

Abstract
Hepatocellular carcinoma (HCC) is a malignant cancer with one of the highest mortality rates. Des‐γ‐carboxyprothrombin (DCP) is an HCC serologic surveillance marker that can complement the low sensitivity of alpha‐fetoprotein (AFP). DCP exists in the blood as a mixture of proteoforms from an impaired carboxylation process at glutamic acid (Glu) residues within the N‐terminal domain. The heterogeneity of DCP may affect the accuracy of measurements because DCP levels are commonly determined using an immunoassay that relies on antibody reactivity to an epitope in the DCP molecule. In this study, we aimed to improve the DCP measurement assay by applying a mass spectrometry (MS)‐based approach for a more inclusive quantification of various DCP proteoforms. We developed a multiple‐reaction monitoring–MS (MRM‐MS) assay to quantify multiple noncarboxylated peptides included in the various des‐carboxylation states of DCP. We performed the MRM‐MS assay in 300 patients and constructed a robust diagnostic model that simultaneously monitored three noncarboxylated peptides. The MS‐based quantitative assay for DCP had reliable surveillance power, which was evident from the area under the receiver operating characteristic curve (AUROC) values of 0.874 and 0.844 for the training and test sets, respectively. It was equivalent to conventional antibody‐based quantification, which had AUROC values at the optimal cutoff (40 mAU/mL) of 0.743 and 0.704 for the training and test sets, respectively. The surveillance performance of the MS‐based DCP assay was validated using an independent validation set consisting of 318 patients from an external cohort, resulting in an AUROC value of 0.793. Conclusion: Due to cost effectiveness and high reproducibility, the quantitative DCP assay using the MRM‐MS method is superior to antibody‐based quantification and has equivalent performance.
Abbreviations
- AFP
alpha‐fetoprotein
- AUROC
area under the receiver operating characteristic curve
- BCLC
Barcelona Clinic Liver Cancer
- CHB
chronic hepatitis B
- CHC
chronic hepatitis C
- CI
confidence interval
- CV
coefficient of variance
- DCP
des‐γ‐carboxyprothrombin
- Gla
γ‐carboxylated glutamic acid
- Glu
glutamic acid
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HPLC
high‐performance liquid chromatography
- LC
liver cirrhosis
- LLOQ
lower limit of quantification
- LOQ
limit of quantification
- MRM‐MS
multiple‐reaction monitoring–mass spectrometry
- MS
mass spectrometry
- PAR
peak area ratio
- ROC
receiver operating characteristic
- SIS
stable isotope‐labeled standard
- ULOQ
upper limit of quantification
- US
ultrasonography
- vol
volume
Liver cancer is the seventh most prevalent cancer worldwide and is the second leading cause of cancer‐related deaths.( 1, 2 ) The most common type of primary liver cancer is hepatocellular carcinoma (HCC), which accounts for approximately 75% of all liver cancer cases.( 2, 3, 4, 5 ) A primary risk factor for HCC is chronic liver cirrhosis due to chronic hepatitis B virus (HBV) or hepatitis C virus (HCV) infection.( 6, 7 )
The prognosis for HCC remains poor, with a 5‐year survival rate of less than 20% in most countries.( 2, 8, 9, 10 ) Consequently, the treatment strategy has shifted toward diagnosing HCC at earlier stages; this strategy has been associated with better survival rates in early stage HCC (70%).( 11, 12, 13 ) Currently, ultrasonography (US) and serum alpha‐fetoprotein (AFP) detection are widely used to surveil at‐risk individuals for the development of HCC( 14, 15, 16 ); however, these methods can often result in misdiagnosis due to the imprecise identification of small tumors in liver cirrhosis backgrounds using US or fluctuations in AFP levels that are caused by benign liver diseases.( 16, 17, 18, 19 ) Further, certain HCCs with normal AFP levels can contribute to the low sensitivity of serum AFP. Therefore, ongoing research has attempted to develop more effective surveillance methods with enhanced sensitivity that can be used independently from or in conjunction with US or serum AFP.( 20, 21, 22, 23 )
Another available marker for HCC surveillance is des‐γ‐carboxyprothrombin (DCP), also known as protein induced by vitamin K absence or antagonist‐II or abnormal prothrombin, which is found at elevated levels in patients with HCC.( 24, 25 ) Several studies have reported that DCP can be used to complement AFP for the early diagnosis of HCC.( 21, 26 ) Normal prothrombin is synthesized as a precursor containing 10 glutamic acid (Glu) residues in the N‐terminal domain (Gla domain), at positions 6, 7, 14, 16, 19, 20, 25, 26, 29, and 32.( 27 ) Under normal conditions, the precursors undergo posttranslational carboxylation of the Glu residues resulting in the conversion of Glu to γ‐carboxylated glutamic acid (Gla) by vitamin K‐dependent glutamyl gamma‐carboxylase in the specific order of 26, 25, 16, 29, 20, 19, 14, 32, 7, and 6.( 28, 29 ) Carboxylation is impaired under conditions of vitamin K deficiency, warfarin treatment, or liver dysfunction,( 30, 31 ) resulting in DCP being released into the bloodstream as a mixture of proteoforms with up to 10 des‐carboxylated Glu residues.( 32 )
DCP concentrations have been determined using a conventional antibody‐based assay featuring a monoclonal antibody produced by the MU3 cell line.( 33 ) The DCP epitope that is recognized by the MU3 antibody is located within the Gla domain at amino acids 17‐27, which includes four Glu residues (19, 20, 25, and 26). Thus, the DCP proteoforms containing some Gla residues at the antibody epitope could have reduced affinity for the MU3 antibody compared with that of the totally noncarboxylated DCP. According to previous studies, the MU3 antibody binds predominantly with DCP molecules containing 9‐10 Glu residues, weakly with those that possess 6‐8 Glu residues, and rarely with those that have less than 5 Glu residues.( 33, 34 ) Recently, several studies aimed to develop discriminative quantification immunoassays for the detection of DCP proteoforms with lower Glu content to overcome this limitation and improve the diagnostic performance of DCP measurement.( 32, 35, 36 ) These studies used additional immunoassays that feature other antibodies, such as 19B7, P‐11, and P‐16, which recognize different epitopes than those that are detected by the MU3 antibody. These studies reported the value of these assays for the detection of DCP proteoforms that contain fewer Glu resides. However, these antibody‐based assays are costly, and because they require the performance of extra and separate immunoassays, they are subject to batch effects.
Multiple‐reaction monitoring–mass spectrometry (MRM‐MS) is a powerful analytical method that can be used to accurately quantify peptides and proteins with high throughput. Recently, the MRM‐MS assay has been shown to be advantageous compared with conventional antibody‐based assays in terms of throughput and the ability to distinguish protein isoforms with common epitopes.( 37, 38, 39 ) In our previous study, we developed an MRM‐MS assay to quantify DCP using a surrogate peptide; this found that the MRM‐MS assay had comparable diagnostic power compared with the conventional immunoassay.( 40, 41 ) However, this MRM‐MS assay remains limited because it only quantifies a surrogate peptide that represents just a small portion of the existing DCP variants.
The objective of the present study was to improve the diagnostic power of the MRM‐MS assay for DCP by inclusively quantifying a wider range of proteoforms with various des‐carboxylation states. In brief, we examined potentially noncarboxylated peptides (referred to as Glu‐peptides) within the Gla domain and developed a robust MRM‐MS assay to quantify multiple Glu‐peptides for the inclusive quantification of DCP proteoforms.
Materials and Methods
Chemicals and Reagents
High‐performance liquid chromatography (HPLC)‐grade solutions, including water, acetonitrile, formic acid, 0.1% formic acid in water, and 0.1% formic acid in acetonitrile, were purchased from Fisher Scientific (Loughborough, United Kingdom). Ammonium bicarbonate (200 mM) solution was purchased from iNtRON Biotechnology (Sungnam, Korea). Dithiothreitol and iodoacetamide were obtained from Merck Co. (Darmstadt, Germany) and Sigma‐Aldrich (St. Louis, MO). RapiGest surfactant was obtained from Waters Corp. (Milford, MA,). Sequencing‐grade chymotrypsin and trypsin were obtained from Promega (Madison, WI). Formic acid was purchased from Fisher Scientific. Stable isotope‐labeled standard (SIS) peptides (heavy peptides) were synthesized by SynPeptide Co., Ltd. (Shanghai, China) (with >99% isotope purity and about >60% purity for individual peptides). Heavy peptides for two tryptic peptides were labeled (13C and 15N) at a C‐terminal arginine (Arg) and lysine (Lys). The SIS peptides for three chymotryptic peptides were labeled (13C and 15N) at C‐terminal tyrosine (Tyr), phenylalanine (Phe,), and leucine (Leu).
