

Abstract
Inflammasomes are intracellular multiprotein complexes that help trigger and maintain the inflammatory response as part of the innate immune system. Recently, it has been increasingly recognized that aberrant inflammasome activation is critically involved in endothelial dysfunction in a variety of human diseases, such as atherosclerosis, acute lung injury (ALI), and type 2 diabetes. The molecular mechanisms underlying endothelial inflammasome activation, however, have not been completely elucidated. In the present study, we identified orphan nuclear receptor Nur77 as a novel regulator in controlling inflammasome activation in vascular endothelial cells (ECs). We demonstrated that LPS-induced inflammasome activation was significantly inhibited by ectopic overexpression of Nur77, predominantly through transcriptional suppression of caspase-1 expression in vascular ECs. Consistent with this observation, we found that LPS-induced inflammasome activation was significantly augmented in lung ECs isolated from Nur77-knockout mice. Mechanistically, we showed that Nur77-induced inhibition of caspase-1 expression was due to an inhibition of IRF1 (IFN regulatory factor 1) expression and its subsequent binding to the caspase-1 promoter. Importantly, in a mouse model of LPS-induced ALI, Nur77 knockout led to a marked activation of caspase-1 in the lung, increased alveolar and circulating IL-1β levels, and exacerbated ALI, all of which were substantially inhibited by administration of caspase-1 inhibitor. Together, our results support the presence of an important role for Nur77 in controlling inflammasome activation in vascular ECs and suggest that Nur77 could be a novel therapeutic target for the treatment of human diseases associated with aberrant inflammasome activation, such as ALI and atherosclerosis.
Keywords: Nur77, inflammasome, caspase-1, IFN regulatory factor 1, acute lung injury
Clinical Relevance
This research identified orphan nuclear receptor Nur77 as an essential regulator controlling inflammasome activation through suppressing caspase-1 expression in lung endothelial cells. Our results suggest that Nur77 could be a novel therapeutic target for the treatment of human diseases associated with aberrant inflammasome activation, such as acute lung injury and atherosclerosis.
Inflammasomes, intracellular multiprotein complexes, serves as signaling platforms for coordinating inflammatory responses and host defense (1). Upon stimulation with inflammatory ligands, upstream sensor proteins are activated and oligomerized to recruit an adaptor protein, ASC, which brings caspase-1 and sensor proteins into proximity. Activation and assembly of inflammasomes leads to autocleavage and activation of caspase-1, which proteolytically triggers the processing and release of proinflammatory IL-1β and IL-18 (2). In addition to the well-characterized functions of inflammasomes in host defense, tumorigenesis, and metabolic disorder, the role of inflammasomes has also been implicated in proinflammatory vascular disorders, such as atherosclerosis and acute lung injury (ALI) (1, 3). For example, previous studies have shown that genetic ablations of inflammasome components and inflammasome-mediated cytokines attenuated the development of atherosclerosis and ALI (4–6). Activation of NLRP3 (Nod-like receptor protein 3) inflammasomes produces high levels of IL-1β, which is critically involved in endothelial-cell (EC) activation, the recruitment of neutrophils, and increased vascular leakage in ALI. Importantly, a recently published article demonstrated that inflammasome activation in lung vascular ECs was proven to be essential in intestinal ischemia/reperfusion–induced ALI (7). However, upstream regulators of inflammasome activation in ECs have been poorly identified and require further investigation.
Nur77, an orphan nuclear receptor belonging to the NR4A (nuclear receptor 4A) family, is an immediate early gene that responds to various stimuli, including inflammatory ligands, proangiogenic factors, and hypoxia (8–10). Nur77 contains an N-terminal transactivation domain, a conserved, central DNA-binding domain, and the C-terminal ligand-binding domain (11). Although endogenous ligands for Nur77 have yet to be identified, accumulated evidence suggests that Nur77 plays a protective role in cardiovascular diseases (12–14). Specifically, the protective effects of Nur77 are largely attributed to its antiinflammatory activities in ECs (15). Previously, we found that Nur77 dampens NF-κB signaling, a master driver of the inflammatory response, by promoting IκBα transcriptional activities in a negative-feedback manner (15, 16). Recently, we and others have shown that Nur77 has a great capacity to ameliorate ALI by suppressing proinflammatory ET-1 (endothelin-1) expression and promoting endothelial barrier functions (17–19). We show that the expression of Nur77 in lung ECs is increased in LPS-treated mice and that Nur77 deficiency promotes lung vascular leakage in ALI, suggesting a potent feedback mechanism of inhibiting excessive EC activation by Nur77 (19). Interestingly, Nur77 expression has been recently shown to be positively correlated with the expression of NLRP3 (20), which is one of the most well-characterized components involved in inflammasome activation (21). Given the well-established antiinflammatory activities of Nur77 in the cardiovascular system, we attempted to determine the role of Nur77 in inflammasome activation in vascular ECs and ALI.
Here, we demonstrate that Nur77 suppresses endotoxin-induced endothelial inflammasome activation and subsequent IL-1β secretion through transcriptionally inhibiting caspase-1 expression. Furthermore, we show that Nur77-induced downregulation of caspase-1 expression is, at least in part, achieved through inhibiting the binding of IRF-1 (IFN regulatory factor-1) to the caspase-1 promoter. By using a murine LPS-induced ALI model, we found that depletion of Nur77 significantly promoted IL-1β secretion and exacerbated lung injury and that, surprisingly, this effect can be reversed by treatment of mice with a caspase-1 inhibitor. Our study provides a novel mechanism for controlling inflammasome activation in vascular ECs by Nur77.
Methods
Mice
Nur77−/− mice were purchased from the Jackson Laboratory (stock no. 006187). Animal protocols were approved by the Institutional Animal Care and Use Committee at Thomas Jefferson University. LPS from Escherichia coli 0111:B4 was purchased from Sigma-Aldrich. The caspase-1 inhibitor (acetyl-tyrosyl-valyl-alanyl-aspartyl-chloromethylketone [Ac-YVAD-CMK]) was purchased from BACHEM. Wild-type (WT) and Nur77-knockout (KO) mice (10–12 wk of age, 20–25 g, five to six per group) were subjected to intraperitoneal injection of LPS (10 mg/kg) with or without the caspase-1 inhibitor Ac-YVAD-CMK (8 mg/kg, i.p.). The lung injury score was determined on the basis of criteria described previously (22).
