

Abstract
Drug‐induced hepatotoxicity limits development of new effective medications. Drugs and numerous endogenous/exogenous agents are metabolized/detoxified by hepatocytes, during which reactive oxygen species (ROS) are generated as a by‐product. ROS has broad adverse effects on liver function and integrity, including damaging hepatocyte proteins, lipids, and DNA and promoting liver inflammation and fibrosis. ROS in concert with iron overload drives ferroptosis. Hepatic nuclear factor kappa B (NF‐κB)‐inducing kinase (NIK) is aberrantly activated in a broad spectrum of liver disease. NIK phosphorylates and activates inhibitor of NF‐κB kinase subunit alpha (IKKα), and the hepatic NIK/IKKα cascade suppresses liver regeneration. However, the NIK/IKKα pathway has not been explored in drug‐induced liver injury. Here, we identify hepatic NIK as a previously unrecognized mediator for acetaminophen (APAP)‐induced acute liver failure. APAP treatment increased both NIK transcription and NIK protein stability in primary hepatocytes as well as in liver in mice. Hepatocyte‐specific overexpression of NIK augmented APAP‐induced liver oxidative stress in mice and increased hepatocyte death and mortality in a ROS‐dependent manner. Conversely, hepatocyte‐specific ablation of NIK or IKKα mitigated APAP‐elicited hepatotoxicity and mortality. NIK increased lipid peroxidation and cell death in APAP‐stimulated primary hepatocytes. Pretreatment with antioxidants or ferroptosis inhibitors blocked NIK/APAP‐induced hepatocyte death. Conclusion: We unravel a previously unrecognized NIK/IKKα/ROS/ferroptosis axis engaged in liver disease progression.
Hepatic NIK is upregulated in response to hepatic toxicants. Ablation of hepatic NIK attenuates, whereas hepatocyte‐specific overexpression of NIK aggravates, APAP‐induced liver injury. NIK promotes hepatic oxidative stress and ferroptosis.

Abbreviations
- 4‐HNE
4 hydroxynonenal
- ALT
alanine aminotransferase
- ANOVA
analysis of variance
- APAP
acetaminophen
- a.u.
arbitrary units
- β‐gal
beta‐galactosidase
- BODIPY
boron‐dipyrromethene
- CCL2
C‐C motif chemokine ligand 2
- CHX
cycloheximide
- Con
control
- CXCL5
C‐X‐C motif ligand 5
- DAPI
4´,6‐diamidino‐2‐phenylindole
- DCF
2',7'‐dichlorofluorescein
- FITC
fluorescein isothiocyanate
- fl
flox
- γ‐H2AX
gamma‐H2A histone family member X
- GATA3
GATA binding protein 3
- GSH
glutathione
- h
hours
- H&E
hematoxylin and eosin
- Hep
hepatocyte
- IKKα
inhibitor of nuclear factor kappa B kinase subunit alpha
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- JNK
c‐Jun N‐terminal kinase
- MPO
myeloperoxidase
- mRNA
messenger RNA
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- NAC
N‐acetyl‐L‐cysteine
- NAPQI
N‐acetyl‐p‐benzo‐quinone imine
- NF‐κB
nuclear factor kappa B
- NIK
nuclear factor kappa B‐inducing kinase
- PBS
phosphate‐buffered saline
- qPCR
quantitative real‐time reverse‐transcription polymerase chain reaction
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- TNFα
tumor necrosis factor alpha
- TUNEL
terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling
Dietary nutrients (glucose, amino acids) and non‐nutrient substances (drugs, xenobiotics) are absorbed from the gastrointestinal tract and transported to the liver. Hepatocytes not only metabolize/process nutrients to maintain metabolic homeostasis but also carry out detoxifications of drugs and xenobiotics to support life.( 1, 2 ) As such, hepatocytes constantly experience metabolic stress, oxidative stress, and/or other types of intracellular stress. Hepatocellular stress increases risk for hepatocyte injury/death, liver inflammation, and fibrosis.( 3, 4 ) Liver oxidative stress is associated with liver disease, as illustrated by increased levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS).( 5, 6, 7, 8 ) ROS and RNS induce modifications on proteins, membrane phospholipids, and/or genomic DNA, leading to cellular dysfunctions, cell injury, and/or death.( 8, 9, 10 ) Oxidative stress‐driven peroxidation of membrane phospholipids in concert with an iron overload underpins ferroptosis.( 11, 12 )
Nuclear factor kappa B (NF‐κB)‐inducing kinase (NIK; also known as mitogen‐activated protein kinase kinase kinase 14) is a serine/threonine kinase that mediates activation of the noncanonical NF‐κB2 pathway.( 13 ) NIK phosphorylates and activates inhibitor of kB (IkB) kinase‐α (IKKα; also referred to as Chuk), and IKKα in turn activates transcription factor NF‐κB2.( 13, 14, 15 ) NIK is activated by a wide range of stimuli, including a subset of cytokines, numerous endogenous metabolites and exogenous substances, and various cellular stress‐inducing agents.( 13, 15, 16 ) Importantly, hepatic NIK is aberrantly activated in liver disease in mice and humans, including alcoholic liver injury, nonalcoholic fatty liver disease, hepatotoxin‐induced liver injury, viral hepatitis, and autoimmune liver disease.( 16, 17, 18, 19 ) We previously reported that a modest elevation of hepatic NIK in obesity augments hepatic glucose production, increasing the risk for type 2 diabetes.( 16, 20 ) Consistently, hepatic NF‐kB2 also increases hepatic glucose production.( 21 ) Additionally, hepatic NIK promotes liver steatosis, presumably by suppressing peroxisome proliferator‐activated receptor alpha and fatty acid β oxidation.( 20, 22 ) Aside from regulating metabolic pathways, hepatic NIK also blocks reparative hepatocyte proliferation, thereby impeding liver regeneration.( 23 ) Furthermore, excessive activation of NIK causes hepatocytes to release mediators that potently stimulate macrophages/Kupffer cells, leading to fatal immune destruction of the liver in mice.( 19 )
Acetaminophen (APAP) overdose is a leading cause for acute liver failure in Europe and North America.( 24 ) APAP is a key constituent of Tylenol, which is commonly used to relieve fever and pain.( 2 ) APAP is converted to N‐acetyl‐p‐benzo‐quinone imine (NAPQI) by hepatocytes, and NAPQI increases hepatic oxidative stress and necrosis.( 2, 25 ) APAP‐induced ROS promotes calcium entry, worsening liver injury.( 26 ) Considering that hepatocellular stress stimulates NIK, we speculated that APAP might activate hepatic NIK. NIK in turn promotes liver oxidative stress, mediating APAP hepatotoxicity. Given that ROS drives ferroptosis, we reasoned that hepatic NIK might augment hepatocyte ferroptosis in APAP‐treated mice. We tested this hypothesis using both hepatocyte‐specific NIK‐knockout and NIK‐overexpressing mice. Our results unveil a previously unrecognized NIK/IKKα/ROS/ferroptosis axis promoting liver disease progression.
Materials and Methods
Animals
NIKflox (fl)/fl , IKKαfl/fl , Albumin‐Cre, hepatocyte (Hep)NIK, and HepControl (Con) mice (C57BL/6J background) have been described.( 17, 19, 27 ) Mice were housed on a 12‐hour light–dark cycle and fed ad libitum a normal chow diet (9% fat; TestDiet, St. Louis, MO).
Ethics Statements
Animal research complied with relevant ethic regulations and was conducted following the protocols approved by the University of Michigan Institutional Animal Care and Use Committee.
APAP and N‐acetyl‐L‐Cysteine Treatments
Mice (8‐10 weeks) were fasted overnight and injected intraperitoneally with a single dose of APAP (200‐300 mg/kg body weight). Mice were pretreated with N‐acetyl‐L‐cysteine (NAC; 300 mg/kg body weight, intraperitoneal) and then treated with APAP 5 minutes later. Blood samples were collected through tail veins. Plasma alanine aminotransferase (ALT) was measured using an ALT reagent set (Pointe Scientific Inc., Canton, MI). Livers were harvested 2‐24 hours after APAP injection for biochemical and histologic analyses.
Immunoblotting and ROS Assays
Liver tissue or hepatocyte cultures were homogenized in ice‐cold lysis buffer (50 mM Tris HCl, pH 7.5, 0.5% Nonidet P‐40, 150 mM NaCl, 2 mM ethylene glycol tetraacetic acid, 1 mM Na3VO4, 100 mM NaF, 10 mM Na4P2O7, 1 mM phenylmethylsulfonyl fluoride, 10 μg/mL aprotinin, 10 μg/mL leupeptin). Tissue or cell extracts were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotted with the indicated antibodies (Supporting Table S1). Liver or cell extracts were incubated for 1 hour at 37°C with 5 µM 2',7'‐dichlorofluorescein (DCF), which is a diacetate fluorescent probe (D6883; Sigma). DCF levels were measured using a BioTek Synergy 2 Multi‐Mode Microplate Reader (485 nm excitation and 527 nm emission).