Clinical Specimens and Study Design
A total of 618 serum samples were obtained from patients with HCC and at‐risk control patients with chronic hepatitis B (CHB), chronic hepatitis C (CHC), or liver cirrhosis (LC). All patients were recruited from two medical centers in Korea (Asan Medical Center and Samsung Medical Center) and provided informed consent before enrollment. This study was approved by the institutional review boards of the Asan (IRB No. 2017‐1049) and Samsung (IRB No. 2017‐08‐164) Medical Centers. The cohort of 300 patients that was used for model construction and assessment was recruited from Asan Medical Center (cohort A) and included 100 cases of HCC and 200 at‐risk controls (Fig. 1). Seventy percent of the patients were randomly defined as the training set (n = 210), and the remaining patients were defined as the test set (n = 90). The training set was used to construct a diagnostic model, while the test set was used to assess model performance. To validate the model performance in an external cohort, an independent validation set was recruited comprising 318 samples collected from Samsung Medical Center (cohort B), including 184 cases of HCC and 134 at‐risk controls.
FIG. 1.
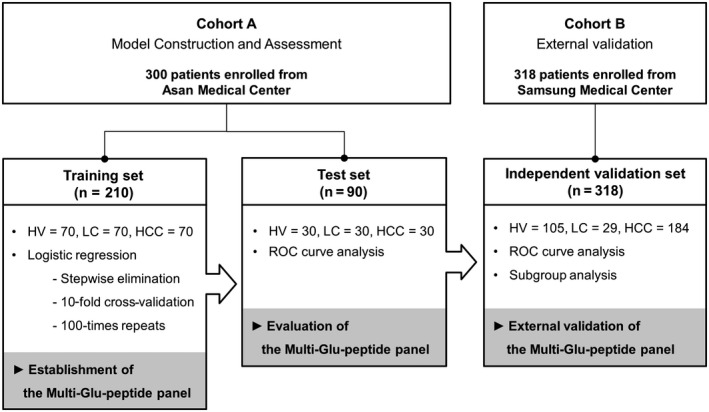
Design of the study. The 618 patients were enrolled in this study from two cohorts (Asan and Samsung Medical Center). The 300 patients from cohort A were randomly divided into the training and test sets to construct the diagnostic model and assess its performance. The diagnostic model was established by stepwise logistic regression in the training set, which consisted of serum samples from 70 patients with HCC and 140 high‐risk controls (70 with HV and 70 with LC). The diagnostic performance of the established model was assessed using the test set, which comprised 90 serum samples from 30 patients with HCC and 60 high‐risk controls (30 with HV and 30 with LC), by ROC curve analysis. The model performance was then validated in an independent validation set from cohort B, including 318 patients consisting of 184 patients with HCC and 134 high‐risk patients (105 with HV and 29 with LC). Additional evaluations for model performance were conducted using subgroups of independent validation sets, such as patients with AFP‐negative and DCP‐negative HCC or very early stage HCC. Abbreviation: HV, patients with hepatitis virus infections.
The HCC diagnosis was confirmed by the results of a histological examination or typical imaging features obtained by US, computed tomography, or magnetic resonance imaging, according to regular clinical practice guidelines.( 42 ) The stages of HCC were defined according to the Barcelona Clinic Liver Cancer (BCLC) classification as follows: very early stage (BCLC stage 0, single nodule <2 cm) and early stage (BCLC stage A, a single 2‐5‐cm lesion or two to three lesions that are each <3 cm). Cirrhosis was defined clinically or radiologically using the following criteria: coarse liver echotexture and nodular liver surface on US, clinical features of portal hypertension (e.g., ascites, splenomegaly, or varices), or thrombocytopenia (platelet count <150 × 1,000/mm3). CHB was defined as the presence of serum hepatitis B surface antigen for more than 6 months. Patients with persistent anti‐HCV and HCV RNA for more than 6 months were defined as CHC. Ages and sex distributions were matched between the control and case groups to the greatest extent possible; the clinical information for each data set and reference values(43) are detailed in Table 1.
Table 1.
Clinical characteristics of the subjects in each cohort
| Characteristics | Cohort A | Cohort B | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Train Set | P Value | Test Set | P Value | Independent Validation Set | P Value | ||||
| (n = 210) | (n = 90) | (n = 318) | |||||||
| Controls | Cases | Controls | Cases | Controls | Cases | ||||
| n | 140 | 70 | 60 | 30 | 134 | 184 | |||
| Age (years) [range] | 53.7 ± 10.0 [28.0‐75.0] | 56.9 ± 9.3 [24.0‐78.0] | 0.0262 | 51.2 ± 8.1 [28.0‐65.0] | 59.7 ± 8.8 [46.0‐74.0] | <0.001 | 50.8 ± 1.7 [24.0‐80.0] | 57.9 ± 9.4 [35.0‐79.0] | <0.001 |
| Sex, n (%) | 0.5926 | 1.0000 | 0.2115 | ||||||
| Male | 108 (77.1%) | 57 (81.4%) | 47 (78.3%) | 23 (76.7%) | 99 (73.9%) | 148 (80.4%) | |||
| Female | 32 (22.9%) | 13 (19.6%) | 13 (21.7%) | 7 (23.3%) | 35 (26.1%) | 36 (19.6%) | |||
| AFP (ng/mL), median [IQR] | 2.6 [1.9‐4.1] | 6.7 [2.8‐32.3] | <0.001 | 2.35 [1.9‐3.3] | 15.85 [4.0‐909.9] | <0.001 | 2.8 [1.8‐3.9] | 8.3 [3.4‐6.1] | <0.001 |
| DCP (mAU/mL), median [IQR] | 15.0 [13.0‐18.5] | 40.0 [24.3‐121.3] | <0.001 | 16.0 [14.0‐18.5] | 36.0 [23.8‐131.9] | <0.001 | 19.5 [15.0‐23.8] | 71.5 [27.8‐268.3] | <0.001 |
| AST (U/L) | 34.5 | 30.5 | 0.2189 | 32.9 | 30.7 | 0.4686 | 44.0 | 156.6 | <0.001 |
| ALT (U/L) | 33.0 | 30.1 | 0.7168 | 36.3 | 27.6 | 0.5403 | 46.8 | 143.8 | <0.001 |
| Prothrombin time (INR) | 1.1 ± 0.1 | 1.0 ± 0.2 | 0.5661 | 1.1 ± 0.2 | 1.0 ± 0.1 | 0.7840 | 1.3 ± 1.9 | 1.2 ± 0.1 | <0.001 |
| Albumin (g/dL) | 4.2 ± 0.4 | 3.9 ± 0.3 | <0.001 | 4.3 ± 0.4 | 3.9 ± 0.4 | <0.001 | 4.5 ± 0.4 | 3.7 ± 0.5 | <0.001 |
| Bilirubin (mg/dL) | 1.2 ± 0.6 | 0.7 ± 0.4 | <0.001 | 1.2 ± 0.9 | 0.6 ± 0.4 | <0.001 | 0.9 ± 0.7 | 1.3 ± 0.6 | <0.001 |
| Creatinine (mg/dL) | 0.8 ± 0.2 | 0.8 ± 0.2 | 0.3185 | 0.9 ± 0.2 | 0.9 ± 0.2 | 0.1606 | 1.0 ± 1.1 | 0.9 ± 0.2 | 0.3119 |
| Etiology, n (%) | 0.8571 | <0.001 | <0.001 | ||||||
| HBV | 96 (68.6%) | 52 (74.3%) | 51 (85.0%) | 20 (66.7%) | 117 (87.3%) | 140 (76.1%) | |||
| HCV | 10 (7.1%) | 4 (5.7%) | 1 (1.7%) | 2 (6.7%) | 15 (11.2%) | 8 (4.3%) | |||
| HBV and HCV | 3 (2.1%) | 1 (1.4%) | 2 (3.3%) | 0 | 0 | 1 (0.5%) | |||
| Others | 31 (22.1%) | 13 (18.6%) | 6 (10.0%) | 8 (26.7%) | 2 (1.5%) | 35 (19.0%) | |||
| Child‐Pugh score, n (%) | 0.0290 | <0.001 | <0.001 | ||||||
| 5 | 121 (86.4%) | 58 (82.9%) | 54 (90.0%) | 22 (73.3%) | 127 (94.8%) | 100 (54.3%) | |||
| 6 | 10 (7.1%) | 12 (17.1%) | 2 (3.3%) | 8 (26.7%) | 6 (4.5%) | 60 (32.6%) | |||
| 7 | 5 (3.6%) | 0 | 3 (5.0%) | 0 | 0 | 22 (12.0%) | |||
| 8 | 4 (2.9%) | 0 | 1 (1.7%) | 0 | 0 | 1 (0.5%) | |||
| 9 | 0 | 0 | 0 | 0 | 1 (0.7%) | 1 (0.5%) | |||
| Child‐Pugh class, n (%) | 0.0476 | 0.3659 | <0.001 | ||||||
| 0 | 1 (0.7%) | 2 (2.9%) | 0 | 0 | 0 | 0 | |||
| A | 130 (92.9%) | 68 (97.1%) | 56 (93.3%) | 30 (100%) | 133 (99.3%) | 160 (87.0%) | |||
| B | 9 (6.4%) | 0 | 4 (6.7%) | 0 | 1 (0.7%) | 24 (13.0%) | |||
| Tumor size (cm) | ‐ | 2.6 ± 1.3 | ‐ | 2.8 ± 1.7 | ‐ | 3.4 ± 2.4 | |||
| Tumor number, n (%) | |||||||||
| 1 | ‐ | 67 (95.7%) | ‐ | 28 (93.3%) | ‐ | 172 (93.5%) | |||
| 2 | ‐ | 2 (2.9%) | ‐ | 2 (6.7%) | ‐ | 12 (6.5%) | |||
| ≥3 | ‐ | 1 (1.4%) | ‐ | 0 | ‐ | 0 | |||
| BCLC stage, n (%) | |||||||||
| 0 | ‐ | 27 (38.6%) | ‐ | 11 (36.7%) | ‐ | 36 (19.6%) | |||
| A | ‐ | 37 (52.9%) | ‐ | 16 (53.3%) | ‐ | 123 (66.8%) | |||
| B | ‐ | 6 (8.6%) | ‐ | 3 (10%) | ‐ | 25 (13.6%) | |||
Except where indicated, data are presented as mean ± SD or median [IQR] (for AFP and DCP). P values for sex, etiology, Child‐Pugh score, Child‐Pugh class were calculated by the chi‐squared test. P values for others were calculated by the t test. Reference ranges for each liver function test are based on laboratory reference values from Massachusetts General Hospital as follows( 43 ): AST, 0‐35 U/L; ALT, 0‐35 U/L; albumin, 3.5‐5.5 g/dL; bilirubin, 0.3‐1.0 mg/dL; creatinine, <1.5 mg/dL; prothrombin time, 11.1‐13.1 seconds. The reference range for prothrombin time as INR is <1.1, per the Mayo Clinic (www.mayoclinic.org).