Cell Culture and Adenovirus Transduction
Human umbilical vein ECs (HUVECs) were purchased from Gibco and cultured in endothelial cell medium (ScienCell) supplemented with 1% penicillin–streptomycin solution, 1% EC growth stimulant, and a 5% FBS BulletKit (Lonza). An adenovirus harboring WT Flag-tagged human Nur77 cDNA (Ad-Nur77) and an adenovirus harboring NOR1 were made by using AdMax (Microbix Biosystems, Inc.) as previously described (15).
Murine Lung EC Isolation
The lungs from WT and Nur77-KO mice (at 7–9 d of age, three per group) were dissected into single lobes and incubated in Dulbecco’s modified Eagle medium containing collagenase solution. The cell suspension was purified with anti-CD31–coated magnetic beads (Invitrogen) and cultured in extracellular matrix medium (ScienCell) supplemented with 1% penicillin–streptomycin solution, 1% EC growth stimulant, and a 20% FBS BulletKit (Lonza) as described previously (23).
Western Blotting
Western blotting was performed for antibodies for Flag (1:1,000 dilution; Sigma-Aldrich), Nur77 (1:1,000 dilution; Novus Biologicals), GAPDH (1:2,000 dilution; Santa Cruz Biotechnology), IRF-1 (1:1,000 dilution; ABclonal Technology), caspase-1 (1:1,000 dilution; ABclonal Technology), cleaved caspase-1 (1:1,000 dilution; Cell Signaling Technology), IL-1β (1:1,000 dilution; ABclonal Technology), and cleaved IL-1β (1:1,000; Cell Signaling Technology).
qRT-PCR
qRT-PCR analysis was performed as described previously (18). The primer sequences are described in Table E1 in the data supplement.
ELISA
Mouse IL-1β levels in supernatant and serum were quantified by using commercially available DuoSet ELISA kits (R&D Systems).
Immunofluorescent Staining
Lungs isolated from mice were fixed in 4% paraformaldehyde and embedded in optimal cutting temperature (OCT) medium. Immunostaining was performed with Nur77 antibody (Novus Biologica, catalog no. NB100-56745; 1:100 dilution) and CD31 (BD Pharmingen, catalog no. 550273; 1:250 dilution), which was followed by incubation with secondary antibodies (Alexa Fluor 555-nm antirabbit and 488-nm antirat secondary antibodies, Invitrogen; 1:1,000 dilution). Images were obtained by using a confocal microscope (Nikon).
Chromatin IP Assays
Chromatin IP (ChIP) assays were performed using a ChIP assay kit (Upstate Biotechnology, catalog no. 17–295), essentially as we described previously (14). The following antibody was used to perform immunoprecipitations: polyclonal IRF-1 (1:1,000 dilution; ABclonal Technology). PCR was performed by using the following primers: human: forward, 5′-TCCCAATACATGTACAGGCCC-3′ and reverse, 5′-CTCCTCCCTTCTTGTGTG ACT-3′; mouse: forward, 5′-TGTATTCAC GCCCTGTTGGA-3′ and reverse, 5′-ATAGAGAAAACTCACCAGCCAT-3′.
Caspase-1 Activation Assay
Caspase-1 activity was determined by using a commercially available caspase-1 colorimetric assay kit (BioVision Inc.).
Statistical Analysis
Data were expressed as the mean (±SD) and analyzed for statistical significance by using a Student’s t test or ANOVA conducted with SPSS software (version 18.0; IBM). A P value less than 0.05 was considered to indicate statistical significance in all experiments.
Results
Nur77 Overexpression Decreases Caspase-1 Levels in HUVECs
Caspase-1 is both an important component and a downstream signal of inflammasomes (24). To determine whether Nur77 regulates caspase-1 expression, we transduced the HUVECs at an increasing multiplicity of infection of Ad-Nur77. At 48 hours after transduction, we determined the expression of caspase-1 by using qRT-PCR analysis and Western blotting, respectively. As shown in Figure 1A, adenovirus-mediated overexpression of Nur77 substantially inhibited caspase-1 protein levels in a dose-dependent manner. Likewise, ectopic overexpression of Nur77 dose-dependently decreased caspase-1 mRNA levels (Figure 1B). Furthermore, we found that overexpression of NOR1, another member of the NR4A family (16), barely affected the expression of caspase-1 (Figure 1C), suggesting that Nur77-mediated inhibition of caspase-1 is specific in vascular ECs.
Figure 1.

Nur77 decreases caspase-1 expression in vascular endothelial cells (ECs). (A) Human umbilical vein ECs (HUVECs) were transfected with an adenovirus harboring wild-type (WT) Flag-tagged human Nur77 cDNA (Ad-Nur77) or an adenovirus harboring LacZ (Ad-LacZ) at a total multiplicity of infection (MOI) of 100. At 48 hours after transfection, the expression of caspase-1 (Casp1) was determined by using Western blotting. *P < 0.05 versus Ad-Nur77 at an MOI = 0. (B) HUVECs were infected with Ad-Nur77, Ad-LacZ, or both at a total MOI of 100; 48 hours after transfection, the expression of caspase-1 was determined by using qRT-PCR analysis. *P < 0.01 versus Ad-Nur77 at an MOI = 0. (C) HUVECs were transfected with Ad-NOR1 or Ad-LacZ at a total MOI of 100. At 48 hours after transfection, the expression of caspase-1 was determined by using Western blotting. Quantitative results are shown as means ± SDs, and the data are representative of three individual experiments. Ad-NOR1 = an adenovirus harboring NOR1.
Nur77 Inhibits LPS-induced Inflammasome Activation in Vascular ECs
Because inflammasome activity is closely related to the abundance of procaspase-1 available for processing, we attempted to determine whether Nur77 suppresses LPS-induced inflammasome activation in vascular ECs. To this end, we examined the effects of overexpression of Nur77 on caspase-1 expression and IL-1β release in HUVECs in response to LPS. As shown in Figures 2A and 2B, LPS stimulation significantly promoted caspase-1 expression at both the mRNA and protein levels. Moreover, transduction of Ad-Nur77 substantially inhibited procaspase-1 expression in a dose-dependent manner. Likewise, increased levels of cleaved caspase-1 and its activity induced by LPS were also markedly suppressed by ectopic expression of Nur77 (Figures 2B and 2C). Accordingly, overexpression of Nur77 inhibited LPS-induced maturation and secretion of IL-1β in vascular ECs (Figure 2D). Together, these data suggest that Nur77 functions as a potent inhibitor of LPS-induced inflammasome activation by downregulating caspase-1 expression in vascular ECs.