Cell Culture, Adenoviral Transduction, and 3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐Diphenyltetrazolium Bromide Assays
Hepa1 and Huh7 hepatocytes were grown in Dulbecco’s modified Eagle’s medium supplemented with 5% fetal bovine serum (FBS), 100 units mL−1 penicillin, and 100 µg mL−1 streptomycin. Primary hepatocytes were isolated from mice using type II collagenase (Worthington Biochem, Lakewood, NJ), as described.( 28 ) Primary hepatocytes were grown on William’s medium E (Sigma) supplemented with 2% FBS, 100 units mL−1 penicillin, and 100 µg mL−1 streptomycin and transduced with β‐galactosidase (β‐gal) or NIK adenoviral vectors. After 12‐14 hours of growth, hepatocytes were treated with APAP for 2‐24 hours in the presence or absence of NAC, ferrostatin‐1 (Item No. 17729, CAS No. 347174‐05‐4; Cayman Chemicals), or liproxstatin‐1 (Item No. 17730 CAS No. 950455‐15‐9, Cayman Chemicals). Hepatocyte viability was measured using colorimetric 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assays (DOT Scientific Inc., Burton, MI).
Immunostaining and Terminal Deoxynucleotidyl Transferase–Mediated Deoxyuridine Triphosphate Nick‐End Labeling Assays
Liver frozen sections were prepared using a Leica cryostat (Leica Biosystems Nussloch GmbH, Nussloch, Germany), fixed in 4% paraformaldehyde for 30 minutes, blocked for 3 hours with 5% normal goat serum (Life Technologies) supplemented with 1% bovine serum albumin (BSA), and incubated overnight at 4°C with the indicated antibodies (Supporting Table S1). Liver sections were stained with terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling (TUNEL) reagents using an in situ cell death detection kit (#11684817910; Roche Diagnostics, Indianapolis, IN). Primary hepatocytes were grown on glass coverslips, transduced with adenoviral vectors, treated with APAP, fixed in 4% paraformaldehyde, blocked with 5% normal goat serum, stained with TUNEL, DCF, or boron‐dipyrromethene (BODIPY) 581/591 C11 probes, and visualized using a fluorescent microscope.
Quantitative Real‐Time Reverse‐Transcription Polymerase Chain Reaction
Total RNA was extracted using TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA). First‐strand complementary DNAs were synthesized using random primers and Moloney Murine leukemia virus (M‐MLV) reverse transcriptase (Promega, Madison, WI). Quantitative real‐time reverse‐transcription polymerase chain reaction (qPCR) was performed using Radiant SYBR Green 2X Lo‐ROX qPCR kits (Alkali Scientific, Pompano Beach, FL), a StepOnePlus Real‐Time PCR System (Life Technologies Corporation, NY), and respective primers (Supporting Table S2).
Flow Cytometry
Mouse primary hepatocyte cultures were transduced with β‐gal or NIK adenoviral vectors for 12 hours and followed by APAP stimulation (1 mM) for 24 hours. Hepatocytes were incubated with BODIPY 581/591 C11 (2.5 µM) at 37°C for 30 minutes, trypsinized, centrifuged for 3 minutes at 200g, suspended in Hank’s balanced salt solution containing 2% FBS, and applied to a Bio‐Rad Ze5 cell analyzer. Hepatocytes were selected based on forward‐scatter and side‐scatter gating and were further analyzed following BODIPY 581/591 C11 staining through fluorescein isothiocyanate (FITC) gating. Data were analyzed using FlowJo software. Mean FITC‐A was calculated on data from six independent experiments.
Statistical Analysis
Data were presented as means ± SEM. Difference was analyzed by the two‐tailed Student t test (two groups) and analysis of variance (ANOVA)/Bonferroni posttest (more than two groups) using GraphPad Prism 8. P < 0.05 was considered statistically significant. Survival rates were calculated using the Kaplan‐Meier method.
Results
APAP Treatment Up‐Regulates Hepatic NIK
To determine whether APAP treatment increases NIK expression and/or stability in the liver, we intraperitoneally injected C57BL/6J male mice with a single dose of APAP. APAP rapidly and substantially increased NIK messenger RNA (mRNA) levels in the liver (Fig. 1A). Increased NIK expression was detected within 2 hours after APAP injection (Supporting Fig. S1A). To determine whether APAP directly increases hepatocyte NIK expression, we stimulated Hepa1 cells (a murine hepatocyte line) with APAP. APAP robustly stimulated NIK expression in a time‐ and dose‐dependent manner (Fig. 1B). To confirm these findings, we isolated primary hepatocytes from C57BL/6J mice and stimulated them with APAP. APAP increased NIK expression time/dose dependently in primary hepatocytes (Fig. 1C). Likewise, APAP treatment also increased NIK expression in Huh7 cells, a human hepatocyte line (Fig. 1D).
FIG. 1.
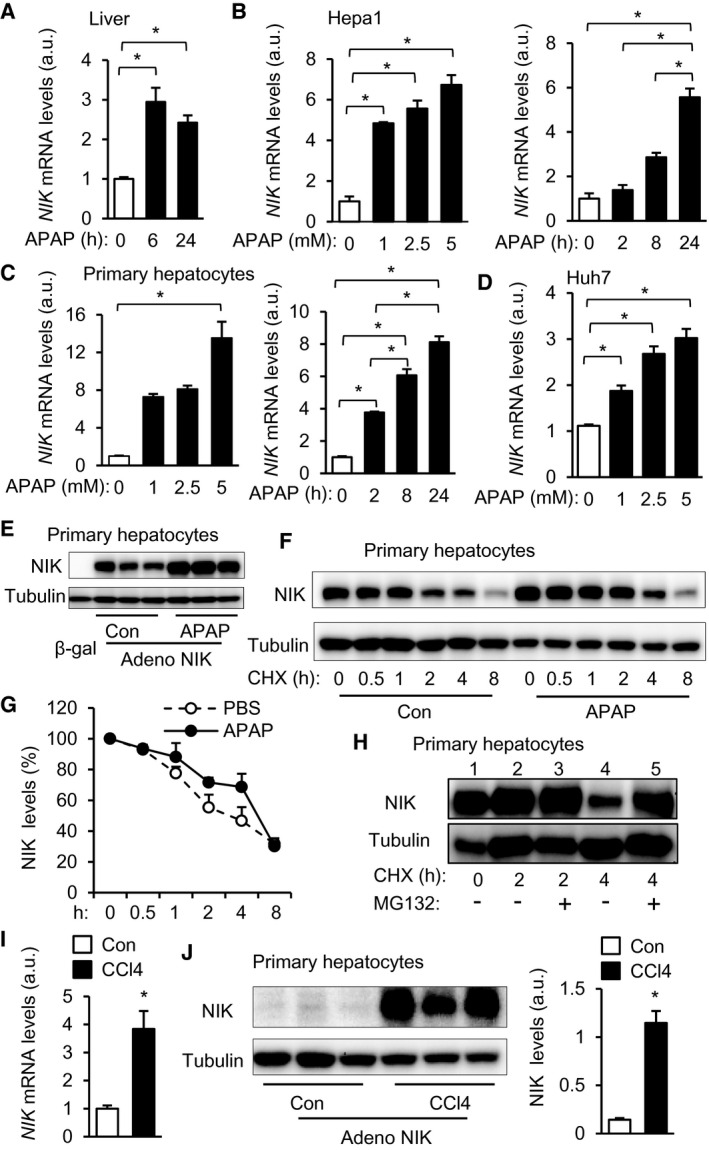
APAP and CCl4 treatments up‐regulate hepatic NIK. (A) C57BL/6J male mice were treated with APAP (300 mg/kg body weight). Liver NIK mRNA levels were measured by qPCR and normalized to 36B4 expression (n = 3‐4 per group). (B) NIK mRNA levels in APAP‐treated Hepa1 cells (normalized to 36B4 expression, n = 3 per group). (C) NIK mRNA levels in mouse primary hepatocytes (normalized to 36B4 expression, n = 3 per group). (D) NIK mRNA levels in Huh7 hepatocytes (normalized to GAPDH expression, n = 3 per group). (E) Mouse primary hepatocytes were transduced with NIK or β‐gal adenoviral vectors and treated with APAP or empty vehicles (Con) for 24 hours. Hepatocyte extracts were immunoblotted with antibodies to NIK and α‐tubulin. (F,G) Mouse primary hepatocytes were transduced with NIK adenoviral vectors, pretreated with CHX, and followed by APAP stimulation. Hepatocyte extracts were immunoblotted with the indicated antibodies. NIK levels were normalized to α‐tubulin levels and presented as percentage of initial values (n = 4 per group). (H) Mouse primary hepatocytes were transduced with NIK adenoviral vectors and treated with CHX and MG132. Hepatocyte extracts were immunoblotted with the indicated antibodies. (I) Mouse primary hepatocytes were stimulated with 2 mM CCl4 for 24 hours. NIK mRNA levels were measured by qPCR (normalized to 36B4 levels, n = 3 per group). (J) Mouse primary hepatocytes were transduced with NIK adenoviral vectors and treated with 2 mM CCl4 for 24 hours. Hepatocyte extracts were immunoblotted with the indicated antibodies. NIK levels were normalized to α‐tubulin levels (n = 3 per group). Data are presented as mean ± SEM. *P < 0.05; (A‐D) one‐way ANOVA/Bonferroni’s multiple comparisons test; (G) two‐way ANOVA/Bonferroni’s multiple comparisons test; (I,J) two‐tailed unpaired Student t test. Abbreviations: adeno, adenovirus; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
NIK is ubiquitously expressed, but its protein levels are low under baseline conditions due to rapid ubiquitination and degradation.( 13, 16, 19, 29 ) To determine whether APAP increases NIK stability, we transduced mouse primary hepatocytes with NIK adenoviral vectors (NIK transcription is under the control of the constitutively active cytomegalovirus [CMV] promoter), followed by APAP stimulation. APAP substantially increased NIK protein levels (Fig. 1E). To directly assess NIK degradation, we transduced mouse primary hepatocytes with NIK adenoviral vectors and stimulated hepatocytes with APAP in the presence of the protein synthesis inhibitor cycloheximide (CHX). To avoid NIK overloading from APAP‐treated cells, we loaded half the cell extracts from APAP‐stimulated hepatocytes relative to phosphate‐buffered saline (PBS)‐treated hepatocytes (control). APAP stimulation considerably suppressed NIK degradation (Fig. 1F,G). To test if 26S proteasomes mediate NIK degradation, we treated primary hepatocytes with the proteasome inhibitor MG132 in conjunction with CHX. NIK degradation was detected at 4 hours but not 2 hours after CHX treatment (Fig. 1H, lanes 1, 2, and 4). MG132 treatment markedly inhibited NIK degradation (Fig. 1H, lane 4 vs. 5). Collectively, these results demonstrate that APAP directly increases both expression and stability of NIK in hepatocytes.
