

Abstract
Patients with pulmonary emphysema often develop locomotor muscle dysfunction, which is independently associated with disability and higher mortality in that population. Muscle dysfunction entails reduced force generation capacity, which partially depends on fibers’ oxidative potential, yet very little mechanistic research has focused on muscle respiration in pulmonary emphysema. Using a recently established animal model of pulmonary emphysema–driven skeletal muscle dysfunction, we found downregulation of SDHC (succinate dehydrogenase subunit C) in association with lower oxygen consumption and fatigue tolerance in locomotor muscles. Reduced SDH activity has been previously observed in muscles from patients with pulmonary emphysema, and we found that SDHC is required to support respiration in cultured muscle cells. Moreover, in vivo gain of SDH function in emphysema animals’ muscles resulted in better oxygen consumption rate and fatigue tolerance. These changes correlated with a larger number of relatively more oxidative type 2-A and 2X fibers and a reduced amount of 2B fibers. Our data suggest that SDHC is a key regulator of respiration and fatigability in pulmonary emphysema–driven skeletal muscles, which could be impactful in developing strategies aimed at attenuating this comorbidity.
Keywords: pulmonary emphysema, succinate dehydrogenase, muscle respiration, COPD
Patients with chronic obstructive pulmonary disease (COPD) often develop locomotor muscle dysfunction, which entails atrophy and reduced force generation capacity (1). Muscle dysfunction, which occurs more often in patients with pulmonary emphysema than in those with chronic bronchitis (2), is strongly associated with higher mortality and other poor outcomes (3–6). These associations persist even after adjusting for the amount of pulmonary disease and other covariables, suggesting that muscle dysfunction could be independently responsible for the worse prognosis (5, 6). Moreover, very few interventions can improve muscle status in pulmonary emphysema (7), and none have shown mortality benefits (8). It is generally accepted that maximal force generation capacity predominantly depends on muscle mass and that submaximal force (or “endurance”) depends on fiber metabolic properties (1, 9–11). Inferential models suggest that fiber metabolic disturbances are associated with higher mortality in these patients, even after correcting for the magnitude of muscle atrophy (12). Although metabolic dysfunction directly impacts fiber contractility and fatigue tolerance (11, 13, 14), very little mechanistic research has been conducted on fiber respiration and its functional repercussions in the context of pulmonary emphysema.
Mechanistic research focused on COPD-driven locomotor muscle dysfunction requires an animal model that ideally should fulfill the following conditions: 1) be inducible to minimize temporal confounders such as muscle development and age-related sarcopenia; 2) be robust enough to reminisce the disease severity shown by the majority of patients with COPD with muscle dysfunction (15); 3) develop the muscle phenotype after (not simultaneously with) the occurrence of pulmonary disease to reflect a secondary COPD comorbidity; 4) recapitulate multidimensional features observed in patients such as morphologic, metabolic, and functional aspects of muscle dysfunction (1, 16, 17); and 5) occur in the context of a pulmonary disease phenotype with airways obstruction, typical histologic changes, and other features demonstrated by humans (18, 19). We have recently described an animal model of pulmonary emphysema–driven skeletal muscle dysfunction that recapitulates many of the features observed in patients with pulmonary emphysema, including dysfunctional fiber respiration and contractility (16, 17, 20). Features characterizing muscle dysfunction do not occur in this animal until the emphysema is established at 8 weeks after induction (16, 17) (Figure E1 in the data supplement), suggesting a similar trajectory to that demonstrated by patients with COPD with muscle dysfunction (1). In an unbiased analysis of that animal’s muscle proteome, we discovered a downregulation of SDH (succinate dehydrogenase) subunit C (SDHC), which is associated with lower oxygen consumption and decreased fatigue tolerance. These associations are all regulated under chronic exercise, which improves SDH expression and activity, oxygen consumption, and fatigue tolerance (16).
SDH is a tetrameric iron–sulfur flavoprotein of the tricarboxylic acid cycle (TCA) (21). The correlation between SDH expression and fiber oxidative capacity was established long ago (22, 23), and indeed the skeletal muscles of patients with COPD have been shown to have a reduced SDH activity (24) associated with lower oxygen consumption (25) even if the substrate availability (succinate) is controlled (26). Moreover, TCA dysfunction caused by lower SDH activity could compromise O2 consumption and ATP generation (27), leading to higher lactate production and fatigability (27–29). Both lactate generation and fatigability are relatively increased in the muscles of patients with COPD under endurance exercise in comparison with those of healthy individuals (30). SDH is the only TCA cycle enzyme that also participates in the electron transport chain as complex II, and previous evidence demonstrates that dysfunction of SDH subunits located at the ubiquinol binding site, such as SDHC, cause increased reactive oxygen species (ROS) formation (31), which occurs in COPD muscles (32, 33).
Although it is not known whether SDH dysfunction causes the clinical manifestations of COPD myopathy, in this study, we hypothesized that downregulation of SDHC directly contributes to reduced oxygen consumption and fatigue tolerance in COPD skeletal muscles and conducted both loss- and gain-of-function studies to mechanistically interrogate that interaction.
Some of the results of these studies have been previously reported in the form of a preprint (34).
Methods
Animals
Experiments were conducted using CC10-rtTA-IL-13 (IL13TG) doxycycline-inducible transgenic mice that develop chronic lung remodeling reminiscent of pulmonary emphysema upon induction (18, 19) (Figure E1). CC10-rtTA-IL-13 heterozygote animals were bred to C57BL/6 mice to obtain IL13TG and wild-type (WT) (IL13WT) littermate control animals. Both IL13TG (emphysema) and WT (nonemphysema, used as control littermates) mice were provided 0.5 g/L doxycycline in their drinking water together with sucrose (0.5 mg/ml), starting at 5 weeks of age for a total dose of 17 weeks. Male and female mice were used for the studies. Food and water were accessible ad libitum, and a 12-hour light/dark cycle was maintained. Sampling of skeletal muscle was performed directly after euthanasia by cervical dislocation. The time elapsed between animal euthanasia and muscle procurement and freezing never exceeded 3 minutes. All the procedures involving animals were approved by the Albany Medical College Institutional Animal Care and Use Committee (07001), and animals were handled according to the National Institutes of Health guidelines; all methods were performed in accordance with the relevant guidelines and regulations as stated by the Journal and public agencies.
Muscle Histology
At 17 weeks after doxycycline initiation (22–23 wk of age), freshly procured extensor digitorum longus (EDL) muscles were placed on saline-moistened gauze in a 60-mm culture dish on ice until freezing. A metal cup containing isopentane was cooled in liquid nitrogen until crystals of isopentane formed at the bottom of the cup. Muscles were transferred to precooled Tissue-Tek embedding cassettes (62520; Electron Microscopy Sciences) which were dropped into the cooled isopentane, submerging the muscle for 1 minute. Muscle samples were then drained and dried on gauze pads at −20°C to remove all isopentane. Frozen muscles were adhered to the sample stage using a small amount of Tissue-Tek optimal cutting temperature compound (62550; Electron Microscopy Sciences), and sections were completed using a Leica CM1860 Cryostat; 10-μm sections were obtained for further analysis.
Muscle Fiber Typing Immunofluorescence
Muscle sections were fixed for 15 minutes in acetone at −20°C and then left at room temperature to dry for 30 minutes. Blocking was performed using mouse-on-mouse blocking reagent (Vector Labs) for 1 hour at room temperature. Sections were then incubated for 45 minutes at 37°C with the primary antibodies indicated in Table E1. Three washes were then performed with PBS. The following secondary antibodies from Jackson ImmunoResearch Laboratories were added (all at 1:250) and incubated for 45 minutes at 37°C: anti-mouse IgG2b-DyLight 405, anti-mouse IgG1-Alexa 488, anti-mouse IgM-Alex594, and anti-rabbit IgG-Alexa 647. Three washes were then performed with PBS. Samples were mounted with Ibidi Mounting Medium. Images were captured the same day using confocal microscopy (Leica SPE).
EM
EDL muscle samples were placed in M. J. Karnovsky fixative immediately after procurement. Samples were dehydrated, embedded, cut on 60-nm–thick sections, mounted on corresponding grids, and imaged by the University of Pittsburgh EM core facility.
RNA Extraction, cDNA Synthesis, and Quantitative Real-Time PCR
RNA from tibialis anterior (TA) muscle was extracted using NucleoSpin RNA kit (Machery-Nagel). The reason TA was used is the higher yield of RNA per sample relative to other muscles. cDNA was synthesized using Quantitect Reverse Transcriptase Kit (Qiagen). Quantitative real-time PCR was performed using iTaq Universal SYBR Green Supermix (Bio-Rad) on a CFX96 real-time PCR detection system (Bio-Rad). Each sample was run in triplicates, and relative expression levels of transcripts of interest were calculated using the comparative Ct (ΔΔCt) method with GAPDH as a housekeeping gene. Primers were purchased from Integrated DNA Technologies, and a list of their sequences is presented in Table E2.
Oxygen Consumption by Muscle Fibers
Oxygen consumption rates (OCRs) from isolated muscle fibers were determined using a Seahorse XFp. At 17 weeks after doxycycline initiation (22–23 wk of age), mouse EDL muscles were isolated tendon to tendon, and respirometry analyses were done as previously established (35). In short, the muscle was bisected using a sterile scalpel, and thin, even sections were placed in Matrigel precoated wells of an eight-well Seahorse plate. The first and last wells were reserved as coating-only controls, and samples were run in triplicates. XF base DMEM media was added to each well including 15 mM glucose and 10 mM sodium pyruvate and were adjusted to pH 7.4 using sodium hydroxide; plates were equilibrated in a CO2-free incubator for 1 hour before running. Rotenone and antimycin A were used as respiratory chain inhibitors following the manufacturer’s suggested concentrations. Data were normalized to the total amount of protein contained in each well as determined by bicinchoninic acid (BCA) assay. This method has the advantage of causing minimal disruption to the cytosolic environment that is intrinsic to the membrane permeabilization step used in conventional protocols (36, 37), does not entail potential mitochondrial respiration biasing due to isolation/differential centrifugation (38), and thus is highly recommendable to specifically interrogate mitochondrial respiration in vivo (35). Interrogation of complex II– and complex IV–specific contribution to oxygen consumption was accomplished following published protocols (39, 40). In short, muscle sections were incubated with 4 mM succinate and 8 μM rotenone for complex II interrogation, and separately with 1 mM N,N,N’,N’-tetramethyl-p-phenylenediamine (TMPD), 1 mM ascorbate, 8 μM rotenone, and 8 μM antimycin A for complex IV interrogation.