Figure 2.

Nur77 overexpression inhibits LPS-induced inflammasome activation in vascular ECs. (A) HUVECs were transfected with Ad-Nur77 or Ad-LacZ at a total MOI of 100; 48 hours after transfection, cells were treated with either LPS (1 μg/ml) or vehicle. Expression of caspase-1 mRNA was determined at 12 hours after LPS treatment by using qRT-PCR analysis. *P < 0.05 versus Ad-Nur77 at an MOI = 0 with LPS and #P < 0.05 versus Ad-Nur77 at an MOI = 0 without LPS. (B) HUVECs were infected with Ad-Nur77, Ad-LacZ, or both at a total MOI of 100; 48 hours after transfection, cells were treated with LPS (1 μg/ml) or vehicle. The expression of procaspase-1 and cleaved-caspase-1 was determined at 12 hours after LPS treatment by using Western blotting. *P < 0.05 versus Ad-Nur77 at an MOI = 0 with LPS and #P < 0.05 versus Ad-Nur77 at an MOI = 0 without LPS. (C) HUVECs were infected with Ad-Nur77, Ad-LacZ, or both at a total MOI of 100; 48 hours after transfection, cells were treated with LPS (1 μg/ml) or vehicle, and the activity of caspase-1 was determined at 12 hours after LPS treatment by using spectrophotometry. *P < 0.05 versus Ad-LacZ without LPS and #P < 0.05 versus Ad-LacZ with LPS. (D) HUVECs were transfected with Ad-Nur77 or Ad-LacZ at a total MOI of 100; 48 hours after transfection, cells were treated with either LPS (1 μg/ml) or vehicle. Expression of mature IL-1β protein was determined at 12 hours after LPS treatment by using Western blotting in both the lysate and the SN. *P < 0.05 versus Ad-LacZ without LPS and #P < 0.05 versus Ad-LacZ with LPS. Quantitative results are shown as means ± SDs, and the data are representative of three individual experiments. SN = supernatant.
Nur77 Deficiency Enhances LPS-induced Inflammasome Activation in Lung Vascular ECs
To further substantiate the role of Nur77 in inflammasome activation, we examined whether Nur77 deficiency exacerbates LPS-induced inflammasome activation in lung vascular ECs. To this end, we isolated murine lung ECs (MLECs) from WT and Nur77−/− mice and stimulated them with LPS. The mRNA and protein levels of caspase-1 and the release of mature IL-1β were then determined by using qRT-PCR analysis, Western blotting, and an ELISA, respectively. Consistent with the results obtained from HUVECs, LPS stimulation significantly increased the expression of caspase-1 in MLECs, whereas Nur77 deficiency substantially enhanced caspase-1 expression at both the mRNA and protein levels, as determined by using qRT-PCR analysis and Western blotting (Figure 3A and 3B). Moreover, the levels of cleaved caspase-1, under both basal and LPS-stimulated conditions, were significantly augmented in Nur77-depleted lung ECs. Accordingly, the concentration of mature IL-1β in the culture supernatant was increased dramatically in Nur77-deficient lung ECs, as compared with WT lung ECs, under both basal and LPS-treated conditions (Figure 3C). The expression of NLRP3 and the expression of the adaptor protein ASC were not significantly altered in Nur77-deficient lung ECs (data not shown). These results suggest that Nur77 deficiency not only enhances the expression of caspase-1 but also promotes LPS-induced inflammasome activation in lung vascular ECs.
Figure 3.

Nur77 deficiency augments LPS-induced inflammasome activation in lung vascular ECs. (A) Murine lung ECs (MLECs) isolated from WT and Nur77-knockout (KO) mice were incubated with 1 μg/ml of LPS or vehicle for 12 hours. The expression of caspase-1 mRNA was determined by using qRT-PCR analysis. *P < 0.05 versus WT without LPS, †P < 0.05 versus KO without LPS, and #P < 0.05 versus WT with LPS. (B) MLECs isolated from WT and Nur77-KO mice were incubated with 1 μg/ml of LPS or vehicle for 12 hours. The expression of procaspase-1 and the expression of cleaved caspase-1 protein were determined by using immunoblotting. *P < 0.05 versus WT without LPS, †P < 0.05 versus KO without LPS, and #P < 0.05 versus WT with LPS. (C) MLECs isolated from WT and Nur77-KO mice were incubated with 1 μg/ml of LPS or vehicle for 12 hours. The amount of secreted IL-1β in the culture medium was determined by using an ELISA. *P < 0.05 versus WT without LPS, †P < 0.05 versus KO without LPS, and #P < 0.05 versus WT with LPS. Quantitative results are shown as means ± SDs, and the data are representative of three individual experiments.
IRF-1 Is Involved in Nur77-mediated Inhibition of Caspase-1 in Vascular ECs
Because IRF-1 has been identified as an important transcriptional factor implicated in the regulation of caspase-1 expression in immune systems (25, 26), we attempted to determine whether Nur77 downregulates caspase-1 expression by inhibiting IRF-1–mediated transcriptional activation of caspase-1. To address the role of IRF-1 in Nur77-mediated inhibition of caspase-1 expression, we first investigated whether Nur77 regulates IRF-1 expression in ECs by transducing HUVECs with either Ad-Nur77 or an adenovirus harboring LacZ in the presence or absence of LPS stimulation. As shown in Figures 4A and 4B, overexpression of Nur77 markedly decreased the basal expression of IRF-1 at both the mRNA and the protein level. In addition, IRF-1 expression was enhanced by LPS stimulation, which was substantially inhibited by Nur77 overexpression in a dose-dependent manner, as determined by using both RT-PCR analysis and Western blotting. Furthermore, we determined the inhibitory effect of Nur77 on IRF-1 expression in MLECs. As shown in Figure 4C and 4D, under basal and LPS-stimulated conditions, IRF-1 expression was significantly enhanced in Nur77-deficient lung ECs compared with WT lung ECs, as determined by using both RT-PCR analysis and Western blotting. Because the caspase-1 promoter has a conserved IRF-1 binding site (Figure 5A), we then investigated the role of IRF-1 in Nur77-mediated inhibition of caspase-1 expression by performing a ChIP assay. In this regard, HUVECs were transfected with Ad-Nur77 or an adenovirus harboring LacZ, and IP of IRF-1 with fragmented chromatin was detected by using PCR analysis with the designed primers targeting the fragment of the human caspase-1 promoter. As shown in Figure 5B, Nur77 overexpression significantly decreased the binding of IRF-1 to the caspase-1 promoter in HUVECs. Similarly, we performed a ChIP assay in MLECs isolated from WT or Nur77-KO mice. As shown in Figure 5C, Nur77 deficiency markedly increased the binding of IRF-1 to the caspase-1 promoter in MLECs. Furthermore, we performed siRNA-mediated knockdown of both Nur77 and IRF-1 in HUVECs to further define the role of IRF-1 in the regulation of caspase-1 expression by Nur77. However, we found that a combinatorial depletion of Nur77 and IRF-1 led to significant cell death despite multiple attempts at optimization (data not shown), which prevented us from further analyzing the data in this study. Nevertheless, our results strongly suggest that inhibition of caspase-1 expression by Nur77 is at least in part achieved through inhibiting IRF-1 binding to the caspase-1 promoter in ECs.