To determine whether NIK up‐regulation is a common signature of the hepatocyte response to hepatotoxins, we stimulated mouse primary hepatocytes with CCl4. Chronic CCl4 treatment is known to increase liver NIK mRNA levels in mice.( 18, 19 ) In accordance, CCl4 directly stimulated expression of endogenous NIK in hepatocytes (Fig. 1I). To test if CCl4 increases NIK stability, we transduced primary hepatocytes with NIK adenoviral vectors and stimulated them with CCl4. CCl4 dramatically increased the levels of recombinant NIK (Fig. 1J). Thus, up‐regulation of hepatic NIK (increased expression and stability) appears to be a common liver response to drugs, xenobiotics, and endogenous insults.
Hepatocyte‐Specific Overexpression of NIK Aggravates APAP‐Induced Liver Injury and Mortality
To examine the role of NIK in acute liver failure, we generated hepatocyte‐specific NIK transgenic mice (HepNIK; genotype, Rosa26‐STOP‐NIK+/−;Cre+/− ) by crossing Rosa26‐STOP‐NIK mice with albumin‐Cre mice. Rosa26‐STOP‐NIK mice (referred to as HepCon hereafter) contain a loxp‐STOP‐loxp‐NIK transgene knocked in the Rosa26 locus, allowing Cre‐dependent expression of recombinant NIK.( 30 ) We previously verified that NIK is overexpressed specifically in the hepatocytes of HepNIK mice.( 19 ) HepNIK mice were grossly normal under baseline conditions. We intraperitoneally injected HepNIK and HepCon mice with a single dose of APAP. Strikingly, survival rates were substantially lower in HepNIK than in HepCon mice (Fig. 2A). Plasma ALT levels were dramatically higher in HepNIK than in HepCon mice (Fig. 2B). To validate liver failure, we examined liver histology. APAP injection caused liver necrosis as expected. Necrotic areas were significantly larger in HepNIK than in HepCon mice (Fig. 2C, hematoxylin and eosin [H&E] staining). Liver cell death was dramatically higher in HepNIK relative to HepCon mice (Fig. 2C, TUNEL staining). Liver neutrophil content was significantly higher in HepNIK relative to HepCon mice, as assessed by staining liver sections with anti‐myeloperoxidase (MPO) antibody (Fig. 2C). Liver expression of cytokines/chemokines (C‐C motif chemokine ligand 2 [CCL2], C‐X‐C motif ligand 5 [CXCL5], interleukin (IL)‐1β, IL6, tumor necrosis factor α [TNFα]) was significantly higher in HepNIK relative to HepCon mice (Fig. 2D). Liver DNA damage was also higher in HepNIK than in HepCon mice, as assessed by staining liver sections with the antibody to gamma‐H2A histone family member X (γ‐H2AX; a DNA damage marker) (Fig. 2C).
FIG. 2.
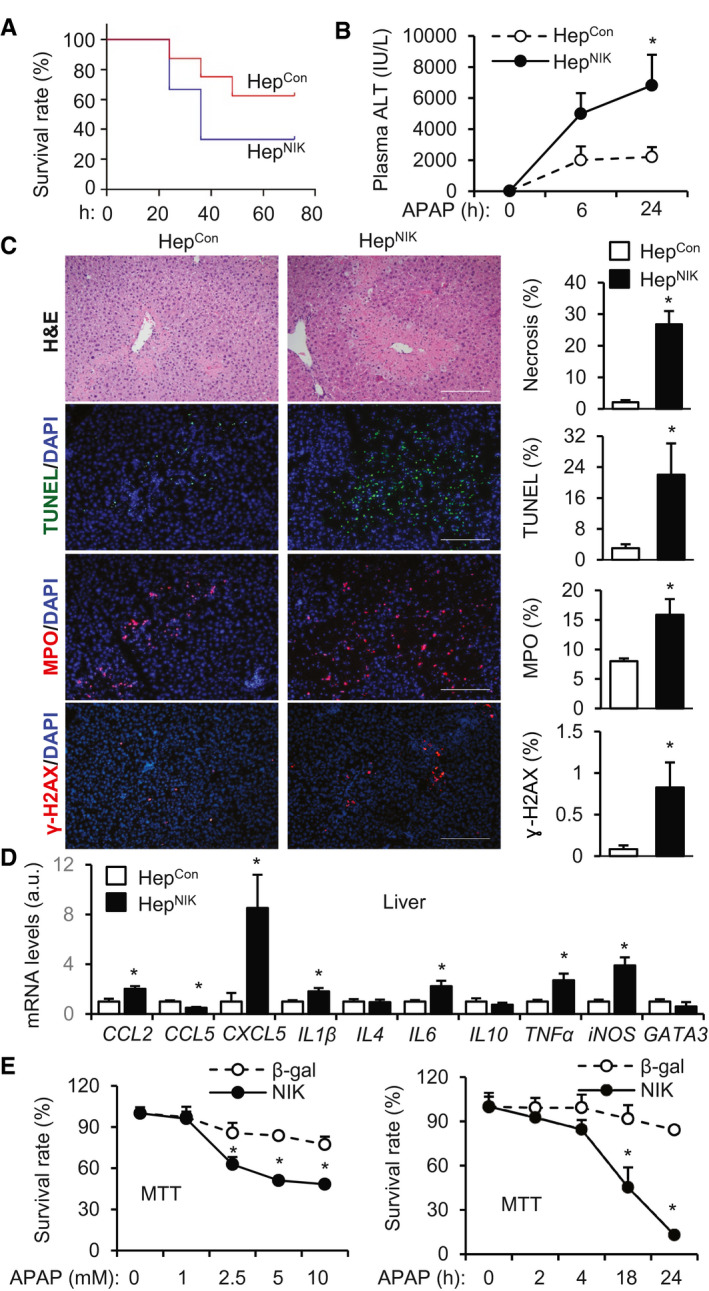
HepNIK mice are prone to APAP‐induced acute liver failure. (A) Survival rates following APAP treatment (300 mg/kg body weight). Male HepNIK mice, n = 6; male HepCon mice, n = 8. (B‐D) HepNIK and HepCon male mice were treated with APAP (200 mg/kg). (B) Plasma ALT levels (n = 8 per group). (C) Liver sections were prepared 24 hours after APAP treatment and stained with the indicated agents. Necrotic area was normalized to total area. TUNEL, MPO, or γ‐H2AX cell numbers were normalized to total cell number (n = 3‐4 per group). Scale bar, 200 μm. (D) Liver gene expression 24 hours after APAP treatment (normalized to 36B4 levels, n = 4‐8 per group). (E) Mouse primary hepatocytes were transduced with NIK or β‐gal adenoviral vectors and treated with APAP. Hepatocyte viability was measured by MTT and normalized to baseline levels (n = 6 per group). Data are presented as mean ± SEM. *P < 0.05; (C,D) two‐tailed unpaired Student t test; (B,E) two‐way ANOVA/Bonferroni’s multiple comparisons test.