SDH Activity
At 17 weeks after doxycycline initiation (22–23 wk of age), SDH activity was determined using a succinate dehydrogenase assay kit (Sigma-Aldrich, MAK197) following the manufacturer’s protocol using the mouse TA muscle lysate. Absorbance measurements were collected using a Cytation 5 plate reader (BioTek).
Isolated Muscle Fatigability
At 17 weeks after doxycycline initiation (22–23 wk of age), muscle fatigability was determined using an isolated muscle contractility platform. Immediately after euthanasia, the EDL muscle was surgically isolated from the mouse carefully, and a suture was tied around the tendon at each end of the muscle before cutting and removal. Special care was taken to assure that no muscle stretching or damage was caused during the procurement process. Once removed, the isolated muscle was placed in ice-cold Ringer’s solution supplemented with 5.5 mM glucose at pH 7.4, adjusted as needed with sodium hydroxide, and bubbled with carbogen. The muscle was then suspended between the isometric force transducer (Harvard Apparatus) and the platinum-stimulating electrode tissue support (160152; Radnoti), lowered into the 25-ml tissue bath (166026; Radnoti) containing the same solution (also bubbled slowly with carbogen but this time at room temperature), and allowed to equilibrate for an additional 15 min. Muscle tension was increased until baseline tension started to increase. A single 1-Hz, 40-V stimulus was delivered with a Grass S-88 electrical stimulator, and the peak contraction was recorded. After a 30-second rest, the voltage was increased by 10 V and delivered again, recording the peak contraction. This process was repeated until no additional increase in the peak contraction force was observed. The optimal length of the muscle was then determined by slowly increasing the muscle tension and delivering a single, maximal voltage stimulus, as previously determined above, while recording the peak force. After a 30-second rest, the tension was slightly increased, and another stimulus was delivered while recording the peak contraction. This process was repeated until maximal peak contraction force was achieved. Subsequent prefatigue stimuli were delivered at 1, 10, 20, 30, 50, 80, 100, and 120 Hz while recording the peak force at each point and allowing for a 1-minute rest between each stimulus. Immediately after prefatigue process, a train of additional stimuli were delivered by stimulating the muscle with 20 Hz (500-ms duration, 1 train/s) for 5 minutes while recording constantly. Fatigue resistance was determined by comparing the initial and the final peak contraction measured during the fatigue program. All data collection and analysis were completed using the PowerLabs 4/20T (ADInstruments) amplifier and LabChart 7 software.
Succinate Quantification
At 17 weeks after doxycycline initiation (22–23 wk of age), the amount of succinate was determined using a succinate colorimetric assay kit (MAK184; Sigma-Aldrich) following the manufacturer’s protocol using the mouse TA muscle lysate. Absorbance measurements were collected using a Cytation 5 plate reader (BioTek).
Cells and siRNA Transfection Protocol
C2C12 cells were cultured in normal conditions with 5% FBS growth medium. Cells were cultured in six-well plates or Seahorse respirometry plates as experimentally required. Cells in each setting were transfected with either 10 nM scramble (control) or 10 nM mouse SDHC siRNA-SMARTpool (L048315–01–0005; Dharmacon) using lipofectamine 3000 (L3000; Life Technologies) following the manufacturer’s recommended protocols. After 48 hours, the same process was repeated before analyzing or collecting cells 48 hours after that.
In Vivo Electroporation
At 9 weeks after doxycycline initiation (14–15 wk of age) the hair on the back legs of animals was removed with Nair while under anesthesia. While still anesthetized, mouse TA/EDL muscles were directly injected with 40 μL hyaluronidase (0.3 U/μL in 0.9% NaCl) in 10-μL aliquots across the length of the muscle and then allowed to incubate without anesthesia for 2 hours. After 2 hours, the animals were anesthetized and 40 μL plasmid with or without SDHC (1 μg/μL in 0.9% NaCl) was injected in 10-μL aliquots across the length of the muscle. The right leg was injected with the SDHC-containing plasmid, and the left leg of the same animal was injected with the empty vector (plasmid with no SDHC). Using an ECM 830 Square Wave Electroporation System (BTX) and Tweezertrode platinum electrodes (BTX), five 25-millisecond pluses of 150 V were delivered through the clamped leg of the mouse. After the electroporation of both sides was completed, the mouse continued receiving doxycycline for 8 additional weeks before collection at 17 weeks after initiation of doxycycline (22–23 wk of age).
We have determined that the plasmid remains upregulated for at least 2 months after electroporation. That timeframe is important for the following two reasons: first, because this is a chronic disease model of COPD-induced muscle dysfunction, and thus gain-of-function is intended to occur gradually after the lung phenotype develops at 8 weeks after doxycycline induction (13 wk of life, see also Figure 1 of recent paper Reference 16). Second, because in vivo electroporation requires hyaluronidase injection followed by electrically driven DNA incorporation 2 hours later, these processes cause muscle injury (41, 42). Rigorous SDH gain-of-function interrogation requires full recovery of fibers integrity, as reflected by reconstitution of histological pattern (43).
Figure 1.
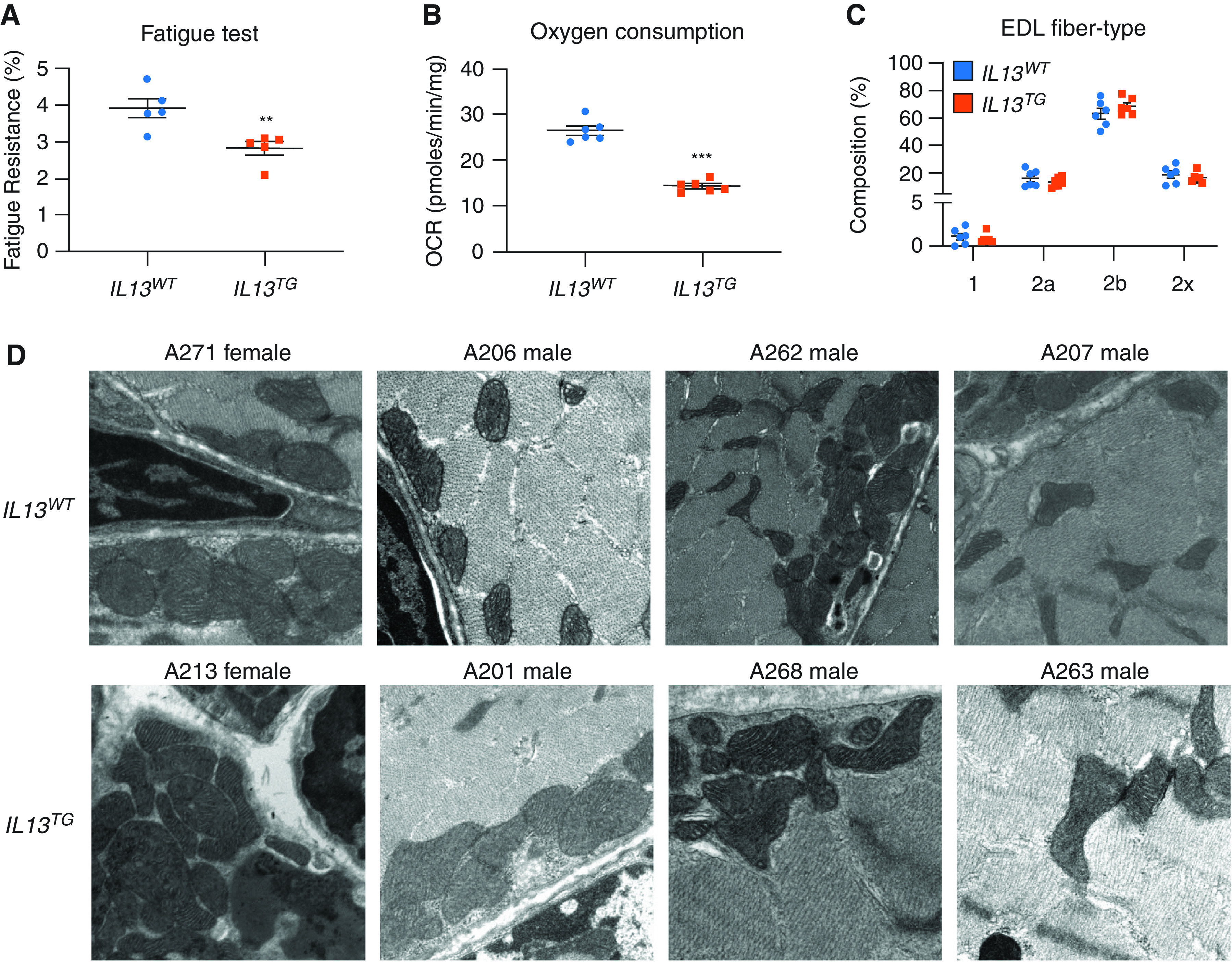
Inducible pulmonary emphysema leads to reduced respiration in skeletal muscle. EDL muscles from IL13TG (emphysema) and IL13WT (wild-type [WT]) mice were (A) assayed in the isolated contractility platform that demonstrated a reduced fatigue tolerance in IL13TG animals’ muscles (n = 5); (B) assayed in the plate respirometry (Seahorse) platform, which demonstrated a reduced oxygen consumption rate (OCR) in IL13TG animals’ muscles (n = 6); (C) immunostained with antibodies to detect MyHC (myosin heavy chain) isoforms to characterize fiber types, showing similar fiber type distribution in IL13TG and WT mice (n = 6); and (D) processed for EM evaluation, which ruled out any conspicuous structural mitochondrial abnormality (n = 4; data above each panel designate the sex and number of the animal producing the micrography). **P < 0.01 and ***P < 0.001. EDL = extensor digitorum longus; TG = transgenic.
