

Abstract
Lysophosphatidic acid (LPA) and sphingosine 1-phosphate (S1P) are structurally related bioactive lipids with growth factor like activities. LPA and S1P are naturally produced in vivo by blood platelets upon platelet aggregation and at least in vitro by fibroblasts, adipocytes and multiple types of tumor cells. Breast cancer cells respond to LPA and S1P. However, their specific actions on breast cancer cell biological functions remain unclear. We therefore conducted an in vitro side by side study of these two lipids on breast cancer cells. LPA mediates human breast cancer MDA-BO2 cell proliferation, migration and invasion through activation of a Gαi/ERK1/2-dependent signalling pathway, whereas activation of Gαi/PI3K predominates upon S1P stimulation. In MDA-BO2 cells, LPA but not S1P activities were dependent on active type 1 insuline-like growth factor and epithelial growth factor receptors. LPA and S1P act directly on endothelial cells to induce angiogenesis. We demonstrate that LPA and S1P have indirect angiogenic properties as judged by induced secretion of angiogenic factors by breast cancer cells primed with these lysophospholipids. S1P, but not LPA, controlled the expression of VEGF-A by breast cancer cells, while LPA, but not S1P, controlled the expression of GM-CSF, Gro-α, MCP-1, IL-6. According to the secretion of these paracrine osteoclastic factors, LPA, but not S1P, stimulate breast cancer cell-induced osteoclastogenesis. These findings suggest that in vivo, LPA and S1P can coordinate their action on tumor and surrounding cells to induce breast cancer progression both at primary and bone metastatic sites.
Keywords: Lysophosphatidic acid, Sphingosine 1-phosphate, Breast cancer cells, Proliferation, Migration, Invasion, Angiogenesis, Osteoclastogenesis
Introduction
Recent studies indicate that the bioactive lipids lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P) present in tumor microenvironment, must be considered as key components during cancer progression and metastatic dissemination (1,2). We have shown that LPA promotes the growth of tumor xenographs and the formation of bone metastases in animals (1). The blockade of LPA signaling by genetic and pharmacological approaches is effective in inhibiting both the formation and the progression of bone metastases (3). A highly specific monoclonal antibody against S1P successfully reduces angiogenesis and the growth of multiple human cancer cell types in animal models (2). LPA and S1P have been shown to exhibit similar growth factor-like activities including the stimulation of cell proliferation, survival, contraction, migration, invasion and matrix assembly (4). They were also described as regulators of angiogenesis, wound healing, and modulators of the immune and cardiovascular systems (5). In the context of breast cancer, the specific mechanisms mobilized by these lysophospholipids are largely unknown because of a lack of a side by side study using the same cell line.
Tumor cells can produce LPA and S1P through the mobilization of specific enzymes such as Autotaxin and Sphingosine kinase 1, respectively (6,7). Consequently, LPA and S1P can act as autocrine and paracine factors. Platelets are the main source of LPA and S1P in the organism (8,9). They also have the unique property of generating high amount of both LPA and S1P upon aggregation (8,9). LPA and S1P act on a class of structurally related cell surface receptors, which possess high sequence homologies with the endothelial differentiation gene 1 (EDG1) (10). Two distinct subgroups have been defined based on their ligand specificity. S1P1,2,3,4,5 respond specifically to S1P whereas LPA1,2,3 are specifically activated by LPA (10). In addition, four additional cell surface receptors unrelated to the members of the EDG family and the transcription factor PPARγ also transduced LPA signals (11–15). LPA and S1P receptors are widely expressed in human tissues (5). Cancer cells frequently express multiple types of LPA and S1P receptors. This suggests specific repertoires of both lysolipid receptors might drive specific biological responses in presence of LPA and S1P in tumor microenvironment.
Direct experimental data are missing to elucidate the role of LPA and S1P in breast cancer growth and metastasis dissemination. In this study we conducted an original side by side comparison of the role of LPA and S1P on breast cancer cell biological functions in vitro. Our results identified overlapping and specific cell signaling activation pathways stimulated by LPA and S1P. These results suggest that the combined activities of LPA and S1P integrate into the mechanisms involving cytokine and growth factor secretion controlling breast cancer development and the formation of metastases.
Materials and Methods
Cell culture
Human breast cancer cells lines, Hs 578T, MCF-7, MDA-MB-231, MDA-MB-435S, SK-BR-3, T-47D, and ZR-75–1 cells were obtained from the American Type Culture Collection. MDA-MB-231/BO2 (MDA-BO2) cells, MDA-BO2 stables transfectants (Sbl 9, Sbl 10 and siLPA1 122, siLPA1 123, siLPA1 132) were described elsewhere (16,17). All cell lines with the exception of SK-BR-3 cells, were routinely cultured in DMEM/NUT.MIX f-12 W/GLUT-1 medium (Life Technologies, Cergy pontoise, France) supplemented with 10% (v/v) fetal bovine serum (FBS, Bio-Media, Issy les moulineaux, France) and 1% penicillin/streptomycin (Life Technologies, Cergy pontoise, France) at 37°C in a 5% CO2 incubator. SK-BR-3 cells were grown in complete McCoy’s 5a medium (Life Technologies, Cergy Pontoise, rance).
Reverse transcription and quantitative PCR
Total RNA from Human breast cancer cell lines was extracted using Total RNA Isolation System (Promega,, Charbonnieres, France). cDNA was synthesized using Moloney murine leukemia virus-1 (Promega, Charbonnieres, France). Primers for human S1P1, S1P2, S1P3, LPA1 and LPA2 were described previously (18). Primers for GAPDH were described before (19). LPA3 primers were designed from the Lpa3 gene (National Center for Biotechnology Information [NCBI] accession number AF127138) using nucleotides 738–756 as the forward primer and nucleotides 994–973 as the reverse primer. PCR reactions were run using a program consisting of 40 cycles of 95°C for 15 s, 53°C for 30 s, and 72°C for 20 s with a preincubation of 95°C for 2 min. Products from standard PCR were separated by electrophoresis on a 2% agarose gel and then visualized with ethidium bromide under ultraviolet light. Human LPA1, LPA2, and LPA3 mRNAs were quantified by real-time PCR using the Master SYBR Green I kit (Roche Diagnostics, Meylan, France). Fluorescence was monitored and analyzed in a Light Cycler (Roche Diagnostics, Meylan, France). The fluorescence data were quantitatively analyzed by using serial dilution of control samples included in each reaction to produce a standard curve. GAPDH mRNA expression was analyzed in parallel to confirm the use of equal amounts of cDNAs in each reaction. Results are expressed as the percentage of gene expression in each cell line compared with that in the parental MDA-BO2 cells.
Cell proliferation assays
Experiments were carried out as previously described (1). Briefly, quiescent tumor cells (5 × 103 cells per well) cultured in 96-well tissue culture plates (BD Biosciences, Franklin Lakes, USA) in medium containing 0.1% (w/v) BSA fatty acid–free (Sigma-Aldrich, Saint Quentin fallavier, France) treated overnight in the presence or absence of increasing concentrations of 1-oleoyl LPA or S1P, and then pulsed with [3H]-thymidine (Sigma-aldrich, Saint Quentin Fallavier, France) for the last 8 hours.
Cell migration and invasion assays
Migration assays were carried out using Bio-Coat cell migration chambers (Becton Dickinson, Franklin Lakes, USA) as described previously (20). Quiescent tumor cells and HUVEC cell culture were incubated in DMEM with or without Wortmannin (10−7 M), PTX (100ng/mL), chelerythrine chloride (10−6 M), PP2 (10−7 M), or PD98059 (10−5 M) (Sigma-Aldrich, Sant Quentin fallavier, France) 3h before stimulation with LPA or S1P (1 μM) in serum-free medium (DMEM) containing 0.1% (wt/vol) fatty acid free BSA. After a 6h incubation at 37°C in a 5% CO2 incubator, cells that had migrated through the filter pores (8 mm pore size) and attached on the below surface of the filter, were fixed and stained. The filter membranes were mounted on glass slides and the cells were counted under microscope. Cell invasion experiments were performed using Bio-Coat cell migration chambers (Becton Dickinson Franklin Lakes, USA) as described above. Filters were previously coated with the basement membrane Matrigel (0.3 mg/ml, Becton Dickinson, Franklin Lakes, USA). After incubation for 48h at 37°C in a 5% CO2 incubator, cells that had migrated through the filters were counted as described above for the migration assay.
ELISA measurements of IL-6 and IL-8 MCP-1, GM-CSF, Gro-α, and VEGF-A in culture media
Conditioned media of cell lines either untreated, or treated with LPA or S1P were collected and analyzed for human cytokine production by ELISA following the manufacturer’s instructions (Bender MedSystems, Vienna, Austria). Concentrations of IL-6, IL-8, MCP-1, GM-CSF, GROa, VEGF-A were expressed in pg/ml per 106 cells.