To verify that NIK directly enhances APAP toxicity, we transduced primary liver cell cultures with NIK or β‐gal adenoviral vectors, followed by APAP stimulation. APAP reduced hepatocyte viability (by MTT assays) in a time‐ and dose‐dependent manner, as expected. Importantly, cell viability was significantly lower in NIK‐ than in β‐gal‐transduced hepatocytes (Fig. 2E). Taken together, these results unveil hepatic NIK as a previously unrecognized inducer for acute liver failure.
Ablation of Hepatic NIK Ameliorates APAP‐Induced Liver Injury and Mortality
To explore the role of endogenous hepatic NIK, we generated hepatocyte‐specific NIK knockout (NIKΔhep ) mice (NIKfl/fl;Cre+/− ) by crossing NIKfl/fl mice with albumin‐Cre drivers. NIKfl/fl mice have been reported.( 20 ) Considering that NIK deficiency might ameliorate liver injury, we increased APAP doses to 250 mg/kg body weight. Survival rates were substantially higher in NIKΔhep than in NIKfl/fl mice after APAP treatments (Fig. 3A). Plasma ALT levels were significantly lower in NIKΔhep than in NIKfl/fl mice (Fig. 3B). Liver necrosis (H&E), hepatocyte death (TUNEL), liver DNA damage (γ‐H2AX), and neutrophil infiltration into the liver (MPO) were all significantly lower in NIKΔhep than in NIKfl/fl mice (Fig. 3C). Liver expression of cytokines/chemokines (CCL2, IL1β, IL10, TNFα) was lower in NIKΔhep relative to NIKfl/fl mice (Fig. 3D). NIKΔhep female mice, like NIKΔhep male mice, were also resistant to APAP‐induced liver injury (Supporting Fig. S1B). To test if hepatic NIK influences APAP metabolism, we measured NAPQI production, as reported previously.( 31 ) We treated mice with APAP for 2 hours and immunoblotted liver extracts with antibodies recognizing NAPQI adducts. The levels of NAPQI protein adducts were comparable between NIKΔhep and NIKfl/fl mice (Supporting Fig. S2A). Likewise, liver NAPQI adduct levels were also comparable between APAP‐treated HepNIK and HepCon mice (Supporting Fig. S2B). Thus, hepatic NIK appears to act downstream of or in parallel to NAPQI to augment APAP hepatotoxicity.
FIG. 3.
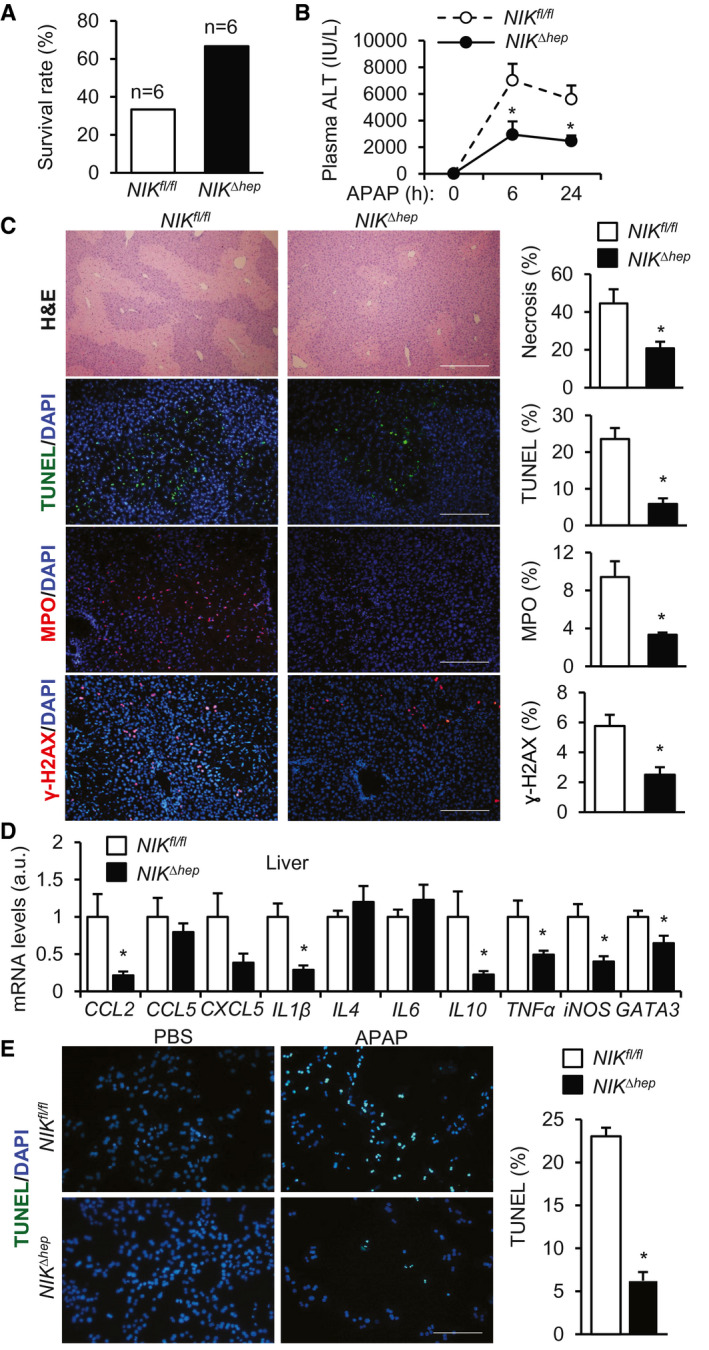
NIKΔhep mice are resistant to APAP‐induced acute liver failure. (A‐D) NIKΔhep and NIKfl/fl male mice were treated with APAP (250 mg/kg). (A) Survival rates 24 hours after APAP treatment (n = 6 per group). (B) Plasma ALT levels. NIKΔhep , n = 15; NIKfl/fl , n = 12. (C) Liver sections were stained with the indicated agents (24 hours after APAP treatment). Necrotic area was normalized to total area. TUNEL, MPO, or γ‐H2AX cell numbers were normalized to total cell number (n = 4‐12 per group). Scale bar, 200 μm. (D) Liver gene expression 24 hours after APAP treatment (normalized to 36B4 levels, n = 9‐12 per group). (E) Liver cells were prepared from NIKΔhep and NIKfl/fl mice, treated with APAP for 24 hours, and stained with TUNEL reagents. TUNEL+ cells were normalized to total cells (n = 3 per group). Data are presented as mean ± SEM. *P < 0.05; (C‐E) two‐tailed unpaired Student t test; (B) two‐way ANOVA/Bonferroni’s multiple comparisons test.
To further validate the role of NIK in APAP toxicity, we prepared primary liver cell cultures from NIKΔhep and NIKfl/fl littermates, stimulated cells with APAP, and measured cell death using TUNEL assays. APAP‐induced liver cell death was significantly lower in the NIKΔhep group relative to the NIKfl/fl group (Fig. 3E). Together, these results demonstrate for the first time that endogenous hepatic NIK plays an important role in drug‐induced liver injury.
Ablation of Hepatic IKKα Blunts APAP‐Induced Liver Injury
We next sought to identify downstream mediators coupling NIK to liver injury. We generated hepatocyte‐specific IKKα‐knockout (IKKαΔhep ) mice by crossing IKKαfl/fl mice with albumin‐Cre mice. IKKαfl/fl mice have been described.( 27 ) We treated IKKαΔhep and IKKαfl/fl littermates with APAP. Plasma ALT levels were significantly lower in IKKαΔhep than in IKKαfl/fl mice (Fig. 4A; Supporting Fig. S1C). Liver necrosis (H&E), hepatocyte death (TUNEL), liver DNA damage (γ‐H2AX), and hepatic neutrophil infiltration (MPO) were substantially lower in IKKαΔhep relative to IKKαfl/fl mice (Fig. 4B). Expression of cytokines/chemokines in the liver (CCL2, CCL5, IL1β) was significantly lower in IKKαΔhep mice (Fig. 4C). These results uncover the hepatic NIK/IKKα pathway as a previously unrecognized player in drug‐induced liver injury.
FIG. 4.