Western Blotting
Muscles were lysed in lysis buffer using a bead beating homogenizer (10158–558; VWR) with 100× protease and phosphatase inhibitor cocktail (78429; Thermo Fisher Scientific). Cells were lysed using cell lysis buffer (9803S; Cell Signaling). All samples were centrifuged at 22,000 × g for 10 minutes at 4˚C and transferred to fresh tubes to remove insoluble debris. Protein concentrations were determined with a BCA assay (23227; Thermo Fisher Scientific) and normalized before gel electrophoresis. Precast 10% or 4–20% mini-PROTEAN TGX 10-well gels (4561034; Bio-Rad) were used for protein separation electrophoresis. Proteins were transferred to nitrocellulose membranes using a wet-transfer system (1703930; Bio-Rad) and blocked in 5% dry milk for 2 hours. IB was performed in 1% dry milk overnight before 1-hour incubation with secondary antibody and visualization with chemiluminescence. Antibodies and specific concentrations used can be found in the Tables E1 and E3.
Statistics
Data are expressed as the mean ± SEM. When results were compared with a reference value, we used a single sample t test; when comparisons were performed between two groups, significance was evaluated by a Student’s t test; and when more than two groups were compared, ANOVA was used followed by the Dunnett test using GraphPad Prism software. Results were considered significant when P < 0.05. For the proteomic analysis, data were processed using MaxQuant software (version 1.6.2.3). Searches were performed against a target-decoy database (Uniprot [mouse]; www.uniprot.org; October 28, 2018). Searches were conducted using a 20-ppm precursor mass tolerance and a 0.04-Da product mass tolerance. A maximum of two missed tryptic cleavages was allowed. The fixed modifications specified were carbamidomethylations of cysteine residues. The variable modifications specified were oxidation of methionine and acetylation of the N-terminus. Within MaxQuant, peptides were filtered to a 1% unique peptide false discovery rate (FDR). Characterized proteins were grouped on the basis of the rules of parsimony and filtered to a 1% FDR. Label-free quantification was performed within MaxQuant using MaxLFQ. Missing values were imputed using the Perseus tool available with MaxQuant. For the proteomic analysis, quantitative data from each experiment were log2-transformed and mean-normalized across all tissues for each given protein. Significantly changing proteins were identified using a two-sided Student’s t test in Excel.
Results
Inducible Pulmonary Emphysema Leads to Reduced Respiration in Skeletal Muscle
We have previously reported that IL13TG (emphysema) mice develop reduced body and muscle weight, decreased muscle fiber cross-sectional area, and force generation capacity in comparison with IL13WT (WT) mice (16, 17). We also observed a significant reduction of fatigue tolerance of EDL muscles from IL13TG mice compared with those from their IL13WT counterparts as evaluated in the isolated contractility platform (Figure 1A). As fatigue tolerance is directly influenced by oxidative capacity (44), we determined the EDL muscle OCR, which was significantly reduced in IL13TG in comparison with IL13WT animals (Figure 1B). Oxidative capacity is sometimes (44), yet not always (13), correlated with the abundance of type II over type I fibers in COPD. However, we found the number of different fiber types to be unaltered in our IL13TG animals in comparison with their WT counterparts (Figure 1C). Our previous data indicate no significant difference in mitochondrial mass between IL13WT and WT animals (16). To investigate whether the observed respiratory changes were associated with conspicuous qualitative mitochondrial changes, we conducted a systematic EM analysis of four IL13TG animals and four WT counterparts. IL13TG animals’ muscles did not demonstrate altered mitochondrial structure as reflected by vacuolization, inclusion bodies, edema, intermembrane separation, or any other obvious morphologic change (Figures 1D and E6). These data suggested that the reduced respiratory capacity of IL13TG animals’ muscles could be due to processes operating at suborganellar level.
Inducible Pulmonary Emphysema Leads to Reduced Expression of Multiple Proteins Related to Cellular Respiration in Skeletal Muscle
To investigate potential mechanisms leading to the respiratory phenotype of IL13TG animals’ muscles, we analyzed a previously published proteomic analysis of EDL muscles (16). Ontology analysis of that dataset revealed a very significant downregulation of the bioenergetics-rich “ATP binding” term (see complete output dataset in the data supplement and Reference 16). We identified multiple dysregulated proteins that could account for the reduced OCR observed in IL13TG mice (Figure 2A). Among them, SDHC (Figure 2B) was found particularly attractive given the previous description of its reduced activity in the muscles of patients with COPD (24). The mRNA level of SDHC, but not that of the other three subunits, was found to be downregulated in IL13TG animals’ muscles (Figure 2C). Moreover, a muscle SDH enzymatic activity assay demonstrated a significant reduction in IL13TG in comparison with WT mice (Figure 2D). As the enzyme activity can be limited by reduced substrate availability, we determined succinate concentration, which was increased in IL13TG versus WT mice (Figure 2E), suggesting that the reduced SDH activity was not substrate dependent. Moreover, as complex I and II parallelly feed the electron transport chain, we specifically interrogated SDH/complex II contribution to respiration by conducting respirometry assays in the presence of the complex I inhibitor rotenone and the SDH/complex II substrate succinate. These experiments showed a significantly reduced OCR in COPD mice muscles in comparison with WT control animals (Figure 2F), suggesting SDH/complex II functional deficiency in the COPD mice. To define the status of oxygen consumption downstream of SDH/complex II, we repeated the respirometry assays supplying electrons to complex IV using TMPD and ascorbate. In this case, we found increased respiratory capacity at complex IV in the COPD mouse muscles (Figure 2G), suggesting that there is respiratory chain dysregulation downstream of SDH/complex II and that complex IV does not appear to contribute to the reduced OCR in COPD animals’ EDL muscles.
Figure 2.
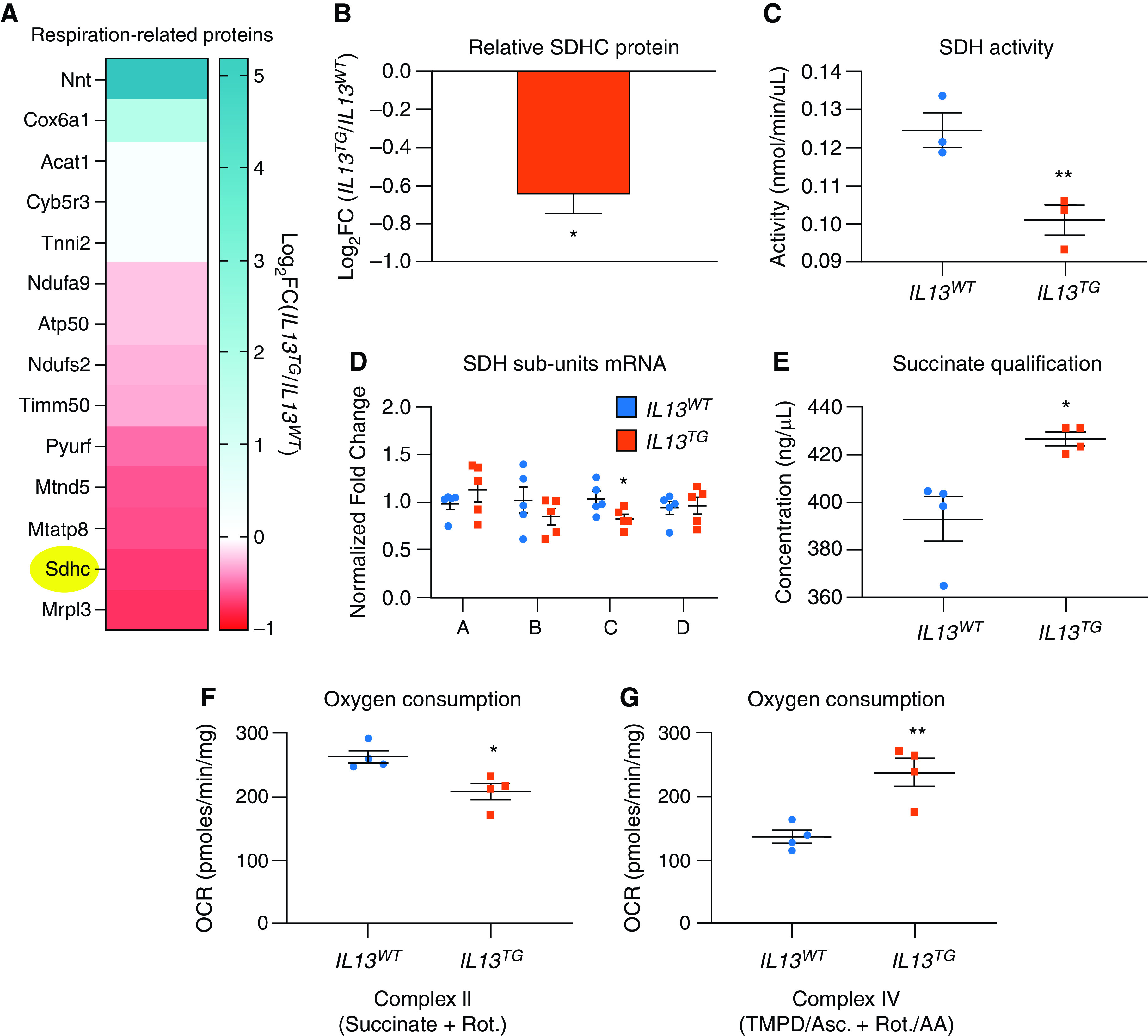
Inducible pulmonary emphysema leads to skeletal muscle reduced expression of multiple proteins related to cellular respiration. (A) Proteomic analysis of EDL muscles identified potentially relevant associations of mitochondrial dysfunction in IL13TG versus WT mice, including SDHC (succinate dehydrogenase subunit C), which is highlighted in yellow; all the described proteins are significantly regulated after correction for multiple comparisons/false discovery rate. The color scale denotes robustness of positive (blue) or negative (red) regulation; see the full dataset in Reference 16. (B) quantification of SDHC downregulation in IL13TG versus WT mice captured in the proteomic analysis. (C) RNA expression of SDH isoforms show that SDHC is the only component of the tetramer SDH/complex II that is significantly downregulated in chronic obstructive pulmonary disease muscles. (D) SDH activity is reduced in tibialis anterior (TA) muscle from IL13TG versus WT mice. (E) succinate is accumulated in TA muscles from IL13TG versus WT mice, indicating that SDH activity is not limited by substrate availability. (F) EDL muscle OCR from IL13TG versus WT mice in the presence of complex I inhibitor rotenone (Rot.) and complex II substrate succinate indicates that complex II–specific oxygen consumption is relatively reduced in IL13TG animals. (G) EDL muscle complex IV–dependent OCR from IL13TG versus WT mice was interrogated with N,N,N’,N’-tetramethyl-p-phenylenediamine (TMPD) and ascorbate the presence of complex I and III inhibitors Rot. and AA. N = 4. *P < 0.05 and **P < 0.01. AA = antimycin A; OCR = oxygen consumption rate.
