

Abstract
Right ventricular (RV) function is the predominant determinant of survival in patients with pulmonary arterial hypertension (PAH). In preclinical models, pharmacological activation of BMP (bone morphogenetic protein) signaling with FK506 (tacrolimus) improved RV function by decreasing RV afterload. FK506 therapy further stabilized three patients with end-stage PAH. Whether FK506 has direct effects on the pressure-overloaded right ventricle is yet unknown. We hypothesized that increasing cardiac BMP signaling with FK506 improves RV structure and function in a model of fixed RV afterload after pulmonary artery banding (PAB). Direct cardiac effects of FK506 on the microvasculature and RV fibrosis were studied after surgical PAB in wild-type and heterozygous Bmpr2 mutant mice. RV function and strain were assessed longitudinally via cardiac magnetic resonance imaging during continuous FK506 infusion. Genetic lineage tracing of endothelial cells (ECs) was performed to assess the contribution of ECs to fibrosis. Molecular mechanistic studies were performed in human cardiac fibroblasts and ECs. In mice, low BMP signaling in the right ventricle exaggerated PAB-induced RV fibrosis. FK506 therapy restored cardiac BMP signaling, reduced RV fibrosis in a BMP-dependent manner independent from its immunosuppressive effect, preserved RV capillarization, and improved RV function and strain over the time course of disease. Endothelial mesenchymal transition was a rare event and did not significantly contribute to cardiac fibrosis after PAB. Mechanistically, FK506 required ALK1 in human cardiac fibroblasts as a BMPR2 co-receptor to reduce TGFβ1-induced proliferation and collagen production. Our study demonstrates that increasing cardiac BMP signaling with FK506 improves RV structure and function independent from its previously described beneficial effects on pulmonary vascular remodeling.
Keywords: right ventricle, pulmonary hypertension, BMPR2, FK506, cardiac fibrosis
Clinical Relevance
This is the first study to report a direct beneficial effect of FK506 on right ventricular structure and function. Pharmacological activation of BMP (bone morphogenetic protein) signaling with FK506 targets the underlying pathological remodeling processes in the pulmonary vasculature of patients with pulmonary arterial hypertension and, at the same time, directly improves right ventricular structure and function. This beneficial effect of FK506 on the right ventricle could be relevant not only for patients with pulmonary arterial hypertension but also in other diseases in which the right ventricle at risk for failure, such as congenital heart disease or group 2–4 pulmonary hypertension.
Pulmonary arterial hypertension (PAH) is a devastating disease characterized by a progressive occlusive pulmonary vasculopathy of small pulmonary arterioles that causes a persistent increase in right ventricular (RV) afterload, RV failure, and ultimately death in most patients (1, 2). A small subgroup of patients with PAH, however, is able to compensate for the persistent increase in afterload with RV hypertrophy and preserved RV function without RV failure (3). Why these patients are capable of adapting to high increases in afterload is unknown, but it suggests that there are genetic mechanisms and signaling pathways that can be pharmacologically targeted to intrinsically strengthen the RV and prevent RV failure in PAH.
Patients with heritable PAH due to germline mutations in the BMPR2 (BMP [bone morphogenetic protein] receptor type 2) gene present with worse RV function than patients with idiopathic PAH despite comparable RV afterload (4), pointing toward intrinsic RV abnormalities caused by reduced BMP signaling. Preclinically, abnormal calcium handling (5) and lipid metabolism in cardiomyocytes (6) were reported in BMPR2 mutants, and increasing BMP signaling with recombinant BMP6 directly reduced cardiac fibrosis in the left ventricle through the inhibition of fibroblast activation and endothelial cell conversion toward a mesenchymal, profibrotic fate, also called endothelial-to-mesenchymal transition (EndMT) (7). We have previously shown that increasing BMP signaling with low-dose FK506 reversed pulmonary vascular remodeling and improved RV function in two complementary PAH animal models (8), stabilized three patients with end-stage PAH (9), and was well tolerated and safe in a larger cohort of patients with stable PAH (10). However, whether FK506 has a direct effect on the RV myocardium remains unknown because all observed improvements in RV function are—at least in part—a direct consequence of the reduced pulmonary vascular resistance in these models.
Identifying ways to improve RV adaptation to an increased afterload would not only be important in PAH but also in other diseases in which the right ventricle is uniquely at risk, such as in congenital heart diseases with right-sided obstructive lesions (tetralogy of Fallot, pulmonary atresia, and pulmonary stenosis), in patients with systemic right ventricles (transposition of great arteries), and in patients with group 2–4 pulmonary hypertension (11–14).
In the current study, we sought to investigate the effects of FK506 on RV structure and function in a mouse model of fixed RV afterload through surgical pulmonary artery banding (PAB) (15), mimicking pulmonary stenosis, in which direct cardiac effects can be studied without interfering afterload alterations in the pulmonary vasculature. We hypothesized that increasing BMP signaling with low-dose FK506 at the time when disease is established improves RV function through the inhibition of pathological remodeling processes directly in the RV myocardium. The data herein are the first to demonstrate a direct cardioprotective effect of FK506 through BMP pathway activation independent of immunosuppression, and they support FK506 as a potentially beneficial therapeutic strategy for RV adaptation in diseases characterized by increased RV afterload, such as pulmonary hypertension and congenital heart disease with right-sided obstructive lesions.
Methods
Additional details on the Methods for making the measurements in this study are provided in a data supplement.
Study Approval
All animal experiments were performed in accordance with the Guide for Care and Use of Laboratory Animals published by the National Research Council. The experimental procedures were approved by local authorities (Administrative Panel on Laboratory Animal Care at Stanford, protocols #27626 and #29539). All experiments were conducted in a blinded fashion whenever possible (i.e., cardiac magnetic resonance [CMR] imaging, hemodynamic measurements, or histological analysis), and interobserver variability was assessed for CMR image analysis.
PAB and Therapeutic Intervention
Male Bmpr2 heterozygous mice (Bmpr2+/–) (16), littermate control mice (Bmpr2+/+), and C57Bl6/J mice (10–14 wk of age) underwent either sham surgery or PAB around a 24-G needle as described previously (15). Only male mice were used to reduce experimental variability after PAB. All mice preemptively received 0.05–0.1 mg/mg body weight buprenorphine subcutaneously before continuous isoflurane (2–3%) anesthesia during surgery. Only animals with moderate pulmonary artery stenosis (peak pressure gradient across the pulmonary band: 20–35 mm Hg at 1 week after surgery by echo Doppler) were included in the study protocol (15). Animals were randomly assigned to either low-dose FK506 (0.05 mg/kg/d), high-dose FK506 (1 mg/kg/d), BMPR2 inhibitor LDN-193189 (2.5 mg/kg/d), or cyclosporine (25 mg/kg/d)—as well as the respective combinations—or vehicle, administered subcutaneously through an osmotic minipump (Alzet) starting either preventively at the day of surgery or therapeutically 1 week after surgery for an additional 6–7 weeks.
CMR Imaging and Intracardiac Catheterization
Cardiac imaging was performed on a 7T magnetic resonance imaging system (Bruker) equipped with a BioSpec 70/30 Ultra Shielded and Refrigerated (USR) Superconducting Magnet System in supine position under continuous isoflurane anesthesia (1–2%). Electrocardiogram and end-expiratory gating were performed during image acquisition, and ventricular volumetric and functional analyses were performed offline using commercially available software. Ventricular end-diastolic and end-systolic contours were manually traced in the axial view for each slice. RV ejection fraction (RVEF) was determined using the modified Simpson’s rule.
Closed-chest intracardiac catheterization was performed in a blinded manner as described elsewhere (17). Terminally, all mice were killed by exsanguination, and the right ventricles were dissected for tissue weight measurements.
Graded Treadmill Exercise
Exercise testing was performed in a blinded manner using a rodent treadmill (Columbus Instruments) and a ramped exercise protocol (18) before intracardiac catheterization and tissue harvest.
Histomorphology
Formalin-fixed, dehydrated, paraffin-embedded, and ultimately sectioned (3-μm) cardiac tissues were stained with either Masson’s trichrome stain, wheat germ agglutinin, or isolectin B4 as described previously (18). RV fibrosis was quantified by ImageJ intensity image analysis software. In cross-sections, cardiomyocyte surface areas were quantified by distance measurements (ImageJ image analysis software), and isolectin B4–positive vessels were assessed as the ratio of counted vessels to cardiomyocyte number. Vimentin expression was determined by either immunohistochemistry or immunofluorescence using a specific antibody (ab92547; Abcam) on paraffin or vibratome (300-μm) sections.
Cell Culture
Human cardiac fibroblasts (hCFs; C-12375; Promocell) were subcultured in a 1:4 ratio in fibroblast growth medium (C-23130; Promocell) and treated with TGFβ1 (5 ng/ml) plus/minus FK506 (15 ng/ml) or the BMPR2 inhibitor LDN-193189 (500 nM) for 24 hours.
Human coronary artery endothelial cells (C-12221; Promocell) were grown in endothelial cell culture growth medium (C-22110; Promocell) supplemented with penicillin/streptomycin on gelatin-coated cell culture dishes and subcultured at a 1:3 ratio. TGFβ1 (10 ng/ml) and IL1β (10 ng/ml) were added to the culture medium for 4 consecutive days, either in the presence or in the absence of FK506 (15 ng/ml) or LDN-193189 (500 nM).
Mouse ventricular cardiomyocytes were freshly isolated from BMPR2+/– and BMPR2+/+ mice as described previously (19). Briefly, animals were anesthetized, and the heart was quickly removed from the chest and retrogradely perfused through the aorta at constant pressure with a Ca2+-free buffer supplemented with collagenase type B (0.5 mg/ml), collagenase type D (0.5 mg/ml) and protease type XIV (0.02 mg/ml) for enzymatic tissue digestion. After cell washing and concentration in HEPES-buffered solution (1 mM CaCl2, 137 mM NaCl, 5.4 mM KCl, 15 mM dextrose, 1.3 mM MgSO4, 1.2 mM NaH2PO4, 20 mM HEPES, pH 7.4), singular isolated cardiomyocytes were loaded with 0.5 μM fura 2-acetoxymethyl ester (Molecular Probes) for 15 minutes for Ca2+ transient measurements. Cells were excited at 340 nm and 380 nm, continuously alternated, with background corrections at 510 nm.
Genetic Endothelial Lineage Labeling and Cell Tracing
Male Tg(Pdgfb-icre/ERT2)1Frut mice were crossed to female mice homozygous for the Cre reporter B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J to generate Pdgfb-CreERT2; tdTomato+/– mice (20). Male Pdgfb-CreERT2; tdTomato+/– mice received 4 mg tamoxifen intraperitoneally on two consecutive days to induce Cre recombinase activation and endothelial cell labeling. Four weeks later, those mice underwent either sham surgery or PAB. PAB-challenged mice were randomly assigned to either placebo or FK506 treatment (0.05 mg/kg/d s.c. through an osmotic pump; Alzet) starting at the day of surgery for an additional 3 weeks.