Figure 4.

Nur77 inhibits IRF-1 (IFN regulatory factor 1) expression in vascular ECs. (A) HUVECs were transduced with Ad-Nur77 or Ad-LacZ at a total MOI of 100. At 48 hours after transduction, cells were treated with LPS (1 μg/ml) or vehicle. The mRNA level of IRF-1 was determined 12 hours after LPS treatment by using qRT-PCR analysis. *P < 0.05 versus Ad-Nur77 at an MOI = 0 with LPS and #P < 0.05 versus Ad-Nur77 at an MOI = 0 without LPS. (B) HUVECs were transduced with Ad-Nur77, Ad-LacZ, or both at a total MOI of 100. At 48 hours after transduction, cells were treated with LPS (1 μg/ml) or vehicle. The expression of IRF-1 protein was determined by using Western blotting. *P < 0.05 versus Ad-Nur77 at an MOI = 0 with LPS and #P < 0.05 versus Ad-Nur77 at an MOI = 0 without LPS. (C) MLECs from WT and Nur77-KO mice were incubated with either 1 μg/ml of LPS or vehicle for 12 hours. The expression of IRF-1 mRNA was determined by using qRT-PCR analysis. *P < 0.05 versus WT without LPS, †P < 0.05 versus KO without LPS, and #P < 0.05 versus WT with LPS. (D) MLECs from WT and Nur77-KO mice were incubated with 1 μg/ml of LPS or vehicle for 12 hours. The protein levels of IRF-1 were determined by using Western blotting. *P < 0.05 versus WT without LPS and #P < 0.05 versus WT with LPS. Quantitative results are shown as means ± SDs, and the data are representative of three individual experiments.
Figure 5.

Nur77 regulates caspase-1 expression through mediating IRF-1 binding to the caspase-1 promoter. (A) Localization of the IRF-1 response element in the promoter region of human, mouse, and rat caspase-1. (B) HUVECs were transduced with either Ad-Nur77 or Ad-LacZ at an MOI of 50. At 48 hours after transduction, the recruitment of IRF-1 to the caspase-1 promoter was determined by using chromatin IP assays. *P < 0.05 versus Ad-LacZ. (C) MLECs isolated from WT and Nur77-KO mice were cultured, and the binding of IRF-1 to the caspase-1 promoter was determined by using chromatin IP assays. *P < 0.05 versus WT. Quantitative results are shown as means ± SDs, and the data are representative of three individual experiments.
Nur77 Deficiency Exacerbates LPS-induced IL-1β Secretion and ALI
Immunofluorescent staining was performed to detect endogenous Nur77 and CD31 protein levels in murine lung tissues. As shown in Figure 6A, under vehicle (saline)-treated conditions, we observed colocalization of Nur77 and CD31, despite the weak signal of Nur77 in the CD31+ area. Importantly, and consistent with our previous findings in isolated lung ECs (19), we observed an increased Nur77 signal in CD31+ vessels in LPS-treated mice compared with vehicle-treated mice (Figure 6A), indicating the pathological significance of endothelial Nur77 in ALI. To determine the functional significance of Nur77 in regulating LPS-induced caspase-1 activation in vivo, WT and Nur77-KO mice were administrated either LPS (10 mg/kg) or vehicle by intraperitoneal injection. At 24 hours after LPS administration, lung samples were harvested for Western blotting to determine the expression of caspase-1. As shown in Figure 6B, LPS administration significantly increased the protein levels of procaspase-1 in lung homogenates by approximately two- to threefold in WT mice. In Nur77-KO mice, the caspase-1 levels were further augmented under both LPS-stimulated and unstimulated conditions. Furthermore, we attempted to determine whether Nur77 deficiency leads to higher levels of circulatory and alveolar IL-1β after administration of LPS and whether this enhancement could be attenuated by inhibition of caspase-1 activity in vivo. To this end, we pretreated WT or Nur77-KO mice with either the caspase-1 inhibitor Ac-YVAD-CMK (10 mg/kg) or vehicle 30 minutes before LPS administration. At 24 hours after LPS treatment, serum and BAL fluid were harvested to determine the levels of IL-1β by using an ELISA. As shown in Figure 6B, administration of LPS resulted in significantly increased levels of serum IL-1β in both WT and Nur77-KO mice. Furthermore, we found that the serum IL-1β levels were increased by ∼40% in Nur77-KO mice as compared with the age-matched control animals. Treatment of mice with the caspase-1 inhibitor Ac-YVAD-CMK significantly attenuated the serum levels of IL-1β in both WT and Nur77-KO mice (Figure 6C). Likewise, the increased alveolar levels of IL-1β were also significantly inhibited in both WT and Nur77-KO mice by administration of the caspase-1 inhibitor (Figure 6D). Likewise, Nur77 depletion caused more severe LPS-induced lung injury than WT Nur77, and this exacerbation was abolished by administration of the caspase-1 inhibitor (Figure 6E). Together, these results further implicate Nur77 as having an important role in regulating caspase-1 activation and IL-1β secretion in mice with ALI.
Figure 6.