Downregulation of SDHC Expression Leads to Reduced Enzymatic Activity and OCR in Cultured Muscle Cells
Although SDH activity is required for cellular respiration, the observed reduction in IL13TG mouse muscles is rather modest (∼25%), making it possible that the remaining SDH activity is enough to support oxidative metabolism. To determine whether the observed magnitude of SDHC downregulation is sufficient to cause reduced oxygen consumption, we used cultured nontransformed immortalized skeletal muscle C2C12 cells (45). Experiments were calibrated on the basis of previous data on SDHC half-life (46) to reach a downregulation level in the range of what IL13TG mice demonstrate (Figure 2B). Transfection of these cells with SDHC-specific siRNA led to a significant reduction of that subunit mRNA product but not of the other three (Figure 3A), which was not associated with evidence of cellular toxicity or altered morphology (Figure 3B). SiRNA also led to decreased protein product (Figure 3C), enzymatic activity (Figure 3D), and OCR (Figure 3E). These data supported further experiments to define whether SDHC downregulation in IL13TG mouse muscles contributes to their altered respiratory profile.
Figure 3.
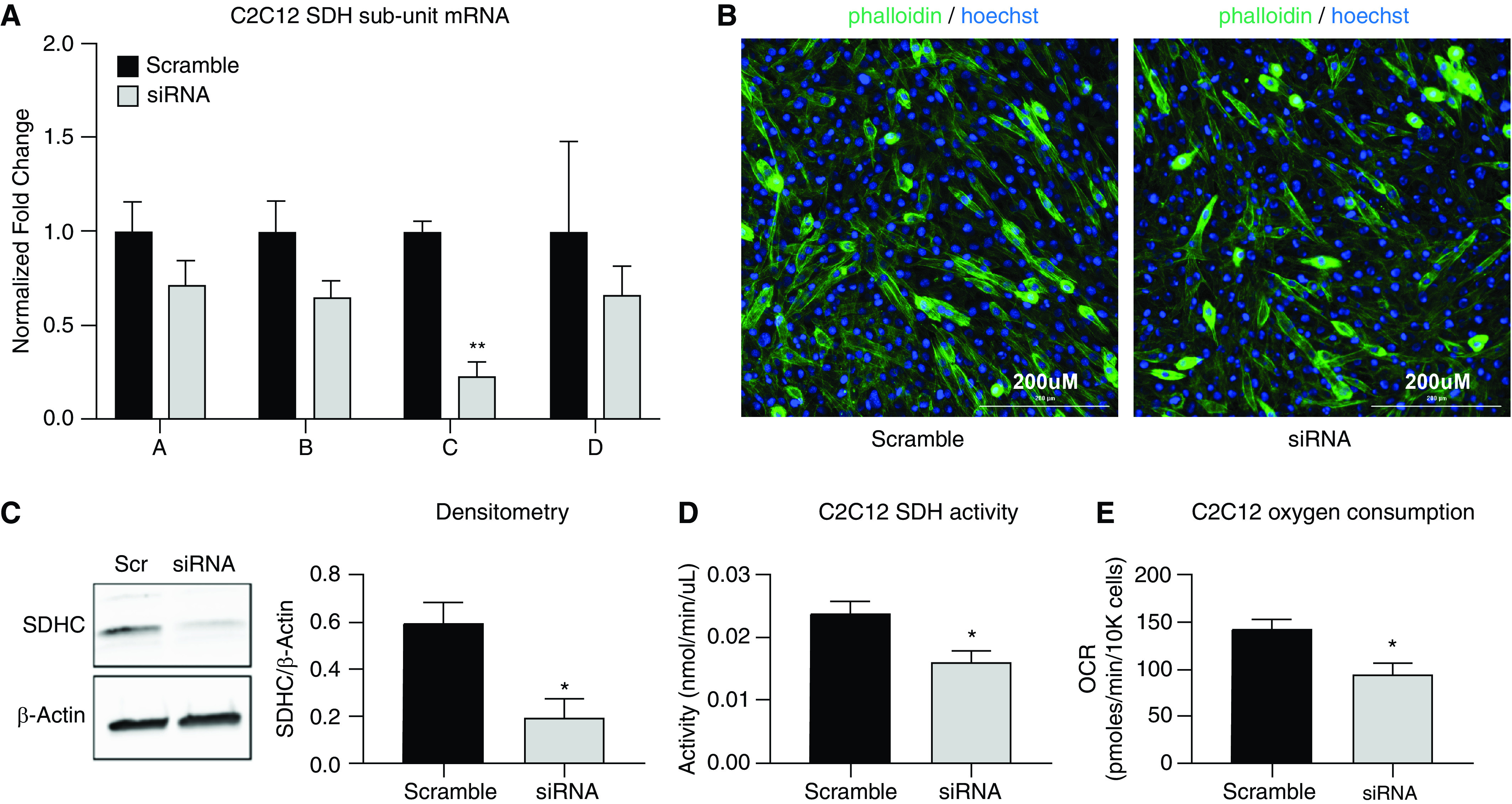
Downregulation of SDHC expression leads to reduced enzymatic activity and OCRs. Cultured muscle (C2C12) cells were transfected with scramble and specific SDHC siRNA. (A) Quantitative PCR demonstrated a significant reduction of SDHC mRNA, whereas the other subunits remained not significantly reduced. (B) Microscopic evaluation of cells stained with phalloidin and Hoechst indicates that SDHC downregulation does not lead to evidence of cellular toxicity. Scale bars, 200 μm. (C) Western blotting using SDHC-specific antibody demonstrates downregulation of the protein product in siRNA-specific transfected cells. (D) SDH enzymatic activity is reduced by ∼35–40% in cells transfected with specific SDHC siRNA. (E) Seahorse platform–determined OCR is reduced in cells previously transfected with specific SDHC siRNA. N = 3. *P < 0.05 and **P < 0.01.
In Vivo Overexpression of SDHC in IL13TG Mouse Skeletal Muscles Leads to Improved Respiration and Fatigue Tolerance
We reasoned that if SDHC downregulation contributes to reduced muscle oxygen consumption and fatigue tolerance, then SDHC restitution should attenuate these deficits. We then performed in vivo muscle electroporation using SDHC-holding or empty (control) constructs. As electroporation causes local muscle injury and the complete myogenic response occurs in approximately 1 month (47), we reasoned that performing experiments at 2 months after electroporation would minimize the possible confounding effect of inadequate muscle repair. Moreover, postinjury muscle repair is known to cause upregulation of slower-twitch fibers and thus more oxidative phenotype (44, 48, 49), which could potentially skew the baseline results. Thus, we performed both gain-of-function and control experiments on the same experimental animals, using their right and left TA muscles, respectively. The plasmid DNA construct was designed with a polypeptide protein tag (FLAG-tag) sequence bound to, and under the control of, the SDHC promoter element and a separate GFP cassette not controlled by SDHC promoter region (42) (see construct map in Figure E2). We determined electroporation efficiency by expression of GFP (Figure 4Aa), which was found on both TA muscles; and we determined SDHC sequence overexpression by FLAG IB, which only happened in the experimental (right) leg (Figure 4Ab). SDHC mRNA overexpression and SDH activity were also significantly elevated compared with empty vector–transfected contralateral muscles (Figures 4B and 4C). Importantly, the succinate accumulation seen in IL13TG mice was not observed after electroporation compared with empty vector–transfected contralateral TA muscle, also indicating better SDH activity (Figure 4C). We then performed TA muscle plate respirometry (Seahorse) assay and found that OCR values were significantly increased in the SDHC-transfected leg when compared with the contralateral muscle (Figure 4D). Moreover, to evaluate the functional effect of improved respiratory capacity, we determined EDL muscle fatigability on the isolated contractility platform, which showed that the reduced fatigue tolerance of IL13TG mice was abrogated after SDHC overexpression (Figure 4F). Finally, we tested the effects of SDHC construct electroporation in WT animals, and, despite the gene’s large overexpression, it did not demonstrate a significant effect on the SDH activity, succinate accumulation, OCR, or fatigability (Figure E3). These data indicate that SDHC gain of function causes improved skeletal muscle oxygen consumption and fatigability in this animal model of pulmonary emphysema.
Figure 4.