Statistics
All data are presented as mean ± SEM. Statistical analyses were performed in GraphPad Prism 8 software using unpaired two-tailed Student’s t test for comparison of two groups, one-way ANOVA for comparison of more than two groups, or two-way ANOVA when comparing two conditions, followed by Tukey’s post hoc analysis. Pearson’s linear correlation was assessed when appropriate. Statistical significance was considered for P < 0.05. Principal component (PC) analysis was performed on the combined dataset of z-score–normalized myocardial mechanics (derived by MR feature tracking) to evaluate clustering between the placebo-treated group and all other groups after a transformation along the first three PCs.
Results
Low BMP Signaling Exaggerates RV Fibrosis in Response to Chronic RV Pressure Overload
We used the PAB mouse model, which is characterized by a fixed RV afterload due to surgical stenosis of the main pulmonary artery. In this model, compensatory RV hypertrophy is established within the first week, followed by a progressive decline in cardiac function with impaired exercise capacity and prominent signs of RV failure, such as septal flattening, RV dilatation, and activation of the fetal gene program, within 7–8 weeks after disease commencement (15, 18). In the PAB model, we first assessed whether low BMP signaling in the right ventricle predisposes to an impaired adaptation to chronic RV pressure overload as data from patients with hereditary PAH and a BMPR2 mutation would suggest (4). Mice heterozygous for Bmpr2 (Bmpr2+/–) (16) and Bmpr2+/+ littermate control mice underwent PAB to induce chronic RV pressure overload or sham control surgery. No disease-related mortality was observed, and all endpoints were assessed 7 weeks after surgery (Figure 1A). Gene expression analyses for Bmpr2 and one of its downstream signaling targets, Id1, confirmed reduced BMP signaling in the right ventricle of Bmpr2+/– mice compared with control mice, suggesting low BMP pathway activity at baseline (Figure 1B). On a histological level, chronic RV pressure overload provoked RV cardiomyocyte hypertrophy (cardiomyocyte surface area, Figure 1C) and reductions in RV capillarization (capillaries/cardiomyocytes, Figure 1D) independent from Bmpr2 expression after PAB. RV fibrosis, characterized by increased collagen amounts (percentage RV collagen, Masson’s trichrome, and Picrosirius red stain, Figures 1E and 1G) and fibroblast accumulation (Vimentin staining, Figure 1F), was increased in Bmpr2+/– animals compared with littermates after PAB (Figures 1E–1G). In addition to fibrosis, mechanisms such as metabolic limitations or changes in calcium cycling have been previously described as contributing factors to functional RV impairment (5, 6). Although we did not observe lipid accumulation in cardiomyocytes or an increased expression of the fatty acid transporter Cd36 in BMPR2+/– mice as previously described in BMPR2 mutant mice (6), we demonstrated that key enzymes regulating fatty acid metabolism, triglyceride (TG) synthases Dgat1 (diacylglycerol O-acyltransferase 1) and Dgat2 (diacylglycerol O-acyltransferase 2) expression were reduced after PAB in WT animals yet not in Bmpr2± animals, suggesting an increased TG synthesis and turnover, which has been linked to impaired myocardial energetics or contractile function (21) (Figure E1 in the data supplement).
Figure 1.
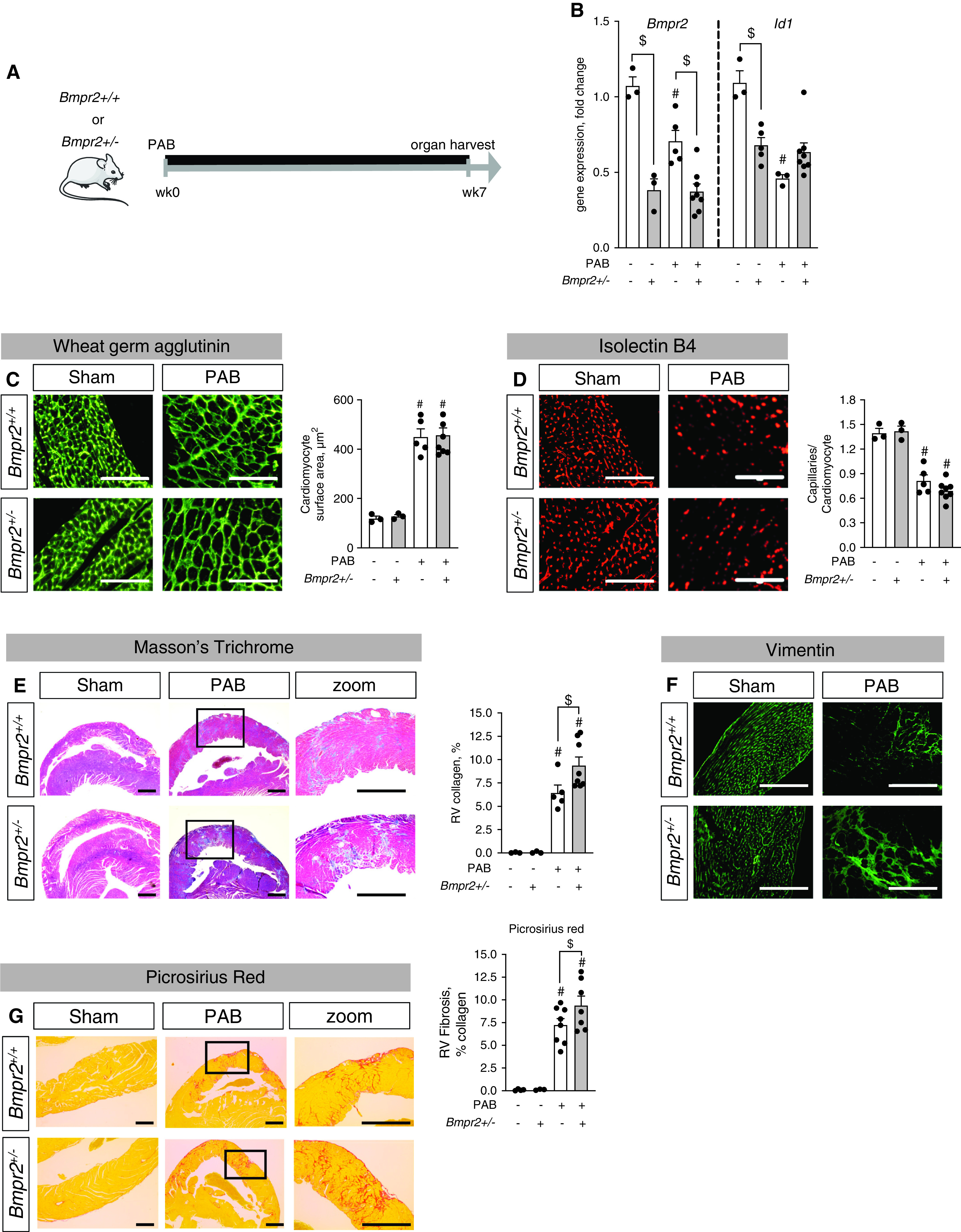
Low BMP (bone morphogenetic protein) signaling exaggerates right ventricular (RV) fibrosis under pressure overload conditions. (A) Male heterozygous Bmpr2 (BMP receptor type 2) mice (BMPR2+/–) and littermate control mice (BMPR2+/+) underwent sham surgery or moderate pulmonary artery banding (PAB) (around a 24-G needle) for 7 weeks. (B) Gene expression analysis of Bmpr2 and Id1 confirmed reduced BMP signaling in the right ventricle in response to PAB, to concentrations comparable with those of BMPR2+/– mice. (C and D) Histological analyses via staining against wheat germ agglutinin and quantification of cardiomyocyte area demonstrated cellular hypertrophy after PAB independent from BMP signaling (C) together with PAB-induced rarefaction of capillaries as assessed by the ratio of capillaries to cardiomyocytes in isolectin B4 staining (D). (E and F) Masson’s trichrome stain demonstrated collagen accumulation in interstitial and perivascular regions (E) together with increased vimentin+ fibroblasts (F). (G) Picrosirius red stain confirms collagen accumulation in interstitium. n = 3 sham-operated animals; n = 5–8 PAB mice. Two-way ANOVA followed by Tukey’s multiple comparison test. #P < 0.05 versus sham and $P < 0.05 versus BMPR2+/+. Scale bars: C and D, 100 μm; E–G, 200 μm.
Furthermore, we observed that Bmpr2+/– mice not only possess an altered response to PAB but also show a reduced exercise capacity in an incremental ramp treadmill exercise protocol at baseline (Figure E2). We provide evidence for impaired cardiomyocyte calcium handling as a consequence of low BMP signaling, evidenced by an increased basal intracellular calcium concentration, reduced calcium uptake during contraction, and delayed calcium release during relaxation (Figure E2) in cardiomyocytes isolated form Bmpr2+/– mice at baseline. Thus, reduced BMP signaling results in an impaired cardiopulmonary response to different external cardiac stresses such as increased RV afterload as well as exercise via multiple pathomechanisms and different cell types.
FK506 Therapy Increases BMP Signaling, Improves RV Capillarization, and Reduces RV Fibrosis in an Animal Model of Chronic RV Pressure Overload
We next assessed whether increasing BMP signaling with FK506 directly affects RV adaptation to chronic RV pressure overload independent from its documented beneficial effects on the pulmonary vasculature (8). Given the fixed, surgically induced pulmonary stenosis in the PAB mouse model, all observed therapeutic effects of FK506 on the RV myocardium would be independent from the previously shown FK506 effects on lowering pulmonary vascular resistance. Treatment with low-dose FK506 (0.05 mg/kg/d s.c. via an osmotic mini pump) was initiated at 1 week after PAB when compensatory hypertrophy was established, and animals were killed after an additional 7 weeks of treatment (Figure 2A). No disease-related mortality within the experimental groups was observed.
Figure 2.