Osteoclastogenesis assays in vitro
Bone marrow cells from mice were collected and seeded in a 48-well tissue culture plate at a density of 2.5 × 103 cells per well in α-MEM medium (Life Technologies, Cergy pontoise, France). This medium was supplemented with the conditioned media of MDA-BO2 cells which had been stimulated for 48h with LPA, S1P (1 μM), FBS, macrophage–CSF (PreproTech, Rocky Hill, NJ), or receptor-activated nuclear receptor factor B ligand (RANK-L, generous gift of Pr. Jurdic, CNRS, Lyon, France). After incubation for 6 days, differentiated osteoclasts were enumerated under a light microscope to quantify multinucleation (more than three nuclei) and TRAP activity (Tartrate-resistant acid phosphatase). Results were expressed as the number of osteoclasts per mm2.
Statistical analysis.
Data were analyzed with the Stat-View 5.0 software using unpaired Student’s t test. P values less than 0.05 were considered statistically significant.
Results
Human breast cancer cells express LPA and S1P receptors.
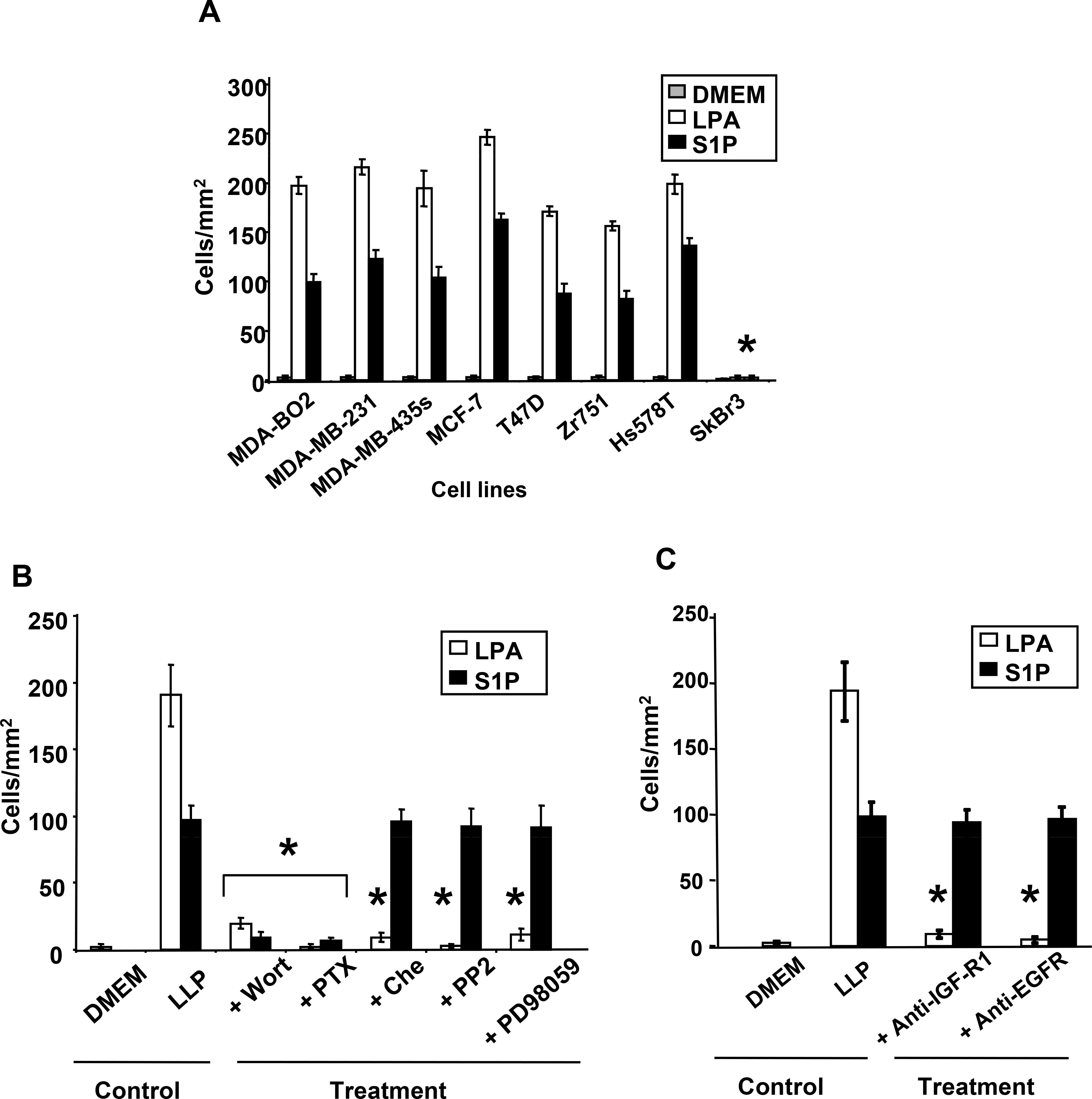
To evaluate the role of LPA and S1P on breast cancer cell activities, we first quantified the expression levels of the receptors specific for LPA (LPA1, LPA2, LPA3) and S1P (S1P1, S1P2, S1P3) in a panel of human breast cancer cell lines (MDA-MB-231, MDA-BO2, MDA-MB-435s, MCF-7, T47D, ZR751, Hs578Ts, SkBr3) (Fig. 1). Expression level of each target gene was normalized to GAPDH mRNA expression. Upon cautions taken due to potential errors in samples tested due to the use of a single housekeeping gene, our real-time PCR experiments indicated that the expression levels of LPA receptors vary strongly among these cell lines (Fig. 1A). In contrast, the receptors for S1P were more homogenously expressed (Fig. 1B). However, we detected elevated expression of S1P1 in MDA-MB-231, MCF-7, and Hs578Ts cell lines. In agreement with previous observations, no expression of LPA receptor was detected in SKBr3 cells (1,21). Interestingly, these cells express S1P receptor mRNA with levels less than 90% compared to other cell lines.
Fig. 1– Expression of LPA and S1P receptors in human breast cancer cells lines.
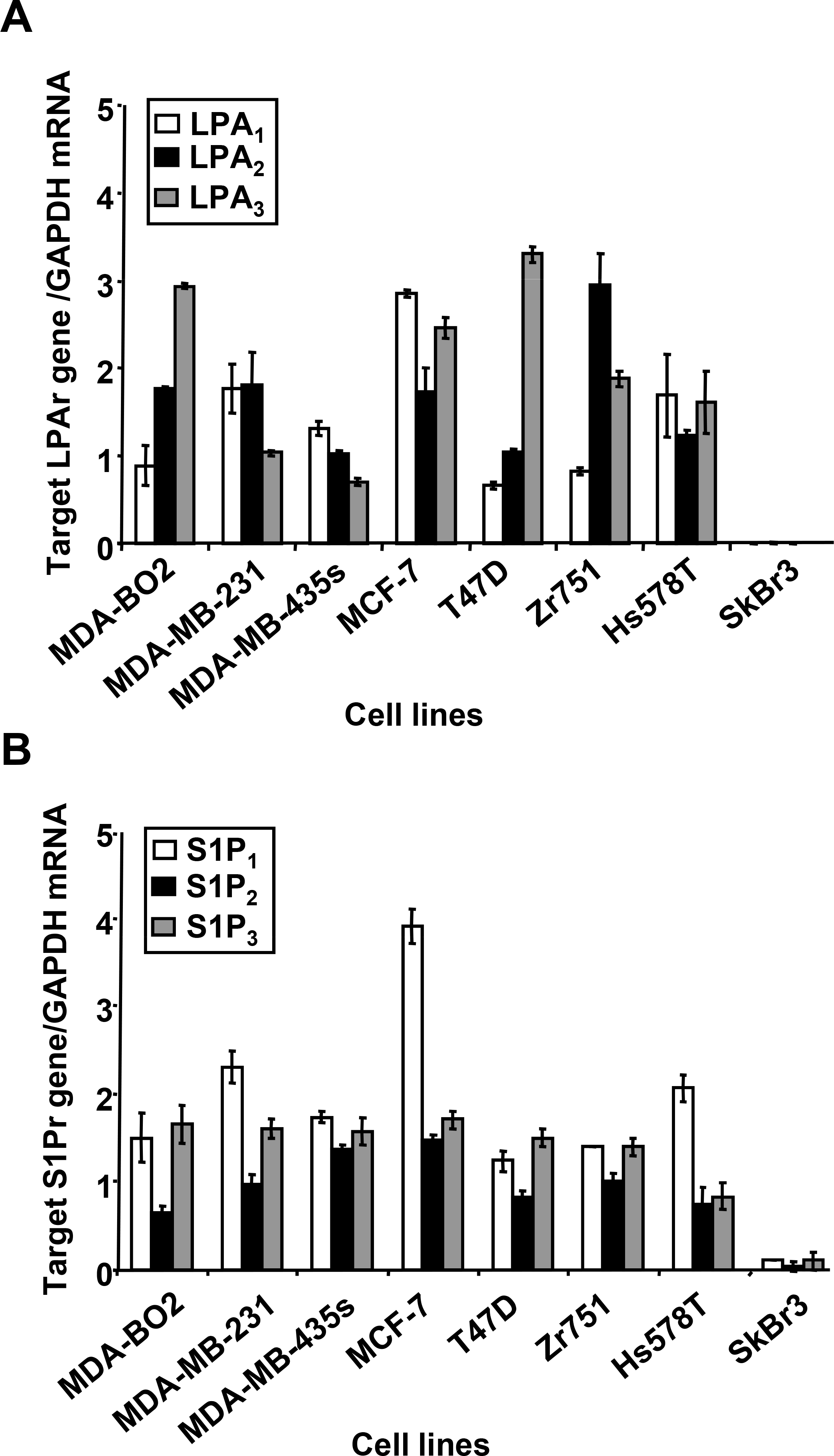
The level of target lysolipid receptor mRNA in breast cancer cell lines was measured using quantitative real-time PCR. Results are expressed as a relative value to GAPDH mRNA. A. Expression levels of LPA receptors (LPA1, LPA2, LPA3). B. Expression levels of S1P receptors (S1P1, S1P2, S1P3). Cell lines are MDA-MB-231, MDA-BO2, MDA-MB-435S, MCF-7, T47D, Zr751, Hs578T and SkBr3. Bars represent means ± SD of at least three experiments.
