

ABSTRACT
Background
Adaptive thermogenesis is an iron-demanding pathway, significantly contributing to whole-body energy expenditure. However, the effects of iron-deficient diets on adaptive thermogenesis and obesity remain unknown.
Objectives
We aimed to determine the impact of dietary iron deficiency on iron homeostasis in adipocytes, adaptive thermogenic capacity, and metabolic consequences in obesity.
Methods
C57BL/6 male mice were assigned to either the iron-adequate (IA, 35 ppm) or the iron-deficient group (ID, 3 ppm) at weaning. Upon 8 wk of age, both IA and ID groups received an isocaloric high-fat diet (45% kcal from fat) for 10 wk, maintaining the same iron content. Mice (n = 8) were used to determine the iron status at the systemic and tissue levels and lipid metabolism and inflammatory signaling in adipose tissue. The same mice were used to evaluate cold tolerance (4°C) for 3 h. For assessing adaptive thermogenesis, mice (n = 5) received an intraperitoneal injection of β3-adrenoceptor agonist CL316243 (CL) for 5 d.
Results
Compared with the IA group, the ID group had nonanemic iron deficiency, lower serum ferritin (42.8%, P < 0.01), and greater weight gain (8.67%, P < 0.05) and insulin resistance (159%, P < 0.01), partly due to reduced AMP-activated protein kinase activation (61.0%, P < 0.05). Upon cold exposure, the ID group maintained a core body temperature 2°C lower than the IA group. The ID group had lower iron content (47.0%, P < 0.01) in the inguinal adipose tissue (iWAT) than the IA group, which was associated with impaired adaptive thermogenesis. In response to CL, ID mice showed decreased heat production (P < 0.01) and defective upregulation of beige adipocyte-specific markers, including uncoupling protein 1 (41.1%, P < 0.001), transferrin receptor 1 (47.5%, P < 0.001), and mitochondrial respiratory chain complexes (P < 0.05) compared with IA mice.
Conclusions
Dietary iron deficiency deregulates iron balance in the iWAT and impairs adaptive thermogenesis, thereby escalating the diet-induced weight gain in C57BL/6 mice.
Keywords: iron deficiency, adaptive thermogenesis, adipose tissue browning, obesity, transferrin receptor 1
Introduction
Adipose tissue plays a vital role in maintaining temperature homeostasis by providing insulation and generating heat through the dissipation of mitochondrial energy. The latter pathway is called adaptive thermogenesis, a part of our body's defense mechanism against external stimuli (e.g., excess energy intake and cold temperature) (1). Humans possess at least 2 different thermogenic fat cells, classical brown and beige adipocytes. Despite the differences in developmental and transcriptional traits (2, 3), once activated, both beige and brown adipocytes perform the same metabolic function: to burn glucose and fatty acids through the enhanced mitochondrial respiration and uncoupling protein 1 (UCP1)–mediated heat release (4). In addition to UCP1 upregulation, thermogenic function of brown/beige fat is proportional to the abundance of mitochondrial mass and activity. Thus, the regulation of mitochondrial biogenesis is a metabolic switch to turn on or off the thermogenic function (5, 6).
Iron is a micronutrient critical for mitochondrial biogenesis and assembly of heme and iron–sulfur (Fe–S) cluster proteins involved in the electron transfer chain. In addition, iron serves as a signaling molecule to regulate the transcription of mitochondrial proteins (7). In recent work, our group reported that beige thermogenesis is an iron-demanding process in which thermogenic stimuli promote 1) the binding of iron-regulatory proteins (IRPs) to the iron-response element (IRE) of their target genes and 2) subsequent iron import into adipocytes via transferrin receptor 1 (TFR1) (8). Conversely, the loss of thermogenic function upon thermoneutral conditions concurred with the exodus of iron along with autophagic degradation of mitochondria (8). Also, the depletion of adipocyte iron by targeted ablation of TFR1 in adipocytes blunted adipose tissue browning (9, 10). These studies indicate that the expansion of iron pool size and resetting new iron balance are essential for cellular makeup in beige adipocytes and the equipment of iron-loaded mitochondria. However, it is largely unknown whether iron deficiency would be a risk factor to compromised adaptive thermogenesis, thereby exacerbating obesity and metabolic dysfunction.
Iron deficiency is the most common nutritional deficiency worldwide and a significant precipitant of anemia. Diet-induced iron deficiency is the primary cause for iron-deficiency anemia (IDA), affecting >1.2 billion individuals, and iron deficiency without anemia is even more prevalent (11). The WHO defines the onset of IDA upon a decrease of hemoglobin concentrations (i.e., <13 g/dL in males, <12 g/dL in nonpregnant females, and <11 g/L in pregnant females) (12). Low serum ferritin concentrations (<30 mg/L are the accepted threshold for mild cases and <10–12 mg/L in IDA) are the hallmark of iron deficiency, reflecting exhausted iron stores in the absence of inflammation (11). In inflammation, the increase of the iron hormone hepcidin suppresses iron mobilization and induces iron sequestration into storage, resulting in systemic iron deficiency without depletion of iron stores (13, 14). Several studies have demonstrated the positive correlation between obesity and iron deficiency via hepcidin (15–17). Hepcidin is a peptide hormone that regulates iron metabolism by inhibiting iron transporter located in the gut enterocytes and macrophages (18). Increased hepcidin concentrations in obese individuals have been proposed as the culprit for causing iron immobilization and systemic iron deficiency (13, 14, 19). However, the reverse mechanism of whether dietary iron deficiency promotes susceptibility to obesity is controversial.
Adaptive thermogenesis is a metabolic process that 1) upsurges iron requirement and 2) counteracts weight gain via thermogenic energy expenditure. Hence, it is rational to hypothesize that dietary iron deficiency induces iron deficiency at the cellular levels in adipocytes, interfering with mitochondrial biogenesis required for beige fat development. We also hypothesized that the diminished thermogenic energy expenditure by chronic low-iron intake exacerbates high-fat (HF) diet-induced obesity. To test these 2 hypotheses simultaneously, we carefully designed a diet formulation that triggers diet-mediated iron deficiency and weight gain in the absence of systemic inflammation in C57BL/6 mice. Our work demonstrated that dietary iron deficiency contributes to obesity by attenuating adaptive thermogenesis.