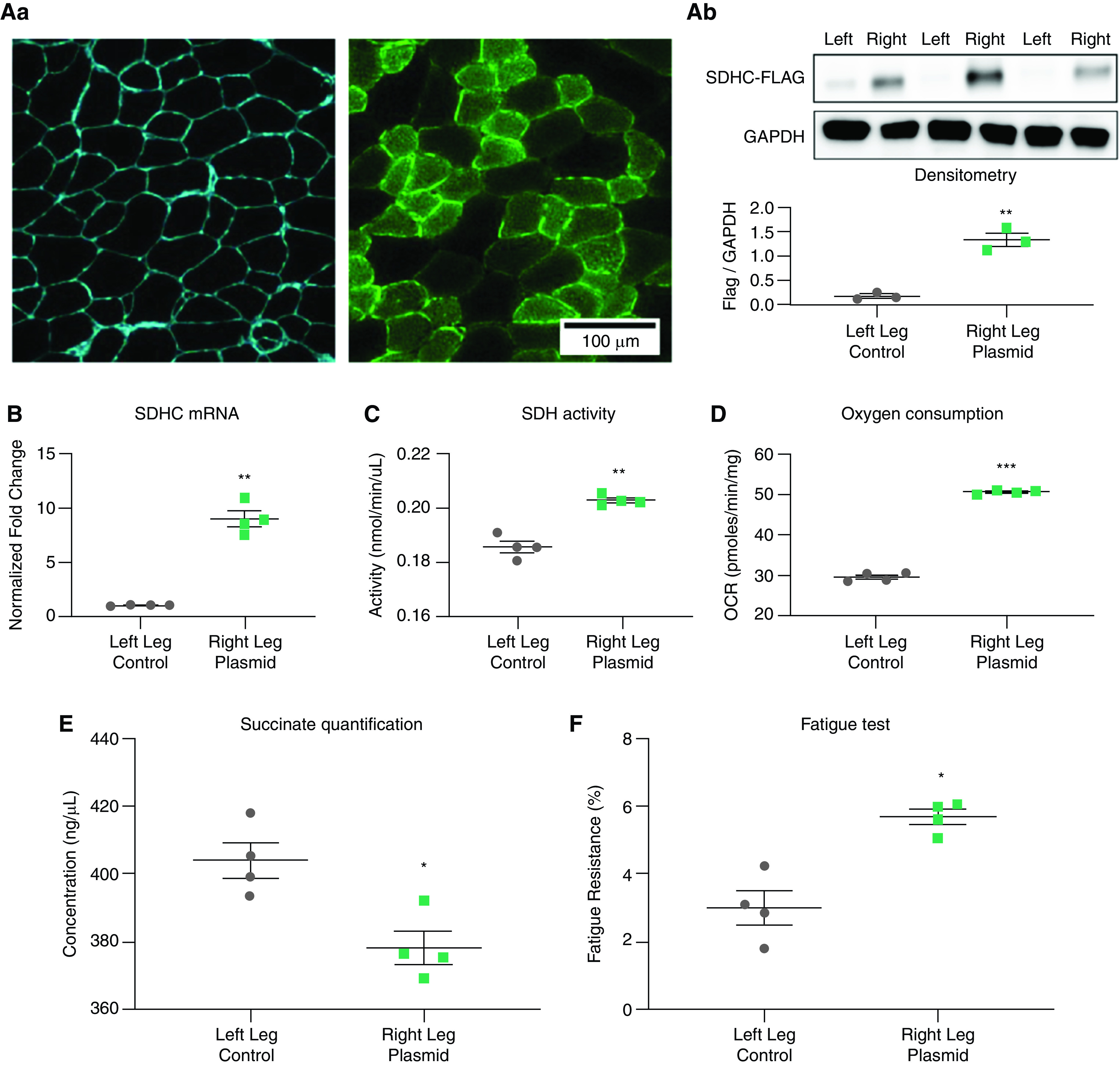
Overexpression of SDHC in IL13TG mice skeletal muscles leads to improved respiration and fatigue tolerance. Muscles from IL13TG (emphysema) and IL13WT (WT) mice were electroporated with a construct holding the SDHC-Flag sequence (right leg) and an SDHC-lacking vector (left leg). Both plasmids hold a GFP sequence as electroporation efficiency reporter. Experiments are performed 2 months after electroporation. (Aa) Microscopic evaluation of muscle section demonstrating preservation of histoarchitecture and positive fluorescence after electroporation. Scale bar, 100 μm. (Ab) Western blotting of muscle samples demonstrating expression of Flag sequence in right leg versus negative expression in left leg. GAPDH was used as a lane loading control. (B) SDHC mRNA normalized to GAPDH mRNA was overexpressed in the right versus left TA muscle after electroporation. (C) SDH activity was significantly elevated in in the right versus left TA muscle after electroporation. (D) OCR was significantly elevated in the right versus left EDL muscle after electroporation. (E) Succinate determination in SDHC-overexpressed leg demonstrates values similar to wild-type mice, suggesting abrogation of substrate accumulation in this setting. (F) fatigue resistance was significantly elevated in the right versus left EDL muscle after electroporation. N = 4. *P < 0.05, **P < 0.01, and ***P < 0.001.
Effect of SDHC Overexpression on Fiber Type Identity in IL13TG Mouse Skeletal Muscles
Fiber type, as defined by the MyHC (myosin heavy chain) isoform expression, correlates with the oxidative capacity and resistance to fatigue (44). These profiles in mice transition in a spectrum that goes from slower to faster twitch and from more to less oxidative capacity and fatigue resistance as follows: type 1 ↔ 2A ↔ 2X ↔ 2B (50, 51). As improved mitochondrial biogenesis and respiratory capacity has been shown to cause fibers to switch from type 2 to type 1 (52), we interrogated whether SDHC overexpression had an effect on IL13TG mouse fiber profile. We found that EDL muscles with overexpressed SDHC-holding constructs had a significant increase in type 2-A and 2X fibers, a reduction of 2B fibers, and no effect on type 1 fibers (Figure 5). These data suggest that the myosin isoform expression, at least in the presented model, could operate downstream of the elevation of oxidative capacity driven by SDHC normalization.
Figure 5.

Effect of SDHC overexpression on fiber type identity in IL13TG mice skeletal muscles. Preelectroporation and postelectroporation EDL muscles from IL13TG mice were immunostained with antibodies to detect MyHC isoforms to characterize fiber types. After electroporation, there was an increase in relatively more oxidative type 2A and 2X fibers and a reduction in relatively glycolytic type 2B fibers without a significant change in the number of type I fibers. Scale bars, 100 μm. N = 4. *P < 0.05.
Discussion
Although COPD locomotor muscles’ reduced endurance, fatigue tolerance, and oxidative capacity have been consistently observed (30, 53), there has been debate on whether intrinsic defects of mitochondrial respiration occur in that setting or not (26, 33, 54–56). In this work, we used a recently described animal model of pulmonary emphysema–induced muscle dysfunction (16, 17, 20) to interrogate the potential mechanisms underpinning abnormal fiber metabolism. The fact that pulmonary emphysema animal muscles demonstrate SDHC downregulation, together with previous observations indicating similarly reduced SDH activity in humans (24, 56, 57), led us to postulate that SDH dysfunction could be partially responsible for a reduced fiber respiration capacity. We confirmed decreased SDH enzymatic activity in the TA muscles, which is associated with succinate accumulation, indicating that substrate availability is not rate-limiting the enzyme work. Functional interrogation of the respiratory chain revealed a decrease in SDH/complex II–dependent respiration in muscles from COPD mice and a downstream increase in complex IV respiratory capacity. The increase in complex IV activity we observed in COPD mouse muscles is not surprising and could represent a compensatory mechanism or reflect a biologically complex interaction, as previously described in multiple models of mitochondrial dysfunction (58–60). Indeed, our analysis of this animal muscle proteome revealed an upregulation of Cox6a1 (Figure 2A), which is a subunit of cytochrome C oxidase/complex IV (61). Whether this finding plays a mechanistic role on the observed increase of complex IV activity will be a matter of future research.
Moreover, siRNA-driven SDHC silencing in cultured muscle cells led to a loss of SDH activity that was of similar magnitude to the one observed in emphysema mouse muscles. This downregulation was associated with a substantial decrease in OCR, supporting the hypothesis that SDHC downregulation in vivo could be partially responsible for the metabolic and functional phenotype observed in emphysema mouse muscles.
Most significantly, in vivo SDH gain of function in emphysema mouse muscles via SDHC overexpression led to improved oxygen consumption and fatigue tolerance and a reduction in succinate accumulation. These data suggest that SDHC downregulation is, at least partially, responsible for the muscle metabolic dysfunction seen in this model of pulmonary emphysema. We believe that a chronic disease model such as IL13TG-induced muscle dysfunction is consistent with the modest, although statistically significant, reduction of SDH activity. Although our data suggest a critical role of SDHC downregulation in the compromised respiratory profile demonstrated by this COPD model, we believe that it is possible that other mitochondrial components or substrate imbalances also contribute to this dysfunction, and these should be addressed in future research. Moreover, the fact that the modest downregulation in SDH activity associates with reduced baseline OCR in EDL muscles suggests that other mechanisms not analyzed here could synergistically undermine cellular respiration in this setting. Future studies using muscle-specific SDHC-knockout mice (46, 62) will facilitate further investigation of this enzyme’s role on baseline respiration.
Although most of the research conducted in the field of COPD-associated muscle dysfunction has so far focused on the conspicuous reduction of muscle mass (1, 4–6, 16, 17, 20, 63–65), to our knowledge, this is the first mechanistic study that disaggregates the investigation of muscle metabolic properties in the context of pulmonary emphysema. Although our study is focused on the fibers’ OCR and its effect on fatigue tolerance, we speculate that other biologically relevant processes could also be involved and should be investigated in the future. Significantly, succinate accumulation could inhibit 2-OGDDs (2-oxoglutrate-dependent dioxygenases) (66), driving DNA and/or chromatin epigenetic changes, including hypermethylation of regulatory elements (67–69), creating a nonpermissive state of critical genes needed to maintain muscle metabolic integrity. For example, it is known that, in SDH-deficient cells, there is an increase in the fraction of methylated DNA (5-mC/5-hmC ratio) and chromatin, which is associated with the silencing of key genes involved in differentiation (66, 70). Succinate accumulation could inhibit PHDs (prolyl-hydroxylases), which are 2-OGDD enzymes that are critical in the regulation of the transcription factor HIF-1 signaling, a master regulator of O2 homeostasis. Succinate has also been involved in post-translational protein modification (succinylation) (71) and in cellular signaling via a specific receptor such as G protein-coupled receptor SUCNR1 (succinate receptor 1) (72), all of which could amplify metabolic dysfunction and other processes. Interestingly, a recent study indicates that exogenous succinate supplementation to cultured muscle cells and mice undermines muscle regeneration after injury (73).