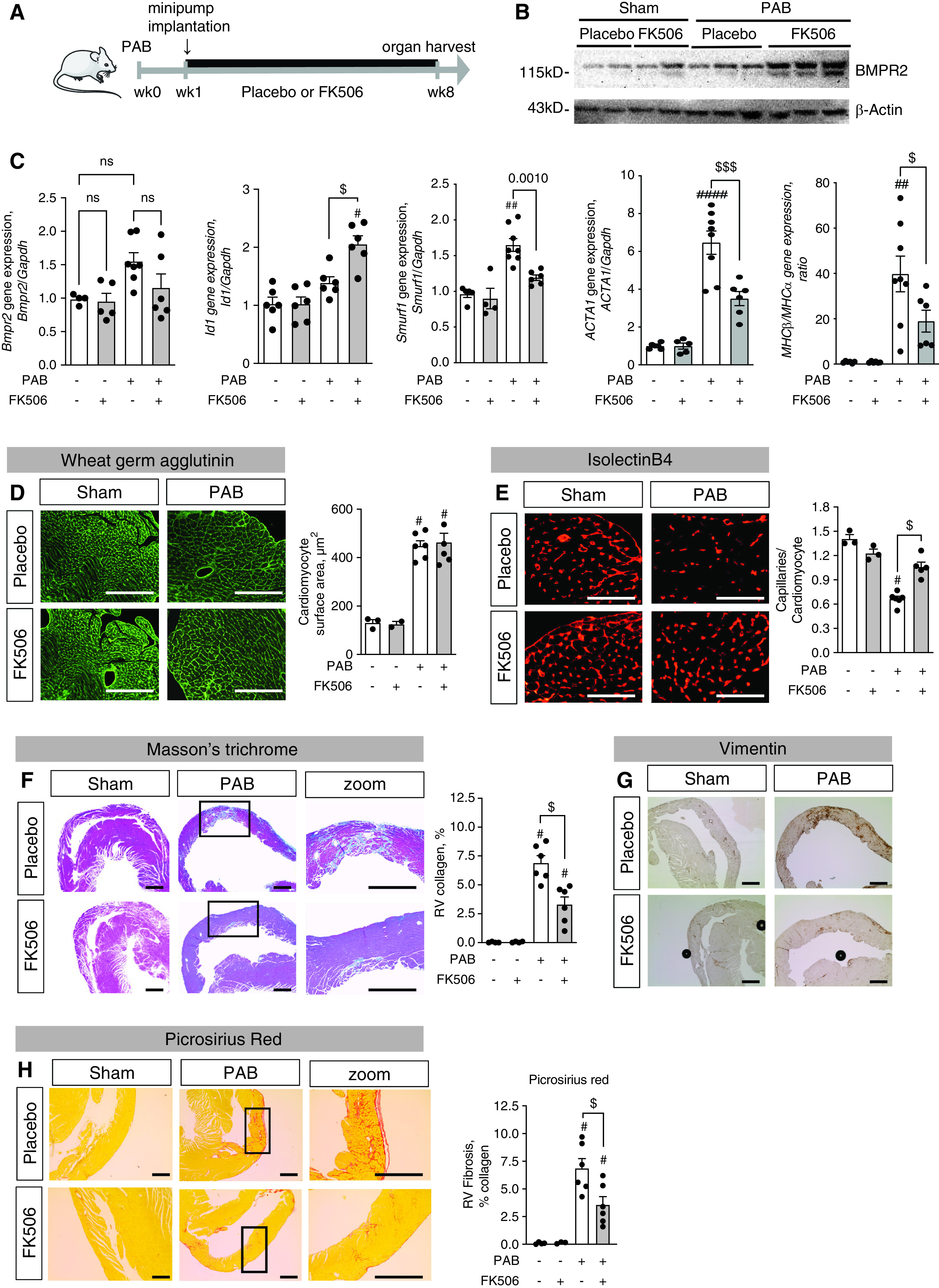
Treatment with FK506 increases BMP signaling, improves RV capillarization, and reduces RV fibrosis under chronic RV pressure overload conditions. (A) Male C57Bl6 mice underwent sham surgery or moderate PAB (around a 24-G needle) and received either placebo or FK506 (0.05 mg/kg/d via osmotic minipump) at Week 1 after surgery for an additional 7 weeks. (B and C) FK506 therapy increased BMPR2 protein expression (B) and Id1, BMPR2, Smurf1 (Smad ubiquitination regulatory factor 1), Acta1, and MHCβ (myosin heavy chain β)/MHCα (myosin heavy chain α) gene expression in the hypertrophied right ventricle (C). (D) Wheat germ agglutinin staining further revealed PAB-induced RV cardiomyocyte hypertrophy independent from FK506 therapy. (E) Isolectin B4 staining of RV capillaries showed an increased capillary:cardiomyocyte ratio, demonstrating that FK506 preserved the RV vasculature after PAB. (F and G) In addition, Masson’s trichrome staining revealed reduced PAB-induced collagen accumulation in the right ventricle (F) together with reduced vimentin+ fibroblasts (G) when animals received FK506 therapy. (H) Picrosirius red stain confirms collagen accumulation in interstitium n = 3 sham-operated mice per group; n = 6 PAB mice per group. Two-way ANOVA followed by Tukey’s multiple comparison post hoc test. #P < 0.05, ##P < 0.01, and ####P < 0.001 versus sham. $P < 0.05 and $$$P < 0.001 versus placebo. Scale bars: F–H, 200 μm; D and E, 100 μm. ns = not significant.
FK506 therapy significantly increased BMPR2 protein concentrations in the right ventricle after PAB (Figures 2B and E9) and increased gene expression of its major downstream signaling target Id1 (Figure 2C) but did not increase BMPR2 gene expression itself. FK506 reduced expression of Smurf1 (Smad ubiquitination regulatory factor 1)—a ubiquitin ligase shown to be a negative regulator of BMP signaling by inducing lysosomal degradation of BMPR2 (22)—thereby pointing to a potential mechanism by which FK506 might increase BMPR2 protein concentrations (by reducing BMPR2 degradation) (Figure 2C). Furthermore, FK506 reduced expression of ACTA1 (actin α 1) and MHCβ (myosin heavy chain β)/MHCα (myosin heavy chain α) ratio, markers of the fetal gene program seen in heart failure (23, 24). On a structural level, increasing BMP signaling with FK506 had no effect on PAB-induced RV cardiomyocyte hypertrophy (cardiomyocyte surface area, Figure 2D) but preserved the RV capillary vasculature (capillaries/cardiomyocytes, Figure 3E) and significantly reduced RV fibrosis as assessed by total RV collagen content (Figures 2F and 2H) and accumulation of fibroblasts (Figure 2G).
Figure 3.
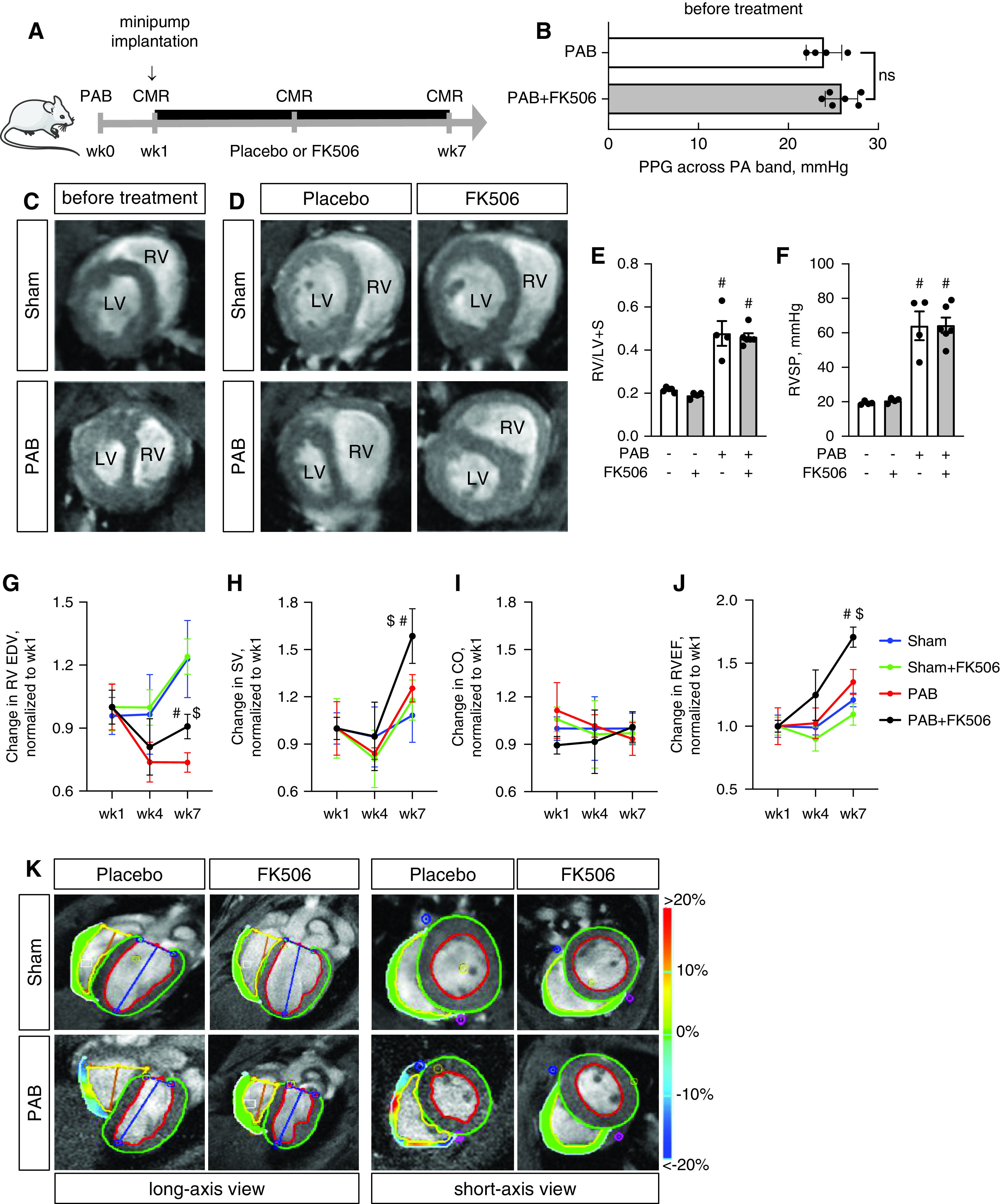
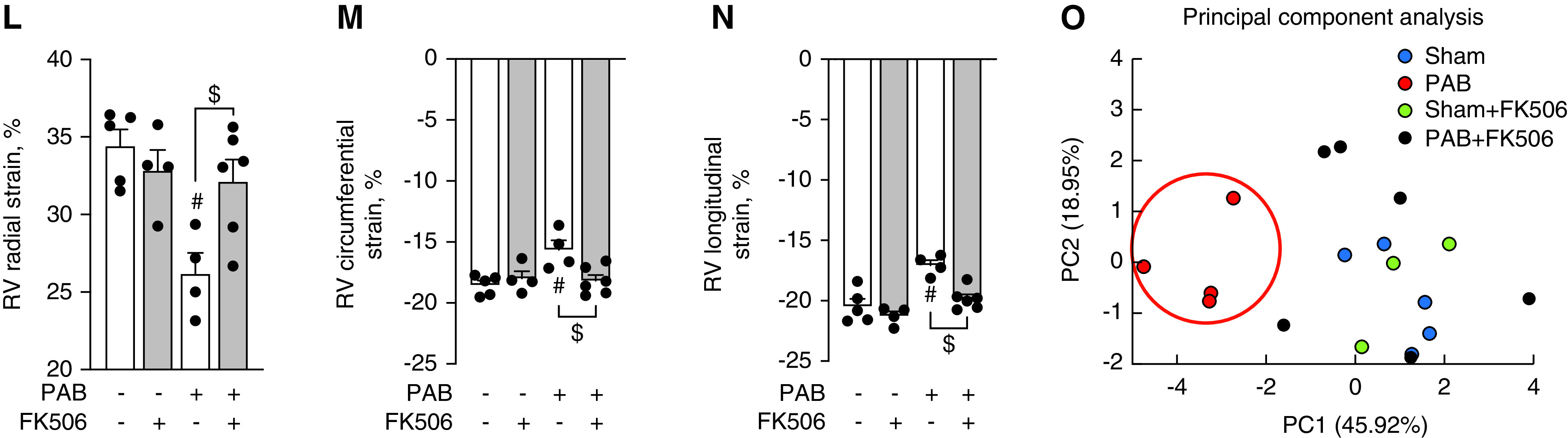
Treatment with FK506 improves RV function under chronic pressure overload conditions. (A) Male C57Bl6 mice underwent sham surgery or moderated PAB (around a 24-G needle) and received either placebo or FK506 (0.05 mg/kg/d via osmotic minipump) at week1 after surgery for additional 6 weeks. (B) Echocardiographic measurement of the PPG across the PA band before treatment commencement demonstrated a comparable degree of stenosis among both PAB groups. (C) CMR imaging confirmed hypertrophic RV remodeling 1 week after PAB. (D–F) FK506 therapy reduced septal flattening and compression of the left ventricle (LV) after PAB as compared with placebo control (D) without changes in RV free-wall hypertrophy, assessed via Fulton’s index (right ventricular [RV]/left ventricular [LV] + septal [S] weight) (E) or RVSP (F). (G–J) During the time course of pressure overload, FK506 attenuated the decrease in RV EDV (G) and increased RV SV (H), CO (I), and RVEF (J). (K–N) Strain imaging (K) further demonstrated increased RV deformation in radial (L), circumferential (M), and longitudinal (N) direction. (O) Principal component (PC) analysis was used to reduce 20 metrics of myocardial strain in both ventricles to three PCs, which showed that FK506-treated PAB animals cluster from placebo-treated PAB mice along PC1 scores, demonstrating that multiple indices of cardiac performance improve with FK506 treatment. n = 4–6 animals per group. Two-way ANOVA followed by Tukey’s multiple comparison post hoc analysis. #P < 0.05 versus sham. $P < 0.05 versus placebo. CMR = cardiac magnetic resonance; CO = cardiac output; EDV = end-diastolic volume; PA = pulmonary artery; PPG = peak pressure gradient; RVEF = RV ejection fraction; RVSP = RV systolic pressure; SV = stroke volume.