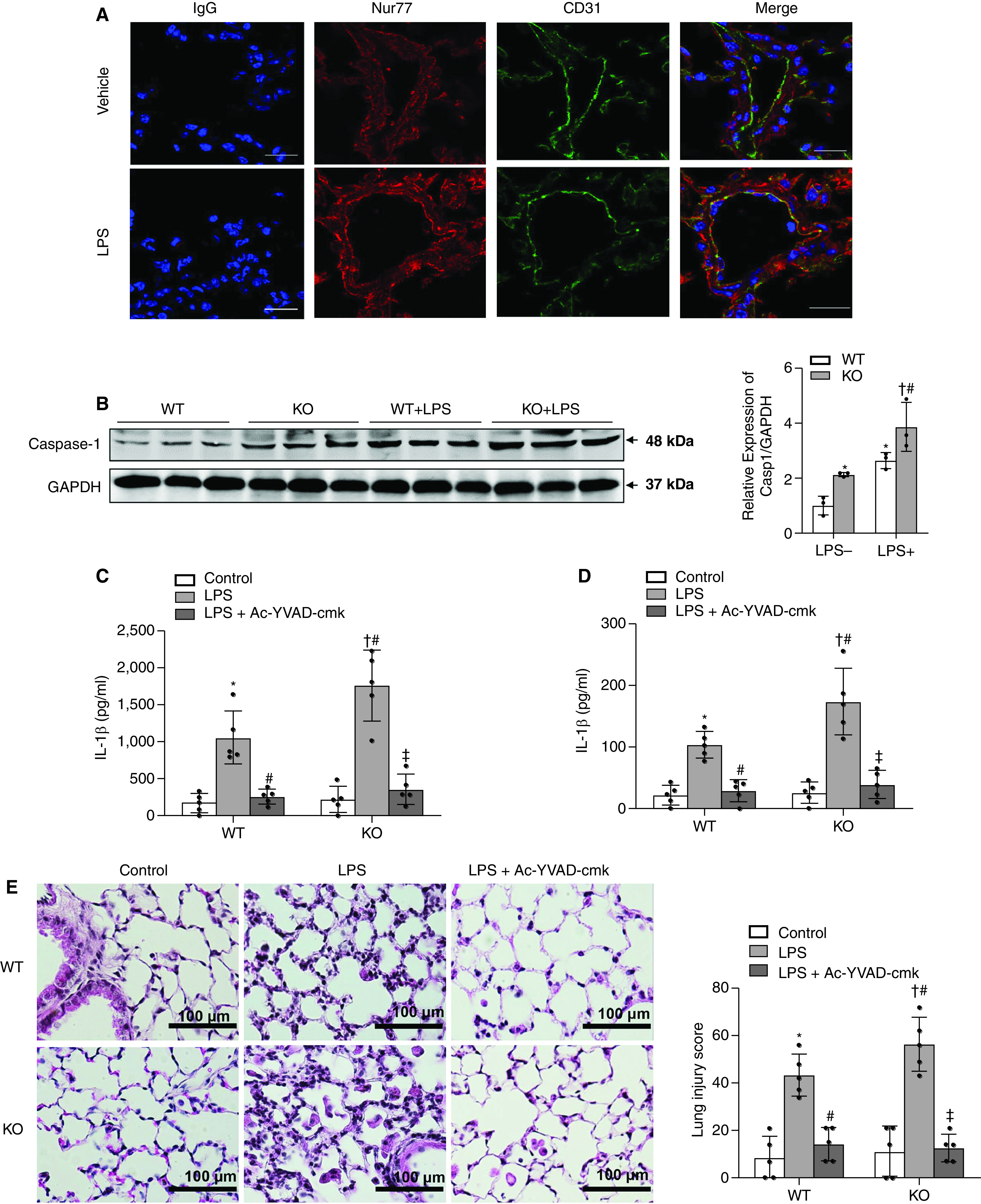
The caspase-1 inhibitor reversed exacerbated LPS-induced IL-1β secretion and lung injury in Nur77−/− mice. (A) Representative images for Nur77 and CD31 colocalization. WT mice were treated with vehicle or LPS (10 mg/kg) by intraperitoneal (i.p.) injection for 24 hours. Lung sections were then stained for Nur77 (red), CD31 (green), and DAPI (blue). Scale bars, 20 μm. (B) WT and Nur77-KO mice were treated with vehicle or LPS (10 mg/kg) by i.p. injection. Lung samples were harvested from mice at 24 hours after treatment. Western blot analysis of the protein expression of caspase-1 in the lung is shown. *P < 0.05 versus WT/vehicle, †P < 0.05 versus Nur77 KO with LPS, and #P < 0.05 versus WT/LPS. n = 3/group. (C) WT and Nur77-KO mice were treated with vehicle or LPS (10 mg/kg) by i.p. injection with or without caspase-1 inhibitor (10 mg/kg) pretreatment. Serum was harvested from mice at 24 hours after treatment. The concentration of IL-1β protein in serum was determined by using an ELISA. *P < 0.05 versus WT/vehicle, #P < 0.05 versus WT/LPS, †P < 0.05 versus Nur77 KO/vehicle, and ‡P < 0.05 versus Nur77 KO/LPS. n = 5/group. (D) WT and Nur77-KO mice were treated with vehicle or LPS (10 mg/kg) by i.p. injection with or without caspase-1 inhibitor (10 mg/kg) pretreatment. BAL fluid was harvested from mice at 24 hours after treatment. The concentration of IL-1β protein in BAL fluid was determined by using an ELISA. *P < 0.05 versus WT/vehicle, #P < 0.05 versus WT/LPS, †P < 0.05 versus Nur77 KO/vehicle, and ‡P < 0.05 versus Nur77 KO/LPS. n = 6/group. (E) WT and Nur77-KO mice were treated with either vehicle or LPS (10 mg/kg) by i.p. injection with or without caspase-1 inhibitor (10 mg/kg) pretreatment. Lung samples was harvested from mice at 24 hours after treatment. Representative hematoxylin and eosin staining (n = 5 mice per group) of lung sections shows marked enhancement of lung inflammatory injury in Nur77-KO mice compared with Nur77-WT mice, and LPS injury is reversed by the caspase-1 inhibitor. Scale bars, 100 μm. *P < 0.05 versus WT/vehicle, #P < 0.05 versus WT/LPS, †P < 0.05 versus Nur77 KO/vehicle, and ‡P < 0.05 versus Nur77 KO/LPS. n = 5/group. Ac-YVAD-cmk = acetyl-tyrosyl-valyl-alanyl-aspartyl-chloromethylketone.