Although SDH catalyzes the TCA cycle conversion of succinate to fumarate, it is also the only TCA enzyme that participates in the electron transport chain as the complex II (74), making it an attractive target to investigate the interaction between bioenergetics and oxidative stress in COPD muscles (75). Importantly, previous evidence indicates that dysfunction of SDH subunits located at the ubiquinol binding site—such as subunit C—leads to an increase in ROS formation, whereas proximal ones—such as SDHA—do not (31). Indeed, complex II–dependent increase of ROS has been demonstrated in the muscles of patients with COPD despite succinate contribution (26). Moreover, in an analysis of this COPD animal model muscle proteome, we recently reported the upregulation of antioxidant ferroproteins, likely indicating ongoing activation of oxidative stress signaling (16, 17) and supporting the role of mitochondria-induced ROS generation in that setting. Future studies focused on the potential ROS-generating role of SDH dysfunction could also be investigated with gain-of-function analyses.
The mechanism leading to SDHC downregulation in this animal model remains to be elucidated. Although the IL13TG mice are hypoxemic (16, 17, 20), we speculate that hypoxia is unlikely to be an upstream regulator of SDH-driven reduced respiration. Indeed, previous data on nonmuscle cells (76) and rat skeletal muscle (77) indicate that hypoxia does not cause significant downregulation of SDH (78). Future studies could investigate the specific interaction between hypoxia and other signals with SDH in the regulation of muscle respiration and fatigability.
We found an increase in oxidative type 2A and 2X and a reduction in the oxidative 2B, in post–SDHC overexpression EDL muscles from IL13TG mice. Although fiber type identity partially depends on the motoneuron innervation profile (44, 79), recent data indicate that it can also be regulated downstream of subcellular processes associated with mitochondrial respiration (52). Moreover, it has been recently reported that myogenesis can contribute to fiber-specific profiles on the basis of the recruitment of twist-2–positive myogenic progenitor cells (80). Interestingly, we have shown that there are no significant baseline differences between fiber types in EDL muscles from IL13TG and IL13WT mice, yet overexpression of SDHC leads to a larger number of types 2A and 2X fibers, suggesting that muscle fiber type profile could be regulated by SDHC independently of its SDH complex enzymatic function. Indeed, previous evidence shows that SDHC has cellular functions that are independent of its canonical catalytic function (21). Future research will be required to elucidate the mechanisms regulating that SDHC-induced fiber change in the context of IL13TG mice.
Although, to our knowledge, this is the first study that mechanistically investigates muscle fiber oxygen consumption in pulmonary emphysema, we realize that this study has limitations. First, the presented model is based on a lung-specific gene overexpression leading to a pulmonary emphysema phenotype. Importantly, IL-13 regulates the pathophysiology of pulmonary emphysema (81) and correlates with disease severity in that setting (82); moreover, IL-13 is not described to cause muscle toxicity independently of the COPD phenotype (1), and, indeed, IL-4/IL-13 signaling has been found to promote muscle trophism, not dysfunction (83, 84), at the expense of higher myogenic capacity. Although this animal model does not demonstrate significant muscle loss up until the pulmonary emphysema develops, at 8 weeks after transgene induction start (16, 17), the possibility of muscle toxicity driven by IL-13 independently of the pulmonary emphysema development cannot be fully excluded, as it has recently been reported on lungs stem cell activity (85).
Second, the present data are based on an animal model that does not necessarily fully reflect the process taking place in patients. For instance, MyHC-2B is not detectable in humans (86), which should be considered when extrapolating the present data to COPD myopathy. Validation of these observations in other animal models of COPD and human samples will be important to address this limitation in the future. Third, the SDH overexpression approach uses a standard in vivo electroporation technique. Although this is a validated method to generate loss- and gain-of-function settings, it cannot prevent biases associated with injury repair and off-target effects of larger-than-physiological protein expressions. Future studies involving a genetic animal model of inducible, muscle-targeted SDHC overexpression could better address this limitation. Moreover, the cellular data we present should be validated with an animal model of muscle-specific SDHC loss of function (46). Fourth, consistent with previous studies (62), we were unable to detect in vivo SDHC overexpression by immunoblot and relied on other surrogates, including FLAG immunoblot, mRNA expression, and SDH activity.
Conclusion
SDHC overexpression abrogates reduced oxygen consumption and fatigue tolerance observed in an animal model of pulmonary emphysema–induced skeletal muscle dysfunction, which is associated with an increase in relatively more oxidative type 2A and X fibers (Figure 6). This mechanism could stimulate research to support SDH function in patients with COPD and skeletal muscle dysfunction (87).
Figure 6.

Cartoon illustrating the proposed mechanism leading to SDHC-driven reduced OCR and fatigue tolerance in this model of pulmonary emphysema.
Acknowledgments
Acknowledgment
The authors thank the National Center for Quantitative Biology of Complex Systems (P41 GM108538) at the University of Wisconsin for graciously providing the high performance liquid chromatography and tandem mass spectrometry (LC-MS/MS) instrument time required to analyze our proteomic samples, and especially Dr. Joshua J. Coon for facilitating sample transport and data acquisition at the National Center for Quantitative Biology of Complex Systems. They also thank Nina Martino and Alejandro P. Adam for their help generating the SDH-holding plasmid used in the in vivo electroporation experiments.
Footnotes
Supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health grants K01-HL130704 (A.J.), 5RO1-HL142807 (D.J.), 5R01-HL049426 (H.A.S.), PO1-HL114501 (J.A.E.), and R01-HL115813 (C.G.L.) and by the Collins Family Foundation Endowment (A.J.).
Author Contributions: J.B., L.A.D., C.E.V., T.C.K., D.V.S., and A.J. designed and performed experiments. J.B. and C.E.V. performed proteomic analyses. L.A.D. performed IB experiments. D.L. performed EM analysis. J.B., D.L., C.G.L., J.A.E., D.J., H.A.S. and A.J. designed the experiments and wrote the current manuscript.
This article has a data supplement, which is accessible from this issue’s table of content online at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0551OC on April 28, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Jaitovich A, Barreiro E. Skeletal Muscle Dysfunction in Chronic Obstructive Pulmonary Disease. What We Know and Can Do for Our Patients. Am J Respir Crit Care Med. 2018;198:175–186. doi: 10.1164/rccm.201710-2140CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vanfleteren LE, Spruit MA, Groenen M, Gaffron S, van Empel VP, Bruijnzeel PL, et al. Clusters of comorbidities based on validated objective measurements and systemic inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;187:728–735. doi: 10.1164/rccm.201209-1665OC. [DOI] [PubMed] [Google Scholar]
- 3. Burtin C, Ter Riet G, Puhan MA, Waschki B, Garcia-Aymerich J, Pinto-Plata V, et al. Handgrip weakness and mortality risk in COPD: a multicentre analysis. Thorax. 2016;71:86–87. doi: 10.1136/thoraxjnl-2015-207451. [DOI] [PubMed] [Google Scholar]
- 4. Marquis K, Debigaré R, Lacasse Y, LeBlanc P, Jobin J, Carrier G, et al. Midthigh muscle cross-sectional area is a better predictor of mortality than body mass index in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166:809–813. doi: 10.1164/rccm.2107031. [DOI] [PubMed] [Google Scholar]
- 5. Shrikrishna D, Patel M, Tanner RJ, Seymour JM, Connolly BA, Puthucheary ZA, et al. Quadriceps wasting and physical inactivity in patients with COPD. Eur Respir J. 2012;40:1115–1122. doi: 10.1183/09031936.00170111. [DOI] [PubMed] [Google Scholar]
- 6. Swallow EB, Reyes D, Hopkinson NS, Man WD, Porcher R, Cetti EJ, et al. Quadriceps strength predicts mortality in patients with moderate to severe chronic obstructive pulmonary disease. Thorax. 2007;62:115–120. doi: 10.1136/thx.2006.062026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vogiatzis I, Terzis G, Stratakos G, Cherouveim E, Athanasopoulos D, Spetsioti S, et al. Effect of pulmonary rehabilitation on peripheral muscle fiber remodeling in patients with COPD in GOLD stages II to IV. Chest. 2011;140:744–752. doi: 10.1378/chest.10-3058. [DOI] [PubMed] [Google Scholar]
- 8. Casaburi R, ZuWallack R. Pulmonary rehabilitation for management of chronic obstructive pulmonary disease. N Engl J Med. 2009;360:1329–1335. doi: 10.1056/NEJMct0804632. [DOI] [PubMed] [Google Scholar]
- 9. Richardson RS, Leek BT, Gavin TP, Haseler LJ, Mudaliar SR, Henry R, et al. Reduced mechanical efficiency in chronic obstructive pulmonary disease but normal peak VO2 with small muscle mass exercise. Am J Respir Crit Care Med. 2004;169:89–96. doi: 10.1164/rccm.200305-627OC. [DOI] [PubMed] [Google Scholar]