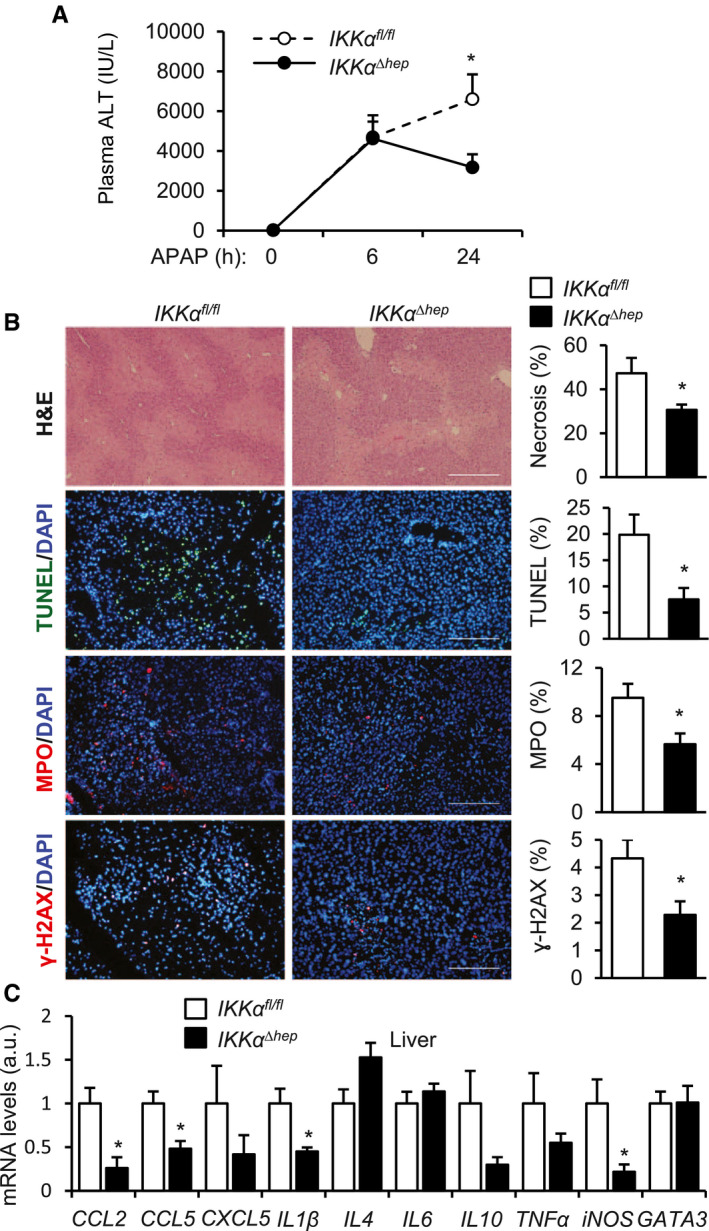
IKKαΔhep mice are resistant to APAP‐induced liver damage. IKKαΔhep and IKKαfl/fl male mice were treated with APAP (250 mg/kg). (A) Plasma ALT levels. IKKαΔhep , n = 9; IKKαfl/fl , n = 10. (B) Liver sections were stained with the indicated agents (24 hours after APAP treatment, n = 3‐8 per group). Scale bar, 200 μm. (C) Liver gene expression 24 hours after APAP treatment (normalized to 36B4 levels, n = 6‐10 per group). Data are presented as mean ± SEM. *P < 0.05; (B,C) two‐tailed unpaired Student t test; (A) two‐way ANOVA/Bonferroni’s multiple comparisons test.
NIK/IKKα Pathway Mediates APAP‐Induced Oxidative Stress in Hepatocytes
We next set out to investigate molecular mechanisms underlying NIK‐promoted liver injury. We confirmed that APAP treatment robustly increased phosphorylation and activation of c‐Jun N‐terminal kinase (JNK)1/2 (Supporting Fig. S3). Liver JNK phosphorylation was slightly lower in NIKΔhep relative to NIKfl/fl mice (Supporting Fig. S3A) and was modestly higher in HepNIK than in HepCon mice 2 hours after APAP treatment (Supporting Fig. S3A). Twenty‐four hours after APAP treatment, liver JNK phosphorylation was comparable both between NIKΔhep and NIKfl/fl mice and between HepNIK and HepCon mice (Supporting Fig. S3).
We reasoned that oxidative stress might couple the hepatic NIK/IKKα pathway to liver failure in APAP‐treated mice. Indeed, liver ROS levels as measured by DCF probes were dramatically lower in APAP‐treated NIKΔhep (relative to NIKfl/fl ) and IKKαΔhep (relative to IKKαfl/fl ) mice (Fig. 5A). Conversely, hepatic ROS was significantly higher in HepNIK than in HepCon mice 24 hours after APAP treatment (Fig. 5A). To assess RNS, we stained liver sections with antibody to nitrotyrosine. Liver nitrotyrosine levels were significantly lower in NIKΔhep (relative to NIKfl/fl ) and IKKαΔhep (relative to IKKαfl/fl ) mice, and were significantly higher in HepNIK than in HepCon mice (Fig. 5B). Considering that APAP induces oxidative stress by depleting the glutathione (GSH) pool,( 2 ) we measured liver GSH levels. GSH levels were significantly higher in NIKΔhep (relative to NIKfl/fl ) and IKKαΔhep (relative to IKKαfl/fl ) mice after APAP treatment (Fig. 5C). Conversely, liver GSH levels were significantly lower in HepNIK than in HepCon mice after APAP treatment, and treatment with the antioxidant NAC completely blocked GSH reduction (Fig. 5C).
FIG. 5.
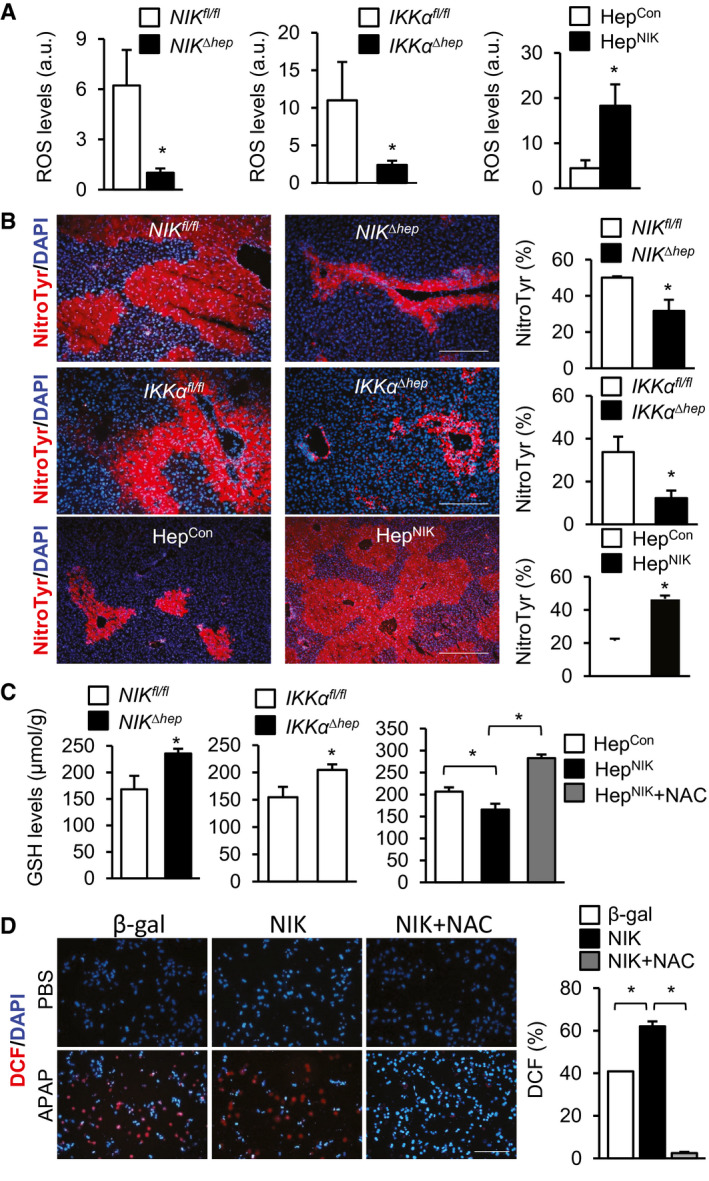
NIK increases oxidative stress in hepatocytes. (A‐C) Male mice were treated with APAP for 24 hours. HepNIK and HepCon, 200 mg/kg body weight; NIKΔhep , NIKfl/fl , IKKαΔhep , and IKKαfl/fl , 250 mg/kg. (A) Liver ROS levels (normalized to liver weight). NIKΔhep , n = 7; NIKfl/fl , n = 9; IKKαΔhep , n = 6; IKKαfl/fl , n = 9; HepNIK, n = 4; and HepCon, n = 4. (B) Liver sections were stained with anti‐nitrotyrosine antibody. Nitrotyrosine area was normalized to total area. NIKΔhep , n = 4; NIKfl/fl , n = 4; IKKαΔhep , n = 3; IKKαfl/fl , n = 3; HepNIK, n = 4; and HepCon, n = 4. Scale bar, 200 μm. (C) Liver GSH levels (normalized to liver weight). NIKΔhep , n = 10; NIKfl/fl , n = 10; IKKαΔhep , n = 9; IKKαfl/fl , n = 7; HepNIK, n = 7; HepCon, n = 7; HepNIK+NAC, n = 4. (D) Mouse primary hepatocytes were transduced with NIK or β‐gal adenoviral vectors, pretreated with 500 μM NAC, and followed by 2.5 mM APAP treatment for 24 hours. Hepatocytes were stained with DCF probes. DCF+ cells were normalized to total cells (n = 3 per group). Data are presented as mean ± SEM. *P < 0.05 (A‐C, left two panels) two‐tailed unpaired Student t test. (C, right panel, D) two‐way ANOVA/Bonferroni’s multiple comparisons test. Abbreviation: NitroTyr, nitrotyrosine.