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; INR, international normalized ratio; IQR, interquartile range.
Candidate Noncarboxylated Peptides Within the Gla Domain of DCP
The Gla domain of prothrombin consists of 46 amino acids at the N‐terminus, including 10 sporadic Gla sequences. We identified potential Glu‐peptides that originated from the Gla domain through an in silico proteolytic digestion using Skyline (McCoss Laboratory, University of Washington). The in silico digestion was performed separately with trypsin and chymotrypsin, and peptides with six to 30 amino acids and without methionine residues were selected to ensure reproducible quantification. Two tryptic peptides (ANTFLEEVR, ECVEETCSYEEAFEALESSTATDVFWAK) and three chymotryptic peptides (EEVRKGNL, ERECVEETCSY, ESSTATDVF) remained as potential targets representing the partially noncarboxylated state of the Gla domain (Supporting Fig. S1). MRM‐MS analysis was then performed to verify the detectability of these five peptides by MS analysis, using corresponding SIS peptides (also referred to as heavy peptides). Among the five Glu‐peptides, the longest tryptic peptide (ECVEETCSYEEAFEALESSTATDVFWAK) did not have any discernable signal due to its long length and hydrophobicity. The other four Glu‐peptides were detected by MRM‐MS and were chosen for further analysis.
Multienzyme Digestion for Sample Preparation
All serum samples were randomized within each set before preparation. The complete sample preparation workflow is shown in Supporting Fig. S2 and detailed in Supporting Table S1. The six most abundant proteins (albumin, immunoglobulin [Ig]G, antitrypsin, IgA, transferrin, and haptoglobin) were depleted using a multiple affinity‐removal system column (Hu‐6, 4.6 × 100 mm; Agilent Technologies, CA) and their exclusive buffers (buffers A and B). The depleted serum was concentrated using a 3‐kDa molecular weight cutoff concentrator (Amicon Ultra‐4 3K; Millipore, MA). The proteins in depleted and concentrated serum samples were quantified by the bicinchoninic acid assay, and 200 µg of proteins were denatured, alkylated, and divided into two equal fractions. Each sample pair was separately digested with trypsin and chymotrypsin to obtain peptides without competing for cleavage sites in a single run while minimizing variations due to prior steps. The incubation was performed at 37°C for 4 hours and was stopped by the addition of formic acid. The supernatant was transferred to clean tubes after centrifugation at 16,602g at 4°C for 1 hour to remove the by‐products of RapiGest‐SF. The individual enzymatic digests were mixed evenly and spiked with corresponding heavy peptides before the MRM‐MS analysis.
Selection of a Quantifier Ion for Each Target Peptide
We experimentally screened the six intense transitions as an initial list of MRM‐MS transitions using the heavy peptides. The best transition was selected as a quantifier ion, considering the results of the reversed response curve analysis and the Automated Detection of Inaccurate and imprecise Transitions (AuDIT) algorithm,( 44 ) according to the following criteria: (1) the best linearity of the response curve (based on the correlation coefficient, R 2); (2) the lower limit of quantification (LLOQ) value was lowest among the transitions; and (3) interference‐free status from AuDIT results.
The background matrix for the response curves was prepared using 100 µg of proteins from depleted pooled hepatitis serum for each enzyme fraction. The calibration points were generated by mixing the background matrix with variable amounts of heavy peptides from 78.13 fmol to 20 pmol, over a 100‐fold range. All calibration points were sequentially analyzed, followed by a blank sample (0.1% formic acid in HPLC water), from zero sample (matrix only) to the highest concentration point in triplicate. The peak area ratio (PAR) was calculated with the peak area of heavy peptides normalized against that of corresponding endogenous (light) peptides existing in a matrix. Linear regression analysis was conducted on the plot in which the PAR of heavy peptides to light peptides was plotted against the nominal concentration of heavy peptides on a log10 scale.
The limit of detection (LOD) and limit of quantification (LOQ) were calculated based on the averaged PAR, plus 3 times and 10 times the SD for a zero sample that was analyzed in triplicate, respectively. The LLOQ was determined as the lowest concentration at which the precision was under 20%, the accuracy was within 20%, and the signal‐to‐noise (S/N) ratio over 5, representing the first point of the response curve. Similarly, the upper LOQ (ULOQ) was defined as the highest concentration on the response curve showing the precision under 20% and the accuracy was within 20%, representing the last point of the response curve. The analytical information and AuDIT results for the quantifier ion used for each peptide and their response curves are shown in Supporting Tables S2 and S3, respectively.
Quantitative MRM‐MS Analysis
The quantification of target peptides for DCP was performed on an Agilent 6490 triple quadrupole MS (Agilent Technologies) with a Jetstream electrospray source coupled with a 1260 Infinity HPLC system (Agilent Technologies). The liquid chromatography–MS system was controlled by MassHunter (vB06.01; Agilent Technologies) software for the establishment of a scheduled MRM‐MS method and data acquisition.
The total liquid chromatography assay was performed over 70 minutes, with a binary gradient consisting of mobile phase A (water 0.1% volume [vol]/vol formic acid) and mobile phase B (acetonitrile 0.1% vol/vol formic acid). Twenty microliters of chymotryptic and tryptic peptides was injected into the guard column (2.1 mm × 30 mm internal diameter [id], 1.8 µm particle size; Agilent Zorbax SB‐C18), which was maintained at 40°C. After online desalting for 10 minutes at 5 µL/minute with 3% B, the peptides were subjected to a reversed‐phase analytical column (150 mm × 0.5 mm id, 3.5 µm particle size; Agilent Zorbax SB‐C18) maintained at 40°C. The separation of the peptides was conducted with a binary gradient of 3% to 35% B through the column for 45 minutes at 40 µL/minute. Equilibration of the column for the next run was performed by raising the gradient to 70% B for 5 minutes and then lowering it to 3% B for 10 minutes.
The ion spray capillary voltage was 2,500 V, and the nozzle voltage was 2,000 V. The drying gas and sheath gas were set to flow at 15 L/minute at 250°C and 12 L/minute at 250°C, respectively. The voltage of the cell accelerator was adjusted to 5 V. The fragment voltage and the delta electron multiplier voltage were set to 380 V and 200 V, respectively. The resolution mode of the first and third quadrupoles was set to unit mode.
Data Analysis
Quantitative analysis after MS analysis was performed using Skyline (McCoss Laboratory), which handled the MRM‐MS raw data files from import to alignment and was used to conduct peak area calculations for transitions. The raw data were processed in Skyline, and each data point was smoothed by the Savitzky‐Golay method. The PAR of the endogenous peptide to the heavy peptide for each peptide was used to represent the relative abundance of the peptide in each sample.
