Cochaperone Aha1. Mol Cell 2010;37(3):344–354. [DOI] [PubMed] [Google Scholar]
- 162.Ihrig V, Obermann WMJ. Identifying Inhibitors of the Hsp90-Aha1 Protein Complex, a Potential Target to Drug Cystic Fibrosis, by Alpha Technology. Slas Discov 2017;22(7):923–928. [DOI] [PubMed] [Google Scholar]
- 163.Sun F, Mi Z, Condliffe SB, Bertrand CA, Gong X, Lu X, Zhang R, Latoche JD, Pilewski JM, Robbins PD, Frizzell RA. Chaperone displacement from mutant cystic fibrosis transmembrane conductance regulator restores its function in human airway epithelia. Faseb J Official Publ Fed Am Soc Exp Biology 2008;22(9):3255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, Gurkan C, Kellner W, Matteson J, Plutner H, Riordan JR, Kelly JW, Yates JR, Balch WE. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 2006;127(4):803–15. [DOI] [PubMed] [Google Scholar]
- 165.Singh JK, Tait B, Guy N, Sivils JC, Culbertson D, Dickey C, Kuo SY, Gestwicki JE, Hutt DM, Dyson JH, Cox MB, Balch WE. Management of Hsp90-Dependent Protein Folding by Small Molecule Targeting the Aha1 Co-Chaperone. Biorxiv 2018;412908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hamos JE, Oblas B, Pulaski-Salo D, Welch WJ, Bole DG, Drachman DA. Expression of heat shock proteins in Alzheimer’s disease. Neurology 1991;41(3):345–50. [DOI] [PubMed] [Google Scholar]
- 167.Harrison PJ, Procter AW, Exworthy T, Roberts GW, Najlerahim A, Barton AJ, Pearson RC. Heat shock protein (hsx70) mRNA expression in human brain: effects of neurodegenerative disease and agonal state. Neuropath Appl Neuro 1993;19(1):10–21. [DOI] [PubMed] [Google Scholar]
- 168.Evans CG, Wisén S, Gestwicki JE. Heat Shock Proteins 70 and 90 Inhibit Early Stages of Amyloid β-(1–42) Aggregation in Vitro. J Biol Chem 2006;281(44):33182–33191. [DOI] [PubMed] [Google Scholar]
- 169.Leverenz JB, Umar I, Wang Q, Montine TJ, McMillan PJ, Tsuang DW, Jin J, Pan C, Shin J, Zhu D, Zhang J. Proteomic identification of novel proteins in cortical lewy bodies. Brain Pathology Zurich Switz 2007;17(2):139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Flower TR, Chesnokova LS, Froelich CA, Dixon C, Witt SN. Heat shock prevents alpha-synuclein-induced apoptosis in a yeast model of Parkinson’s disease. J Mol Biol 2005;351(5):1081–100. [DOI] [PubMed] [Google Scholar]
- 171.Shen H-Y, He J-C, Wang Y, Huang Q-Y, Chen J-F. Geldanamycin induces heat shock protein 70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J Biological Chem 2005;280(48):39962–9. [DOI] [PubMed] [Google Scholar]
- 172.Quigney DJ, Gorman AM, Samali A. Heat shock protects PC12 cells against MPP+ toxicity. Brain Res 2003;993(1–2):133–9. [DOI] [PubMed] [Google Scholar]
- 173.Lindersson E, Beedholm R, Højrup P, Moos T, Gai W, Hendil KB, Jensen PH. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biological Chem 2004;279(13):12924–34. [DOI] [PubMed] [Google Scholar]
- 174.Dedmon MM, Christodoulou J, Wilson MR, Dobson CM. Heat shock protein 70 inhibits alpha-synuclein fibril formation via preferential binding to prefibrillar species. J Biological Chem 2005;280(15):14733–40. [DOI] [PubMed] [Google Scholar]
- 175.Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ. Hsp70 Reduces alpha-Synuclein Aggregation and Toxicity. J Biological Chem 2004;279(24):25497–502. [DOI] [PubMed] [Google Scholar]