To further validate the role of NIK in oxidative stress, we transduced mouse primary hepatocytes with NIK or β‐gal adenoviral vectors, followed by APAP stimulation. Overexpression of NIK significantly increased ROS levels in APAP‐treated hepatocytes, and NAC treatment completely blocked ROS elevations (Fig. 5D). Liver expression of inducible nitric oxide synthase (iNOS) was significantly lower in NIKΔhep (relative to NIKfl/fl ) (Fig. 3D) and IKKαΔhep (relative to IKKαfl/fl ) mice (Fig. 4C). Conversely, liver iNOS expression was significantly higher in HepNIK than in HepCon mice (Fig. 2D). To verify that NIK directly increases iNOS expression in hepatocytes, we overexpressed NIK in mouse primary hepatocytes using NIK adenoviral vectors. NIK profoundly increased iNOS expression under both baseline and APAP‐stimulated conditions (Supporting Fig. S4). These results show for the first time that the hepatic NIK/IKKα pathway augments liver oxidative stress in response to hepatotoxins, at least in part by increasing iNOS expression.
ROS is Involved in NIK‐Induced Liver Damage
To determine whether ROS mediates NIK action, we treated HepNIK mice with antioxidant NAC, followed by APAP stimulation. NAC dramatically decreased plasma ALT, liver ROS, and liver nitrotyrosine levels in APAP‐treated mice (Fig. 6A‐C). Importantly, NAC also drastically reduced liver necrosis (H&E), hepatocyte death (TUNEL), and liver neutrophil content (MPO) (Fig. 6C). Expression of CXCL5, IL1β, TNFα, and iNOS in the liver was significantly lower in NAC‐treated relative to empty vehicle‐treated HepNIK mice (Fig. 6D). To corroborate these results, we transduced mouse primary hepatocytes with NIK or β‐gal adenoviral vectors and treated hepatocytes with APAP in the presence or absence of NAC. Overexpression of NIK increased APAP‐stimulated hepatocyte death as expected, and NAC completely reversed NIK/APAP‐induced hepatocyte death (Fig. 6E). Collectively, these results suggest that oxidative stress plays an important role in NIK‐induced liver injury.
FIG. 6.
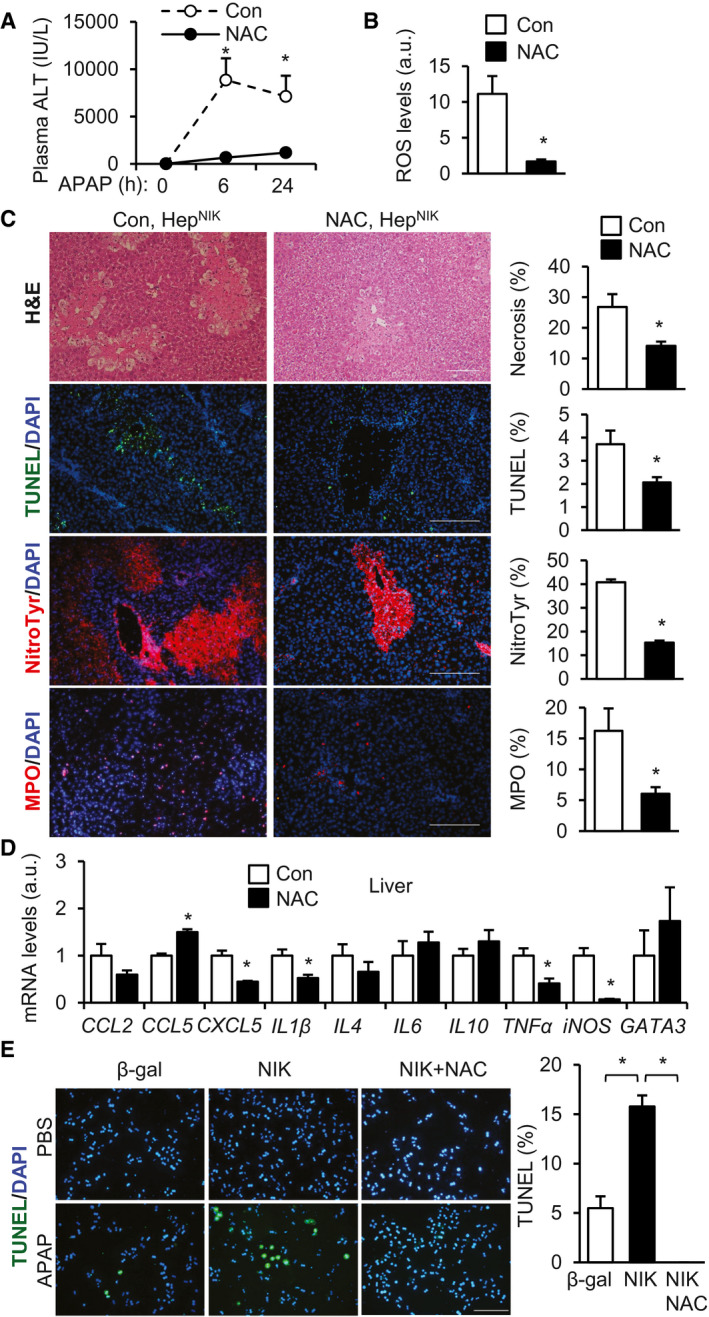
Oxidative stress mediates NIK‐induced liver injury. (A‐D) HepNIK male mice were pretreated with NAC (300 mg/kg body weight) or empty vehicles (Con) and treated with APAP (200 mg/kg) for 24 hours. (A) Plasma ALT levels (n = 4 per group). (B) Liver ROS levels (normalized to liver weight, n = 4 per group). (C) Liver sections were stained with the indicated agents. Necrosis area, TUNEL+, MPO+, and nitrotyrosine cells were normalized to total area (n = 3‐4 mice per group). Scale bar, 200 μm. (D) Liver gene expression (normalized to 36B4 levels, n = 4 per group). (E) Mouse primary hepatocyte cultures were transduced with NIK or β‐gal adenoviral vectors, pretreated with 500 μM NAC, and followed by 2.5 mM APAP stimulation for 24 hours. Hepatocyte death was assessed by TUNEL assays (normalized to total cells, n = 3 per group). Data are presented as mean ± SEM. *P < 0.05; (B‐D) two‐tailed unpaired Student t test; (A,E) two‐way ANOVA/Bonferroni’s multiple comparisons test). Abbreviation: NitroTyr, nitrotyrosine.
NIK Acts Cell Autonomously to Promote Hepatocyte Ferroptosis
Given that ROS stimulates lipid peroxidation and ferroptosis, we speculated that NIK might augment hepatocyte ferroptosis. We transduced mouse primary hepatocytes with NIK or β‐gal adenoviral vectors, followed by APAP stimulation. We blocked ferroptosis by treating hepatocytes with ferrostatin‐1 or liproxstatin‐1, two chemically distinct ferroptosis inhibitors. Overexpression of NIK markedly decreased the viability of APAP‐treated hepatocytes, as assessed by MTT assays (Fig. 7A; β‐gal vs. NIK). Treatment with either ferrostatin‐1 or liproxstatin‐1 markedly increased viabilities of both β‐gal‐expressing and NIK‐expressing hepatocytes after APAP treatment (Fig. 7A). Liproxstatin‐1 fully abrogated the ability of NIK to enhance APAP hepatotoxicity (Fig. 7A, right panels). Overexpression of NIK increased APAP‐stimulated hepatocyte death (by TUNEL assays), and ferrostatin‐1 or liproxstatin‐1 profoundly inhibited hepatocyte death induced by NIK/APAP (Fig. 7B; Supporting Fig. S5).
FIG. 7.