In the training set, a DCP multi‐Glu‐peptide panel was constructed to discriminate cases from controls by stepwise backward logistic regression with 10‐fold cross‐validation (100 times repeated). The stepwise backward elimination strategy was used to maximize the opportunity to identify the best combination of Glu‐peptides for the discriminative quantification of DCP proteoforms between cases and control groups. The 10‐fold cross‐validation approach was used to avoid the overfitting of the model. A receiver operating characteristic (ROC) curve was used to generate area under the ROC curve (AUROC) values to evaluate the predictive ability of the DCP multi‐Glu‐peptide panel in each data set. The cut‐off point was identified by calculating the Youden Index (J = max [sensitivity + specificity – 1]) for the training set. The relative differences in abundance for each peptide in the panel were compared between the control and case groups using the Mann‐Whitney U test. DeLong`s tests were conducted to compare the AUROC values. All reported P values are from two‐sided tests, and two‐tailed P < 0.05 was considered significant.
All statistical analyses were performed using R (version 3.6.3; R Foundation, Vienna, Austria), IBM SPSS (version 25.0; IBM, Chicago, IL), and GraphPad Prism (version 6.0; GraphPad, San Diego, CA).
Results
Reversed Response Curves for Four Candidate Glu‐Peptides in Depleted Human Serum
The reversed response curves for four DCP Glu‐peptides are shown in Supporting Fig. S3. Each curve satisfied the U.S. Food and Drug Administration guidelines for validating response curves( 22 ); more than six calibration points were composed in each curve, and the coefficients of variance (CVs) of the measurements (n = 3) at all points in the curve were below 20% (Supporting Table S3). All correlation coefficients (R 2) of the response curve were above 0.99, except that for the ESSTATDVF peptide, which had an R 2 value that was slightly lower than the others (0.9872). The LOD, LOQ, LLOQ, and ULOQ values for the quantifier ion in each of the four Glu‐peptides are summarized in Supporting Fig. S3, and the results of the linear regression analyses for the response curve for each peptide are summarized in Supporting Table S4.
The analytical sensitivities of the target peptides at the LLOQ concentration met the requirements for precision, accuracy, and S/N criteria described in the Materials and Methods section (Supporting Table S5). The potential interferences of the analytes in the biological samples were inspected as the analytical specificity of individual serum samples from 6 patients with hepatitis. The interference values of peptides satisfied the standard criteria in all samples (interference <20%), as shown in Supporting Table S6. The average interference values of six matrices for the ANTFLEEVR, ERECVEETCSY, ESSTATDVF, and EEVRKGNL peptides were 6.4%, 3.2%, 7.5%, and 6.8%, respectively. The carryovers were inspected to ensure that the ULOQ samples would not affect the subsequent sequential analysis of specimens. The average carryover of the four analytes ranged from 3.28% to 12.10%, which met the criteria (carryover <20%; Supporting Table S7).
Reproducibility of the MRM‐MS Assay Using the Multienzyme Digestion Workflow
We evaluated the reproducibility of the total MRM‐MS assay that used the multienzyme digestion workflow with depleted pooled HCC serum. The serum was prepared over 5 days and analyzed daily in triplicate. The average CV values of each target peptide were under 20% in both the intra‐assay and interassay analyses, as shown in Supporting Table S8. The average CV values of the intra‐assay analysis ranged from 7.03% to 17.35%. The corresponding values for the interassay analysis ranged from 14.81% to 17.67%. These results demonstrate that the total MRM‐MS assay using the multienzyme digestion workflow is stable for the quantitation of four peptides over several days.
Construction of the DCP Multi‐Glu‐Peptide Panel
As a result of the logistic regression analysis performed on the training set, a multi‐Glu‐peptide panel containing three Glu‐peptides (ANTFLEEVR, ERECVEETCSY, and ESSTATDVF) was established as the best panel for predicting HCC, eliminating a nonsignificant Glu‐peptide (EEVRKGNL). The three Glu‐peptides contributed significantly to the panel (P < 0.005), as indicated by the final logistic model, which is detailed in Table 2. The three‐Glu‐peptide panel obtained an AUROC of 0.873 (95% confidence interval [CI], 0.818‐0.928), with a sensitivity of 71.4% and a specificity of 90.0% at the optimal cut‐off value in the training set (Fig. 2A). The constructed panel achieved a greater AUROC value than each individual Glu‐peptide (AUROC values: 0.801 for ERECVEETCSY, 0.734 for ESSTATDVF, and 0.561 for ANTFLEEVR; Supporting Fig. S4). The predictive performance of the three‐Glu‐peptide panel was consistent in the test set, with an AUROC value of 0.844 (95% CI, 0.761‐0.928), which was equivalent to the AUROC value of 0.873 obtained for the training set (DeLong`s test, P = 0.5722; Fig. 2A).
Table 2.
Summary of the DCP three‐Glu‐peptide logistic regression model
| Estimate | Standard Error | z Value | P Value | |
|---|---|---|---|---|
| (Intercepts) | −5.202 | 1.0066 | −5.168 | 2.37E‐07 |
| ANTFLEEVR | −0.9199 | 0.3111 | −2.957 | 0.00311 |
| ERECVEETCSY | 11.1108 | 1.8697 | 5.942 | 2.81E‐09 |
| ESSTATDVF | 2.1785 | 0.5023 | 4.337 | 1.44E‐05 |
A logistic regression model to predict the probability of having HCC (P) was built with the following equation: logit(P) = log(P/[1 − P]) = −5.202 0.9199 × ANTFLEEVR + 11.1108 × ERECVEETCSY + 2.1785 × ESSTATDVF. The numeric values of each peptide in the equation were raw values for relative concentrations (PAR of endogenous light peptides to heavy SIS peptides). The optimal cut‐off value for the above equation is 0.432.
FIG. 2.
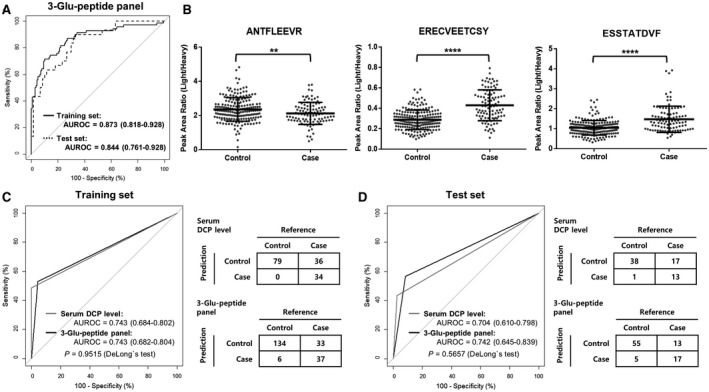
Assessment of the diagnostic performance of the DCP three‐Glu‐peptide panel in the training and test sets and comparison with the immunoassay at the optimal cut‐off point. (A) ROC curves for the DCP three‐Glu‐peptide panel in the training (solid black line) and test sets (dotted black line). The AUROC of the DCP three‐Glu‐peptide panel was 0.873 in the training set, which was consistent with the test set, which had an AUROC of 0.844 (DeLong`s test, P = 0.5722). (B) The relative concentrations of three Glu‐peptides in the training and test sets. The relative concentrations of ANTFLEEVR peptide were significantly lower in cases than in controls, whereas those for ERECVEETCSY and ESSTATDVF were significantly higher in cases than in controls. The comparisons of relative concentrations separately plotted for the training and test sets are shown in Supporting Fig. S5. (C,D) Comparison of the ROC curve and the corresponding confusion matrix at the optimal cutoff for the DCP three‐Glu‐peptide panel and the serum DCP level from the immunoassay, respectively, in the (C) training set and (D) test set. The optimal cut‐off value for the serum DCP level (gray lines) was 40 mAU/mL and that of the three‐Glu‐peptide panel (black lines) was 0.432, presented by the Youden Index for the training set (shown in Supporting Fig. S4). The AUROC of the DCP three‐Glu‐peptide panel was statistically equivalent to that of the immunoassay at the optimal cut‐off value in the training and test sets (DeLong`s test, P > 0.05). All AUROC values are summarized with 95% CI for ROC curves. Relative concentrations of three Glu‐peptides were plotted as PARs of light peptides to heavy SIS peptides for individual patients. Middle horizon lines and error bars indicate the mean and SD, respectively. P values were calculated with the Mann‐Whitney U test to compare the relative concentrations of each peptide. **P < 0.01, ****P < 0.0001.
The levels of three peptides in the 300 individual samples of both the training and test sets were plotted as scatter dot plots, with lines showing the mean and SD (Fig. 2B); these were plotted separately for the training and test sets (Supporting Fig. S5). By Mann‐Whitney U test, we found that the levels of the three Glu‐peptides were significantly altered in cases compared with controls (P < 0.05). Notably, the level of the ANTFLEEVR peptide was significantly decreased in the HCC case group compared with that in the control group, whereas the levels of the other two peptides were significantly elevated in the HCC case group.