Methods
Animals
All protocols and procedures were approved by the Institutional Animal Care and Use Committee of the University of Massachusetts–Amherst. C57BL/6J male mice were randomly assigned to either the iron-adequate (IA) or the iron-deficient (ID) diet group (n = 13/group) at weaning. The mice were fed an AIN-93G diet formulated with a standard mineral mix containing an adequate amount of iron (35–50 ppm, IA group) or iron-depleted mineral mix (3–5 ppm iron, ID group). To avoid metabolic disturbance due to early exposure to an HF diet (20, 21), we implemented the HF feeding after mice reached sexual maturity at 8 wk of age (upon sexual maturity). The mice received an HF diet (45% of calories from fat) for 10 wk and maintained the same dietary iron content (diet compositions in Supplemental Table 1). Also, to minimize the confounding effects of HF diet–induced inflammation on iron metabolism, diets were devoid of cholesterol, which instigates HF diet–induced inflammation (22). All the mice were fed ad libitum and provided with distilled water to prevent iron intake from drinking water. The body weight and food intake were monitored weekly. After 10 wk of dietary iron modulation (ID or IA) with HF feeding, mice were analyzed to determine the iron status, metabolic outcomes, cold tolerance, and adaptive thermogenesis. For acute cold treatment, the mice (n = 8/group) were placed in a rodent incubator at 4°C (Powers Scientific) for 3 h. To stimulate adipose tissue browning, the mice (n = 5/group) received an intraperitoneal injection of β3-adrenoceptor (β3AR) agonist CL316243 (CL, 1 mg/kg BW; Santa Cruz Technology) for 5 consecutive days. We used a digital thermometer (Thermocouple Meter) combined with a thermocouple rectal probe (Kent Scientific Corp.) to measure the core body temperature. For the detection of heat release, we used an infrared camera (A655sc; FLIR Systems), as we described previously (15). Each mouse was fully perfused with 25 mL ice-cold saline at necropsy to avoid potential contamination of blood iron. The collected tissues were snap-frozen in liquid nitrogen and kept at −80°C until analysis.
Serum biochemistry and hematologic analyses
Whole blood was collected from the submandibular vein and stored in a heparinized tube. For the hematocrit measurement, the whole blood was transferred to a microhematocrit capillary tube (CNT-ZPC7-40HE; LW Scientific), and the red blood cells were separated using the hematocrit centrifuge at 10,000 × g for 3 min at room temperature. After centrifugation, the separation line was measured with a microhematocrit reader card (LW Scientific). The blood hemoglobin (Hb) concentrations were quantified using a hemoglobin colorimetric assay kit purchased from Cayman (700,540). To prepare the serum, whole blood without an anticoagulant was allowed to clot for 30 min at 25°C, and the top serum layer was collected after centrifugation at 2,000 × g for 15 min at 4°C. The serum ferritin concentration was measured using a mouse ELISA kit from Abcam (ab157713).
The blood glucose concentrations were measured using a glucometer (Contour; Bayer) after overnight fasting (12 h), and serum insulin concentrations were analyzed using a mouse ELISA kit purchased from Crystal Chem. HOMA-IR, an index for insulin resistance, was calculated as described previously (23). The serum concentrations of total cholesterol (TC) and triglycerides (TGs) were quantified using colorimetric enzymatic methods according to the manufacturer's protocol provided by Abcam (ab65390) and BioVision (K622), respectively. Nonesterified free fatty acids (NEFAs) were assayed in serum using commercial kits purchased from Cell Biolabs (STA-618).
Iron determination in tissue
For iron measurement in the tissue, ∼100 mg adipose tissue [inguinal white adipose tissue (iWAT) and brown adipose tissue (BAT)] was digested in 70% nitric acid for 2 h at 75°C and diluted with distilled water. Inductively coupled plasma mass spectrometry (NexION 300X; Perkin Elmer) was used to determine total iron content, and the data were normalized by wet tissue weight.
Hematoxylin and eosin staining
iWAT and BAT were fixed in 10% buffered formalin. Paraffin-embedded tissues were cut into 5-μm sections and processed for hematoxylin and eosin (H&E) staining as described previously (15).
Real-time qPCR
Total RNA was extracted with TRIzol reagent (Invitrogen) from homogenized tissues following the manufacturer's instruction. Isolated total RNA was treated with DNase I (Bio-Rad) to avoid potential genomic DNA contamination. Then, 2 μg RNA was reverse-transcribed for cDNA synthesis (iScript; Bio-Rad), and qPCR was carried out on a CFX96 real-time PCR system (Bio-Rad) using SYBR Green. The relative gene expression was calculated based on the 2−ΔΔCT method with normalization of the raw Ct values by 36b4. Primer sequences are available in Supplemental Table 2.
Western blot analysis
Tissue samples were homogenized in RIPA buffer containing protease inhibitors (MilliporeSigma) and phosphatase inhibitors (2 mM Na3VO4, 20 mM β-glycerophosphate, and 10 mM NaF). The proteins were separated by 10% SDS-PAGE, blotted onto polyvinylidene difluoride membranes, and incubated with the relevant antibodies (Supplemental Table 3). The chemiluminescence from ECL solution (Western Lightning) was detected using an Odyssey FC Imaging System (Li-Cor). The band intensity was analyzed with Image Studio Lite Software Ver 5.2.5 (Li-Cor).
Statistical analysis
Data were expressed as mean ± SD. The Student t test was used for comparison between 2 groups. Multigroup comparisons were performed by a 1-factor ANOVA followed by Tukey multiple comparison test. The effects of the diet (IA compared with ID) and cold exposure time (0–3 h) on the core body temperature were analyzed by a 2-factor repeated-measures ANOVA followed by Bonferroni post hoc test. Differences were considered significant at P < 0.05. All statistical analyses were performed with GraphPad Prism 8.0 (Version 8.4.2; GraphPad Software).
Results
Changes in iron status, body weight, serum lipids, and insulin sensitivity
The intake of the ID diet for 10 wk did not affect hematocrit or whole-blood Hb concentrations compared with the IA diet (Figure 1A). Also, the mice in the ID group did not develop IDA according to the guideline of anemia published by the WHO (12). The serum ferritin concentration was lower in the ID group compared with the IA group (P < 0.01), reflecting iron deficiency at the tissue level (Figure 1B). In addition, we examined the expression of hepatic genes integral to the regulation of systemic iron homeostasis, such as hepcidin and TFR1. There was a drastic suppression of hepcidin (Hamp1) gene expression (P < 0.001) but a significant increase of Tfr1 (P < 0.001) in the ID group compared with the IA group, which is indicative of a drop in the organismal iron status by the ID diet. IL-6 and bone morphogenetic protein 6 (BMP6) are 2 crucial signaling molecules to increase hepcidin transcription in the liver (18, 24). The mRNA expression concentrations of IL-6 (Il6) were almost identical between IA and ID, indicating that IL-6 is not the primary cause of altering the hepcidin concentrations (Figure 1C). We found that transcripts of genes with BMP-responsive elements and thus targets of BMP6, such as Atoh8, Smad7, and Id1 (25, 26), were consistently downregulated in the ID group (Figure 1D). Collectively, we defined the iron status of the ID mice as a nonanemic iron deficiency compared with the mice in the IA group.