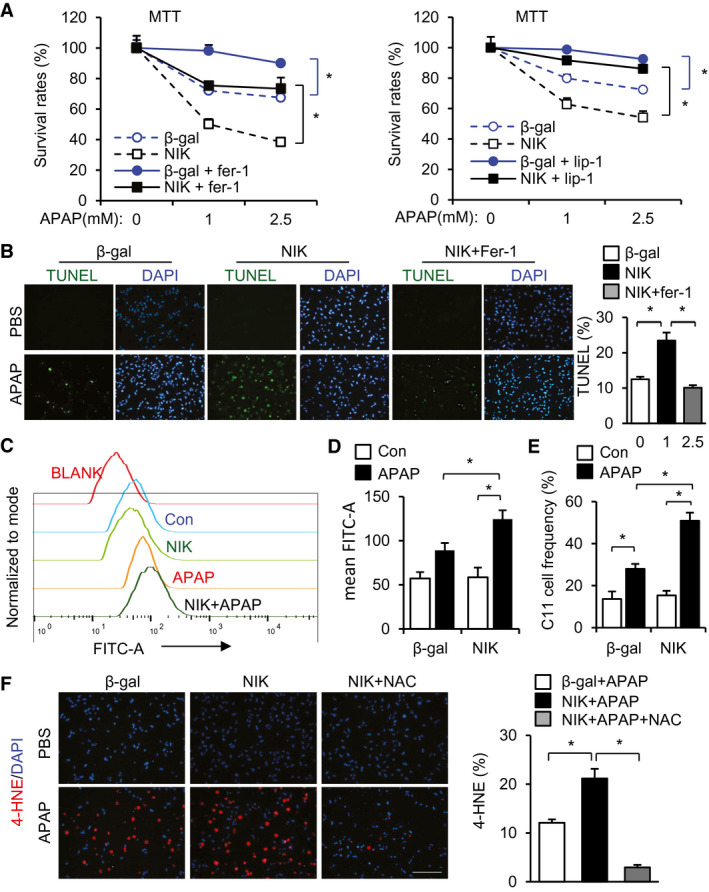
NIK enhances ferroptosis of APAP‐treated hepatocytes. (A,B) Mouse primary hepatocytes were transduced with NIK or β‐gal adenoviral vectors, pretreated with 2 μM ferrostatin‐1 or 2 μM liproxstatin‐1, and followed by APAP stimulation for 24 hours. (A) MTT assays (n = 3 per group). (B) TUNEL assays (normalized to total cells, n = 3 per group).( C,D) Mouse primary hepatocytes were transduced with NIK or β‐gal adenoviral vectors, treated with 2.5 mM APAP for 24 hours, stained with BODIPY 581/591 C11 probe, and analyzed by flow cytometry. (C) Representative C11 tracing. (D) Mean C11 density (n = 6 per group). (E) C11high hepatocyte frequency (normalized to total hepatocytes, n = 6 per group). (F) Mouse primary hepatocytes were transduced with NIK or β‐gal adenoviral vectors, treated with APAP for 24 hours in the presence or absence of 500 μM NAC, and stained with anti‐4‐HNE antibody (normalized to total cells, n = 3 per group). Scale bar, 200 μm. Data are presented as mean ± SEM. *P < 0.05; two‐way ANOVA/Bonferroni’s multiple comparisons test. Abbreviations: Fer‐1, ferrostatin‐1; Lip‐1, liproxstatin‐1.
To further validate ferroptosis, we examined the effects of NIK and APAP on production of lipid peroxides (ferroptosis marker), using the lipid peroxidation probe BODIPY 581/591 C11.( 32 ) We transduced mouse primary hepatocytes with NIK or β‐gal adenoviral vectors, stimulated cells with APAP, stained hepatocytes with BODIPY 581/591 C11, and assessed hepatocyte lipid peroxidation levels using flow cytometry. APAP increased BODIPY 581/591 C11 levels in β‐gal adenoviral transduced hepatocytes (Fig. 7C,D). Overexpression of NIK considerably augmented the ability of APAP to increase BODIPY 581/591 C11 levels in hepatocytes (Fig. 7C,D). The frequency of C11high hepatocytes was significantly higher in the NIK than in the β‐gal groups after APAP stimulation (Fig. 7E). Using fluorescent microscopy, we confirmed that NIK overexpression augmented the ability of APAP to increase the C11high hepatocyte number (Supporting Fig. S6).
We further validated ferroptosis using an antibody to 4 hydroxynonenal (4‐HNE). We transduced mouse primary hepatocytes with NIK or β‐gal adenoviral vectors, stimulated cells with APAP, and immunostained hepatocytes with an antibody to 4‐HNE. APAP increased the number of 4‐HNE+ hepatocytes; overexpression of NIK further increased 4‐HNE+ hepatocytes (Fig. 7F). Treatment with antioxidant NAC abrogated the ability of NIK/APAP to increase 4‐HNE+ hepatocytes (Fig. 7F). Taken together, these results unravel a previously unrecognized NIK/ROS/lipid peroxidation/ferroptosis axis.
Discussion
In this work, we have uncovered hepatic NIK and IKKα as previously unrecognized risk factors for acute liver failure. We observed that APAP, CCl4, and possibly other hepatotoxins directly increase NIK expression and stability in hepatocytes. It is likely that these agents inhibit the abilities of tumor necrosis factor receptor–associated factor (TRAF)2, TRAF3, cellular inhibitor of apoptosis protein 1/2 (cIAP1/2), carboxy‐terminus of Hsc70 interacting protein (CHIP), and/or related ubiquitin E3 ligases to promote the ubiquitination and degradation of NIK. Hepatocyte‐specific overexpression of NIK substantially increased liver injury and mortality in mice treated with APAP. Hepatocyte death, DNA damage, and liver inflammation were markedly elevated in NIK‐overexpressing mice after APAP treatment. Conversely, hepatocyte‐specific ablation of NIK markedly decreased APAP‐triggered acute liver failure and mortality. APAP‐treated IKKαΔhep mice phenocopied NIKΔhep mice. These findings reveal the hepatic NIK/IKKα pathway as an important player in drug‐induced liver toxicity.
Liver‐specific overexpression of NIK markedly increased liver ROS and RNS levels in APAP‐treated HepNIK mice. Conversely, ablation of either hepatic NIK or IKKα markedly decreased liver ROS and RNS levels in APAP‐treated NIKΔhep or IKKαΔhep mice, respectively. Liver GSH levels were also higher in NIKΔhep and IKKαΔhep mice. These results indicate that endogenous hepatic NIK and IKKα are required for APAP and possibly other hepatotoxic drugs and agents to induce pathogenic oxidative stress in the liver. In line with this notion, in primary hepatocytes, overexpression of NIK increased ROS levels cell autonomously following APAP stimulation. Of note, overexpression of NIK increased whereas ablation of NIK decreased expression of iNOS in hepatocytes. iNOS deficiency mitigates alcoholic liver injury.( 33 ) Hence, iNOS mediates, at least in part, NIK‐promoted oxidative stress and liver injury.
Antioxidant NAC treatment reversed APAP/NIK‐induced liver failure in HepNIK mice. NAC also blocked APAP/NIK‐induced death in primary hepatocyte cultures. These results suggest that oxidative stress plays a pivotal role in NIK/APAP‐induced liver failure. In line with this notion, DNA damage, presumably caused by ROS, was lower in NIKΔhep and IKKαΔhep mice and higher in HepNIK mice relative to respective control mice after APAP treatment. DNA damage is likely to be involved in liver necrosis. ROS is well known to fuel lipid peroxidation. Accordingly, we found that overexpression of NIK increased the ability of APAP to stimulate production of lipid peroxides in hepatocytes (assessed by BODIPY 581/591 C11). 4‐HNE levels were higher in NIK‐overexpressing hepatocytes after APAP stimulation. Cell membrane lipid peroxidation in conjunction with iron overload is known to drive ferroptosis.( 11, 12 ) Treatment with the chemically distinct ferroptosis inhibitors ferrostatin‐1 and liproxstatin‐1 blocked the ability of NIK/APAP to induce hepatocyte death. In accordance with these results, two groups recently reported that APAP induces ferroptosis in the liver.( 34, 35 ) We acknowledge that some groups consider ferroptosis not important for APAP hepatotoxicity.( 36 ) We argue that the outcomes of APAP overdose are influenced by multiple genetic and nongenetic cofactors. Aberrant activation of the hepatic NIK/IKKα/ROS/lipid peroxidation pathway likely augments hepatocyte ferroptosis. Of note, hepatic NIK modestly enhanced liver JNK activation in mice treated with APAP for 2 hours, raising the possibility that JNK may be involved in mediating NIK/APAP‐induced liver injury.
NIK has been extensively examined in lymph organ development and the immune system.( 13 ) Global NIK knockout results in thymus atrophy and autoimmune disorders (e.g., autoimmune hepatitis) in mice.( 17 ) Medullary thymic epithelial‐specific deletion of NIK or IKKα impairs thymic medulla development and central T‐cell tolerance, resulting in fatal autoimmune hepatitis in mice.( 37 ) A modest elevation of hepatocyte NIK increases gluconeogenesis and suppresses fatty acid β oxidation in obesity, promoting metabolic disorders.( 16, 20, 22 ) Hepatic NIK and IKKα inhibit reparative hepatocyte proliferation by suppressing the JAK2/signal transducer and activator of transcription 3 pathway, impeding liver regeneration.( 23 ) Excessive hepatic NIK promotes hepatocytes to release mediators that potently activate macrophages/Kupffer cells, and activated macrophages/Kupffer cells launch fatal immune destruction against the liver.( 19 ) Here, we found that hepatic NIK promotes liver oxidative stress and ferroptosis. Given that hepatic NIK is highly activated in a broad spectrum of liver injuries, we speculate that in both acute and chronic liver diseases, hepatic NIK serves as a common route connecting liver stress/inflammation to hepatocyte oxidative stress, ferroptosis, and liver injury. We further postulate that the pathogenic outcomes of hepatic NIK are influenced by NIK dosages and cofactors. Additional studies are warranted to delineate NIK dosage effects and identify cofactors regulating NIK responses.