Discussion
Aberrant inflammasome activation has been linked to a variety of proinflammatory and autoimmune diseases, including neurodegenerative diseases, atherosclerosis, type 2 diabetes, and ALI (1). Identification of novel regulators controlling inflammasome activation is essential for developing novel therapies against these diseases. In the present study, we identified Nur77 as a novel regulator for controlling LPS-induced inflammasome activation in vascular ECs. We showed that Nur77 suppresses inflammasome activation by downregulating caspase-1 expression. Furthermore, we found that Nur77-induced downregulation of caspase-1 expression is caused by decreased binding of IRF-1 to the caspase-1 promoter. Importantly, genetic depletion of Nur77 increased both alveolar and systemic IL-1β levels and exacerbated sepsis-induced lung injury. Importantly, these effects could be reversed by a caspase-1 inhibitor, further indicating that the Nur77/caspase-1 axis has an essential role in vascular inflammation.
Inflammasomes act as an integral part of the innate immune system and can be activated by various pathological factors, such as endotoxins (1). Although inflammasome activation is a protective host-defense measure to control pathogen replication, it can also function as a platform to trigger caspase-1 activation and the subsequent release of a large number of inflammatory cytokines, eventually resulting in a secondary damage to organs, as seen in the pathology of acute respiratory distress syndrome (ARDS). Indeed, previous studies have shown that both pharmacological inhibition and genetic loss of inflammasome components and products, including NLRP3, caspase-1, IL-1β, and IL-18, lead to significant attenuation of ALI (4–6, 27). One of the mechanisms of inflammasome-enhanced ALI is demonstrated to be the increased endothelial permeability caused by IL-1β (28, 29). However, inflammasome-mediated production of proinflammatory cytokines can occur in various cell types, including innate immune cells and ECs (7, 30). Nevertheless, Ito and colleagues (7) reported that lung vascular ECs are primary targets for NLRP3 inflammasome activation and IL-1β production in intestinal ischemia/reperfusion–induced ALI. Moreover, CIRP (cold-inducible RNA-binding protein), a damage-associated molecular pattern molecule secreted in hemorrhage and sepsis, was shown to activate NLRP3 inflammasomes in lung vascular ECs (31). These studies suggest the lung vascular endothelium as a promising target for studying inflammasome activation in the pathogenesis of ALI. In this study, we showed that inflammasome activation and the subsequent release of mature IL-1β are triggered by LPS in both HUVECs and MLECs. Importantly, we demonstrated that pulmonary caspase-1 expression is significantly upregulated in response to LPS. In addition, administration of the caspase-1 inhibitor Ac-YVAD-CMK decreased alveolar and circulating IL-1β levels and alleviated lung injury. Our results provide further evidence supporting the importance of inflammasomes in the pathogenesis of ALI and prove that the lung endothelium is indeed a potential site of inflammasome activation in ALI.
NR4A orphan nuclear receptors are immediate early genes that respond to various cell stressors, including proinflammatory ligands and hypoxia. Although all three members in the NR4A family possess antiinflammatory roles, only the expression of Nur77 was reported to be dynamically regulated in lungs in response to LPS, indicating the pathophysiological significance of Nur77 in ALI (16, 15, 19, 32). Indeed, Nur77 has been demonstrated to play a protective role in ALI. Jiang and colleagues (17) reported that Nur77 attenuates ARDS in rats by suppressing epithelial ET-1 expression via downregulation of NF-κB and p38 MAPK signaling. Moreover, our previous studies also demonstrated that Nur77 preserves endothelial barrier function by stabilizing β-catenin (19). Significantly, pharmacological activation of Nur77 was shown to dramatically attenuate lung injury in various ALI models (17, 19), suggesting that targeting Nur77 may provide a therapeutic benefit in ARDS treatment. Our findings show that Nur77 inhibits both basal and LPS-induced upregulation of caspase-1 in ECs. More importantly, as the procaspase-1 protein levels were decreased, the actively cleaved caspase-1 was concomitantly decreased, which in turn attenuated the caspase-1–induced release of inflammatory cytokines. Reciprocally, depletion of Nur77 resulted in increased caspase-1 activation and mature IL-1β secretion. Using an LPS-induced ALI model, we found that Nur77 depletion exacerbated LPS-induced lung injury and further promoted alveolar and circulating IL-1β levels, which were substantially inhibited by coadministration of a specific caspase-1 inhibitor. In this regard, our results highlight the importance of Nur77 in regulating inflammasome activation both in vitro and in vivo and provide a novel mechanism for the protective effects of Nur77 in ALI. However, at this time, we cannot exclude the possibility that Nur77 deficiency in other cells, such as alveolar epithelial cells, airway epithelial cells, and alveolar macrophages, may also contribute to activation of inflammasomes, leading to increased BAL fluid and serum IL-1β and exacerbated lung injury. Further studies using endothelium-specific Nur77-KO mice will help to delineate the specific role of endothelial Nur77 in ALI.
Initially identified as transcriptional regulators of type I IFN systems, IRFs have been shown to possess antiviral and proinflammatory functions in response to various stimuli, including IFN and endotoxins (33). IRFs mainly bind to ISRE (IFN-stimulated response element; 5′-AANNGAAA-3′) to exert their roles as transcriptional factors (25). Importantly, Jain and colleagues (34) found that IRF-1 can enhance caspase-1 promoter activity in response to TNF-α. In addition, it has also been reported that IRF-1 is transcriptionally upregulated through activation of the NF-κB pathway. Because Nur77 is recognized as a negative regulator of NF-κB signaling because of its blocking p65 binding to the κB element and transcriptionally upregulating IκBα (15, 35, 36), we investigated whether regulation of caspase-1 expression by Nur77 is IRF-1 dependent. Our data show that Nur77 downregulates both basal and LPS-induced IRF-1 expression in human and mouse ECs. Accordingly, depletion of Nur77 in MLECs upregulates both basal and LPS-induced IRF-1 expression. Significantly, we also identified a well-conserved ISRE on the caspase-1 promoter across species. In results consistent with previous findings (37), we demonstrated that IRF-1 directly binds to caspase-1 promoters in both human and mouse ECs by using ChIP assays. TLR4 was previously reported to be increased in macrophages isolated from Nur77-KO mice fed with a high-fat diet (12). In this study, we found that the mRNA levels of TLR4 were not significantly altered in lung ECs isolated from Nur77-KO mice compared with those isolated from WT mice under either basal or LPS-stimulated conditions (data not shown). However, at this point, we cannot entirely exclude the possibility that other mechanisms could contribute to Nur77-mediated downregulation of caspase-1 expression. For example, IRF-8 has been shown to directly bind to caspase-1 promoter and enhance its transcriptional activity. Therefore, it will be interesting to investigate whether IRF-8 is also involved in Nur77-mediated inhibition of caspase-1 expression (38). Furthermore, activated caspase-1 has been shown to be encapsulated in microparticles released from monocytes and mediate pulmonary endothelial pyroptosis, which has been shown to contribute to disease progression in ALI (24, 39). Because Nur77 downregulates caspase-1 expression as well as its activation, whether Nur77 affects lung endothelial pyroptosis warrants further investigation.