FIGURE 1.
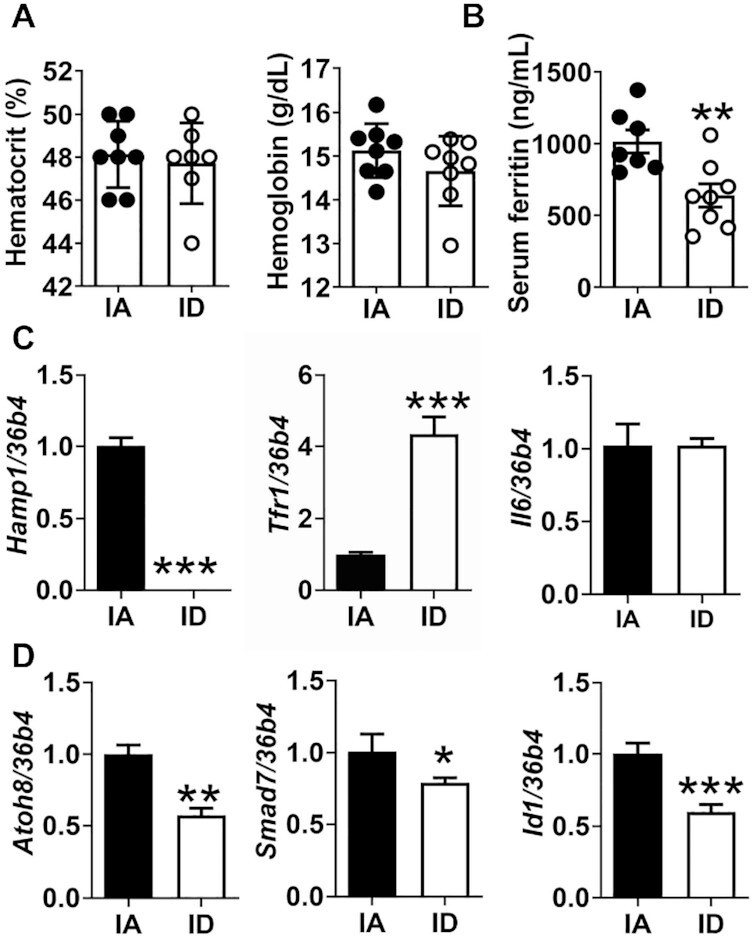
Chronic low-iron intake induces nonanemic iron deficiency. (A) Hematocrit and hemoglobin values in whole blood; (B) serum soluble ferritin; (C) mRNA expression concentrations of hepcidin (Hamp1), transferrin receptor (Tfr1), and IL6 (Il6) in the liver; and (D) mRNA expression concentrations of target genes of BMP6 in the liver. Values are mean ± SD, n = 8. In C and D, 36b4 was the reference gene. Asterisks indicate the difference from IA, *P < 0.05, **P < 0.01, and ***P < 0.001, by Student t test. Atoh8, atonal BHLH transcription factor 8; BMP6; bone morphogenetic protein 6, Hamp1, hepcidin; IA, iron adequate; ID, iron deficient; Id1, inhibitor of DNA binding protein 1; Il6, interleukin 6; Tfr1, transferrin receptor 1.
Despite reduced food intake compared with the isocaloric IA diet (Figure 2A), the ID diet induced a small but significant increase in body weight (P < 0.05). The ID mice showed a significant increase in white adipose tissue mass, both epididymal WAT (eWAT) and inguinal WAT (iWAT) (Figure 2B). In addition, the ID diet reduced the spleen mass while posing no significant impacts on the liver and BAT mass compared with the IA diet (Figure 2B). Given the increased body weight in the ID mice, we also examined insulin sensitivity. There was no change in the fasting serum glucose concentrations in ID mice, but the serum insulin concentrations were significantly higher in the ID mice than in IA mice (Figure 2C). Therefore, the HOMA-IR score was higher in ID mice than IA mice (Figure 2C), indicating the earlier onset of insulin resistance in ID mice. However, dietary iron concentrations showed no effect on serum lipid indices, including NEFA, TG, and TC concentrations (Figure 2C).
FIGURE 2.
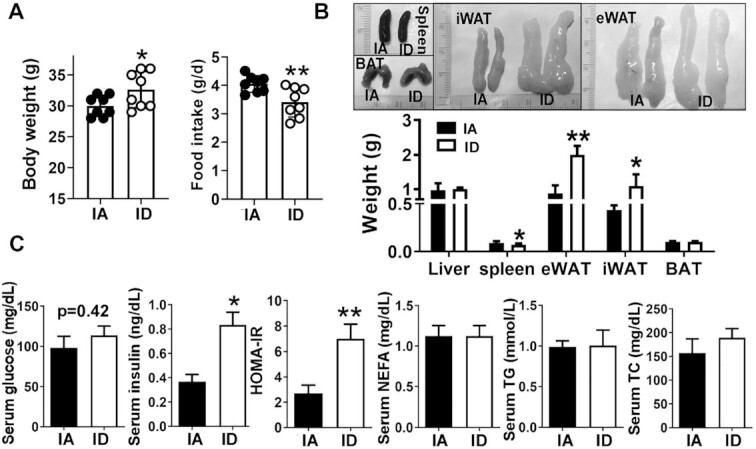
Diet-induced iron deficiency promotes visceral adiposity and insulin resistance. (A) Body weight and food intake. (B) Gross images of different adipose depots (upper) and mass (lower). (C) Fasting blood glucose and insulin concentrations in serum, HOMA-IR index, and lipid profiles in serum; NEFA, TG, and TC. Values are mean ± SD, n = 8. Asterisks indicate the difference from IA, *P < 0.05 and **P < 0.01, by Student t test. BAT, brown adipose tissue; eWAT, epididymal white adipose tissue; IA, iron adequate; ID, iron deficient; iWAT, inguinal white adipose tissue; NEFA, nonesterified fatty acid; TC, total cholesterol; TG, triglyceride.