LPA and S1P control breast cancer cell proliferation.
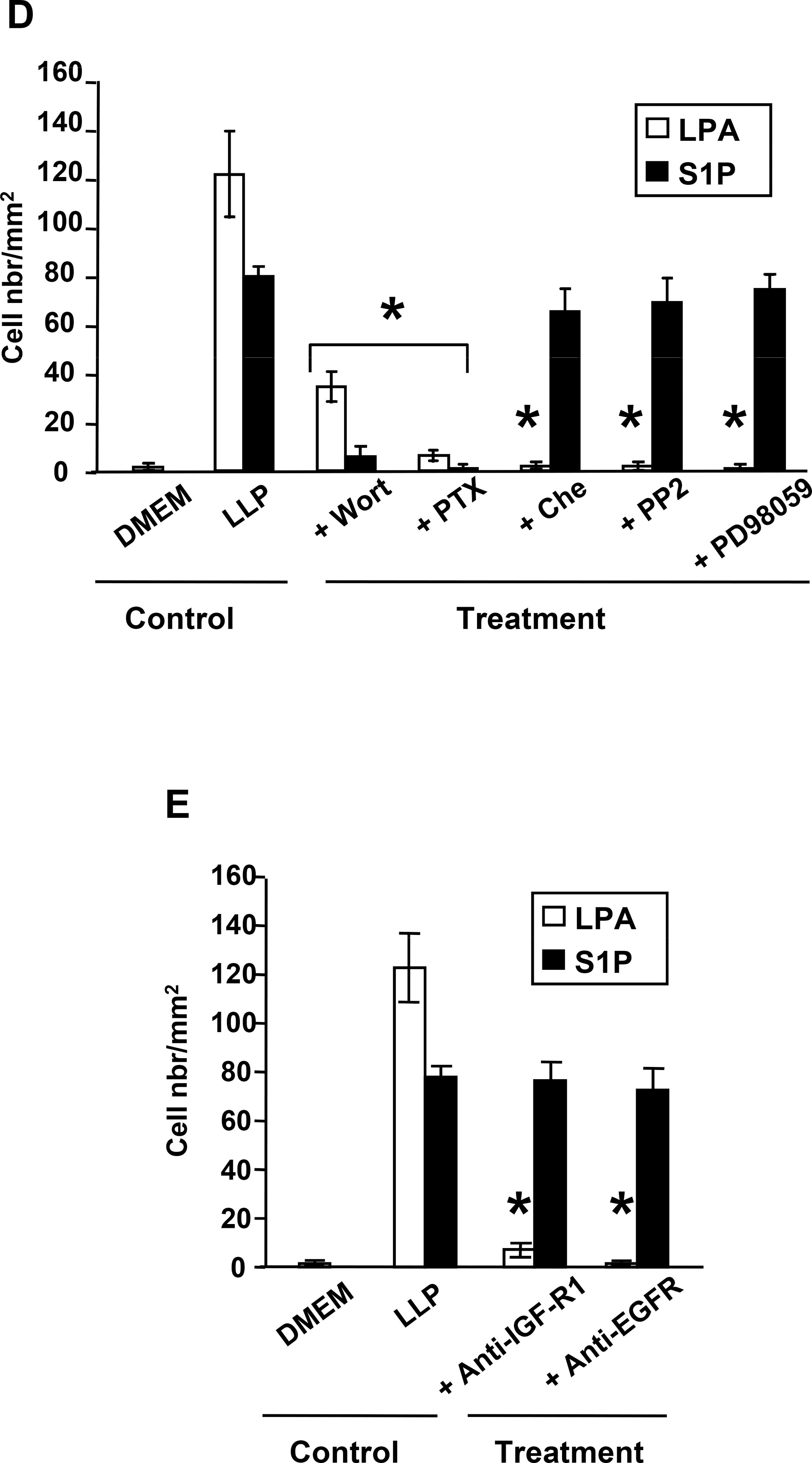
We have previously observed that all breast cancer cell lines used in this study that express LPA receptors responded to LPA stimulation in a dose-dependent manner and further demonstrated that LPA is highly mitogenic on breast cancer cells both in vitro (Fig. 2A) and in vivo (1). To analyse the effect of S1P on cell proliferation, we stimulated our series of human breast cancer cells with increasing concentrations of S1P. All the cells that expressed S1P receptors responded to S1P stimulation in a dose-dependent manner (Fig. 2B). SKBr3 cells did not respond to S1P stimulation. MDA-BO2 cells which express both lysolipid receptors were more sensitive to S1P than LPA stimulation, as shown by a significant 1.8 fold increase of H3-thymidine incorporation using equimolar concentrations of lysolipids (Fig. 2C). We have previously shown that LPA-induced MDA-BO2 cell proliferation was dependent on activation of the MAP Kinase ERK1/2 (3). We found that S1P-induced H3-thymidine incorporation was totally blocked when cells were treated with inhibitors for PI3K, Gi protein, MEK1, PKC and Src family kinases (Fig. 2C). Previous reports have shown that anti-IGF-II and anti-IGFR1 antibodies partially inhibited proliferation of breast cancer cells to LPA and S1P (19). Additionally, LPA and S1P were shown to transactivate EGFR in cancer cells (22,23). We found that LPA-induced but not S1P-mediated proliferation of MDA-BO2 cells was totally inhibited by anti-IGFR1 and anti-EGFR antibodies (Fig. 2D).
Fig. 2– LPA and S1P induce breast cancer cell proliferation through distinct signaling pathways.
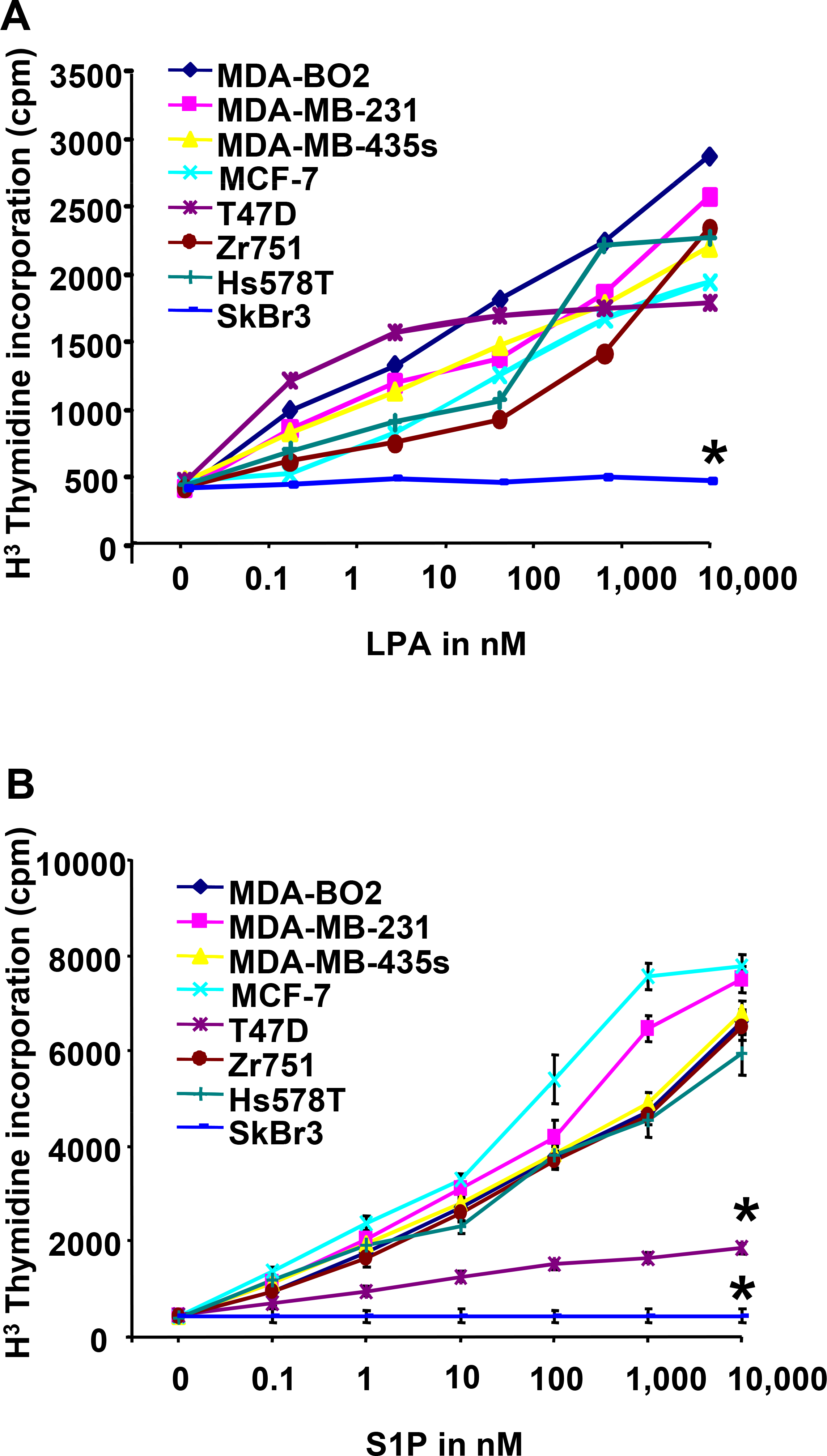
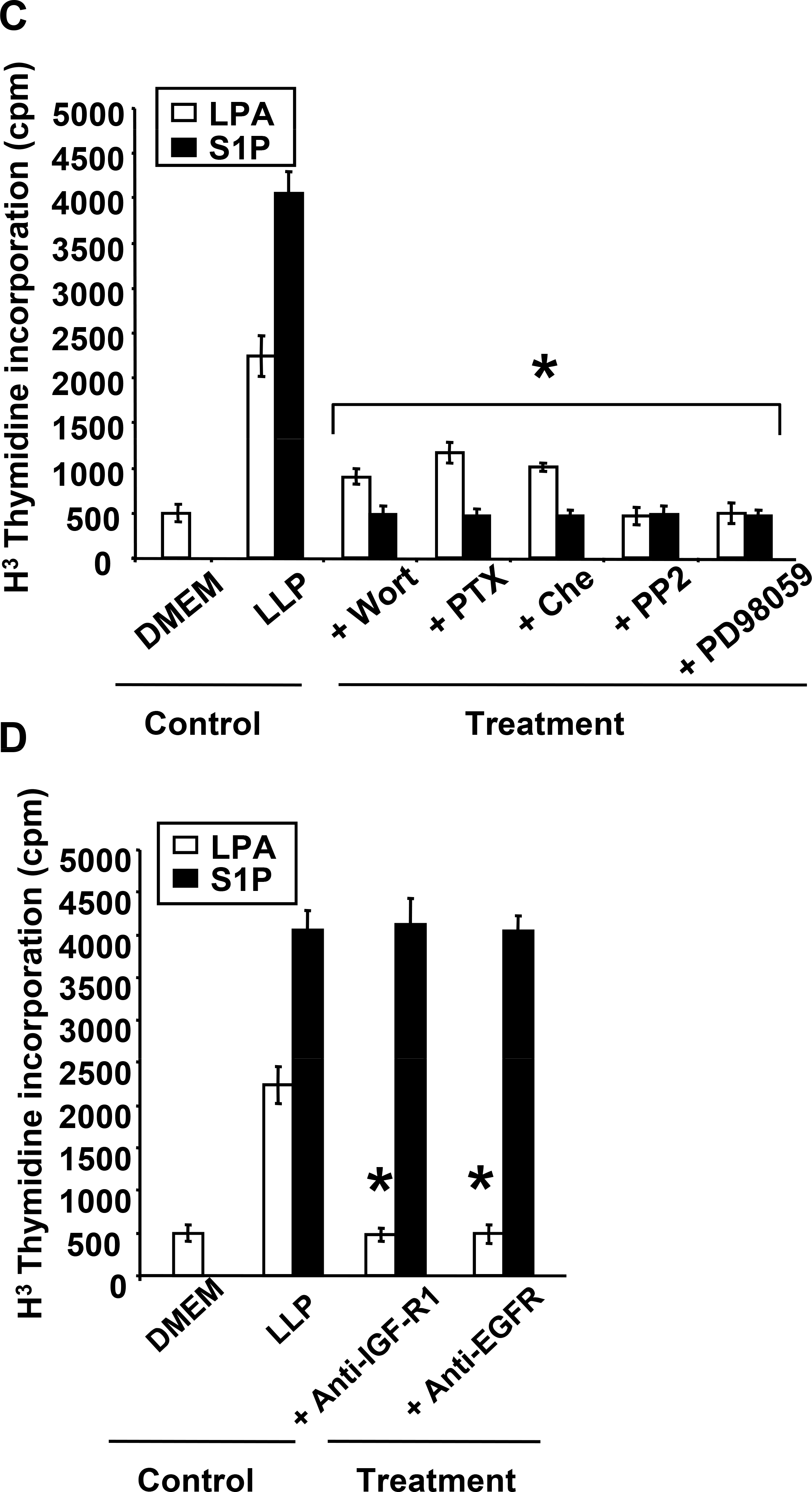
A. Mitogenic activity of LPA and B. S1P were analyzed on quiescent tumor cell lines treated overnight in the presence of increasing concentrations of either lysolipid (0 to 10−5 M) and pulsed with [3H]-thymidine for the last 8 hours of lysolipid treatment. *, P<0.001 compared to MDA-BO2 cell line. C. Effect of inhibitors PI3K (Wortmannin, 10−7 M), Gαi (PTX, 100ng/mL), PKC (chelerythrine chloride, 10−6 M), Src kinase (PP2, 10−7 M), or ERK1/2 phosphorylation (PD98059, 10−5 M) on the proliferation of MDA-BO2 cells stimulated with either lysolipids (LLP), LPA or S1P (10−7 M). D. Effect of anti-IGFR1 and EGFR antibodies (1μg/ml) on the mitogenic activity of LPA and S1P (10−7 M) on MDA-BO2 cells. Data are expressed as specific H3Thymidine incorporation and represent means ± SD of at least three independent experiments. *, P<0.001 compared to corresponding lysolipid treatment.