Comparison of the MRM‐MS Assay and the Immunoassay
To assess whether the diagnostic performance of the DCP three‐Glu‐peptide panel based on the MRM‐MS assay was comparable to the diagnostic performance of measuring serum DCP levels using the immunoassay, we conducted ROC curve analysis at the optimal cut‐off value. The optimal cutoff of the serum DCP level is 40 mAU/mL, whereas that of the three‐Glu‐peptide panel was 0.432, as determined by the Youden Index of the training set. The AUROC values for the quantitative MRM‐MS assay were 0.743 and 0.742 in the training and test sets, respectively, whereas those for the immunoassay were 0.743 and 0.704 (Fig. 2C,D). The AUROCs of the DCP three‐Glu‐peptide panel were statistically equivalent to those of the immunoassay for each set, based on DeLong`s test (P > 0.05). Moreover, the MRM‐MS assay provided higher accuracy than the immunoassay. The accuracies of the MRM‐MS assay were 0.814 (95% CI, 0.755‐0.865) and 0.800 (95% CI, 0.703‐0.877) for the training and test sets, respectively, whereas those for the immunoassay were 0.758 (95% CI, 0.682‐0.825) and 0.739 (95% CI, 0.619‐0.838). Further, the DCP levels by immunoassay and the logit(P) values from the DCP three‐Glu‐peptide panel correlated weakly (Pearson`s correlation, R = 0.24; P = 2.3e‐05), as shown in Supporting Fig. S6.
Combined Model of the DCP Three‐Glu‐Peptide Panel and the Serum AFP Levels
We conducted further logistic regression analyses to determine whether combining the DCP panel with serum AFP levels could enhance the predictive power for HCC detection. The combined model using both the three‐Glu‐peptide panel and serum AFP levels increased the AUROC values to 0.903 (95% CI, 0.855‐0.952) for the training set (Fig. 3A). The combined model outperformed serum AFP levels (AUROC, 0.770; 95% CI, 0.698‐0.842) for the training set, based on DeLong`s test (P < 0.05). Similarly, the AUROC value of the combined model significantly increased from 0.844 to 0.913 (95% CI, 0.851‐0.974) for the test set. Notably, the combined model had greater sensitivity in both the training and test sets (68.9% and 76.7%, respectively) compared with the low sensitivity of serum AFP levels alone (35.7% and 56.7%, respectively), as shown in Supporting Fig. S7C.
FIG. 3.
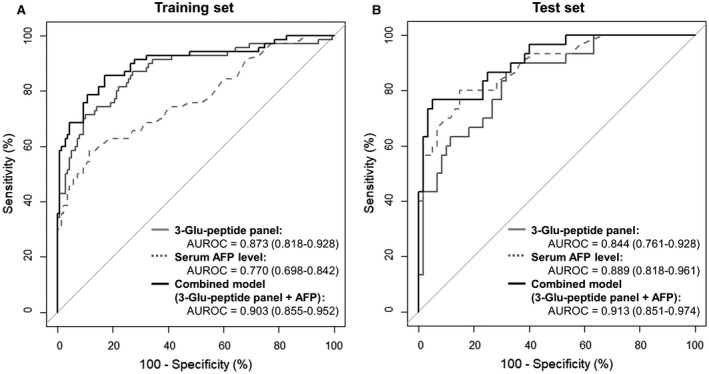
Comparison of the diagnostic performances of the DCP three‐Glu‐peptide panel, serum AFP levels, and a combined model (DCP three‐Glu‐peptide panel and serum AFP level) in the training and test sets. AUROC values are presented with 95% CI for ROC curves. The combined model (black lines) had a higher AUROC value than the DCP three‐Glu‐peptide panel (solid gray lines) or serum AFP levels (dotted gray lines) alone, for all data sets. (A) In the training set, the combined model had a higher AUROC value (0.903) than either AFP levels (0.770) or DCP three‐Glu‐peptide panel (0.873) alone. The AUROC value for the combined model was statistically different from that for AFP levels (DeLong`s test, P < 0.05) but not for the DCP three‐Glu‐peptide panel (DeLong`s test, P = 0.079). (B) In the test set, the combined model also had a higher AUROC value (0.913) than both the AFP levels (0.889) and the DCP three‐Glu‐peptide panel (0.844). The AUROC value for the combined model was statistically different from that for the DCP three‐Glu‐peptide panel (DeLong`s test, P < 0.05) but not from AFP levels (DeLong`s test, P = 0.484).
Validation of the DCP Three‐Glu‐Peptide Panel and the Combined Model With AFP Levels in an External Cohort
We analyzed another 318 samples from an external cohort as an independent validation set; this consisted of 134 controls and 184 cases (Table 1). The AUROC values of the three‐Glu‐peptide panel and the combined model with AFP levels were 0.793 (95% CI, 0.745‐0.842) and 0.863 (95% CI, 0.822‐0.903), respectively, for the independent validation set (Fig. 4A). The AUROC values for the three‐Glu‐peptide panel and the combined model were statistically equivalent to those identified in the test set (0.844 and 0.913, respectively) based on the results of DeLong`s test (P > 0.05). Moreover, the Mann‐Whitney U test revealed that the levels of each peptide were also significantly different between the control and case groups, as shown in Fig. 4B (P < 0.0001). The combined model had greater sensitivity compared with serum AFP levels alone in the independent validation set (45.1% to 64.1%), whereas the other diagnostic performances of the panel remained equivalent, as detailed in Table 3 and Supporting Fig. S7.
FIG. 4.
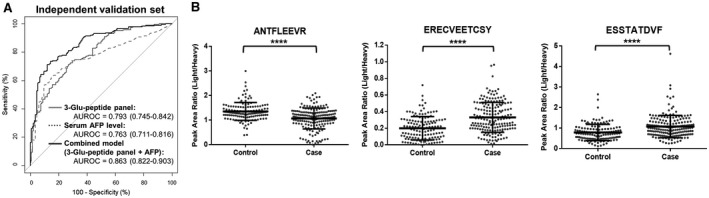
Validation of the diagnostic performance of the DCP three‐Glu‐peptide panel, serum AFP levels, and the combined model (DCP three‐Glu‐peptide panel and serum AFP level) in the independent validation set. (A) ROC curves for the DCP three‐Glu‐peptide panel (solid gray lines), serum AFP levels (dotted gray lines), and the combined model (black lines) for the independent validation set. AUROC values are presented with 95% CI for ROC curves. Differences between the AUROC value for the combined model and those for the DCP three‐Glu‐peptide panel and serum AFP levels were significant for the independent validation set (DeLong`s test, P < 0.005). The diagnostic performance of the DCP panel was consistent in the independent validation set, although the performance was slightly higher in the training set (AUROC = 0.873) than in the independent validation set (DeLong`s test, P = 0.0336). (B) Relative concentrations of three Glu‐peptides in cohort B. The tendencies for the relative concentrations of each peptide were consistent in the independent validation set with those observed in the training set. The relative concentrations of three Glu‐peptides were plotted as PARs of the light peptides to heavy SIS peptides for individual patients. Middle horizon lines and error bars indicate the mean and SD, respectively. P values were calculated using the Mann‐Whitney U test to compare the relative concentrations of each peptide. ****P < 0.0001.
Table 3.
Comparison of diagnostic performance of the three‐Glu‐peptide panel, AFP level, and combined model in each data set
| Diagnostic performance | AUROC (95% CI) | Sensitivity (%) | Specificity (%) | PPV (%) | NPV (%) | Accuracy (95% CI) | ||
|---|---|---|---|---|---|---|---|---|
| Training set | P value | |||||||
| DCP three‐Glu‐peptide panel | 0.873 | 52.9 | 95.7 | 86.1 | 80.2 | 0.814 | ||
| (0.818‐0.928) | (0.755‐0.865) | |||||||
| AFP level | 0.770 | <0.05 | * | 35.7 | 97.9 | 89.3 | 75.3 | 0.771 |
| (0.698‐0.842) | (0.709‐0.826) | |||||||
| DCP three‐Glu‐peptide panel + AFP level | 0.903 | 0.079 | † | 68.9 | 94.3 | 85.7 | 85.7 | 0.857 |
| (0.855‐0.952) | (0.802‐0.902) | |||||||
| Test set | P value | |||||||
| DCP three‐Glu‐peptide panel | 0.844 | 56.7 | 91.7 | 77.3 | 80.9 | 0.800 | ||
| (0.761‐0.928) | (0.703‐0.877) | |||||||
| AFP level | 0.889 | 0.393 | * | 56.7 | 98.3 | 94.4 | 81.9 | 0.844 |
| (0.818‐0.961) | (0.7528‐0.912) | |||||||
| DCP three‐Glu‐peptide panel + AFP level | 0.913 | <0.05 | † | 76.7 | 90.0 | 79.3 | 88.5 | 0.856 |
| (0.851‐0.974) | (0.766‐0.921) | |||||||
| Independent validation set | P value | |||||||
| DCP three‐Glu‐peptide panel | 0.793 | 37.0 | 95.5 | 91.9 | 52.5 | 0.616 | ||
| (0.745‐0.842) | (0.561‐0.670) | |||||||
| AFP level | 0.764 | 0.408 | * | 45.1 | 91.8 | 88.3 | 54.9 | 0.648 |
| (0.711‐0.816) | (0.593‐0.700) | |||||||
| DCP three‐Glu‐peptide panel + AFP level | 0.862 | <0.05 | † | 64.1 | 90.3 | 90.1 | 64.7 | 0.752 |
| (0.822‐0.903) | (0.700‐0.798) | |||||||
DCP three‐Glu‐peptide panel versus AFP level.