Intriguingly, peroxisome proliferator-activated receptor γ (Pparγ), stearoyl-CoA desaturase 1 (Scd1), and sterol regulatory element-binding protein 1c (Srebp1c) were not significantly different between groups (Figure 3A). The mRNA expression concentrations that regulate fatty acid (FA) oxidation, such as peroxisome proliferator-activated receptor α (Pparα), carnitine-palmitoyl transferase 1 (Cpt1), and acyl-CoA oxidase 1 (Acox1), were significantly lower in ID mice than IA mice (Figure 3A). The reduced CPT1 protein expression with no differences in lipogenic regulators of PPARγ and SCD1 in ID were validated by Western blot analysis (Figure 3B). The phosphorylation concentrations of AMP-activated protein kinase (AMPK) were significantly lower in the ID mice than IA mice (Figure 3B), indicating that reduced catabolic pathways, but not augmented lipogenesis, cause weight gain in the ID mice.
FIGURE 3.

Diet-induced iron deficiency reduces catabolic pathways and promotes inflammation in eWAT. (A) mRNA expression of genes related to lipogenesis (Pparγ, Scd1, and Srebp1c) and fatty acid oxidation (Pparα, Cpt1, and Acox1). (B) Protein expression of lipid metabolism (PPARγ, SCD1, CPT1, p-AMPK, and t-AMPK). (C) mRNA expression of genes related to inflammation (Mcp1, F4/80, and Cd11c). (D) Protein expression of macrophage marker and inflammatory signaling (CD11c, F4/80, IκBα, p-JNK, p-p38, p-ERK, and t-ERK). (E) mRNA expression of genes related to mitochondrial biogenesis (Sirt1 and Pgc1α). (F) Protein patterns of TFR1 (iron importer), FTL (storage iron), and OxPhos. Values are mean ± SD, n = 8. In qPCR analysis, 34b4 was used as a reference gene. In Western blot analysis (n = 6), β-actin was used as a loading control except for t-AMPK and t-ERK to normalize p-AMPK (in B) and p-ERK (in D). Quantitation is shown next to the representative Western blot images. Asterisks indicate the difference from IA, *P < 0.05, **P < 0.01, and ***P < 0.001, by Student t test. Acox1, peroxisomal acyl-coenzyme A oxidase 1; Cd11c, cluster of differentiation 11c; Cpt1, carnitine palmitoyltransferase; ERK, extracellular-signal-regulated kinase; FTL, ferritin light; IA, iron adequate; ID, iron deficient; IκBα, NF-κB inhibitor α; JNK, c-Jun N-terminal kinase; Mcp1, monocyte chemoattractant protein; OxPhos, oxidative phosphorylation; Pgc1α, Pparγ coactivator 1α; Pparα, peroxisome proliferator-activated receptor α; Pparγ, peroxisome proliferator-activated receptor γ; Scd1, stearoyl-CoA desaturase 1; Sirt1, sirtuin 1; Srebp1c, sterol regulatory element-binding transcription factor 1; TFR1, transferrin receptor 1.
We also observed elevated mRNA expression concentrations of monocyte chemoattractant protein 1 (Mcp1), monocyte-macrophage (Mϕ) marker F4/80, and proinflammatory M1 Mϕ marker Cd11c in the ID mice compared with IA mice (Figure 3C). The significant increase of F4/80 and CD11c was confirmed at the protein level in the ID mice (Figure 3D). Unexpectedly, we examined no significant degradation of IκBα protein in both groups, which excludes NF-κB activation (Figure 3D). Among the family members of MAP kinases, the ID diet promoted phosphorylation concentrations of c-JUN N-terminal kinase (JNK) and extracellular signal–regulated kinase (ERK) but not p38, suggesting that the ID diet triggers inflammatory signaling involved ERK/JNK signaling pathways (Figure 3D).
There was a substantial decrease in transcriptional regulators for mitochondrial biogenesis, such as sirtuin 1 (Sirt1) and PPARγ coactivator 1α (Pgc1α), in ID-fed eWAT compared with IA-fed eWAT (Figure 3E). To determine whether the ID diet triggers changes in iron homeostasis–related genes in the eWAT, we examined the expression concentrations of iron-handling proteins. TFR1 protein expression concentrations were significantly higher (P < 0.05) in the ID-fed eWAT than in the IA-fed eWAT. However, expression concentrations of ferritin light (FTL), a cytosolic iron storage protein, were not different between groups. Among the mitochondrial oxidative phosphorylation (OxPhos) protein complexes, the expression concentrations of complexes I, II, and III, but not IV and V, were significantly reduced with the ID diet (Figure 3F). These results collectively suggest that nonanemic iron deficiency contributes to visceral obesity and insulin resistance via reduced FA catabolism and mitochondrial function.
Adipocyte iron balance and acute cold tolerance
Next, we examined the correlation between iron content and basal thermogenic function. The decrease of iron content by ID diet was not significant in the BAT but evident in the iWAT (P < 0.01) (Figure 4A). Reflecting a reduction in iron content, the ID diet significantly decreased the FTL and increased the TFR1 expression concentrations in the iWAT and BAT. Notably, these responses occurred without an increase in IRP2 protein abundance (Figure 4B).
FIGURE 4.

Diet-induced iron deficiency attenuates thermogenic function upon acute cold treatment. (A) The iron content of iWAT and BAT by inductively coupled plasma mass spectrometry. (B) Protein expression patterns of iron homeostasis–related proteins. Quantification of protein expression (n = 4, normalized to β-actin) is shown below to the representative Western blot images. (C) Kinetic changes of core body temperature upon acute cold treatment for 3 h. Values are expressed as mean ± SD, n = 8. In A and B, asterisks indicate the difference from IA, *P < 0.05 and **P < 0.01, by Student t test. In C, asterisks indicate the difference from IA at a given time, **P < 0.01 and ***P < 0.001, by 2-factor repeated-measures ANOVA (diet and cold exposure time) with Bonferroni post hoc test. FTL, ferritin light; IA, iron adequate; ID, iron deficient; IRP2, iron-regulatory protein 2; iWAT, inguinal white adipose tissue; TFR1, transferrin receptor 1; UCP1, uncoupling protein 1.