LPA and S1P control breast cancer cell migration.
Motility is a hallmark of metastatic cancer cells. S1P and LPA have been shown to control the migration of many cell types including immune cells (24), endothelial cells (25) and cancer cells (26). To analyze the effect of LPA and S1P on the motility of breast cancer cells, we used transwell migration chambers wherein MDA-BO2 cells were challenged to migrate throughout a membrane with 8 μm pores. Cells were incubated in the absence or presence of either a positive or a negative gradient of lysolipid concentration. Each lysolipid enhanced the migration of MDA-BO2 cells when added to the lower chamber (Fig. 3). In contrast to S1P, LPA induced cell migration when added in the upper chamber only, or in both the upper and the lower chambers (Fig. 3). Then, we challenged our series of cell lines with increasing concentrations of S1P or LPA added in the lower chamber. We observed that S1P and LPA induced migration of all cell lines that express cognates receptors (Fig. 4A, 4B). In contrast to S1P, the action of LPA followed a bell-shape curve of concentrations with a maximum activity at 10−7 M and a complete inhibition of cell migration at 10−5 M. Recent reports indicate that LPA receptor LPA1 was the motility factor for neoplastic and non-neoplastic cells in response to LPA (26). Therefore, we silenced the expression of LPA1 in MDA-BO2 cells using a RNA interference strategy (siRNA) (3), and tested the ability of these cells to migrate in response to LPA. We observed that silencing of LPA1 expression almost totally abolished the LPA-induced migration of MDA-BO2 cells (Fig. 4C). To determine the signalling pathways involved in LPA- and S1P-induced breast cancer cell migration we challenged MDA-BO2 cells to migrate in response to the lysolipids in the presence of specific inhibitors or anti-growth factor receptor blocking antibodies. We observed that S1P activity was totally inhibited with inhibitors Wortmannin and PTX (Fig. 4D). Interestingly, migratory activity of LPA on MDA-BO2 cells was partially (66%) inhibited with Wortmannin and totally blocked with inhibitors of ERK1/2, PKC, Src family kinases, GαI and blocking antibodies against IGF1 and EGF receptors (Fig. 4C and 4D).
Fig. 3– LPA and S1P exhibit differential migratory activities on breast cancer cells.