DCP three‐Glu‐peptide panel with versus without AFP level.
Abbreviations: NPV, negative predictive value; PPV, positive predictive value.
Diagnostic Performance for Subgroups of the Independent Validation Set
We examined the diagnostic abilities of the panel that was constructed using three DCP Glu‐peptides to distinguish patients with HCC in various subgroups of the independent validation set. First, we examined the performance of the three‐Glu‐peptide panel in the AFP‐negative and DCP‐negative subgroup, consisting of 127 patients at risk and 39 patients with HCC, with AFP and DCP levels below the reference values. The AUROC values of the three‐Glu‐peptide panel and the combined model with AFP levels were 0.803 (95% CI, 0.726‐0.880) and 0.821 (95% CI, 0.739‐0.903), respectively, for the AFP‐negative and DCP‐negative subgroup (Fig. 5A). Notably, our three‐Glu‐peptide panel could discriminate 18 patients with HCC, corresponding to approximately half of the 39 patients with HCC with AFP and DCP levels below the reference values, reducing the false‐negative rate.
FIG. 5.
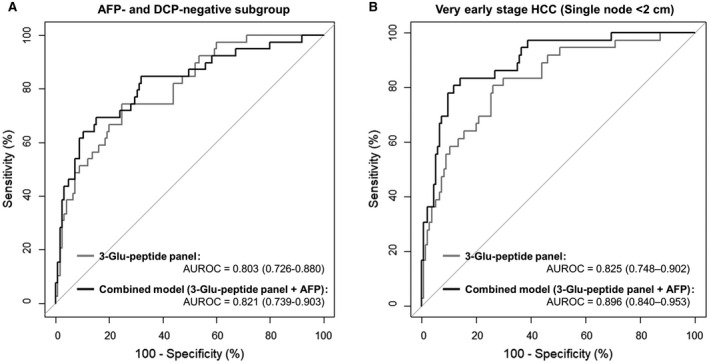
HCC surveillance abilities of the DCP three‐Glu‐peptide panel and the combined model (DCP three‐Glu‐peptide panel and serum AFP level) in subgroups of the independent validation set. (A) ROC curves for the AFP‐negative and DCP‐negative subgroup, consisting of 127 at‐risk patients and 39 patients with HCC with <20 ng/mL for AFP and <40 mAU/mL for DCP. (B) ROC curves for the discrimination of very early stage HCC (tumor size <2 cm) cases, consisting of 36 patients from 134 at‐risk controls. All AUROC values are summarized with 95% CI for the ROC curves. The three‐Glu‐peptide panel (gray lines) and the combined model (black lines) with serum AFP levels showed reliable surveillance performance for patients with very early stage HCC and patients with HCC with serum AFP and DCP values under the corresponding reference values, as indicated by AUROC values over 0.8.
We also analyzed the diagnostic performance of the DCP 3‐Gu‐peptide panel for detecting very early stage HCC (BCLC 0, single lesion <2 cm; Fig. 5B). The very early stage HCC subgroup in the independent validation set consisted of 36 cases. According to the ROC analysis results for the detection of very early stage patients with HCC from among at‐risk controls, the AUROC value of the three‐Glu‐peptide panel was 0.825 (95% CI, 0.748‐0.902). Further, a combined model using both serum AFP levels and the three‐Glu‐peptide panel had significantly greater diagnostic power for very early stage HCC, with an AUROC of 0.896 (95% CI, 0.840‐0.953). The diagnostic power of the three‐Glu‐peptide panel and combined model in discriminating very early stage HCC was comparable with the overall performance in the original validation set by DeLong`s test (P > 0.05). Further, no significant differences were observed between the AUROC values of each panel for distinguishing very early stage HCC from control subgroups that were stratified by etiology and the entire control group (DeLong`s test, P > 0.05), as shown in Table 4. This tendency was maintained when assessing discriminating early stage HCC (DeLong`s test, P > 0.05).
Table 4.
Comparison of diagnostic performance of the three‐Glu‐peptide panel, AFP level, and combined model for discriminating early stage HCC from stratified controls
| Diagnostic Performance | Very Early Stage HCC (BCLC 0) | Early Stage HCC (BCLC 0 and A) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (n = 36) | (n = 159) | |||||||||||||||
| AUROC (95% CI) | Sensitivity (%) | Specificity (%) | PPV (%) | NPV (%) | Accuracy (95% CI) | AUROC (95% CI) | Sensitivity (%) | Specificity (%) | PPV (%) | NPV (%) | Accuracy (95% CI) | |||||
| Controls (n = 134) | P value | P value | ||||||||||||||
| DCP three‐Glu‐peptide panel | 0.825 | 36.1 | 95.5 | 68.4 | 84.8 | 0.829 | 0.794 | 38.4 | 95.5 | 91.0 | 56.6 | 0.645 | ||||
| (0.748‐0.902) | (0.764‐0.883) | (0.744‐0.845) | (0.587‐0.700) | |||||||||||||
| AFP value | 0.764 | 0.376 | * | 50.0 | 91.8 | 62.1 | 87.2 | 0.829 | 0.754 | 0.293 | * | 44.7 | 91.8 | 86.6 | 58.3 | 0.662 |
| (0.659‐0.869) | (0.764‐0.883) | (0.698‐0.811) | (0.605‐0.716) | |||||||||||||
| DCP three‐Glu‐peptide panel + AFP value | 0.896 | <0.05 | † | 69.4 | 90.3 | 65.8 | 91.7 | 0.859 | 0.864 | <0.05 | † | 64.8 | 90.3 | 88.8 | 68.4 | 0.765 |
| (0.840‐0.953) | (0.797‐0.907) | (0.823‐0.906) | (0.712‐0.812) | |||||||||||||
| CHB with no LC (n = 92) | P valve | P value | ||||||||||||||
| DCP three‐Glu‐peptide panel | 0.835 | 36.1 | 97.8 | 86.7 | 79.7 | 0.805 | 0.803 | 38.4 | 97.8 | 96.8 | 47.9 | 0.602 | ||||
| (0.755‐0.915) | (0.725‐0.869) | (0.750‐0.857) | (0.538‐0.663) | |||||||||||||
| AFP value | 0.793 | 0.547 | * | 50.0 | 93.5 | 75.0 | 82.7 | 0.813 | 0.787 | 0.686 | * | 44.7 | 93.5 | 92.2 | 49.4 | 0.626 |
| (0.731‐0.844) | (0.734‐0.876) | (0.731‐0.844) | (0.562‐0.686) | |||||||||||||
| DCP three‐Glu‐peptide panel + AFP value | 0.915 | <0.05 | † | 69.4 | 94.6 | 83.3 | 88.8 | 0.875 | 0.884 | <0.05 | † | 64.8 | 94.6 | 95.4 | 60.8 | 0.757 |
| (0.861‐0.969) | (0.805‐0.927) | (0.842‐0.926) | (0.699‐0.809) | |||||||||||||
| CHC with no LC (n = 13) | P value | P value | ||||||||||||||
| DCP three‐Glu‐peptide panel | 0.729 | 36.1 | 84.6 | 86.7 | 32.4 | 0.490 | 0.687 | 38.4 | 84.6 | 96.8 | 10.1 | 0.419 | ||||
| (0.557‐0.900) | (0.344‐0.637) | (0.536‐0.837) | (0.344‐0.496) | |||||||||||||
| AFP value | 0.682 | 0.683 | * | 50.0 | 100.0 | 100.0 | 41.9 | 0.813 | 0.663 | 0.799 | * | 44.7 | 100.0 | 100.0 | 12.9 | 0.488 |
| (0.537‐0.846) | (0.734‐0.876) | (0.574‐0.753) | (0.412‐0.566) | |||||||||||||
| DCP three‐Glu‐peptide panel + AFP value | 0.840 | <0.05 | † | 69.4 | 84.6 | 92.6 | 50.0 | 0.735 | 0.801 | <0.05 | † | 64.8 | 84.6 | 98.1 | 16.4 | 0.663 |
| (0.714‐0.965) | (0.589‐0.851) | (0.691‐0.911) | (0.587‐0.733) | |||||||||||||
| LC (n = 29) | P value | P value | ||||||||||||||
| DCP three‐Glu‐peptide panel | 0.835 | 36.1 | 93.1 | 86.7 | 54.0 | 0.615 | 0.816 | 38.4 | 93.1 | 96.8 | 21.6 | 0.468 | ||||
| (0.731‐0.939) | (0.486‐0.734) | (0.723‐0.909) | (0.723‐0.909) | |||||||||||||
| AFP value | 0.707 | 0.140 | * | 50.0 | 82.8 | 78.3 | 57.1 | 0.646 | 0.691 | 0.074 | * | 44.7 | 82.8 | 93.4 | 21.4 | 0.505 |
| (0.576‐0.838) | (0.518‐0.761) | (0.591‐0.791) | (0.432‐0.579) | |||||||||||||
| DCP three‐Glu‐peptide panel + AFP value | 0.861 | 0.533 | † | 69.4 | 79.3 | 80.7 | 67.7 | 0.739 | 0.831 | 0.650 | † | 64.8 | 79.3 | 94.5 | 29.1 | 0.670 |
| (0.769‐0.953) | (0.615‐0.840) | (0.749‐0.914) | (0.598‐0.737) | |||||||||||||
DCP three‐Glu‐peptide panel versus AFP level.