At an ambient temperature, the UCP1 protein was undetectable in the iWAT regardless of iron status. The UCP1 expression concentrations of BAT were not affected by the ID diet in the BAT (Figure 4B). Also, UCP1 expression concentrations were not different between groups in the BAT (Figure 4B). When the mice were exposed to the cold temperature (4°C), the ID mice maintained a core body temperature significantly lower than the IA mice (P < 0.001). Upon acute cold exposure for 3 h, the core body temperature was 2°C lower in the ID mice than in the IA mice (Figure 4C). Collectively, these results suggest that reduced iron content in BAT attenuates the thermogenic function without altering UCP1 expression.
Adipose tissue browning and adaptive thermogenesis
When the mice were administered with a β3AR agonist CL, the CL-induced heat release from iWAT and BAT depots was substantially lower in the ID mice than in the IA mice (Figure 5A). Consistent with our previous report (8), IA + CL showed signs of increased IRP activity in the iWAT, including 1) accumulation of IRP2 and degradation of F-box and leucine-rich repeat protein 5 (FBXL5), 2) upregulation of TFR1, and 3) downregulation of FTL, along with the rapid emergence of UCP1 expression (Figure 5B). However, the aforementioned iron homeostasis–related protein expression (i.e., increased IRP2, TFR1, and decrease in FBXL5 expression) was significantly lower in the ID + CL mice along with compromised UCP1 upregulation (Figure 5B). We note that FTL expression was almost absent in the iWAT of ID + CL, suggesting a substantial reduction in cytosolic iron storage in the iWAT. The CL treatment posed no significant impacts on UCP1 expression and other iron homeostasis–related proteins in the BAT in both groups. Intriguingly, ID + CL mice showed an aberrantly high expression of FTL, implicating atypical IRP/IRE responses in the BAT (Figure 5C). In agreement with the reduced UCP1 expression, H&E staining revealed a paucity of beige-specific multilocular morphology in the iWAT of ID + CL mice compared with IA + CL mice (Figure 5D). Likewise, there were no significant differences in BAT-specific morphology between the 2 groups (Figure 5D).
FIGURE 5.

Diet-induced iron deficiency attenuates adipose tissue browning, thermogenic heat release, and mitochondrial development. (A) Study design (upper), images of heat release caught by infrared camera upon CL injection (middle), and average surface temperature (bottom). Protein concentrations of UCP1 and iron-responsive proteins (IRP2, FBXL5, TFR1, FTL, and CYT C) in iWAT (B) and BAT (C). (D) H&E staining images of BAT (upper) and iWAT (lower). The mitochondrial OxPhos protein concentrations in the iWAT (E) and BAT (F). (G) Graphic images of mitochondrial proteins (OxPhos and Cytc) containing Fe–S clusters and heme groups. Values are expressed as mean ± SD, n = 5. Quantitation of protein expression (normalized to β-actin) is shown below to the representative Western blot images. In B and E, 1-factor ANOVA was conducted with Tukey post hoc correction. Labeled means without a common letter differ, P < 0.05. In C and F, asterisks indicate the difference from IA + CL, *P < 0.05 and **P < 0.01, by Student t test. BAT, brown adipose tissue; CL, CL316243; CYT C, cytochrome c; FTL, ferritin light; FBXL5, F-box leucine-rich repeat protein 5; IA, iron adequate; ID, iron deficient; IRP2, iron-regulatory protein 2; iWAT, inguinal white adipose tissue; OxPhos, oxidative phosphorylation; TFR1, transferrin receptor 1; UCP1, uncoupling protein 1.
In response to CL stimulation, the enhanced expression of all of the OxPhos protein complexes (CI–V) (Figure 5E) and cytochrome C (Figure 5B) was manifest in the iWAT from IA + CL mice, which was blunted significantly in the ID + CL mice. In particular, the defective expression of CI and CII, which possess multiple Fe–S cluster moieties, was more evident with the ID diet (Figure 5E). However, the CL treatment exerted no significant impact on OxPhos protein complexes in the BAT regardless of dietary iron content. Collectively, these results demonstrate that dietary iron deficiency substantially diminishes adaptive thermogenesis in the iWAT, presumably due to defective mitochondrial development.
Discussion
We have recently reported that metabolic plasticity of white to beige conversion (from energy depositing to energy emancipating) is governed by modulation of iron metabolism, underlining the metabolic significance of adipocyte iron regulation for thermogenic activation. Iron deficiency is one of the most common nutrient-related suboptimal health conditions and is routinely associated with obesity. However, the relative contribution of iron deficiency to obesity and thermogenic energy expenditure remains unknown. This study aimed to evaluate the impacts of dietary iron deficiency on obesity by analyzing the iron-regulatory patterns in different adipose depots and thermogenic capacity. We demonstrated that chronic low-iron/HF intake triggers nonanemic iron deficiency at systemic levels and iron deficiency at tissue levels, including adipose tissue. Iron deficiency in the eWAT augments adiposity via reduced AMPK signaling, whereas iron deficiency in the iWAT attenuates the thermogenic heat release and beige fat development. Collectively, our work demonstrated that iron deficiency reduces the activation of catabolic pathways and thermogenic energy expenditure in adipose tissue, thereby escalating the risk of obesity and type 2 diabetes.
An accumulating body of evidence suggests that iron imbalance alters energy metabolism in adipose tissue, serving as a risk factor for type 2 diabetes (27). Intriguingly, iron deficiency, irrespective of anemic or nonanemic conditions, impaired insulin sensitivity in rodents and humans. Numerous studies demonstrated that chronic intake of low iron elevated plasma glucose concentrations in rodents (28, 29). In addition, the severity of iron deficiency was proportional to the HbA1c concentrations in patients with diabetes (30, 31). Notably, the improvement of iron-deficient conditions in premenopausal women decreased the HOMA-IR values (30). These studies unanimously support that iron deficiency is a critical determinant of energy metabolism and insulin sensitivity. However, little is known about the contribution of adipocyte iron regulation to iron deficiency–mediated metabolic dysfunction. Our work demonstrated that adipocyte iron deficiency could occur in the absence of systemic anemia, which significantly diminishes metabolic activities and insulin sensitivity.