Treatment with FK506 Directly Improves RV Function in an Animal Model of Chronic RV Pressure Overload
Next, we assessed the effects of FK506 on RV function using gold-standard, noninvasive CMR imaging in a longitudinal fashion, followed by terminal intracardiac catheterization (Figure 3A). Peak pressure gradient measurements across the pulmonary artery band revealed a comparable degree of stenosis among both PAB groups before treatment initiation (Figure 3B). Signs of remodeling, including RV free-wall hypertrophy and dilatation, were prevalent at 1 week after PAB, confirming established remodeling before treatment commencement (Figure 3C). Using this timeline, we therefore aimed to assess the effect of FK506 on preventing progression from RV hypertrophy to RV failure in this experiment. At Week 7, PAB-challenged placebo-treated animals presented with compression of the left ventricle, septal flattening, and severe RV dilatation, which was attenuated by FK506 therapy (Figure 3D). FK506 had no effect on RV hypertrophy, assessed ex vivo via Fulton’s index (the Fulton index is defined as: RV/LV [left ventricular] + S [septal] weight, Figure 3E), and RV systolic pressure, measured via closed-chest RV catheterization (Figure 3F), both important to maintain RV coupling against the fixed increased afterload. Longitudinal CMR imaging uncovered improvements in RV end-diastolic volume, RV stroke volume, and RVEF over the time course of maintained RV pressure overload when PAB-challenged animals received FK506 therapy, whereas cardiac output showed only a trend toward improvement (Figures 3G–3J). As animals had variable baseline right ventricular end-diastolic volume (RVEDV), stroke volume (SV), and RVEF at 1 week, we chose to present the change of parameters compared with Week 1 to reflect an FK506 treatment response. The absolute values, as documented in the supplement (Figure E3), confirm an increase in RV stroke volume as well as RVEF in the PAB+FK506 versus PAB mice, whereas the absolute RVEDVs were unchanged in both groups. These findings were accompanied by regional deformation improvements in radial, circumferential, and longitudinal directions of strain and displacement, confirming an improved RV performance in these mice (Figures 3K–3N). PC analysis was used to reduce 20 metrics of myocardial strain in both ventricles to three PCs, which showed that FK506-treated PAB animals clustered from placebo-treated PAB mice along PC1 scores (Figure 3O), whereas PAB+FK506, sham, and sham+FK506 animals clustered together. This demonstrated that PAB changed interventricular cardiac contractile mechanics relative to sham control animals, which was improved by FK506 therapy.
FK506 Administration Reduces RV Fibrosis through Increasing BMP Signaling Rather Than Immunosuppression
Given that FK506 at higher doses has prominent immunosuppressive potential, we tested whether the observed beneficial effects of low-dose FK506 therapy on RV structure and function are BMP dependent or secondarily caused by systemic immunosuppression. As a tool to selectively inhibit BMP signaling, we treated mice with the BMP inhibitor LDN-193189 (25, 26). Chronic BMP pathway inhibition with LDN-193189 in the healthy heart had neither an effect on RV capillarization nor on RV fibrosis (Figure E4). As previously shown, low-dose FK506 therapy (0.05 mg/kg/d) improved RV capillarization, yet concomitant LDN-193189 administration (2.5 mg/kg/d) did not reduce the beneficial effect, suggesting either that FK506 improves RV capillarization in a BMP-independent manner or that inhibition of BMP signaling with LDN-193189 was incomplete (Figures 4A–4D). Immunosuppression with cyclosporine (25 mg/kg/d), a calcineurin inhibitor that differs from FK506 in that it does not bind to FKBP12 (8) and therefore does not target BMP signaling, did not improve PAB-induced vascular rarefaction (Figures 4A–4D). Low-dose FK506 significantly reduced the development of RV fibrosis, whereas high-dose FK506 administration (1 mg/kg/d) was less effective in reducing RV fibrosis (Figures 4C and 4E). The reduced efficacy of FK506 on RV fibrosis with additional LDN-193189 treatment suggests that the antifibrotic effect of FK506 is partly BMP dependent rather than being a result of FK506-induced immunosuppression. This is supported by the finding that chronic immunosuppression with cyclosporine further increased RV fibrosis compared with placebo-treated PAB animals.
Figure 4.
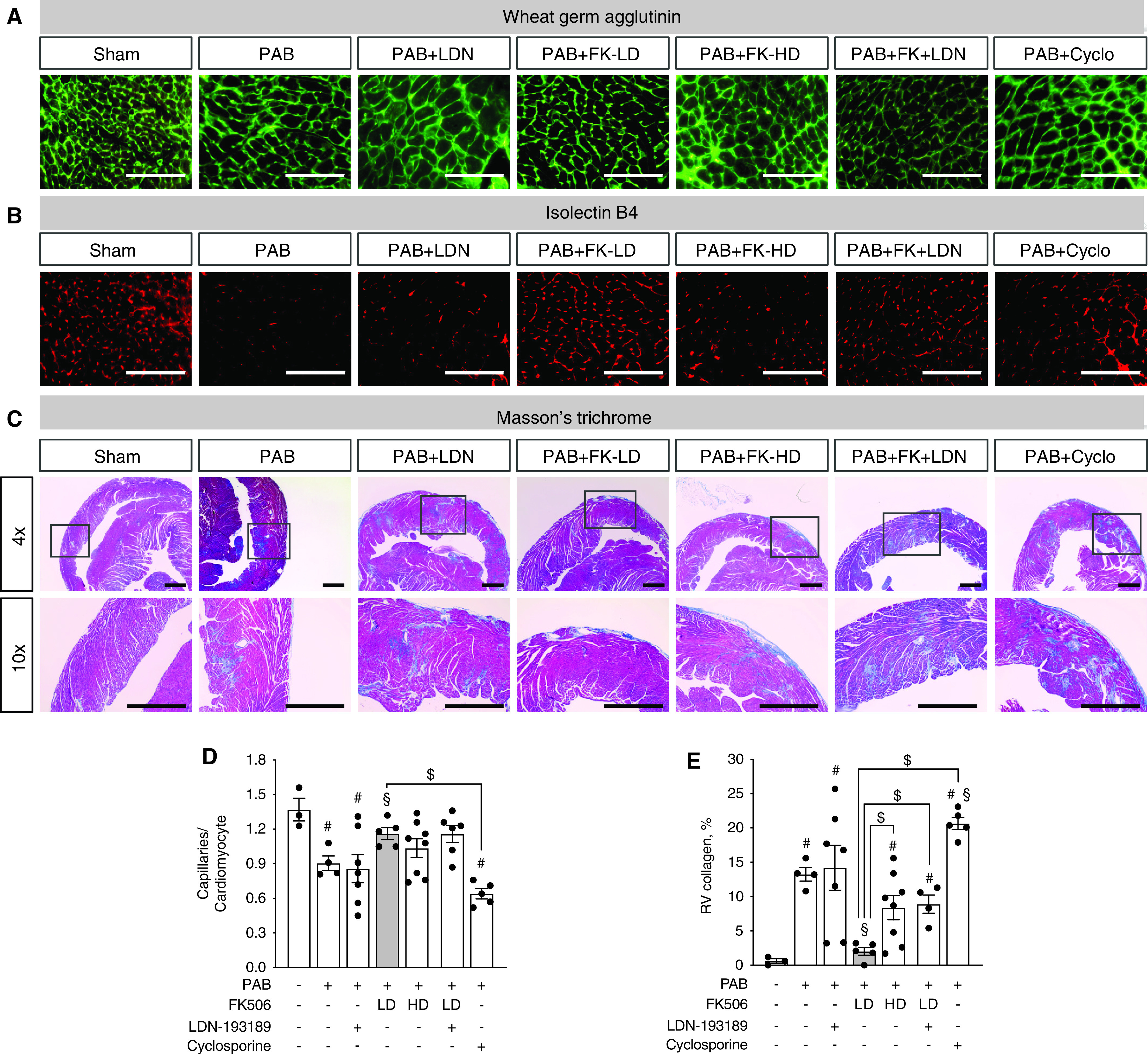
Low-dose FK506 reduces RV fibrosis by increasing BMP signaling, whereas high-dose FK506 is less effective, and cyclosporine worsens RV fibrosis in response to chronic RV pressure overload. (A–D) Histological analysis of wheat germ agglutinin (A), isolectin B4 (B), and Masson’s trichrome (C) staining in RV tissue sections from male C57Bl6 mice after sham surgery or PAB and treatment with either placebo, low-dose FK506 (0.05 mg/kd/d) (gray bar), high-dose FK506 (1 mg/kd/d), low-dose FK506+ LDN-193189 (2.5 mg/kd/d), or cyclosporine (25 mg/kg/d) demonstrated that PAB-induced reductions in capillarization are attenuated by low-dose FK506 therapy independent from LDN-193189 coadministration and high-dose FK506 but not cyclosporine treatment (D). (E) PAB-induced RV fibrosis assessed as RV collagen amount is reduced by low-dose (gray bar) but not high-dose FK506 administration and lost after concomitant LDN-193189 treatment, whereas immunosuppression with cyclosporine worsens RV fibrosis. n = 3 sham-operated animals; n = 4–6 PAB mice per group. One-way ANOVA followed by Tukey’s multiple comparison post hoc analysis. #P < 0.05 versus sham. §P < 0.05 versus placebo. $P < 0.05 versus low-dose FK506. Scale bars, 100 μm.