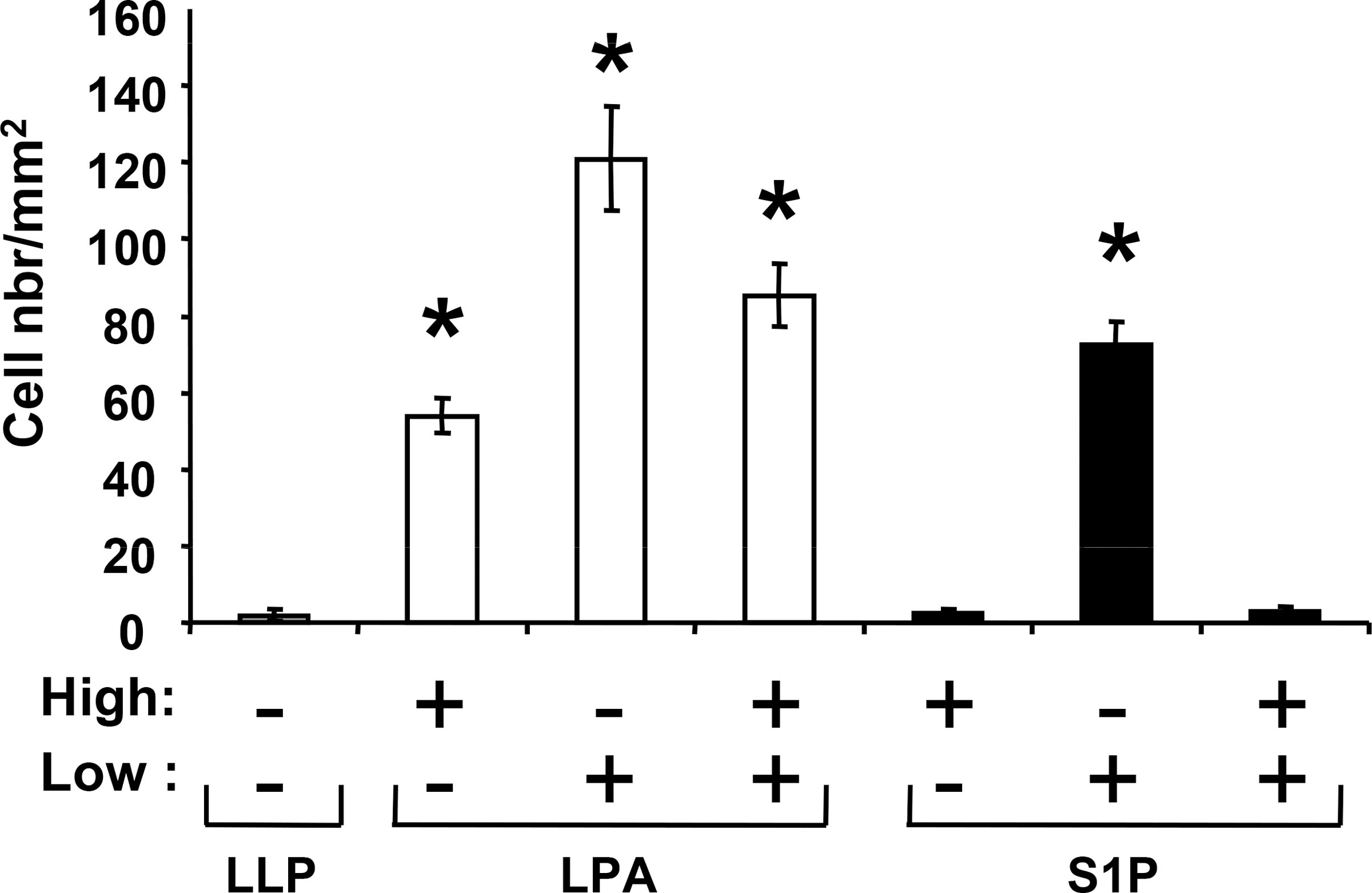
MDA-BO2 cells were seeded on higher (High) and/or lower (Low) chambers of transwell migration chambers. No lysolipid (LLP) or S1P (10−7M) or LPA (10−7M) were added on indicated chambers. LPA exhibits both chemotactic and chemokinetic activities, whereas S1P acts only as a chemo-attractant factor on MDA-BO2 cells. Cells that have passed through the 8 μm membrane were enumerated after 6 hours of treatment. Data are expressed as number of cells/mm2. Bars represent means ± SD of at least three experiments. *, P<0.001 compared to control condition in absence of lysolipid.
Fig. 4– LPA and S1P induce migration of breast cancer cells through specific signaling pathways.
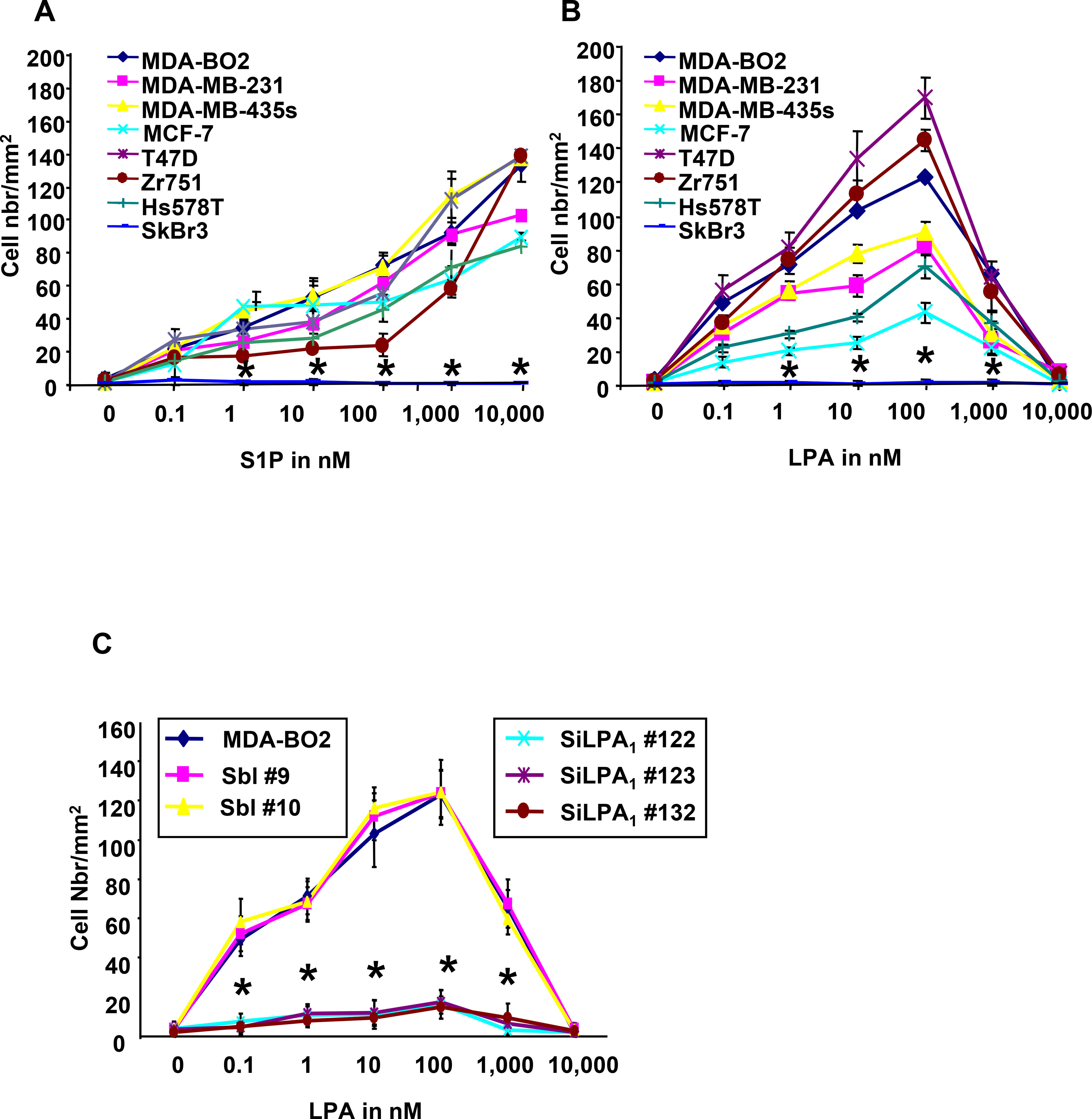
A and. B. LPA and S1P stimulate dose-dependently the migration of human breast cancer cells that express cognate lysolipid receptors. Increased concentrations (0 to 10−5 M) of S1P (A) or LPA (B) were added in the lower compartments of transwell plate migration chambers. Indicated cell lines were seeded on the higher chamber and challenged to migrate through the filter membrane for 6h. While S1P exhibits a continuous chemotactic activity throughout the range of concentrations, LPA action follows a belt shape curve, with a pic of maximum activity at 10−7M. *, P<0.001 compared to MDA-BO2 cells. C. Silencing of LPA1 expression inhibits LPA-induced migration of MDA-BO2 cells. Three independent SiLPA1 transfectants, two scramble-SiRNA control clones (Sbl) and MDA-BO2 parental cells (3) were incubated with increased concentrations of LPA as described above. *, P<0.001 compared to MDA-BO2 cells. D. Effect of inhibition of signaling pathways on migration of MDA-BO2 cells stimulated with LPA or S1P (10−7M). Inhibitors used were identical as described in Fig. 2B. E. Effect of anti-IGFR1 and EGFR antibodies (1μg/ml) on the migration of MDA-BO2 cells induced by LPA and S1P (10−7 M). Data are expressed in cell number/mm2. Bars represent means ± SD of at least three experiments. *, P<0.001 compared to corresponding lysolipid treatment.
LPA and S1P control tumor cell invasion in vitro.
Invasion activity is crucial for tumor cells to escape from their primary site and to disseminate to secondary target tissues (27). To analyze the effects of LPA and S1P on invasion of breast cancer cells, we used transwell migration chambers with membranes coated with matrigel which mimics an artificial basal membrane. All cell lines that express LPA and S1P receptors responded to LPA and S1P, respectively (Fig. 5A). Treatments with specific blockers of GαI and PI3K totally inhibited S1P activity on cell invasion (Fig. 5B). Treatments of all breast cancer cell lines with inhibitors of ERK1/2, PKC, Src family kinases, Gαi or PI3K totally inhibited LPA activity. Also, coincubation of breast cancer cells with anti-IGFR1 or anti-EGFR antibodies totally inhibited cell invasion induced by LPA but not S1P (Fig. 5C).