The relation between iron deficiency and defective temperature control has been recognized for a long time. Animal models of IDA exhibited insufficient thermoregulatory capacity upon cold exposure (32, 33). Several studies have proposed that iron deficiency inhibits the conversion of thyroxine to triiodothyronine, required for thermal activation (5, 6, 34). Supporting this notion, the restoration of iron status recovered triiodothyronine conversion and thermogenic capacity (34). Although these studies contributed to establishing the endocrine function of iron balance to activate thermogenic hormones, the molecular events of how iron is involved in thermogenic heat release are uncertain. For the past decade, we have witnessed the advancement of adipocyte biology, revealing the existence of metabolically active brown and beige adipocytes in adult humans (35). The relief of energy burden through thermogenic energy expenditure is a promising strategy to curb obesity and type 2 diabetes in humans (36, 37). Therefore, these earlier studies on iron deficiency and hypothermia should be revisited in a new biological standpoint of “beige fat development and adaptive thermogenesis.” Our work definitively demonstrated that adipocyte iron deficiency impedes beige fat development and adaptive heat release due to a lack of available iron for mitochondrial biogenesis.
The cellular levels of iron homeostasis are under the control of IRPs, which sense iron deficiency and regulate the iron import, utilization, and storage in a coordinated manner (38). We and others showed that white adipocytes maintain higher cytosolic iron storage (ferritin) than brown adipocytes, and the iron demands during white adipocyte differentiation are relatively low (8, 39). In contrast, thermogenic fat (i.e., brown/beige adipocytes) maintains lower cytosolic ferritin concentrations, allowing constitutive IRP binding to IRE, TFR1-mediated iron import, and iron redistribution into mitochondria (8). The role of adipocyte iron import during beige fat development was confirmed in mice with targeted deletion of TFR1 in adipocytes; Li et al. (9) demonstrated that the genetic ablation of adipocyte TFR1 resulted in defective beige fat development in the iWAT and early onset of insulin resistance (10). It is worth noting that metabolic sequelae of adipocyte-specific deletion of Tfr1 (9, 10) resemble those of adipocyte iron deficiency induced by chronic low-iron intake (Figures 2 and 5). These studies underline the metabolic significance of adipocyte iron homeostasis, implicating that disruption of adipocyte iron homeostasis per se without modifying systemic iron status could promote metabolic insult and insulin resistance.
We showed that chronic low-iron intake (3 ppm) with HF feeding induces nonanemic and stage 1 iron deficiency (normal Hb, reduced serum ferritin, and the compensatory increase in iron absorption) (40). It is surprising to find that stage 1 iron deficiency alters the iron balance in all adipose depots (i.e., eWAT, iWAT, and BAT) and instigates whole-body insulin resistance (Figure 2A). Unexpectedly, BAT that features the highest iron requirement among the fat depots (BAT > iWAT > eWAT) (8) was affected the least by the iron-deficiency diet upon β3AR-induced thermogenic activation (Figure 4A). The systemic iron deficiency modulated iron homeostasis–related proteins in the iWAT, including increased TFR1 and decreased cytosolic ferritin storage (Figure 4B). However, the iron-compensatory signal solicited by low-iron intake seems to be different from the β3AR-mediated canonical IRP/IRE signaling (Figure 5E). The primary difference would be the absence of IRP2 accumulation in the iWAT in response to iron deficiency conditions (Figure 4B), whereas cytosolic IRP2 accumulation is a reliable marker upon β3AR signaling for adaptive thermogenesis (8). The preexisting adipocyte iron-deficiency signaling hinders β3AR-induced IRP/IRE signaling in the iWAT, indispensable for beige conversion and thermogenic heat release. The molecular details of how adipose tissue differentially handles the 2 iron-demanding signals (i.e., β3AR signal compared with ID signal) via IRP2 regulation are under investigation.
The most common denominator to counteract thermogenic activity would be inflammation. A growing body of evidence suggests that inflammation directly alters thermogenic activity by interfering with insulin signaling and glucose uptake, leading to type 2 diabetes (17). Conversely, inflammation could be one of the mechanisms to cause adipocyte iron deficiency. Paradoxically, obesity and metabolic syndrome are associated with increased total iron content within adipose tissue due to massive macrophage infiltration (41). On closer examination, adipose tissue macrophages (ATMs) are the primary cells sequestering iron in adipose tissue, whereas adipocytes remain relatively iron deficient (42, 43). Consistent with this notion, Winn et al. (43) proposed that resident macrophages attract iron (serve as so-called ferrostat), limiting the local iron availability. These studies open up a potential mechanism that obesity-mediated macrophage infiltration into adipose tissue would result in 1) iron accumulation and immobilization within ATM, 2) iron deficiency in adipocytes, 3) diminished adipocyte catabolism and mitochondrial biogenesis, and 4) dysfunctional thermogenic function.
In agreement with this scenario, obese individuals and patients with type 2 diabetes are hypothermic. A higher BMI is associated with less active BAT in adults, even at an ambient temperature (44). The chronic cold treatment significantly activated the thermogenic function of BAT in lean adults, whereas thermogenic activation was substantially reduced in obese individuals (16). Also, patients with diabetes showed blunted thermogenic activity after exposure to cold temperature (36) and reduced glucose uptake into BAT (45). Moreover, overweight or obese individuals are more prone to anemia than lean individuals (46, 47), and individuals with type 2 diabetes pose a higher risk for IDA (48, 49). Despite these multiple hints, current evidence is insufficient to draw a direct correlation between adipocyte iron regulation and thermogenesis in obesity and inflammation, especially in humans. Clinical trials that establish a solid relation between adipocyte iron regulation and thermogenic capacity in lean and obese individuals would be the next step in research.
In summary, our work demonstrated that iron deficiency without overt anemia could disrupt whole-body energy metabolism, partly through compromised thermogenesis. We also showed that adipocyte iron is a crucial factor in governing adiposity and thermogenic energy expenditure. Our work suggests that adipocyte iron metabolism could be a potential therapeutic target to augment thermogenic energy expenditure.
Supplementary Material
Acknowledgments
The authors’ responsibilities were as follows—J-SY and SC: conceived of the project, designed the experiments, and wrote the manuscript together; J-SY, SST, AMT, and MY: performed the experiments and analyzed the data; Y-CK and ZL: helped interpret data and provided critical comments; JL: performed inductively coupled plasma mass spectrometry and provided critical comments; SC: supervised the project, interpreted data, and edited the manuscript; and all authors: read and approved the final manuscript.
Notes
Supported by National Institutes of Health (1R21HD094273).
Author disclosures: The authors report no conflicts of interest.
Supplemental Tables 1–3 are available from the “Supplementary data” link in the online posting of the article and from the same link in the online table of contents at https://academic.oup.com/jn/.