FK506 Reduces Cardiac Fibroblast Proliferation and Collagen Production and Preserves Endothelial Cell Function in a BMP-Dependent Manner
On a cellular level, we provide evidence in primary hCFs that FK506 blocked TGFβ1-induced proliferation (Figure 5A), reduced extracellular matrix molecules COL1A1 (Figure 5B) and COL3A1 (Figure 5C), and decreased ACTA2 expression, which is required for contractile function (Figure 5D), all in a BMP-dependent manner. We show that FK506 increased BMP signaling (even under conditions of TGFβ pathway activation) independent from BMPR2 expression levels, as evidenced by increased ID1 gene expression (Figures 5E and 5F). Mechanistically, siRNA-mediated gene silencing revealed that ALK1 is the BMPR2 co-receptor required to mediate the antifibrotic effects of FK506 in hCFs (Figures E5A, E5B and E5E), as FK506 was not able to reduce TGFβ-induced COL1A1 expression in ALK1-deficient cells. We also showed that FK506 was able to reduce IL4-induced COL3A1 expression in hCFs—with IL4 being a BMP/TGFβ independent stimulus—again in an ALK1-dependent manner (Figures E5C and E5D).
Figure 5.

FK506 reduces cardiac fibroblast proliferation and collagen production and preserves endothelial cell function in a BMP pathway–dependent manner. (A–D) Human cardiac fibroblasts were activated by TGFβ1 (10 ng/ml) treatment to induce hyperproliferation (A), collagen 1A1 and 3A1 production (B and C), and procontractile function, which was assessed via quantification of ACTA2 expression (D). Cotreatment with FK506 (15 ng/ml) blocked hyperproliferation and collagen production and decreased contractile function in a BMP-dependent manner, as coadministration of LDN-193189 attenuated the FK506 effects. (E and F) FK506 had no effect on BMPR2 gene and protein expression (E) but restored ID1 gene expression in TGFβ1-treated cells (F), demonstrating effective BMP pathway activation. n = 3 experiments. One-way ANOVA followed by Tukey’s multiple comparison post hoc test. #P < 0.05 versus unstimulated control. *P < 0.05 versus TGFβ1. $P < 0.05 versus LDN. (G–I) Stimulation of human coronary artery endothelial cells (hCAECs) with TGFβ1 and proinflammatory IL-1β (10 ng/ml) decreased endothelial cell tube formation capacity (G), increased collagen production (Col1A1) (H), and reduced endothelial marker gene expression (Cd31) (I). FK506 treatment preserved tube formation capacity in a BMP-dependent fashion, reduced collagen production, and maintained endothelial cell fate. (J and K) In hCAECs, FK506 administration increased BMPR2 (J) and ID1 expression (K), confirming its potency to activate BMP signaling in this cell type. n = 3 experiments. One-way ANOVA followed by Tukey’s multiple comparison post hoc test. #P < 0.05 versus unstimulated control. $P < 0.05 versus TGFβ1+ IL-1β. Scale bars, 100 μm. TGFβ1 = transforming growth factor β-1.
In human cardiac endothelial cells, FK506 improved tube formation capacity after TGFβ1+IL-1β treatment (Figure 5G), similar to our in vivo observations of preserved capillary numbers and reduced COL1A1 expression (Figure 5H). This was accompanied by maintained endothelial CD31 expression (Figure 5I), suggesting preservation of endothelial cell fate. This notion was further supported by morphologic analyses and immunostaining for endothelial (CD31 and vWF) and mesenchymal (FN1) markers (Figure E6). In contrast to hCFs, FK506 treatment increased BMP signaling in human cardiac endothelial cells by increasing both BMPR2 and ID1 expression (Figures 5J and 5K), pointing toward cell type–specific regulatory mechanisms of FK506 that control BMP pathway activity in the heart.
FK506 Inhibits Cardiac Endothelial Cell Transition toward Fibroblast-like Cells That Minimally Contribute to Pressure Overload–induced RV Fibrosis In Vivo
To address whether endothelium-derived cells in the right ventricle transition toward a mesenchymal profibrotic fate upon PAB, as our in vitro findings and data focusing on the pressure-overloaded left ventricle suggest (7), and thereby contribute to vascular rarefaction and fibrosis, we performed genetic endothelial lineage labeling followed by cell fate mapping using Pdgfb-CreERT2; tdTomato+/– transgenic mice. In these animals, cardiac endothelial cells and their progeny were genetically marked through tdTomato expression upon tamoxifen-induced recombination 4 weeks before sham surgery or PAB (Week −4) to exclude de novo cell labeling caused by residual tamoxifen at the time of surgery. FK506 therapy was initiated directly after disease induction to increase BMP signaling in the right ventricle at the time when resident fibroblasts are activated and fibrosis develops (Week 0) and was continued for additional 3 weeks (Week 3) until RV fibrosis was established (Figure 6A). Cell sorting at the time before surgery revealed tdTomato labeling in >98% of endothelial cells (CD31+CD45−) isolated from the right ventricle, demonstrating effective genetic endothelial cell labeling via this approach (Figure 6B). This finding was further supported by endothelium-specific costaining of isolectin B4 and tdTomato in capillaries and larger blood vessels of the RV free wall without labeling of epicardium or endocardium, excluding epicardial and endocardial contributions to endothelial cell conversion (Figure 6C). Three weeks after PAB, patchy areas of fibroblast accumulation, defined as Vim+IB4− cells, in interstitial and perivascular regions of the RV free wall were apparent without obvious tdTomato colabeling (Figures 6D and E7), suggesting a nonendothelial cell origin of the Vim+IB4− mesenchymal cells. Quantification of EndMT events in perivascular and interstitial fibrotic areas (Figure 6E) further supported this finding, as only a minority of cells (<5% of lineage-labeled endothelial cells, Figure 6F) transitioned toward a mesenchymal fate or acquired a mesenchymal expression profile, evidenced by Vim+IB4-tdTomato+ labeling. Of note, EndMT events occurred predominantly in cells located at the branching point of capillaries in nonfibrotic interstitial regions (Figures 6E and E8). These rare transitioned cells at the observed time point did not contribute to capillary tube formation. Treatment with FK506 further reduced rare EndMT events (Figure 6F) and, importantly, reduced type 1 collagen accumulation in the pressure-overloaded right ventricle (Figure 6G), again confirming the antifibrotic potential of FK506 therapy.
Figure 6.
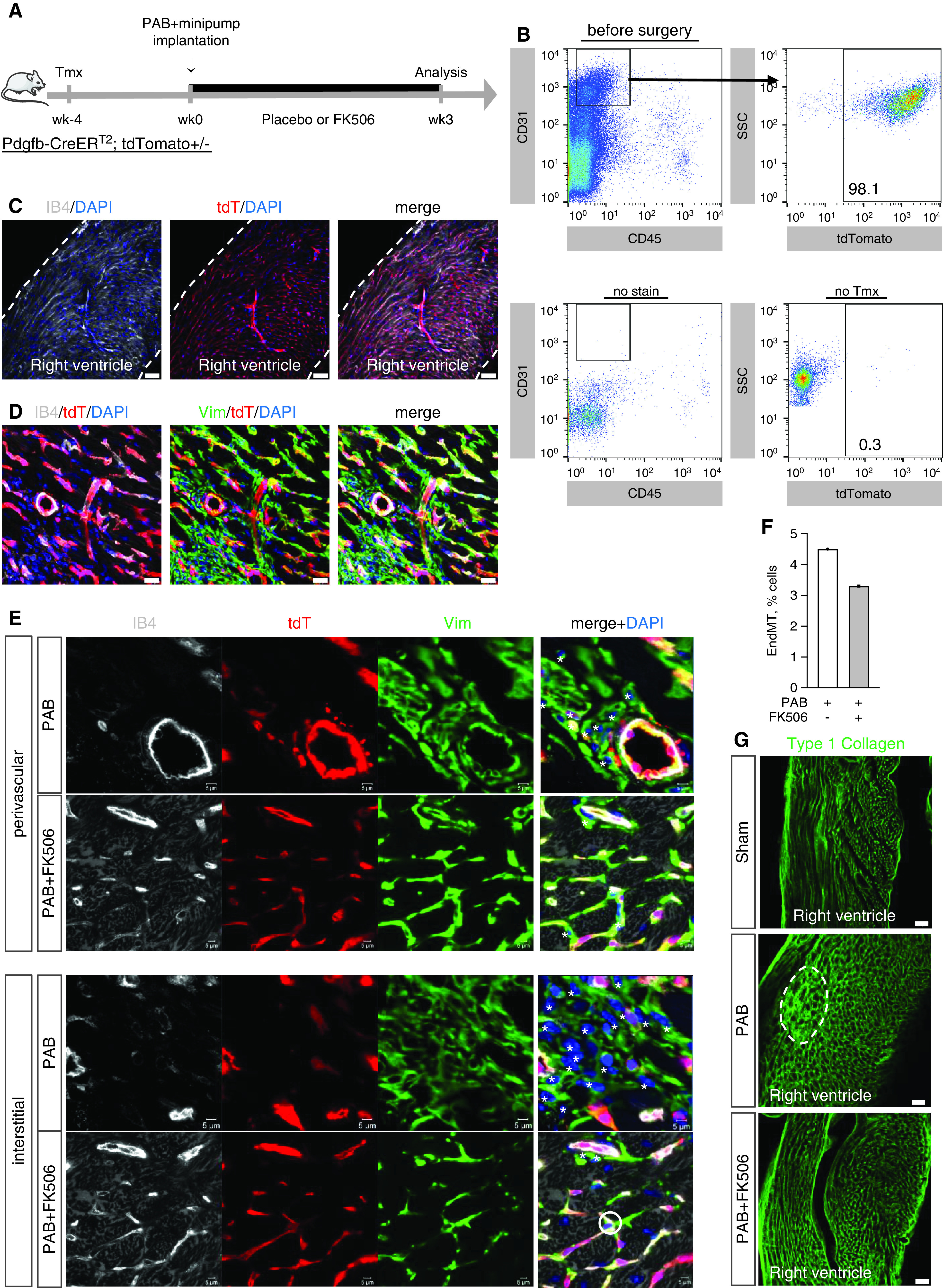
Minority of cardiac endothelial cells transition toward fibroblast-like cells and contribute to pressure overload–induced RV fibrosis, which is reduced by FK506 therapy. Permanent genetic labeling of cardiac endothelial cells and their progeny was performed through tamoxifen (Tmx) administration (2 × 4 mg i.p.) to Pdgfb-CreERT2 mice driving tdTomato (tdT) reporter expression (Week −4). (A) Four weeks after the initial labeling pulse, mice underwent sham surgery or moderate PAB (around 24-G needle) with subsequent placebo or FK505 (0.05 mg/kg/d via osmotic minipump) treatment (Week 0) beginning at the day of surgery for an additional 3 weeks of cell fate chasing. Fluorescent activated cell sorting at the time before surgery revealed tdT reporter labeling in right ventricle endothelial cells (CD31+CD45−), demonstrating efficient endothelial lineage labeling (>98%). (B) Cell gating was defined in control experiments with either lack of CD31 staining (no stain) or lack of tdT labeling (no Tmx). (C) Immunostaining confirmed endothelial IB4 (isolectin B4) costaining with tdT in capillaries and larger blood vessels but neither epicardium nor endocardium (dashed lines), excluding a contribution of epicardial or endocardial cells to EndMT (maximum intensity projections). (D) PAB caused patchy interstitial and perivascular fibrosis with fibroblast (vimentin+ IB4−) accumulation in fibrotic regions within the RV free wall without prevalent tdT colabeling. (E and F) Tissue analysis further revealed that only a minority (<5%) of fibroblasts in interstitial and perivascular fibrotic regions were tdT lineage labeled and derived from an endothelial origin (*: Vim+IB4-tdT-; o: Vim+IB4-tdT+). (G) Type 1 collagen staining demonstrated that patchy collagen accumulation (dashed line) in the PAB-challenged hypertrophic right ventricle is attenuated by preventive administration of FK506. Scale bars: C and G, 50 μm; D, 10 μm; E, 5 μm. EndMT = endothelial-to-mesenchymal transition; SSC = side scatter; Vim = vimentin.