In summary, this study is the first to demonstrate that Nur77 is an important regulator controlling caspase-1 expression and LPS-induced inflammasome activation in vascular ECs. Collectively, our findings suggest that Nur77 activators, such as cytosporone B (40), hyperoside (41), and others, might have therapeutic potential for the treatment of human diseases associated with aberrant inflammasome activation, such as ALI and atherosclerosis.
Footnotes
Supported by National Heart, Lung, and Blood Institute grant R01HL103869 (J.S.), National Institute of General Medical Sciences grant R01GM123047 (J.S.), American Heart Association Established Investigator Award 16EIA27710023 (J.S.), National Natural Science Foundation of China grant 81770352 (R.D.), China Scholarship Council award 201803170014 (R.D.), and National Natural Science Foundation of China grant 81672927 (X.X.).
Author Contributions: D.A.D., C.L., and J.S. conceived the project. R.D., X.X., and J.S. obtained funding for the project. R.D., X.S., B.Y., X.X., D.A.D., C.L., and J.S. coordinated the research. R.D., X.S., B.Y., W.L., K.K., X.X., C.L., and J.S. contributed to the research protocol development. R.D., X.S., B.Y., and W.L. performed the practical research. R.D., X.S., B.Y., W.L., K.K., X.X., D.A.D., and J.S. reviewed the data. R.D., X.S., and J.S. wrote the manuscript. All authors reviewed the manuscript and approved the final version to be published.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0524OC on May 10, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 3. Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med. 2012;185:1225–1234. doi: 10.1164/rccm.201201-0003OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fukumoto J, Fukumoto I, Parthasarathy PT, Cox R, Huynh B, Ramanathan GK, et al. NLRP3 deletion protects from hyperoxia-induced acute lung injury. Am J Physiol Cell Physiol. 2013;305:C182–C189. doi: 10.1152/ajpcell.00086.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mizushina Y, Karasawa T, Aizawa K, Kimura H, Watanabe S, Kamata R, et al. Inflammasome-independent and atypical processing of IL-1β contributes to acid aspiration-induced acute lung injury. J Immunol. 2019;203:236–246. doi: 10.4049/jimmunol.1900168. [DOI] [PubMed] [Google Scholar]
- 7. Ito H, Kimura H, Karasawa T, Hisata S, Sadatomo A, Inoue Y, et al. NLRP3 inflammasome activation in lung vascular endothelial cells contributes to intestinal ischemia/reperfusion-induced acute lung injury. J Immunol. 2020;205:1393–1405. doi: 10.4049/jimmunol.2000217. [DOI] [PubMed] [Google Scholar]
- 8. Pei L, Castrillo A, Chen M, Hoffmann A, Tontonoz P. Induction of NR4A orphan nuclear receptor expression in macrophages in response to inflammatory stimuli. J Biol Chem. 2005;280:29256–29262. doi: 10.1074/jbc.M502606200. [DOI] [PubMed] [Google Scholar]
- 9. Ismail H, Mofarrahi M, Echavarria R, Harel S, Verdin E, Lim HW, et al. Angiopoietin-1 and vascular endothelial growth factor regulation of leukocyte adhesion to endothelial cells: role of nuclear receptor-77. Arterioscler Thromb Vasc Biol. 2012;32:1707–1716. doi: 10.1161/ATVBAHA.112.251546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. To SK, Zeng WJ, Zeng JZ, Wong AS. Hypoxia triggers a Nur77-β-catenin feed-forward loop to promote the invasive growth of colon cancer cells. Br J Cancer. 2014;110:935–945. doi: 10.1038/bjc.2013.816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kurakula K, Koenis DS, van Tiel CM, de Vries CJ. NR4A nuclear receptors are orphans but not lonesome. Biochim Biophys Acta. 2014;1843:2543–2555. doi: 10.1016/j.bbamcr.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 12. Hanna RN, Shaked I, Hubbeling HG, Punt JA, Wu R, Herrley E, et al. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ Res. 2012;110:416–427. doi: 10.1161/CIRCRESAHA.111.253377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen J, Jia J, Ma L, Li B, Qin Q, Qian J, et al. Nur77 deficiency exacerbates cardiac fibrosis after myocardial infarction by promoting endothelial-to-mesenchymal transition. J Cell Physiol. 2021;236:495–506. doi: 10.1002/jcp.29877. [DOI] [PubMed] [Google Scholar]
- 14. Yan G, Zhu N, Huang S, Yi B, Shang X, Chen M, et al. Orphan nuclear receptor Nur77 inhibits cardiac hypertrophic response to beta-adrenergic stimulation. Mol Cell Biol. 2015;35:3312–3323. doi: 10.1128/MCB.00229-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. You B, Jiang YY, Chen S, Yan G, Sun J. The orphan nuclear receptor Nur77 suppresses endothelial cell activation through induction of IκBα expression. Circ Res. 2009;104:742–749. doi: 10.1161/CIRCRESAHA.108.192286. [DOI] [PubMed] [Google Scholar]
- 16. Bonta PI, Pols TW, de Vries CJ. NR4A nuclear receptors in atherosclerosis and vein-graft disease. Trends Cardiovasc Med. 2007;17:105–111. doi: 10.1016/j.tcm.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 17. Jiang Y, Zeng Y, Huang X, Qin Y, Luo W, Xiang S, et al. Nur77 attenuates endothelin-1 expression via downregulation of NF-κB and p38 MAPK in A549 cells and in an ARDS rat model. Am J Physiol Lung Cell Mol Physiol. 2016;311:L1023–L1035. doi: 10.1152/ajplung.00043.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qin Q, Chen M, Yi B, You X, Yang P, Sun J. Orphan nuclear receptor Nur77 is a novel negative regulator of endothelin-1 expression in vascular endothelial cells. J Mol Cell Cardiol. 2014;77:20–28. doi: 10.1016/j.yjmcc.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhu N, Zhang GX, Yi B, Guo ZF, Jang S, Yin Y, et al. Nur77 limits endothelial barrier disruption to LPS in the mouse lung. Am J Physiol Lung Cell Mol Physiol. 2019;317:L615–L624. doi: 10.1152/ajplung.00425.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Licandro G, Ling Khor H, Beretta O, Lai J, Derks H, Laudisi F, et al. The NLRP3 inflammasome affects DNA damage responses after oxidative and genotoxic stress in dendritic cells. Eur J Immunol. 2013;43:2126–2137. doi: 10.1002/eji.201242918. [DOI] [PubMed] [Google Scholar]