Fig. 5– LPA and S1P stimulate invasion of human breast cancer cells through specific signaling pathways.
Transwell migration chambers were previously coated with Matrigel. A. Cells were incubated in absence (DMEM) or presence of S1P (10−7M) or LPA (10−7M) added in the lower chamber. After 48h hours of lysolipid treatment cells passed thought the matrigel barrier were enumerated. *, P<0.001 compared to MDA-BO2 cells. B. MDA-BO2 cells pretreated with Wortmannin (10−7M), PTX (100ng/mL), chelerythrine chloride (10−6M), PP2 (10−7M), or PD98059 (10−5M) were seeded on the higher chamber and challenged for 48 hours, to invade and migrate through the filter membrane precoated with matrigel, upon stimulation with lysolipids (LPL) added in the lower chamber. C. Effect of anti-IGFR1 and EGFR antibodies (1μg/ml) on invasion of MDA-BO2 cells induced by LPA and S1P (10−7 M). Data are expressed in cell number/mm2. Bars represent means ± SD of at least three experiments. *, P<0.001 compared to corresponding lysolipid treatment.
LPA and S1P control angiogenesis through direct and indirect tumor cell-induced stimulation of endothelial cells (HUVEC).
It is known for more than a decade that the angiogenesis switch is a prequisite for tumors to expend over a volume of 5 mm3 and to achieve the dissemination of distant metastases (28). Many reports indicate that LPA and S1P act directly on endothelial cell to stimulate proliferation, survival and migration (25). Here, we observed that S1P induced an 80% folds increase of migration (Fig. 6A) and proliferation (data not shown) of human vascular endothelial cells (HUVEC) than equimolar concentrations of LPA. We have shown recently that, in the context of bone metastasis formation, LPA acts on tumor cells to induce the secretion of paracrine factors that stimulate osteoclats and bone destruction (1). Therefore, we sought to determine whether LPA and S1P induced the secretion of paracrine factors by breast cancer cells that stimulate endothelial cells. We primed our series of breast cancer cell lines with LPA or S1P for two days. Then, cell-conditioned media were collected and used to stimulate HUVEC migration. We observed that culture media collected from all cell lines, excepted SKBr3 cells, sustained HUVEC migration (Fig. 6A).
Fig. 6– Commune induction of tumor cell-mediated angiogenesis by LPA and S1P and specific secretion of VEGF-A by S1P.
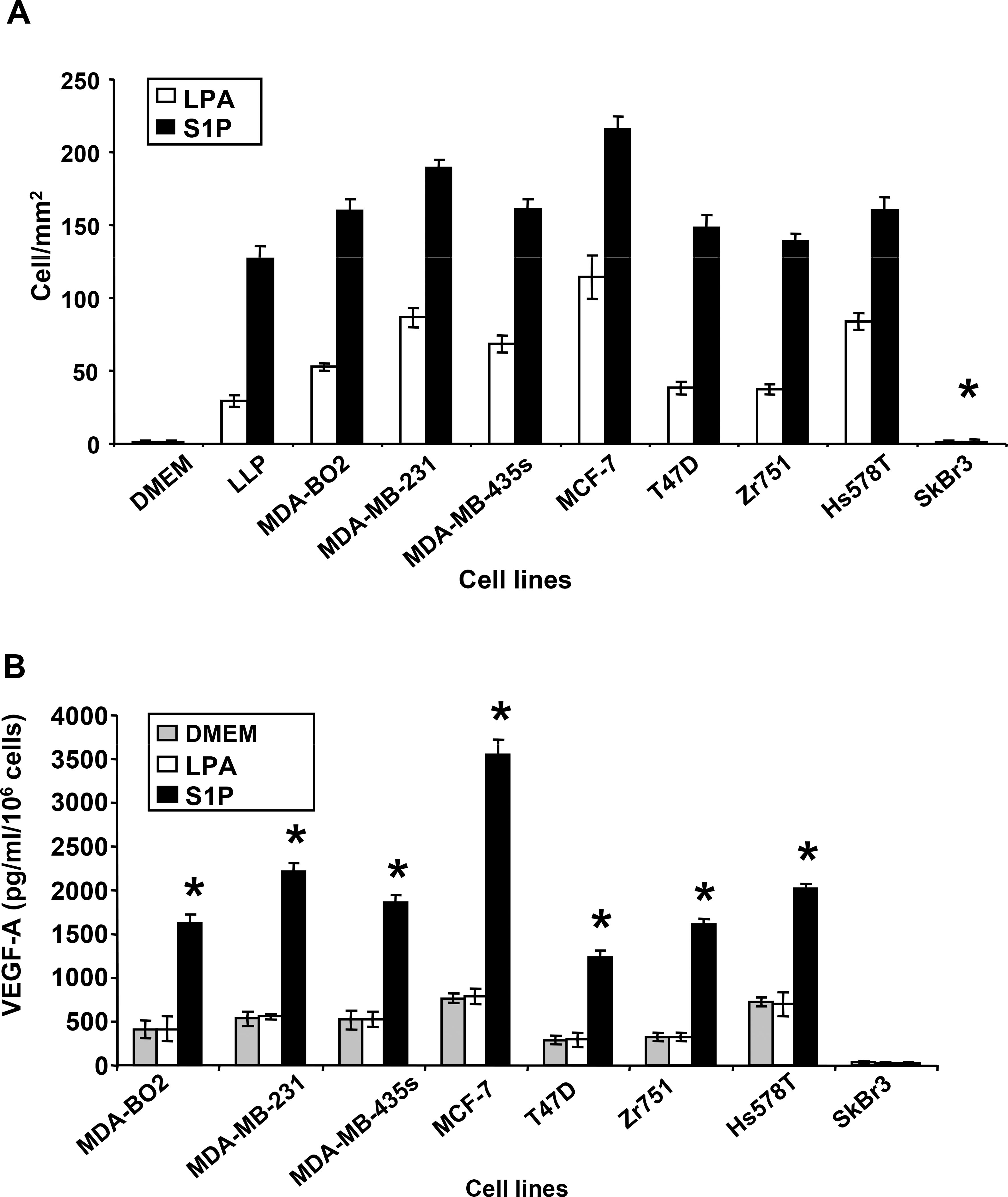
A. Conditioned media from human breast cancer cell lines primed with LPA or S1P (10−6M) for 48h, were tested for their capacity to induce HUVEC cell migration. HUVEC cells were seeded in the higher chamber of transwell migration chambers and challenged to migrate through the filter membrane for 6h. Data are expressed as the number of cells per mm2. *, P<0.001 compared to MDA-BO2 cell conditioned medium B. The secretion of VEGF-A was quantified by ELISA from conditioned media collected from indicated breast cancer cell lines stimulated with LPA (10−7 M) or S1P (10−7 M) for 48h. Data are expressed in pg/ml/106 cells. All bars and curves represent means ± SD of at least three experiments. *, P<0.001 compared to DMEM treated cells.
S1P, but not LPA, stimulates the secretion of VEGF-A by breast cancer cells.
It has been shown that LPA induces the release of vascular endothelial growth factor (VEGF) from ovarian cancer cells (29). Therefore, we ask to know whether LPA and S1P induced the secretion of VEGF-A by breast cancer cells. Except for SKBr3 cells, all cell lines tested here, secreted high levels of VEGF-A at steady-state (Fig. 6B). Consistent with our previous observation on MDA-BO2 cells (3), LPA did not promote the secretion of VEGF-A on this series of breast cancer cell lines. In contrast to LPA, S1P increased the secretion of VEGF-A by these cells from 3 to 6 folds (Fig. 6B). These results suggested that LPA and S1P might drive tumor-induced angiogenesis through distinct pathways involving a S1P-dependent and LPA-independent secretion of VEGF-A.
LPA, but not S1P, stimulates breast cancer cell osteoclastic activity.
Bone is a frequent metastatic site for many cancers including breast cancers (30). We have recently shown that LPA, derived from activated platelets, plays a central role in the progression of bone metastases by controlling both tumor growth and tumor cell-induced osteoclast activity (1). Because platelets produce both LPA and S1P during platelet aggregation (8,31,32) we investigated the potential activity of S1P on breast cancer cells to stimulate osteoclast formation. LPA induced the secretion of pro-osteclastic cytokines and growth factors, IL-6, IL-8, granulocyte-macrophage colony-stimulating factor (GM-CSF), growth-related oncogene (Gro-α), and macrophage chemoattractant protein 1 (MCP-1) by MDA-BO2 cells (3). We observed that S1P also enhanced the secretion of IL-8 (Fig. 7A). However, in contrast to LPA, S1P did not promote the secretion of IL-6, GM-CSF, GRO-α and MCP-1 by MDA-BO2 cells (Fig. 7A) and did not induce osteoclast formation in vitro (Fig. 7B).
Fig. 7– LPA but not S1P induces pro-osteoclastogenic activity of MDA-BO2 cells.