- 10. Sala E, Roca J, Marrades RM, Alonso J, Gonzalez De Suso JM, Moreno A, et al. Effects of endurance training on skeletal muscle bioenergetics in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;159:1726–1734. doi: 10.1164/ajrccm.159.6.9804136. [DOI] [PubMed] [Google Scholar]
- 11. van den Borst B, Slot IGM, Hellwig VACV, Vosse BAH, Kelders MCJM, Barreiro E, et al. Loss of quadriceps muscle oxidative phenotype and decreased endurance in patients with mild-to-moderate COPD. J Appl Physiol (1985) 2013;114:1319–1328. doi: 10.1152/japplphysiol.00508.2012. [DOI] [PubMed] [Google Scholar]
- 12. Patel MS, Natanek SA, Stratakos G, Pascual S, Martínez-Llorens J, Disano L, et al. Vastus lateralis fiber shift is an independent predictor of mortality in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;190:350–352. doi: 10.1164/rccm.201404-0713LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maltais F, LeBlanc P, Whittom F, Simard C, Marquis K, Bélanger M, et al. Oxidative enzyme activities of the vastus lateralis muscle and the functional status in patients with COPD. Thorax. 2000;55:848–853. doi: 10.1136/thorax.55.10.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gosker HR, Zeegers MP, Wouters EF, Schols AM. Muscle fibre type shifting in the vastus lateralis of patients with COPD is associated with disease severity: a systematic review and meta-analysis. Thorax. 2007;62:944–949. doi: 10.1136/thx.2007.078980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kwan HY, Maddocks M, Nolan CM, Jones SE, Patel S, Barker RE, et al. The prognostic significance of weight loss in chronic obstructive pulmonary disease-related cachexia: a prospective cohort study. J Cachexia Sarcopenia Muscle. 2019;10:1330–1338. doi: 10.1002/jcsm.12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Balnis J, Korponay TC, Vincent CE, Singer DV, Adam AP, Lacomis D, et al. IL-13-driven pulmonary emphysema leads to skeletal muscle dysfunction attenuated by endurance exercise. J Appl Physiol (1985) 2020;128:134–148. doi: 10.1152/japplphysiol.00627.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Balnis J, Vincent CE, Jones AJ, Drake LA, Coon JJ, Lee CG, et al. Established Biomarkers of Chronic Obstructive Pulmonary Disease Reflect Skeletal Muscle Integrity’s Response to Exercise in an Animal Model of Pulmonary Emphysema. Am J Respir Cell Mol Biol. 2020;63:266–269. doi: 10.1165/rcmb.2019-0439LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ, Jr, et al. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest. 2000;106:1081–1093. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Campbell EJ. Animal models of emphysema: the next generations. J Clin Invest. 2000;106:1445–1446. doi: 10.1172/JCI11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Balnis J, Lee CG, Elias JA, Jaitovich A. Hypercapnia-Driven Skeletal Muscle Dysfunction in an Animal Model of Pulmonary Emphysema Suggests a Complex Phenotype. Front Physiol. 2020;11:600290. doi: 10.3389/fphys.2020.600290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Satoh N, Yokoyama C, Itamura N, Miyajima-Nakano Y, Hisatomi H. Alternative splicing isoform in succinate dehydrogenase complex, subunit C causes downregulation of succinate-coenzyme Q oxidoreductase activity in mitochondria. Oncol Lett. 2015;9:330–334. doi: 10.3892/ol.2014.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kugelberg E, Edström L. Differential histochemical effects of muscle contractions on phosphorylase and glycogen in various types of fibres: relation to fatigue. J Neurol Neurosurg Psychiatry. 1968;31:415–423. doi: 10.1136/jnnp.31.5.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reichmann H, Pette D. A comparative microphotometric study of succinate dehydrogenase activity levels in type I, IIA and IIB fibres of mammalian and human muscles. Histochemistry. 1982;74:27–41. doi: 10.1007/BF00495049. [DOI] [PubMed] [Google Scholar]
- 24. Gosker HR, van Mameren H, van Dijk PJ, Engelen MP, van der Vusse GJ, Wouters EF, et al. Skeletal muscle fibre-type shifting and metabolic profile in patients with chronic obstructive pulmonary disease. Eur Respir J. 2002;19:617–625. doi: 10.1183/09031936.02.00762001. [DOI] [PubMed] [Google Scholar]
- 25. Puente-Maestu L, Pérez-Parra J, Godoy R, Moreno N, Tejedor A, González-Aragoneses F, et al. Abnormal mitochondrial function in locomotor and respiratory muscles of COPD patients. Eur Respir J. 2009;33:1045–1052. doi: 10.1183/09031936.00112408. [DOI] [PubMed] [Google Scholar]
- 26. Picard M, Godin R, Sinnreich M, Baril J, Bourbeau J, Perrault H, et al. The mitochondrial phenotype of peripheral muscle in chronic obstructive pulmonary disease: disuse or dysfunction? Am J Respir Crit Care Med. 2008;178:1040–1047. doi: 10.1164/rccm.200807-1005OC. [DOI] [PubMed] [Google Scholar]
- 27. Chandel N, Budinger GR, Kemp RA, Schumacker PT. Inhibition of cytochrome-c oxidase activity during prolonged hypoxia. Am J Physiol. 1995;268:L918–L925. doi: 10.1152/ajplung.1995.268.6.L918. [DOI] [PubMed] [Google Scholar]
- 28. Berchtold MW, Brinkmeier H, Müntener M. Calcium ion in skeletal muscle: its crucial role for muscle function, plasticity, and disease. Physiol Rev. 2000;80:1215–1265. doi: 10.1152/physrev.2000.80.3.1215. [DOI] [PubMed] [Google Scholar]
- 29. Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11:102. doi: 10.1038/s41467-019-13668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Casaburi R, Patessio A, Ioli F, Zanaboni S, Donner CF, Wasserman K. Reductions in exercise lactic acidosis and ventilation as a result of exercise training in patients with obstructive lung disease. Am Rev Respir Dis. 1991;143:9–18. doi: 10.1164/ajrccm/143.1.9. [DOI] [PubMed] [Google Scholar]
- 31. Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008;28:718–731. doi: 10.1128/MCB.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barreiro E, Schols AM, Polkey MI, Galdiz JB, Gosker HR, Swallow EB, et al. ENIGMA in COPD project. Cytokine profile in quadriceps muscles of patients with severe COPD. Thorax. 2008;63:100–107. doi: 10.1136/thx.2007.078030. [DOI] [PubMed] [Google Scholar]
- 33. Puig-Vilanova E, Rodriguez DA, Lloreta J, Ausin P, Pascual-Guardia S, Broquetas J, et al. Oxidative stress, redox signaling pathways, and autophagy in cachectic muscles of male patients with advanced COPD and lung cancer. Free Radic Biol Med. 2015;79:91–108. doi: 10.1016/j.freeradbiomed.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 34.Balnis J, Drake LA, Vincent CE, Korponay TC, Singer DV, Lacomis D, et al. Succinate Dehydrogenase (SDH)-subunit C regulates muscle oxygen consumption and fatigability in an animal model of pulmonary emphysema [preprint] bioRxiv 2021. [accessed 2021 Jan 22]. Available from: https://www.biorxiv.org/content/10.1101/2021.01.22.427763v1. [DOI] [PMC free article] [PubMed]
- 35. Shintaku J, Guttridge DC. Analysis of Aerobic Respiration in Intact Skeletal Muscle Tissue by Microplate-Based Respirometry. Methods Mol Biol. 2016;1460:337–343. doi: 10.1007/978-1-4939-3810-0_23. [DOI] [PubMed] [Google Scholar]
- 36. Kay L, Nicolay K, Wieringa B, Saks V, Wallimann T. Direct evidence for the control of mitochondrial respiration by mitochondrial creatine kinase in oxidative muscle cells in situ. J Biol Chem. 2000;275:6937–6944. doi: 10.1074/jbc.275.10.6937. [DOI] [PubMed] [Google Scholar]
- 37. Milner DJ, Mavroidis M, Weisleder N, Capetanaki Y. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J Cell Biol. 2000;150:1283–1298. doi: 10.1083/jcb.150.6.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Piper HM, Sezer O, Schleyer M, Schwartz P, Hütter JF, Spieckermann PG. Development of ischemia-induced damage in defined mitochondrial subpopulations. J Mol Cell Cardiol. 1985;17:885–896. doi: 10.1016/s0022-2828(85)80102-4. [DOI] [PubMed] [Google Scholar]
- 39. Salabei JK, Gibb AA, Hill BG. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat Protoc. 2014;9:421–438. doi: 10.1038/nprot.2014.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boutagy NE, Rogers GW, Pyne ES, Ali MM, Hulver MW, Frisard MI. Using Isolated Mitochondria from Minimal Quantities of Mouse Skeletal Muscle for High throughput Microplate Respiratory Measurements. J Vis Exp. 2015:e53216. doi: 10.3791/53216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. DiFranco M, Quinonez M, Capote J, Vergara J. DNA transfection of mammalian skeletal muscles using in vivo electroporation. J Vis Exp. 2009;(32):1520. doi: 10.3791/1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sokołowska E, Błachnio-Zabielska AU. A Critical Review of Electroporation as A Plasmid Delivery System in Mouse Skeletal Muscle. Int J Mol Sci. 2019;20:2776. doi: 10.3390/ijms20112776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol Rev. 2013;93:23–67. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schiaffino S, Reggiani C. Fiber types in mammalian skeletal muscles. Physiol Rev. 2011;91:1447–1531. doi: 10.1152/physrev.00031.2010. [DOI] [PubMed] [Google Scholar]
- 45. Silberstein L, Webster SG, Travis M, Blau HM. Developmental progression of myosin gene expression in cultured muscle cells. Cell. 1986;46:1075–1081. doi: 10.1016/0092-8674(86)90707-5. [DOI] [PubMed] [Google Scholar]