Abbreviations used: AMPK, AMP-activated protein kinase; ATM, adipose tissue macrophage; BAT, brown adipose tissue; BMP6, bone morphogenetic protein 6; CL, CL316243; CPT1, carnitine-palmitoyl transferase 1; ERK, extracellular signal–regulated kinase; eWAT, epididymal white adipose tissue; FA, fatty acid; FBXL5, F-box and leucine-rich repeat protein 5; Fe–S, iron–sulfur; FTL, ferritin light; Hb, hemoglobin; H&E, hematoxylin and eosin; HF, high fat; IA, iron adequate; ID, iron deficient; IDA, iron-deficiency anemia; IRE, iron response element; IRP, iron-regulatory protein; JNK, c-JUN N-terminal kinase; iWAT, inguinal white adipose tissue; Mϕ, macrophage; NEFA, nonesterified free fatty acid; OxPhos, oxidative phosphorylation; PPARγ, peroxisome proliferator-activated receptor γ; SCD1, stearoyl-CoA desaturase 1; TC, total cholesterol; TG, triglyceride; TFR1, transferrin receptor 1; UCP1, uncoupling protein 1; β3AR, β3-adrenoceptor.
Contributor Information
Jin-Seon Yook, Department of Nutrition, University of Massachusetts, Amherst, MA, USA.
Shalom Sara Thomas, Department of Nutrition, University of Massachusetts, Amherst, MA, USA.
Ashley Mulcahy Toney, Department of Nutrition and Health Sciences, University of Nebraska–Lincoln, Lincoln, NE, USA.
Mikyoung You, Department of Nutrition, University of Massachusetts, Amherst, MA, USA.
Young-Cheul Kim, Department of Nutrition, University of Massachusetts, Amherst, MA, USA.
Zhenhua Liu, Department of Nutrition, University of Massachusetts, Amherst, MA, USA.
Jaekwon Lee, Department of Biochemistry, University of Nebraska–Lincoln, Lincoln, NE, USA.
Soonkyu Chung, Department of Nutrition, University of Massachusetts, Amherst, MA, USA; Department of Nutrition and Health Sciences, University of Nebraska–Lincoln, Lincoln, NE, USA.
References
- 1.Lowell BB, Spiegelman BM. Towards a molecular understanding of adaptive thermogenesis. Nature. 2000;404(6778):652–60. [DOI] [PubMed] [Google Scholar]
- 2.Kajimura S, Spiegelman BM, Seale P. Brown and beige fat: physiological roles beyond heat generation. Cell Metab. 2015;22(4):546–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Min SY, Desai A, Yang Z, Sharma A, DeSouza T, Genga RM, Kucukural A, Lifshitz LM, Nielsen S, Scheele C. Diverse repertoire of human adipocyte subtypes develops from transcriptionally distinct mesenchymal progenitor cells. Proc Natl Acad Sci. 2019;116(36):17970–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cedikova M, Kripnerova M, Dvorakova J, Pitule P, Grundmanova M, Babuska V, Mullerova D, Kuncova J. Mitochondria in white, brown, and beige adipocytes. Stem Cells Int. 2016;2016:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dillman E, Gale C, Green W, Johnson DG, Mackler B, Finch C. Hypothermia in iron deficiency due to altered triiodothyronine metabolism. Am J Physiol. 1980;239(5):R377–81. [DOI] [PubMed] [Google Scholar]
- 6.Mackler B, Person R, Grace R. Iron deficiency in the rat: effects on energy metabolism in brown adipose tissue. Pediatr Res. 1985;19(10):989–91. [DOI] [PubMed] [Google Scholar]
- 7.Xu W, Barrientos T, Andrews NC. Iron and copper in mitochondrial diseases. Cell Metab. 2013;17(3):319–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yook JS, You M, Kim Y, Zhou M, Liu Z, Kim YC, Lee J, Chung S. The thermogenic characteristics of adipocytes are dependent on the regulation of iron homeostasis. J Biol Chem. 2021;296:100452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Pan X, Pan G, Song Z, He Y, Zhang S, Ye X, Yang X, Xie E, Wang X. Transferrin receptor 1 regulates thermogenic capacity and cell fate in brown/beige adipocytes. Adv Sci. 2020;7(12):1903366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiu J, Zhang Z, Wang S, Chen Y, Liu C, Xu S, Wang D, Su J, Ni M, Yu J. Transferrin receptor functionally marks thermogenic adipocytes. Front Cell Dev Biol. 2020;8:572459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camaschella C. Iron deficiency. Blood. 2019;133(1):30–9. [DOI] [PubMed] [Google Scholar]
- 12.WHO . Iron deficiency anaemia assessment, prevention, and control: a guide for programme managers. [Internet]. 2021; [cited 2021 May 1]. Available from: https://www.who.int/nutrition/publications/en/ida_assessment_prevention_control.pdf. [Google Scholar]
- 13.Stoffel NU, El-Mallah C, Herter-Aeberli I, Bissani N, Wehbe N, Obeid O, Zimmermann MB. The effect of central obesity on inflammation, hepcidin, and iron metabolism in young women. Int J Obes. 2020;44(6):1291–300. [DOI] [PubMed] [Google Scholar]
- 14.Vaquero MP, Martínez-Suárez M, García-Quismondo Á, Del Cañizo FJ, Sánchez-Muniz FJ. Diabesity negatively affects transferrin saturation and iron status. The DICARIVA study. Diabetes Res Clin Pract. 2021;172:108653. [DOI] [PubMed] [Google Scholar]
- 15.Okla M, Wang W, Kang I, Pashaj A, Carr T, Chung S. Activation of Toll-like receptor 4 (TLR4) attenuates adaptive thermogenesis via endoplasmic reticulum stress. J Biol Chem. 2015;290(44):26476–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360(15):1500–8. [DOI] [PubMed] [Google Scholar]
- 17.Omran F, Christian M. Inflammatory signaling and brown fat activity. Front Endocrinol. 2020;11:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sangkhae V, Nemeth E. Regulation of the iron homeostatic hormone hepcidin. Adv Nutr. 2017;8(1):126–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Longo DL, Camaschella C. Iron-deficiency anemia. N Engl J Med. 2015;372(19):1832–43. [DOI] [PubMed] [Google Scholar]
- 20.Haysom SS, Vickers MH, Yu LH, Reynolds CM, Firth EC, McGlashan SR. Post-weaning high-fat diet results in growth cartilage lesions in young male rats. PLoS One. 2017;12(11):e0188411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maejima Y, Yokota S, Horita S, Shimomura K. Early life high-fat diet exposure evokes normal weight obesity. Nutr Metab. 2020;17(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subramanian S, Han CY, Chiba T, McMillen TS, Wang SA, Haw A III, Kirk EA, O'Brien KD, Chait A. Dietary cholesterol worsens adipose tissue macrophage accumulation and atherosclerosis in obese LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28(4):685–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matthews D, Hosker J, Rudenski A, Naylor B, Treacher D, Turner R. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28(7):412–9. [DOI] [PubMed] [Google Scholar]