- 21. Chen Z, Martin M, Li Z, Shyy JY. Endothelial dysfunction: the role of sterol regulatory element-binding protein-induced NOD-like receptor family pyrin domain-containing protein 3 inflammasome in atherosclerosis. Curr Opin Lipidol. 2014;25:339–349. doi: 10.1097/MOL.0000000000000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, et al. Acute Lung Injury in Animals Study Group. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44:725–738. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fehrenbach ML, Cao G, Williams JT, Finklestein JM, Delisser HM. Isolation of murine lung endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;296:L1096–L1103. doi: 10.1152/ajplung.90613.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest. 2017;127:4124–4135. doi: 10.1172/JCI94495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 26. Tamura T, Ishihara M, Lamphier MS, Tanaka N, Oishi I, Aizawa S, et al. An IRF-1-dependent pathway of DNA damage-induced apoptosis in mitogen-activated T lymphocytes. Nature. 1995;376:596–599. doi: 10.1038/376596a0. [DOI] [PubMed] [Google Scholar]
- 27. Li Y, Li H, Liu S, Pan P, Su X, Tan H, et al. Pirfenidone ameliorates lipopolysaccharide-induced pulmonary inflammation and fibrosis by blocking NLRP3 inflammasome activation. Mol Immunol. 2018;99:134–144. doi: 10.1016/j.molimm.2018.05.003. [DOI] [PubMed] [Google Scholar]
- 28. Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. Transient expression of IL-1β induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest. 2001;107:1529–1536. doi: 10.1172/JCI12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ganter MT, Roux J, Miyazawa B, Howard M, Frank JA, Su G, et al. Interleukin-1β causes acute lung injury via αvβ5 and αvβ6 integrin-dependent mechanisms. Circ Res. 2008;102:804–812. doi: 10.1161/CIRCRESAHA.107.161067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fan EKY, Fan J. Regulation of alveolar macrophage death in acute lung inflammation. Respir Res. 2018;19:50. doi: 10.1186/s12931-018-0756-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang WL, Sharma A, Wang Z, Li Z, Fan J, Wang P. Cold-inducible RNA-binding protein causes endothelial dysfunction via activation of Nlrp3 inflammasome. Sci Rep. 2016;6:26571. doi: 10.1038/srep26571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oh M, Kim SY, Gil JE, Byun JS, Cha DW, Ku B, et al. Nurr1 performs its anti-inflammatory function by regulating RasGRP1 expression in neuro-inflammation. Sci Rep. 2020;10:10755. doi: 10.1038/s41598-020-67549-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao GN, Jiang DS, Li H. Interferon regulatory factors: at the crossroads of immunity, metabolism, and disease. Biochim Biophys Acta. 2015;1852:365–378. doi: 10.1016/j.bbadis.2014.04.030. [DOI] [PubMed] [Google Scholar]
- 34. Jain N, Sudhakar Ch, Swarup G. Tumor necrosis factor-α-induced caspase-1 gene expression: role of p73. FEBS J. 2007;274:4396–4407. doi: 10.1111/j.1742-4658.2007.05969.x. [DOI] [PubMed] [Google Scholar]
- 35. Antonczyk A, Krist B, Sajek M, Michalska A, Piaszyk-Borychowska A, Plens-Galaska M, et al. Direct inhibition of IRF-dependent transcriptional regulatory mechanisms associated with disease. Front Immunol. 2019;10:1176. doi: 10.3389/fimmu.2019.01176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li L, Liu Y, Chen HZ, Li FW, Wu JF, Zhang HK, et al. Impeding the interaction between Nur77 and p38 reduces LPS-induced inflammation. Nat Chem Biol. 2015;11:339–346. doi: 10.1038/nchembio.1788. [DOI] [PubMed] [Google Scholar]
- 37. Bowie ML, Troch MM, Delrow J, Dietze EC, Bean GR, Ibarra C, et al. Interferon regulatory factor-1 regulates reconstituted extracellular matrix (rECM)-mediated apoptosis in human mammary epithelial cells. Oncogene. 2007;26:2017–2026. doi: 10.1038/sj.onc.1210013. [DOI] [PubMed] [Google Scholar]
- 38. Lv DW, Zhang K, Li R. Interferon regulatory factor 8 regulates caspase-1 expression to facilitate Epstein-Barr virus reactivation in response to B cell receptor stimulation and chemical induction. PLoS Pathog. 2018;14:e1006868. doi: 10.1371/journal.ppat.1006868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang J, Zhao Y, Zhang P, Li Y, Yang Y, Yang Y, et al. Hemorrhagic shock primes for lung vascular endothelial cell pyroptosis: role in pulmonary inflammation following LPS. Cell Death Dis. 2016;7:e2363. doi: 10.1038/cddis.2016.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhan Y, Du X, Chen H, Liu J, Zhao B, Huang D, et al. Cytosporone B is an agonist for nuclear orphan receptor Nur77. Nat Chem Biol. 2008;4:548–556. doi: 10.1038/nchembio.106. [DOI] [PubMed] [Google Scholar]
- 41. Huo Y, Yi B, Chen M, Wang N, Chen P, Guo C, et al. Induction of Nur77 by hyperoside inhibits vascular smooth muscle cell proliferation and neointimal formation. Biochem Pharmacol. 2014;92:590–598. doi: 10.1016/j.bcp.2014.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]