- 46. Smestad J, Hamidi O, Wang L, Holte MN, Khazal FA, Erber L, et al. Characterization and metabolic synthetic lethal testing in a new model of SDH-loss familial pheochromocytoma and paraganglioma. Oncotarget. 2017;9:6109–6127. doi: 10.18632/oncotarget.23639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu N, Nelson BR, Bezprozvannaya S, Shelton JM, Richardson JA, Bassel-Duby R, et al. Requirement of MEF2A, C, and D for skeletal muscle regeneration. Proc Natl Acad Sci USA. 2014;111:4109–4114. doi: 10.1073/pnas.1401732111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jerkovic R, Argentini C, Serrano-Sanchez A, Cordonnier C, Schiaffino S. Early myosin switching induced by nerve activity in regenerating slow skeletal muscle. Cell Struct Funct. 1997;22:147–153. doi: 10.1247/csf.22.147. [DOI] [PubMed] [Google Scholar]
- 49. Jerkovic R, Vitadello M, Kelly R, Buckingham M, Schiaffino S. Fibre type-specific and nerve-dependent regulation of myosin light chain 1 slow promoter in regenerating muscle. J Muscle Res Cell Motil. 1997;18:369–373. doi: 10.1023/a:1018630311208. [DOI] [PubMed] [Google Scholar]
- 50. Pette D, Staron RS. Cellular and molecular diversities of mammalian skeletal muscle fibers. Rev Physiol Biochem Pharmacol. 1990;116:1–76. doi: 10.1007/3540528806_3. [DOI] [PubMed] [Google Scholar]
- 51. Bottinelli R, Betto R, Schiaffino S, Reggiani C. Maximum shortening velocity and coexistence of myosin heavy chain isoforms in single skinned fast fibres of rat skeletal muscle. J Muscle Res Cell Motil. 1994;15:413–419. doi: 10.1007/BF00122115. [DOI] [PubMed] [Google Scholar]
- 52. Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 53. Allaire J, Maltais F, Doyon JF, Noël M, LeBlanc P, Carrier G, et al. Peripheral muscle endurance and the oxidative profile of the quadriceps in patients with COPD. Thorax. 2004;59:673–678. doi: 10.1136/thx.2003.020636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Couillard A, Prefaut C. From muscle disuse to myopathy in COPD: potential contribution of oxidative stress. Eur Respir J. 2005;26:703–719. doi: 10.1183/09031936.05.00139904. [DOI] [PubMed] [Google Scholar]
- 55. Willis-Owen SAG, Thompson A, Kemp PR, Polkey MI, Cookson WOCM, Moffatt MF, et al. COPD is accompanied by co-ordinated transcriptional perturbation in the quadriceps affecting the mitochondria and extracellular matrix. Sci Rep. 2018;8:12165. doi: 10.1038/s41598-018-29789-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Konokhova Y, Spendiff S, Jagoe RT, Aare S, Kapchinsky S, MacMillan NJ, et al. Failed upregulation of TFAM protein and mitochondrial DNA in oxidatively deficient fibers of chronic obstructive pulmonary disease locomotor muscle. Skelet Muscle. 2016;6:10. doi: 10.1186/s13395-016-0083-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jakobsson P, Jorfeldt L, Henriksson J. Metabolic enzyme activity in the quadriceps femoris muscle in patients with severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1995;151:374–377. doi: 10.1164/ajrccm.151.2.7842194. [DOI] [PubMed] [Google Scholar]
- 58. Suthammarak W, Yang YY, Morgan PG, Sedensky MM. Complex I function is defective in complex IV-deficient Caenorhabditis elegans. J Biol Chem. 2009;284:6425–6435. doi: 10.1074/jbc.M805733200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Havlíčková Karbanová V, Cížková Vrbacká A, Hejzlarová K, Nůsková H, Stránecký V, Potocká A, et al. Compensatory upregulation of respiratory chain complexes III and IV in isolated deficiency of ATP synthase due to TMEM70 mutation. Biochim Biophys Acta. 2012;1817:1037–1043. doi: 10.1016/j.bbabio.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 60. Perez-Gomez R, Magnin V, Mihajlovic Z, Slaninova V, Krejci A. Downregulation of respiratory complex I mediates major signalling changes triggered by TOR activation. Sci Rep. 2020;10:4401. doi: 10.1038/s41598-020-61244-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Taanman JW, Hall RE, Tang C, Marusich MF, Kennaway NG, Capaldi RA. Tissue distribution of cytochrome c oxidase isoforms in mammals. Characterization with monoclonal and polyclonal antibodies. Biochim Biophys Acta. 1993;1225:95–100. doi: 10.1016/0925-4439(93)90128-n. [DOI] [PubMed] [Google Scholar]
- 62. Al Khazal F, Holte MN, Bolon B, White TA, LeBrasseur N, Maher LJ., III A conditional mouse model of complex II deficiency manifesting as Leigh-like syndrome. FASEB J. 2019;33:13189–13201. doi: 10.1096/fj.201802655RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Balnis J, Korponay TC, Jaitovich A. AMP-Activated Protein Kinase (AMPK) at the Crossroads Between CO2 Retention and Skeletal Muscle Dysfunction in Chronic Obstructive Pulmonary Disease (COPD) Int J Mol Sci. 2020;21:955. doi: 10.3390/ijms21030955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jaitovich A, Angulo M, Lecuona E, Dada LA, Welch LC, Cheng Y, et al. High CO2 levels cause skeletal muscle atrophy via AMP-activated kinase (AMPK), FoxO3a protein, and muscle-specific Ring finger protein 1 (MuRF1) J Biol Chem. 2015;290:9183–9194. doi: 10.1074/jbc.M114.625715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Korponay TC, Balnis J, Vincent CE, Singer DV, Chopra A, Adam AP, et al. High CO2 Downregulates Skeletal Muscle Protein Anabolism via AMP-activated Protein Kinase α2-mediated Depressed Ribosomal Biogenesis. Am J Respir Cell Mol Biol. 2020;62:74–86. doi: 10.1165/rcmb.2019-0061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Letouzé E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 67. Ansó E, Weinberg SE, Diebold LP, Thompson BJ, Malinge S, Schumacker PT, et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat Cell Biol. 2017;19:614–625. doi: 10.1038/ncb3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Singer BD. A Practical Guide to the Measurement and Analysis of DNA Methylation. Am J Respir Cell Mol Biol. 2019;61:417–428. doi: 10.1165/rcmb.2019-0150TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139–1142. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell. 2013;50:919–930. doi: 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lu YT, Li LZ, Yang YL, Yin X, Liu Q, Zhang L, et al. Succinate induces aberrant mitochondrial fission in cardiomyocytes through GPR91 signaling. Cell Death Dis. 2018;9:672. doi: 10.1038/s41419-018-0708-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Arneson-Wissink PC, Hogan KA, Ducharme AM, Samani A, Jatoi A, Doles JD. The wasting-associated metabolite succinate disrupts myogenesis and impairs skeletal muscle regeneration. JCSM Rapid Commun. 2020;3:56–69. doi: 10.1002/rco2.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chandel NS. Evolution of Mitochondria as Signaling Organelles. Cell Metab. 2015;22:204–206. doi: 10.1016/j.cmet.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 75. DeBerardinis RJ, Chandel NS. We need to talk about the Warburg effect. Nat Metab. 2020;2:127–129. doi: 10.1038/s42255-020-0172-2. [DOI] [PubMed] [Google Scholar]
- 76. McGee SL, Walder KR. Exercise and the Skeletal Muscle Epigenome. Cold Spring Harb Perspect Med. 2017;7:a029876. doi: 10.1101/cshperspect.a029876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Punkt K, Unger A, Welt K, Hilbig H, Schaffranietz L. Hypoxia-dependent changes of enzyme activities in different fibre types of rat soleus and extensor digitorum longus muscles. A cytophotometrical study. Acta Histochem. 1996;98:255–269. doi: 10.1016/S0065-1281(96)80017-1. [DOI] [PubMed] [Google Scholar]
- 78. Powell CS, Jackson RM. Mitochondrial complex I, aconitase, and succinate dehydrogenase during hypoxia-reoxygenation: modulation of enzyme activities by MnSOD. Am J Physiol Lung Cell Mol Physiol. 2003;285:L189–L198. doi: 10.1152/ajplung.00253.2002. [DOI] [PubMed] [Google Scholar]
- 79. Ciciliot S, Rossi AC, Dyar KA, Blaauw B, Schiaffino S. Muscle type and fiber type specificity in muscle wasting. Int J Biochem Cell Biol. 2013;45:2191–2199. doi: 10.1016/j.biocel.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 80. Liu N, Garry GA, Li S, Bezprozvannaya S, Sanchez-Ortiz E, Chen B, et al. A Twist2-dependent progenitor cell contributes to adult skeletal muscle. Nat Cell Biol. 2017;19:202–213. doi: 10.1038/ncb3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Doyle AD, Mukherjee M, LeSuer WE, Bittner TB, Pasha SM, Frere JJ, et al. Eosinophil-derived IL-13 promotes emphysema. Eur Respir J. 2019;53:1801291. doi: 10.1183/13993003.01291-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lee JS, Rosengart MR, Kondragunta V, Zhang Y, McMurray J, Branch RA, et al. Inverse association of plasma IL-13 and inflammatory chemokines with lung function impairment in stable COPD: a cross-sectional cohort study. Respir Res. 2007;8:64. doi: 10.1186/1465-9921-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell. 2013;153:376–388. doi: 10.1016/j.cell.2013.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Leavy O. Type 2 immunity: regenerating muscles the type 2 way. Nat Rev Immunol. 2013;13:395. doi: 10.1038/nri3460. [DOI] [PubMed] [Google Scholar]
- 85. Glisinski KM, Schlobohm AJ, Paramore SV, Birukova A, Moseley MA, Foster MW, et al. Interleukin-13 disrupts type 2 pneumocyte stem cell activity. JCI Insight. 2020;5:e131232. doi: 10.1172/jci.insight.131232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Smerdu V, Karsch-Mizrachi I, Campione M, Leinwand L, Schiaffino S. Type IIx myosin heavy chain transcripts are expressed in type IIb fibers of human skeletal muscle. Am J Physiol. 1994;267:C1723–C1728. doi: 10.1152/ajpcell.1994.267.6.C1723. [DOI] [PubMed] [Google Scholar]
- 87. Fan F, Sam R, Ryan E, Alvarado K, Villa-Cuesta E. Rapamycin as a potential treatment for succinate dehydrogenase deficiency. Heliyon. 2019;5:e01217. doi: 10.1016/j.heliyon.2019.e01217. [DOI] [PMC free article] [PubMed] [Google Scholar]