Supporting information
Supplementary Material
Acknowledgment
We thank Dr. Klaus Rajewsky (Immune Disease Institute, Harvard Medical School, Boston, MA) for providing Rosa26‐STOP‐NIK mice; Dr. Lei Yin (University of Michigan) for providing Hepa1 cells; Dr. Cynthia Ju (University of Texas Health Science Center at Houston) for providing antibodies recognizing NAPQI adducts (originally from Dr. Lance R. Pohl, the National Heart, Lung, and Blood Institute, National Institutes of Health); Rashi Singhal, Xin Tong, and Lei Yin (University of Michigan) for helpful discussions; and Yankai Wen and Constance L. Atkins (University of Texas Health Science Center at Houston) for assistance in performing NAPQI adduct experiments.
Supported by the National Institutes of Health (NIH) (grants RO1 DK114220, RO1 DK115646, and R21 AA025945 to L.R. and DK095201, CA148828, and CA245546 to Y.S.). This work used the cores supported by the Michigan Diabetes Research and Training Center (NIH DK020572), Michigan Metabolomics and Obesity Center (NIH DK089503), and the University of Michigan Gut Peptide Research Center (NIH DK34933).
Potential conflict of interest: Nothing to report.
Contributor Information
Xue‐Gong Fan, Email: xgfan@hotmail.com.
Liangyou Rui, Email: ruily@umich.edu.
References
- 1.Rui L. Energy metabolism in the liver. Compr Physiol 2014;4:177‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan M, Huo Y, Yin S, Hu H. Mechanisms of acetaminophen‐induced liver injury and its implications for therapeutic interventions. Redox Biol 2018;17:274‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008;134:1655‐1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol 2013;59:583‐594. [DOI] [PubMed] [Google Scholar]
- 5.Bhattacharyya S, Sinha K, Sil PC. Cytochrome P450s: mechanisms and biological implications in drug metabolism and its interaction with oxidative stress. Curr Drug Metab 2014;15:719‐742. [DOI] [PubMed] [Google Scholar]
- 6.Masarone M, Rosato V, Dallio M, Gravina AG, Aglitti A, Loguercio C, et al. Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxid Med Cell Longev 2018;2018:9547613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol 2015;12:231‐242. [DOI] [PubMed] [Google Scholar]
- 8.Cichoz‐Lach H, Michalak A. Oxidative stress as a crucial factor in liver diseases. World J Gastroenterol 2014;20:8082‐8091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sohal RS. Role of oxidative stress and protein oxidation in the aging process. Free Radic Biol Med 2002;33:37‐44. [DOI] [PubMed] [Google Scholar]
- 10.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature 2000;408:239‐247. [DOI] [PubMed] [Google Scholar]
- 11.Conrad M, Kagan VE, Bayir H, Pagnussat GC, Head B, Traber MG, et al. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev 2018;32:602‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galaris D, Barbouti A, Pantopoulos K. Iron homeostasis and oxidative stress: an intimate relationship. Biochim Biophys Acta Mol Cell Res 2019;1866:118535. [DOI] [PubMed] [Google Scholar]
- 13.Thu YM, Richmond A. NF‐kappaB inducing kinase: a key regulator in the immune system and in cancer. Cytokine Growth Factor Rev 2010;21:213‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao G, Harhaj EW, Sun SC. NF‐kappaB‐inducing kinase regulates the processing of NF‐kappaB2 p100. Mol Cell 2001;7:401‐409. [DOI] [PubMed] [Google Scholar]
- 15.Sun SC. The noncanonical NF‐kappaB pathway. Immunol Rev 2012;246:125‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheng L, Zhou Y, Chen Z, Ren D, Cho KW, Jiang L, et al. NF‐kappaB‐inducing kinase (NIK) promotes hyperglycemia and glucose intolerance in obesity by augmenting glucagon action. Nat Med 2012;18:943‐949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen H, Sheng L, Xiong Y, Kim YH, Jiang L, Chen Z, et al. Thymic NF‐kappaB‐inducing kinase (NIK) regulates CD4+ T cell‐elicited liver injury and fibrosis in mice. J Hepatol 2017;67:100‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren X, Li X, Jia L, Chen D, Hou H, Rui L, et al. A small‐molecule inhibitor of NF‐kappaB‐inducing kinase (NIK) protects liver from toxin‐induced inflammation, oxidative stress, and injury. FASEB J 2017;31:711‐718. [DOI] [PubMed] [Google Scholar]
- 19.Shen H, Sheng L, Chen Z, Jiang L, Su H, Yin L, et al. Mouse hepatocyte overexpression of NF‐kappaB‐inducing kinase (NIK) triggers fatal macrophage‐dependent liver injury and fibrosis. Hepatology 2014;60:2065‐2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Sheng L, Xiong Y, Shen H, Liu Y, Rui L. Liver NF‐kappaB‐inducing kinase (NIK) promotes liver steatosis and glucose counterregulation in male mice with obesity. Endocrinology 2017;158:1207‐1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang WS, Pan A, Zhang X, Ying A, Ma G, Liu BL, et al. Inactivation of NF‐kappaB2 (p52) restrains hepatic glucagon response via preserving PDE4B induction. Nat Commun 2019;10:4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Chen M, Zhou Y, Tang C, Zhang W, Zhong Y, et al. NIK links inflammation to hepatic steatosis by suppressing PPARalpha in alcoholic liver disease. Theranostics 2020;10:3579‐3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiong YI, Torsoni AS, Wu F, Shen H, Liu Y, Zhong X, et al. Hepatic NF‐kB‐inducing kinase (NIK) suppresses mouse liver regeneration in acute and chronic liver diseases. Elife 2018;7.e34152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernal W, Wendon J. Acute liver failure. N Engl J Med 2013;369:2525‐2534. [DOI] [PubMed] [Google Scholar]
- 25.Raucy JL, Lasker JM, Lieber CS, Black M. Acetaminophen activation by human liver cytochromes P450IIE1 and P450IA2. Arch Biochem Biophys 1989;271:270‐283. [DOI] [PubMed] [Google Scholar]
- 26.Kheradpezhouh E, Ma L, Morphett A, Barritt GJ, Rychkov GY. TRPM2 channels mediate acetaminophen‐induced liver damage. Proc Natl Acad Sci U S A 2014;111:3176‐3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu B, Xia X, Zhu F, Park E, Carbajal S, Kiguchi K, et al. IKKalpha is required to maintain skin homeostasis and prevent skin cancer. Cancer Cell 2008;14:212‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Y, Jiang L, Rui L. Identification of MUP1 as a regulator for glucose and lipid metabolism in mice. J Biol Chem 2009;284:11152‐11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang B, Shen H, Chen Z, Yin L, Zan L, Rui L. Carboxyl terminus of HSC70‐interacting protein (CHIP) down‐regulates NF‐kappaB‐inducing kinase (NIK) and suppresses NIK‐induced liver injury. J Biol Chem 2015;290:11704‐11714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sasaki Y, Calado DP, Derudder E, Zhang B, Shimizu Y, Mackay F, et al. NIK overexpression amplifies, whereas ablation of its TRAF3‐binding domain replaces BAFF:BAFF‐R‐mediated survival signals in B cells. Proc Natl Acad Sci U S A 2008;105:10883‐10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao RY, Wang M, Liu Q, Feng D, Wen Y, Xia Y, et al. Hypoxia‐inducible factor‐2alpha reprograms liver macrophages to protect against acute liver injury through the production of interleukin‐6. Hepatology 2020;71:2105‐2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pap EH, Drummen GP, Winter VJ, Kooij TW, Rijken P, Wirtz KW, et al. Ratio‐fluorescence microscopy of lipid oxidation in living cells using C11‐BODIPY(581/591). FEBS Lett 1999;453:278‐282. [DOI] [PubMed] [Google Scholar]
- 33.McKim SE, Gäbele E, Isayama F, Lambert JC, Tucker LM, Wheeler MD, et al. Inducible nitric oxide synthase is required in alcohol‐induced liver injury: studies with knockout mice. Gastroenterology 2003;125:1834‐1844. [DOI] [PubMed] [Google Scholar]
- 34.Wang M, Liu C‐Y, Wang T, Yu H‐M, Ouyang S‐H, Wu Y‐P, et al. (+)‐Clausenamide protects against drug‐induced liver injury by inhibiting hepatocyte ferroptosis. Cell Death Dis 2020;11:781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamada N, Karasawa T, Kimura H, Watanabe S, Komada T, Kamata R, et al. Ferroptosis driven by radical oxidation of n‐6 polyunsaturated fatty acids mediates acetaminophen‐induced acute liver failure. Cell Death Dis 2020;11:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaeschke H, Adelusi OB, Ramachandran A. Ferroptosis and acetaminophen hepatotoxicity ‐ are we going down another rabbit hole? Gene Expr 2021; 10.3727/105221621X16104581979144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen H, Ji Y, Xiong Y, Kim H, Zhong X, Jin MG, et al. Medullary thymic epithelial NF‐kB‐inducing kinase (NIK)/IKKalpha pathway shapes autoimmunity and liver and lung homeostasis in mice. Proc Natl Acad Sci U S A 2019;116:19090‐19097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material