- 24.Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol. 2006;2(8):406–14. [DOI] [PubMed] [Google Scholar]
- 25.Parrow NL, Fleming RE. Bone morphogenetic proteins as regulators of iron metabolism. Annu Rev Nutr. 2014;34(1):77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kautz L, Meynard D, Monnier A, Darnaud V, Bouvet R, Wang RH, Deng C, Vaulont S, Mosser J, Coppin Het al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood. 2008;112(4):1503–9. [DOI] [PubMed] [Google Scholar]
- 27.Shah SV, Fonseca VA. Iron and diabetes revisited. Am Diabetes Assoc. 2011;112(4):1503–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brooks GA, Henderson SA, Dallman PR. Increased glucose dependence in resting, iron-deficient rats. Am J Physiol. 1987;253(4):E461–6. [DOI] [PubMed] [Google Scholar]
- 29.Yamagishi H, Komabayashia T. Alteration of glucose metabolism and increased fructosamine in iron-deficiency anemic rats. Nutr Res. 2003;23(11):1547–53. [Google Scholar]
- 30.Kim C, Bullard KM, Herman WH, Beckles GL. Association between iron deficiency and A1C levels among adults without diabetes in the National Health and Nutrition Examination Survey, 1999–2006. Diabetes Care. 2010;33(4):780–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christy AL, Manjrekar PA, Babu RP, Hegde A, Rukmini MS. Influence of iron deficiency anemia on hemoglobin A1c levels in diabetic individuals with controlled plasma glucose levels. Iran Biomed J. 2014;18(2):88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicolas G, Bennoun M, Porteu A, Mativet S, Beaumont C, Grandchamp B, Sirito M, Sawadogo M, Kahn A, Vaulont S. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci. 2002;99(7):4596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dillmann E, Johnson DG, Martin J, Mackler B, Finch C. Catecholamine elevation in iron deficiency. Am J Physiol. 1979;237(5):R297–300. [DOI] [PubMed] [Google Scholar]
- 34.Beard JL, Borel M, Derr J. Impaired thermoregulation and thyroid function in iron-deficiency anemia. Am J Clin Nutr. 1990;52(5):813–9. [DOI] [PubMed] [Google Scholar]
- 35.Nishimoto Y, Tamori Y. CIDE family-mediated unique lipid droplet morphology in white adipose tissue and brown adipose tissue determines the adipocyte energy metabolism. J Atheroscler Thromb. 2017;24(10):989–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanssen MJ, Hoeks J, Brans B, van der Lans AA, Schaart G, Van Den Driessche JJ, Jörgensen JA, Boekschoten MV, Hesselink MK, Havekes B. Short-term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nat Med. 2015;21(8):863. [DOI] [PubMed] [Google Scholar]
- 37.Hanssen MJ, van der Lans AA, Brans B, Hoeks J, Jardon KM, Schaart G, Mottaghy FM, Schrauwen P, van Marken Lichtenbelt WD. Short-term cold acclimation recruits brown adipose tissue in obese humans. Diabetes. 2016;65(5):1179–89. [DOI] [PubMed] [Google Scholar]
- 38.Anderson CP, Shen M, Eisenstein RS, Leibold EA. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta. 2012;1823(9):1468–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Festa M, Ricciardelli G, Mele G, Pietropaolo C, Ruffo A, Colonna A. Overexpression of H ferritin and up-regulation of iron regulatory protein genes during differentiation of 3T3-L1 pre-adipocytes. J Biol Chem. 2000;275(47):36708–12. [DOI] [PubMed] [Google Scholar]
- 40.Al-Naseem A, Sallam A, Choudhury S, Thachil J. Iron deficiency without anaemia: a diagnosis that matters. Clin Med. 2021;21(2):107–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caslin HL, Bhanot M, Bolus WR, Hasty AH. Adipose tissue macrophages: unique polarization and bioenergetics in obesity. Immunol Rev. 2020;295(1):101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma X, Pham VT, Mori H, MacDougald OA, Shah YM, Bodary PF. Iron elevation and adipose tissue remodeling in the epididymal depot of a mouse model of polygenic obesity. PLoS One. 2017;12(6):e0179889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winn NC, Volk KM, Hasty AH. Regulation of tissue iron homeostasis: the macrophage “ferrostat” JCI Insight. 2020;5(2):e132964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria Aet al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360(15):1509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blondin DP, Labbé SM, Noll C, Kunach M, Phoenix S, Guérin B, Turcotte ÉE, Haman F, Richard D, Carpentier AC. Selective impairment of glucose but not fatty acid or oxidative metabolism in brown adipose tissue of subjects with type 2 diabetes. Diabetes. 2015;64(7):2388–97. [DOI] [PubMed] [Google Scholar]
- 46.Nead KG, Halterman JS, Kaczorowski JM, Auinger P, Weitzman M. Overweight children and adolescents: a risk group for iron deficiency. Pediatrics. 2004;114(1):104–8. [DOI] [PubMed] [Google Scholar]
- 47.Cepeda-Lopez AC, Osendarp SJ, Melse-Boonstra A, Aeberli I, Gonzalez-Salazar F, Feskens E, Villalpando S, Zimmermann MB. Sharply higher rates of iron deficiency in obese Mexican women and children are predicted by obesity-related inflammation rather than by differences in dietary iron intake. Am J Clin Nutr. 2011;93(5):975–83. [DOI] [PubMed] [Google Scholar]
- 48.Miranda MA, Lawson HA. Ironing out the details: untangling dietary iron and genetic background in diabetes. Nutrients. 2018;10(10):1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guralnik JM, Eisenstaedt RS, Ferrucci L, Klein HG, Woodman RC. Prevalence of anemia in persons 65 years and older in the United States: evidence for a high rate of unexplained anemia. Blood. 2004;104(8):2263–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.