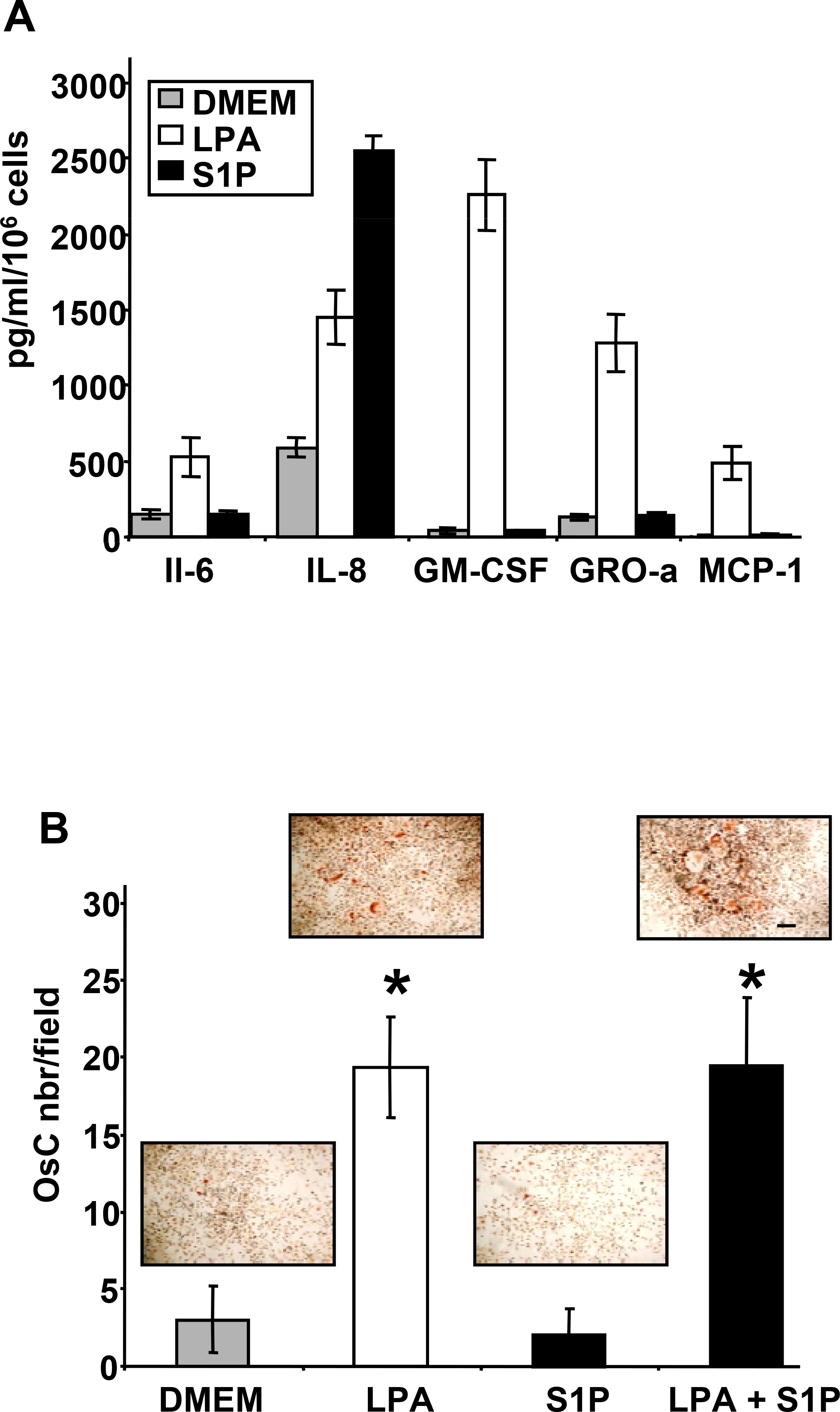
A. pro-osteoclastogenic factors (IL-6, IL-8, GM-CSF, GRO-a, MCP-1) were quantified by ELISA from conditioned media generated from MDA-BO2 cells stimulated with LPA (10−6M) or S1P (10−6M). B. LPA but not S1P, stimulates tumor cell-induced formation of mature osteoclasts. Bone marrow cells were cultured in the presence of conditioned media collected from MDA-BO2 cells treated with LPA (10−6M) or S1P (10−6M), or with a combination of LPA and S1P (10−6M). Mature osteoclasts, which are TRAP positive multinucleated cells, were enumerated under microscope. Data are expressed as the number of osteoclasts per field. Bars represent means ± SD of at least three experiments. Insets: representative picture of osteoclasts differentiated in each condition. Size ladder 100 μm. *, P<0.001 compared to DMEM treated cells.
Discussion
LPA and S1P act on two distinct, but structurally related, seven-transmembrane spanning domain receptors coupled to G proteins (5). LPA and S1P receptors share similar down-stream signalling pathways suggesting potential redundant activities of each receptor subtype (5). In agreement with previous reports, we observed that breast cancer cells express both LPA and S1P receptors. The role of S1P receptors in breast cancers is largely unknown since gene expression analyses for these receptors at the primary site have not been conducted yet. Expression of LPA2 was consistently found elevated at the primary site of different cancers, including ductal breast carcinoma (33). This suggests that LPA2 might play a preferential role in carcinogenesis. However, direct proofs are still needed as genes encoding either LPA1, LPA2, or LPA4 support the transformation of MEF cells overexpressing c-Myc and Tbx2, indicating that these genes behave as oncogenes(34). Cell motility is controlled by both LPA and S1P. LPA, but not S1P, exerted a chemokinetic activity on tumor cells. In the context of primary tumor growth this suggests that LPA might be a metastatic promoting factor. However, we found that LPA activity, but not S1P, followed a bell-shape curve which suggested that high local concentrations of LPA in vivo might preferentially drive tumor cells to proliferate rather than to induce migration. This assumption was supported by the increase of xenograft tumor growth of MDA-BO2 cells previously sensitized to LPA consequently to over-expression of LPA1 after cell transfection (3).
EGFR is known as a central transducer of a wide range of different agonists that mobilized G protein-coupled receptors, receptor with thyrosine kinase activity, and sensing receptors (35). Activation of LPA receptor, LPA1, induces gastric cancer cell migration through SPHK1/S1P formation and transactivation of EGFR (23). Similar “criss-cross” signal transduction was reported between EGF and S1P receptors to mediate estrogen-dependent breast cancer cell proliferation (22). Strikingly, we observed that biological activities of LPA, but not S1P, on MDA-BO2 breast cancer cells were dependent on EGFR and IGFR1 transactivations. Gαi/ERK1/2 and Gαi/PI3K signalling pathways control specifically LPA and S1P biological activities in these cells, respectively. In Cos7 and mammary epithelial cells, transactivation of EGFR is required for LPA- and IGF1-induced phosphorylation of ERK1/2 (36,37). As observed in MDA-BO2 cells, S1P-induced migration of vascular smooth muscle cells is mediated through a pathway dependent on PI3K but not on EGFR (38). Specific activation of Gαi/ERK1/2 and Gαi/PI3K pathways might explain discrepancies in the central role of receptors for EGF and IGF1 as signal transducers for LPA and S1P. This suggests that, in the context of cancer development, LPA and S1P could have overlapped synergistic activities through the mobilization of specific signalling pathways to mediate tumor growth and distant metastasis formation.
LPA and S1P are known to exhibit pro-angiogenic activities, as they induce the proliferation and migration of endothelial cells (25). Besides these direct angiogenesis activities, we found that LPA and S1P also elicited indirect tumor cell-induced angiogenesis. S1P activity correlated with its capacity to stimulate the secretion of VEGF-A by breast cancer cell lines. LPA also induced indirect tumor cell-driven angiogenesis. However, in contrast to what was shown in ovarian cancer cells (29), LPA did not enhance the secretion of VEGF-A from our series of breast cancer cell lines. VEGF-A is highly expressed at steady state by the cell lines used in this study in normoxic cell culture conditions. LPA-induced expression of VEGF-A was shown to be dependent on HIF1α expression which occurs when cells are placed under hypoxic conditions (39). Thus, our observation does not exclude that in vivo LPA might stimulate the secretion of VEGF-A upon hypoxia. Additionally, the indirect tumor cell-driven angiogenesis elicited by LPA is likely to be achieved through enhanced secretion of cytokines and growth factors from tumor cells. These include IL-6 and IL-8 which are known to promote angiogenesis (40,41).
IL-6 and IL-8 are also known to mediate the differentiation of mature osteoclasts from circulating precursors and their miss-regulation in breast cancers were associated with increased formation of bone metastasis (42,43). We showed previously that LPA exhibited an indirect action on osteoclasts by inducing the secretion of pro-osteoclastic cytokines (IL-6, IL-8, GM-CSF, Gro-α and MCP-1) (1,3). Interestingly, S1P also enhanced the secretion of IL-6 on MDA-BO2 cells, but not of the other pro-osteoclastic cytokines. As a marked difference with LPA, S1P was not effective in eliciting a full osteoclast activity by these cells in vitro (Fig. 7B). This observation suggests that, at the bone metastatic site, S1P might sustain the action of LPA on tumor cell proliferation, but not on bone destruction.
Altogether, our data provide new insights on the role of LPA and S1P during breast cancer cell progression and indicate that LPA and S1P exhibit additive and complementary activities to mediate tumor growth, to the escape of the tumor cells from their primary site, and to support bone metastasis formation. Together, these new findings suggest that LPA and S1P are prime targets for the development of new therapies for cancer patients.