Taken together, we propose that FK506 rebalances TGFβ/BMP signaling directly in the pressure-overloaded right ventricle by increasing BMP signaling, reduces collagen production and proliferation of cardiac fibroblasts, and inhibits the rare event of endothelial fate conversion, leading to reduced RV fibrosis, preserved RV capillarization, and, most important, better RV function (Figure 7).
Figure 7.

Schematic representation of the FK506 effects on cardiac fibroblasts and endothelial cells under chronic pressure overload conditions. We propose that FK506 rebalances TGFβ/BMP signaling directly in the pressure-overloaded right ventricle by increasing BMP signaling, efficiently reduces collagen production and proliferation of cardiac fibroblasts, and inhibits the rare event of endothelial fate conversion, leading to reduced RV fibrosis, preserved RV capillarization, and, most importantly, better RV function. Red arrows pointing up mean increased. Red arrows pointing down mean decreased.
Discussion
The results of the present study demonstrate that FK506 reduces RV fibrosis in a BMP-dependent manner independent from immunosuppression, preserves the RV vasculature, and, importantly, improves RV function over the time course of maintained RV pressure overload. Mechanistically, we show that FK506 requires ALK1 as a BMPR2 co-receptor to rebalance TGFβ/BMP signaling and thereby reduces hyperproliferation and decreases collagen production of human cardiac fibroblasts. In human cardiac endothelial cells, FK506 improves endothelial cell function and reduces collagen production and EndMT. Despite these findings in cell culture, EndMT in response to chronic RV pressure overload in vivo is a rather rare event and does not significantly contribute to RV fibrosis. To our knowledge, this study is the first description of a direct beneficial effect of FK506 on RV structure and function by increasing BMP signaling. We provide evidence that therapies that increase BMP signaling are a promising strategy to help the right ventricle adapt in the presence of a persistent RV afterload, such as in untreated PAH as well as in congenital heart disease with right-sided obstructive lesions, especially in diseases in which the right ventricle is not the primary cause of heart failure.
Previous data from our laboratory have shown that low-dose FK506 stabilizes RV function in patients with end-stage PAH who were treated on a compassionate basis (9), yet no efficacy trial has been performed to support the benefit of FK506 in patients with PAH up to this point. A recent safety and tolerability trial, however, demonstrated that low-dose FK506 was well tolerated, with a good safety profile (10). This is encouraging, as antifibrotic drugs intended for heart failure therapy often had limited clinical use in the past because effective cardiac dosing often caused noncardiac toxic events (27). Here, we report that continuous subcutaneous FK506 infusion to mice increased BMPR2 protein expression directly in the RV myocardium together with evidence of increased BMP downstream signaling through its major signaling target Id1, demonstrating effective cardiac dosing in mice, without noncardiac toxicity signals. This is in line with our finding that low-dose FK506 effectively increases BMP signaling in the diseased pulmonary vasculature and in peripheral blood mononuclear cells (PBMCs) (8).
In our PAB model, we describe RV fibrosis of up to 7–10% collagen accumulation in interstitial and perivascular regions of the RV free wall—similar to that of patients with end-stage PAH (28, 29)—and document that low-dose FK506 is able to reduce RV fibrosis. The physiological and pathophysiological relevance of cardiac fibrosis in the pressure-overloaded right ventricle is unknown (30), and, therefore, cardiac fibrosis as a therapeutic target is controversially discussed (31–33). Although the development of fibrosis initially serves as RV adaptation to normalize wall stress, excessive fibrosis in the long run leads to diastolic dysfunction that frequently progresses to combined systolic/diastolic dysfunction (34). Interestingly, patients with congenital heart disease and Eisenmenger physiology present with less cardiac fibrosis at the time of transplantation compared with patients with PAH, which may be causal for a better diastolic function in this patient group (30). Along these lines, increased miR-21 concentrations in the right ventricle and peripheral blood correlated with the degree of fibrosis and reduced RV function in children with combined pulmonary insufficiency and fibrosis (33). On the other hand, progressive clinical RV failure in rats after PAB was correlated with a lesser degree of fibrosis, whereas profibrotic signaling (TGFβ1 and osteopontin) was similar in compensated and decompensated RV adaptation, pointing toward a higher degradation of collagen in the PAB RV failure group (35). Excessive fibrosis not only increases stiffness of the RV free wall but also disrupts the myocardial architecture; causes mechanic, electrical, and vasomotor dysfunction; hampers excitation–contraction coupling of the cardiomyocytes; and impairs the exchange of oxygen and nutrients (36, 37). Cardiac fibrosis is also known to increase arrhythmogenesis (38), and targeting cardiac fibrosis might therefore be of particular importance in patients with end-stage PAH who have an increased burden of arrhythmias, which increases the risk of mortality in this patient group (39). Evidence from patients with nonischemic left heart failure linked histologically proven myocardial fibrosis to a higher long-term mortality (32, 40). However, clinical trials of antifibrotic therapies in cardiovascular diseases, such as in LV heart failure, have been disappointing in the past, as they often failed to replicate successful results of animal studies; and preclinical findings focusing on the role of RV fibrosis in the hypertrophied right ventricle propose a disconnect between RV fibrosis and function (31). As the extent of RV fibrosis is often less severe when compared with the diseased left ventricle, RV failure might represent a more promising target for antifibrotic therapies. This is supported by the greater capacity of the pressure-overloaded right ventricle to reverse remodel compared with the left ventricle, which may be explained by the smaller extent of fibrosis (41, 42).
Compared with mice, fibrosis in humans likely develops over decades and requires long-term treatment (43). We therefore infused FK506 continuously over 6–7 weeks as an experimental long-term treatment approach. We report an improvement in RV function over the time course of disease progression in the PAB model with FK506 therapy via repetitive gold-standard CMR imaging, in line with preclinical findings focusing on pharmacological inhibition of profibrotic signaling pathways (17, 44). CMR imaging was further used for myocardial deformation (“strain”) analysis, which has emerged as a sensitive and accurate geometry-independent method to identify RV dysfunction (45). Strain refers to the change in the length of the myocardium relative to its resting length, and reduced RV global longitudinal strain has been shown to predict survival in adult patients with PAH (46).
Whether our observed improvements in RV function with FK506 are solely caused by reductions in RV fibrotic remodeling is unknown, however, because FK506 and other repurposed drugs and antifibrotic compounds propagate a variety of cellular and molecular effects on the heart, including preservation of the vasculature and cardiomyocyte changes in energy metabolism or calcium handling, which could all play a role in RV adaptation.
This study focused primarily on the effect of FK506 on cardiac fibrosis, a structural hallmark of the failing right ventricle that is nonhomogeneously distributed throughout the RV free wall, which we confirmed with conventional tissue sampling and preparation methods. We therefore did not use unbiased stereology and random sampling approaches as recently suggested (47), as we would have likely missed the extent of fibrosis given its inhomogeneous distribution. Therefore, our results on a second feature of RV maladaptation, RV capillarization, have to be interpreted with caution because in this situation, a random tissue sampling would have been beneficial (48). Impaired compensatory angiogenesis was recently highlighted as a potential contributing factor to RV maladaptation in experimental pulmonary hypertension (47), yet whether rarefaction is a pivotal driver of RV failure in different mouse models (49) and patients with end-stage PAH (48) is controversially discussed. Recent data focusing on the relationship between capillary rarefaction and fibrosis development suggest a role of compensatory angiogenesis in preventing cardiac fibrosis or vice versa (50).
The antifibrotic potential of FK506 on skin (51), lung (52), and cardiac fibroblasts (53) is well described. Mechanistically, FK506 was linked to calcineurin inhibition in cardiac fibroblasts (53). Because FK506 (tacrolimus) is clinically approved as an immunosuppressant targeting calcineurin/NFATc signaling, we used low-dose FK506 in the current study, which has previously been shown to result in FK506 plasma concentrations of 0.2 ng/ml, 50-fold below the targeted immunosuppressive range in humans (10–15 ng/ml), yet capable of increasing BMP signaling via an FKBP12 (FK-binding protein 12)-mediated mechanism (8, 54, 55). In rats, low-dose FK506 therapy did not reduce pulmonary IL-2 or IFNγ expression, major downstream targets of NFATc signaling, pointing toward inefficient immunosuppression (8) at the low dose. In addition, calcineurin inhibition with cyclosporine worsened RV fibrosis in our PAB model and did not affect RV capillarization, supporting a potential beneficial antifibrotic role of T-cell activation that is inhibited by cyclosporine in the pressure-overloaded right ventricle (56). We therefore conclude that low-dose FK506 does not mediate its beneficial antiremodeling effects on the right ventricle by calcineurin inhibition and its immunosuppressive properties. However, we acknowledge the limitations in claiming that a repurposed drug only acts through one specific pathway (such as BMP signaling), given the multiple (and often unknown) treatment targets of a drug and the inability to pharmacologically block all off-target effects. The fact that the BMP inhibitor LDN 193189 only partially prevented the effect of low-dose FK506 on reducing fibrosis reveals, on the one hand, the limitations of pharmacological BMP inhibition versus different mouse BMPR2-deficient mutants but further documents that the beneficial effects of FK506 might not be fully explained by its activating effect on BMP signaling.