DCP three‐Glu‐peptide panel with versus without AFP level. No significant differences were found between the AUROC values of each panel for distinguishing very early stage or early stage HCC from stratified control subgroups and the entire control group (DeLong`s test, P > 0.05).
Abbreviations: NPV, negative predictive value; PPV, positive predictive value.
Discussion
According to recent studies, the γ‐carboxylation of the 10 Glu residues in the N‐terminal Gla domain of DCP occurs in a specific order, resulting in blood DCP populations consisting of a heterogeneous mixture of 10 possible proteoforms (Supporting Fig. S8).( 28, 29 ) To quantify the DCP proteoforms inclusively, we constructed a quantitative assay for DCP measurement to simultaneously monitor three noncarboxylated peptides within the Gla domain, using the MRM‐MS method. The three monitored Glu‐peptides could be obtained from different subgroups of DCP proteoforms (Supporting Table S9). The ANTFLEEVR peptide contains the ninth and tenth carboxylated residues and represents the subgroup of DCP containing more than two Glu residues (two to 10 Glu residues). Similarly, the ESSTATDVF peptide contains the eighth carboxylated Glu residue and represents the subgroup of DCP with more than three Glu residues (three to 10 Glu residues). The ERECVEETCSY peptide (referred to as ERE peptide) contains the third, fifth, sixth, and seventh carboxylated Glu residues and represents the subgroup of DCP with more than eight Glu residues (eight to 10 Glu residues). Therefore, our quantification assay is able to detect both substantially des‐carboxylated forms and less des‐carboxylated forms within the same batch, requiring lower cost and labor than immunoassay detection methods and with minimized potential for variations due to batch effects.
In the present study, the ERECVEETCSY peptide quantity significantly increased in the HCC group, implying that DCP proteoforms with more than eight Glu residues (eight to 10 Glu residues) are elevated in patients with HCC relative to the control group (fold change, 1.70; P < 0.0001). The increasing tendency in DCP proteoforms observed for the HCC group was consistent in the subgroup containing a wider range of DCP proteoforms, as represented by the ESSTATDVF peptide quantity (three to 10 Glu residues; fold change, 1.35; P < 0.0001). However, the ANTFLEEVR peptide was elevated in control patients (fold change, 0.85; P < 0.0001). When considering that the ANTFLEEVR peptide targets the DCP with two Glu residues as well as the DCP proteoforms that were targeted by the ESSTATDVF peptide (Supporting Table S9), the DCP with two Glu residues appeared to constitute a higher proportion of the DCP population in the control versus case group. Meanwhile, the DCP variants with two Glu residues have approximately half the activity of normal prothrombin.( 45, 46 ) Presumably, a larger portion of DCP variants with two Glu residues would be beneficial for benign liver diseases than for the HCC group, although the direct impact of prothrombin activity in the progression of HCC remains unknown. The combination of the relatively lower level of ANTFLEEVR peptides and a higher level of ERECVEETCSY or ESSTATDVF peptide could be used to characterize the DCP proteoforms that are synthesized during HCC rather than benign liver diseases.
Our work has some limitations. First, our assay was unable to cover the three Glu residues at positions 25, 26, and 29, which are located within either a long tryptic peptide with poor ionization or a short chymotryptic peptide with fewer than six amino acids. If alternative proteolytic enzymes are available to generate an appropriate peptide length for stable MS analysis, the quantification and discernment of additional DCP proteoforms might be possible. Further, the cohorts in this study consisted solely of individuals of Korean ethnicity, primarily with an HBV etiology. Therefore, additional studies are needed to validate our assay using different populations consisting of other ethnicities and etiologies. Analytical validations to confirm the robustness and reproducibility of the MRM‐MS assay should be evaluated using larger cohorts in future studies.
In conclusion, the quantitative MRM‐MS assay for DCP measurement that was designed in this study shows equivalent diagnostic performance as the antibody‐based DCP immunoassay. Our MRM‐MS assay to quantify three Glu‐peptides enabled not only the extensive detection of DCP proteoforms but also a detailed comparison of the DCP proteoform compositions between HCC and benign liver diseases. This study indicates that the comprehensive measurement of DCP proteoforms using the MRM‐MS assay has great potential as a surveillance test for the detection of HCC at the very early stage, even among patients with AFP and DCP levels under the corresponding cut‐off values. Further, this assay is advantageous compared with the DCP immunoassay because it facilitates the high‐throughput analysis of large cohorts while requiring lower costs and sample volumes. The multiplexing ability of the MS‐based quantification approach has the potential to develop an HCC surveillance assay that simultaneously analyzes the DCP proteoforms in combination with hundreds of existing serological biomarkers in a high‐throughput format that would be suitable for routine check‐ups.
Supporting information
Supplementary Material
Acknowledgment
We thank Lisa Giles, Ph.D., from Blue Pencil Science (http://www.bluepencilscience.com/) for editing an English draft of this manuscript.
Supported by the Industrial Strategic Technology Development Program (Funding number: 20000134 throughout the whole 2020.), the Korea Health Industry Development Institute (Funding numbers: HL19C0020 throughout the whole 2020, HI19C1132 from May 2020 for 1 year.), Seoul National University Hospital (grant 2021), and the National Research Foundation of Korea BK21‐Plus Education Program (scholarship to J.L. and D.S.).
Potential conflict of interest: Dr. Lim consults for, advises, is on the speakers’ bureau of, and received grants from Gilead. The other authors have nothing to report.
Contributor Information
Taesung Park, Email: taesungp@gmail.com.
Youngsoo Kim, Email: biolab@snu.ac.kr, Email: taesungp@gmail.com.
References
Author names in bold designate shared co‐first authorship.