Acknowledgements
This study was supported by grants from INSERM (O.P. and P.C.), the Comité Départemental de la Loire de la Ligue Nationale contre le Cancer (O.P.). We thank Dr Harald Hirling and Dr Ruth Luthi-Carter from the Faculté des Sciences de la Vie, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland for generous help.
References
- 1.Boucharaba A; Serre CM; Gres S; Saulnier-Blache JS; Bordet JC; Guglielmi J; Clezardin P; and Peyruchaud OJ Clin. Invest. 114: 1714–1725. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Visentin B; Vekich JA; Sibbald BJ; Cavalli AL; Moreno KM; Matteo RG; Garland WA; Lu Y; Yu S; Hall HS; Kundra V; Mills GB; and Sabbadini RA Cancer Cell 9: 225–238. 2006. [DOI] [PubMed] [Google Scholar]
- 3.Boucharaba A; Serre CM; Guglielmi J; Bordet JC; Clezardin P; and Peyruchaud O Proc Natl Acad Sci U S A 103: 9643–9648. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishii I; Fukushima N; Ye X; and Chun J Annu Rev Biochem 73: 321–354. 2004. [DOI] [PubMed] [Google Scholar]
- 5.Birgbauer E; and Chun J Cell. Mol. Life Sci. 63: 2695–2701. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Umezu-Goto M; Kishi Y; Taira A; Hama K; Dohmae N; Takio K; Yamori T; Mills GB; Inoue K; Aoki J; and Arai H J Cell. Biol. 158: 227–233. 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maceyka M; Sankala H; Hait NC; Le Stunff H; Liu H; Toman R; Collier C; Zhang M; Satin LS; Merrill AH Jr.; Milstien S; and Spiegel SJ Biol. Chem. 280: 37118–37129. 2005. [DOI] [PubMed] [Google Scholar]
- 8.Eichholtz T; Jalink K; Fahrenfort I; and Moolenaar WH Biochem J. 291: 677–680. 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yatomi Y; Ohmori T; Rile G; Kazama F; Okamoto H; Sano T; Satoh K; Kume S; Tigyi G; Igarashi Y; and Ozaki Y Blood 96: 3431–3438. 2000. [PubMed] [Google Scholar]
- 10.Meyer Zu Heringdorf DJ Cell. Biochem. 92: 937–948. 2004. [DOI] [PubMed] [Google Scholar]
- 11.Kotarsky K; Boketoft A; Bristulf J; Nilsson NE; Norberg A; Hansson S; Owman C; Sillard R; Leeb-Lundberg LM; and Olde BJ Pharmacol Exp. Ther. 318: 619–628. 2006. [DOI] [PubMed] [Google Scholar]
- 12.Noguchi K; Ishii S; and Shimizu TJ Biol. Chem. 278: 25600–25606. 2003. [DOI] [PubMed] [Google Scholar]
- 13.Tabata K; Baba K; Shiraishi A; Ito M; and Fujita N Biochem. Biophys. Res. Commun. 363: 861–866. 2007. [DOI] [PubMed] [Google Scholar]
- 14.Siess W; and Tigyi GJ Cell. Biochem. 92: 1086–1094. 2004. [DOI] [PubMed] [Google Scholar]
- 15.Pasternack SM; von Kugelgen I; Aboud KA; Lee YA; Ruschendorf F; Voss K; Hillmer AM; Molderings GJ; Franz T; Ramirez A; Nurnberg P; Nothen MM; and Betz RC Nat. Genet. 40: 329–334. 2008. [DOI] [PubMed] [Google Scholar]
- 16.Pecheur I; Peyruchaud O; Serre CM; Guglielmi J; Voland C; Bourre F; Margue C; Cohen-Solal M; Buffet A; Kieffer N; and Clezardin P Faseb J. 16: 1266–1268. 2002. [DOI] [PubMed] [Google Scholar]
- 17.Peyruchaud O; Winding B; Pecheur I; Serre CM; Delmas P; and Clezardin PJ Bone Miner. Res. 16: 2027–2034. 2001. [DOI] [PubMed] [Google Scholar]
- 18.Peyruchaud O; and Mosher DF Cell. Mol. Life Sci. 57: 1109–1116. 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goetzl EJ; Dolezalova H; Kong Y; and Zeng L Cancer Res. 59: 4732–4737. 1999. [PubMed] [Google Scholar]
- 20.Boissier S; Ferreras M; Peyruchaud O; Magnetto S; Ebetino FH; Colombel M; Delmas P; Delaisse JM; and Clezardin P Cancer Res. 60: 2949–2954. 2000. [PubMed] [Google Scholar]
- 21.Fang X; Yu S; Bast RC; Liu S; Xu HJ; Hu SX; LaPushin R; Claret FX; Aggarwal BB; Lu Y; and Mills GB J. Biol. Chem. 279: 9653–9661. 2004. [DOI] [PubMed] [Google Scholar]
- 22.Sukocheva O; Wadham C; Holmes A; Albanese N; Verrier E; Feng F; Bernal A; Derian CK; Ullrich A; Vadas MA; and Xia PJ Cell. Biol. 173: 301–310. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shida D; Fang X; Kordula T; Takabe K; Lepine S; Alvarez SE; Milstien S; and Spiegel S Cancer Res. 68: 6569–6577. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graler MH; and Goetzl EJ Biochim. Biophys. Acta. 1582: 168–174. 2002. [DOI] [PubMed] [Google Scholar]
- 25.Panetti TS Biochim. Biophys. Acta. 1582: 190–196. 2002. [DOI] [PubMed] [Google Scholar]
- 26.Hama K; Aoki J; Fukaya M; Kishi Y; Sakai T; Suzuki R; Ohta H; Yamori T; Watanabe M; Chun J; and Arai HJ Biol. Chem. 279: 17634–17639. 2004. [DOI] [PubMed] [Google Scholar]
- 27.Pantel K; and Brakenhoff RH Nat. Rev. Cancer 4, 448–456. 2004. [DOI] [PubMed] [Google Scholar]
- 28.Hanahan D; and Folkman J Cell. 86: 353–364. 1996. [DOI] [PubMed] [Google Scholar]
- 29.Hu YL; Tee MK; Goetzl EJ; Auersperg N; Mills GB; Ferrara N; and Jaffe RB J. Natl. Cancer Inst. 93: 762–768. 2001. [DOI] [PubMed] [Google Scholar]
- 30.Coleman RE Cancer 80: 1588–1594. 1997. [DOI] [PubMed] [Google Scholar]
- 31.Aoki J; Taira A; Takanezawa Y; Kishi Y; Hama K; Kishimoto T; Mizuno K; Saku K; Taguchi R; and Arai HJ Biol. Chem. 277, 48737–48744. 2002. [DOI] [PubMed] [Google Scholar]
- 32.Yatomi Y; Igarashi Y; Yang L; Hisano N; Qi R; Asazuma N; Satoh K; Ozaki Y; and Kume SJ Biochem. (Tokyo) 121: 969–973. 1997. [DOI] [PubMed] [Google Scholar]
- 33.Mills GB; and Moolenaar WH Nat. Rev. Cancer 3: 582–591. 2003. [DOI] [PubMed] [Google Scholar]
- 34.Taghavi P; Verhoeven E; Jacobs JJ; Lambooij JP; Stortelers C; Tanger E; Moolenaar WH; and van Lohuizen M Oncogene. 27:6806–16. 2008. [DOI] [PubMed] [Google Scholar]
- 35.Zwick E; Hackel PO; Prenzel N; and Ullrich A Trends Pharmacol. Sci. 20: 408–412. 1999. [DOI] [PubMed] [Google Scholar]
- 36.Roudabush FL; Pierce KL; Maudsley S; Khan KD; and Luttrell LM J. Biol. Chem. 275: 22583–22589. 2000. [DOI] [PubMed] [Google Scholar]
- 37.Rodland KD; Bollinger N; Ippolito D; Opresko LK; Coffey RJ; Zangar R, and Wiley HS J. Biol. Chem. 283:31477–87. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fegley AJ; Tanski WJ; Roztocil E; and Davies MG J. Surg. Res. 113: 32–41. 2003. [DOI] [PubMed] [Google Scholar]
- 39.Lee J; Park SY; Lee EK; Park CG; Chung HC; Rha SY; Kim YK; Bae GU; Kim BK; Han JW; and Lee HY Clin. Cancer Res. 12: 6351–6358; 2006. [DOI] [PubMed] [Google Scholar]
- 40.Motro B; Itin A; Sachs L; and Keshet E Proc. Natl. Acad. Sci. U S A 87: 3092–3096. 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koch AE; Polverini PJ; Kunkel SL; Harlow LA; DiPietro LA; Elner VM; Elner SG; and Strieter RM Science 258: 1798–1801.1992. [DOI] [PubMed] [Google Scholar]
- 42.de la Mata J; Uy HL; Guise TA; Story B; Boyce BF; Mundy GR; and Roodman GD J. Clin. Invest. 95: 2846–2852. 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bendre MS; Gaddy-Kurten D; Mon-Foote T; Akel NS; Skinner RA; Nicholas RW; and Suva LJ Cancer Res. 62: 5571–5579. 2002. [PubMed] [Google Scholar]