On a cellular level, endothelial cell fate conversion after PAB minimally contributes to the observed RV fibrosis in vivo. We therefore propose that the therapeutic value of any such inhibition in vivo would be minimal. Our findings are in contrast to those of Zeisberg and colleagues, who proposed a 30% contribution of EndMT to pressure overload–induced LV fibrosis, yet the group used a non–fibroblast-selective, constitutively active labeling system, which may have confounded the results (7). The minimal contribution of EndMT to RV fibrosis in vivo suggests that the antifibrotic FK506 effect is predominantly caused by a direct effect on cardiac fibroblasts.
Mechanistically, we had previously shown that FK506 binds FKBP12 and thereby increased BMP downstream signaling through its main target, ID1, in pulmonary arterial endothelial cells (8). Here, we add data to show that ALK1 is required as a BMPR2 co-receptor for FK506 to reduce collagen production in cardiac fibroblasts. FKBP12 itself is associated with the calcium release channel of cardiac muscle, and pharmacological dissociation of this complex alters its gating characteristics (57, 58), causing increased calcium accumulation in the cardiomyocyte and a potential additional positive inotropic effect for FK506—in line with our findings in isolated cardiomyocytes from Bmpr2+/– mice. Although the role of dysfunctional BMP signaling in cardiomyocyte function has yet to be determined, evidence from Hemnes and colleagues suggests a BMPR2-dependent lipid toxicity in BMPR2 mutant cardiomyocytes that causes reductions in contractility (59).
Although we recognize sex differences in PAH as well as RV adaptation (60, 61), this study focused on the mechanism of action for FK506 in improving RV structure and function after PAB. The PAB surgery already caused a variable degree of pulmonary stenosis, with a number of animals not meeting our inclusion criterium (peak pressure gradient of 20–35 mm Hg at 1 week after surgery by echo Doppler). To focus on the effect of FK506 in animals with a comparable degree of RV pressure overload and with statistical power sufficient to draw conclusions from the experiments, we precluded sex as an additional variable at this point.
In summary, this is the first study to report a direct beneficial effect of FK506 on RV structure (fibrosis) and function by increasing BMP signaling. Our data associate reduced cardiac BMP signaling with increased RV fibrosis under pressure overload conditions and give mechanistic insights into the development of RV fibrosis. Despite multiple available vasodilatory therapies, PAH remains a serious life-threatening disease with unacceptably high mortality. Pharmacological activation of BMP signaling with FK506 targets the underlying pathological remodeling processes in the pulmonary vasculature—as we had previously demonstrated (8)—and, at the same time, directly improves RV structure and function in the presence of a fixed RV pressure overload. This beneficial effect of FK506 on the right ventricle could be relevant not only for PAH but also for other diseases in which the right ventricle is at risk for failure, such as in congenital heart disease or group 2–4 pulmonary hypertension, for which therapies that reduce the RV afterload are not readily available.
Footnotes
Supported by a postdoctoral research fellowship of the Max Kade Foundation, Inc., (M.B.) and grant money from the Stanford Cardiovascular Institute, National Heart, Lung, and Blood Institute (R01 HL128734), Wall Center for Pulmonary Vascular Disease Stanford, a Pulmonary Hypertension Association career development grant, and Department of Defense grant (PR161256).
Author Contributions: M.B. and E.S. designed and performed the experiments, analyzed and interpreted data, and wrote the manuscript. R.J.M. designed the experiments and analyzed and interpreted data. X.T., M.K.A., Y.M., K.I., M.Z., K.K., S.D.P., G.F., M.J.D., and X.Q. performed the experiments and analyzed data. V.O.K., D.B., S.R., R.T.Z., and F.H. analyzed and interpreted data. All authors were involved in critically revising the manuscript.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0528OC on October 31, 2019
This article has an data supplement, which is accessible from this issue’s table of content online at www.atsjournals.org.
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest. 2009;135:794–804. doi: 10.1378/chest.08-0492. [DOI] [PubMed] [Google Scholar]
- 2. Naeije R, Manes A. The right ventricle in pulmonary arterial hypertension. Eur Respir Rev. 2014;23:476–487. doi: 10.1183/09059180.00007414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rich S, Pogoriler J, Husain AN, Toth PT, Gomberg-Maitland M, Archer SL. Long-term effects of epoprostenol on the pulmonary vasculature in idiopathic pulmonary arterial hypertension. Chest. 2010;138:1234–1239. doi: 10.1378/chest.09-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van der Bruggen CE, Happé CM, Dorfmüller P, Trip P, Spruijt OA, Rol N, et al. Bone morphogenetic protein receptor type 2 mutation in pulmonary arterial hypertension: a view on the right ventricle. Circulation. 2016;133:1747–1760. doi: 10.1161/CIRCULATIONAHA.115.020696. [DOI] [PubMed] [Google Scholar]
- 5. Hautefort A, Mendes-Ferreira P, Sabourin J, Manaud G, Bertero T, Rucker-Martin C, et al. Bmpr2 mutant rats develop pulmonary and cardiac characteristics of pulmonary arterial hypertension. Circulation. 2019;139:932–948. doi: 10.1161/CIRCULATIONAHA.118.033744. [DOI] [PubMed] [Google Scholar]
- 6. Talati MH, Brittain EL, Fessel JP, Penner N, Atkinson J, Funke M, et al. Mechanisms of lipid accumulation in the bone morphogenetic protein receptor type 2 mutant right ventricle. Am J Respir Crit Care Med. 2016;194:719–728. doi: 10.1164/rccm.201507-1444OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 8. Spiekerkoetter E, Tian X, Cai J, Hopper RK, Sudheendra D, Li CG, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest. 2013;123:3600–3613. doi: 10.1172/JCI65592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Spiekerkoetter E, Sung YK, Sudheendra D, Bill M, Aldred MA, van de Veerdonk MC, et al. Low-dose FK506 (tacrolimus) in end-stage pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192:254–257. doi: 10.1164/rccm.201411-2061LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Spiekerkoetter E, Sung YK, Sudheendra D, Scott V, Del Rosario P, Bill M, et al. Randomised placebo-controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur Respir J. 2017;50:1602449. doi: 10.1183/13993003.02449-2016. [DOI] [PubMed] [Google Scholar]
- 11. Reddy S, Bernstein D. The vulnerable right ventricle. Curr Opin Pediatr. 2015;27:563–568. doi: 10.1097/MOP.0000000000000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stout KK, Broberg CS, Book WM, Cecchin F, Chen JM, Dimopoulos K, et al. American Heart Association Council on Clinical Cardiology, Council on Functional Genomics and Translational Biology, and Council on Cardiovascular Radiology and Imaging. Chronic heart failure in congenital heart disease: a scientific statement from the American Heart Association. Circulation. 2016;133:770–801. doi: 10.1161/CIR.0000000000000352. [DOI] [PubMed] [Google Scholar]
- 13. Reddy S, Bernstein D. Molecular mechanisms of right ventricular failure. Circulation. 2015;132:1734–1742. doi: 10.1161/CIRCULATIONAHA.114.012975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vonk Noordegraaf A, Bogaard HJ. Restoring the right ventricle. Chest. 2020;157:251–252. doi: 10.1016/j.chest.2019.10.022. [DOI] [PubMed] [Google Scholar]
- 15. Urashima T, Zhao M, Wagner R, Fajardo G, Farahani S, Quertermous T, et al. Molecular and physiological characterization of RV remodeling in a murine model of pulmonary stenosis. Am J Physiol Heart Circ Physiol. 2008;295:H1351–H1368. doi: 10.1152/ajpheart.91526.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Song Y, Jones JE, Beppu H, Keaney JF, Jr, Loscalzo J, Zhang YY. Increased susceptibility to pulmonary hypertension in heterozygous BMPR2-mutant mice. Circulation. 2005;112:553–562. doi: 10.1161/CIRCULATIONAHA.104.492488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Budas GR, Boehm M, Kojonazarov B, Viswanathan G, Tian X, Veeroju S, et al. ASK1 inhibition halts disease progression in preclinical models of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2018;197:373–385. doi: 10.1164/rccm.201703-0502OC. [DOI] [PubMed] [Google Scholar]
- 18. Boehm M, Tian X, Mao Y, Ichimura K, Dufva MJ, Ali K, et al. Delineating the molecular and histological events that govern right ventricular recovery using a novel mouse model of pulmonary artery de-banding. Cardiovasc Res. 2020;116:1700–1709. doi: 10.1093/cvr/cvz310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fajardo G, Zhao M, Urashima T, Farahani S, Hu DQ, Reddy S, et al. Deletion of the β2-adrenergic receptor prevents the development of cardiomyopathy in mice. J Mol Cell Cardiol. 2013;63:155–164. doi: 10.1016/j.yjmcc.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Claxton S, Kostourou V, Jadeja S, Chambon P, Hodivala-Dilke K, Fruttiger M. Efficient, inducible Cre-recombinase activation in vascular endothelium. Genesis. 2008;46:74–80. doi: 10.1002/dvg.20367. [DOI] [PubMed] [Google Scholar]
- 21. Roe ND, Handzlik MK, Li T, Tian R. The role of diacylglycerol acyltransferase (DGAT) 1 and 2 in cardiac metabolism and function. Sci Rep. 2018;8:4983. doi: 10.1038/s41598-018-23223-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murakami K, Etlinger JD. Role of SMURF1 ubiquitin ligase in BMP receptor trafficking and signaling. Cell Signal. 2019;54:139–149. doi: 10.1016/j.cellsig.2018.10.015. [DOI] [PubMed] [Google Scholar]