- 1.Villanueva A. Hepatocellular carcinoma. N Engl J Med 2019;380:1450‐1462. [DOI] [PubMed] [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394‐424.Erratum in: CA Cancer J Clin 2020;70:313.. [DOI] [PubMed] [Google Scholar]
- 3.Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet 2018;391:1301‐1314. [DOI] [PubMed] [Google Scholar]
- 4.Petrick JL, Florio AA, Znaor A, Ruggieri D, Laversanne M, Alvarez CS, et al. International trends in hepatocellular carcinoma incidence, 1978‐2012. Int J Cancer 2020;147:317‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGlynn KA, Petrick JL, El‐Serag HB. Epidemiology of hepatocellular carcinoma. Hepatology 2021;73(Suppl. 1):4‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sartorius K, Sartorius B, Aldous C, Govender PS, Madiba TE. Global and country underestimation of hepatocellular carcinoma (HCC) in 2012 and its implications. Cancer Epidemiol 2015;39:284‐290. [DOI] [PubMed] [Google Scholar]
- 7.de Martel C, Maucort‐Boulch D, Plummer M, Franceschi S. World‐wide relative contribution of hepatitis B and C viruses in hepatocellular carcinoma. Hepatology 2015;62:1190‐1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yin Z, Dong C, Jiang K, Xu Z, Li R, Guo K, et al. Heterogeneity of cancer‐associated fibroblasts and roles in the progression, prognosis, and therapy of hepatocellular carcinoma. J Hematol Oncol 2019;12:101.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erstad DJ, Tanabe KK. Prognostic and therapeutic implications of microvascular invasion in hepatocellular carcinoma. Ann Surg Oncol 2019;26:1474‐1493. [DOI] [PubMed] [Google Scholar]
- 10.Sarveazad A, Agah S, Babahajian A, Amini N, Bahardoust M. Predictors of 5 year survival rate in hepatocellular carcinoma patients. J Res Med Sci 2019;24:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Njei B, Rotman Y, Ditah I, Lim JK. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology 2015;61:191‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singal AG, Pillai A, Tiro J. Early detection, curative treatment, and survival rates for hepatocellular carcinoma surveillance in patients with cirrhosis: a meta‐analysis. PLoS Med 2014;11:e1001624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sangiovanni A, Del Ninno E, Fasani P, De Fazio C, Ronchi G, Romeo R, et al. Increased survival of cirrhotic patients with a hepatocellular carcinoma detected during surveillance. Gastroenterology 2004;126:1005‐1014. [DOI] [PubMed] [Google Scholar]
- 14.Ryder SD; British Society of Gastroenterology . Guidelines for the diagnosis and treatment of hepatocellular carcinoma (HCC) in adults. Gut 2003;52(Suppl. 3):iii1‐iii8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kansagara D, Papak J, Pasha AS, O'Neil M, Freeman M, Relevo R, et al. Screening for hepatocellular carcinoma in chronic liver disease: a systematic review. Ann Intern Med 2014;161:261‐269. [DOI] [PubMed] [Google Scholar]
- 16.Tzartzeva K, Obi J, Rich NE, Parikh ND, Marrero JA, Yopp A, et al. Surveillance imaging and alpha fetoprotein for early detection of hepatocellular carcinoma in patients with cirrhosis: a meta‐analysis. Gastroenterology 2018;154:1706‐1718.e1701.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.European Association for the Study of the Liver; European Organisation For Research and Treatment of Cancer . EASL‐EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol 2012;56:908‐943.Erratum in: J Hepatol 2012;56:1430.. [DOI] [PubMed] [Google Scholar]
- 18.Di Bisceglie AM, Hoofnagle JH. Elevations in serum alpha‐fetoprotein levels in patients with chronic hepatitis B. Cancer 1989;64:2117‐2120. [DOI] [PubMed] [Google Scholar]
- 19.Di Bisceglie AM, Sterling RK, Chung RT, Everhart JE, Dienstag JL, Bonkovsky HL, et al.; HALT‐C Trial Group . Serum alpha‐fetoprotein levels in patients with advanced hepatitis C: results from the HALT‐C Trial. J Hepatol 2005;43:434‐441. [DOI] [PubMed] [Google Scholar]
- 20.Marrero JA, Feng Z, Wang Y, Nguyen MH, Befeler AS, Roberts LR, et al. Alpha‐fetoprotein, des‐gamma carboxyprothrombin, and lectin‐bound alpha‐fetoprotein in early hepatocellular carcinoma. Gastroenterology 2009;137:110‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lok AS, Sterling RK, Everhart JE, Wright EC, Hoefs JC, Di Bisceglie AM, et al.; HALT‐C Trial Group . Des‐gamma‐carboxy prothrombin and alpha‐fetoprotein as biomarkers for the early detection of hepatocellular carcinoma. Gastroenterology 2010;138:493‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim H, Sohn A, Yeo I, Yu SJ, Yoon JH, Kim Y. Clinical assay for AFP‐L3 by using multiple reaction monitoring‐mass spectrometry for diagnosing hepatocellular carcinoma. Clin Chem 2018;64:1230‐1238. [DOI] [PubMed] [Google Scholar]
- 23.Wang T, Zhang KH. New blood biomarkers for the diagnosis of AFP‐negative hepatocellular carcinoma. Front Oncol 2020;10:1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liebman HA, Furie BC, Tong MJ, Blanchard RA, Lo KJ, Lee SD, et al. Des‐gamma‐carboxy (abnormal) prothrombin as a serum marker of primary hepatocellular carcinoma. N Engl J Med 1984;310:1427‐1431. [DOI] [PubMed] [Google Scholar]
- 25.Weitz IC, Liebman HA. Des‐gamma‐carboxy (abnormal) prothrombin and hepatocellular carcinoma: a critical review. Hepatology 1993;18:990‐997. [DOI] [PubMed] [Google Scholar]
- 26.Tsai SL, Huang GT, Yang PM, Sheu JC, Sung JL, Chen DS. Plasma des‐gamma‐carboxyprothrombin in the early stage of hepatocellular carcinoma. Hepatology 1990;11:481‐488. [DOI] [PubMed] [Google Scholar]
- 27.Borowski M, Furie BC, Furie B. Distribution of gamma‐carboxyglutamic acid residues in partially carboxylated human prothrombins. J Biol Chem 1986;261:1624‐1628. [PubMed] [Google Scholar]
- 28.Uehara S, Gotoh K, Handa H, Honjo K, Hirayama A. Process of carboxylation of glutamic acid residues in the gla domain of human des‐gamma‐carboxyprothrombin. Clin Chim Acta 1999;289:33‐44. [DOI] [PubMed] [Google Scholar]
- 29.Liska DJ, Suttie JW. Location of gamma‐carboxyglutamyl residues in partially carboxylated prothrombin preparations. Biochemistry 1988;27:8636‐8641. [DOI] [PubMed] [Google Scholar]
- 30.Huisse MG, Leclercq M, Belghiti J, Flejou JF, Suttie JW, Bezeaud A, et al. Mechanism of the abnormal vitamin K‐dependent gamma‐carboxylation process in human hepatocellular carcinomas. Cancer 1994;74:1533‐1541. [DOI] [PubMed] [Google Scholar]
- 31.Friedman PA, Przysiecki CT. Vitamin K‐dependent carboxylation. Int J Biochem 1987;19:1‐7. [DOI] [PubMed] [Google Scholar]
- 32.Uehara S, Gotoh K, Handa H, Tomita H, Senshuu M. Distribution of the heterogeneity of des‐gamma‐carboxyprothrombin in patients with hepatocellular carcinoma. J Gastroenterol Hepatol 2005;20:1545‐1552. [DOI] [PubMed] [Google Scholar]
- 33.Naraki T, Kohno N, Saito H, Fujimoto Y, Ohhira M, Morita T, et al. gamma‐Carboxyglutamic acid content of hepatocellular carcinoma‐associated des‐gamma‐carboxy prothrombin. Biochim Biophys Acta 2002;1586:287‐298. [DOI] [PubMed] [Google Scholar]
- 34.Sugo T, Watanabe K, Naraki T, Matsuda M. Chemical modification of gamma‐carboxyglutamic acid residues in prothrombin elicits a conformation similar to that of abnormal (des‐gamma‐carboxy)prothrombin. J Biochem 1990;108:382‐387. [DOI] [PubMed] [Google Scholar]
- 35.Tameda M, Shiraki K, Sugimoto K, Ogura S, Inagaki Y, Yamamoto N, et al. Des‐gamma‐carboxy prothrombin ratio measured by P‐11 and P‐16 antibodies is a novel biomarker for hepatocellular carcinoma. Cancer Sci 2013;104:725‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagaoka S, Yatsuhashi H, Hamada H, Yano K, Matsumoto T, Daikoku M, et al. The des‐gamma‐carboxy prothrombin index is a new prognostic indicator for hepatocellular carcinoma. Cancer 2003;98:2671‐2677. [DOI] [PubMed] [Google Scholar]
- 37.Shi T, Fillmore TL, Sun X, Zhao R, Schepmoes AA, Hossain M, et al. Antibody‐free, targeted mass‐spectrometric approach for quantification of proteins at low picogram per milliliter levels in human plasma/serum. Proc Natl Acad Sci U S A 2012;109:15395‐15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bros P, Vialaret J, Barthelemy N, Delatour V, Gabelle A, Lehmann S, et al. Antibody‐free quantification of seven tau peptides in human CSF using targeted mass spectrometry. Front Neurosci 2015;9:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yocum AK, Gratsch TE, Leff N, Strahler JR, Hunter CL, Walker AK, et al. Coupled global and targeted proteomics of human embryonic stem cells during induced differentiation. Mol Cell Proteomics 2008;7:750‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sohn A, Kim H, Yu SJ, Yoon JH, Kim Y. A quantitative analytical method for PIVKA‐II using multiple reaction monitoring‐mass spectrometry for early diagnosis of hepatocellular carcinoma. Anal Bioanal Chem 2017;409:2829‐2838. [DOI] [PubMed] [Google Scholar]
- 41.Sohn A, Kim H, Yeo I, Kim Y, Son M, Yu SJ, et al. Fully validated SRM‐MS‐based method for absolute quantification of PIVKA‐II in human serum: clinical applications for patients with HCC. J Pharm Biomed Anal 2018;156:142‐146. [DOI] [PubMed] [Google Scholar]
- 42.Heimbach JK, Kulik LM, Finn RS, Sirlin CB, Abecassis MM, Roberts LR, et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018;67:358‐380. [DOI] [PubMed] [Google Scholar]
- 43.Kratz A, Ferraro M, Sluss PM, Lewandrowski KB. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Laboratory reference values. N Engl J Med 2004;351:1548‐1563.Erratum in: N Engl J Me 2004;351:2461. [DOI] [PubMed] [Google Scholar]
- 44.Abbatiello SE, Mani DR, Keshishian H, Carr SA. Automated detection of inaccurate and imprecise transitions in peptide quantification by multiple reaction monitoring mass spectrometry. Clin Chem 2010;56:291‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malhotra OP, Nesheim ME, Mann KG. The kinetics of activation of normal and gamma‐carboxyglutamic acid‐deficient prothrombins. J Biol Chem 1985;260:279‐287. [PubMed] [Google Scholar]
- 46.Ratcliffe JV, Furie B, Furie BC. The importance of specific gamma‐carboxyglutamic acid residues in prothrombin. Evaluation by site‐specific mutagenesis. J Biol Chem 1993;268:24339‐24345. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material