- 23. Nakao K, Minobe W, Roden R, Bristow MR, Leinwand LA. Myosin heavy chain gene expression in human heart failure. J Clin Invest. 1997;100:2362–2370. doi: 10.1172/JCI119776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann N Y Acad Sci. 2010;1188:191–198. doi: 10.1111/j.1749-6632.2009.05100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Malhotra R, Burke MF, Martyn T, Shakartzi HR, Thayer TE, O’Rourke C, et al. Inhibition of bone morphogenetic protein signal transduction prevents the medial vascular calcification associated with matrix Gla protein deficiency. PLoS One. 2015;10:e0117098. doi: 10.1371/journal.pone.0117098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mayeur C, Kolodziej SA, Wang A, Xu X, Lee A, Yu PB, et al. Oral administration of a bone morphogenetic protein type I receptor inhibitor prevents the development of anemia of inflammation. Haematologica. 2015;100:e68–e71. doi: 10.3324/haematol.2014.111484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Herum KM, Lunde IG, McCulloch AD, Christensen G. The soft- and hard-heartedness of cardiac fibroblasts: mechanotransduction signaling pathways in fibrosis of the heart. J Clin Med. 2017;6:53. doi: 10.3390/jcm6050053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rain S, Handoko ML, Trip P, Gan CT-J, Westerhof N, Stienen GJ, et al. Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation. 2013;128:2016–2025, 2001–2010. doi: 10.1161/CIRCULATIONAHA.113.001873. [DOI] [PubMed] [Google Scholar]
- 29. Omura J, Habbout K, Shimauchi T, Wu W-H, Breuils-Bonnet S, Tremblay E, et al. Identification of long noncoding RNA H19 as a new biomarker and therapeutic target in right ventricular failure in pulmonary arterial hypertension. Circulation. 2020;142:1464–1484. doi: 10.1161/CIRCULATIONAHA.120.047626. [DOI] [PubMed] [Google Scholar]
- 30. Gomez-Arroyo J, Santos-Martinez LE, Aranda A, Pulido T, Beltran M, Muñoz-Castellanos L, et al. Differences in right ventricular remodeling secondary to pressure overload in patients with pulmonary hypertension. Am J Respir Crit Care Med. 2014;189:603–606. doi: 10.1164/rccm.201309-1711LE. [DOI] [PubMed] [Google Scholar]
- 31. Crnkovic S, Egemnazarov B, Damico R, Marsh LM, Nagy BM, Douschan P, et al. Disconnect between fibrotic response and right ventricular dysfunction. Am J Respir Crit Care Med. 2019;199:1550–1560. doi: 10.1164/rccm.201809-1737OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Azevedo CF, Nigri M, Higuchi ML, Pomerantzeff PM, Spina GS, Sampaio RO, et al. Prognostic significance of myocardial fibrosis quantification by histopathology and magnetic resonance imaging in patients with severe aortic valve disease. J Am Coll Cardiol. 2010;56:278–287. doi: 10.1016/j.jacc.2009.12.074. [DOI] [PubMed] [Google Scholar]
- 33. Reddy S, Hu DQ, Zhao M, Blay E, Jr, Sandeep N, Ong SG, et al. miR-21 is associated with fibrosis and right ventricular failure. JCI Insight. 2017;2:e91625. doi: 10.1172/jci.insight.91625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heymans S, González A, Pizard A, Papageorgiou AP, López-Andrés N, Jaisser F, et al. Searching for new mechanisms of myocardial fibrosis with diagnostic and/or therapeutic potential. Eur J Heart Fail. 2015;17:764–771. doi: 10.1002/ejhf.312. [DOI] [PubMed] [Google Scholar]
- 35. Borgdorff MA, Koop AM, Bloks VW, Dickinson MG, Steendijk P, Sillje HH, et al. Clinical symptoms of right ventricular failure in experimental chronic pressure load are associated with progressive diastolic dysfunction. J Mol Cell Cardiol. 2015;79:244–253. doi: 10.1016/j.yjmcc.2014.11.024. [DOI] [PubMed] [Google Scholar]
- 36. Gyöngyösi M, Winkler J, Ramos I, Do QT, Firat H, McDonald K, et al. Myocardial fibrosis: biomedical research from bench to bedside. Eur J Heart Fail. 2017;19:177–191. doi: 10.1002/ejhf.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rother J, Richter C, Turco L, Knoch F, Mey I, Luther S, et al. Crosstalk of cardiomyocytes and fibroblasts in co-cultures. Open Biol. 2015;5:150038. doi: 10.1098/rsob.150038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nguyen MN, Kiriazis H, Gao XM, Du XJ. Cardiac fibrosis and arrhythmogenesis. Compr Physiol. 2017;7:1009–1049. doi: 10.1002/cphy.c160046. [DOI] [PubMed] [Google Scholar]
- 39. Olsson KM, Nickel NP, Tongers J, Hoeper MM. Atrial flutter and fibrillation in patients with pulmonary hypertension. Int J Cardiol. 2013;167:2300–2305. doi: 10.1016/j.ijcard.2012.06.024. [DOI] [PubMed] [Google Scholar]
- 40. Aoki T, Fukumoto Y, Sugimura K, Oikawa M, Satoh K, Nakano M, et al. Prognostic impact of myocardial interstitial fibrosis in non-ischemic heart failure: -comparison between preserved and reduced ejection fraction heart failure.- Circ J. 2011;75:2605–2613. doi: 10.1253/circj.cj-11-0568. [DOI] [PubMed] [Google Scholar]
- 41. McCann GP, Gan CT, Beek AM, Niessen HW, Vonk Noordegraaf A, van Rossum AC. Extent of MRI delayed enhancement of myocardial mass is related to right ventricular dysfunction in pulmonary artery hypertension. AJR Am J Roentgenol. 2007;188:349–355. doi: 10.2214/AJR.05.1259. [DOI] [PubMed] [Google Scholar]
- 42. Shehata ML, Lossnitzer D, Skrok J, Boyce D, Lechtzin N, Mathai SC, et al. Myocardial delayed enhancement in pulmonary hypertension: pulmonary hemodynamics, right ventricular function, and remodeling. AJR Am J Roentgenol. 2011;196:87–94. doi: 10.2214/AJR.09.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fang L, Murphy AJ, Dart AM. A clinical perspective of anti-fibrotic therapies for cardiovascular disease. Front Pharmacol. 2017;8:186. doi: 10.3389/fphar.2017.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kojonazarov B, Novoyatleva T, Boehm M, Happe C, Sibinska Z, Tian X, et al. p38 MAPK inhibition improves heart function in pressure-loaded right ventricular hypertrophy. Am J Respir Cell Mol Biol. 2017;57:603–614. doi: 10.1165/rcmb.2016-0374OC. [DOI] [PubMed] [Google Scholar]
- 45. Valsangiacomo Buechel ER, Mertens LL. Imaging the right heart: the use of integrated multimodality imaging. Eur Heart J. 2012;33:949–960. doi: 10.1093/eurheartj/ehr490. [DOI] [PubMed] [Google Scholar]
- 46. Park JH, Park MM, Farha S, Sharp J, Lundgrin E, Comhair S, et al. Impaired global right ventricular longitudinal strain predicts long-term adverse outcomes in patients with pulmonary arterial hypertension. J Cardiovasc Ultrasound. 2015;23:91–99. doi: 10.4250/jcu.2015.23.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Frump AL, Bonnet S, de Jesus Perez VA, Lahm T. Emerging role of angiogenesis in adaptive and maladaptive right ventricular remodeling in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2018;314:L443–L460. doi: 10.1152/ajplung.00374.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Graham BB, Koyanagi D, Kandasamy B, Tuder RM. Right ventricle vasculature in human pulmonary hypertension assessed by stereology. Am J Respir Crit Care Med. 2017;196:1075–1077. doi: 10.1164/rccm.201702-0425LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kolb TM, Peabody J, Baddoura P, Fallica J, Mock JR, Singer BD, et al. Right ventricular angiogenesis is an early adaptive response to chronic hypoxia-induced pulmonary hypertension. Microcirculation. 2015;22:724–736. doi: 10.1111/micc.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Noly PE, Haddad F, Arthur-Ataam J, Langer N, Dorfmüller P, Loisel F, et al. The importance of capillary density-stroke work mismatch for right ventricular adaptation to chronic pressure overload. J Thorac Cardiovasc Surg. 2017;154:2070–2079. doi: 10.1016/j.jtcvs.2017.05.102. [DOI] [PubMed] [Google Scholar]
- 51. Que J, Cao Q, Sui T, Du S, Kong D, Cao X. Effect of FK506 in reducing scar formation by inducing fibroblast apoptosis after sciatic nerve injury in rats. Cell Death Dis. 2013;4:e526. doi: 10.1038/cddis.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nagano J, Iyonaga K, Kawamura K, Yamashita A, Ichiyasu H, Okamoto T, et al. Use of tacrolimus, a potent antifibrotic agent, in bleomycin-induced lung fibrosis. Eur Respir J. 2006;27:460–469. doi: 10.1183/09031936.06.00070705. [DOI] [PubMed] [Google Scholar]
- 53. White M, Montezano AC, Touyz RM. Angiotensin II signalling and calcineurin in cardiac fibroblasts: differential effects of calcineurin inhibitors FK506 and cyclosporine A. Ther Adv Cardiovasc Dis. 2012;6:5–14. doi: 10.1177/1753944711432901. [DOI] [PubMed] [Google Scholar]
- 54. Donners MM, Bot I, De Windt LJ, van Berkel TJ, Daemen MJ, Biessen EA, et al. Low-dose FK506 blocks collar-induced atherosclerotic plaque development and stabilizes plaques in ApoE-/- mice. Am J Transplant. 2005;5:1204–1215. doi: 10.1111/j.1600-6143.2005.00821.x. [DOI] [PubMed] [Google Scholar]
- 55. Bai L, Gabriels K, Wijnands E, Rousch M, Daemen MJ, Tervaert JW, et al. Low- but not high-dose FK506 treatment confers atheroprotection due to alternative macrophage activation and unaffected cholesterol levels. Thromb Haemost. 2010;104:143–150. doi: 10.1160/TH09-07-0502. [DOI] [PubMed] [Google Scholar]
- 56. Leitner J, Drobits K, Pickl WF, Majdic O, Zlabinger G, Steinberger P. The effects of Cyclosporine A and azathioprine on human T cells activated by different costimulatory signals. Immunol Lett. 2011;140:74–80. doi: 10.1016/j.imlet.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mayrleitner M, Timerman AP, Wiederrecht G, Fleischer S. The calcium release channel of sarcoplasmic reticulum is modulated by FK-506 binding protein: effect of FKBP-12 on single channel activity of the skeletal muscle ryanodine receptor. Cell Calcium. 1994;15:99–108. doi: 10.1016/0143-4160(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 58. Ahern GP, Junankar PR, Dulhunty AF. Single channel activity of the ryanodine receptor calcium release channel is modulated by FK-506. FEBS Lett. 1994;352:369–374. doi: 10.1016/0014-5793(94)01001-3. [DOI] [PubMed] [Google Scholar]
- 59. Hemnes AR, Brittain EL, Trammell AW, Fessel JP, Austin ED, Penner N, et al. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med. 2014;189:325–334. doi: 10.1164/rccm.201306-1086OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ventetuolo CE, Hess E, Austin ED, Barón AE, Klinger JR, Lahm T, et al. Sex-based differences in veterans with pulmonary hypertension: results from the veterans affairs-clinical assessment reporting and tracking database. PLoS One. 2017;12:e0187734. doi: 10.1371/journal.pone.0187734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tello K, Richter MJ, Yogeswaran A, Ghofrani HA, Naeije R, Vanderpool R, et al. Sex differences in right ventricular-pulmonary arterial coupling in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2020;202:1042–1046. doi: 10.1164/rccm.202003-0807LE. [DOI] [PMC free article] [PubMed] [Google Scholar]